 Mengsi Liu
Mengsi Liu Zhen Zhang
Zhen Zhang Yating Chen
Yating Chen Ting Feng1,2,3,4
Ting Feng1,2,3,4 Xuefei Tian
Xuefei Tian
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 22 December 2023
Sec. Cancer Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1292011
This article is part of the Research TopicCircadian Rhythm in Metabolism and EndocrinologyView all 5 articles
Recent research has emphasized the interaction between the circadian clock and lipid metabolism, particularly in relation to tumors. This review aims to explore how the circadian clock regulates lipid metabolism and its impact on carcinogenesis. Specifically, targeting key enzymes involved in fatty acid synthesis (SREBP, ACLY, ACC, FASN, and SCD) has been identified as a potential strategy for cancer therapy. By disrupting these enzymes, it may be possible to inhibit tumor growth by interfering with lipid metabolism. Transcription factors, like SREBP play a significant role in regulating fatty acid synthesis which is influenced by circadian clock genes such as BMAL1, REV-ERB and DEC. This suggests a strong connection between fatty acid synthesis and the circadian clock. Therefore, successful combination therapy should target fatty acid synthesis in addition to considering the timing and duration of drug use. Ultimately, personalized chronotherapy can enhance drug efficacy in cancer treatment and achieve treatment goals
Cancer is a major global public health issue due to its high incidence and the metabolic reprogramming, immune evasion, proliferative signaling, growth suppression avoidance, cell death resistance, replication immortality, angiogenesis induction, invasion and metastasis activation it entails (1). Tumor cells undergo lipid metabolic reprogramming through de novo lipogenesis involving key transcription factors such as sterol regulatory element binding protein (SREBP), ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN) and stearoyl-CoA desaturase 1 (SCD1). Unhealthy lifestyle habits like shift work disrupt circadian rhythm which can contribute to the development of diabetes and cancer. Shift work is classified as a potential human carcinogen due to its disruption of circadian rhythms that regulate crucial biological processes leading to abnormal cell proliferation, gene mutation, and resistance to apoptosis (1–3). Research has also demonstrated that circadian clock genes and lipid metabolism play a role in cancer development by regulating signaling pathways and metabolites.
In this article, we have provided a comprehensive analysis of lipid metabolism and clock genes, with focus on SREBP as key factor. We have summarized the impact of transcription factors involved in de novo lipogenesis on lipid metabolism, as well as their mechanism of intersection with circadian clock genes. Furthermore, we have examined the interaction relationship between transcription factors responsible for de novo fatty acid synthesis, clock genes and cancer. Lastly, we have reviewed the advancements in utilizing circadian clock and lipid metabolism in cancer treatment and discussed their potential application in clinical therapies.
The Circadian clock is a regulatory system consisting of clock genes and controlled genes, which regulate the rhythmic movement of physiological and metabolic activities in organisms and synchronizes it with the changes in the environment. This regulation is mainly controlled by core transcription genes and their downstream regulated genes, which control the metabolic rhythm by regulating protein synthesis and degradation. The central clock system is located in the suprachiasmatic nucleus (SCN) of the hypothalamus, while the peripheral clock system is found in organs such as liver, spleen, lung, kidney, colon, adipose tissue and muscle. Light signals are converted into electrical signals by retinal ganglion cells and transmitted to the SCN through optic nerve tract to form rhythmic periodic activities that maintain organism homeostasis. Additionally, light signals can also be transmitted to the peripheral clock via neural and humoral pathways to synchronize them with external environment forming circadian rhythms (4). However, factors like body temperature, diet, and hormone levels also influence circadian oscillations (5–8). Before exploring the relationship between the circadian clock and lipid metabolism disorders as well as impact on cancer development, it’s important to understand how molecular mechanisms produce circadian oscillations within the circadian clock system (Figure 1).
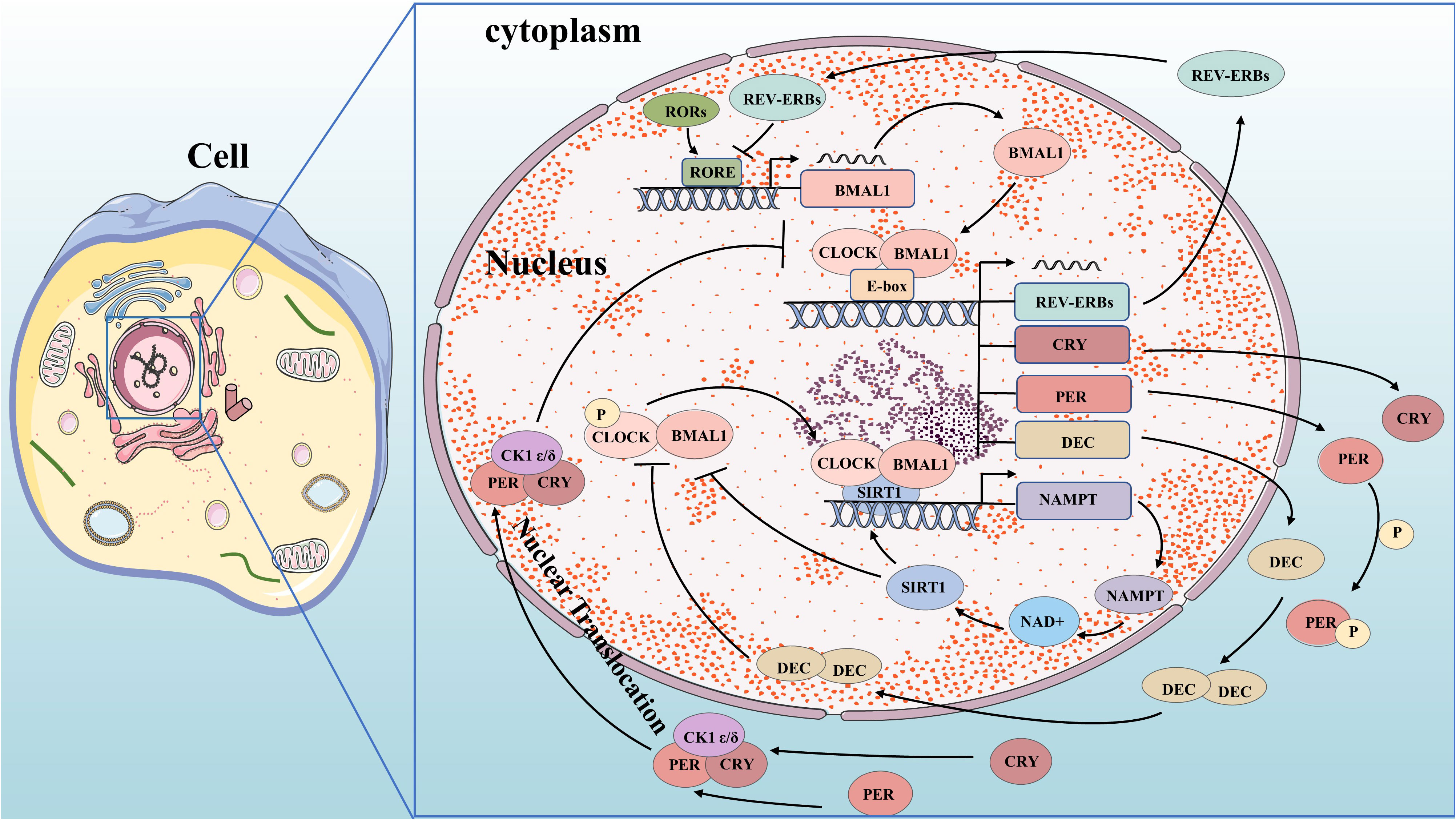
Figure 1 Molecular mechanisms of circadian clock. BMAL1 and CLOCK complex bind to the E-box and control gene promoters, including the genes for REV-ERBs, CRY, PER, and DEC. PER and CRY bind to CK1δ or CK1ϵ and translocate to the nucleus, where they inhibit their own transcription. DEC inhibits E-box promoters by directly binding to BMAL1 protein or CLOCK-BMAL1 complex and competing to suppress their expression. REV-ERBs compete for RORE in the promoter region of BMAL1 to inhibit its transcription. CLOCK-BMAL1 isoform can directly promote the circadian expression of NAMPT through the E-box of its promoter, thereby improving the circadian level of NAD+ and increasing the activity of SIRT1. Increased SIRT1 activity inhibits CLOCK-BMAL1 complex expression, forming a feedback loop that controls the circadian expression of core clock genes.
The circadian oscillation is a self-regulating transcription-translation feedback loop (TTFL) that occurs periodically through the expression of circadian clock genes. Core clock genes, including ARNTL/BMAL1 and CLOCK as activators, and PER1,2,3 and CRY1,2 as repressors (9), are involved in this process. Transcription and translation of clock genes require cis-regulatory elements such as E-box, D-box, and RORE (ROR-elements). BMAL1 forms a heterodimer with CLOCK or NPAS2 to bind to the E-box regulatory elements on PER and CRY genes for transcription initiation (10–12). PER and CRY proteins form heterodimers in the cytoplasm which interact with casein kinase 1δ (CK1δ) and CK1ϵ kinases to undergo phosphorylation before translocating into the nucleus (13). In the nucleus, they combine with BMAL1-CLOCK heterodimers to form a quaternary complex that inhibits CLOCK-BMAL1 activity. This leads to reduced transcriptional activation of PER and CRY genes forming a negative feedback loop (14, 15). As CLOCK-BMAL1 complex activity is inhibited by this interaction, levels of PER and CRY proteins decrease. Once bound to F-box regulatory elements, specific E3 ubiquitin ligase complexes ubiquitinate PER and CRY proteins, leading to their degradation by the proteasome. This relieves inhibition on CLOCK-BMAL1 complex allowing for initiation of new rounds of transcription (15–17). The activity of BMAL1-CLOCK heterodimer is influenced by both ROR and REV-ERB proteins. BMAL1 and CLOCK activate REV-ERB by binding to its promoter region. In response, REV-ERB accumulates and competes with ROR for ROREs in the promoter region of BMAL1, inhibiting its transcription (18). Meanwhile, RORs compete to activate BMAL1 transcription, forming a secondary feedback loop. Additionally, the clock-controlled gene DEC (Deleted in Esophageal Cancer) forms an autoregulatory feedback loop through encoding basic helix-loop-helix (bHLH) transcription factors. DEC directly binds to the BMAL1 protein or CLOCK-BMAL1 complex and competes to suppress their expression by inhibiting E-box promoters (19–24). Clock genes play a crucial role in tumorigenesis by regulating oncogenes and tumor suppressors. We have started exploring the key signaling pathways in carcinogenesis based on the molecular mechanism of circadian clock genes.
Lipids maintain cell structure, provide energy, and regulate cellular signaling. Lipid metabolism is regulated by various signaling pathways and produces different intermediates. Disturbances in lipid metabolism affect lipid levels, cell membrane composition, and permeability, thereby impacting the regulation of signaling pathways that contribute to cancer progression. Lipids consist of triacylglycerols (TAG), which are composed of fatty acids (FA) and glycerol, as well as adipoid. The majority of fatty acids in the body come from exogenous sources through food intake, while de novo FA synthesis contributes only a small proportion (25). Tumor cells enhance the de novo FA synthesis pathway to promote cancer cell biofilm formation and increase membrane lipid saturation. This alteration affects essential life processes such as cellular signaling and gene expression, supporting rapid cell proliferation and promoting cancer progression (26–28). Therefore, maintaining lipid homeostasis is crucial for human health, making research on lipid metabolism disorders an important focus in oncology. Understanding its molecular mechanism is necessary (Figure 2).
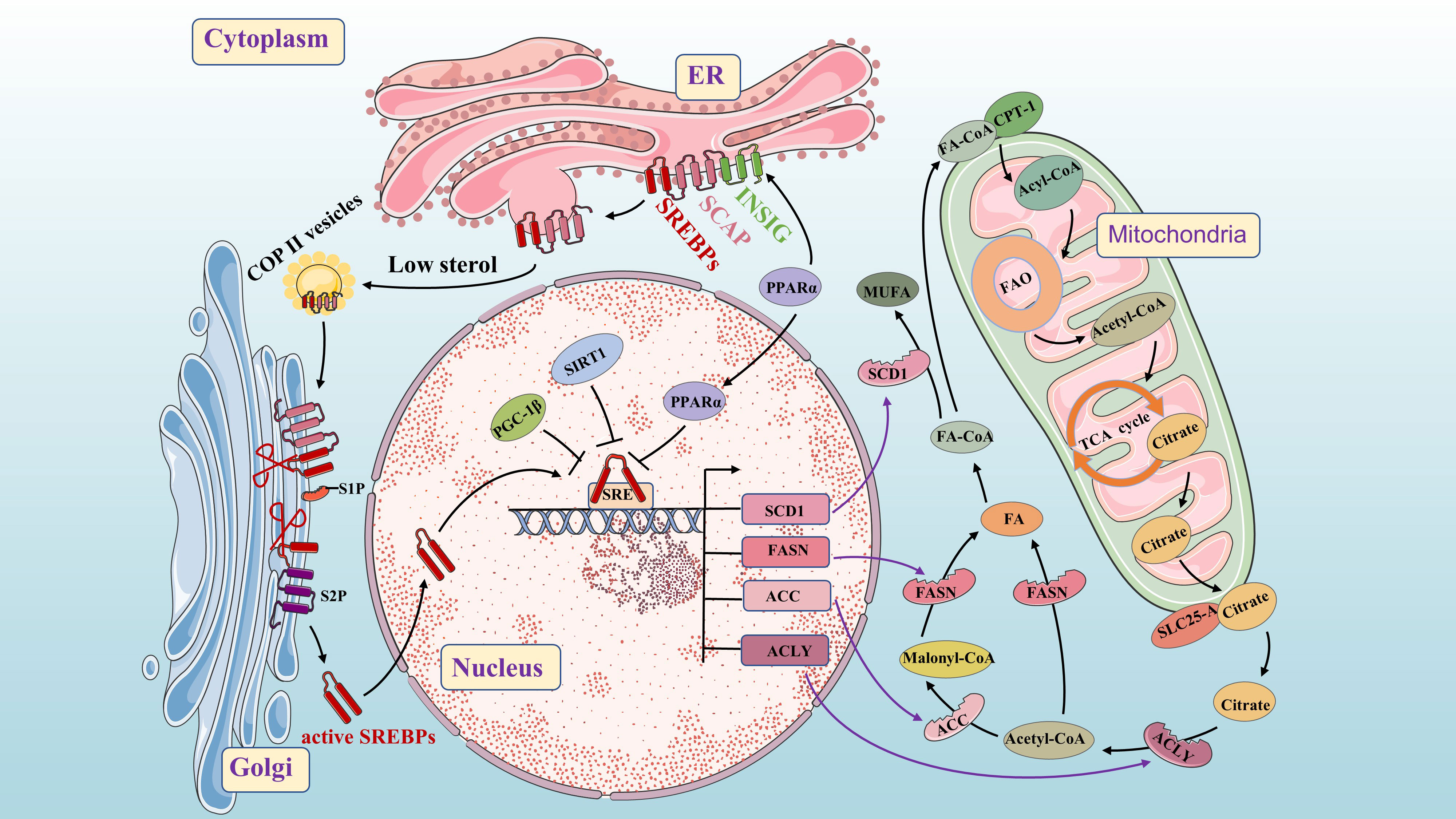
Figure 2 Molecular mechanisms of de novo fatty acid synthesis. As a key lipid source gene, SREBP is an important transcription factor throughout the process. The INSIG/SCAP/SREBPs complex is stable in the ER. When sterol levels decrease, SCAP dissociates from INSIG and facilitates the inclusion of SCAP/SREBP into COPII-coated vesicles for subsequent transport to the Golgi. PPAR α up-regulates the expression of the INSIG gene by binding to the PPRE in the promoter region and inhibits the nuclear translocation of the transcription factor SREBPs. SREBPs undergoes proteolysis under the action of S1P and S2P, and then translocate to the nucleus to activate SREBP target genes (such as FASN, SCD1, ACC, and ACLY). Citrate is acted upon by ACLY to form acetyl-CoA and oxaloacetate. Acetyl-CoA is converted into FA under the action of FASN. Acetyl-CoA is converted into Malonyl-CoA under the action of ACC, and then Malonyl-CoA is converted into FA under the action of FASN (Conversion of FA to MUFA by SCD). SIRT1 and PGC-1β can inhibit SREBP1 in the nucleus.
From previous studies, we know that Fatty acyl-CoA is transported across the mitochondrial membrane by carnitine palmitoyl transferase 1 (CPT1) and enter mitochondria for fatty acid β-oxidation (FAO), producing acetyl-CoA. Acetyl-CoA then enters the tricarboxylic acid (TCA) cycle to generate adenosine triphosphate (ATP) for energy production and combines with oxaloacetate to form citrate. Citrate is transported into the cytoplasm by a solute carrier family 25 member protein (SLC25-A), which is activated by SREBP1 (29). In the cytoplasm, citrate is converted into acetyl-CoA and oxaloacetate by ACLY. Under the action of ACLY, ACC and FASN, acetyl-CoA generates fatty acids in a closed circuit that regulate fatty acid metabolism in the body. The conversion of FA to monounsaturated fatty acids (MUFA) occurs through SCD (30). Cancer cells increase lipogenesis to promote their proliferation, and SREBP, a key transcription factor in FA synthesis, plays a central role in this process. It can affect the expression of ACLY, ACC, FASN and SCD1 at multiple stages of lipid biosynthesis (31, 32). SREBP consists of three subtypes: SREBP1a, SREBP1c and SREBP2 which regulate the expression of different lipid synthesis-related genes (33). SREBP1a and SREBP1c are encoded by the unigenes SREBF1 and mainly control the expression of lipogenic genes, while SREBP2, encoded by the unigenes SREBF2, primarily regulates cholesterol biosynthesis genes (34). Here, we focus on SREBP1. Previous studies have found that SREBP cleavage-activating protein (SCAP) binds to INSIGs protein in a sterol-dependent manner to form INSIGs/SCAP/SREBP1 complex stored in the endoplasmic reticulum (ER). At low sterol levels, SCAP dissociates from INSIG proteins and forms a complex with SCAP-SREBP1 that binds to coat protein complex–II (COP II) in the ER for translocation to the Golgi. In the Golgi, under the action of site 1 protease (S1P) and Site 2 protease (S2P), proteolysis occurs resulting in activation of nuclear translocation for increased FA biosynthesis through activation of target genes (35). When sterol content increases, changes occur in SCAP’s structure due to activation of its sterol sensing domain. This prevents entry into ER affecting vesicular transport from ER to Golgi and subsequently halts transcriptional regulation of FA synthesis (33, 36).
Peroxisome proliferator-activated receptor alpha (PPARα) is considered to be a crucial FA sensor (37). Its natural ligands include various FAs such as linoleic acid, oxidized fatty acids, and prostaglandin J2 (38, 39). PPARα mRNA is mainly expressed in tissues that oxidize fatty acids, such as the liver, heart, brown adipose tissue, kidney, and intestine (40). It regulates key transcriptional pathways involved in mitochondria, peroxisome and microsomal FAO as well as other lipolysis processes. Additionally, it plays a role in cellular functions like proliferation and metabolism. PPAR α mRNA is essential for maintaining nutrient homeostasis and lipid metabolism (38), while also regulating energy balance through activation of FA catabolism and stimulating of gluconeogenesis (41). Activation of PPARα increases the expression of acyl-CoA oxidase, CPT1, malonyl-CoA decarboxylase to enhance FA and triglycerides oxidation. Furthermore, PPARα activation down-regulates FASN and SREBP1 expression to impact de novo fatty acid synthesis (38, 42, 43). By binding to the PPAR response element (PPRE) in the promoter region, PPAR α up-regulates INSIG gene expression while inhibiting nuclear transfer of transcription factor SREBP1c to suppress FA production (44). Moreover, peroxisome proliferator-activated receptor γ coactivator β (PGC-1β), a transcription cofactor interacting with SREBP1c, can induce lipid synthesis-related gene transcription (45).
The activation of SREBP1c, a key transcription factor regulating lipid metabolism, in cancer cells and its impact on FA synthesis make it an important research focus for understanding tumorigenesis.
The hypothalamus regulates the circadian rhythms of diet and energy metabolism, which in turn affect lipid metabolism through the active expression of metabolic enzymes and transport systems (46, 47). Lipids are primarily stored as TAG in the human body. Core clock genes control adipose lipolysis by regulating TAG levels (48). Deletion of both REV-ERBα and REV-ERBβ has been found to cause severe defects in lipid metabolism, including significant increased TAG levels in the liver and hepatic steatosis (49, 50). Numerous studies have shown that disruption in circadian clock function can lead to disorders in lipid metabolism (51). The circadian clock plays a crucial role in maintaining lipid homeostasis by rhythmically activating and regulating proteins involved in lipid transport, synthesis, and degradation (Figure 3).
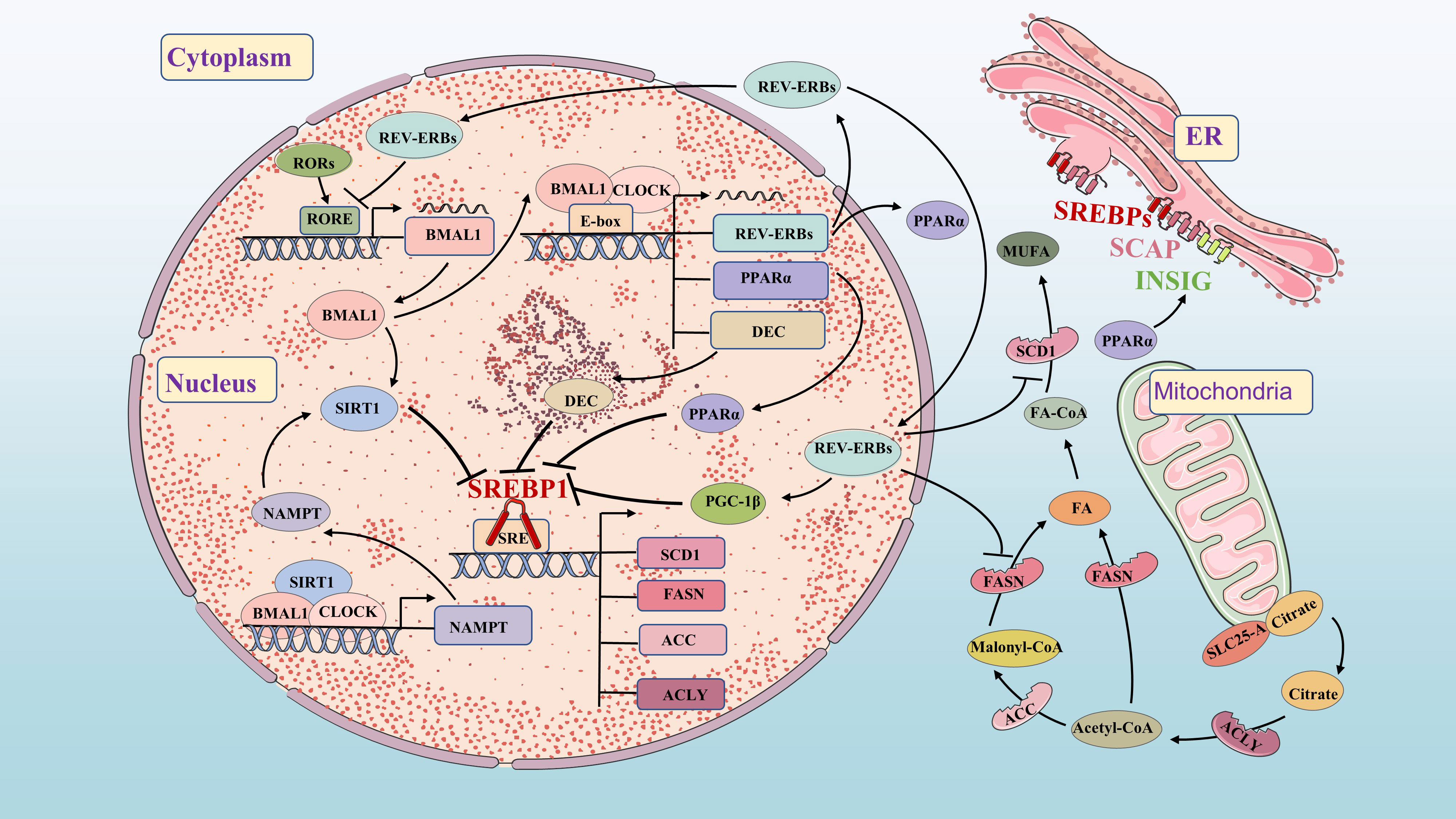
Figure 3 Molecular mechanisms of FA synthetic and circadian clock. FA synthetic and circadian clock networks have multiple interwoven negative feedback loops, such as BMAL1/DEC/SREBP1, PPARα/INSIG/SREBP1, BMAL1/SIRT1/SREBP1 and REV-ERBs/PGC-1β/SREBP1. The circadian network regulates the circadian expression of FA synthesis genes and metabolites through feedback loops.
Circadian variation in FA synthesis is partly mediated by the circadian clock’s effects on SREBP1c and its downstream targets (52, 53). Sirtuin 1 (SIRT1) deacetylates SREBP1c, inhibiting its activity and reducing occupancy on the promoter of lipogenesis genes. Increasing SIRT1 expression can enhance deacetylation of SREBP1c and suppress its expression, thereby inhibiting FA synthesis (54). SIRT1 is a NAD+-dependent histone deacetylase (HDAC) and a member of the HDAC family. Nicotinamide phosphoribosyltransferase (NAMPT) is the key enzyme of in NAD+ biosynthesis. The CLOCK-BMAL1 isoform directly promotes circadian expression of NAMPT, increasing the level of NAD+ and activating SIRT1. Increased SIRT1 activity inhibits CLOCK-BMAL1 complex expression, forming a feedback loop that controls circadian expression of core clock genes (55–57). DEC1 and DEC2 clock-controlled gene, regulate FA synthesis by inhibiting lipogenesis in the liver through binding to the promoter region of SREBP1c (58, 59). FASN and ACC also exhibit circadian fluctuations in adipose tissue and liver regulated by clock genes (60, 61). REV-ERBs effectively inhibit the expression of FASN, SCD1 and PGC-1β to impact FA synthesis (32, 62, 63).
PPARα directly binds to the circadian clock promoter, maintaining circadian oscillations of the BMAL1 gene in brain and muscle (64). BMAL1 regulates PPARα expression (65), while REV-ERBs is also a target gene of PPARα (66). The BMAL1-CLOCK complex activates PPARα by binding to the E-box (67). In turn, the response element of PPARα binds to the promoters of BMAL1 and REV-ERBs genes, activating their expression (65, 66). This highlights the close relationship between the circadian clock and FA metabolism. Previous studies have shown that both circadian clock and lipid metabolism disorders significantly impact carcinogenesis. Further research aims to understand key signaling pathways connecting circadian clock and lipid metabolism disorders in tumorigenesis, providing new directions for future investigations.
Although cancer types and genetic alterations vary, cancer is characterized by abnormal cell growth and proliferation, resulting in increased energy and macromolecules demands. These demands necessitate the synthesis of cellular building blocks such as nucleic acids, proteins, and lipids (68). Lipid synthesis is particularly enhanced in malignant tumors (68, 69), contributing to their aggressiveness. In fact, lipid synthesis is generally upregulated in human cancers (70), with 95% of tumor cell fat derived from endogenous de novo lipogenesis compared to normal cells (71). Notably, the key transcription factor SREBP1 and its downstream lipid-derived genes ALCY, ACC, FASN, and SCD1 play crucial roles across various tumors (Table 1).
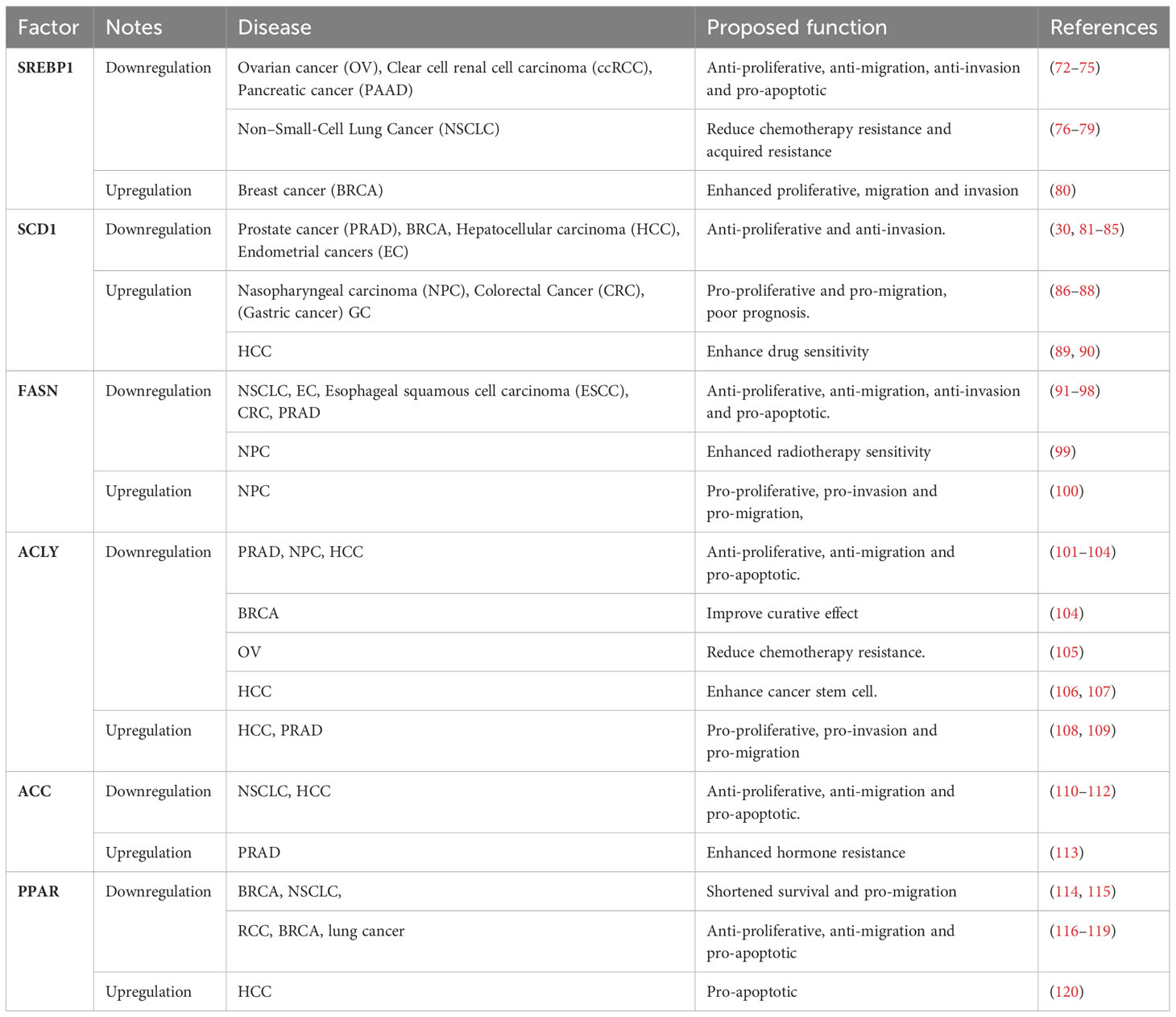
Table 1 Association of de novo fat synthesis genes with tumor development.
SREBP1 primarily controls the expression of lipogenic genes (121). It acts as a key transcription factor regulating de novo lipogenesis and lipid homeostasis. In tumor cells, SREBP1 plays a crucial role in promoting growth, survival, proliferation, apoptosis, invasion and metastasis (72, 73, 122, 123). Overexpression of SREBP1 is closely associated with cancer progression and metastasis. Conversely, knockdown of SREBP1 reduces the expression of FA synthesis genes, inhibits cancer cell proliferation, and suppresses tumor growth (124). SREBP1 plays a significant role in promoting lipogenesis and tumor growth in breast cancer (BRCA), hepatocellular carcinoma (HCC), Esophageal squamous cell carcinoma (ESCC), pancreatic cancer (PAAD), and gastric cancer (GC) (74, 80, 125). Inhibition of SREBP1 can hinder tumor progression and metastasis. SREBP1 also plays a role in chemotherapy resistance among cancer cells which has been associated with reduced patient survival rates. Targeting SREBP1 can inhibit lipid synthesis, gemcitabine-induced CSCs, increases sensitivity to gemcitabine, reducing chemical resistance, and ultimately improving patient survival rates (75, 126, 127). In addition to chemical resistance, Osimertinib facilitated degradation of mature form of SREBP1 and reduced protein levels regulated by genes in EGFR-mutant NSCLC. This inhibition leads to decreased lipogenesis, cancer cells apoptosis, and reduced acquired resistance to Osimertinib (76). Moreover, inhibition of SREBP1 results in lipid peroxidation. Patients with EGFR-mutated NSCLC developed resistance to gefitinib following targeted therapy. However, SREBP1 restoration restores their sensitivity to gefitinib and exhibits synergistic effects on anti-proliferation and pro-apoptosis (77–79). In conclusion, SREBP1 is generally up-regulated in CRC, HCC, PRAD, BRCA, THCA, NSCLC and PAAD, which aligns with its role in regulating lipid homeostasis in human cells.
SCD1, a rate-limiting enzyme in MUFA biosynthesis, is overexpressed in lung cancer, BRCA and OV (128–132). Inhibiting SCD-induced accumulation of MUFA leads to cancer cell death (133), and SCD1 has also been identified as a marker of CSCs in CRC (134). Increased expression of SCD1 is associated with tumor progression and poor prognosis (30, 135). Its expression is regulated by factors such as the millet bran (a total polyphenol consisting of 12 compounds extracted from foxtail millet bran, BPIS), Hypoxia-inducible factor-1α (HIF-1α), and miR-433-3p. Targeting SCD1 may be a potential therapeutic strategy for different cancers (81, 82, 86). Cancer cells rely on regulating SREBP1 expression as well as activating both SCD1 and FASN to promote lipogenesis and proliferation (87, 88). In genetic and pharmacological studies, inhibiting SCD1 alters cellular lipid composition which disrupts plasma membrane fluidity leading to inhibiting of HCC cell invasion in vitro, which could serve as a biomarker for HCC aggressiveness (30, 83). Studies on FA metabolism in EC have shown that reducing SCD1 expression can inhibit EC growth (84). SCD1 has been associated with drug resistance acquisition. The multikinase inhibitor sorafenib targets SCD1 through the ATP-AMPK-mTOR-SREBP1 pathway to suppress MUFA synthesis, disrupt lipogenesis, induce liver cancer cell death, and enhance sensitivity to sorafenib (89, 90) Docetaxel effectively impedes the tumor progress in PRAD by down-regulating mRNA and protein levels of SREBP1 and SCD1. This enhances docetaxel’s anti-proliferation, anti-migration and anti-invasion capabilities (85).
FASN up-regulation promotes cancer progression by enhancing lipid synthesis and signaling pathways, while its inhibition can impede tumor development and indicate poor prognosis. Compared to normal human tissues, increased FASN expression promotes endogenous FA synthesis in various cancer tissues (91–94, 136–138). FASN is associated with tumor invasiveness, and increased expression of FASN is positively correlated with lipid droplet formation and enhanced cancer cell activity. FASN regulates metabolic disorders and can indicate poor prognosis (139–142). Various compounds have been found to reduce FASN expression and inhibit lipogenesis, demonstrating potential anticancer activity such as extract of eriobotrya japonica and Davallia formosana, Oridonin (95–97). Additionally, Long intergene non-coding RNA2570(LINC02570) promotes NPC progression by up-regulation SREBP1 and FASN through miR-4649-3p (100). In the reprogramming of lipid metabolism that occurs in cancer-associated fibroblasts (CAF), FASN is significantly increased in CAF, enhancing colorectal cancer cell migration (98, 143). Moreover, FASN inhibitors can enhance the effects of chemotherapy drugs, restore tumor sensitivity to treatment, and inhibit tumor growth in resistant cancer cells (144, 145). Combination therapy with FASN inhibitors and other targeted therapies or radiotherapy shows promising therapeutic effects (146). Additionally, some studies have demonstrated that inhibiting the FASN gene significantly increases nasopharyngeal carcinoma cell sensitivity to radiotherapy (99).
ACLY is up-regulated in various tumors and plays a crucial role in cancer cell proliferation, growth, migration and apoptosis (147–149). Its overexpression provides energy for malignant proliferation of tumor cells and promotes their progression (101). Increased expression of ACLY and SCD1 mRNA in HCC leads to enhanced FA synthesis, resulting in cell proliferation and deterioration of HCC (108). Modulating the expression of ACLY and FASN through SREBP1 affects de novo lipogenesis production in PRAD cells, promoting cell proliferation (109), which is consistent with the inhibitory effects observed upon ACLY knockdown in NPC and PRAD that inhibit tumor cell migration and growth (102, 103). Targeting ACLY can synergistically enhance the efficacy of targeted therapy and chemotherapy while reducing drug resistance. The ACLY inhibitor BMS-303141 induces apoptosis in HCC cells when combined with sorafenib to improve therapeutic efficacy (101). Competitive inhibition of ACLY by Hydroxycitric acid enhances tamoxifen’s toxic effect on breast cancer cells, improving its efficacy (104). Down-regulation of ACLY promotes apoptosis in ovarian cancer cells while attenuating cisplatin resistance (105). Increased levels of FASN and ACLY contribute to cancer stem cell-like properties, self-renewal induction, cellular steatosis, affecting HCC progression (106, 107). Additionally, ACLY has emerged as a potential biomarker for predicting breast cancer recurrence (150).
ACC plays a crucial role in de novo lipogenesis and its inhibition can hinder tumor nutrient supply and development, making it a potential target for cancer treatment. It has been reported that FASN utilizes ACC for de novo lipogenesis (151). The regulation of de novo lipogenesis affects the tumor’s energy supply and is divided into two phenotypes: ACC1 (ACCα or ACACA) and ACC2 (ACCβ or ACACB) (152). ACC1 is highly enriched in lipid tissues, while ACC2 occurs in oxidized tissues and has distinct metabolic effects due to their different locations (153). Malonyl-CoA produced by ACC1 serves as a substrate for lipogenesis, whereas malonyl-CoA produced by ACC2 inhibits CPT1, thereby preventing FA degradation. Upregulation of ACC1 has been observed in various tumors, likely promoting lipogenesis to meet the demands of rapid growth and proliferation (70, 154, 155). Therefore, it can be speculated that inhibiting ACC expression can hinder tumor nutrient supply and tumor development. Knockdown of SREBP1-associated genes such as ACLY and both isoforms of ACC in NSCLC cell lines promote cell apoptosis and differentiation (110, 111). Moreover, reduced phosphorylation of ACC was found to increase HCC genesis in mice and the proliferation of liver cancer cells. Inhibitors targeting allosteric sites on ACC can alleviate HCC deterioration and improve survival rate (112). Other studies have highlighted the significance of ACC and FASN expression in hormone resistance and cancer prevention (113).
PPARα plays a crucial role in lipid metabolism and is highly expressed in organs with significant FA catabolism, such as the liver (156). It acts as key regulator of lipid and glucose metabolism, controlling FA catabolism and lipoprotein metabolism (157). As a ligand-activated transcription factor, PPAR is involved in cellular processes like cell differentiation, proliferation, and apoptosis (158–160). When activated, PPARα exhibits antiangiogenic and anti-inflammatory effects, thereby inhibiting tumor development (161). PPARα agonists like fenofibrate and WY-14,643 have been found to inhibit tumor growth in multiple cancer studies and are potential targets for treating various malignancies (38). Knockdown of PPARα has been associated with reduced breast cancer-specific survival (114). The chemical sensitivity of breast cancer cells to the PPARα agonist clofibrate was high. Clofibrate significantly affected the PPARα DNA-binding activity and free FA production while effectively reducing levels of SREBP-1c and FASN. It targeted lipogenic and inflammatory pathways in invasive breast cancer (162). The PPARα agonist clofibrate induced apoptosis in HepG2 cells in both time-dependent and concentration-dependent manners (120).
Interestingly, PPARα regulates FAO activity to meet the higher energy requirements of high-grade renal cell carcinoma (RCC) compared with low-grade RCC (163). Additionally, in PAAD and CRC, the PPARα signaling pathway ensures high lipid turnover in cancer stem cells, maintaining their high energy requirements and self-renewal (164). The levels of PPARα protein were associated with RCC invasiveness in two renal cell carcinoma cell lines (AKI-1 and 786-O) (116). Furthermore, loss of PPARα expression in host animals inhibited tumor growth in lung cancer cells according to a study using a mouse xenograft model (117). Studies have demonstrated that regulating the PPAR/NF-κB signaling pathway can promote multi-organ distant metastasis of NSCLC (115). Mice lacking PPARα showed resistance to increased DNA synthesis and liver tumorigenesis induced by the agonist WY-14, 643, further supporting the involvement of PPARα in HCC (165). In breast cancer stem cells, the PPARα antagonist GW6471 has anti-proliferation and pro-apoptotic effects while the PPARα agonist Wy14643 promotes clonal expansion through NF-κB/IL-6 axis signaling activity promotion (118, 119). Clofibrate was also found to promote OV and PRAD progression without correlation with PPARα (166). Therefore, it is evident that the influence of PPARα on tumor progression varies depending on tissue type and difference in its ligand. This highlights the potential for developing specific synthetic ligand targeting PPARα as a novel approach for cancer treatment.
Circadian rhythm disorders are independent risk factors for cancer, as disruption of the circadian clock may be associated with cancer cell proliferation, senescence, metabolism and DNA damage (167–169). However, the exact molecular mechanisms underlying this effect have yet to be elucidated. Studies have shown that disturbance or dysregulation of circadian rhythm is associated with poor prognosis of various tumors. CLOCK genes such as BMAL1 and REV-ERB mainly play an anticancer role while DEC has both anticancer and cancer-promoting effects in relevant literature (Table 2).
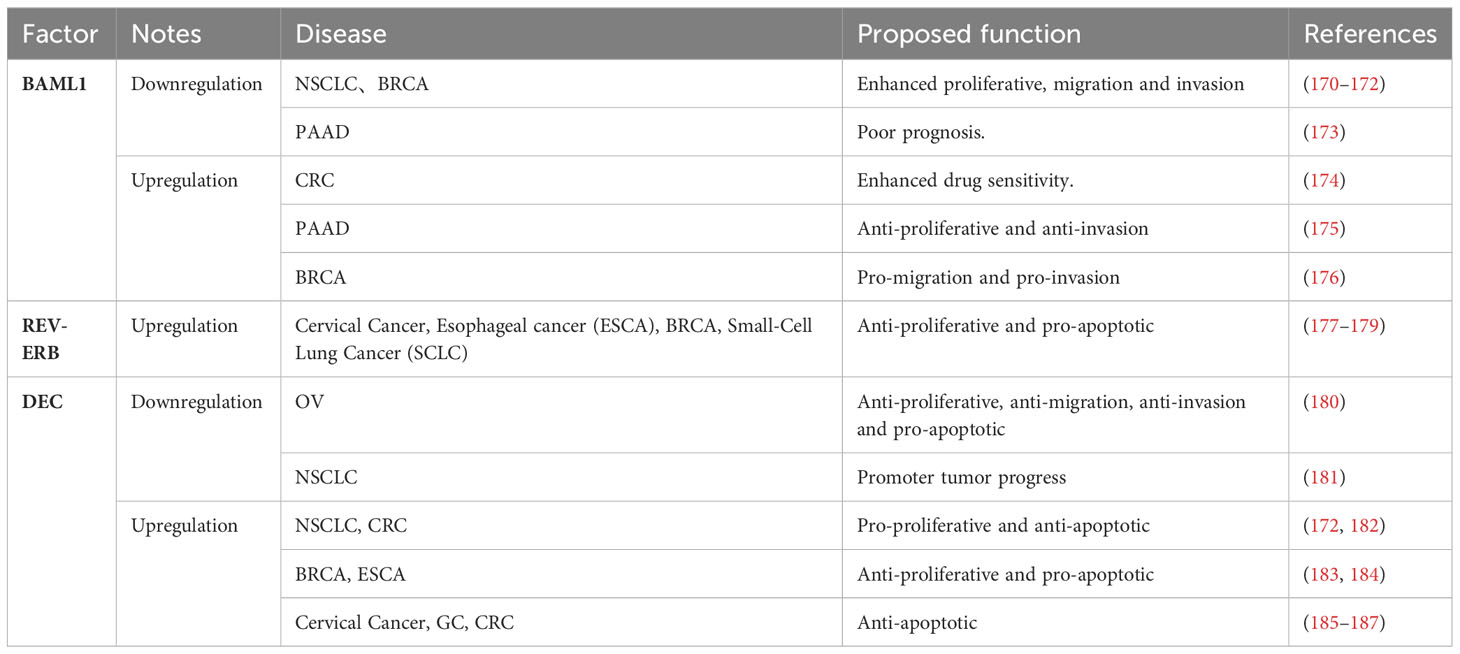
Table 2 Association of circadian rhythm genes with cancer development.
The circadian clock is composed of rhythmic genes and gene products that regulate the expression of clock control genes, generating a distinct circadian rhythm. BMAL1 serves as a crucial transcription factor within the transcription-translation feedback loop of the circadian clock. Its functions are associated with cellular processes such as metabolism, proliferation, metastasis, and cell cycle regulation. Additionally, it plays a significant role in modulating oncogenes and tumor suppressor genes (175, 188). Analysis conducted on GSE39582, GSE21510 and Cancer Genome Atlas (TCGA) pan-cancer datasets revealed notable down-regulation of BMAL1 expression in BRCA and CRC tumor samples when compared to normal tissues. However, in tumors exhibiting TGFβ activation or BRAF mutations, BMAL1 showed slight up-regulation (189, 190). Knockdown of BMAL1 disrupts circadian rhythm and enhances migration or invasion in lung cancer, breast cancer, and glioma cells (170, 171). Loss of BMAL1 may contribute to lung cancer development by activating KRAS and cancer-regulating genes such as P53 and c-Myc (172). Overexpression of BMAL1 inhibits proliferation and increases sensitivity to oxaliplatin in CRC cell lines and HCT116 cell models in vivo (174). Immunohistochemical analysis reveals that low Bmal1 expression in tumor tissues significantly impact tumor progression and prognosis compared to adjacent non-tumor tissues (173). In vitro experiments demonstrated that BMAL1 overexpression suppresses proliferation and invasion of pancreatic cancer cells through the activation of P53 pathway (175). Interestingly, on the contrary, BMAL1 overexpression promotes invasion and metastasis of breast cancer cells by upregulation matrix metalloproteinase 9 (MMP9), a mediator of local tumor invasion and distant metastasis (176). Additionally, CLOCK and BMAL1 overexpression can promote cancer cells growth by affecting F-actin formation (191), indicating that regulation of BMAL1 has diverse effects on tumor proliferation, invasion and metastasis across different oncogenic pathways.
The nuclear hormone receptors REV-ERBα and REV-ERBβ (REV-ERBs) are important components of the circadian clock (49, 50). Abnormal expression of REV-ERBs has been observed in various cancer types and is involved in tumor metabolism, proliferation, as well as regulation of plasma glucose levels, lipid and energy metabolism (192–195). Treatment with agonists SR9011 and SR1078 specifically enhances the function of REV-ERBα and RORα receptors in cells. After 72 hours of agonist treatment, cervical cancer and esophageal cancer cells exhibit dose-dependent decreases in proliferation accompanied by induced apoptosis. Moreover, these agonists have minimal impact on the viability of normal cells or tissues (177). In breast cancer cells, SR9011 remains unaffected by ER and HER2 expression while inhibiting the proliferation of breast cancer cell lines (178). The REV-ERB agonist SR9009 selectively induces cell death in both chemosensitive and chemoresistant SCLC cells. REV-ERBα demonstrates antitumor effects in SCLC cells (179). Activation of REV-ERBα eliminates oncogene-induced senescent cells, mediates chemotherapy resistance and relapse, thus making REV-ERBα agonists potential therapeutic options for different types of cancer (62).
The DEC family genes, including DEC1 and DEC2, are expressed by differentiated embryonic chondrocytes. Physiologically, DEC1 and DEC2 are both expressed in a circadian manner and are regulators of the mammalian circadian clock, forming the fifth clock gene family that regulates circadian and metabolic functions (19, 196, 197). Abnormal expression of DEC1 is associated with tumor development and invasiveness, making it a potential predictor for cancer prognosis after treatment (183, 198, 199). Dysregulated expression of DEC may alter normal circadian rhythms and significantly contribute to the development of various diseases, including cancer. It has been observed that DEC1 expression is increased in various cancers and promotes cell proliferation and survival, while DEC2 expression is low in lung cancer. The impact of DEC on tumor development varies depending on the specific cancer type (23, 172, 180–182, 185, 200). Furthermore, it has been found that the role of DEC1 in apoptosis may differ among different type of cancer tissues. DEC1 exhibits a pro-apoptotic effect in BRCA and ESCA (183, 184). But it has an anti-apoptotic effect in cervical cancer, GC, and colon cancer (185–187). Currently, the effect of DEC on tumor remains controversial; however, there is no doubt that alterations in the level of the DEC gene significantly influence tumor occurrence and progression.
Circadian clock genes have a dual role in cancer, acting as tumor suppressors in most cases but possibly serving as catalysts in specific cancer cells. The precise mechanisms and factors governing their roles as oncogenes or tumor suppressors are still unknown, posing both challenges and opportunities for future research.
Circadian clock disorders strongly affect tumor transformation and tumor growth by altering a variety of cancer regulatory pathways, such as lipid metabolism. Multiple organs of the human body are involved in lipid metabolism, and these organs are controlled by the circadian clock to regulate the body’s metabolic functions. In metabolic organs such as white adipose tissue, liver, gut, pancreas and muscle, the circadian clock drives the rhythmic expression of export genes involved in metabolism, biosynthesis, signal transduction, and cell-cycle pathways. It also coordinates glucose, lipid, and protein metabolism (201). Researchers have found a link between the circadian clock regulated by chromatin remodeling and cellular metabolism, suggesting that metabolic disorders in cancer may be the result of circadian clock disturbances (169, 202). Cancer cells use metabolic reprogramming to meet the energy requirements for rapid cell proliferation and survival, indicating metabolic plasticity. Mitochondria contain metabolic centers that catabolize fatty acids, amino acids, and glucose to provide energy for cell growth. Related studies have found that circadian gene expression and mitochondrial activity seem to interact and balance each other, but the detailed underlying mechanisms are still unclear. Previous studies have found a link between cancer cells’ metabolic disorders and disruptions to the circadian clock, which may play an important role in cancer progression (203).
50% of liver metabolites in mice are regulated by circadian rhythm (204). The circadian rhythms of 50% of metabolites in liver, muscle, adipose tissue, and serum were phase shifted in mice fed a high-fat diet (61, 205–207). The disruption of the circadian system leads to the dysregulation of chronic jet lag driver genes, resulting in hepatic metabolic dysfunction and changes in circulating energy consumption, lipid metabolism, insulin, and glucose signaling to promote the occurrence of liver cancer (204, 208). Systemic and liver-specific knockdown of BMAL1 can cause metabolic disorders, leading to hyperlipidemia and increased lipoprotein production (209). In untreated MDA-MB-231 cells and chronically insulin-treated MDA-MB-231 cells, BMAL1 knockdown inhibited the utilization of glutamine and FA by increasing their oxidation (189). Disruption of circadian clock genes can cause disorders of lipid metabolism. BMAL1 can play a role as a tumor suppressor in obese mice, inhibiting the growth of BRCA and lung metastases, and the down-regulation of BMAL1 was associated with an increased risk of breast cancer metastasis (189). It has been reported that BMAL1 can regulate metabolic reprogramming and affect the expression of PD-L1 in macrophages (210), suggesting that the circadian clock influences tumor development by regulating metabolic pathways.
REV-ERBα is a transcription factor that plays an important role in a series of physiological processes, including the regulation of glucose, lipid metabolism, and circadian rhythm, as a core inhibitory component of the cell autonomous clock and a regulator of metabolic genes (194, 211–213). REV-ERBα and β are present on several key lipid and bile acid regulatory genes, including Insig2 and SREBP, providing mechanisms for rhythmic lipid and bile acid metabolism molecules (50). Coordination of REV-ERBα and REV-ERBβ activities is required for normal hepatic clock gene expression and lipid metabolism (49). The hepatic circadian clock regulates the transcription function of the circadian transcription suppressor REV-ERBα, thereby controlling the production of SREBP-dependent fatty acids, cholesterol, and bile acids (53). REV-ERBs can also inhibit lipid producing enzymes including FASN and SCD1, and strictly control lipid metabolism (63). When comparing metabolic parameters between tamoxifen-treated controls and REV-ERBα and β double knockout animals, it was found that the double knockout mice showed increased circulating glucose and triglyceride levels and decreased free fatty acid levels (50). Cancer cells are highly dependent on de novo lipogenesis, which plays a central role in meeting the metabolic needs of cancer cells and is one of the important cancer markers involved in the anticancer activity of REV-ERB agonists (1). REV-ERB agonists SR9009 and SR9011 play a key role in regulating autophagy and de novo lipogenesis to induce an apoptotic response in malignant cells. REV-ERB agonists can act as novel inhibitors of de novo lipogenesis and have selective activity against malignant and benign tumors (62).
SIRT1 affects the circadian expression of core clock genes BMAL1 and ROR γ, which in turn is controlled by NAD-dependent mammalian sirtuins (214–217). The levels of NAD, NADP, NADH, and NADPH affect the ability of CLOCK-BMAL1 heterodimer to bind to E-box elements (218). Meanwhile, the circadian clock can regulate the rhythmic activity of niacinamide phosphoribosyl transferase (NAMPT), thus regulating the cyclic availability of NAD and forming a closed pathway (56, 57). SIRT1 can not only modify the expression rhythm of circadian clock genes (CLOCK and BMAL1) but also affect the expression rhythm of clock-controlled genes related to lipid metabolism (PPARα, SREBP-1c, ACC1, and FASN) in high-fat diet mice (219). SRT1720 (a chemical activator of SIRT1) inhibits the expression of SREBP target genes, and orthologs of SIRT1 inhibit lipid synthesis and fat storage by downregulating SREBP orthologs during fasting (220). SIRT1 has also been shown to alter cellular metabolism and responses to stress, thereby affecting the progression of direct transcription, apoptosis, autophagy, DNA damage repair, and senescence (221–223). SIRT1 overexpression has been found in BRCA, PRAD, GC, CRC, and liver cancer (224–228). Significant upregulation of SIRT1 promotes tumor proliferation, migration, and invasion by targeting SREBP1 and lipogenesis in EC (229). SIRT1 expression has been found to be significantly associated with distant metastatic recurrence and reduced survival in BRCA (224). Down-regulation of SIRT1 can continuously inhibit the proliferation of HCC and prostate cancer cells by inducing senescence or apoptosis (226). SIRT1 can inhibit PRCA invasion and enhance chemical sensitivity (230).
Through the above studies, it has been found that SIRT1 functions as an oncogene, and the inhibition of SIRT1 expression can inhibit tumor development. However, some studies have found that SIRT1 acts as a tumor suppressor gene in cancer tissues. After the downregulation of NAMPT expression, the activity of SIRT1 deacetylase significantly decreased, and the gene expression of two key lipid factors, FASN and SREBP1c, significantly increased, promoting the accumulation of triglycerides in HepG2 cells (231). SIRT1 mRNA was found to be down-regulated in GC (232), which is significantly related to shortened overall survival and relapse-free survival in gastric cancer (233). SIRT1 depletion enhanced proliferation and metastasis, promoting the growth of GC. SIRT1 may play a role as a tumor suppressor (228). However, SIRT1 overexpression inhibits lipid metabolism in prostate cancer cells by activating AMPK phosphorylation and inhibiting SREBP1 expression and nuclear translocation. Additionally, astragalus polysaccharide has been found to inhibit tumor progression and lipid metabolism by regulating the miR-138-5p/SIRT1/SREBP1 pathway (234). The present results suggest that SIRT1 has dual roles as a tumor promoter and tumor suppressor (235).
PPAR is involved in circadian clock control independently of the suprachiasmatic nucleus (236). CLOCK and BMAL1 play important roles in lipid homeostasis by regulating the circadian activation of controlled target genes of potential PPAR response elements (237). PPARα deficiency disrupts the normal circadian regulation of certain SREBP-sensitive genes in the liver (238). Furthermore, studies have shown that the PPARα-SCD1 axis promotes CSC properties in HCC cells. Inhibition of the PPARα pathway or SCD1 decreases the expression of CSC-related markers, leading to the loss of CSC properties (239).
From the above studies, it is reasonable to speculate that the circadian clock may be related to increased de novo fatty acid synthesis in tumors, and tumor-dependent metabolites may be secreted in a temporal manner, which indicates that targeted pharmacological studies can be conducted on the daily peak of fatty acid metabolism pathway. Although the specific mechanism of the occurrence and development of cancer is still unknown, a large number of studies have been conducted on the related role of circadian clock and lipid metabolism in the process of tumor development. How circadian clock affects tumors by regulating lipid metabolism has been explored, but still needs to be further studied. Collectively, there is a key signaling axis involved that coordinates the central pacemaker and peripheral circadian transcription with lipid metabolism, although the implications of these findings for humans remain unclarified.
In the reprogramming of lipid metabolism in cancer cells, endogenous FA are usually up-regulated and are essential for maintaining cell proliferation, division, and ATP energy (240). FA are substrates for producing lipid signaling molecules, and the mutual adjustment between lipid metabolic factors and oncogenic signals affects tumor proliferation, migration, and apoptosis. There is a close relation between the abnormal increase of de novo fatty acid synthesis and the growth and differentiation of cancer cells. SREBP1, ACLY, ACC, FASN, SCD and PPARα have been widely studied as key lipid metabolic factors. SREBP regulates lipid homeostasis by controlling the expression of a series of enzymes required for the synthesis of endogenous FA, cholesterol, triacylglycerol, and phospholipids. SREBP inhibitors reduce cell membrane fluidity, which leads to decreased tyrosine phosphorylation of EGFR and enhances the sensitivity of gefitinib in lung cancer cells (79). ACLY provides energy for cancer cells to function properly during catabolism and biosynthesis. ACLY overexpression in a variety of cancers indirectly destroys citrate binding by altering the citrate binding site of the enzyme, which is one of the options for cancer treatment. The ACLY inhibitor SB-204990 was found to inhibit the growth of tumors, such as lung cancer and PRAD (241). In addition, the functional polymorphism of ACLY can also be used as a prognostic marker to predict the recurrence of CRC (242). ACC is a key enzyme in the process of tumor lipid metabolism. It was found that ACC inhibitor ND-654 inhibited the proliferation and differentiation of liver cancer cells by inhibiting the production of nascent FA (112). Preclinical animal studies have shown that ACC inhibitors ND-646 and ND-654 significantly inhibit the growth of mouse lung tumors and rat HCC, respectively (110, 112). Additionally, ND-646 also inhibited the growth of NSCLC (243). TVB2640, a FASN inhibitor, has been observed to have a significant inhibitory effect on tumor growth in cancer cell lines and xenograft models, but due to its pharmacological nature, its clinical transformation and application are limited (244). In ovarian cancer models, the SCD1 inhibitor BZ36 reduces SCD1 expression and increases cancer cell sensitivity to ferroptosis inducers, thereby inducing tumor cell apoptosis (245). One study found that activators of PPARα could be used to prevent or treat CRC (246). In a variety of cancer types, a considerable number of drugs that can inhibit the synthesis of SREBP, ACLY, ACC, FASN, SCD, and PPARα have been tested for anticancer effects in preclinical and clinical studies. Targeting lipid metabolism has been confirmed to have positive anti-tumor effects, which may become a new therapeutic strategy for cancer.
The circadian clock regulates the absorption, distribution, metabolism, and elimination of drugs (247). Circadian timing of drugs may be an important parameter in the treatment of diseases, therefore, chronotherapy has received a lot of attention. Chronotherapy is a strategy that uses the natural rhythms and cycles of the physiological and biochemical processes of an organism to treat disease (248–250). Prior to the discovery of a more detailed mechanism of the core clock, therapeutic strategies to reduce toxic side effects and improve efficacy during cancer treatment by controlling the duration of treatment have been used in clinical practice (251–253). It has also been found through mechanistic studies that many anticancer drugs have been shown to increase cytotoxicity at specific stages of cell division (247, 254). Chronotherapy was also found to improve survival and quality of life in cancer patients by minimizing the cytotoxicity of anticancer drugs (255–257). This suggests that optimizing the timing of treatment administration by predicting the drug properties associated with circadian rhythms can translate into desired clinical outcomes. It has also been shown that for anticancer drugs that are limited by their ability to cause side effects on healthy host tissues, regulating the timing of administration in accordance with their circadian characteristics not only helps to produce beneficial effects, but also avoids adverse effects (247, 258, 259). Multiple phase I to III clinical trials have demonstrated the effectiveness of chronotherapy in various tumors (260, 261). More than 30 chemotherapeutic drugs were found to differ in efficacy by more than 50% due to the different time of administration (259). One study, which included 186 cases of patients with metastatic colorectal cancer in a randomized multicenter phase III trial, found that compared with the constant rate of infusion, adjusting the delivery time of oxaliplatin, 5-fluorouracil (5-FU), and folic acid reduced the incidence of severe mucosal toxicity by about 5 times. At the same time, the rate of peripheral nerve lesions caused by dysfunction decreased by about 50% (251). Additionally, it was found that the maximum plasma concentration and optimal tolerance time were at 4:00 a.m. in cancer patients treated with 5-FU for 5 days (262, 263). In addition, timed irinotecan therapy in 31 cancer patients showed that time-mediated irinotecan infusion caused less diarrhea and patient-to-patient variability than the traditional 30-minute morning infusion (264). In a study of 41 patients with NSCLC, lower gastrointestinal toxicity was observed in patients treated with cisplatin at 6 p.m. or 6 a.m. compared with conventional chemotherapy, and a higher clearance rate was observed in patients treated with cisplatin at 6 p.m (265).. The REV-ERBs agonists SR9009 and SR9011 have been reported to have anticancer activity in different tumor types, including leukemia, brain, colon, breast, and melanoma, without significant side effects on normal cells or tissues (62, 178, 179, 266). In addition, autophagy and de novo lipogenesis were identified as key events in initiating apoptotic responses in malignant cells treated with SR9009 and SR9011 (62). SR9011 and SR9009 also reduce the proliferation of glioblastoma stem cells by inhibiting the expression of TCA cycle enzymes and inhibiting autophagy, and are lethal to chemoresistant small cell lung cancer cells (179, 267). However, paradoxically, the study found that SR9009 can exert an effect on cell proliferation independently of REV-ERB proteins, questioning the role of REV-ERB in the action of the drug specifically in cancer treatment (268).
A comparison of the morning and evening doses of cisplatin in patients with prostate, breast, cervical and ovarian cancer showed significant differences in the efficacy, indicating that chronotherapy can reduce the toxicity ratio of cisplatin treatment and enhance the efficacy (269). In addition, other chemotherapeutic agents showed optimal timing of administration to improve outcomes in bladder, colorectal, endometrial, and renal cancers (270–273). These studies found that the activity of several anticancer drugs may be limited by their side effects and toxicity on healthy cells, thus proving that temporal therapy offers the potential to optimize drug dosage and treatment duration to effectively eliminate cancer cells and reduce adverse effects, thereby preventing early drug resistance. Therefore, chronotherapy aims to optimize drug administration time to improve drug efficacy and safety without increasing drug dosage or changing drug type, maximize the antitumor effect of cancer chemotherapy by minimizing toxicity and adverse side effects, and increase tolerance to improve survival of patients.
The data reviewed here demonstrate an overlap between the circadian clock and de novo FA synthesis in tumor lipid metabolism. Disorders of circadian clock can affect de novo FA synthesis, while dysregulation of this process can affect tumor development. The interaction between circadian clock and lipid metabolism plays a crucial role in tumor occurrence and progression, representing a potential mechanism influencing tumor development. However, several questions remain unanswered regarding the role of lipid metabolism in carcinogenesis, including the possible beneficial or harmful effects on cancer development that hinder the development of new therapeutic strategies. Our review establishes a connection between a disrupted circadian clock and key regulators of dysregulated lipid metabolism, as well as the molecular mechanisms underlying their effects on cancer. Nevertheless, to date, research has primarily focused on how the circadian clock regulates transcription factors involved in novo FA synthesis without thoroughly investigating key pathways through which both systems jointly influence tumor development. Although current studies have shown a strong association between circadian disruption, dysregulation of lipid metabolism, and cancer progression, there is still no systematic establishment of the mechanisms linking these factors together; thus future clinical and research efforts are necessary in this area. Future hypothesis-oriented studies should concentrate on specific interactions between circadian clock genes and lipid metabolism mechanisms to fully realize the clinical potential of their link in tumors.
To gain a deeper comprehension of how specific cancer genes obliterate tumor cells, targeting lipid metabolism and integrating it with the biological clock may offer a potent tool for optimizing cancer treatment. This approach also aims to enhance patients’ quality of life and survival rates by individualizing treatment time.
ML: Writing – original draft. ZZ: Writing – original draft. YC: Software, Writing – original draft. TF: Methodology, Writing – original draft. QZ: Writing – review & editing. XT: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors gratefully acknowledge the financial support from the Key projects supported by, National Natural Sciences Foundation of China (82074450), Key scientific research project of Hunan Education Department (grant number 21A0243), Key project of academician workstation guidance project (grant number 21YS003), National Natural Science Foundation of China (U20A20408).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
2. Ward. EM, Germolec. D, Kogevinas. M, McCormick. D, Vermeulen. R, Anisimov. VN, et al. Carcinogenicity of night shift work. Lancet Oncol (2019) 20(8):1058–9. doi: 10.1016/S1470-2045(19)30455-3
3. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9
4. Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci (2012) 35:445–62. doi: 10.1146/annurev-neuro-060909-153128
5. Zmrzljak UP, Rozman D. Circadian regulation of the hepatic endobiotic and xenobitoic detoxification pathways: the time matters. Chem Res Toxicol (2012) 25(4):811–24. doi: 10.1021/tx200538r
6. Buhr ED, Yoo SH, Takahashi JS. Temperature as a universal resetting cue for mammalian circadian oscillators. Sci (New York NY) (2010) 330(6002):379–85. doi: 10.1126/science.1195262
7. Challet E. The circadian regulation of food intake. Nat Rev Endocrinol (2019) 15(7):393–405. doi: 10.1038/s41574-019-0210-x
8. Chaix A, Lin T, Le HD, Chang MW, Panda S. Time-restricted feeding prevents obesity and metabolic syndrome in mice lacking a circadian clock. Cell Metab (2019) 29(2):303–19.e304. doi: 10.1016/j.cmet.2018.08.004
9. Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet (2017) 18(3):164–79. doi: 10.1038/nrg.2016.150
10. Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Sci (New York NY) (1998) 280(5369):1564–9. doi: 10.1126/science.280.5369.1564
11. Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, et al. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell (1999) 98(2):193–205. doi: 10.1016/S0092-8674(00)81014-4
12. Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B, et al. Interacting molecular loops in the mammalian circadian clock. Sci (New York NY) (2000) 288(5468):1013–9. doi: 10.1126/science.288.5468.1013
13. Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol (2007) 8(2):139–48. doi: 10.1038/nrm2106
14. Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell (2001) 107(7):855–67. doi: 10.1016/S0092-8674(01)00610-9
15. Lowrey PL, Takahashi JS. Genetics of circadian rhythms in Mammalian model organisms. Adv Genet (2011) 74:175–230. doi: 10.1016/B978-0-12-387690-4.00006-4
16. Preußner M, Heyd F. Post-transcriptional control of the mammalian circadian clock: implications for health and disease. Pflugers Archiv Eur J Physiol (2016) 468(6):983–91. doi: 10.1007/s00424-016-1820-y
17. Siepka SM, Yoo SH, Park J, Song W, Kumar V, Hu Y, et al. Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell (2007) 129(5):1011–23. doi: 10.1016/j.cell.2007.04.030
18. Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, et al. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell (2002) 110(2):251–60. doi: 10.1016/S0092-8674(02)00825-5
19. Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M, et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature (2002) 419(6909):841–4. doi: 10.1038/nature01123
20. Sato F, Kawamoto T, Fujimoto K, Noshiro M, Honda KK, Honma S, et al. Functional analysis of the basic helix-loop-helix transcription factor DEC1 in circadian regulation. Interaction with BMAL1. Eur J Biochem (2004) 271(22):4409–19. doi: 10.1111/j.1432-1033.2004.04379.x
21. Kawamoto T, Noshiro M, Sato F, Maemura K, Takeda N, Nagai R, et al. A novel autofeedback loop of Dec1 transcription involved in circadian rhythm regulation. Biochem Biophys Res Commun (2004) 313(1):117–24. doi: 10.1016/j.bbrc.2003.11.099
22. Hamaguchi H, Fujimoto K, Kawamoto T, Noshiro M, Maemura K, Takeda N, et al. Expression of the gene for Dec2, a basic helix-loop-helix transcription factor, is regulated by a molecular clock system. Biochem J (2004) 382(Pt 1):43–50. doi: 10.1042/BJ20031760
23. Li Y, Xie M, Song X, Gragen S, Sachdeva K, Wan Y, et al. DEC1 negatively regulates the expression of DEC2 through binding to the E-box in the proximal promoter. J Biol Chem (2003) 278(19):16899–907. doi: 10.1074/jbc.M300596200
24. St-Pierre B, Flock G, Zacksenhaus E, Egan SE. Stra13 homodimers repress transcription through class B E-box elements. J Biol Chem (2002) 277(48):46544–51. doi: 10.1074/jbc.M111652200
25. Korshunov DA, Kondakova IV, Shashova EE. Modern perspective on metabolic reprogramming in Malignant neoplasms. Biochem Biokhimiia (2019) 84(10):1129–42. doi: 10.1134/S000629791910002X
26. Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res (2012) 72(15):3709–14. doi: 10.1158/0008-5472.CAN-11-4112.
27. Hall Z, Ament Z, Wilson CH, Burkhart DL, Ashmore T, Koulman A, et al. Myc expression drives aberrant lipid metabolism in lung cancer. Cancer Res (2016) 76(16):4608–18. doi: 10.1158/0008-5472.CAN-15-3403.
28. Louie SM, Roberts LS, Mulvihill MM, Luo K, Nomura DK. Cancer cells incorporate and remodel exogenous palmitate into structural and oncogenic signaling lipids. Biochim Biophys Acta (2013) 1831(10):1566–72. doi: 10.1016/j.bbalip.2013.07.008.
29. Damiano F, Gnoni GV, Siculella L. Functional analysis of rat liver citrate carrier promoter: differential responsiveness to polyunsaturated fatty acids. Biochem J (2009) 417(2):561–71. doi: 10.1042/BJ20081082.
30. Budhu A, Roessler S, Zhao X, Yu Z, Forgues M, Ji J, et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology (2013) 144(5):1066–75.e1061. doi: 10.1053/j.gastro.2013.01.054.
31. Shimomura I, Shimano H, Korn BS, Bashmakov Y, Horton JD. Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. J Biol Chem (1998) 273(52):35299–306. doi: 10.1074/jbc.273.52.35299
32. Sangineto M, Villani R, Cavallone F, Romano A, Loizzi D, Serviddio G. Lipid metabolism in development and progression of hepatocellular carcinoma. Cancers (2020) 12(6):1419. doi: 10.3390/cancers12061419
33. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell (1997) 89(3):331–40. doi: 10.1016/S0092-8674(00)80213-5
34. Hua X, Wu J, Goldstein JL, Brown MS, Hobbs HH. Structure of the human gene encoding sterol regulatory element binding protein-1 (SREBF1) and localization of SREBF1 and SREBF2 to chromosomes 17p11.2 and 22q13. Genomics (1995) 25(3):667–73. doi: 10.1016/0888-7543(95)80009-B
35. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest (2002) 109(9):1125–31. doi: 10.1172/JCI0215593
36. Goldstein JL, Rawson RB, Brown MS. Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch Biochem biophysics (2002) 397(2):139–48. doi: 10.1006/abbi.2001.2615
37. Wahli W, Braissant O, Desvergne B. Peroxisome proliferator activated receptors: transcriptional regulators of adipogenesis, lipid metabolism and more. Chem Biol (1995) 2(5):261–6. doi: 10.1016/1074-5521(95)90045-4
38. Rakhshandehroo M, Knoch B, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res (2010) 2010:612089. doi: 10.1155/2010/612089
39. Pahan K. Lipid-lowering drugs. Cell Mol Life Sci (2006) 63(10):1165–78. doi: 10.1007/s00018-005-5406-7
40. Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A (1994) 91(15):7355–9. doi: 10.1073/pnas.91.15.7355
41. Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest (2006) 116(3):571–80. doi: 10.1172/JCI27989
42. Watts GF, Dimmitt SB. Fibrates, dyslipoproteinaemia and cardiovascular disease. Curr Opin lipidology (1999) 10(6):561–74. doi: 10.1097/00041433-199912000-00011
43. Ozasa H, Miyazawa S, Osumi T, Hashimoto T. Induction of peroxisomal beta-oxidation enzymes in primary cultured rat hepatocytes by clofibric acid. J Biochem (1985) 97(5):1273–8. doi: 10.1093/oxfordjournals.jbchem.a135178
44. König B, Koch A, Spielmann J, Hilgenfeld C, Hirche F, Stangl GI, et al. Activation of PPARalpha and PPARgamma reduces triacylglycerol synthesis in rat hepatoma cells by reduction of nuclear SREBP-1. Eur J Pharmacol (2009) 605(1-3):23–30. doi: 10.1016/j.ejphar.2009.01.009
45. Lin J, Yang R, Tarr PT, Wu PH, Handschin C, Li S, et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell (2005) 120(2):261–73. doi: 10.1016/j.cell.2004.11.043
46. Froy O. The relationship between nutrition and circadian rhythms in mammals. Front Neuroendocrinol (2007) 28(2-3):61–71. doi: 10.1016/j.yfrne.2007.03.001
47. Kohsaka A, Bass J. A sense of time: how molecular clocks organize metabolism. Trends Endocrinol Metab (2007) 18(1):4–11. doi: 10.1016/j.tem.2006.11.005
48. Shostak A, Meyer-Kovac J, Oster H. Circadian regulation of lipid mobilization in white adipose tissues. Diabetes (2013) 62(7):2195–203. doi: 10.2337/db12-1449
49. Bugge A, Feng D, Everett LJ, Briggs ER, Mullican SE, Wang F, et al. Rev-erbα and Rev-erbβ coordinately protect the circadian clock and normal metabolic function. Genes Dev (2012) 26(7):657–67. doi: 10.1101/gad.186858.112
50. Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature (2012) 485(7396):123–7. doi: 10.1038/nature11048
51. Gooley JJ, Chua EC. Diurnal regulation of lipid metabolism and applications of circadian lipidomics. J Genet Genomics = Yi Chuan xue bao (2014) 41(5):231–50. doi: 10.1016/j.jgg.2014.04.001
52. Gilardi F, Migliavacca E, Naldi A, Baruchet M, Canella D, Le Martelot G, et al. Genome-wide analysis of SREBP1 activity around the clock reveals its combined dependency on nutrient and circadian signals. PLoS Genet (2014) 10(3):e1004155. doi: 10.1371/journal.pgen.1004155
53. Le Martelot G, Claudel T, Gatfield D, Schaad O, Kornmann B, Lo Sasso G, et al. REV-ERBalpha participates in circadian SREBP signaling and bile acid homeostasis. PLoS Biol (2009) 7(9):e1000181. doi: 10.1371/journal.pbio.1000181.
54. Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao J, Zang M, et al. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J Biol Chem (2010) 285(44):33959–70. doi: 10.1074/jbc.M110.122978
55. Hirano A, Braas D, Fu YH, Ptáček LJ. FAD regulates CRYPTOCHROME protein stability and circadian clock in mice. Cell Rep (2017) 19(2):255–66. doi: 10.1016/j.celrep.2017.03.041
56. Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Sci (New York NY) (2009) 324(5927):654–7. doi: 10.1126/science.1170803
57. Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Sci (New York NY) (2009) 324(5927):651–4. doi: 10.1126/science.1171641
58. Choi SM, Cho HJ, Cho H, Kim KH, Kim JB, Park H. Stra13/DEC1 and DEC2 inhibit sterol regulatory element binding protein-1c in a hypoxia-inducible factor-dependent mechanism. Nucleic Acids Res (2008) 36(20):6372–85. doi: 10.1093/nar/gkn620
59. Shen L, Cui A, Xue Y, Cui Y, Dong X, Gao Y, et al. Hepatic differentiated embryo-chondrocyte-expressed gene 1 (Dec1) inhibits sterol regulatory element-binding protein-1c (Srebp-1c) expression and alleviates fatty liver phenotype. J Biol Chem (2014) 289(34):23332–42. doi: 10.1074/jbc.M113.526343
60. Kudo T, Tamagawa T, Kawashima M, Mito N, Shibata S. Attenuating effect of clock mutation on triglyceride contents in the ICR mouse liver under a high-fat diet. J Biol rhythms (2007) 22(4):312–23. doi: 10.1177/0748730407302625
61. Kohsaka A, Laposky AD, Ramsey KM, Estrada C, Joshu C, Kobayashi Y, et al. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab (2007) 6(5):414–21. doi: 10.1016/j.cmet.2007.09.006
62. Sulli G, Rommel A, Wang X, Kolar MJ, Puca F, Saghatelian A, et al. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature (2018) 553(7688):351–5. doi: 10.1038/nature25170
63. Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature (2012) 485(7396):62–8. doi: 10.1038/nature11030
64. Duszka K, Wahli W. Peroxisome proliferator-activated receptors as molecular links between caloric restriction and circadian rhythm. Nutrients (2020) 12(11):3476. doi: 10.3390/nu12113476
65. Canaple L, Rambaud J, Dkhissi-Benyahya O, Rayet B, Tan NS, Michalik L, et al. Reciprocal regulation of brain and muscle Arnt-like protein 1 and peroxisome proliferator-activated receptor alpha defines a novel positive feedback loop in the rodent liver circadian clock. Mol Endocrinol (Baltimore Md) (2006) 20(8):1715–27. doi: 10.1210/me.2006-0052
66. Gervois P, Chopin-Delannoy S, Fadel A, Dubois G, Kosykh V, Fruchart JC, et al. Fibrates increase human REV-ERBalpha expression in liver via a novel peroxisome proliferator-activated receptor response element. Mol Endocrinol (Baltimore Md) (1999) 13(3):400–9. doi: 10.1210/mend.13.3.0248
67. Oishi K, Shirai H, Ishida N. CLOCK is involved in the circadian transactivation of peroxisome-proliferator-activated receptor alpha (PPARalpha) in mice. Biochem J (2005) 386(Pt 3):575–81. doi: 10.1042/BJ20041150
68. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab (2013) 18(2):153–61. doi: 10.1016/j.cmet.2013.05.017
69. Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discovery (2012) 2(10):881–98. doi: 10.1158/2159-8290.CD-12-0345
70. Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care (2006) 9(4):358–65. doi: 10.1097/01.mco.0000232894.28674.30
71. Vazquez-Martin A, Colomer R, Brunet J, Lupu R, Menendez JA. Overexpression of fatty acid synthase gene activates HER1/HER2 tyrosine kinase receptors in human breast epithelial cells. Cell proliferation (2008) 41(1):59–85. doi: 10.1111/j.1365-2184.2007.00498.x
72. Lee JH, Jeon YG, Lee KH, Lee HW, Park J, Jang H, et al. RNF20 suppresses tumorigenesis by inhibiting the SREBP1c-PTTG1 axis in kidney cancer. Mol Cell Biol (2017) 37(22):e00265-17. doi: 10.1128/MCB.00265-17
73. Nie LY, Lu QT, Li WH, Yang N, Dongol S, Zhang X, et al. Sterol regulatory element-binding protein 1 is required for ovarian tumor growth. Oncol Rep (2013) 30(3):1346–54. doi: 10.3892/or.2013.2575
74. Zhou C, Qian W, Li J, Ma J, Chen X, Jiang Z, et al. High glucose microenvironment accelerates tumor growth via SREBP1-autophagy axis in pancreatic cancer. J Exp Clin Cancer Res (2019) 38(1):302. doi: 10.1186/s13046-019-1288-7
75. Zhou C, Qian W, Ma J, Cheng L, Jiang Z, Yan B, et al. Resveratrol enhances the chemotherapeutic response and reverses the stemness induced by gemcitabine in pancreatic cancer cells via targeting SREBP1. Cell proliferation (2019) 52(1):e12514. doi: 10.1111/cpr.12514
76. Chen Z, Yu D, Owonikoko TK, Ramalingam SS, Sun SY. Induction of SREBP1 degradation coupled with suppression of SREBP1-mediated lipogenesis impacts the response of EGFR mutant NSCLC cells to osimertinib. Oncogene (2021) 40(49):6653–65. doi: 10.1038/s41388-021-02057-0
77. Xu C, Zhang L, Wang D, Jiang S, Cao D, Zhao Z, et al. Lipidomics reveals that sustained SREBP-1-dependent lipogenesis is a key mediator of gefitinib-acquired resistance in EGFR-mutant lung cancer. Cell Death Discovery (2021) 7(1):353. doi: 10.1038/s41420-021-00744-1
78. Pan Z, Wang K, Chen Q, Zheng X, Song Z, Ding X. SFI enhances therapeutic efficiency of gefitinib: an insight into reversal of resistance to targeted therapy in non-small cell lung cancer cells. J Cancer (2020) 11(2):334–44. doi: 10.7150/jca.32989
79. Li J, Yan H, Zhao L, Jia W, Yang H, Liu L, et al. Inhibition of SREBP increases gefitinib sensitivity in non-small cell lung cancer cells. Oncotarget (2016) 7(32):52392–403. doi: 10.18632/oncotarget.10721
80. Zhang N, Zhang H, Liu Y, Su P, Zhang J, Wang X, et al. SREBP1, targeted by miR-18a-5p, modulates epithelial-mesenchymal transition in breast cancer via forming a co-repressor complex with Snail and HDAC1/2. Cell Death differentiation (2019) 26(5):843–59. doi: 10.1038/s41418-018-0158-8
81. Lounis MA, Péant B, Leclerc-Desaulniers K, Ganguli D, Daneault C, Ruiz M, et al. Modulation of de novo lipogenesis improves response to enzalutamide treatment in prostate cancer. Cancers (2020) 12(11):3339. doi: 10.3390/cancers12113339
82. Zhang L, La X, Tian J, Li H, Li A, Liu Y, et al. The phytochemical vitexin and syringic acid derived from foxtail fillet bran inhibit breast cancer cells proliferation via GRP78/SREBP-1/SCD1 signaling axis. J Funct Foods (2021). 85:104620. doi: 10.1016/j.jff.2021.104620
83. Huang GM, Jiang QH, Cai C, Qu M, Shen W. SCD1 negatively regulates autophagy-induced cell death in human hepatocellular carcinoma through inactivation of the AMPK signaling pathway. Cancer Lett (2015) 358(2):180–90. doi: 10.1016/j.canlet.2014.12.036
84. Li W, Bai H, Liu S, Cao D, Wu H, Shen K, et al. Targeting stearoyl-CoA desaturase 1 to repress endometrial cancer progression. Oncotarget (2018) 9(15):12064–78. doi: 10.18632/oncotarget.24304
85. Chen J, Wu Z, Ding W, Xiao C, Zhang Y, Gao S, et al. SREBP1 siRNA enhance the docetaxel effect based on a bone-cancer dual-targeting biomimetic nanosystem against bone metastatic castration-resistant prostate cancer. Theranostics (2020) 10(4):1619–32. doi: 10.7150/thno.40489
86. Yin H, Qiu X, Shan Y, You B, Xie L, Zhang P, et al. HIF-1α downregulation of miR-433-3p in adipocyte-derived exosomes contributes to NPC progression via targeting SCD1. Cancer Sci (2021) 112(4):1457–70. doi: 10.1111/cas.14829
87. Wang H, Chen Y, Liu Y, Li Q, Luo J, Wang L, et al. The lncRNA ZFAS1 regulates lipogenesis in colorectal cancer by binding polyadenylate-binding protein 2 to stabilize SREBP1 mRNA. Mol Ther Nucleic Acids (2022) 27:363–74. doi: 10.1016/j.omtn.2021.12.010
88. Wang C, Shi M, Ji J, Cai Q, Zhao Q, Jiang J, et al. Stearoyl-CoA desaturase 1 (SCD1) facilitates the growth and anti-ferroptosis of gastric cancer cells and predicts poor prognosis of gastric cancer. Aging (2020) 12(15):15374–91. doi: 10.18632/aging.103598
89. Liu G, Kuang S, Cao R, Wang J, Peng Q, Sun C. Sorafenib kills liver cancer cells by disrupting SCD1-mediated synthesis of monounsaturated fatty acids via the ATP-AMPK-mTOR-SREBP1 signaling pathway. FASEB J (2019) 33(9):10089–103. doi: 10.1096/fj.201802619RR
90. Ma MKF, Lau EYT, Leung DHW, Lo J, Ho NPY, Cheng LKW, et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J Hepatol (2017) 67(5):979–90. doi: 10.1016/j.jhep.2017.06.015
91. He Y, Xu S, Qi Y, Tian J, Xu F. Long noncoding RNA SNHG25 promotes the Malignancy of endometrial cancer by sponging microRNA-497-5p and increasing FASN expression. J Ovarian Res (2021) 14(1):163. doi: 10.1186/s13048-021-00906-w
92. Wang X, Tian J, Zhao Q, Yang N, Ying P, Peng X, et al. Functional characterization of a low-frequency V1937I variant in FASN associated with susceptibility to esophageal squamous cell carcinoma. Arch Toxicol (2020) 94(6):2039–46. doi: 10.1007/s00204-020-02738-x
93. Sun L, Yao Y, Pan G, Zhan S, Shi W, Lu T, et al. Small interfering RNA-mediated knockdown of fatty acid synthase attenuates the proliferation and metastasis of human gastric cancer cells via the mTOR/Gli1 signaling pathway. Oncol Lett (2018) 16(1):594–602. doi: 10.3892/ol.2018.8648
94. Zhu M, Shi W, Chen K, Hu H, Ye X, Jiang Y. Pulsatilla saponin E suppresses viability, migration, invasion and promotes apoptosis of NSCLC cells through negatively regulating Akt/FASN pathway via inhibition of flotillin-2 in lipid raft. J receptor Signal transduction Res (2022) 42(1):23–33. doi: 10.1080/10799893.2020.1839764
95. Hsieh PF, Jiang WP, Basavaraj P, Huang SY, Ruangsai P, Wu JB, et al. Cell suspension culture extract of Eriobotrya japonica attenuates growth and induces apoptosis in prostate cancer cells via targeting SREBP-1/FASN-driven metabolism and AR. Phytomedicine (2021) 93:153806. doi: 10.1016/j.phymed.2021.153806
96. Hsieh PF, Jiang WP, Huang SY, Basavaraj P, Wu JB, Ho HY, et al. Emerging Therapeutic Activity of Davallia formosana on Prostate Cancer Cells through Coordinated Blockade of Lipogenesis and Androgen Receptor Expression. Cancers (2020) 12(4):914. doi: 10.3390/cancers12040914
97. Kwan HY, Yang Z, Fong WF, Hu YM, Yu ZL, Hsiao WL. The anticancer effect of oridonin is mediated by fatty acid synthase suppression in human colorectal cancer cells. J Gastroenterol (2013) 48(2):182–92. doi: 10.1007/s00535-012-0612-1
98. Gong J, Lin Y, Zhang H, Liu C, Cheng Z, Yang X, et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis (2020) 11(4):267. doi: 10.1038/s41419-020-2434-z
99. Chen J, Zhang F, Ren X, Wang Y, Huang W, Zhang J, et al. Targeting fatty acid synthase sensitizes human nasopharyngeal carcinoma cells to radiation via downregulating frizzled class receptor 10. Cancer Biol Med (2020) 17(3):740–52. doi: 10.20892/j.issn.2095-3941.2020.0219
100. Liu F, Wei J, Hao Y, Lan J, Li W, Weng J, et al. Long intergenic non-protein coding RNA 02570 promotes nasopharyngeal carcinoma progression by adsorbing microRNA miR-4649-3p thereby upregulating both sterol regulatory element binding protein 1, and fatty acid synthase. Bioengineered (2021) 12(1):7119–30. doi: 10.1080/21655979.2021.1979317
101. Zheng Y, Zhou Q, Zhao C, Li J, Yu Z, Zhu Q. ATP citrate lyase inhibitor triggers endoplasmic reticulum stress to induce hepatocellular carcinoma cell apoptosis via p-eIF2α/ATF4/CHOP axis. J Cell Mol Med (2021) 25(3):1468–79. doi: 10.1111/jcmm.16235
102. Tian H, Shi S, You B, Zhang Q, Gu M, You Y. ER resident protein 44 promotes Malignant phenotype in nasopharyngeal carcinoma through the interaction with ATP citrate lyase. J Trans Med (2021) 19(1):77. doi: 10.1186/s12967-020-02694-1
103. Gao Y, Islam MS, Tian J, Lui VW, Xiao D. Inactivation of ATP citrate lyase by Cucurbitacin B: A bioactive compound from cucumber, inhibits prostate cancer growth. Cancer Lett (2014) 349(1):15–25. doi: 10.1016/j.canlet.2014.03.015
104. Ismail A, Doghish AS, EME B, Salama SA, Mariee AD. Hydroxycitric acid potentiates the cytotoxic effect of tamoxifen in MCF-7 breast cancer cells through inhibition of ATP citrate lyase. Steroids (2020) 160:108656. doi: 10.1016/j.steroids.2020.108656
105. Wei X, Shi J, Lin Q, Ma X, Pang Y, Mao H, et al. Targeting ACLY attenuates tumor growth and acquired cisplatin resistance in ovarian cancer by inhibiting the PI3K-AKT pathway and activating the AMPK-ROS pathway. Front Oncol (2021) 11:642229. doi: 10.3389/fonc.2021.642229
106. Husain A, Chiu YT, Sze KM, Ho DW, Tsui YM, Suarez EMS, et al. Ephrin-A3/EphA2 axis regulates cellular metabolic plasticity to enhance cancer stemness in hypoxic hepatocellular carcinoma. J Hepatol (2022) 77(2):383–96. doi: 10.1016/j.jhep.2022.02.018
107. Yu W, Ma Y, Shrivastava SK, Srivastava RK, Shankar S. Chronic alcohol exposure induces hepatocyte damage by inducing oxidative stress, SATB2 and stem cell-like characteristics, and activating lipogenesis. J Cell Mol Med (2022) 26(7):2119–31. doi: 10.1111/jcmm.17235
108. Yang Y, Cai J, Yang X, Wang K, Sun K, Yang Z, et al. Dysregulated m6A modification promotes lipogenesis and development of non-alcoholic fatty liver disease and hepatocellular carcinoma. Mol Ther (2022) 30(6):2342–53. doi: 10.1016/j.ymthe.2022.02.021
109. Li L, Hua L, Fan H, He Y, Xu W, Zhang L, et al. Interplay of PKD3 with SREBP1 promotes cell growth via upregulating lipogenesis in prostate cancer cells. J Cancer (2019) 10(25):6395–404. doi: 10.7150/jca.31254
110. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med (2016) 22(10):1108–19. doi: 10.1038/nm.4181
111. Hanai J, Doro N, Sasaki AT, Kobayashi S, Cantley LC, Seth P, et al. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J Cell Physiol (2012) 227(4):1709–20. doi: 10.1002/jcp.22895
112. Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, et al. Inhibition of acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab (2019) 29(1):174–182.e175. doi: 10.1016/j.cmet.2018.08.020
113. Karantanos T, Karanika S, Wang J, Yang G, Dobashi M, Park S, et al. Caveolin-1 regulates hormone resistance through lipid synthesis, creating novel therapeutic opportunities for castration-resistant prostate cancer. Oncotarget (2016) 7(29):46321–34. doi: 10.18632/oncotarget.10113
114. Baker BG, Ball GR, Rakha EA, Nolan CC, Caldas C, Ellis IO, et al. Lack of expression of the proteins GMPR2 and PPARα are associated with the basal phenotype and patient outcome in breast cancer. Breast Cancer Res Treat (2013) 137(1):127–37. doi: 10.1007/s10549-012-2302-3
115. Chen J, Liu A, Lin Z, Wang B, Chai X, Chen S, et al. Downregulation of the circadian rhythm regulator HLF promotes multiple-organ distant metastases in non-small cell lung cancer through PPAR/NF-κb signaling. Cancer Lett (2020) 482:56–71. doi: 10.1016/j.canlet.2020.04.007
116. Abu Aboud O, Wettersten HI, Weiss RH. Inhibition of PPARα induces cell cycle arrest and apoptosis, and synergizes with glycolysis inhibition in kidney cancer cells. PLoS One (2013) 8(8):e71115. doi: 10.1371/journal.pone.0071115
117. Kaipainen A, Kieran MW, Huang S, Butterfield C, Bielenberg D, Mostoslavsky G, et al. PPARalpha deficiency in inflammatory cells suppresses tumor growth. PLoS One (2007) 2(2):e260. doi: 10.1371/journal.pone.0000260
118. Castelli V, Catanesi M, Alfonsetti M, Laezza C, Lombardi F, Cinque B, et al. PPARα-selective antagonist GW6471 inhibits cell growth in breast cancer stem cells inducing energy imbalance and metabolic stress. Biomedicines (2021) 9(2):127. doi: 10.3390/biomedicines9020127
119. Papi A, Guarnieri T, Storci G, Santini D, Ceccarelli C, Taffurelli M, et al. Nuclear receptors agonists exert opposing effects on the inflammation dependent survival of breast cancer stem cells. Cell Death differentiation (2012) 19(7):1208–19. doi: 10.1038/cdd.2011.207
120. Maggiora M, Oraldi M, Muzio G, Canuto RA. Involvement of PPARα and PPARγ in apoptosis and proliferation of human hepatocarcinoma HepG2 cells. Cell Biochem Funct (2010) 28(7):571–7. doi: 10.1002/cbf.1691
121. Edwards PA, Tabor D, Kast HR, Venkateswaran A. Regulation of gene expression by SREBP and SCAP. Biochim Biophys Acta (2000) 1529(1-3):103–13. doi: 10.1016/S1388-1981(00)00140-2
122. Li W, Tai Y, Zhou J, Gu W, Bai Z, Zhou T, et al. Repression of endometrial tumor growth by targeting SREBP1 and lipogenesis. Cell Cycle (Georgetown Tex) (2012) 11(12):2348–58. doi: 10.4161/cc.20811
123. Siqingaowa, Sekar S, Gopalakrishnan V, Taghibiglou C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem Biophys Res Commun (2017) 488(1):136–40. doi: 10.1016/j.bbrc.2017.05.023
124. Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther (2014) 13(4):855–66. doi: 10.1158/1535-7163.MCT-13-0797
125. Zhu T, Wang Z, Zou T, Xu L, Zhang S, Chen Y, et al. SOAT1 promotes gastric cancer lymph node metastasis through lipid synthesis. Front Pharmacol (2021) 12:769647. doi: 10.3389/fphar.2021.769647
126. Shen W, Xu T, Chen D, Tan X. Targeting SREBP1 chemosensitizes colorectal cancer cells to gemcitabine by caspase-7 upregulation. Bioengineered (2019) 10(1):459–68. doi: 10.1080/21655979.2019.1676485
127. Wen YA, Xiong X, Zaytseva YY, Napier DL, Vallee E, Li AT, et al. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis (2018) 9(3):265. doi: 10.1038/s41419-018-0330-6
128. Noto A, De Vitis C, Pisanu ME, Roscilli G, Ricci G, Catizone A, et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene (2017) 36(32):4573–84. doi: 10.1038/onc.2017.75
129. El Helou R, Pinna G, Cabaud O, Wicinski J, Bhajun R, Guyon L, et al. miR-600 Acts as a Bimodal Switch that Regulates Breast Cancer Stem Cell Fate through WNT Signaling. Cell Rep (2017) 18(9):2256–68. doi: 10.1016/j.celrep.2017.02.016
130. Igal RA. Stearoyl CoA desaturase-1: New insights into a central regulator of cancer metabolism. Biochim Biophys Acta (2016) 1861(12 Pt A):1865–80. doi: 10.1016/j.bbalip.2016.09.009
131. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, et al. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell (2017) 20(3):303–314.e305. doi: 10.1016/j.stem.2016.11.004
132. Angelucci C, Maulucci G, Colabianchi A, Iacopino F, D'Alessio A, Maiorana A, et al. Stearoyl-CoA desaturase 1 and paracrine diffusible signals have a major role in the promotion of breast cancer cell migration induced by cancer-associated fibroblasts. Br J Cancer (2015) 112(10):1675–86. doi: 10.1038/bjc.2015.135
133. Ariyama H, Kono N, Matsuda S, Inoue T, Arai H. Decrease in membrane phospholipid unsaturation induces unfolded protein response. J Biol Chem (2010) 285(29):22027–35. doi: 10.1074/jbc.M110.126870
134. Tracz-Gaszewska Z, Dobrzyn P. Stearoyl-CoA desaturase 1 as a therapeutic target for the treatment of cancer. Cancers (2019) 11(7):948. doi: 10.3390/cancers11070948
135. Holder AM, Gonzalez-Angulo AM, Chen H, Akcakanat A, Do KA, Fraser Symmans W, et al. High stearoyl-CoA desaturase 1 expression is associated with shorter survival in breast cancer patients. Breast Cancer Res Treat (2013) 137(1):319–27. doi: 10.1007/s10549-012-2354-4
136. Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer (2016) 16(11):732–49. doi: 10.1038/nrc.2016.89
137. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer (2007) 7(10):763–77. doi: 10.1038/nrc2222
138. Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutr (Burbank Los Angeles County Calif) (2000) 16(3):202–8. doi: 10.1016/S0899-9007(99)00266-X
139. Jiang W, Xing XL, Zhang C, Yi L, Xu W, Ou J, et al. MET and FASN as prognostic biomarkers of triple negative breast cancer: A systematic evidence landscape of clinical study. Front Oncol (2021) 11:604801. doi: 10.3389/fonc.2021.604801
140. Shurbaji MS, Kalbfleisch JH, Thurmond TS. Immunohistochemical detection of a fatty acid synthase (OA-519) as a predictor of progression of prostate cancer. Hum Pathol (1996) 27(9):917–21. doi: 10.1016/S0046-8177(96)90218-X
141. Rossi S, Graner E, Febbo P, Weinstein L, Bhattacharya N, Onody T, et al. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol Cancer Res (2003) 1(10):707–15.
142. Xu W, Hu X, Anwaier A, Wang J, Liu W, Tian X, et al. Fatty acid synthase correlates with prognosis-Related abdominal adipose distribution and metabolic disorders of clear cell renal cell carcinoma. Front Mol Biosci (2020) 7:610229. doi: 10.3389/fmolb.2020.610229
143. Jia J, Che L, Cigliano A, Wang X, Peitta G, Tao J, et al. Pivotal role of fatty acid synthase in c-MYC driven hepatocarcinogenesis. Int J Mol Sci (2020) 21(22):8467. doi: 10.3390/ijms21228467
144. Che L, Paliogiannis P, Cigliano A, Pilo MG, Chen X, Calvisi DF. Pathogenetic, prognostic, and therapeutic role of fatty acid synthase in human hepatocellular carcinoma. Front Oncol (2019) 9:1412. doi: 10.3389/fonc.2019.01412
145. Rae C, Fragkoulis GI, Chalmers AJ. Cytotoxicity and radiosensitizing activity of the fatty acid synthase inhibitor C75 is enhanced by blocking fatty acid uptake in prostate cancer cells. Adv Radiat Oncol (2020) 5(5):994–1005. doi: 10.1016/j.adro.2020.06.022
146. Papaevangelou E, Almeida GS, Box C, deSouza NM, Chung YL. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int J Cancer (2018) 143(4):992–1002. doi: 10.1002/ijc.31392
147. Singh M, Richards EG, Mukherjee A, Srere PA. Structure of ATP citrate lyase from rat liver. Physicochemical studies and proteolytic modification. J Biol Chem (1976) 251(17):5242–50.
148. Granchi C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J medicinal Chem (2018) 157:1276–91. doi: 10.1016/j.ejmech.2018.09.001
149. Chypre M, Zaidi N, Smans K. ATP-citrate lyase: a mini-review. Biochem Biophys Res Commun (2012) 422(1):1–4. doi: 10.1016/j.bbrc.2012.04.144
150. Chen Y, Li K, Gong D, Zhang J, Li Q, Zhao G, et al. ACLY: A biomarker of recurrence in breast cancer. Pathology Res Pract (2020) 216(9):153076. doi: 10.1016/j.prp.2020.153076
151. Abu-Elheiga L, Matzuk MM, Kordari P, Oh W, Shaikenov T, Gu Z, et al. Mutant mice lacking acetyl-CoA carboxylase 1 are embryonically lethal. Proc Natl Acad Sci U S A (2005) 102(34):12011–6. doi: 10.1073/pnas.0505714102
152. Tong L. Acetyl-coenzyme A carboxylase: crucial metabolic enzyme and attractive target for drug discovery. Cell Mol Life Sci (2005) 62(16):1784–803. doi: 10.1007/s00018-005-5121-4
153. Munday MR. Regulation of mammalian acetyl-CoA carboxylase. Biochem Soc Trans (2002) 30(Pt 6):1059–64. doi: 10.1042/bst0301059
154. Milgraum LZ, Witters LA, Pasternack GR, Kuhajda FP. Enzymes of the fatty acid synthesis pathway are highly expressed in in situ breast carcinoma. Clin Cancer Res (1997) 3(11):2115–20.
155. Yahagi N, Shimano H, Hasegawa K, Ohashi K, Matsuzaka T, Najima Y, et al. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur J Cancer (Oxford Engl 1990) (2005) 41(9):1316–22. doi: 10.1016/j.ejca.2004.12.037
156. Braissant O, Foufelle F, Scotto C, Dauça M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology (1996) 137(1):354–66. doi: 10.1210/endo.137.1.8536636
157. Almeida PE, Carneiro AB, Silva AR, Bozza PT. PPARγ Expression and function in mycobacterial infection: roles in lipid metabolism, immunity, and bacterial killing. PPAR Res (2012) 2012:383829. doi: 10.1155/2012/383829
158. Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer (2004) 4(1):61–70. doi: 10.1038/nrc1254
159. Michalik L, Wahli W. PPARs mediate lipid signaling in inflammation and cancer. PPAR Res (2008) 2008:134059. doi: 10.1155/2008/134059
160. Ward JE, Tan X. Peroxisome proliferator activated receptor ligands as regulators of airway inflammation and remodelling in chronic lung disease. PPAR Res (2007) 2007:14983. doi: 10.1155/2007/14983
161. Lakshmi SP, Reddy AT, Banno A, Reddy RC. PPAR agonists for the prevention and treatment of lung cancer. PPAR Res (2017) 2017:8252796. doi: 10.1155/2017/8252796
162. Chandran K, Goswami S, Sharma-Walia N. Implications of a peroxisome proliferator-activated receptor alpha (PPARα) ligand clofibrate in breast cancer. Oncotarget (2016) 7(13):15577–99. doi: 10.18632/oncotarget.6402
163. Perroud B, Ishimaru T, Borowsky AD, Weiss RH. Grade-dependent proteomics characterization of kidney cancer. Mol Cell Proteomics MCP (2009) 8(5):971–85. doi: 10.1074/mcp.M800252-MCP200
164. Kuramoto K, Yamamoto M, Suzuki S, Togashi K, Sanomachi T, Kitanaka C, et al. Inhibition of the lipid droplet-peroxisome proliferator-activated receptor α Axis suppresses cancer stem cell properties. Genes (2021) 12(1):99. doi: 10.3390/genes12010099
165. Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis (1997) 18(11):2029–33. doi: 10.1093/carcin/18.11.2029
166. Wang S, Hannafon BN, Zhou J, Ding WQ. Clofibrate induces heme oxygenase 1 expression through a PPARα-independent mechanism in human cancer cells. Cell Physiol Biochem (2013) 32(5):1255–64. doi: 10.1159/000354524
167. Fu L, Kettner NM. The circadian clock in cancer development and therapy. Prog Mol Biol Trans Sci (2013) 119:221–82. doi: 10.1016/B978-0-12-396971-2.00009-9
168. Kinouchi K, Sassone-Corsi P. Metabolic rivalry: circadian homeostasis and tumorigenesis. Nat Rev Cancer (2020) 20(11):645–61. doi: 10.1038/s41568-020-0291-9
169. Sahar S, Sassone-Corsi P. Metabolism and cancer: the circadian clock connection. Nat Rev Cancer (2009) 9(12):886–96. doi: 10.1038/nrc2747
170. Jung CH, Kim EM, Park JK, Hwang SG, Moon SK, Kim WJ, et al. Bmal1 suppresses cancer cell invasion by blocking the phosphoinositide 3-kinase-Akt-MMP-2 signaling pathway. Oncol Rep (2013) 29(6):2109–13. doi: 10.3892/or.2013.2381
171. Korkmaz T, Aygenli F, Emisoglu H, Ozcelik G, Canturk A, Yilmaz S, et al. Opposite carcinogenic effects of circadian clock gene BMAL1. Sci Rep (2018) 8(1):16023. doi: 10.1038/s41598-018-34433-4
172. Zhang H, Liu R, Zhang B, Huo H, Song Z. Advances in the study of circadian genes in non-small cell lung cancer. Integr Cancer therapies (2022) 21:15347354221096080. doi: 10.1177/15347354221096080
173. Li W, Liu L, Liu D, Jin S, Yang Y, Tang W, et al. Decreased circadian component Bmal1 predicts tumor progression and poor prognosis in human pancreatic ductal adenocarcinoma. Biochem Biophys Res Commun (2016) 472(1):156–62. doi: 10.1016/j.bbrc.2016.02.087
174. Zeng ZL, Luo HY, Yang J, Wu WJ, Chen DL, Huang P, et al. Overexpression of the circadian clock gene Bmal1 increases sensitivity to oxaliplatin in colorectal cancer. Clin Cancer Res (2014) 20(4):1042–52. doi: 10.1158/1078-0432.CCR-13-0171
175. Jiang W, Zhao S, Jiang X, Zhang E, Hu G, Hu B, et al. The circadian clock gene Bmal1 acts as a potential anti-oncogene in pancreatic cancer by activating the p53 tumor suppressor pathway. Cancer Lett (2016) 371(2):314–25. doi: 10.1016/j.canlet.2015.12.002
176. Wang J, Li S, Li X, Li B, Li Y, Xia K, et al. Circadian protein BMAL1 promotes breast cancer cell invasion and metastasis by up-regulating matrix metalloproteinase9 expression. Cancer Cell Int (2019) 19:182. doi: 10.1186/s12935-019-0902-2
177. van der Watt PJ, Roden LC, Davis KT, Parker MI, Leaner VD. Circadian oscillations persist in cervical and esophageal cancer cells displaying decreased expression of tumor-suppressing circadian clock genes. Mol Cancer Res MCR (2020) 18(9):1340–53. doi: 10.1158/1541-7786.MCR-19-1074
178. Wang Y, Kojetin D, Burris TP. Anti-proliferative actions of a synthetic REV-ERBα/β agonist in breast cancer cells. Biochem Pharmacol (2015) 96(4):315–22. doi: 10.1016/j.bcp.2015.06.010
179. Shen W, Zhang W, Ye W, Wang H, Zhang Q, Shen J, et al. SR9009 induces a REV-ERB dependent anti-small-cell lung cancer effect through inhibition of autophagy. Theranostics (2020) 10(10):4466–80. doi: 10.7150/thno.42478
180. Yi Y, Liao B, Zheng Z, Yang X, Yang Y, Zhou Y, et al. Downregulation of DEC1 inhibits proliferation, migration and invasion, and induces apoptosis in ovarian cancer cells via regulation of Wnt/β-catenin signaling pathway. Exp Ther Med (2021) 21(4):372. doi: 10.3892/etm.2021.9803
181. Liu Y, Wang L, Lin XY, Wang J, Yu JH, Miao Y, et al. The transcription factor DEC1 (BHLHE40/STRA13/SHARP-2) is negatively associated with TNM stage in non-small-cell lung cancer and inhibits the proliferation through cyclin D1 in A549 and BE1 cells. Tumour Biol (2013) 34(3):1641–50. doi: 10.1007/s13277-013-0697-z
182. Shan E, Huo Y, Wang H, Zhang Z, Hu J, Wang G, et al. Differentiated embryonic chondrocyte expressed gene-1 (DEC1) enhances the development of colorectal cancer with an involvement of the STAT3 signaling. Neoplasia (New York NY) (2022) 27:100783. doi: 10.1016/j.neo.2022.100783
183. Seino H, Wu Y, Morohashi S, Kawamoto T, Fujimoto K, Kato Y, et al. Basic helix-loop-helix transcription factor DEC1 regulates the cisplatin-induced apoptotic pathway of human esophageal cancer cells. Biomed Res (Tokyo Japan) (2015) 36(2):89–96. doi: 10.2220/biomedres.36.89
184. Wu Y, Sato F, Bhawal UK, Kawamoto T, Fujimoto K, Noshiro M, et al. Basic helix-loop-helix transcription factors DEC1 and DEC2 regulate the paclitaxel-induced apoptotic pathway of MCF-7 human breast cancer cells. Int J Mol Med (2011) 27(4):491–5. doi: 10.3892/ijmm.2011.617
185. Li Y, Zhang H, Xie M, Hu M, Ge S, Yang D, et al. Abundant expression of Dec1/stra13/sharp2 in colon carcinoma: its antagonizing role in serum deprivation-induced apoptosis and selective inhibition of procaspase activation. Biochem J (2002) 367(Pt 2):413–22. doi: 10.1042/bj20020514
186. Jia Y, Hu R, Li P, Zheng Y, Wang Y, Ma X. DEC1 is required for anti-apoptotic activity of gastric cancer cells under hypoxia by promoting Survivin expression. Gastric Cancer (2018) 21(4):632–42. doi: 10.1007/s10120-017-0780-z
187. Sato F, Bhawal UK, Sugiyama N, Osaki S, Oikawa K, Muragaki Y. Potential role of DEC1 in cervical cancer cells involving overexpression and apoptosis. Clocks sleep (2020) 2(1):26–38. doi: 10.3390/clockssleep2010004
188. Mullenders J, Fabius AW, Madiredjo M, Bernards R, Beijersbergen RL. A large scale shRNA barcode screen identifies the circadian clock component ARNTL as putative regulator of the p53 tumor suppressor pathway. PLoS One (2009) 4(3):e4798. doi: 10.1371/journal.pone.0004798
189. Ramos CA, Ouyang C, Qi Y, Chung Y, Cheng CT, LaBarge MA, et al. A non-canonical function of BMAL1 metabolically limits obesity-promoted triple-negative breast cancer. iScience (2020) 23(2):100839. doi: 10.1016/j.isci.2020.100839
190. Sahar N-e, Qadir J, Riaz SK, Sultan A, Arif A. Malik MFA: Dysregulation of core circadian genes, BMAL1 and CLOCK, in colorectal cancer. Biol Rhythm Res (2022) 53(9):1400–13. doi: 10.1080/09291016.2021.1940623
191. Ma TJ, Zhang ZW, Lu YL, Zhang YY, Tao DC, Liu YQ, et al. CLOCK and BMAL1 stabilize and activate RHOA to promote F-actin formation in cancer cells. Exp Mol Med (2018) 50(10):1–15. doi: 10.1038/s12276-018-0156-4
192. Everett LJ, Lazar MA. Nuclear receptor Rev-erbα: up, down, and all around. Trends Endocrinol metabolism: TEM (2014) 25(11):586–92. doi: 10.1016/j.tem.2014.06.011
193. Woldt E, Sebti Y, Solt LA, Duhem C, Lancel S, Eeckhoute J, et al. Rev-erb-α modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat Med (2013) 19(8):1039–46. doi: 10.1038/nm.3213
194. Zhang Y, Fang B, Emmett MJ, Damle M, Sun Z, Feng D, et al. GENE REGULATION. Discrete functions of nuclear receptor Rev-erbα couple metabolism to the clock. Sci (New York NY) (2015) 348(6242):1488–92. doi: 10.1126/science.aab3021
195. Kojetin DJ, Burris TP. REV-ERB and ROR nuclear receptors as drug targets. Nat Rev Drug Discovery (2014) 13(3):197–216. doi: 10.1038/nrd4100
196. Mao T, Xiong H, Hu X, Hu Y, Wang C, Yang L, et al. DEC1: a potential biomarker of Malignant transformation in oral leukoplakia. Braz Oral Res (2020) 34:e052. doi: 10.1590/1807-3107bor-2020.vol34.0052
197. Sato F, Kohsaka A, Bhawal UK, Muragaki Y. Potential roles of Dec and Bmal1 genes in interconnecting circadian clock and energy metabolism. Int J Mol Sci (2018) 19(3):781. doi: 10.3390/ijms19030781
198. Bi H, Li S, Qu X, Wang M, Bai X, Xu Z, et al. DEC1 regulates breast cancer cell proliferation by stabilizing cyclin E protein and delays the progression of cell cycle S phase. Cell Death Dis (2015) 6(9):e1891. doi: 10.1038/cddis.2015.247
199. Peng Y, Liu W, Xiong J, Gui HY, Feng XM, Chen RN, et al. Down regulation of differentiated embryonic chondrocytes 1 (DEC1) is involved in 8-methoxypsoralen-induced apoptosis in HepG2 cells. Toxicology (2012) 301(1-3):58–65. doi: 10.1016/j.tox.2012.06.022
200. Giatromanolaki A, Koukourakis MI, Sivridis E, Turley H, Wykoff CC, Gatter KC, et al. DEC1 (STRA13) protein expression relates to hypoxia- inducible factor 1-alpha and carbonic anhydrase-9 overexpression in non-small cell lung cancer. J Pathol (2003) 200(2):222–8. doi: 10.1002/path.1330
201. Sinturel F, Petrenko V, Dibner C. Circadian clocks make metabolism run. J Mol Biol (2020) 432(12):3680–99. doi: 10.1016/j.jmb.2020.01.018
202. Masri S, Sassone-Corsi P. The emerging link between cancer, metabolism, and circadian rhythms. Nat Med (2018) 24(12):1795–803. doi: 10.1038/s41591-018-0271-8
203. Altman BJ, Hsieh AL, Sengupta A, Krishnanaiah SY, Stine ZE, Walton ZE, et al. MYC disrupts the circadian clock and metabolism in cancer cells. Cell Metab (2015) 22(6):1009–19. doi: 10.1016/j.cmet.2015.09.003
204. Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, et al. Circadian homeostasis of liver metabolism suppresses hepatocarcinogenesis. Cancer Cell (2016) 30(6):909–24. doi: 10.1016/j.ccell.2016.10.007
205. Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, et al. Reprogramming of the circadian clock by nutritional challenge. Cell (2013) 155(7):1464–78. doi: 10.1016/j.cell.2013.11.034
206. Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab (2012) 15(6):848–60. doi: 10.1016/j.cmet.2012.04.019
207. Dyar KA, Lutter D, Artati A, Ceglia NJ, Liu Y, Armenta D, et al. Atlas of circadian metabolism reveals system-wide coordination and communication between clocks. Cell (2018) 174(6):1571–85.e1511. doi: 10.1016/j.cell.2018.08.042
208. Krishnaiah SY, Wu G, Altman BJ, Growe J, Rhoades SD, Coldren F, et al. Clock regulation of metabolites reveals coupling between transcription and metabolism. Cell Metab (2017) 25(4):961–74.e964. doi: 10.1016/j.cmet.2017.03.019
209. Pan X, Bradfield CA, Hussain MM. Global and hepatocyte-specific ablation of Bmal1 induces hyperlipidaemia and enhances atherosclerosis. Nat Commun (2016) 7:13011. doi: 10.1038/ncomms13011
210. Deng W, Zhu S, Zeng L, Liu J, Kang R, Yang M, et al. The circadian clock controls immune checkpoint pathway in sepsis. Cell Rep (2018) 24(2):366–78. doi: 10.1016/j.celrep.2018.06.026
211. Burris TP. Nuclear hormone receptors for heme: REV-ERBalpha and REV-ERBbeta are ligand-regulated components of the mammalian clock. Mol Endocrinol (Baltimore Md) (2008) 22(7):1509–20. doi: 10.1210/me.2007-0519
212. Duez H, Staels B. Rev-erb-alpha: an integrator of circadian rhythms and metabolism. J Appl Physiol (Bethesda Md 1985) (2009) 107(6):1972–80. doi: 10.1152/japplphysiol.00570.2009
213. Yin L, Wu N, Lazar MA. Nuclear receptor Rev-erbalpha: a heme receptor that coordinates circadian rhythm and metabolism. Nucl receptor Signaling (2010) 8:e001. doi: 10.1621/nrs.08001
214. Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell (2008) 134(2):317–28. doi: 10.1016/j.cell.2008.06.050
215. Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell (2008) 134(2):329–40. doi: 10.1016/j.cell.2008.07.002
216. Masri S, Rigor P, Cervantes M, Ceglia N, Sebastian C, Xiao C, et al. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell (2014) 158(3):659–72. doi: 10.1016/j.cell.2014.06.050
217. Aguilar-Arnal L, Katada S, Orozco-Solis R, Sassone-Corsi P. NAD(+)-SIRT1 control of H3K4 trimethylation through circadian deacetylation of MLL1. Nat Struct Mol Biol (2015) 22(4):312–8. doi: 10.1038/nsmb.2990
218. Rutter J, Reick M, Wu LC, McKnight SL. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Sci (New York NY) (2001) 293(5529):510–4. doi: 10.1126/science.1060698
219. Sun L, Wang Y, Song Y, Cheng XR, Xia S, Rahman MR, et al. Resveratrol restores the circadian rhythmic disorder of lipid metabolism induced by high-fat diet in mice. Biochem Biophys Res Commun (2015) 458(1):86–91. doi: 10.1016/j.bbrc.2015.01.072
220. Walker AK, Yang F, Jiang K, Ji JY, Watts JL, Purushotham A, et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev (2010) 24(13):1403–17. doi: 10.1101/gad.1901210
221. Powell MJ, Casimiro MC, Cordon-Cardo C, He X, Yeow WS, Wang C, et al. Disruption of a Sirt1-dependent autophagy checkpoint in the prostate results in prostatic intraepithelial neoplasia lesion formation. Cancer Res (2011) 71(3):964–75. doi: 10.1158/0008-5472.CAN-10-3172
222. Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer (2009) 9(2):123–8. doi: 10.1038/nrc2562
223. Liu T, Liu PY, Marshall GM. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res (2009) 69(5):1702–5. doi: 10.1158/0008-5472.CAN-08-3365
224. Lee H, Kim KR, Noh SJ, Park HS, Kwon KS, Park BH, et al. Expression of DBC1 and SIRT1 is associated with poor prognosis for breast carcinoma. Hum Pathol (2011) 42(2):204–13. doi: 10.1016/j.humpath.2010.05.023
225. Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, Elgavish A, et al. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res (2007) 67(14):6612–8. doi: 10.1158/0008-5472.CAN-07-0085
226. Chen J, Zhang B, Wong N, Lo AW, To KF, Chan AW, et al. Sirtuin 1 is upregulated in a subset of hepatocellular carcinomas where it is essential for telomere maintenance and tumor cell growth. Cancer Res (2011) 71(12):4138–49. doi: 10.1158/0008-5472.CAN-10-4274
227. Kriegl L, Vieth M, Kirchner T, Menssen A. Up-regulation of c-MYC and SIRT1 expression correlates with Malignant transformation in the serrated route to colorectal cancer. Oncotarget (2012) 3(10):1182–93. doi: 10.18632/oncotarget.628
228. Zhang S, Yang Y, Huang S, Deng C, Zhou S, Yang J, et al. SIRT1 inhibits gastric cancer proliferation and metastasis via STAT3/MMP-13 signaling. J Cell Physiol (2019) 234(9):15395–406. doi: 10.1002/jcp.28186
229. Lin L, Zheng X, Qiu C, Dongol S, Lv Q, Jiang J, et al. SIRT1 promotes endometrial tumor growth by targeting SREBP1 and lipogenesis. Oncol Rep (2014) 32(6):2831–5. doi: 10.3892/or.2014.3521
230. Zhao G, Cui J, Zhang JG, Qin Q, Chen Q, Yin T, et al. SIRT1 RNAi knockdown induces apoptosis and senescence, inhibits invasion and enhances chemosensitivity in pancreatic cancer cells. Gene Ther (2011) 18(9):920–8. doi: 10.1038/gt.2011.81
231. Ilbeigi D, Nourbakhsh M, Pasalar P, Meshkani R, Shokri Afra H, Panahi GH, et al. Nicotinamide phosphoribosyltransferase knockdown leads to lipid accumulation in HepG2 cells through the SIRT1-AMPK pathway. Cell J (2020) 22(Suppl 1):125–32. doi: 10.22074/cellj.2020.7013
232. Yang Q, Wang B, Gao W, Huang S, Liu Z, Li W, et al. SIRT1 is downregulated in gastric cancer and leads to G1-phase arrest via NF-κB/Cyclin D1 signaling. Mol Cancer Res MCR (2013) 11(12):1497–507. doi: 10.1158/1541-7786.MCR-13-0214
233. Cha EJ, Noh SJ, Kwon KS, Kim CY, Park BH, Park HS, et al. Expression of DBC1 and SIRT1 is associated with poor prognosis of gastric carcinoma. Clin Cancer Res (2009) 15(13):4453–9. doi: 10.1158/1078-0432.CCR-08-3329
234. Guo S, Ma B, Jiang X, Li X, Jia Y. Astragalus polysaccharides inhibits tumorigenesis and lipid metabolism through miR-138-5p/SIRT1/SREBP1 pathway in prostate cancer. Front Pharmacol (2020) 11:598. doi: 10.3389/fphar.2020.00598
235. Song NY, Surh YJ. Janus-faced role of SIRT1 in tumorigenesis. Ann New York Acad Sci (2012) 1271(1):10–9. doi: 10.1111/j.1749-6632.2012.06762.x
236. Shirai H, Oishi K, Kudo T, Shibata S, Ishida N. PPARalpha is a potential therapeutic target of drugs to treat circadian rhythm sleep disorders. Biochem Biophys Res Commun (2007) 357(3):679–82. doi: 10.1016/j.bbrc.2007.04.002
237. Inoue I, Shinoda Y, Ikeda M, Hayashi K, Kanazawa K, Nomura M, et al. CLOCK/BMAL1 is involved in lipid metabolism via transactivation of the peroxisome proliferator-activated receptor (PPAR) response element. J Atheroscl Thromb (2005) 12(3):169–74. doi: 10.5551/jat.12.169
238. Patel DD, Knight BL, Wiggins D, Humphreys SM, Gibbons GF. Disturbances in the normal regulation of SREBP-sensitive genes in PPAR alpha-deficient mice. J Lipid Res (2001) 42(3):328–37. doi: 10.1016/S0022-2275(20)31655-2
239. Ma XL, Sun YF, Wang BL, Shen MN, Zhou Y, Chen JW, et al. Sphere-forming culture enriches liver cancer stem cells and reveals Stearoyl-CoA desaturase 1 as a potential therapeutic target. BMC Cancer (2019) 19(1):760. doi: 10.1186/s12885-019-5963-z
240. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer (2020) 122(1):4–22. doi: 10.1038/s41416-019-0650-z
241. Wei J, Leit S, Kuai J, Therrien E, Rafi S, Harwood HJ Jr., et al. An allosteric mechanism for potent inhibition of human ATP-citrate lyase. Nature (2019) 568(7753):566–70. doi: 10.1038/s41586-019-1094-6
242. Xie S, Zhou F, Wang J, Cao H, Chen Y, Liu X, et al. Functional polymorphisms of ATP citrate lyase gene predicts clinical outcome of patients with advanced colorectal cancer. World J Surg Oncol (2015) 13:42. doi: 10.1186/s12957-015-0440-x
243. Li EQ, Zhao W, Zhang C, Qin LZ, Liu SJ, Feng ZQ, et al. Synthesis and anti-cancer activity of ND-646 and its derivatives as acetyl-CoA carboxylase 1 inhibitors. Eur J Pharm Sci (2019) 137:105010. doi: 10.1016/j.ejps.2019.105010
244. Jones SF, Infante JR. Molecular pathways: fatty acid synthase. Clin Cancer Res (2015) 21(24):5434–8. doi: 10.1158/1078-0432.CCR-15-0126
245. Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, et al. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res (2019) 79(20):5355–66. doi: 10.1158/0008-5472.CAN-19-0369
246. Luo Y, Xie C, Brocker CN, Fan J, Wu X, Feng L, et al. Intestinal PPARα Protects against colon carcinogenesis via regulation of methyltransferases DNMT1 and PRMT6. Gastroenterology (2019) 157(3):744–59.e744. doi: 10.1053/j.gastro.2019.05.057
247. Dallmann R, Okyar A, Lévi F. Dosing-time makes the poison: circadian regulation and pharmacotherapy. Trends Mol Med (2016) 22(5):430–45. doi: 10.1016/j.molmed.2016.03.004
248. Sancar A, Van Gelder RN. Clocks, cancer, and chronochemotherapy. Sci (New York NY) (2021) 371(6524):eabb0738. doi: 10.1126/science.abb0738
249. Yang Y, Lindsey-Boltz LA, Vaughn CM, Selby CP, Cao X, Liu Z, et al. Circadian clock, carcinogenesis, chronochemotherapy connections. J Biol Chem (2021) 297(3):101068. doi: 10.1016/j.jbc.2021.101068
250. Ballesta A, Innominato PF, Dallmann R, Rand DA, Lévi FA. Systems chronotherapeutics. Pharmacol Rev (2017) 69(2):161–99. doi: 10.1124/pr.116.013441
251. Lévi F, Zidani R, Misset JL. Randomised multicentre trial of chronotherapy with oxaliplatin, fluorouracil, and folinic acid in metastatic colorectal cancer. International Organization for Cancer Chronotherapy. Lancet (London England) (1997) 350(9079):681–6. doi: 10.1016/S0140-6736(97)03358-8
252. Sandor V. Chronotherapy with 5-fluorouracil, oxaliplatin, and folinic acid in colorectal cancer. Lancet (London England) (1997) 350(9087):1325–6. doi: 10.1016/S0140-6736(05)62486-5
253. Efficace F, Innominato PF, Bjarnason G, Coens C, Humblet Y, Tumolo S, et al. Validation of patient's self-reported social functioning as an independent prognostic factor for survival in metastatic colorectal cancer patients: results of an international study by the Chronotherapy Group of the European Organisation for Research and Treatment of Cancer. J Clin Oncol (2008) 26(12):2020–6. doi: 10.1200/JCO.2007.12.3117
254. Dong D, Yang D, Lin L, Wang S, Wu B. Circadian rhythm in pharmacokinetics and its relevance to chronotherapy. Biochem Pharmacol (2020) 178:114045. doi: 10.1016/j.bcp.2020.114045
255. Bernard S, Cajavec Bernard B, Lévi F, Herzel H. Tumor growth rate determines the timing of optimal chronomodulated treatment schedules. PLoS Comput Biol (2010) 6(3):e1000712. doi: 10.1371/journal.pcbi.1000712
256. Hesse J, Martinelli J, Aboumanify O, Ballesta A, Relógio A. A mathematical model of the circadian clock and drug pharmacology to optimize irinotecan administration timing in colorectal cancer. Comput Struct Biotechnol J (2021) 19:5170–83. doi: 10.1016/j.csbj.2021.08.051
257. Dulong S, Ballesta A, Okyar A, Lévi F. Identification of circadian determinants of cancer chronotherapy through in vitro chronopharmacology and mathematical modeling. Mol Cancer Ther (2015) 14(9):2154–64. doi: 10.1158/1535-7163.MCT-15-0129
258. Lévi F, Okyar A, Dulong S, Innominato PF, Clairambault J. Circadian timing in cancer treatments. Annu Rev Pharmacol Toxicol (2010) 50:377–421. doi: 10.1146/annurev.pharmtox.48.113006.094626
259. Innominato PF, Roche VP, Palesh OG, Ulusakarya A, Spiegel D, Lévi FA. The circadian timing system in clinical oncology. Ann Med (2014) 46(4):191–207. doi: 10.3109/07853890.2014.916990
260. Bajetta E, Pietrantonio F, Buzzoni R, Ferrario E, Valvo F, Mariani L, et al. Chronomodulated capecitabine and adjuvant radiation in intermediate-risk to high-risk rectal cancer: a phase II study. Am J Clin Oncol (2014) 37(6):545–9. doi: 10.1097/COC.0b013e31827ecd1d
261. Akgun Z, Saglam S, Yucel S, Gural Z, Balik E, Cipe G, et al. Neoadjuvant chronomodulated capecitabine with radiotherapy in rectal cancer: a phase II brunch regimen study. Cancer chemotherapy Pharmacol (2014) 74(4):751–6. doi: 10.1007/s00280-014-2558-x
262. Levi F, Schibler U. Circadian rhythms: mechanisms and therapeutic implications. Annu Rev Pharmacol Toxicol (2007) 47:593–628. doi: 10.1146/annurev.pharmtox.47.120505.105208
263. Altinok A, Lévi F, Goldbeter A. Identifying mechanisms of chronotolerance and chronoefficacy for the anticancer drugs 5-fluorouracil and oxaliplatin by computational modeling. Eur J Pharm Sci (2009) 36(1):20–38. doi: 10.1016/j.ejps.2008.10.024
264. Innominato PF, Karaboué A, Focan C, Chollet P, Giacchetti S, Bouchahda M, et al. Efficacy and safety of chronomodulated irinotecan, oxaliplatin, 5-fluorouracil and leucovorin combination as first- or second-line treatment against metastatic colorectal cancer: Results from the International EORTC 05011 Trial. Int J Cancer (2020) 148(10):2512–21. doi: 10.1002/ijc.33422
265. Li J, Chen R, Ji M, Zou SL, Zhu LN. Cisplatin-based chronotherapy for advanced non-small cell lung cancer patients: a randomized controlled study and its pharmacokinetics analysis. Cancer chemotherapy Pharmacol (2015) 76(3):651–5. doi: 10.1007/s00280-015-2804-x
266. Wagner PM, Monjes NM, Guido ME. Chemotherapeutic effect of SR9009, a REV-ERB agonist, on the human glioblastoma T98G cells. ASN Neuro (2019) 11:1759091419892713. doi: 10.1177/1759091419892713
267. Dong Z, Zhang G, Qu M, Gimple RC, Wu Q, Qiu Z, et al. Targeting glioblastoma stem cells through disruption of the circadian clock. Cancer Discovery (2019) 9(11):1556–73. doi: 10.1158/2159-8290.CD-19-0215
268. Dierickx P, Emmett MJ, Jiang C, Uehara K, Liu M, Adlanmerini M, et al. SR9009 has REV-ERB-independent effects on cell proliferation and metabolism. Proc Natl Acad Sci U S A (2019) 116(25):12147–52. doi: 10.1073/pnas.1904226116
269. Kobayashi M, Wood PA, Hrushesky WJ. Circadian chemotherapy for gynecological and genitourinary cancers. Chronobiology Int (2002) 19(1):237–51. doi: 10.1081/CBI-120002600
270. Lévi F. Circadian chronotherapy for human cancers. Lancet Oncol (2001) 2(5):307–15. doi: 10.1016/S1470-2045(00)00326-0
271. Lévi F, Giacchetti S, Zidani R, Brezault-Bonnet C, Tigaud JM, Goldwasser F, et al. Chronotherapy of colorectal cancer metastases. Hepato-gastroenterology (2001) 48(38):320–2.
272. Chan S, Rowbottom L, McDonald R, Bjarnason GA, Tsao M, Danjoux C, et al. Does the time of radiotherapy affect treatment outcomes? A review of the literature. Clin Oncol (Royal Coll Radiologists (Great Britain)) (2017) 29(4):231–8. doi: 10.1016/j.clon.2016.12.005
Keywords: circadian clock, lipid metabolism, cancer, fatty acid synthetic, SREBP
Citation: Liu M, Zhang Z, Chen Y, Feng T, Zhou Q and Tian X (2023) Circadian clock and lipid metabolism disorders: a potential therapeutic strategy for cancer. Front. Endocrinol. 14:1292011. doi: 10.3389/fendo.2023.1292011
Received: 10 September 2023; Accepted: 30 November 2023;
Published: 22 December 2023.
Edited by:
Paraskevi Xekouki, University of Crete, GreeceReviewed by:
Tony Lefebvre, Lille University of Science and Technology, FranceCopyright © 2023 Liu, Zhang, Chen, Feng, Zhou and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuefei Tian, MDAzNjQwQGhudWNtLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.