 Manoocher Soleimani
Manoocher Soleimani
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 05 December 2023
Sec. Renal Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1321338
This article is part of the Research Topic Endocrine Regulation of Homeostasis of Water, Electrolytes and Organic Solutes View all 4 articles
The Syndrome of Inappropriate ADH secretion (SIADH) presents with excess ADH release caused by a range of conditions; including pneumonia, brain tumors, certain lung cancers, and diseases of the hypothalamus. It presents with significant reduction in both sodium and chloride concentrations in the blood. However, reports examining the acid base status indicate a normal serum bicarbonate concentration and systemic acid base homeostasis. The mechanisms for the absence of abnormalities in acid base homeostasis remain speculative. This mini review is highlighting the recent advances in renal molecular physiology to provide answers for the maintenance of acid base status and serum bicarbonate in a physiological range.
The Syndrome of Inappropriate Anti-Diuretic Hormone secretion (SIADH) is a frequent clinical encounter with significant morbidity and mortality in hospital admissions. At its core, this disorder presents with excess ADH due to the inability of the body to suppress its secretion. It is caused by a range of conditions, including pneumonia, brain tumors, certain lung cancers, and diseases of the hypothalamus (1–3).
There are hardly any studies examining the mechanism of acid base homeostasis in SIADH despite the significant reduction in both sodium and chloride concentrations in the blood. All reports refer to normal serum bicarbonate levels and by inference a balanced acid base status in SIADH. In this short commentary, we have discussed the physiological and molecular pathways that may be playing a significant role in maintaining a normal acid base homeostasis in SIADH.
A 62-year-old man was brought to an emergency room with altered mental status. Patient has a history of a stable lung mass, but has refused any workup.
Blood chemistries on admission showed the following: Na+112 meq/l, Cl- 79 meq and BUN 5 mg/dl. The urine Na+ was 65, urine K+ was 42 and the urine osmolality was 420 mosm/l. Serum bicarbonate concentration was 26 mEq/l.
The above presentation is typical of SIADH-induced hyponatremia. Excess ADH causes the water absorbing channel, AQP-2, to be translocated to the apical membrane of kidney principal cells; therefore, enhancing water absorption and impairing water excretion (4). This results in the dilution of serum Na+ and Cl- and the concentration of urine osmolality, respectively. Patients can present with profound hyponatremia, hypochloremia, and frequently with low uric acid.
Schematic diagram in Figure 1 depicts the binding of ADH with the vasopressin receptor isoform 2 (V2R) which results in enhanced intracellular cAMP and AQP-2 insertion into the apical membrane of principal cells; therefore, increasing water absorption.

Figure 1 The schematic diagram exhibits the binding of ADH (AVP) with V2R resulting in enhanced intracellular cAMP which leads to the insertion of AQP-2 water channel into the apical membrane of principal cells; therefore, increasing water absorption. The absorbed water exits the cell via the basolateral AQP-3 and AQP-4 water channels.
Despite a significant reduction in serum Na+ and Cl- concentration, all clinical studies demonstrate a normal serum bicarbonate level consistent with the absence of any acid/base disturbance in individuals with SIADH (5). The exception to this statement is found in SIADH associated with Addison Disease which can present with hyponatremia and metabolic acidosis (6).
While there are no concrete studies to address the mechanism of static acid base homeostasis in SIADH-induced hyponatremia, possibilities such as an initial dilutional (volume expansion) acidosis stimulating a restorative acid excretion is plausible. However, there are no studies available that point to an initial metabolic acidosis in SIADH-induced hyponatremia.
A reappraisal of the published studies, as well as recent investigations on the role of ADH on acid base balance, may provide some strong clues on the mechanism of stable serum HCO3- concentration in SIADH.
Aside from the V2 receptor (AVPR2) which is expressed on the basolateral membrane of principal cells (Figure 1) and is inhibited by tolvaptan (5), recent studies demonstrated the presence of V1Ra (AVPR1a) receptor on the basolateral membrane of A-intercalated cells. See Figure 2 for detail. These studies further indicate that ADH addition to the interstitial compartment of perfused collecting duct enhances the translocation of H+-ATPase to the apical membrane and stimulation of H+ secretion into the lumen of the collecting duct (7, 8).
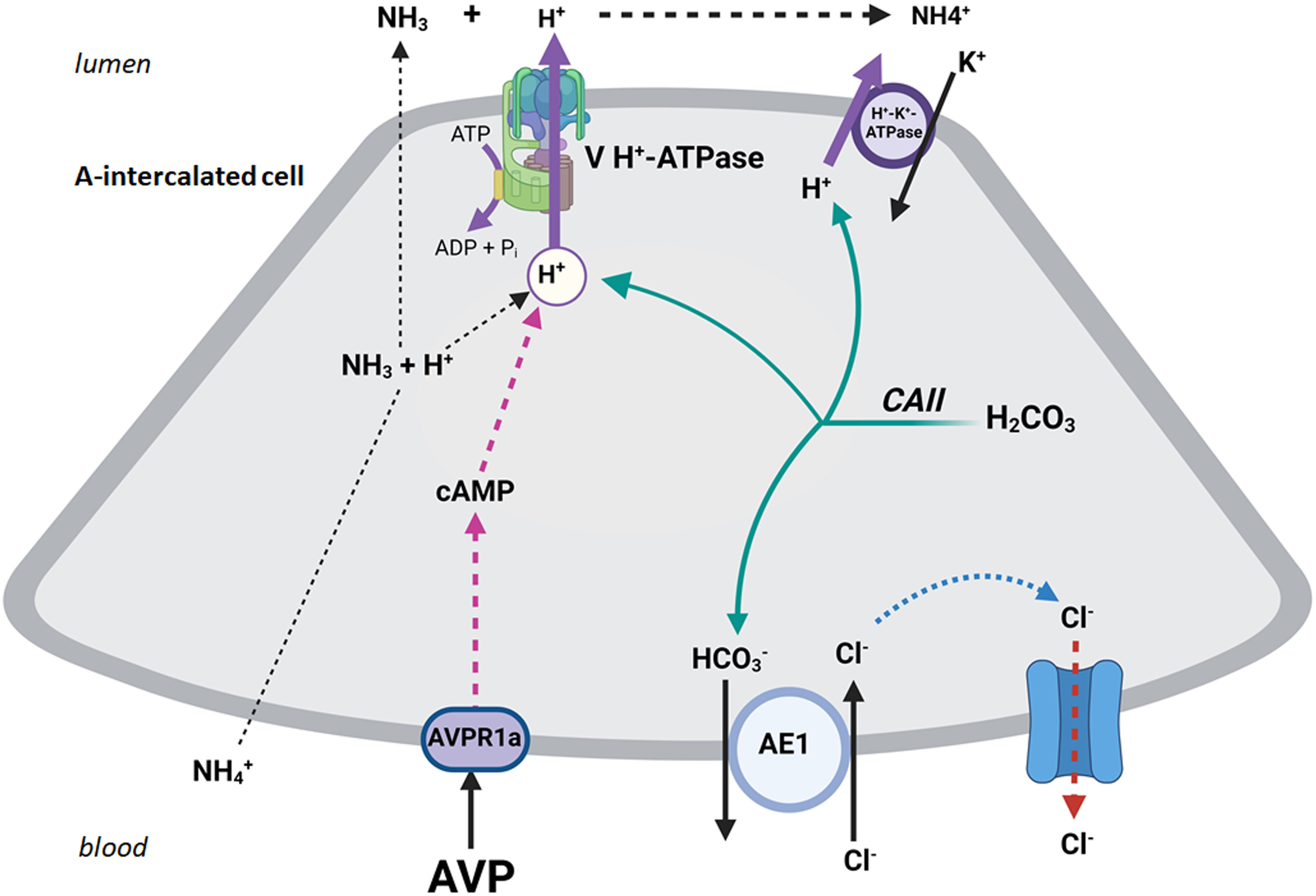
Figure 2 The schematic diagram exhibits the binding of ADH (AVP) with the basolateral V1R (AVPR1a) in A-IC cells resulting in the translocation of H+-ATPase to the apical membrane and stimulation of H+ secretion into the lumen of the collecting duct.
Independent of the excess ADH-mediated effect, hypotonicity can directly stimulate H+-ATPase in cultured kidney IMCD cells (9). These studies demonstrate a direct stimulatory effect of hypotonicity on H+ secretion via H+-ATPase in cultured kidney collecting duct cells (9). The effect is rapid in onset (less than few minutes) and is likely mediated through the activation of existing H+-ATPase molecules in the plasma membrane of cultured kidney cells.
Activation of H+-ATPase by itself may not significantly enhance bicarbonate absorption in the collecting duct or increase blood bicarbonate concentration (specifically if the urine pH is already less than 6). However, H+-ATPase activation is necessary for retaining the new bicarbonate which is generated consequent to enhanced ammoniagenesis.
Studies by Halperin and Ching show that hyponatremia can enhance ammoniagenesis in mammalian kidneys (10). It is worth mentioning that there are no studies examining urinary NH4+ excretion in patients with SIADH and hyponatremia.
In addition to the collecting duct, ADH and osmolality are two critical factors capable of regulating bicarbonate absorption in the medullary thick ascending limb (MTAL). Detailed studies in perfused tubules demonstrated that hypertonicity markedly inhibits (11, 12); whereas, hypotonicity stimulates bicarbonate absorption in the MTAL, the latter is due to activation of the apical Na+/H+ exchanger isoform NHE3 (13). The presence of vasopressin inhibited the hypotonicity-stimulated bicarbonate absorption via NHE3 in MTAL (13). These results strongly indicate that enhanced luminal H+ secretion via NHE3 and the consequent bicarbonate absorption in MTAL by hypotonicity do not contribute to maintaining acid base homeostasis in patients with SIADH since both are inhibited by AVP.
Lastly, it is worth mentioning that the circulating aldosterone level is not suppressed in individuals with SIADH and hyponatremia. Studies examining the effect of aldosterone on acid secretion in the kidney OMCD indicate that the stimulatory effect of aldosterone on H+ secretion (presumably via H+-ATPase and/or H+-K+ ATPase) requires a functional V1a receptor in the intercalated cells (14).
Hypotonicity and ADH can stimulate H+-ATPase and in coordination with enhanced ammoniagenesis maintain the systemic acid base status and serum bicarbonate concentration at a normal level in hypotonic SIADH states. Examining urine NH4+ excretion in individuals with SIADH and hyponatremia, as well as determining the expression of ammoniagenesis enzymes and H+-ATPase in rodents with ADH injection and excess water consumption (mimicking SIADH-induced hyponatremia), can shed light on this issue.
MS: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Resources, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. These studies were supported by Merit Review Award 5I01BX001000-11 (Department of Veterans Administration).
Figures were created by Sharon Barone on BioRender. PI is Senior Clinician Scientist Investigator at the Veterans Health Administration Department.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med (2007) 356(20):2064–72. doi: 10.1056/NEJMcp066837
2. Sterns RH, Silver SM. Cerebral salt wasting versus SIADH: what difference? J Am Soc Nephrol (2008) 19(2):194–6. doi: 10.1681/ASN.2007101118
3. Warren AM, Grossmann M, Christ-Crain M, Russell N. Syndrome of inappropriate antidiuresis: from pathophysiology to management. Endocr Rev (2023) 44(5):819–61. doi: 10.1210/endrev/bnad010
4. Nielsen S, Frør J, Knepper MA. Renal aquaporins: key roles in water balance and water balance disorders. Curr Opin Nephrol Hypertens (1998) 7(5):509–16. doi: 10.1097/00041552-199809000-00005
5. Stern RH. Pathophysiology and etiology of the syndrome of inappropriate antidiuretic hormone secretion (2021). Available at: https://www.uptodate.com/contents/pathophysiology-and-etiology-of-the-syndrome-of-inappropriate-antidiuretic-hormone-secretion-siadh.
6. Almiani M, Gorthi J, Subbiah S, Firoz M. Quiz page November 2012: an unusual case of acute hyponatremia and normal anion gap metabolic acidosis. Am J Kidney Dis (2012) 60(5):xxxiii–xxxvi. doi: 10.1053/j.ajkd.2012.05.026
7. Giesecke T, Himmerkus N, Leipziger J, Bleich M, Koshimizu TA, Fähling M, et al. Vasopressin increases urinary acidification via V1a receptors in collecting duct intercalated cells. J Am Soc Nephrol (2019) 30(6):946–61. doi: 10.1681/ASN.2018080816
8. Yasuoka Y, Kobayashi M, Sato Y, Zhou M, Abe H, Okamoto H, et al. The intercalated cells of the mouse kidney OMCD (is) are the target of the vasopressin V1a receptor axis for urinary acidification. Clin Exp Nephrol (2013) 17(6):783–92. doi: 10.1007/s10157-013-0783-y
9. Amlal H, Goel A, Soleimani M. Activation of H+-ATPase by hypotonicity: a novel regulatory mechanism for H+ secretion in IMCD cells. Am J Physiol (1998) 275(4):F487–501. doi: 10.1152/ajprenal.1998.275.4.F487
10. Halperin ML, Ching BC. Influence of acute hyponatremia on renal ammoniagenesis in dogs with chronic metabolic acidosis. Am J Physiol (1990) 258(2 Pt 2):F328–32. doi: 10.1152/ajprenal.1990.258.2.F328
11. Good DW. Inhibition of bicarbonate absorption by peptide hormones and cyclic adenosine monophosphate in rat medullary thick ascending limb. J Clin Invest (1990) 85(4):1006–13. doi: 10.1172/JCI114530
12. Good DW. Effects of osmolality on bicarbonate absorption by medullary thick ascending limb of the rat. J Clin Invest (1992) 89(1):184–90. doi: 10.1172/JCI115560
13. Good DW, Di Mari JF, Watts BA 3rd. Hyposmolality stimulates Na(+)/H(+) exchange and HCO(3)(-) absorption in thick ascending limb via PI 3-kinase. Am J Physiol Cell Physiol (2000) 279(5):C1443–54. doi: 10.1152/ajpcell.2000.279.5.C1443
Keywords: SIADH, hyponatremia, acid base balance, bicarbonate, collecting duct, AVPR1a, intercalated cells, H+-ATPase
Citation: Soleimani M (2023) Acid base homeostasis and serum bicarbonate concentration in syndrome of inappropriate anti-diuretic hormone secretion (SIADH) with hyponatremia. Front. Endocrinol. 14:1321338. doi: 10.3389/fendo.2023.1321338
Received: 13 October 2023; Accepted: 16 November 2023;
Published: 05 December 2023.
Edited by:
Kamel Kamel, St. Michael’s Hospital, CanadaReviewed by:
George Seki, The University of Tokyo, JapanCopyright © 2023 Soleimani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manoocher Soleimani, TVNvbGVpbWFuaUBzYWx1ZC51bm0uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.