 Zijiao Tian
Zijiao Tian Xinlin Li
Xinlin Li Xue Yu
Xue Yu Shuxin Yan1
Shuxin Yan1 Wenxin Ma
Wenxin Ma Xiaoyun Zhu
Xiaoyun Zhu Yang Tang
Yang Tang- 1College of Traditional Chinese Medicine of Beijing University of Chinese Medicine, Beijing, China
- 2Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
Primary cilia (PC) are non-motile and microtube-based organelles protruding from the surface of almost all thyroid follicle cells. They maintain homeostasis in thyrocytes and loss of PC can result in diverse thyroid diseases. The dysfunction of structure and function of PC are found in many patients with common thyroid diseases. The alterations are associated with the cause, development, and recovery of the diseases and are regulated by PC-mediated signals. Restoring normal PC structure and function in thyrocytes is a promising therapeutic strategy to treat thyroid diseases. This review explores the function of PC in normal thyroid glands. It summarizes the pathology caused by PC alterations in thyroid cancer (TC), autoimmune thyroid diseases (AITD), hypothyroidism, and thyroid nodules (TN) to provide comprehensive references for further study.
1 Introduction
Primary cilia (PC) are immobile, rod-shaped micro-organelles that extend as a solitary unit from the basal body in most cell types of vertebrates (1–3). Initially, follicular cells with PC were discovered in some embryos and mature vertebrate species (4). PC are the signal center of multiple signaling transmission pathways. Specific lipids and receptors on PC enable them to sense various extracellular chemical and mechanical signals and transmit them to the intracellular level, invoking responses that regulate varying cellular developmental and physiological processes (1, 5). The PC structure consists of a basal body, a transition zone, and an axoneme (6). The basal body defines cell polarity and initiates ciliogenesis. Dysfunctions in ciliogenesis can lead to a variety of ciliopathies (7), which may display clinical phenotypes of congenital hypothyroidism (8). The transition zone connects the basal body and axoneme backbone and acts as a docking medium for intraflagellar transport (IFT) particles, which are responsible for transporting all proteins into and along the ciliary compartment (9). Proteins are transported along PC from the base to the tip by anterograde IFT, which is catalyzed by cytoplasmic dynein 2/1b motor, and from the tip to the base of PC by IFT catalyzed by kinesin-2 motor (10). Alpha and beta tubulin form the axoneme, a microtubule structure modified post-translationally to avoid depolymerization. PC length ranges from 5.0 mm to 10.7 mm and has a mean length of 7.3 ± 1.2 mm (4). PC length is associated with cilia-mediated signaling function and altered by agonists for receptors (11), transcriptional regulation of IFT genes, and actin depolymerization (12). Defects in PC length and morphology can lead to dysregulation of signaling transduction and cellular functionality, which contribute to developing diseases termed ciliopathies (13). An experiment showed that the frequency of PC in normal human thyroid follicles is 67.53 ± 3.62% (5). Considering the potential of concentrating signal cascade proteins within ciliary lumens and membranes, PC are an ideal mediator for regulating endocrine pathways (14).
The mammalian thyroid gland regulates the synthesis and secretion of thyroid hormone (TH). TH plays a crucial role in normal development, growth, neural differentiation, and metabolic regulation in mammals (15–17). Furthermore, it can regulate adult hippocampal neurogenesis, which is responsible for learning, memory, and mood by acting through thyroid hormone receptorα (TRα) (18, 19). As the basic unit of the thyroid gland, globular-shaped, vascular, encircled, and colloid-filled follicles are essential for its functional integrity. The colloid-facing membrane of follicular epithelial cells functions as an active exchange interface in hormone production (5), and follicular cells are a prerequisite for TH synthesis (20). Thyroid cancer (TC), Graves’ disease (GD), Hashimoto’s thyroiditis (HT), hypothyroidism, and thyroid nodules (TN) are common thyroid diseases worldwide. Studies show that thyroid illnesses emerge due to oncogenic mutations and abnormal changes in some intracellular downstream signaling pathways and cytokines (21–23). Previous research revealed that as a key mediator, PC influences the pathogenesis of thyroid abnormalities in function and structure (6). However, there are no inductive studies that describe the systematic role of PC in thyroid diseases.
The purpose of this article is to reveal the role that PC in follicular epithelial play in thyroid diseases and provide novel ideas for further research.
2 Primary cilia and thyroid cancer
Thyroid cancer is one of the endocrine cancers with the highest morbidity and a rapidly and steadily rising incidence (24).
In patients with TC, the length and frequency of PC change when responding to extracellular and intracellular stimuli (6). However, there are differences in the length and frequency changes in PC among different TC phenotypes. A previous study found that compared to individuals with normal thyroid glands, patients with follicular thyroid cancer (FTC) and papillary thyroid cancer (PTC) display PC with significantly increased length and invariable frequency, and oncocytic PTC variants exhibit PC with decreased length and frequency (25). PC frequency is observably lower in anaplastic thyroid carcinoma (ATC) (5) and is associated with TC tumorigenesis and TC progression (26). Researchers believe that the absence of PC in PTCs leads to increased apoptosis and is associated with reduced tumor aggressiveness and malignant potential (16).
A mouse model with PC loss mediated by intraflagellar transport 88 (IFT88) showed an irregularly dilated thyroid gland with destroyed follicles, malignant properties, and progressively differentiated thyroid cancer (PDTC) (5), which reflected the pathogenicity of PC mutations in the thyroid gland. As mentioned in the literature review, these findings confirm that abnormal changes in PC structure are closely related to TC tumorigenesis and progression. Thus, maintaining normal ciliogenesis is probably a therapeutic target for TC.
As the mediator of signaling pathways, PC play an essential role in regulating the development of various subtypes of TC. Communication and interflow between tumor microenvironments and TC cells are affected by alterations in PC and influence the prognosis and therapeutic effect on TC (27–30).
IFT88 plays a critical role in anterograde transport in ciliary proteins (31, 32), which maintains bidirectional motility along the axonemes and is indispensable for ciliogenesis and functional proficiency (33). Therefore, dysfunctional mutations of IFT88 lead to severe defects in ciliogenesis (26). Many studies have examined IFT88 function and demonstrated PC loss in many types of cancer (34–37) associated with higher cancer aggressiveness (5, 38, 39). A previous experiment showed that thyroid-specific IFT88-deficient mice developed TC without additional activation of thyroid oncogenic kinases. Clinical studies have provided evidence that IFT88/PC dysfunction causes abnormalities in cellular metabolism, such as oxidative phosphorylation (OxPhos), decreased mitochondrial membrane potential, reduced ATP synthesis, and increased aerobic glycolysis with increased fatty acid synthesis, loss of mitochondrial function, and even mitochondrial fragmentation in rare cases (26). The mechanism of TC resulting from loss of function (LOF) of IFT88/PC is inconsistent in different studies. LOF of IFT88 prevents the PC-mediated Hedgehog pathway from being used to promote carcinogenesis caused by SmoM2. Nevertheless, lesions resembling basal cell carcinoma can be accelerated by LOF of IFT88 through a tumorigenic pathway independent of the PC (34). Therefore, the specific role of LOF of IFT88/PC in TC needs further exploration.
The Hedgehog (Hh) pathway is crucial for vertebrate embryonic development, and misregulation is responsible for many cancers (40). Studies have reported abnormal Hedgehog (Hh) pathways activated by PC (41). Sonic Hh (Shh) (one of the secreted proteins) belongs to the Hedgehog family (42). Immunolocalization tests demonstrate that scores of core components of Sonic hedgehog (Shh) signal transformation localize on PC, and PC is fundamentally important for canonical Shh signaling in vertebrates (29). Shh ligand stimulates Gli (including Gli1, Gli2, and Gli3) transport from PC to the nucleus, where they activate Hh target genes (41). Hh pathway is considered a bona fide ciliary pathway presently (1). PC mediate the interaction between stroma and cancer cells, the defects of which may interfere with the interaction that mediates the aberrant activation of Hh pathways. This finding proves that the Hh pathways mediated by PC are likely to affect TC tumorigenesis (6). In the early stage of TC development, PC-mediated growth factor binding to RTKs triggers the activation of the MAPK and PI3K-AKT cascades, which regulate TC cell proliferation. Increased RTK activity promotes RET/RAS/BRAF mutations (43, 44), which influence Hh pathway activation without ligands in tumor cells (45), implying that PC carry the signaling proteins above. Hh and RTK signaling crosstalk are integrated by PC, which coordinate the synthesis and development of TH. Therefore, PC mutations can influence pathways related to tumorigenesis and the development of TC directly.
NIMA-related kinases (Nek), LKB1, Aurora kinase A (AURKA), and polo-like kinase (Plk1) are PC-related proteins identified as crucial for PC regulation.
Situated in PC and centrosomes, Nek may help to coordinate cell cycle progression and ciliogenesis (46). Studies have shown that cells lacking NIMA or some NIMA-related kinases suffer from chromosome segregation and mitotic errors and then undergo apoptosis (47), manifesting as bizarre and heterogeneous PC (48). In both the classical and follicular variants of PTC, overexpression of Nek1 is frequently associated with aggressiveness, which is highly specific and sensitive. Therefore, Nek 1 may affect the identification of malignant features during TC diagnosis (49).
The LKB1, a tumor suppressor kinase located in PC on epithelial cells, is known to inhibit mTOR activation by inhibiting AMPK signaling, which represses tumor cell polarization and metastasis (47, 50). Therefore, LKB1 expression negatively correlates with increased tumor aggressiveness and is assumed as a prospective therapeutic target for TC (51, 52).
AURKA is located in the basal body, centrosome, and radial microtubules of PC, participates in cellular responsiveness to growth factors, and regulates ciliary disassembly (53, 54). AURKA gene amplification or overexpression is linked to malignancies, such as colon, liver, pancreatic, breast, and gastric cancers (39, 55). Previous studies suggested that AURKA could physiologically cause lung cancer and breast cancer through weakening LKB1/AMPK signaling pathways and mediating resistance to autophagic cell death, respectively (56, 57). Research reveals that the genes encoding the Aurora kinases induce malignant transformation of thyrocytes, and some TC-derived cell lines and tissues exhibit the detection of their overexpression, implying a poor prognosis (6). Although the evidence that AURKA influences TC is insufficient, it can be inferred from the mechanism of diverse cancers. Thus, it is probably a novel and valuable therapeutic target of TC at present (58–60). In preclinical research and clinical trials, MLN8237, a conformation-changing AURKA inhibitor, demonstrated exceptional anticancer activity by inhibiting AURKA (61).
Considered a cellular proliferation marker, Plk1 is localized in the PC transition zone in epithelial cells (62). The small-molecule inhibitor of Plk1 activity can limit the first two phases of ciliary disassembly, while induced PC disassembly can evoke the activity of Plk1 kinase (63). Therefore, this interrelationship between Plk1 and ciliary disassembly can serve as one of the arguments that Plk1 is a PC-related protein critical for PC regulation (6). The findings of several studies show that PLK1 is unlikely to contribute directly to the mitosis of papillary carcinoma cells. One of the explanations is that PLK1 plays an oncogenic role and is constitutively required in papillary carcinoma during the early phase, while it is less necessary for the development of this carcinoma during the advanced stage (64–66). Plk1 is only expressed occasionally in normal thyrocytes while overexpressed in the bulk of microcarcinomas, smaller PTC, ATCs, and incidental carcinomas, which may support the assumption (67) that Plk1 makes a constitutive effect on PTC in the early stage. Furthermore, several tests related to PLK1 inhibitors show promising results in preclinical settings (68–71). Yet, clinical investigations reveal that hematologic toxicity (neutropenia) frequently causes therapeutic action to occur at or beyond the maximum tolerated dosage (72–74).
Cysteine cathepsins are a group of proteases that are proteolytically active to different extents (75). Secreted from Nthy-ori 3-1 cells in vitro (76) and from human thyrocytes in situ (77), cysteine cathepsins contribute to tissue homeostasis in the thyroid gland (78). Cysteine cathepsins B, K, L, and S are involved in the proteolytic processing and degradation of thyroglobulin (Tg) in the follicular lumen for initial TH liberation (77, 79). The production of prohormone Tg and its proteolytic processing are essential for thyroid function. A previous study by Alara Gaye Doğru et al. discovered the presence of cysteine cathepsins B and L at the PC of Fisher rat thyroid (FRT) cells, indicating their localization, and showed that inhibiting these cysteine cathepsins leads to the elimination of the PC (80). An interesting possibility is that different substrates of truncated cathepsin B and V variants are present in the nuclei of thyroid carcinoma cells (75). Several studies implicate the involvement of cysteine cathepsins in malignancies and cancer progression due to an increased expression and activity in cancer cells and tumor-associated tissue. As deduced from co-localization studies and in vitro degradation assays, a study suggested that nuclear variants of cathepsins are involved in the development of thyroid malignancies (75).
Thyroid-stimulating hormone (TSH) receptor activation increases cytosolic calcium levels, stimulating the release of cysteine cathepsins. These cysteine cathepsins then enzymatically break down Tg to release TH. The ultimate result of Tg processing is the production of Tg fragments that function as thyropins, which effectively suppress the activity of cysteine cathepsins (81). This process affects thyroid autoregulation and contributes to thyroid diseases.
Although the mechanisms of PC loss in TC cells remain uncertain, the survival of TC cells is determined by proper regulation of PC proteins between ciliogenesis and the cell cycle (82). Considering these PC proteins as targets for treating TC is a relatively new idea.
Studies on the drug effects on PC in human cells are limited. Notably, specific drugs, for example, U0126 and ganetespib (83, 84), affect disassembling PC, restoring ciliogenesis, shortening PC, preventing Smo accumulation in PC, and inhibiting the proliferation of TC cells in different histological types (6, 85, 86). Many drugs are shown to be effective in TC treatment. For example, docetaxel lowers PC levels in olfactory cells, paclitaxel produces PC elongation in the quail oviduct, doxorubicin increases PC synthesis in breast fibroblasts (87), and carboplatin causes PC disassembly in sensory cells (88). Several RTK inhibitors, such as sorafenib, lenvatinib, vandetanib, and cabozantinib, were approved for clinical practice for treating differentiated thyroid cancer (DTC), PDTC, and metastatic thyroid cancer (MTC) (89). However, there is no information on the effects of RTK inhibitors on PC. They have been evaluated in preclinical and clinical investigations in TC, and taking these aspects into account will help develop logical therapy methods for TC. Therefore, restoring PC in TC may be a promising therapeutic strategy (90). Thus, there is a broad scientific prospect of developing drugs with PC as a target for thyroid cancer therapy.
3 Primary cilia and autoimmune thyroid disease
As the most common autoimmune disease, autoimmune thyroid disease (AITD) is prevalent in approximately 5% of the population (91, 92). GD and HT are two main clinical presentations of AITD, both characterized by lymphocytic infiltration of the thyroid parenchyma (93). The pathological process of GD is influenced by a defective Taar1 located in the PC. The shedding of PC on kidney epithelial cells indicates that oxidative stress may damage PC in thyroid epithelial cells, while the pathological process of HT is regulated by PC, which mediate apoptosis and affect the normal expression of miRNA. A summary of past research confirms that PC play an essential role in AITD, especially GD and HT.
3.1 Primary cilia and Graves’ disease
Thyrotoxicosis is an autoimmune response characterized by the presence of autoantibodies targeting the TSH receptor (TSHR-Ab), resulting in goiter and hyperthyroidism (93, 94). GD is one of the most common causes of thyrotoxicosis. Moreover, the thyroid gland of GD is infiltrated by autoreactive lymphocytes and circulating thyroid antibodies (23).
Although the precise pathogenic mechanisms remain unknown, researchers have revealed a close correlation between morbidity and sex and susceptibility genes (95, 96). Most current research shows an increase in the length and frequency of PC, whereas studies exploring the mechanisms underlying the influence of PC on GD are rare. Therefore, the central links (Taar1, oxidative stress) that may be related to PC in GD pathogenesis are summarized here to fill the gap in this field.
Recent research shows that GD results from structural and functional defects in PC, also known as ciliopathies (9, 97, 98). Defects in ciliogenesis should appear in GD, characterized by follicle integrity changes, deregulation of hormone synthesis, and altered proliferation rate (99). Until now, the role of PC has not been demonstrated directly in the thyroid, while it can be inferred from existing work. In previous experiments, GD follicular cells were characterized by a convex surface adorned with microvilli, and small cilia that resembled microvilli were infrequently seen near the cell core (23). In follicular cells of GD, PC length and frequency decreased significantly compared to the normal group, and the absence of PC intensified the alterable presence of follicles in apoptosis (99). The findings proved that ciliogenesis plays a key role in sustaining the normal physical condition of follicular cells and directly impacts the functional abnormalities of the thyroid gland (6, 99). Moreover, GD thyroid samples with shorter axonemal and lower ciliary frequencies had a worse response to antithyroid medication therapy (99). Therefore, the therapeutic effect may be enhanced by maintaining the normal length and frequency of the PC, providing a novel idea in clinical research and treatment.
The hypothalamic–pituitary–thyroid (HPT) axis is a vital regulatory pathway of the thyroid gland, in which low concentrations of TH trigger negative feedback, resulting in the release of thyrotropin-releasing hormone (TRH) from the hypothalamus and TSH from the pituitary gland (100–102). Evidence suggests that Taar1, a putative receptor of thyronamines localized at PC (103), is vital to maintaining canonical regulation of the HPT axis (100) and might contribute to hyperthyroidism through its effect on the HPT axis. An experiment revealed that the serum of male mice with Taar1 knocked out showed mild TSH receptor resistance and elevated TSH concentrations (100). Inversely, serum TSH concentrations revealed a decline in serum TSH concentrations in Taar1 knockout male mice with older age, while the increase in serum TSH concentrations was significant when comparing young adult Taar1 knockout male mice (100). These varying results might be attributable to the heterogeneity of the thyroid tissue. That is, Taar1-deficient mice are hyperthyrotropinemic, which is the characteristic of GD. Therefore, there is a high probability that the defect of Taar1 caused by alterations in PC structure and function is related to the pathogenesis of GD.
Moreover, there is evidence that oxidative processes play a role in GD pathogenesis (104). Abalovich et al. (105) found that hyperthyroid and GD patients showed an increase in oxidative stress markers and a decrease in markers of the antioxidant system. Previous studies have also revealed that hyperthyroid patients demonstrate elevated levels of oxidative stress markers in plasma and other tissues (106–108). Presently, there is insufficient evidence on the effect of oxidative stress on PC, hence eventually causing GD. Studies on ischemia/reperfusion injury have shown that, to some degree, excessive ROS production and oxidative stress lead to the shedding of PC on kidney epithelial cells (67), implying that oxidative stress may damage PC in thyroid epithelial cells as well and contribute to thyroid disease pathogenesis accordingly.
3.2 Primary cilia and Hashimoto’s thyroiditis
Autoimmune thyroiditis, or HT, is a chronic inflammatory autoimmune illness that uses the thyroid tissue as an antigen. The incidence has increased considerably in recent years (109, 110). Autoimmune thyroiditis is a cellular autoimmune illness with apparent inflammatory infiltration that destroys the thyroid gland (23). In children and teenagers, it is the most prevalent cause of goiter and acquired hypothyroidism.
Researchers proved that the frequency and length of PC decreased significantly in experimental specimens compared to controls. Follicular cells in HT exhibited a variety of forms and uneven borders, which occasionally created difficulty in distinguishing between different cells. Each cell core was invaded by hypomorphic cilia, which occasionally resembled “volcano-like” formations (23).
The loss of thyroid epithelial cells is a hallmark of HT. An experiment revealed that the percentage of in situ apoptotic thyrocytes increases in HT; thus, the apoptosis of thyroid follicular cells plays a vital role in the pathogenesis of HT (111). Apoptosis can be initiated in various ways and affects several cellular processes (112). Junguee Lee et al. believed that HT is usually associated with mitochondrial dysfunction (25). As crucial regulators of cell death, mitochondria act by an intrinsic process called the mitochondria-dependent pathway of apoptosis. The intrinsic apoptotic route is normally mediated by mitochondrial outer membrane permeabilization (MOMP), which includes the voltage-dependent anion channel (VDAC). Moreover, VDACs concentrate in the basal body of PC, among which VDAC1 and VDAC3 regulate ciliogenesis negatively (113). A study found that mice lacking PC in thyroid follicular cells showed apoptotic cell death, resulting in altered follicular structure. Furthermore, inhibiting ciliogenesis in thyroid cancer cell lines resulted in VDAC1 oligomerization following VDAC1 overexpression, leading ultimately to apoptosis. Consequently, the LOF of PC is probably one of the potential factors that stimulate apoptogenesis (114).
To summarize, HT samples showed loss of thyroid epithelial cells caused by mitochondrial dysfunction. Furthermore, defects in the structure and function of PC affect mitochondrial dysfunction and apoptosis accordingly. PC, a core link in the entire process, is closely related to HT. Although no relevant reports show that HT may be affected by apoptosis caused by PC abnormality, it is a likely potential mechanism that needs investigation.
As the microenvironment of AITD is clearly pro-inflammatory, cytokines and chemokines are considered important in its pathogenesis (109, 115). As a marker of AITD, thyroid peroxidase antibodies (ATPO) are present in nearly all HT patients (115), which may trigger the synthesis of autoantibodies by MHC expression on thyrocytes and lymphocyte infiltration (116). Especially in HT, cytokines stimulated by T and B lymphocytes enhance the inflammatory response and production of antibodies, which results in thyroid tissue damage by apoptosis (117). Experiments reveal that pro-inflammatory cytokines IFN-γ and TNF-a increase the expression of the genes mentioned above while decreasing the number of PC (23). Previous studies have shown that IL-1 causes cilia to elongate through a mechanism dependent on protein kinase A, indicating the significance of inflammation in controlling PC shape and function (118). Studies validated a decrease in miR-141 expression and associated this decrease with the transforming growth factor beta 1 (TGF-β1) signaling pathway in HT (119); furthermore, they showed that decreased expression of miR146b, miR-221, and miR-222 could play a role in the development of papillary thyroid carcinoma (120). A proposed model of the mechanisms underlying ciliary defects in AITD showed abnormal antigen presentation in the thyroid and breakdown of tolerance to self-antigens, leading to complex and interrelated intrathyroidal immune processes. A combination of a pro-inflammatory environment and increased miRNA expression may repress the expression of ciliary-related genes, leading to impaired ciliogenesis (23). Moreover, miRNA transfection affects ciliary growth in thyrocytes. More critically, studies show that aberrant miRNA expression is the foundation of immune cell differentiation and activation (121), and the regulation between cytokine activities and miRNA expression is bidirectional (122, 123). Moreover, the potential utilization of this pathway as a novel therapeutic approach to treat these disorders arises from the involvement of pro-inflammatory cytokines and miRNA targeting in the formation of PC in thyroid cells (23).
Epithelial–mesenchymal transition (EMT) is a process that occurs under both physiological and pathological conditions. It is characterized by the loss of epithelial characteristics and the acquisition of mesenchymal features by epithelial cells (124). Recent research shows that PC deficiency triggers EMT under resting conditions and exacerbates it under the influence of fibrotic signals such as TGF-β (125, 126). Research data indicate an increase in the acquisition of mesenchymal markers by thyroid follicle cells in AITD that may contribute to the pathogenesis of these diseases. Furthermore, EMT induction by TGF-β in thyroid cells suggests the potential usefulness of this pathway as a novel therapeutic strategy to treat AITD (22).
4 Primary cilia and hypothyroidism
Hypothyroidism, which may be acquired or congenital, is related to elevated serum TSH levels and low serum free T4 and/or free T3 levels (127). The prevalence of hypothyroidism rises with age and is higher in women, patients with other autoimmune diseases, and those with Down syndrome and Turner syndrome (128). As an essential micro-organelle mediating signaling pathways and maintaining thyroid homeostasis, PC are speculated to be related to the pathological mechanisms underlying hypothyroidism. However, the association has not received the attention of researchers. The probable mechanism by which PC influence hypothyroidism via protein and hormone levels is discussed below.
Recent research indicates that PC play a significant role in regulating the processing and Gli protein function (129). Presently, the Hh pathway is considered a bona fide ciliary pathway, and PC mediate the interaction between stroma and cancer cells, the defects of which may interfere with the interaction that mediates the aberrant activation of Hh pathways. Gli3 is stimulated by the Shh ligand for transport from PC to the nucleus, where Hh target genes are activated. Therefore, Glis3 may be part of the signal transduction pathway mediated by PC, which requires activation before translocation to the cytoplasm and nucleus (130). Known as an anterograde IFT motor, KIF3a is necessary for Gli3 activator formation and proteolysis (131). Glis3 expression is restricted to thyroid follicular cells. A previous study clarified that TH biosynthesis depends on the expression of a specific group of genes directly regulated by Glis3, particularly the two iodide transporter genes, NIS and PDS (132). In Glis3 mutant mice, TSH were found increased, while TH were observed decreased (133), and nkx2.4b and pax2a expression, which are important transcription factors regulating thyroid development, exhibited a decrease in Glis3-deficient zebrafish (134). Humans and mice with loss of Glis3 develop congenital or neonatal hypothyroidism and have a reduced life span (8, 135, 136). Recent studies have identified a link between Glis3 missense mutations and thyroid dysgenesis, as well as the association between the Glis3 variation, rs1571583, and thyroid malfunction regulation (133). Several findings revealed that thyroid dyshormonogenesis, rather than thyroid dysgenesis, was responsible for hypothyroidism in animals lacking Glis3 (137). Although the present research on the mechanism underlying the defects in PC that influence thyroid function and cause hypothyroidism is limited, it can be inferred that PC interact with Glis3 via the signaling pathway and result in thyroid dysfunction.
The ciliary pocket located on PC is an invagination of cytoplasm, where the receptor-mediated endocytosis takes place (138). Tg endocytosis, which has a critical effect on TH release, is primarily mediated by low-density lipoprotein receptor protein 2 (LRP2)/megalin situated at PC and regulated by TSH (139–141). Tg uptake triggered by TSH is a key factor of TH and Tg release into the blood circulation (142–144). Hypothyroidism in megalin knockout mice is accompanied by reduced free T4 (fT4) and serum Tg levels and noticeably higher serum TSH levels (145). A study showed that defective PC cause a significant loss of LRP2/megalin with high TSH levels and colloid Tg depletion (5). Consequently, damaged PC may affect the normal function of LRP2/megalin and result in Tg abnormality, causing hypothyroidism.
There has been no research on the length and frequency alterations in PC. Thus, restoring normal PC formation and shape is probably a promising treatment strategy for hypothyroidism, which requires further study.
5 Primary cilia and thyroid nodule
TN is a common clinical disease with one or more structurally abnormal masses in the thyroid gland. It is usually benign (146), with palpable nodules in 5% of the population, especially older patients (147, 148); 5%–15% is proved malignant (149, 150), while the data increase to 35% by positron emission tomography scanning (151).
Thyroid follicular cells are the primary source of TN (152). The follicles in nodular hyperplastic thyroid tissue are highly heterogeneous in size and morphology, ranging from small follicles with little colloid and high columnar thyrocyte linings to very large follicles with abundant colloid and flat epithelium, which probably is a typical pattern of nodular hyperplasia (153, 154).
Fernández-Santos elucidated that trace amine-associated receptor 1 (Taar1), the putative receptor of thyronamines, is located in PC on thyroid follicles and might play a role in regulating cathepsin-mediated proteolysis of Tg and, consequently, TH synthesis (100). They found that, in TN cells, the average PC length and frequency in follicular cells decreased considerably. Although follicles had noticeable size disparities, there was no relation between follicular size and PC mutations (99).
The varied follicular patterns also showed differences in ciliogenesis, with a decreased PC frequency in areas with changed follicles compared to those with predominant normal-appearance follicles. In murine thyroid follicles, LOF of PC causes aberrant and irregular follicles that finally give rise to papillary and solid proliferative nodules (5).
We acknowledge the various discussions on the relationship and interactions between PC and TN; however, researchers have still not verified this hypothesis due to insufficient experimental and clinical evidence to support the conjecture at present. Therefore, the relationship between TN and PC needs further investigation.
6 Conclusions and prospect
Based on the exposition described above, PC play an essential role in mediating signaling pathways and sustaining thyrocyte homeostasis in various thyroid diseases. Impaired function and structure of PC affect pathways and proteins associated with TC aggressiveness. Furthermore, it mediates GD by impairing Taar1 and HT by disrupting apoptosis and affecting normal miRNA expression. Defective PC also result in abnormal secretion of TSH and TH, causing hypothyroidism. Further studies are necessary due to insufficient experimental and clinical evidence on the relationship between TN and PC. Thus, there is a strong possibility that PC is a potential target in thyroid diseases, and studies on the probable effects of impaired functionality and structure of PC on proteins, signaling pathways, and genes may reveal the mechanisms underlying thyroid disease pathogenesis in the future (Figure 1). Restoring PC is probably a promising therapeutic strategy in clinical settings. To assess the role of PC in the incidence and progression of thyroid disorders, preclinical and clinical research is necessary. Thus, further studies are required to explore the pathological mechanism underlying impaired function and structure of PC, provide new research directions for thyroid disease treatment, and develop a broad idea for other diseases related to PC.
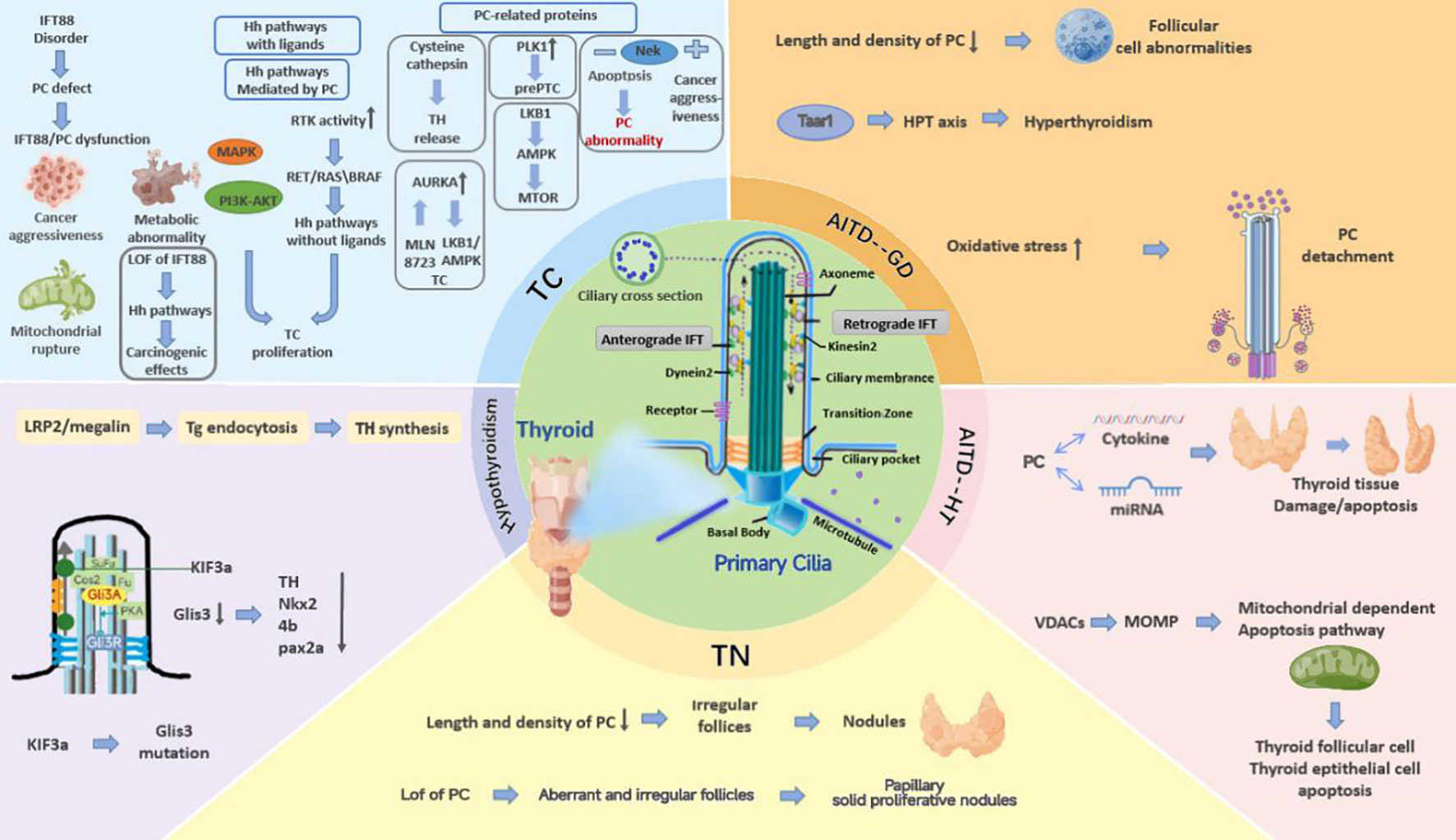
Figure 1 The mechanisms of PC mutations that cause thyroid diseases. PC play an essential role in mediating signaling pathways and sustaining thyrocyte homeostasis in various thyroid diseases. Impaired function and structure of PC affect pathways and proteins associated with TC aggressiveness, and it mediates GD by impairing Taar1 and HT by disrupting apoptosis and affecting normal miRNA expression. Defective PC also result in abnormal secretion of TSH and TH, causing hypothyroidism. Further studies are necessary due to insufficient experimental and clinical evidence on the relationship between TN and PC.
Author contributions
ZT: Writing – original draft, Writing – review & editing. XL: Writing – original draft, Writing – review & editing. XY: Writing – original draft, Writing – review & editing. SY: Writing – review & editing. JS: Writing – review & editing. WM: Writing – review & editing. XZ: Supervision, Writing – review & editing. YT: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China, Youth Project (#82004337), and Guang’anmen Hospital Research Project (#2023103).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB, Christensen ST. Cellular signalling by primary cilia in development, organ function and disease. Nat Rev Nephrol (2019) 15(4):199–219. doi: 10.1038/s41581-019-0116-9
2. Christensen ST, Pedersen LB, Schneider L, Satir P. Sensory cilia and integration of signal transduction in human health and disease. Traffic (2007) 8(2):97–109. doi: 10.1111/j.1600-0854.2006.00516.x
3. Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol (2007) 69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236
4. Utrilla JC, Gordillo-Martínez F, Gómez-Pascual A, Fernández-Santos JM, Garnacho C, Vázquez-Román V, et al. Comparative study of the primary cilia in thyrocytes of adult mammals. J Anat (2015) 227(4):550–60. doi: 10.1111/joa.12360
5. Lee J, Yi S, Chang JY, Kim JT, Sul HJ, Park KC, et al. Loss of primary cilia results in the development of cancer in the murine thyroid gland. Mol Cells (2019) 42(2):113–22. doi: 10.14348/molcells.2018.0430
6. Ma CX, Ma XN, Li YD, Fu SB. The role of primary cilia in thyroid cancer: from basic research to clinical applications. Front Endocrinol (Lausanne) (2021) 12:685228. doi: 10.3389/fendo.2021.685228
7. Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol (2017) 18(9):533–47. doi: 10.1038/nrm.2017.60
8. Senée V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec JC, et al. Mutations in glis3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet (2006) 38(6):682–7. doi: 10.1038/ng1802
9. Wheway G, Nazlamova L, Hancock JT. Signaling through the primary cilium. Front Cell Dev Biol (2018) 6:8. doi: 10.3389/fcell.2018.00008
10. Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol (2002) 3(11):813–25. doi: 10.1038/nrm952
11. Hamamoto A, Yamato S, Katoh Y, Nakayama K, Yoshimura K, Takeda S, et al. Modulation of primary cilia length by melanin-concentrating hormone receptor 1. Cell Signal (2016) 28(6):572–84. doi: 10.1016/j.cellsig.2016.02.018
12. Kang GM, Han YM, Ko HW, Kim J, Oh BC, Kwon I, et al. Leptin elongates hypothalamic neuronal cilia via transcriptional regulation and actin destabilization. J Biol Chem (2015) 290(29):18146–55. doi: 10.1074/jbc.M115.639468
13. Ocbina PJ, Eggenschwiler JT, Moskowitz I, Anderson KV. Complex interactions between genes controlling trafficking in primary cilia. Nat Genet (2011) 43(6):547–53. doi: 10.1038/ng.832
14. O'Toole SM, Chapple JP. Primary cilia: A link between hormone signalling and endocrine-related cancers? Biochem Soc Trans (2016) 44(5):1227–34. doi: 10.1042/bst20160149
15. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev (2010) 31(2):139–70. doi: 10.1210/er.2009-0007
16. Williams GR. Neurodevelopmental and neurophysiological actions of thyroid hormone. J Neuroendocrinol (2008) 20(6):784–94. doi: 10.1111/j.1365-2826.2008.01733.x
17. Tata JR. The road to nuclear receptors of thyroid hormone. Biochim Biophys Acta (2013) 1830(7):3860–6. doi: 10.1016/j.bbagen.2012.02.017
18. Desouza LA, Ladiwala U, Daniel SM, Agashe S, Vaidya RA, Vaidya VA. Thyroid hormone regulates hippocampal neurogenesis in the adult rat brain. Mol Cell Neurosci (2005) 29(3):414–26. doi: 10.1016/j.mcn.2005.03.010
19. Kapoor R, van Hogerlinden M, Wallis K, Ghosh H, Nordstrom K, Vennstrom B, et al. Unliganded thyroid hormone receptor alpha1 impairs adult hippocampal neurogenesis. FASEB J (2010) 24(12):4793–805. doi: 10.1096/fj.10-161802
20. Nunez J, Pommier J. Formation of thyroid hormones. Vitam Horm (1982) 39:175–229. doi: 10.1016/s0083-6729(08)61137-1
21. Miller KA, Yeager N, Baker K, Liao XH, Refetoff S, Di Cristofano A. Oncogenic kras requires simultaneous pi3k signaling to induce erk activation and transform thyroid epithelial cells in vivo. Cancer Res (2009) 69(8):3689–94. doi: 10.1158/0008-5472.Can-09-0024
22. Sacristán-Gómez P, Serrano-Somavilla A, Castro-Espadas L, Sampedro-Núñez M, Muñoz-De-Nova JL, et al. Evaluation of epithelial-mesenchymal transition markers in autoimmune thyroid diseases. Int J Mol Sci (2023) 24(4):3359. doi: 10.3390/ijms24043359
23. Martinez-Hernandez R, Serrano-Somavilla A, Ramos-Levi A, Sampedro-Nunez M, Lens-Pardo A, De Nova JLM, et al. Integrated mirna and mrna expression profiling identifies novel targets and pathological mechanisms in autoimmune thyroid diseases. Ebiomedicine (2019) 50:329–42. doi: 10.1016/j.ebiom.2019.10.061
24. Cabanillas ME, McFadden DG, Durante C. Thyroid cancer. Lancet (2016) 388(10061):2783–95. doi: 10.1016/s0140-6736(16)30172-6
25. Lee J, Yi S, Kang YE, Chang JY, Kim JT, Sul HJ, et al. Defective ciliogenesis in thyroid hurthle cell tumors is associated with increased autophagy. Oncotarget (2016) 7(48):79117–30. doi: 10.18632/oncotarget.12997
26. Lee J, Yi S, Won M, Song YS, Yi HS, Park YJ, et al. Loss-of-function of ift88 determines metabolic phenotypes in thyroid cancer. Oncogene (2018) 37(32):4455–74. doi: 10.1038/s41388-018-0211-6
27. Berbari NF, O'Connor AK, Haycraft CJ, Yoder BK. The primary cilium as a complex signaling center. Curr Biol (2009) 19(13):R526–35. doi: 10.1016/j.cub.2009.05.025
28. Gerdes JM, Katsanis N. Ciliary function and wnt signal modulation. Curr Top Dev Biol (2008) 85:175–95. doi: 10.1016/s0070-2153(08)00807-7
29. Goetz SC, Anderson KV. The primary cilium: A signalling centre during vertebrate development. Nat Rev Genet (2010) 11(5):331–44. doi: 10.1038/nrg2774
30. Grisanti L, Revenkova E, Gordon RE, Iomini C. Primary cilia maintain corneal epithelial homeostasis by regulation of the notch signaling pathway. Development (2016) 143(12):2160–71. doi: 10.1242/dev.132704
31. Katoh Y, Terada M, Nishijima Y, Takei R, Nozaki S, Hamada H, et al. Overall architecture of the intraflagellar transport (Ift)-B complex containing cluap1/ift38 as an essential component of the ift-B peripheral subcomplex. J Biol Chem (2016) 291(21):10962–75. doi: 10.1074/jbc.M116.713883
32. Taschner M, Weber K, Mourão A, Vetter M, Awasthi M, Stiegler M, et al. Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin-binding ift-B2 complex. EMBO J (2016) 35(7):773–90. doi: 10.15252/embj.201593164
33. Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, et al. Chlamydomonas ift88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol (2000) 151(3):709–18. doi: 10.1083/jcb.151.3.709
34. Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH Jr., et al. Primary cilia can both mediate and suppress hedgehog pathway-dependent tumorigenesis. Nat Med (2009) 15(9):1055–61. doi: 10.1038/nm.2011
35. Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene (2009) 28(40):3513–25. doi: 10.1038/onc.2009.220
36. Schraml P, Frew IJ, Thoma CR, Boysen G, Struckmann K, Krek W, et al. Sporadic clear cell renal cell carcinoma but not the papillary type is characterized by severely reduced frequency of primary cilia. Mod Pathol (2009) 22(1):31–6. doi: 10.1038/modpathol.2008.132
37. Yuan K, Frolova N, Xie Y, Wang D, Cook L, Kwon YJ, et al. Primary cilia are decreased in breast cancer: analysis of a collection of human breast cancer cell lines and tissues. J Histochem Cytochem (2010) 58(10):857–70. doi: 10.1369/jhc.2010.955856
38. Sánchez I, Dynlacht BD. Cilium assembly and disassembly. Nat Cell Biol (2016) 18(7):711–7. doi: 10.1038/ncb3370
39. Seeger-Nukpezah T, Little JL, Serzhanova V, Golemis EA. Cilia and cilia-associated proteins in cancer. Drug Discovery Today Dis Mech (2013) 10(3-4):e135–e42. doi: 10.1016/j.ddmec.2013.03.004
40. McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol (2003) 53:1–114. doi: 10.1016/s0070-2153(03)53002-2
41. Hassounah NB, Bunch TA, McDermott KM. Molecular pathways: the role of primary cilia in cancer progression and therapeutics with a focus on hedgehog signaling. Clin Cancer Res (2012) 18(9):2429–35. doi: 10.1158/1078-0432.Ccr-11-0755
42. Martinez-Chinchilla P, Riobo NA. Purification and bioassay of hedgehog ligands for the study of cell death and survival. Methods Enzymol (2008) 446:189–204. doi: 10.1016/s0076-6879(08)01611-x
43. Abdullah MI, Junit SM, Ng KL, Jayapalan JJ, Karikalan B, Hashim OH. Papillary thyroid cancer: genetic alterations and molecular biomarker investigations. Int J Med Sci (2019) 16(3):450–60. doi: 10.7150/ijms.29935
44. Christensen ST, Morthorst SK, Mogensen JB, Pedersen LB. Primary cilia and coordination of receptor tyrosine kinase (Rtk) and transforming growth factor B (Tgf-B) signaling. Cold Spring Harb Perspect Biol (2017) 9(6). doi: 10.1101/cshperspect.a028167
45. Lei J, Ma J, Ma Q, Li X, Liu H, Xu Q, et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol Cancer (2013) 12:66. doi: 10.1186/1476-4598-12-66
46. Mahjoub MR, Trapp ML, Quarmby LM. Nima-related kinases defective in murine models of polycystic kidney diseases localize to primary cilia and centrosomes. J Am Soc Nephrol (2005) 16(12):3485–9. doi: 10.1681/asn.2005080824
47. Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, et al. Primary cilia regulate mtorc1 activity and cell size through lkb1. Nat Cell Biol (2010) 12(11):1115–22. doi: 10.1038/ncb2117
48. Chen Y, Chiang HC, Litchfield P, Pena M, Juang C, Riley DJ. Expression of nek1 during kidney development and cyst formation in multiple nephron segments in the nek1-deficient kat2j mouse model of polycystic kidney disease. J BioMed Sci (2014) 21(1):63. doi: 10.1186/s12929-014-0063-5
49. Melo-Hanchuk TD, Martins MB, Cunha LL, Soares FA, Ward LS, Vassallo J, et al. Expression of the nek family in normal and cancer tissue: an immunohistochemical study. BMC Cancer (2020) 20(1):23. doi: 10.1186/s12885-019-6408-4
50. Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. Hef1-dependent aurora a activation induces disassembly of the primary cilium. Cell (2007) 129(7):1351–63. doi: 10.1016/j.cell.2007.04.035
51. Kou B, Wang XD, Sun XP, Qi Q, Yang M, Yun YN, et al. Lkb1 inhibits proliferation, metastasis and angiogenesis of thyroid cancer by upregulating sik1. J Cancer (2022) 13(9):2872–83. doi: 10.7150/jca.72021
52. Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, et al. Lkb1 is a master kinase that activates 13 kinases of the ampk subfamily, including mark/par-1. EMBO J (2004) 23(4):833–43. doi: 10.1038/sj.emboj.7600110
53. Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, et al. A homologue of drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J (1998) 17(11):3052–65. doi: 10.1093/emboj/17.11.3052
54. Plotnikova OV, Nikonova AS, Loskutov YV, Kozyulina PY, Pugacheva EN, Golemis EA. Calmodulin activation of aurora-a kinase (Aurka) is required during ciliary disassembly and in mitosis. Mol Biol Cell (2012) 23(14):2658–70. doi: 10.1091/mbc.E11-12-1056
55. Zou Z, Yuan Z, Zhang Q, Long Z, Chen J, Tang Z, et al. Aurora kinase a inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy (2012) 8(12):1798–810. doi: 10.4161/auto.22110
56. Zheng X, Chi J, Zhi J, Zhang H, Yue D, Zhao J, et al. Aurora-a-mediated phosphorylation of lkb1 compromises lkb1/ampk signaling axis to facilitate nsclc growth and migration. Oncogene (2018) 37(4):502–11. doi: 10.1038/onc.2017.354
57. Xu LZ, Long ZJ, Peng F, Liu Y, Xu J, Wang C, et al. Aurora kinase a suppresses metabolic stress-induced autophagic cell death by activating mtor signaling in breast cancer cells. Oncotarget (2014) 5(17):7498–511. doi: 10.18632/oncotarget.2241
58. Baldini E, Sorrenti S, D'Armiento E, Prinzi N, Guaitoli E, Favoriti P, et al. Aurora kinases: new molecular targets in thyroid cancer therapy. Clin Ter (2012) 163(6):e457–62.
59. Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, et al. Vx-680, a potent and selective small-molecule inhibitor of the aurora kinases, suppresses tumor growth in vivo. Nat Med (2004) 10(3):262–7. doi: 10.1038/nm1003
60. Lok W, Klein RQ, Saif MW. Aurora kinase inhibitors as anti-cancer therapy. Anticancer Drugs (2010) 21(4):339–50. doi: 10.1097/CAD.0b013e3283350dd1
61. Dauch D, Rudalska R, Cossa G, Nault JC, Kang TW, Wuestefeld T, et al. A myc-aurora kinase a protein complex represents an actionable drug target in P53-altered liver cancer. Nat Med (2016) 22(7):744–53. doi: 10.1038/nm.4107
62. Park JE, Soung NK, Johmura Y, Kang YH, Liao C, Lee KH, et al. Polo-box domain: A versatile mediator of polo-like kinase function. Cell Mol Life Sci (2010) 67(12):1957–70. doi: 10.1007/s00018-010-0279-9
63. Seeger-Nukpezah T, Liebau MC, Hopker K, Lamkemeyer T, Benzing T, Golemis EA, et al. The centrosomal kinase plk1 localizes to the transition zone of primary cilia and induces phosphorylation of nephrocystin-1. PloS One (2012) 7(6):e38838. doi: 10.1371/journal.pone.0038838
64. Ito Y, Miyoshi E, Sasaki N, Kakudo K, Yoshida H, Tomoda C, et al. Polo-like kinase 1 overexpression is an early event in the progression of papillary carcinoma. Br J Cancer (2004) 90(2):414–8. doi: 10.1038/sj.bjc.6601540
65. Salvatore G, Nappi TC, Salerno P, Jiang Y, Garbi C, Ugolini C, et al. A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res (2007) 67(21):10148–58. doi: 10.1158/0008-5472.Can-07-1887
66. Zhang XG, Lu XF, Jiao XM, Chen B, Wu JX. Plk1 gene suppresses cell invasion of undifferentiated thyroid carcinoma through the inhibition of Cd44v6, Mmp-2 and Mmp-9. Exp Ther Med (2012) 4(6):1005–9. doi: 10.3892/etm.2012.729
67. Han SJ, Jang HS, Seu SY, Cho HJ, Hwang YJ, Kim JI, et al. Hepatic ischemia/reperfusion injury disrupts the homeostasis of kidney primary cilia via oxidative stress. Biochim Biophys Acta Mol Basis Dis (2017) 1863(7):1817–28. doi: 10.1016/j.bbadis.2017.05.004
68. Degenhardt Y, Greshock J, Laquerre S, Gilmartin AG, Jing J, Richter M, et al. Sensitivity of cancer cells to plk1 inhibitor Gsk461364a is associated with loss of P53 function and chromosome instability. Mol Cancer Ther (2010) 9(7):2079–89. doi: 10.1158/1535-7163.Mct-10-0095
69. Hikichi Y, Honda K, Hikami K, Miyashita H, Kaieda I, Murai S, et al. Tak-960, a novel, orally available, selective inhibitor of polo-like kinase 1, shows broad-spectrum preclinical antitumor activity in multiple dosing regimens. Mol Cancer Ther (2012) 11(3):700–9. doi: 10.1158/1535-7163.Mct-11-0762
70. Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krssák M, et al. Bi 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol (2007) 17(4):316–22. doi: 10.1016/j.cub.2006.12.037
71. Ito Y, Uruno T, Nakano K, Takamura Y, Miya A, Kobayashi K, et al. An observation trial without surgical treatment in patients with papillary microcarcinoma of the thyroid. Thyroid (2003) 13(4):381–7. doi: 10.1089/105072503321669875
72. Hofheinz RD, Al-Batran SE, Hochhaus A, Jäger E, Reichardt VL, Fritsch H, et al. An open-label, phase I study of the polo-like kinase-1 inhibitor, Bi 2536, in patients with advanced solid tumors. Clin Cancer Res (2010) 16(18):4666–74. doi: 10.1158/1078-0432.Ccr-10-0318
73. Sebastian M, Reck M, Waller CF, Kortsik C, Frickhofen N, Schuler M, et al. The efficacy and safety of bi 2536, a novel Plk-1 inhibitor, in patients with stage Iiib/Iv non-small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open-label, randomized phase Ii clinical trial. J Thorac Oncol (2010) 5(7):1060–7. doi: 10.1097/JTO.0b013e3181d95dd4
74. Olmos D, Barker D, Sharma R, Brunetto AT, Yap TA, Taegtmeyer AB, et al. Phase I study of Gsk461364, a specific and competitive polo-like kinase 1 inhibitor, in patients with advanced solid Malignancies. Clin Cancer Res (2011) 17(10):3420–30. doi: 10.1158/1078-0432.Ccr-10-2946
75. Tedelind S, Poliakova K, Valeta A, Hunegnaw R, Yemanaberhan EL, Heldin NE, et al. Nuclear cysteine cathepsin variants in thyroid carcinoma cells. Biol Chem (2010) 391(8):923–35. doi: 10.1515/bc.2010.109
76. Al-Hashimi A, Venugopalan V, Sereesongsaeng N, Tedelind S, Pinzaru AM, Hein Z, et al. Significance of nuclear cathepsin V in normal thyroid epithelial and carcinoma cells. Biochim Biophys Acta Mol Cell Res (2020) 1867(12):118846. doi: 10.1016/j.bbamcr.2020.118846
77. Jordans S, Jenko-Kokalj S, Kühl NM, Tedelind S, Sendt W, Brömme D, et al. Monitoring compartment-specific substrate cleavage by cathepsins B, K, L, and S at physiological ph and redox conditions. BMC Biochem (2009) 10:23. doi: 10.1186/1471-2091-10-23
78. Brix K, Szumska J, Weber J, Qatato M, Venugopalan V, Al-Hashimi A, et al. Auto-regulation of the thyroid gland beyond classical pathways. Exp Clin Endocrinol Diabetes (2020) 128(6-7):437–45. doi: 10.1055/a-1080-2969
79. Friedrichs B, Tepel C, Reinheckel T, Deussing J, von Figura K, Herzog V, et al. K and L. J Clin Invest (2003) 111(11):1733–45. doi: 10.1172/jci15990
80. Szumska J, Batool Z, Al-Hashimi A, Venugopalan V, Skripnik V, Schaschke N, et al. Treatment of rat thyrocytes in vitro with cathepsin B and L inhibitors results in disruption of primary cilia leading to redistribution of the trace amine associated receptor 1 to the endoplasmic reticulum. Biochimie (2019) 166:270–85. doi: 10.1016/j.biochi.2019.07.010
81. Doğru AG, Rehders M, Brix K. Investigations on primary cilia of nthy-ori 3-1 cells upon cysteine cathepsin inhibition or thyrotropin stimulation. Int J Mol Sci (2023) 24(11):9292. doi: 10.3390/ijms24119292
82. Albee AJ, Kwan AL, Lin H, Granas D, Stormo GD, Dutcher SK. Identification of cilia genes that affect cell-cycle progression using whole-genome transcriptome analysis in chlamydomonas reinhardtti. G3 (Bethesda) (2013) 3(6):979–91. doi: 10.1534/g3.113.006338
83. Kim JI, Kim J, Jang HS, Noh MR, Lipschutz JH, Park KM. Reduction of oxidative stress during recovery accelerates normalization of primary cilia length that is altered after ischemic injury in murine kidneys. Am J Physiol Renal Physiol (2013) 304(10):F1283–94. doi: 10.1152/ajprenal.00427.2012
84. Nikonova AS, Deneka AY, Kiseleva AA, Korobeynikov V, Gaponova A, Serebriiskii IG, et al. Ganetespib limits ciliation and cystogenesis in autosomal-dominant polycystic kidney disease (Adpkd). FASEB J (2018) 32(5):2735–46. doi: 10.1096/fj.201700909R
85. Kim JB, Yang EY, Woo J, Kwon H, Lim W, Moon BI. Sodium selenite enhanced the anti-proliferative effect of mek-erk inhibitor in thyroid cancer cells. In Vivo (2020) 34(1):185–90. doi: 10.21873/invivo.11760
86. Lin SF, Lin JD, Hsueh C, Chou TC, Yeh CN, Chen MH, et al. Efficacy of an hsp90 inhibitor, ganetespib, in preclinical thyroid cancer models. Oncotarget (2017) 8(25):41294–304. doi: 10.18632/oncotarget.17180
87. Kavoi BM, Plendl J, Makanya AN, Ochieng S, Kiama SG. Effects of anticancer drug docetaxel on the structure and function of the rabbit olfactory mucosa. Tissue Cell (2014) 46(3):213–24. doi: 10.1016/j.tice.2014.04.005
88. Mount RJ, Takeno S, Wake M, Harrison RV. Carboplatin ototoxicity in the chinchilla: lesions of the vestibular sensory epithelium. Acta Otolaryngol Suppl (1995) 519:60–5. doi: 10.3109/00016489509121871
89. Viola D, Valerio L, Molinaro E, Agate L, Bottici V, Biagini A, et al. Treatment of advanced thyroid cancer with targeted therapies: ten years of experience. Endocr Relat Cancer (2016) 23(4):R185–205. doi: 10.1530/erc-15-0555
90. Peixoto E, Richard S, Pant K, Biswas A, Gradilone SA. The primary cilium: its role as a tumor suppressor organelle. Biochem Pharmacol (2020) 175:113906. doi: 10.1016/j.bcp.2020.113906
91. Hwangbo Y, Park YJ. Genome-wide association studies of autoimmune thyroid diseases, thyroid function, and thyroid cancer. Endocrinol Metab (Seoul) (2018) 33(2):175–84. doi: 10.3803/EnM.2018.33.2.175
92. Saevarsdottir S, Olafsdottir TA, Ivarsdottir EV, Halldorsson GH, Gunnarsdottir K, Sigurdsson A, et al. Flt3 stop mutation increases flt3 ligand level and risk of autoimmune thyroid disease. Nature (2020) 584(7822):619–23. doi: 10.1038/s41586-020-2436-0
93. Weetman AP. Autoimmune thyroid disease. Autoimmunity (2004) 37(4):337–40. doi: 10.1080/08916930410001705394
94. Ramos-Levi AM, Marazuela M. Pathogenesis of thyroid autoimmune disease: the role of cellular mechanisms. Endocrinol Nutr (2016) 63(8):421–9. doi: 10.1016/j.endonu.2016.04.003
95. Manji N, Carr-Smith JD, Boelaert K, Allahabadia A, Armitage M, Chatterjee VK, et al. Influences of age, gender, smoking, and family history on autoimmune thyroid disease phenotype. J Clin Endocrinol Metab (2006) 91(12):4873–80. doi: 10.1210/jc.2006-1402
96. Vanderpump MP, Tunbridge WM, French JM, Appleton D, Bates D, Clark F, et al. The incidence of thyroid disorders in the community: A twenty-year follow-up of the whickham survey. Clin Endocrinol (Oxf) (1995) 43(1):55–68. doi: 10.1111/j.1365-2265.1995.tb01894.x
97. Luo N, Conwell MD, Chen X, Kettenhofen CI, Westlake CJ, Cantor LB, et al. Primary cilia signaling mediates intraocular pressure sensation. Proc Natl Acad Sci U.S.A. (2014) 111(35):12871–6. doi: 10.1073/pnas.1323292111
98. Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med (2011) 364(16):1533–43. doi: 10.1056/NEJMra1010172
99. Fernandez-Santos JM, Utrilla JC, Vazquez-Roman V, Villar-Rodriguez JL, Gutierrez-Aviles L, Martin-Lacave I. Primary cilium in the human thyrocyte: changes in frequency and length in relation to the functional pathology of the thyroid gland. Thyroid (2019) 29(4):595–606. doi: 10.1089/thy.2018.0401
100. Qatato M, Szumska J, Skripnik V, Rijntjes E, Köhrle J, Brix K. Canonical tsh regulation of cathepsin-mediated thyroglobulin processing in the thyroid gland of male mice requires taar1 expression. Front Pharmacol (2018) 9:221. doi: 10.3389/fphar.2018.00221
101. Fekete C, Lechan RM. Central regulation of hypothalamic-pituitary-thyroid axis under physiological and pathophysiological conditions. Endocr Rev (2014) 35(2):159–94. doi: 10.1210/er.2013-1087
102. Ortiga-Carvalho TM, Chiamolera MI, Pazos-Moura CC, Wondisford FE. Hypothalamus-pituitary-thyroid axis. Compr Physiol (2016) 6(3):1387–428. doi: 10.1002/cphy.c150027
103. Szumska J, Qatato M, Rehders M, Fuhrer D, Biebermann H, Grandy DK, et al. Trace amine-associated receptor 1 localization at the apical plasma membrane domain of fisher rat thyroid epithelial cells is confined to cilia. Eur Thyroid J (2015) 4(Suppl 1):30–41. doi: 10.1159/000434717
104. Zarković M. The role of oxidative stress on the pathogenesis of graves' Disease. J Thyroid Res (2012) 2012:302537. doi: 10.1155/2012/302537
105. Abalovich M, Llesuy S, Gutierrez S, Repetto M. Peripheral parameters of oxidative stress in graves' Disease: the effects of methimazole and 131 iodine treatments. Clin Endocrinol (Oxf) (2003) 59(3):321–7. doi: 10.1046/j.1365-2265.2003.01850.x
106. Komosinska-Vassev K, Olczyk K, Kucharz EJ, Marcisz C, Winsz-Szczotka K, Kotulska A. Free radical activity and antioxidant defense mechanisms in patients with hyperthyroidism due to graves' Disease during therapy. Clin Chim Acta (2000) 300(1-2):107–17. doi: 10.1016/s0009-8981(00)00306-5
107. Bianchi G, Solaroli E, Zaccheroni V, Grossi G, Bargossi AM, Melchionda N, et al. Oxidative stress and anti-oxidant metabolites in patients with hyperthyroidism: effect of treatment. Horm Metab Res (1999) 31(11):620–4. doi: 10.1055/s-2007-978808
108. Mezosi E, Szabo J, Nagy EV, Borbely A, Varga E, Paragh G, et al. Nongenomic effect of thyroid hormone on free-radical production in human polymorphonuclear leukocytes. J Endocrinol (2005) 185(1):121–9. doi: 10.1677/joe.1.05968
109. Antonelli A, Ferrari SM, Corrado A, Di Domenicantonio A, Fallahi P. Autoimmune thyroid disorders. Autoimmun Rev (2015) 14(2):174–80. doi: 10.1016/j.autrev.2014.10.016
110. Leese GP, Flynn RV, Jung RT, Macdonald TM, Murphy MJ, Morris AD. Increasing prevalence and incidence of thyroid disease in tayside, scotland: the thyroid epidemiology audit and research study (Tears). Clin Endocrinol (Oxf) (2008) 68(2):311–6. doi: 10.1111/j.1365-2265.2007.03051.x
111. Lin JD. The role of apoptosis in autoimmune thyroid disorders and thyroid cancer. Bmj (2001) 322(7301):1525–7. doi: 10.1136/bmj.322.7301.1525
112. Arscott PL, Baker JR Jr. Apoptosis and thyroiditis. Clin Immunol Immunopathol (1998) 87(3):207–17. doi: 10.1006/clin.1998.4526
113. Majumder S, Cash A, Fisk HA. Non-overlapping distributions and functions of the vdac family in ciliogenesis. Cells (2015) 4(3):331–53. doi: 10.3390/cells4030331
114. Lee J, Park KC, Sul HJ, Hong HJ, Kim KH, Kero J, et al. Loss of primary cilia promotes mitochondria-dependent apoptosis in thyroid cancer. Sci Rep (2021) 11(1):4181. doi: 10.1038/s41598-021-83418-3
115. Mikoś H, Mikoś M, Obara-Moszyńska M, Niedziela M. The role of the immune system and cytokines involved in the pathogenesis of autoimmune thyroid disease (Aitd). Endokrynol Pol (2014) 65(2):150–5. doi: 10.5603/ep.2014.0021
116. Prummel MF, Wiersinga WM. Thyroid peroxidase autoantibodies in euthyroid subjects. Best Pract Res Clin Endocrinol Metab (2005) 19(1):1–15. doi: 10.1016/j.beem.2004.11.003
117. Palazzo FF, Hammond LJ, Goode AW, Mirakian R. Death of the autoimmune thyrocyte: is it pushed or does it jump? Thyroid (2000) 10(7):561–72. doi: 10.1089/thy.2000.10.561
118. Wann AK, Knight MM. Primary cilia elongation in response to interleukin-1 mediates the inflammatory response. Cell Mol Life Sci (2012) 69(17):2967–77. doi: 10.1007/s00018-012-0980-y
119. Dorris ER, Smyth P, O'Leary JJ, Sheils O. Mir141 expression differentiates hashimoto thyroiditis from ptc and benign thyrocytes in irish archival thyroid tissues. Front Endocrinol (Lausanne) (2012) 3:102. doi: 10.3389/fendo.2012.00102
120. Zhu J, Zhang Y, Zhang W, Zhang W, Fan L, Wang L, et al. Microrna-142-5p contributes to hashimoto's thyroiditis by targeting cldn1. J Transl Med (2016) 14(1):166. doi: 10.1186/s12967-016-0917-6
121. Wang B, Shao X, Song R, Xu D, Zhang JA. The emerging role of epigenetics in autoimmune thyroid diseases. Front Immunol (2017) 8:396. doi: 10.3389/fimmu.2017.00396
122. Benes V, Collier P, Kordes C, Stolte J, Rausch T, Muckentaler MU, et al. Identification of cytokine-induced modulation of microrna expression and secretion as measured by a novel microrna specific Qpcr assay. Sci Rep (2015) 5:11590. doi: 10.1038/srep11590
123. Ando Y, Yang GX, Kenny TP, Kawata K, Zhang W, Huang W, et al. Overexpression of microrna-21 is associated with elevated pro-inflammatory cytokines in dominant-negative Tgf-B Receptor type ii mouse. J Autoimmun (2013) 41:111–9. doi: 10.1016/j.jaut.2012.12.013
124. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139(5):871–90. doi: 10.1016/j.cell.2009.11.007
125. Han SJ, Jung JK, Im SS, Lee SR, Jang BC, Park KM, et al. Deficiency of primary cilia in kidney epithelial cells induces epithelial to mesenchymal transition. Biochem Biophys Res Commun (2018) 496(2):450–4. doi: 10.1016/j.bbrc.2018.01.079
126. Ehnert S, Sreekumar V, Aspera-Werz RH, Sajadian SO, Wintermeyer E, Sandmann GH, et al. Tgf-B(1) impairs mechanosensation of human osteoblasts via hdac6-mediated shortening and distortion of primary cilia. J Mol Med (Berl) (2017) 95(6):653–63. doi: 10.1007/s00109-017-1526-4
127. Thvilum M, Brandt F, Brix TH, Hegedus L. A review of the evidence for and against increased mortality in hypothyroidism. Nat Rev Endocrinol (2012) 8(7):417–24. doi: 10.1038/nrendo.2012.29
128. Chaker L, Bianco AC, Jonklaas J, Peeters RP. Hypothyroidism. Lancet (2017) 390(10101):1550–62. doi: 10.1016/s0140-6736(17)30703-1
129. Nozawa YI, Lin C, Chuang PT. Hedgehog signaling from the primary cilium to the nucleus: an emerging picture of ciliary localization, trafficking and transduction. Curr Opin Genet Dev (2013) 23(4):429–37. doi: 10.1016/j.gde.2013.04.008
130. Kang HS, Beak JY, Kim YS, Herbert R, Jetten AM. Glis3 is associated with primary cilia and wwtr1/taz and implicated in polycystic kidney disease. Mol Cell Biol (2009) 29(10):2556–69. doi: 10.1128/mcb.01620-08
131. Huangfu D, Anderson KV. Cilia and hedgehog responsiveness in the mouse. Proc Natl Acad Sci U.S.A. (2005) 102(32):11325–30. doi: 10.1073/pnas.0505328102
132. Jetten AM. Glis1-3 transcription factors: critical roles in the regulation of multiple physiological processes and diseases. Cell Mol Life Sci (2018) 75(19):3473–94. doi: 10.1007/s00018-018-2841-9
133. Porcu E, Medici M, Pistis G, Volpato CB, Wilson SG, Cappola AR, et al. A meta-analysis of thyroid-related traits reveals novel loci and gender-specific differences in the regulation of thyroid function. PloS Genet (2013) 9(2):e1003266. doi: 10.1371/journal.pgen.1003266
134. Rurale G, Marelli F, Duminuco P, Persani L. Glis3 as a critical regulator of thyroid primordium specification. Thyroid (2020) 30(2):277–89. doi: 10.1089/thy.2019.0196
135. Taha D, Barbar M, Kanaan H, Williamson Balfe J. Neonatal diabetes mellitus, congenital hypothyroidism, hepatic fibrosis, polycystic kidneys, and congenital glaucoma: A new autosomal recessive syndrome? Am J Med Genet A (2003) 122a(3):269–73. doi: 10.1002/ajmg.a.20267
136. Watanabe N, Hiramatsu K, Miyamoto R, Yasuda K, Suzuki N, Oshima N, et al. A murine model of neonatal diabetes mellitus in glis3-deficient mice. FEBS Lett (2009) 583(12):2108–13. doi: 10.1016/j.febslet.2009.05.039
137. Kang HS, Kumar D, Liao G, Lichti-Kaiser K, Gerrish K, Liao XH, et al. Glis3 is indispensable for tsh/tshr-dependent thyroid hormone biosynthesis and follicular cell proliferation. J Clin Invest (2017) 127(12):4326–37. doi: 10.1172/jci94417
138. Molla-Herman A, Ghossoub R, Blisnick T, Meunier A, Serres C, Silbermann F, et al. The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J Cell Sci (2010) 123(Pt 10):1785–95. doi: 10.1242/jcs.059519
139. Lee J, Sul HJ, Kim KH, Chang JY, Shong M. Primary cilia mediate tsh-regulated thyroglobulin endocytic pathways. Front Endocrinol (Lausanne) (2021) 12:700083. doi: 10.3389/fendo.2021.700083
140. Marinò M, Zheng G, Chiovato L, Pinchera A, Brown D, Andrews D, et al. Role of megalin (Gp330) in transcytosis of thyroglobulin by thyroid cells. A novel function in the control of thyroid hormone release. J Biol Chem (2000) 275(10):7125–37. doi: 10.1074/jbc.275.10.7125
141. Marinò M, Pinchera A, McCluskey RT, Chiovato L. Megalin in thyroid physiology and pathology. Thyroid (2001) 11(1):47–56. doi: 10.1089/10507250150500667
142. Romagnoli P, Herzog V. Transcytosis in thyroid follicle cells: regulation and implications for thyroglobulin transport. Exp Cell Res (1991) 194(2):202–9. doi: 10.1016/0014-4827(91)90355-x
143. Herzog V. Transcytosis in thyroid follicle cells. J Cell Biol (1983) 97(3):607–17. doi: 10.1083/jcb.97.3.607
144. Herzog V. Pathways of endocytosis in thyroid follicle cells. Int Rev Cytol (1984) 91:107–39. doi: 10.1016/s0074-7696(08)61315-7
145. Lisi S, Segnani C, Mattii L, Botta R, Marcocci C, Dolfi A, et al. Thyroid dysfunction in megalin deficient mice. Mol Cell Endocrinol (2005) 236(1-2):43–7. doi: 10.1016/j.mce.2005.03.009
146. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin (2012) 62(1):10–29. doi: 10.3322/caac.20138
147. Walsh JP. Managing thyroid disease in general practice. Med J Aust (2016) 205(4):179–84. doi: 10.5694/mja16.00545
148. Burman KD, Wartofsky L. Clinical practice. Thyroid nodules. N Engl J Med (2015) 373(24):2347–56. doi: 10.1056/NEJMcp1415786
149. Gharib H, Papini E, Paschke R, Duick DS, Valcavi R, Hegedüs L, et al. American association of clinical endocrinologists, associazione medici endocrinologi, and european thyroid association medical guidelines for clinical practice for the diagnosis and management of thyroid nodules: executive summary of recommendations. Endocr Pract (2010) 16(3):468–75. doi: 10.4158/ep.16.3.468
150. Yassa L, Cibas ES, Benson CB, Frates MC, Doubilet PM, Gawande AA, et al. Long-term assessment of a multidisciplinary approach to thyroid nodule diagnostic evaluation. Cancer (2007) 111(6):508–16. doi: 10.1002/cncr.23116
151. Soelberg KK, Bonnema SJ, Brix TH, Hegedüs L. Risk of Malignancy in thyroid incidentalomas detected by 18f-fluorodeoxyglucose positron emission tomography: A systematic review. Thyroid (2012) 22(9):918–25. doi: 10.1089/thy.2012.0005
152. Wong R, Farrell SG, Grossmann M. Thyroid nodules: diagnosis and management. Med J Aust (2018) 209(2):92–8. doi: 10.5694/mja17.01204
153. Studer H, Ramelli F. Simple goiter and its variants: euthyroid and hyperthyroid multinodular goiters. Endocrine Rev (1982) 3(1):40–61. doi: 10.1210/edrv-3-1-40
Keywords: primary cilia, thyroid diseases, thyroid cancer, graves’ disease, Hashimoto’s thyroiditis, hypothyroidism, thyroid nodule
Citation: Tian Z, Li X, Yu X, Yan S, Sun J, Ma W, Zhu X and Tang Y (2024) The role of primary cilia in thyroid diseases. Front. Endocrinol. 14:1306550. doi: 10.3389/fendo.2023.1306550
Received: 04 October 2023; Accepted: 05 December 2023;
Published: 08 January 2024.
Edited by:
Noriyuki Koibuchi, Gunma University, JapanReviewed by:
Soledad Bárez-López, Spanish National Research Council (CSIC), SpainJeehee Yoon, Chonnam National University Bitgoeul Hospital, Republic of Korea
Copyright © 2024 Tian, Li, Yu, Yan, Sun, Ma, Zhu and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Tang, dGFuZ3lhbmdAYnVjbS5lZHUuY24=; Xiaoyun Zhu, cWllYmVuYmVuQDE2My5jb20=; Wenxin Ma, bWF3ZW54aW4xMDIzNjI3QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship