 Tian Wang
Tian Wang Shuhua Chen1
Shuhua Chen1 Yuan Shang
Yuan Shang Roberta Diaz Brinton
Roberta Diaz Brinton
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 22 December 2023
Sec. Neuroendocrine Science
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1286931
This article is part of the Research Topic Neuroprotective and Therapeutic Effects of Progesterone, Allopregnanolone, and Their Derivatives View all 3 articles
Objective: Allopregnanolone (Allo) is a neurosteroid with pleiotropic action in the brain that includes neurogenesis, oligogenesis, human and rodent neural stem cell regeneration, increased glucose metabolism, mitochondrial respiration and biogenesis, improved cognitive function, and reduction of both inflammation and Alzheimer’s disease (AD) pathology. Because the breadth of Allo-induced responses requires activation of multiple systems of biology in the absence of an Allo-specific nuclear receptor, analyses were conducted in both neurons and astrocytes to identify unifying systems and signaling pathways.
Methods: Mechanisms of Allo action were investigated in embryonic hippocampal neurons and astrocytes cultured in an Aging Model (AM) media. Cellular morphology, mitochondrial function, and transcriptomics were investigated followed by mechanistic pathway analyses.
Results: In hippocampal neurons, Allo significantly increased neurite outgrowth and synaptic protein expression, which were paralleled by upregulated synaptogenesis and long-term potentiation gene expression profiles. Mechanistically, Allo induced Ca2+/CREB signaling cascades. In parallel, Allo significantly increased maximal mitochondrial respiration, mitochondrial membrane potential, and Complex IV activity while reducing oxidative stress, which required both the GABAA and L-type Ca2+ channels. In astrocytes, Allo increased ATP generation, mitochondrial function and dynamics while reducing oxidative stress, inflammasome indicators, and apoptotic signaling. Mechanistically, Allo regulation of astrocytic mitochondrial function required both the GABAA and L-type Ca2+ channels. Furthermore, Allo activated NRF1-TFAM signaling and increased the DRP1/OPA1 protein ratio, which led to increased mitochondrial biogenesis and dynamics.
Conclusion: Collectively, the cellular, mitochondrial, transcriptional, and pharmacological profiles provide evidence in support of calcium signaling as a unifying mechanism for Allo pleiotropic actions in the brain.
Allopregnanolone (Allo, 3α-hydroxy-5α-pregnan-20-one) is an endogenous neurosteroid derived from progesterone (1–6). Despite the lack of a specific nuclear transcriptional receptor, Allo functions as a pleiotropic systems biology regulator (1–3, 7–12). The systems of biology activated by Allo in the brain include neural stem cell regeneration, neurogenesis, oligogenesis and increased white matter (3, 7, 13–19), mitochondrial respiration and bioenergetics (10, 20), cholesterol homeostasis (21, 22), immune regulation (12, 21, 23–25), reduced beta amyloid and pTau (21), restoration of synaptic transmission, and cognition (9, 26, 27).
The mechanisms of action of Allo have been extensively studied in mature neurons and neural stem cells (3, 13, 28–30). In both cell types, Allo prolongs the opening of the GABAA receptor complex (30). However, modulation of the GABAA receptor complex by Allo results in hyperpolarization in mature neurons and the opposite, depolarization, in neural stem cells (3, 13). Mature neurons, although expressing Na-K-Cl cotransporter (NKCC1), maintain a low intracellular Cl− level due to the upregulation of the neuron-specific K-Cl cotransporter 2 (KCC2) during their maturation (31). Allo prolongation of the GABAA receptor complex open time increases chloride influx, hyperpolarization and potentiation of the inhibitory post-synaptic potential (30). In contrast, neural stem cells exhibit a high-intracellular Cl− level attributed to NKCC1 expression (3). Allo prolongs the GABAA receptor complex open time, which results in depolarization of the plasma membrane due to efflux of intracellular chloride (3, 32). This Allo-induced membrane depolarization leads to activation of voltage-dependent L-type calcium (Ca2+) channels (28). The subsequent rise in intracellular Ca2+ activates a Ca2+-dependent kinase, CaMK IV, which then phosphorylates and activates the transcription factor cyclic AMP-responsive element-binding protein 1 (CREB1) (3). Through CREB1 activation, Allo upregulates the expression of cell cycle genes that promote mitosis, leading to increased neural stem cell regeneration (3, 13, 28).
The breadth of Allo regulation of systems of biology requires multiple cell types. Although the effects of Allo have been reported in multiple neural cell types (33), a unifying mechanism of action across multiple cell types remains unresolved. Herein, we investigated the impact of Allo on a common set of responses in embryonic hippocampal neurons and astrocytes, both of which express GABAA receptors and have a high-intracellular chloride concentration (31, 34, 35). Transcriptional analyses were conducted to identify both unique and common pathways relevant to Allo-induced outcomes.
Results reported herein indicate that, in embryonic neurons, Allo activates the Ca2+/CREB signaling pathway that promotes neural plasticity and mitochondrial function. In parallel, Allo activated the Ca2+ signaling pathway in astrocytes, which was associated with increased mitochondrial biogenesis and dynamics through an increase in the NRF1-TFAM signaling cascade and DRP1/OPA1 protein ratio. These mitochondrial outcomes were associated with reduced inflammasome activation and oxidative stress. Notably, Allo’s systems of biology actions rely on a high-intracellular Cl− concentration, mediated by expression of the NKCC1, that enables efflux of intracellular Cl− through the GABAA receptor and thus membrane depolarization that activates the L-type Ca2+ channels enabling an influx of Ca2+ and activation of the Ca2+ signaling pathway (3). Collectively, findings reported herein provide insights into common Ca2+-mediated and mitochondria-related mechanisms that underlie the pleiotropic action of Allo to activate systems of biology enabling integration of signaling and function in neurons and astrocytes.
Primary embryonic hippocampal neurons and astrocytes were isolated from day 18 (E18) fetuses of Sprague Dawley rats (Envigo) as previously described (36, 37). Briefly, hippocampi were dissected from the brains of E18 fetuses and digested by incubation with 0.02% trypsin in HBSS (Invitrogen,14170-112) for 3 min at 37°C. The digested tissue was dissociated by repeated passing through a series of fire-polished constricted Pasteur pipettes, then filtered through a 40-µm Nylon cell strainer into 50-ml conical centrifuge tubes and centrifuged at 1,000 rpm for 3 min.
Neurons were cultured in Neurobasal Medium (Gibco, 12348017) supplemented with 2%B-27 (Gibco, 17504044), 25 μM glutamate (MP Biomedicals, 02101800), 0.5 mM L-glutamine (Gibco, 25030081) and 10U/mL penicillin/streptomycin (Gibco, 15140122). Mixed glia was cultured in Dulbecco’s Modified Eagle Medium (DMEM)/F-12 (Gibco, 11039021) supplemented with 10% FBS (ATCC, 30-2020) and 20 U/mL penicillin/streptomycin (Gibco, 15140122) in T75 flasks. Upon 80% confluency, cultures were shaken on an orbital shaker overnight and then washed with cold Phosphate buffer saline (PBS). The supernatants containing microglia and oligodendrocytes were discarded, and the remaining enriched astrocytes were then trypsinized and re-plated onto poly-D-lysine coated wells or coverslips.
For imaging or immunocytochemistry studies, 20,000 cells were plated onto each poly-D-lysine (50 μg/mL) coated 22-mm coverslip. For Western blotting, 106 cells were seeded on poly-D-lysine coated six well plates. Cells were incubated at 37°C in a humidified 5% CO2 incubator.
Because aging is associated with decreased levels of hormone, growth, and trophic factors (38, 39), a supplement-reduced media condition was used as an Aging Model (AM) to investigate the effect of Allo on neurons and astrocytes under an aging condition. Neurons (Figure 1A) were cultured in Neurobasal media with a reduced amount of B27 (0.2% and 10% Supplement-deprived medium, AM) at day 10 for 4h, then treated with 100 nM Allo (SAFC Inc, UCDQR-001) or vehicle (0.001% EtOH) for 16h in AM medium (40). Astrocytes were cultured in DMEM/F-12 supplemented with 10% charcoal-stripped FBS (Gibco, 12676029, AM) for 24h, then treated with 100 nM Allo or vehicle for 24h in AM medium (41). Allo (100 nM) and the treatment duration were based on previously established analyses across multiple cell types (11, 13, 28). Allo at 100 nM is comparable to physiological pregnancy levels of Allo, and thus, is translationally relevant (42, 43).
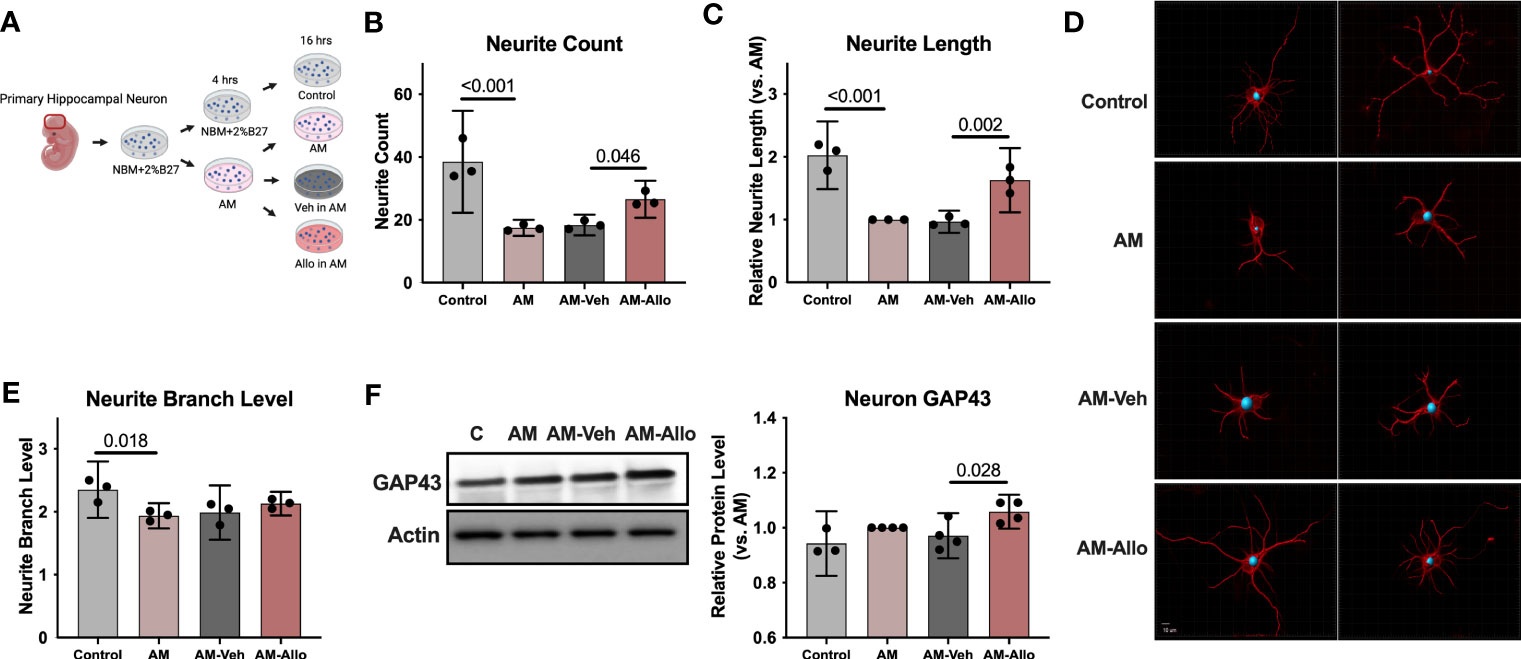
Figure 1 Allo treatment significantly reversed AM-induced deficits in neurite outgrowth. (A) The schematic diagram of Allo treatment (Created with BioRender.com). Allo treatment (100 nM) reversed AM-induced deficits in neurite count (B) (n = 3, one-way analysis of variance (ANOVA), F (3, 8) = 22.28, p < 0.001, η2 = 0.893) and neurite length (C) (n = 3, one-way ANOVA, F (3, 8) = 33.38, p < 0.001, η2 = 0.926). (D) Representative neuron images labeled with anti–β-tubulin antibody and processed with Imaris. (E) The significant decrease in neurite branch levels induced by AM (n = 3, one-way ANOVA, F (3, 8) = 5.491, p = 0.024, η2 = 0.673) was not affected by Allo treatment. (F) Relative to AM-Veh group, AM-Allo treatment significantly increased GAP43 protein levels (n = 3–4, one-way ANOVA, F (3, 11) = 5.763, p = 0.013, η2 = 0.611). All bar graphs are presented as mean ± 95% CI with individual data points. Statistical significance was calculated using one-way ANOVA, followed by Holm-Sidak multiple comparisons specifically between three selected groups: AM versus Control, AM-Veh versus AM, AM-Allo versus AM-Veh.
Neurons were cultured in AM medium at day 10 for 4h, then pre-treated with 10 μM bicuculline (GABAA receptor antagonist, Sigma-Aldrich, St. Louis, United States, 14343) in AM medium for 1h, followed by co-incubation of 100 nM Allo and bicuculline in AM medium for 16h. Sixteen-hour nifedipine (Ca2+ channel blocker, Sigma-Aldrich, N7634) (28) treatment triggered neuronal cell death in AM medium. Therefore, neurons were cultured in AM medium for 4h, then co-treated with 100 nM Allo and 10 μM nifedipine for 3h.
Astrocytes were cultured in AM medium for 24h, then pre-treated with 10 μM bicuculline or 10 μM nifedipine in AM medium for 1h, followed by co-incubation of 100 nM Allo and either bicuculline or nifedipine in AM medium for 24h.
Cellular respiratory capacity was determined by XF96e metabolic flux analyzer as previously described (44–46). Hippocampal primary neurons and astrocytes were seeded on poly-D-lysine–coated Seahorse XF96 well plates at a density of 25,000 cells per well and cultured and treated as described above. On the day of assay, cell culture media was changed to unbuffered DMEM medium (pH 7.4, Sigma-Aldrich, D5030) supplemented with 25 mM glucose, 1 mM sodium pyruvate, 31 mM NaCl, and 2 mM glutamine (Gibco, 25030081). Cells were then incubated at 37°C in a non‐CO2 incubator for 1h. Oxygen consumption rate (OCR) was used as an indicator of mitochondrial oxidative phosphorylation. Mitochondrial basal respiration, spare and maximal respiratory capacity were determined by sequential acute injection of mitochondrial electron transport chain inhibitors and un-couplers: oligomycin (4 μM, MP Biomedicals, 02151786), FCCP (1 μM, carbonyl cyanide4(trifluoromethoxy)-phenylhydrazone) (TOCRIS Bioscience, 0453), rotenone (1 μM, MP Biomedicals, 02150154), and antimycin (1 μM, Sigma-Aldrich, A-8674). OCR results were normalized to protein concentrations.
For neurons, 5 μM CellROX Green Reagent (Invitrogen, C10444) and 10 μg/ml Hoechst-33342 (nuclear counter staining) were added to cell medium. Cells were then incubated for 30 min at 37°C in a humidified 5% CO2 incubator, followed by washing 3 times with pre-warmed PBS. The fluorescence intensity was measured using Cytation 5 (BioTek) at 520 nM for CellROX and 461 nm for Hoechst-33342. The individual ROS level was calculated by normalizing CellROX intensity (ROS level) to corresponding Hoechst-33342 intensity (overall cell density).
Astrocytes were incubated with 5 μM CellROX Green Reagent (Invitrogen, C10444) for 30 min at 37°C in a humidified 5% CO2 incubator. After three washes with pre-warmed PBS, astrocytes were trypsinized then pelleted by centrifuging and resuspended in 150 μl of PBS with 4′,6-diamidino-2-phenylindole (DAPI). Fluorescence signal was detected by MACSQuant Analyzer Flow Cytometry (Mitenyi Biotec) and analyzed with FlowLogic software.
Cells were fixed with 4% paraformaldehyde for 15 min and blocked in blocking buffer (5% normal goat serum + 0.3% Triton X-100 in PBS) for 60 min at room temperature. The following primary antibodies were then applied for overnight at 4°C: mouse anti-MAP2 (1:1000, Sigma-Aldrich, M4403), rabbit anti-TOM20 (1:250, Abcam, ab186734), mouse anti-GFAP (1:1000, Chemicon, MAB360), and mouse anti-beta tubulin (1:300, Millipore, MAB1637). Cells were then incubated with Alexa Fluor 488 or 555 goat anti-mouse or anti-rabbit secondary antibodies for 60 min at room temperature. Nuclei were stained with DAPI. Images were captured with Zeiss LSM 880 Airyscan Confocal Microscope or Axiovert 200 M Marianas Digital Fluorescence Microscopy Workstation (Intelligent Imaging Innovations, Denver, CO).
Neurite quantification was conducted using the filament function of Imaris 9.5 software. All analyses were performed with the experimenter blinded to the condition. Fifteen to 20 images were randomly obtained per group and processed with Imaris. The mean neurite count, length, and branch level were calculated per group. The whole procedure was repeated three times, and the mean values from these three independent experiments were plotted and used for statistical analysis.
Quantification of mitochondrial morphological subtypes was performed using MitoMorph software (47, 48). Mitochondria were labeled using anti-TOM20 antibody (section 2.5), and individual mitochondria were categorized according to area, morphology, and length into six distinct types: small globules, swollen globules, straight tubules, twisted tubules, branched tubules, and loops. The proportion of fused mitochondria (fusion products) was calculated as the sum of straight tubule, twisting tubule, loop, and branch tubule mitochondrial populations. The numbers of small globules and swollen globules were combined and classified as segregated mitochondria (fission product).
Cells were lysed in M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, 78501) with 1% Pierce Protease and Phosphatase inhibitor (Thermo Fisher Scientific, A32961). Protein concentrations were then determined by using Protein Assay Dye Reagent (Bio-Rad). Equal amounts of protein (10 μg) were loaded in each well of 12% SDS-PAGE gels (Bio-Rad Laboratories), electrophoresed with a tris/glycine running buffer, and transferred to a polyvinylidine difluoride membrane (0.45 µm PVDF). The following primary antibodies were incubated overnight: rabbit anti-GAP43 (1:1000, Abcam, ab16053), rabbit anti-pERK42/44 (1:1000, Cell Signaling, 4370s), rabbit anti-ERK42/44 (1:1000, Cell Signaling, 4695s), rabbit anti–c-Fos (1:1000, Cell Signaling, 2250), rabbit anti–c-Jun (1:1000, Cell Signaling, 9165), mouse total OXPHOS antibody cocktail (1:1000, Abcam, 110413), rabbit anti-DRP1 (1:1000, Cell Signaling, 8570s), rabbit anti-OPA1 (1:1000, Cell Signaling, 80471s), and mouse anti-Actin (1:10000, Millipore, MAB 1501). HRP-conjugated anti-rabbit or anti-mouse secondary antibody (1:5000, Vector Laboratories, Burlingame, CA) were then applied. The signal was visualized by Pierce SuperSignal Chemiluminescent Substrates or SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, IL, Waltham, MA) and captured by ChemiDoc MP Imaging System (BioRad, Hercules, CA). All band intensities were quantified using Image Lab 6.0.1 (Bio-Rad, Hercules, CA) and normalized to corresponding actin intensity. Relative fold change was calculated in comparison to the AM group.
Total DNA was isolated from cells with QIAamp DNA mini kit (Qiagen, Valencia, CA) and analyzed by quantitative PCR. Relative mtDNA/nDNA ratio was calculated as the relative fold change of mt-CO2 (mtDNA) content to β-Globin (nDNA) content. Primers were as follows: mt-CO2 forward: 5′- AAACCAGGTGAACTTCGTCTAT-3′; mt-CO2 reverse: 5′- GGACGTCTTCGGATGAGATTAG -3′; β-Globin forward: 5′- GCTTTCCTGCTCAAATTCCTATC -3′; and β-Globin reverse: 5′- AACACTCCACAGGGCATATC -3′.
Mitochondrial complex IV activity was measured by Complex IV Rodent Enzyme Activity Microplate Assay Kit (Abcam, ab109911) following the manufacturer’s instructions. Mitochondrial proteins were extracted from sample homogenates with the concentration of 5.5 mg/ml. Sample (50 μg/200 μl) was added to each well and incubated for 3h at room temperature. The reduced Cytochrome C reagent was then added and the OD550 was measured by Cytation 5 Cell Imaging Reader (BioTek) at 1-min intervals for 2h at 30°C. The initial rate of oxidation of Cytochrome C was calculated within the linear range.
Mitochondrial membrane potential was assessed by MITO-ID Membrane Potential Detection Kit (Enzo, ENZ-51018-0025) following the manufacturer’s instructions. Cells were washed with 1X assay buffer, then incubated with MITO-ID MP Detection Reagent for 15 min at room temperature. The MMP ratio was detected by Cytation 5 (BioTek). Fluorescence signals were collected at 530/20 nm with 485/20 nm excitation (green) and 570/10 nm with 540/10 nm excitation (orange). The intensity ratio of orange/green was then calculated.
Cellular ATP levels were measured by ATP determination kit (Molecular Probes, A22066) following the manufacturer’s instructions. Luminescence was measured by Cytation 5 (BioTek).
Cells were lysed in TRIzol® Reagent, followed by chloroform extraction at a volume ratio of 1:5 to that of the TRIzol® Reagent. Ethanol was then used to precipitate nucleic acids from the aqueous phase. RNA was further purified using PureLink™ RNA Mini Kit (Invitrogen, 12183018A) following manufacturer’s instructions. Purelink™ DNase (Invitrogen, 12185010) was used to eliminate DNA contamination. Purified RNA was eluted in RNase-free diH2O. RNA concentration and quality were checked by NanoDrop™ One (Thermo Fisher Scientific).
Purified RNA was then reverse transcribed to cDNA using SuperScript VILO Master MIX (Invitrogen, 11755). cDNA (20 ng) was used for rt-PCR with TaqMan Universal PCR Master Mix (Applied Biosystems, 4304437). The following TaqMan primers were used: Nrf1 (Rn01455958), Tfam (Rn00580051), Ppargc1a (Rn00580241), and Actin (Rn00667869). Target cDNA was amplified and detected using Applied Biosystems QuantStudio 6 Flex system (Thermo Fisher Scientific). Relative gene expression level (fold change) to reference group was calculated by the comparative Ct method.
RNA-seq was conducted at Vanderbilt Technologies for Advanced Genomics (VANTAGE). Only RNA samples with an acceptable RNA quality indicator score (RQI > 7) were used for sequencing. mRNA enrichment and cDNA library preparation were done using a stranded mRNA (poly(A)–selected) sample preparation kit. Sequencing was performed at 150 bp paired-end on NovaSeq6000, targeting 30 million reads per sample. Transcripts were mapped to the rat genome (ensemble release 95) using Salmon 0.14. Tximport V1.6.0 (49) was used to generate a counts table, and DESeq2 V1.18.1 (50) was used to calculate normalized read counts for each gene and/or transcript and to perform expression analysis. Variance stabilizing transformation normalized expression was used for visualization.
RNA-seq data were processed by the core analysis function of IPA using a p-value cutoff of 0.05. The canonical pathways were identified based on enrichment of qualified genes. The upstream regulator analysis predicted activation or inhibition of regulatory molecules based on expression of respective downstream genes and networks compiled from literature and IPA’s Ingenuity knowledge base.
Statistical significance was calculated using one-way analysis of variance (ANOVA), followed by Holm-Sidak multiple comparisons specifically between three selected groups: AM versus Control, AM-Veh versus AM, and AM-Allo versus AM-Veh. To assess the effect of pathway inhibitors, a two-way ANOVA was utilized, followed by Holm-Sidak multiple comparisons. Statistical significance and effect size (fold change) for RNA-seq data were calculated by DESeq2 V1.18.1. Comparisons with a p-value smaller than 0.05 were considered statistically significant.
The gene expression data are available through the Gene Expression Omnibus (GEO) repository (GSE229627, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE229627).
Morphological complexity of neurons has long been a correlate of information processing and storage capacity (51). Thus, the impact of Allo on morphological indicators of neuronal plasticity was assessed in hippocampal neurons (Figure 1A). Relative to control neurons, neurite outgrowth was significantly inhibited in the neurons treated with AM medium (Figures 1B–E). The morphological phenotype of AM neurons was characterized by decreased neurite count (p < 0.001 vs. Control), neurite length (p < 0.001 vs. Control), and neurite branch level (p = 0.018 vs. Control). Allo (16h treatment) significantly increased neurite count (p = 0.046 vs. AM-Veh) and neurite length (p = 0.002 vs. AM-Veh), without affecting neurite branch level. Consistent with the morphological impact, protein level of GAP43 (Growth Associated Protein 43, a marker of synaptic growth) was significantly increased in Allo-treated neurons (Figure 1F, p = 0.028 vs. AM-Veh). No differences in neurite growth were observed between AM and AM-Veh groups, confirming the specificity of the Allo effect.
To investigate the impact of Allo on transcriptional pathways relevant to neuroplasticity and to identify mechanistic pathways, unbiased exploratory transcriptomic analysis was conducted in Veh and Allo-treated (16h treatment) hippocampal neurons in parallel to morphological analyses. As shown in Table 1, transcriptomic analysis revealed that Allo significantly upregulated gene expression associated with synaptogenesis and synaptic long-term potentiation, which is consistent with neuroplasticity. Mechanistically, Ca2+ and CREB (cAMP-responsive element-binding protein) signaling were identified as the top pathways activated by Allo (Table 1), which replicated our previous findings (28). Transcriptomic profiling indicated significant increases in the gene expression of key activators of Ca2+-CREB signaling, including Mapk1 (Mitogen-activated protein kinase 1, ERK2, Figure 2A, p = 0.008, fold change = 1.074), Mapk3 (Mitogen-activated protein kinase 3, ERK1, Figure 2B, p < 0.001, fold change = 1.101), Elk1 (ETS transcription factor ELK1, Figure 2C, p < 0.001, fold change = 1.181), Camk2a (Calcium/Calmodulin dependent protein kinase II Alpha, Figure 2D, p = 0.006, fold change = 1.250), and Camk2g (Calcium/Calmodulin dependent protein kinase II gamma, Figure 2E, p = 0.016, fold change = 1.072). Consistent with transcriptional analyses, Allo (16h treatment) significantly increased protein levels of pERK1/2 (Figure 2F, p = 0.029 vs. AM-Veh) and induced a trend toward increased ERK1/2 protein levels (p = 0.077 vs. AM-Veh). Further, protein level of c-Jun, another effector of Ca2+ signaling, was also significantly increased by Allo treatment (16h treatment, Figure 2G, p = 0.017 vs. AM-Veh).
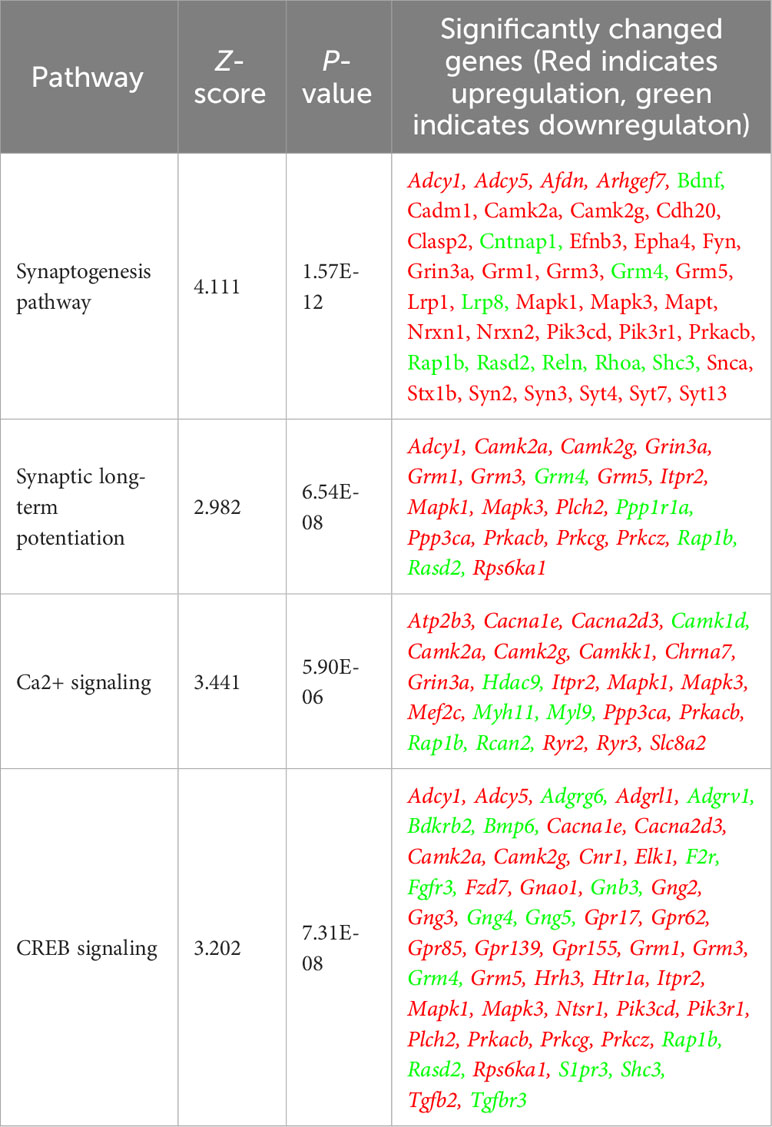
Table 1 Allo-activated neuroplasticity and calcium signaling pathways.
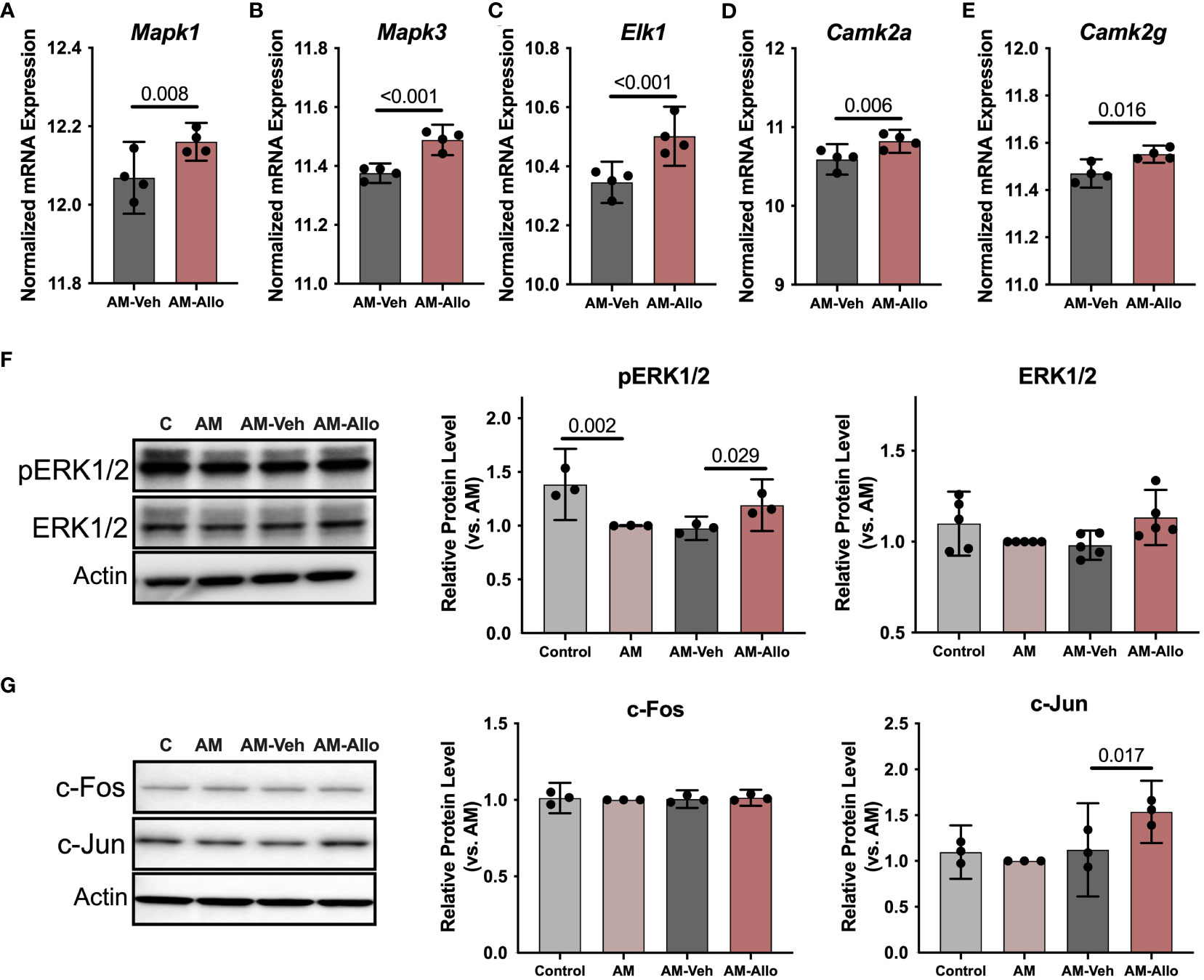
Figure 2 Allo activated Ca2+ signaling cascades associated with neuroplasticity in hippocampal neurons. Allo induced significant increases in gene expression levels of Mapk1 (A), Mapk3 (B), Elk1 (C), Camk2a (D) and Camk2g (E) (n = 4). (F) Allo treatment significantly restored protein levels of pERK1/2 (n = 3, one-way analysis of variance (ANOVA), F (3, 8) = 14.95, p = 0.001, η2 = 0.849) and induced a trend toward increased ERK1/2 protein levels (n = 5, one-way ANOVA, F (3, 16) = 2.847, p = 0.071, η2 = 0.348). (G) Protein levels of c-Jun were significantly increased by Allo treatment (n = 3, one-way ANOVA, F (3, 8) = 9.113, p = 0.006, η2 = 0.774). All bar graphs are presented as mean ± 95% CI with individual data points. Statistical significance for RNA-seq data was calculated by DESeq2 V1.18.1. Other data were analyzed using one-way ANOVA, followed by Holm-Sidak multiple comparisons specifically between three selected groups: AM versus Control, AM-Veh versus AM, AM-Allo versus AM-Veh.
Collectively, these data indicate that Allo-induced neuroplasticity was associated with activation of the Ca2+/CREB signaling pathway.
Our previous in-vivo analyses indicated that Allo promoted brain mitochondrial respiration (10). Herein, we investigated the cellular contribution of the Allo-induced increase in mitochondrial respiration from neurons. Neuronal mitochondrial maximal respiration (Figures 3A, B, p = 0.041 vs. Control) was reduced in the AM group. In contrast, Allo treatment (16h treatment) significantly increased mitochondrial maximal respiration (Figures 3A, B, p = 0.046 vs. AM-Veh). Furthermore, Allo treatment reversed the AM-induced decrease in Complex IV (COX) activity (Figure 3C, p = 0.023 vs. AM-Veh). A significant increase in the ratio of high membrane potential mitochondria was also observed in Allo-treated group compared to Veh-treated group (Figure 3D, p = 0.002 vs. AM-Veh), indicative of improved mitochondrial efficiency. Furthermore, Allo (16h treatment) significantly decreased ROS levels (p = 0.009 vs. AM-Veh) relative to the rise in cellular oxidative status observed in the AM group (Figure 3E, p = 0.001 vs. Control). Consistent with the transcriptomic profiles, inhibiting the GABAA receptor with bicuculline (Figure 3F, p = 0.048 vs. Allo) or inhibiting the L-type Ca2+ channel with nifedipine (Figure 3G, p = 0.005 vs. Allo) significantly blocked this Allo-induced increase of mitochondrial maximal respiration.
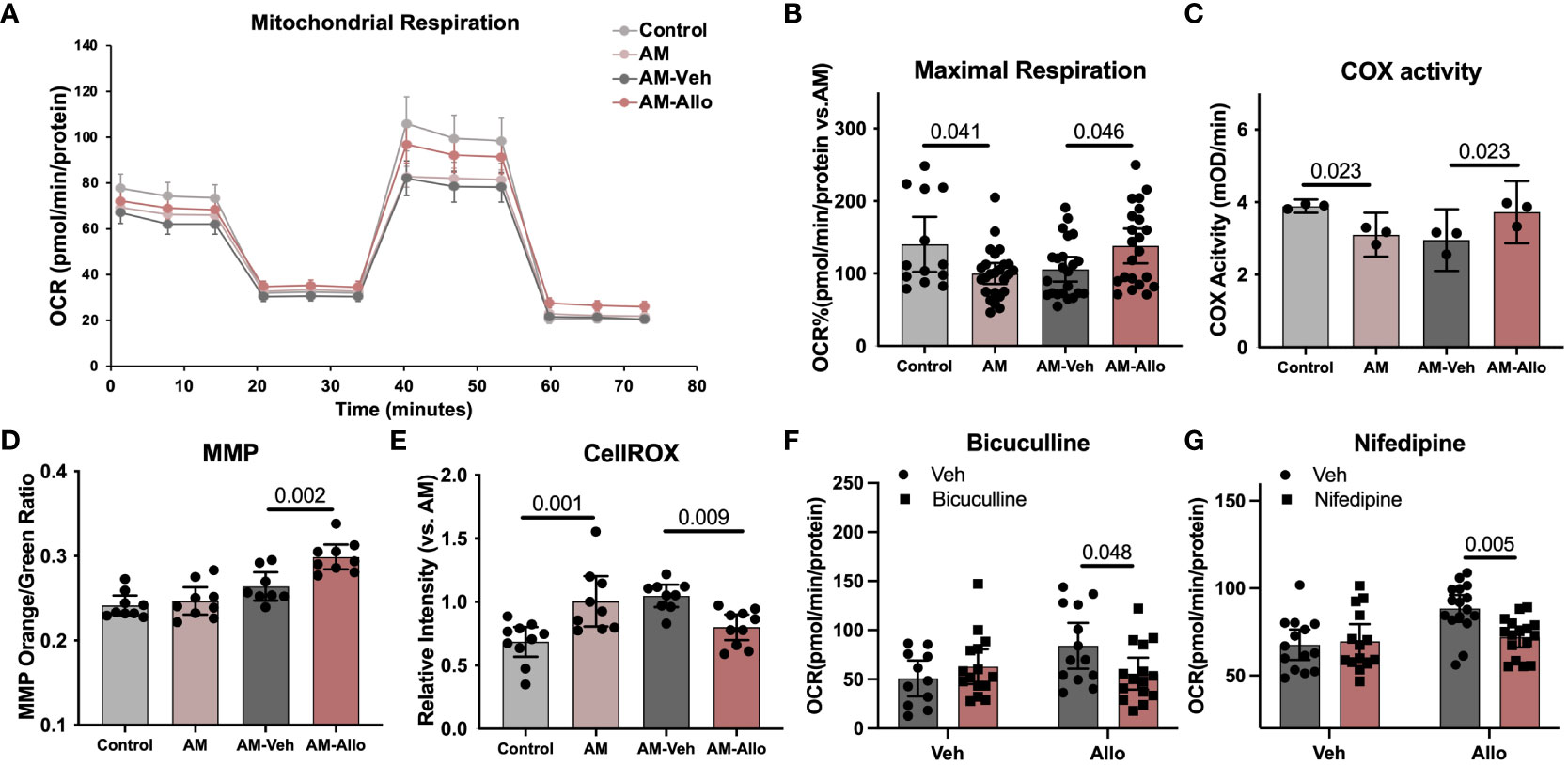
Figure 3 Allo restored mitochondrial respiration and oxidative phosphorylation via GABAA and L-type calcium channel activation while preventing free radical generation in hippocampal neurons. (A) Representative mitochondrial stress test results using the Seahorse XF96 Extracellular Flux Analyzer (mean ± S.E.M.). (B) Allo treatment restored AM-induced decrease in maximal mitochondrial respiration. (n = 13–25 wells per group, one-way analysis of variance (ANOVA), F (3, 79) = 4.096, p = 0.009, η2 = 0.135). (C) AM-impaired COX activity was restored by Allo treatment (n = 3, one-way ANOVA, F (3, 8) = 8.386, p = 0.008, η2 = 0.759). (D) Allo treatment significantly increased the ratio of mitochondria with high MMP (n = 8–9, one-way ANOVA, F (3, 31) = 16.78, p < 0.001, η2 = 0.619). (E) Allo treatment significantly reversed AM-induced increase in CellROX levels (n = 9–10, one-way ANOVA, F (3, 34) = 8.973, p < 0.001, η2 = 0.442). (F) Bicuculline blocked the effect of Allo on promoting mitochondrial maximal respiration (n = 11–15 wells per group, two-way ANOVA, interaction: F (1, 50) = 5.213, p = 0.027, 9.003% of total variation; Allo: F (1, 50) = 2.198, p = 0.145, 3.795% of total variation; bicuculline: F (1, 50) = 0.8641, p = 0.357, 1.492% of total variation). (G) Nifedipine blocked the effect of Allo on promoting mitochondrial maximal respiration (n = 14–16 wells per group, two-way ANOVA, interaction: F (1, 56) = 5.924, p = 0.018, 7.808% of total variation; Allo: F (1, 56) = 9.577, p = 0.003, 12.62% of total variation; nifedipine: F (1, 56) = 3.704, p = 0.059, 4.882% of total variation). All bar graphs are presented as mean ± 95% CI with individual data points. Statistical significance was calculated using one-way ANOVA, followed by Holm-Sidak multiple comparisons specifically between three selected groups: AM versus Control, AM-Veh versus AM, and AM-Allo versus AM-Veh. The inhibitor’s effect was analyzed by two-way ANOVA, followed by Holm-Sidak multiple comparisons.
Collectively, the data indicate that Allo significantly increased neuronal mitochondrial respiration and oxidative phosphorylation while preventing free radical generation via GABAA and L-type Ca2+ channel-dependent Ca2+ signaling activation in hippocampal neurons.
Improved mitochondrial function could be coupled with mitochondrial biogenesis and dynamic restructuring (52). To assess whether the observed Allo-induced rise in neuronal mitochondrial function (section 3.2 above) required mitochondrial biogenesis or dynamics, Allo regulation of mitochondrial DNA copy number, complex protein levels, and mitochondrial subtype distribution was determined (16h treatment). In hippocampal neurons, Allo did not impact mitochondrial DNA copy number (Figure 4A). Consistent with this outcome, mitochondrial complex IV (COX) protein levels remained unchanged by Allo treatment (Figure 4B). AM significantly reduced mitochondrial fission/fusion ratio (Figures 4C, D, p = 0.031 vs. Control), without changing the protein expression ratio of DRP1 (Dynamin-1-like protein, an essential protein for mitochondrial fission) to OPA1 (OPA1 mitochondrial dynamin like GTPase, an essential protein for mitochondrial fusion) (Figure 4E). Allo did not affect the overall mitochondrial fission/fusion ratio (Figures 4C, D) nor the balance between DRP1 and OPA1 (Figure 4E). Together, Allo-induced improvement of mitochondrial function was not mediated via the direct regulation of mitochondrial biogenesis or dynamics but rather through promoting COX activity and increasing the ratio of high membrane potential mitochondria that promote mitochondrial efficiency (section 3.2 above).
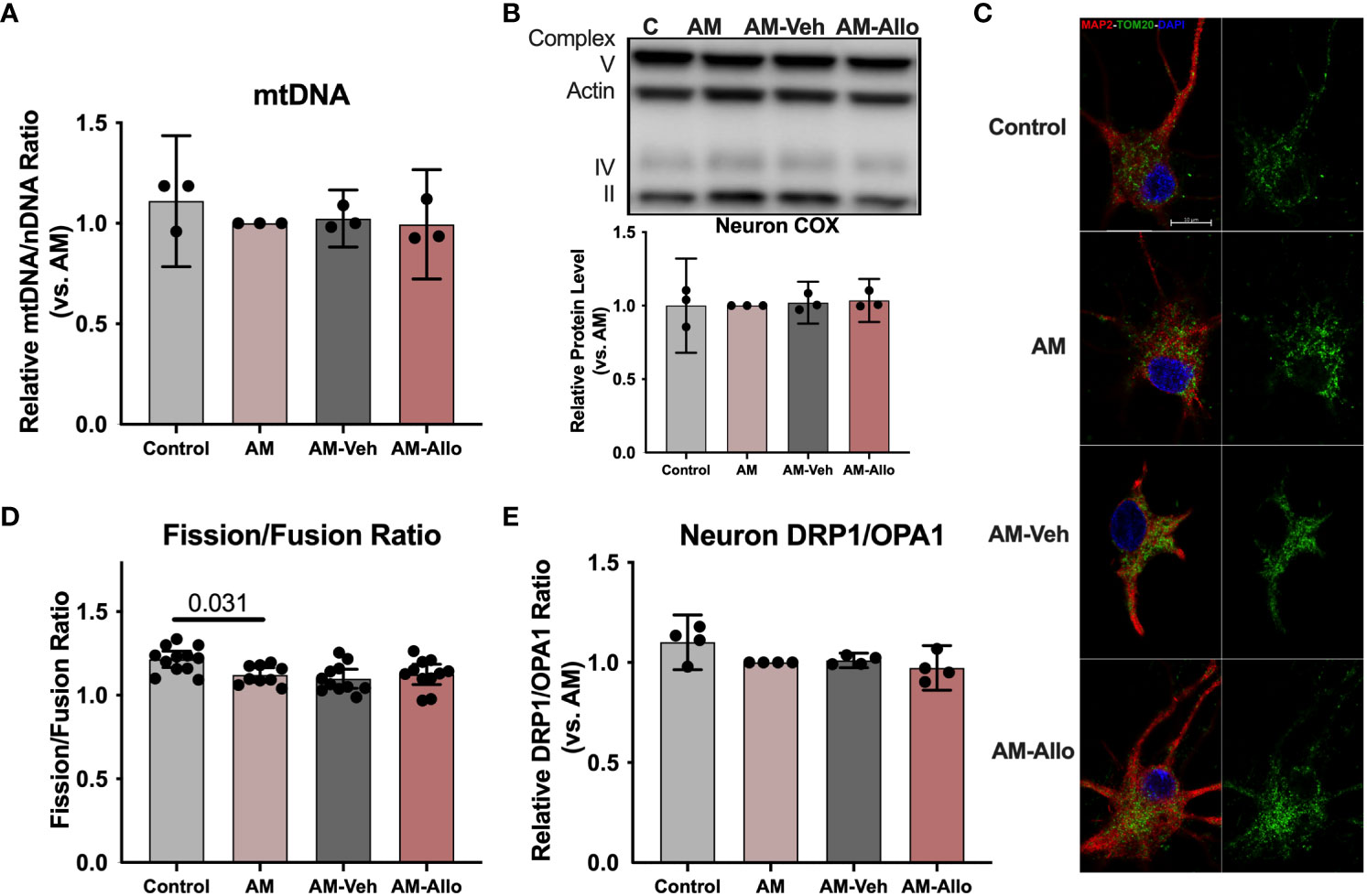
Figure 4 In hippocampal neurons, Allo does not affect mitochondrial biogenesis or dynamics. (A) Mitochondrial DNA copy number remained unchanged following Allo treatment (n = 3). (B) Mitochondrial complex IV protein levels were not changed by Allo treatment (n = 3). (C) Representative neuron images labeled using anti-MAP2 and anti-TOM20 antibodies and DAPI. (D) The overall mitochondrial fission/fusion ratio was significantly reduced in the AM group (Mitochondria were labeled using the anti-TOM20 antibody, n = 9–12, one-way ANOVA, F (3, 39) = 4.848, p = 0.006, η2 = 0.272) and was not reversed by Allo treatment. (E) The DRP1/OPA1 protein ratio (n = 4) was not affected by Allo treatment. All bar graphs are presented as mean ± 95% CI with individual data points. Statistical significance was calculated using one-way ANOVA, followed by Holm-Sidak multiple comparisons specifically between three selected groups: AM versus Control, AM-Veh versus AM, and AM-Allo versus AM-Veh.
Astrocyte-related neuroinflammation and oxidative stress are observed in and can contribute to Alzheimer’s disease (AD) pathology (53, 54). Therefore, the effect of Allo (24h treatment, Figure 5A) on astrocytic oxidative stress status and inflammatory activation was investigated. As shown in Figure 5B, AM induced an increase in cellular ROS levels (p < 0.001 vs. Control). Allo significantly decreased ROS levels (p = 0.020 vs. AM-Veh), which was accompanied by restored cellular ATP levels (Figure 5C, p = 0.002 vs. AM-Veh). These results indicated that Allo relieved astrocytic oxidative stress and promoted ATP generation. Consistent with these findings, broader transcriptomic analyses revealed that Allo treatment significantly activated stress response EIF2 (eukaryotic initiation factor-2) signaling and inhibited apoptosis signaling (Table 2). Notably, the inflammasome signaling pathway was inhibited by Allo treatment. Relative to Veh-treated group, a significant decrease in gene expression was detected for S100b (Figure 5D, p = 0.049, fold change = 0.940), Nlrc4 (NLR family CARD domain-containing protein 4, Figure 5E, p = 0.038, fold change = 0.614) and Pycard (PYD and CARD domain containing, Figure 5F, p = 0.039, fold change = 0.615). In addition, IPA upstream regulator analysis predicted that Allo treatment would activate TGFB1 (Transforming growth factor b1), a key factor in preventing inflammation in neurological disorders (Figure 5G) (55). Together, these results indicated that Allo prevented astrocytic oxidative stress and inhibited apoptosis and pro-inflammation activation.
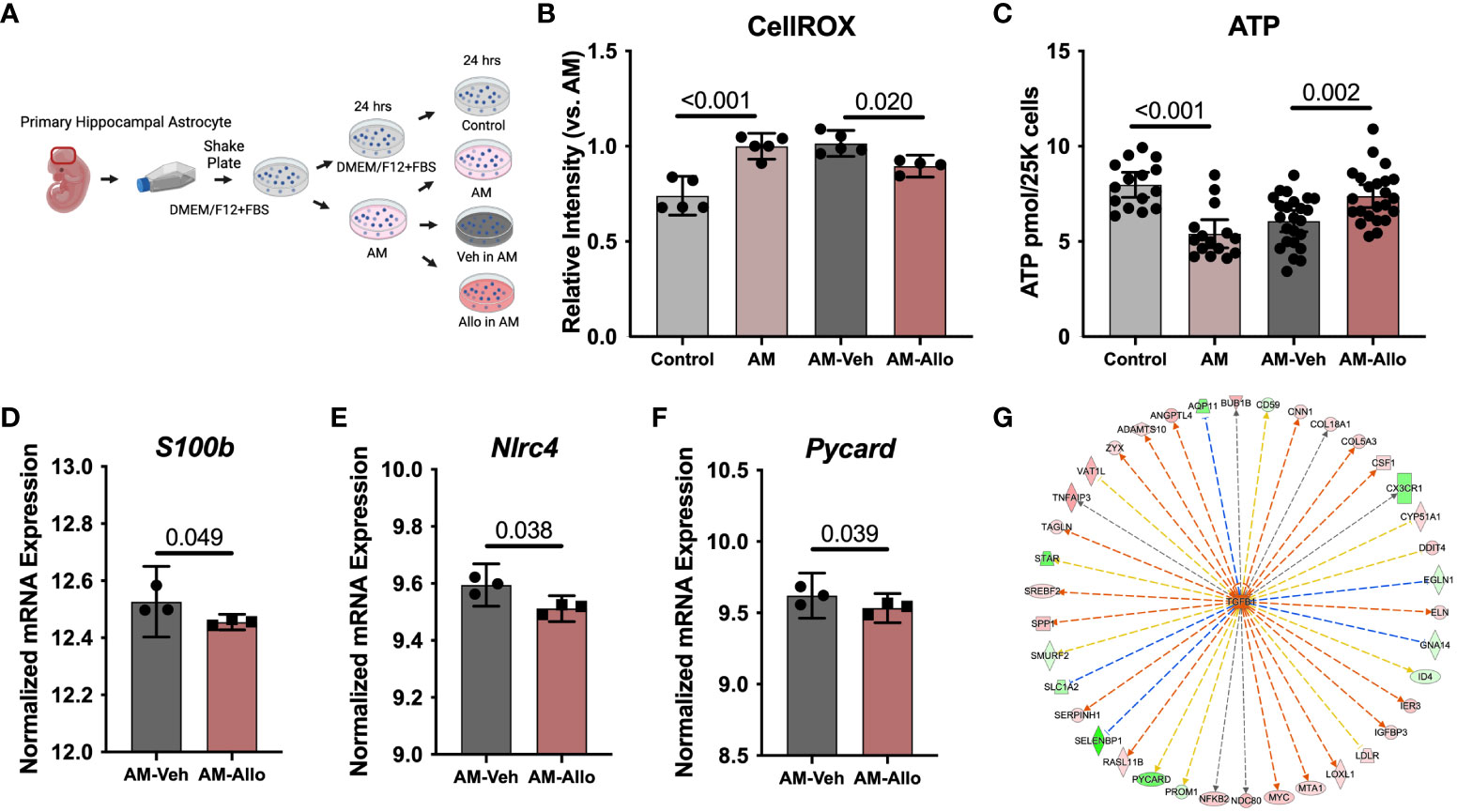
Figure 5 Allo decreased AM-induced oxidative stress and pro-inflammatory signaling in astrocytes. (A) The schematic diagram of Allo treatment (Created with BioRender.com). (B) Allo treatment significantly reversed AM-induced increase in CellROX levels (n = 4–5, one-way ANOVA, F (3, 15) = 22.03, p < 0.001, η2 = 0.815). (C) ATP levels were significantly restored by Allo treatment (n = 15–24, one-way ANOVA, F (3, 73) = 13.25, p < 0.001, η2 = 0.353). Relative to AM-Veh group, Allo treatment decreased the RNA levels of S100b (D) (n = 3), Nlrc4 (E) (n = 3) and Pycard (F) (n = 3). (G) IPA predicted TGFB1 would be activated by Allo treatment. All bar graphs are presented as mean ± 95% CI with individual data points. Statistical significance for RNA-seq data was calculated by DESeq2 V1.18.1. Other data were analyzed using one-way ANOVA, followed by Holm-Sidak multiple comparisons specifically between three selected groups: AM versus Control, AM-Veh versus AM, and AM-Allo versus AM-Veh.
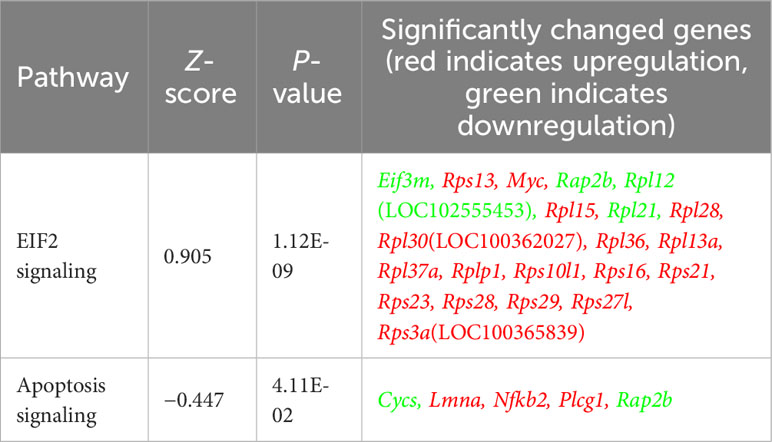
Table 2 Allo significantly increased stress response EIF2 signaling and inhibited apoptosis signaling in astrocytes.
Allo-induced reduction in oxidative stress and increased ATP production is consistent with improved mitochondrial function. Furthermore, because the inflammasome can be activated by ROS-generating mitochondria (56), enhanced mitochondria function would lead to inhibition of inflammasome activation. Therefore, we hypothesized that the effect of Allo to reverse astrocytic oxidative stress, apoptosis, and pro-inflammation activation was associated with its regulation of mitochondrial function. To address this hypothesis, mitochondrial respiration and membrane potential were assessed. AM significantly reduced astrocytic mitochondrial spare (Figures 6A, B, p < 0.001 vs. Control) and maximal respiration (Figures 6A, C, p < 0.001 vs. Control), which was accompanied by a significant decrease in the ratio of mitochondria with high membrane potential (Figure 6D, p < 0.001 vs. Control). Allo treatment (24h treatment) induced a trend toward increased spare respiration (Figure 6B, p = 0.077 vs. AM-Veh) and increased maximal respiration significantly (Figure 6C, p = 0.049 vs. AM-Veh), without affecting mitochondrial membrane potential (Figure 6D). Furthermore, bicuculline (Figure 6E, p < 0.001 vs. Allo) and nifedipine (Figure 6F, p = 0.006 vs. Allo) significantly blocked the Allo effect on promoting mitochondrial maximal respiration. Together, these results indicate that Allo restored astrocytic mitochondrial function, which required both GABAA and L-type Ca2+ channel activation.
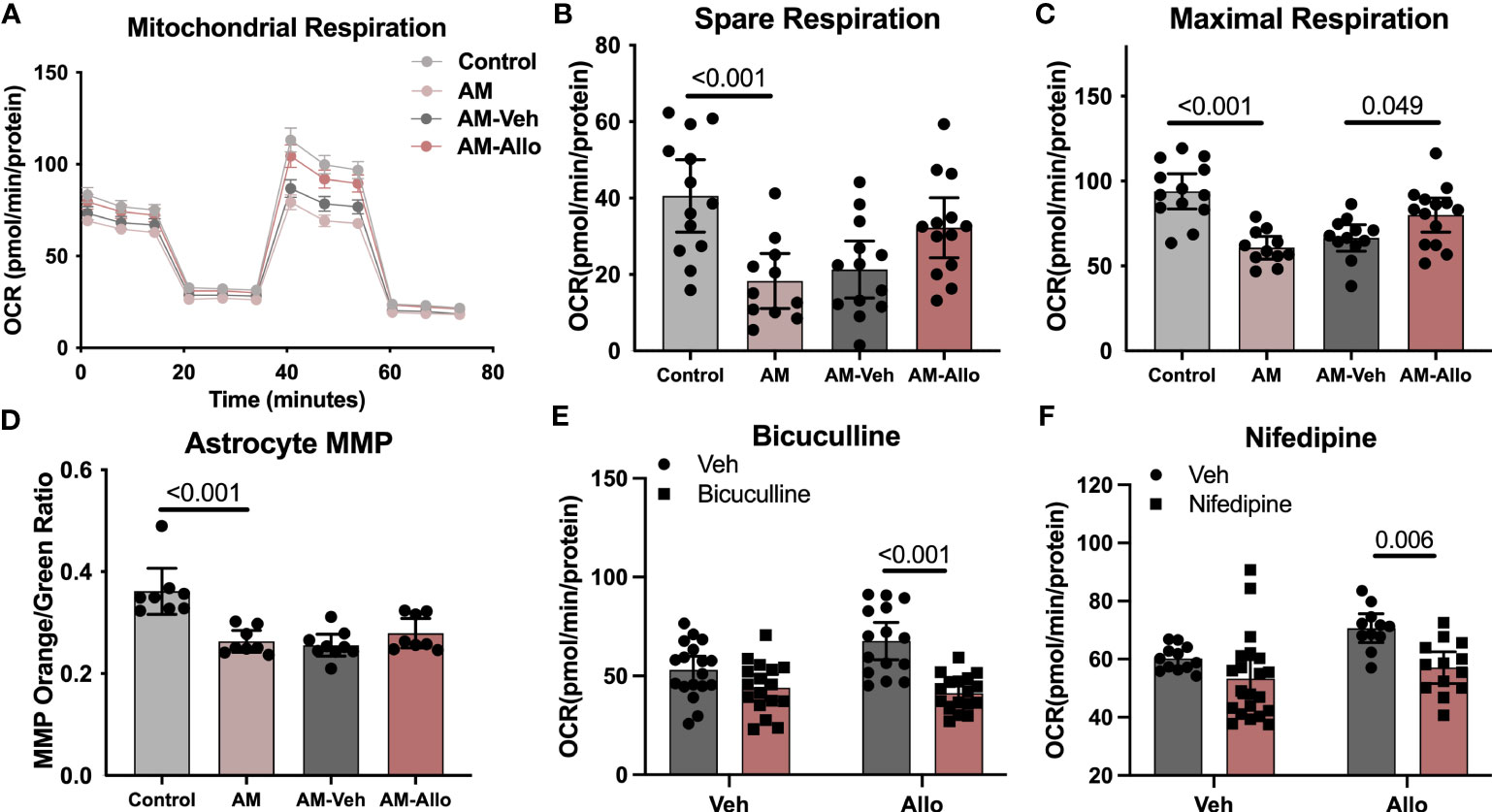
Figure 6 Allo restored astrocytic mitochondrial function via GABAA and L-type calcium channels. (A) Representative mitochondrial stress test results using the Seahorse XF96 Extracellular Flux Analyzer (mean ± S.E.M.). AM significantly reduced astrocytic mitochondrial spare (B) (One-way analysis of variance (ANOVA), F (3, 46) = 7.483, p < 0.001, η2 = 0.328) and maximal respiration (C) (one-way ANOVA, F (3, 46) = 12.26, p < 0.001, η2 = 0.444). Allo treatment induced a trend toward increased spare respiration and significantly increased maximal respiration (B, C) (n = 11–14 wells per group). (D) The AM-induced significant decrease in astrocytic mitochondrial membrane potential (n = 8–9, one-way ANOVA, F (3, 29) = 14.03, p < 0.001, η2 = 0.592) was not reversed by Allo treatment. (E) Bicuculline blocked Allo’s effect on promoting mitochondrial maximal respiration (n = 15–18 wells per group, two-way ANOVA, interaction: F (1, 63) = 7.005, p = 0.010, 7.05% of total variation; Allo: F (1, 63) = 3.256, p = 0.076, 3.277% of total variation; bicuculline: F (1, 63) = 28.29, p < 0.001, 28.47% of total variation). (F) Nifedipine blocked Allo’s effect on promoting mitochondrial maximal respiration (n = 11–21 wells per group, two-way ANOVA, interaction: F (1, 53) = 1.340, p = 0.252, 1.834% of total variation; Allo: F (1, 53) = 6.115, p = 0.017, 8.367% of total variation; nifedipine: F (1, 53) = 12.53, p < 0.001, 17.14% of total variation). All bar graphs are presented as mean ± 95% CI with individual data points. Statistical significance was calculated using one-way ANOVA, followed by Holm-Sidak multiple comparisons specifically between three selected groups: AM versus Control, AM-Veh versus AM, and AM-Allo versus AM-Veh. The inhibitor’s effect was analyzed by two-way ANOVA, followed by Holm-Sidak multiple comparisons.
To investigate whether the Allo-induced increase in astrocytic mitochondrial function was associated with regulation of mitochondrial biogenesis or dynamics, quantitative transcriptomic analyses of key mitochondrial biogenesis regulators, mitochondria DNA copy number, and protein levels were conducted (24h treatment). Real-time PCR outcomes indicated that Allo significantly increased both Nrf1 (nuclear respiratory factor 1, Figure 7B, p = 0.023 vs. AM-Veh) and Tfam (mitochondrial transcription factor A, Figure 7C, p = 0.026 vs. AM-Veh) mRNA levels without impacting Ppargc1a (PGC1α, PPARG coactivator 1 alpha, Figure 7A) mRNA expression. Consistent with transcriptional data, upstream regulator pathway analysis predicted that Allo treatment would activate NRF1 signaling (Figure 7D). Allo treatment significantly increased OXPHOS gene expression (Figures 7E, F), including Ndufa4, Ndufab1, Ndufb2, Uqcr10, Uqcrq, Cox6b1, Cox6c, Cox7a2, Cox7b, Cox8a, Atp5f1d, Atp5mg, and Atp5pf, and significantly increased astrocytic mitochondrial DNA copy number (Figure 7G, p = 0.044 vs. AM-Veh). Consistent with the transcriptomic profile, Allo significantly increased mitochondrial COX protein level (Figure 7H, p = 0.015 vs. AM-Veh) and activity (Figure 7I, p = 0.048 vs. AM-Veh). Inhibiting either the GABAA receptor with bicuculline or the L-type Ca2+ channel with nifedipine (Figure 7J, p = 0.023 and p = 0.023 vs. Allo, respectively) blocked the Allo-induced increase in mitochondrial DNA copy number. These data indicated that Allo regulation of mitochondrial biogenesis required both GABAA and L-type Ca2+ channel activation.
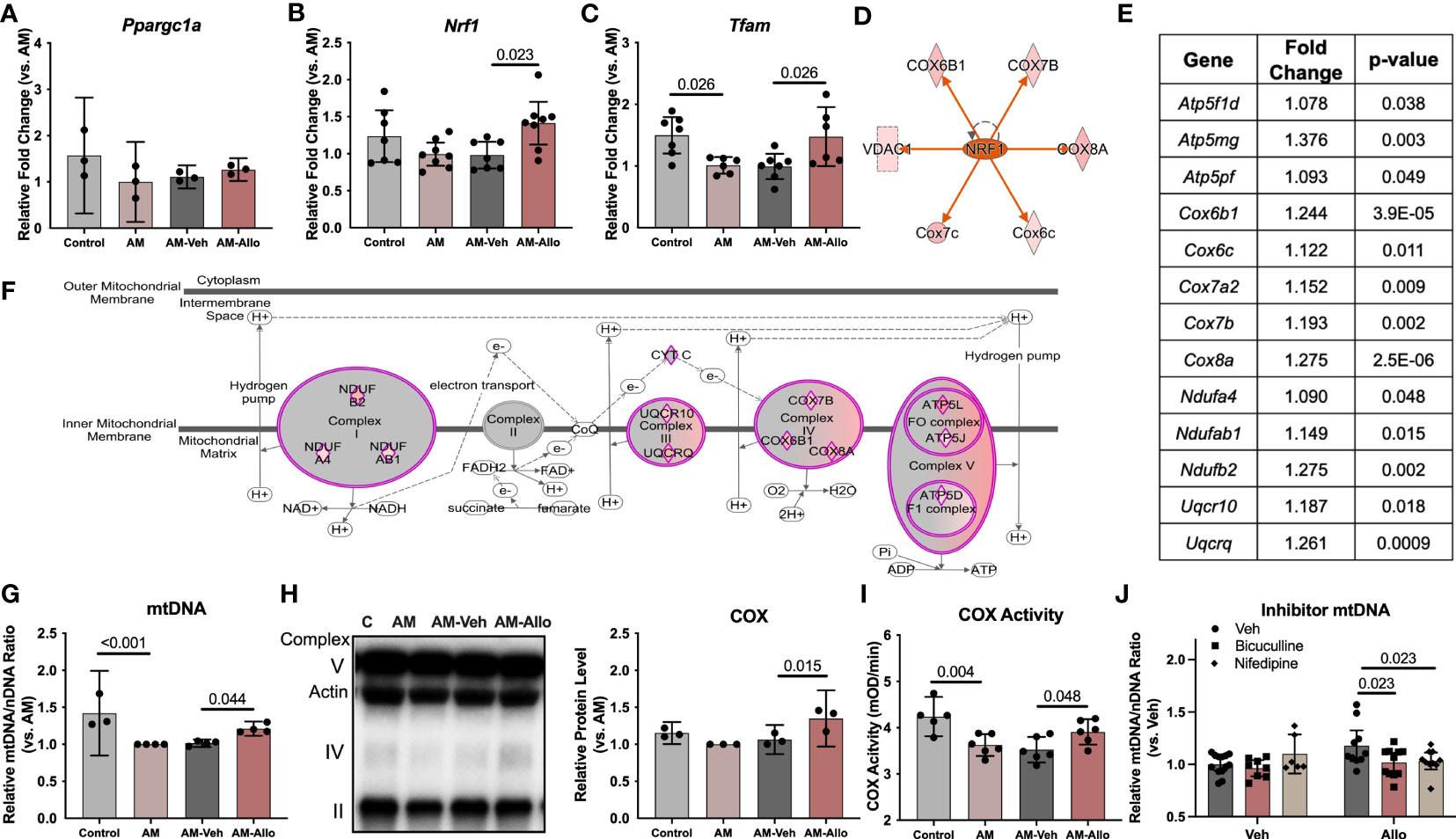
Figure 7 Allo promoted astrocytic mitochondrial biogenesis via NRF1-TFAM signaling. (A) Ppargc1a (PGC1alpha) mRNA level was not affected by Allo treatment (n = 3). Allo treatment significantly increased Nrf1 (B) (n = 7–8, one-way ANOVA, F (3, 26) = 3.985, p = 0.018, η2 = 0.315) and Tfam (C) (n = 6–7, one-way ANOVA, F (3, 22) = 5.577, p = 0.005, η2 = 0.432) mRNA levels compared to AM-Veh group. (D) Upstream regulator analysis predicted NRF1 would be activated by Allo treatment. (E, F). Allo significantly increased nuclear-encoded gene expression for mitochondrial complex I, III, IV, and V (in purple diamonds, IPA). (G) Allo treatment significantly increased mitochondrial DNA copy number (n = 3–4, one-way ANOVA, F (3, 11) = 12.13, p < 0.001, η2 = 0.768). Allo treatment specifically and significantly increased COX protein levels (H), (n = 3, one-way ANOVA, F (3, 8) = 8.328, p = 0.008, η2 = 0.758) which was coupled with a significant increase in COX activity (I) (n = 5–6, one-way ANOVA, F (3, 19) = 7.539, p = 0.002, η2 = 0.544). (J) Bicuculline and nifedipine significantly inhibited Allo effect on promoting mitochondrial biogenesis (n = 6–13, two-way ANOVA, interaction: F (2, 52) = 3.540, p = 0.036, 10.64% of total variation; Allo: F (1, 52) = 2.112, p = 0.152, 3.174% of total variation; inhibitor: F (2, 52) = 2.630, p = 0.082, 7.906% of total variation). All bar graphs are presented as mean ± 95% CI with individual data points. Statistical significance was calculated using one-way ANOVA, followed by Holm-Sidak multiple comparisons specifically between three selected groups: AM versus Control, AM-Veh versus AM, and AM-Allo versus AM-Veh. The inhibitor’s effect was analyzed by two-way ANOVA, followed by Holm-Sidak multiple comparisons.
At the structural level, AM induced hyperfused mitochondrial networks in astrocytes as indicated by a significantly decreased mitochondrial fission fraction (p < 0.001 vs. Control) and fission/fusion ratio (p < 0.001 vs. Control) (Figures 8A–C). Allo treatment (24h treatment) significantly relieved mitochondrial hyperfusion (Figure 8A) by increasing the fraction of mitochondrial fission product (Figure 8B, p = 0.028 vs. AM-Veh), which led to the corresponding restoration of the fission/fusion ratio in the Allo-treated group (Figure 8C, p = 0.048 vs. AM-Veh). Consistent with the mitochondrial restructuring, the protein expression ratio of DRP1 to OPA1 was also significantly increased by Allo treatment (Figure 8D, p = 0.037 vs. AM-Veh).
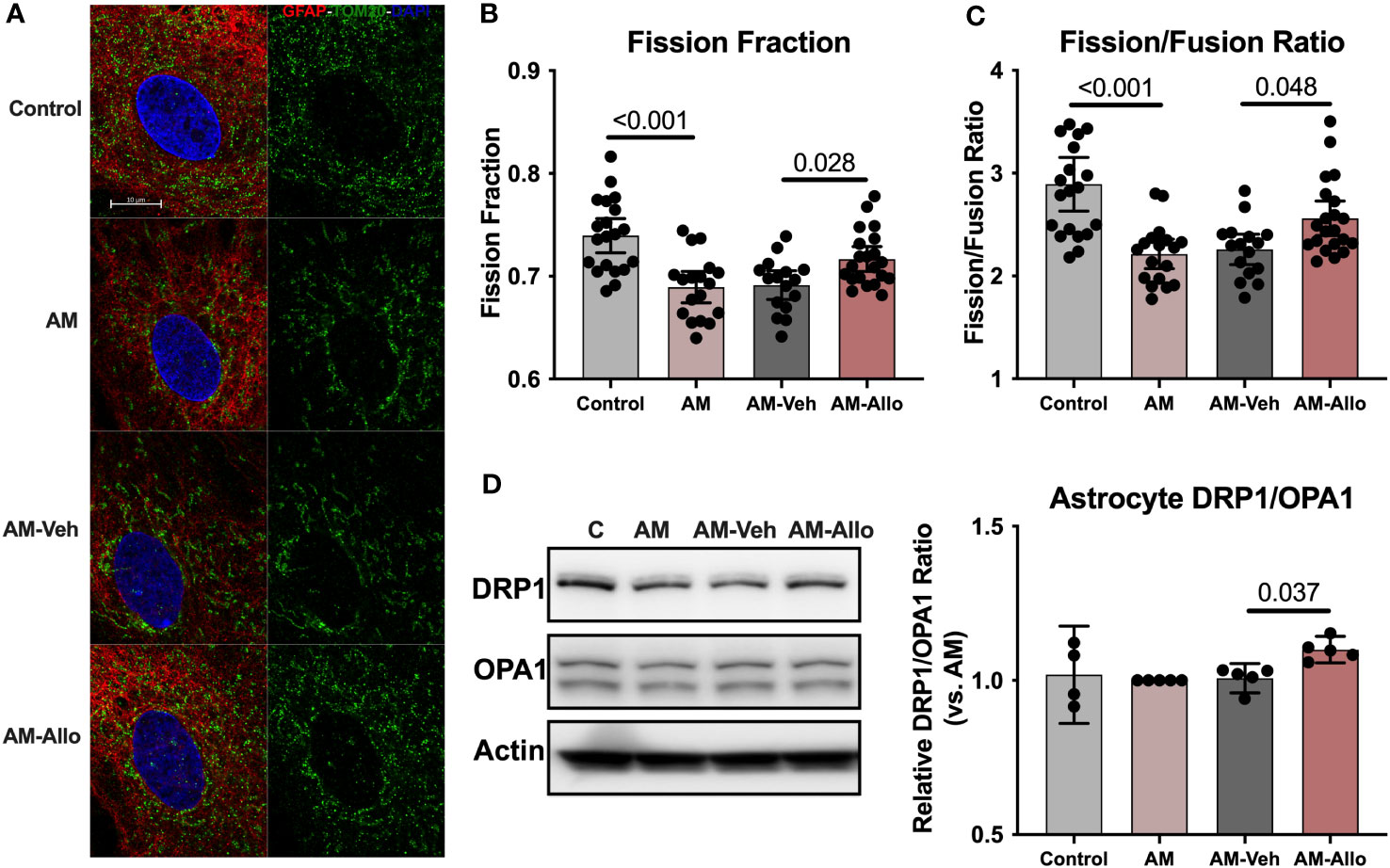
Figure 8 Allo relieved AM-induced astrocytic mitochondrial hyperfusion by increasing DRP1/OPA1 protein ratio. (A) Representative astrocyte images labeled with anti-GFAP and anti-TOM20 antibodies and DAPI. Allo treatment significantly increased mitochondrial fission product fraction (B) (Mitochondria were labeled using the anti-TOM20 antibody, one-way ANOVA, F (3, 71) = 11.56, p < 0.001, η2 = 0.328) and restored the mitochondrial fission/fusion ratio compared to AM-Veh groups (C), (n = 16–21, one-way ANOVA, F (3, 71) = 11.79, p < 0.001, η2 = 0.333). (D) The DRP1/OPA1 protein ratio was significantly increased by Allo treatment (n = 4–5, one-way ANOVA, F (3, 15) = 3.976, p = 0.029, η2 = 0.443). All bar graphs are presented as mean ± 95% CI with individual data points. Statistical significance was calculated using one-way ANOVA, followed by Holm-Sidak multiple comparisons specifically between three selected groups: AM versus Control, AM-Veh versus AM, and AM-Allo versus AM-Veh.
Collectively, the data indicate that Allo promoted astrocytic mitochondrial biogenesis through NRF1-TFAM activation and increased DRP1/OPA1 protein ratio to reverse stress-induced hyperfused mitochondrial network to restore mitochondrial and bioenergetic homeostasis (57).
Our previous analyses demonstrated that Allo exerted pleiotropic actions in the brain that included neurogenesis, oligogenesis, human and rodent neural stem cell regeneration, improved cognitive function, enhanced glucose metabolism, increased mitochondrial respiration and biogenesis, and reduced burden of AD pathology (1–3, 9, 10, 21, 26, 58). Herein, we investigated the potential of a common mechanistic signaling pathway that could serve to unify the pleiotropic outcomes induced by Allo in neural cells. Outcomes of those analyses provide strong support that Allo’s pleiotropic actions activate and are dependent upon Ca2+ signaling. Allo activation of Ca2+ signaling in both neurons and astrocytes serves to integrate multiple systems of biology critical to sustained brain function and health.
Key to calcium signaling as a unifying mechanism of Allo action is the expression of the NKCC1 chloride transporter (Figure 9, created with BioRender.com). Hippocampal immature neurons and astrocytes express NKCC1 and consequently harbor a high-intracellular chloride concentration. In NKCC1-expressing immature neurons, Allo activation of GABAA channels results in Cl− efflux and subsequent depolarization of the membrane potential and activation of the voltage-dependent L-type Ca2+ channel, which in turn results in an influx of Ca2+ (3). The Allo-induced rise in intracellular Ca2+ activates CREB/ELK1/c-Jun signaling through CaMKII and ERK1/2 activation. In neurons, activation of CREB/ELK1/c-Jun signaling is a well-established neural plasticity pathway (61, 62), which is consistent with the significant increases in both neuronal morphology and activation of transcriptional networks required for synaptic plasticity. The rise in intracellular Ca2+ results in sequestration of Ca2+ into the mitochondria, increasing mitochondrial membrane potential and promoting mitochondrial respiration (63).
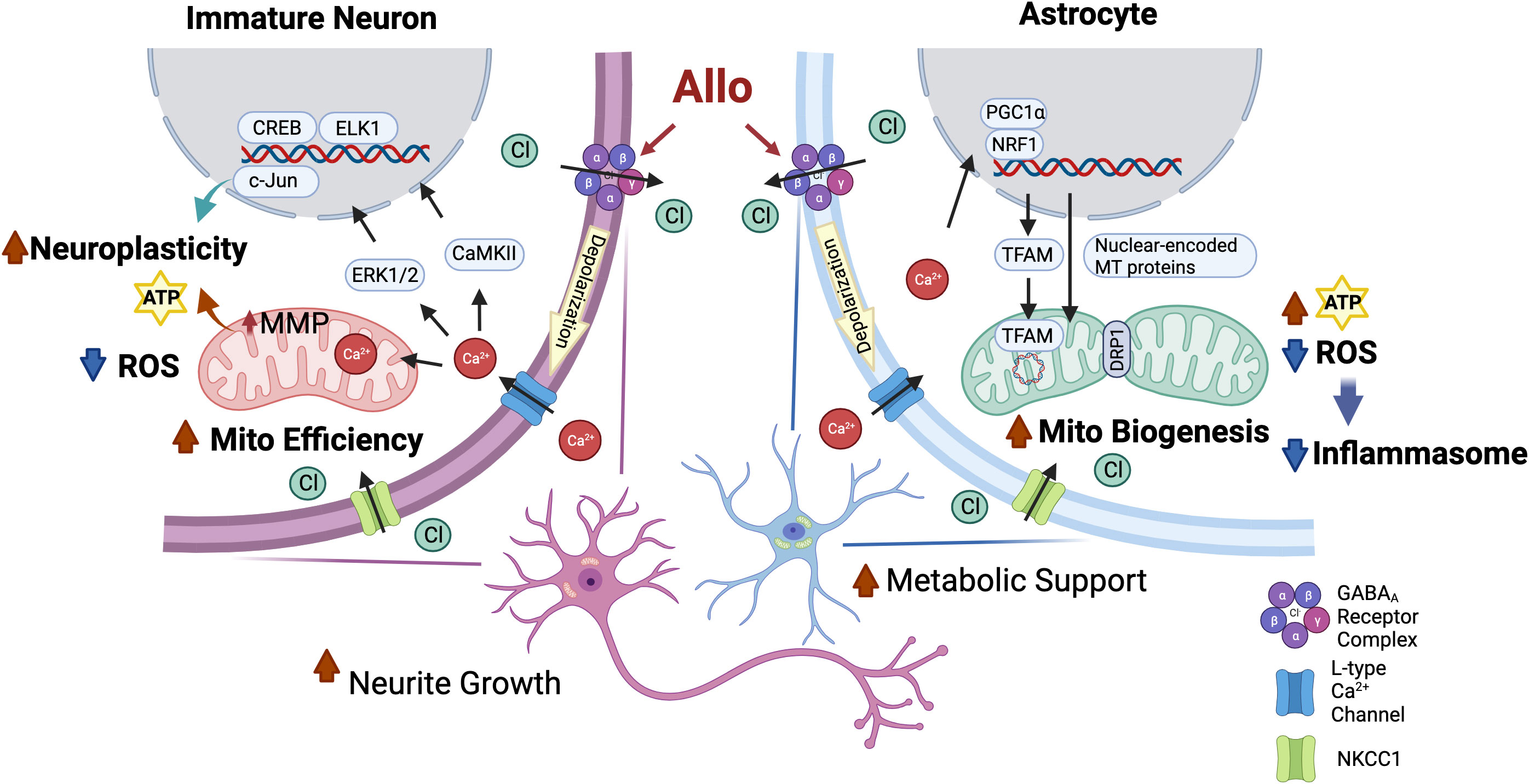
Figure 9 Common mechanisms of action of Allo to activate neuronal and astrocytic functions required for brain regeneration. Both hippocampal immature neurons and astrocytes exhibit a high-intracellular chloride concentration (31, 34). Allo activation of GABAA channels results in Cl- efflux and subsequent depolarization of the membrane potential and activation of the voltage-dependent L-type Ca2+ channel, which in turn results in an influx of Ca2+ (3, 35). In immature neurons, increased intracellular Ca2+ levels activate downstream CREB/ELK1/c-Jun signaling through CaMKII and ERK1/2 activation, which in turn lead to increased neuroplasticity and reduced oxidative stress. The rise in intracellular Ca2+ results in sequestration of Ca2+ into the mitochondria that promotes mitochondrial respiration and mitochondrial membrane potential. In astrocytes, GABAA and L-type calcium channel activation triggers the activation of NRF1-TFAM signaling and increases the DRP1/OPA1 protein ratio to increase mitochondrial biogenesis and dynamics and consequent ATP production, potentially via calcium-induced PGC-1α activation (59, 60). Furthermore, Allo reduced oxidative stress, thereby reducing activation of inflammasome and apoptosis signaling.
Astrocytes also express both GABAA receptors and NKCC1 transporters and produce and release GABA (64). Allo activation of GABAA receptors in astrocytes results in activation of the voltage-dependent L-type Ca2+ channel (35) and consequent activation of NRF1-TFAM signaling and increased DRP1/OPA1 protein ratio, promoting mitochondrial biogenesis and dynamics, potentially via Ca2+-induced PGC-1α activation (59, 60). Furthermore, Allo reduced oxidative stress and enhanced mitochondrial function, leading to increased ATP concentration and reduced oxidative stress, thereby reducing activation of inflammasome and apoptosis signaling.
These results were consistent with our previous findings demonstrating that Allo induced a rapid and transient increase in cytosolic Ca2+ by GABAA and L-type calcium channel activation in embryonic hippocampal neurons from days in vitro 3–10 (28) and increased phosphorylation of CREB in vivo (65).
The quality and capacity of mitochondria decline with aging, and this is linked to the development of age-related diseases (66). To maintain an adequate and functional mitochondrial population, mitochondrial biogenesis and dynamics are highly regulated. Herein, our results demonstrated that in-vitro Allo treatment promoted mitochondrial function in hippocampal neurons through increased mitochondrial efficiency whereas in astrocytes Allo promoted mitochondrial biogenesis and dynamics.
Allo regulation of mitochondria in hippocampal neurons targeted mitochondrial respiratory efficiency without affecting mitochondrial number, potentially through direct stimulation of mitochondrial oxidative phosphorylation due to increased sequestration of Ca2+ into the mitochondria (63). Although mitochondrial dynamics overall were not affected by Allo, the restoration of neurite growth in neurons upon Allo treatment suggested improved axonal transport of mitochondria, which is required for synaptic plasticity (67). In astrocytes, Allo activated mitochondrial biogenesis through the NRF1-TFAM signaling pathway and attenuated the fused mitochondrial phenotype by upregulating DRP1/OPA1 protein ratio.
Consistent with our previous in-vivo findings that Allo improved mitochondrial function and reduced expression of mitochondrial fusion-related genes in ovariectomized 3xTgAD females (10), in-vitro results reported herein indicate that Allo promotes mitochondrial function in both immature neurons and astrocytes through cell-specific mechanisms. Thus, Allo-mediated potentiation of mitochondrial function observed in vivo is likely the result of promoting mitochondrial respiration in immature neurons and mitochondrial biogenesis in astrocytes.
Transcriptomic pathway analysis revealed that, in hippocampal immature neurons, Allo activated the Ca2+/CREB signaling pathway which resulted in the activation of synaptogenesis and long-term potentiation pathways. Furthermore, a rise in mitochondrial Ca2+ via activation of L-type calcium channels stimulates mitochondrial oxidative respiration, which is essential for induction and maintenance of neuroplasticity (68). This Allo-induced calcium signaling depends on a high-intracellular chloride concentration, making it mechanistically inapplicable to mature neurons. However, our previous analyses demonstrated that Allo promotes neurogenesis in the 3xTgAD and normal-aged mouse hippocampus (13, 21, 26, 28). Furthermore, Allo promotes neuronal and oligodendrocyte differentiation of neural stem cells (11). These findings collectively support the potential of Allo to promote regeneration in the developing, adult and AD.
In astrocytes, Allo promoted mitochondrial biogenesis that increased mitochondrial respiratory capacity, which was dependent upon L-type calcium channel activation. Furthermore, Allo significantly reduced inflammasome activation by downregulating the expression of Nlrc4 and Pycard. Additionally, Allo activated the TGFB1 signaling pathway, consistent with Allo’s effect on TGFB1 signaling in cancer cell models (69). NLRC4 has been reported to mediate inflammasome activation in microglia and astrocytes (70), which play an important role in neuroinflammation associated with AD (71). Furthermore, TGFB1 has been reported to exert neuroprotective effects in AD, and its deficiency is associated with Aβ pathology and neurofibrillary tangle formation (72). Therefore, Allo’s anti-inflammation effect, manifested as inhibition of NLRC4-mediated inflammasome activation and activation of TGFB1 pathway, could contribute to its therapeutic effect in reducing Aβ burden observed in vivo (21).
In conclusion, Allo is a neurosteroid with pleiotropic actions in the brain that span neural regeneration, bioenergetics, and immune systems of biology, which collectively impact synaptic plasticity, cognition, burden of neurodegenerative disease, and recovery of brain function. Unlike classic steroids, Allo exerts pleiotropic effects through non-nuclear receptors. Outcomes of analyses reported herein advance the breadth of knowledge regarding Allo’s pleiotropic action in both immature neurons and astrocytes. Allo’s regulation of mitochondrial function in both immature neurons and astrocytes is dependent on both GABAA and L-type calcium activation, suggesting the calcium signaling pathway as a common mechanism of action. The calcium signaling cascade provides a unifying mechanistic pathway that bridges multiple systems of biology in multiple cell types in the brain.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
All animal studies were performed following National Institutes of Health guidelines on the use of laboratory animals and all procedures were approved by the University of Arizona Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
TW: Conceptualization, Investigation, Writing – original draft, Data curation, Writing – review & editing, Formal analysis. SC: Investigation, Writing – original draft. ZM: Investigation, Writing – original draft. YS: Writing – original draft, Formal analysis. RB: Conceptualization, Supervision, Writing – review & editing, Funding acquisition.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by NIA grants U01-AG031115, U01-AG047222, and R01AG063826 to RB.
We thank James Brett Stanton, Francesca Vitali and John W. McLean for their contributions.
Author RB is the founder and president of NeuTherapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Irwin RW, Brinton RD. Allopregnanolone as regenerative therapeutic for Alzheimer’s disease: translational development and clinical promise. Prog Neurobiol (2014) 113:40–55. doi: 10.1016/j.pneurobio.2013.08.004
2. Irwin RW, Solinsky CM, Brinton RD. Frontiers in therapeutic development of allopregnanolone for Alzheimer’s disease and other neurological disorders. Front Cell Neurosci (2014) 8:203. doi: 10.3389/fncel.2014.00203
3. Brinton RD. Neurosteroids as regenerative agents in the brain: therapeutic implications. Nat Rev Endocrinol (2013) 9(4):241–50. doi: 10.1038/nrendo.2013.31
4. Brinton RD, Yao J, Yin F, Mack WJ, Cadenas E. Perimenopause as a neurological transition state. Nat Rev Endocrinol (2015) 11(7):393–405. doi: 10.1038/nrendo.2015.82
5. Diviccaro S, Cioffi L, Falvo E, Giatti S, Melcangi RC. Allopregnanolone: An overview on its synthesis and effects. J Neuroendocrinol (2022) 34(2):e12996. doi: 10.1111/jne.12996
6. Belelli D, Brown AR, Mitchell SJ, Gunn BG, Herd MB, Phillips GD, et al. Endogenous neurosteroids influence synaptic GABA(A) receptors during postnatal development. J Neuroendocrinol (2018) 30(2). doi: 10.1111/jne.12537
7. Irwin RW, Wang JM, Chen S, Brinton RD. Neuroregenerative mechanisms of allopregnanolone in Alzheimer’s disease. Front Endocrinol (Lausanne) (2011) 2:117. doi: 10.3389/fendo.2011.00117
8. Hernandez GD, Brinton RD. Allopregnanolone: Regenerative therapeutic to restore neurological health. Neurobiol Stress (2022) 21:100502. doi: 10.1016/j.ynstr.2022.100502
9. Singh C, Liu L, Wang JM, Irwin RW, Yao J, Chen S, et al. Allopregnanolone restores hippocampal-dependent learning and memory and neural progenitor survival in aging 3xTgAD and nonTg mice. Neurobiol Aging (2012) 33(8):1493–506. doi: 10.1016/j.neurobiolaging.2011.06.008
10. Wang T, Yao J, Chen S, Mao Z, Brinton RD. Allopregnanolone reverses bioenergetic deficits in female triple transgenic Alzheimer’s mouse model. Neurotherapeutics (2020) 17(1):178–88. doi: 10.1007/s13311-019-00793-6
11. Chen S, Wang T, Yao J, Brinton RD. Allopregnanolone promotes neuronal and oligodendrocyte differentiation in vitro and in vivo: therapeutic implication for Alzheimer’s disease. Neurotherapeutics (2020) 17(4):1813–24. doi: 10.1007/s13311-020-00874-x
12. Boero G, Porcu P, Morrow AL. Pleiotropic actions of allopregnanolone underlie therapeutic benefits in stress-related disease. Neurobiol Stress (2020) 12:100203. doi: 10.1016/j.ynstr.2019.100203
13. Wang JM, Johnston PB, Ball BG, Brinton RD. The neurosteroid allopregnanolone promotes proliferation of rodent and human neural progenitor cells and regulates cell-cycle gene and protein expression. J Neurosci (2005) 25(19):4706–18. doi: 10.1523/JNEUROSCI.4520-04.2005
14. Raikes AC, Hernandez GD, Matthews DC, Lukic AS, Law M, Shi Y, et al. Exploratory imaging outcomes of a phase 1b/2a clinical trial of allopregnanolone as a regenerative therapeutic for Alzheimer’s disease: Structural effects and functional connectivity outcomes. Alzheimers Dement (N Y) (2022) 8(1):e12258. doi: 10.1002/trc2.12258
15. Sun C, Ou X, Farley JM, Stockmeier C, Bigler S, Brinton RD, et al. Allopregnanolone increases the number of dopaminergic neurons in substantia nigra of a triple transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res (2012) 9(4):473–80. doi: 10.2174/156720512800492567
16. Leonelli E, Bianchi R, Cavaletti G, Caruso D, Crippa D, Garcia-Segura LM, et al. Progesterone and its derivatives are neuroprotective agents in experimental diabetic neuropathy: a multimodal analysis. Neuroscience (2007) 144(4):1293–304. doi: 10.1016/j.neuroscience.2006.11.014
17. Melcangi RC, Magnaghi V, Cavarretta I, Zucchi I, Bovolin P, D’Urso D, et al. Progesterone derivatives are able to influence peripheral myelin protein 22 and P0 gene expression: possible mechanisms of action. J Neurosci Res (1999) 56(4):349–57. doi: 10.1002/(SICI)1097-4547(19990515)56:4<349::AID-JNR3>3.0.CO;2-H
18. Kalakh S, Mouihate A. Enhanced remyelination during late pregnancy: involvement of the GABAergic system. Sci Rep (2019) 9(1):7728. doi: 10.1038/s41598-019-44050-4
19. Noorbakhsh F, Baker GB, Power C. Allopregnanolone and neuroinflammation: a focus on multiple sclerosis. Front Cell Neurosci (2014) 8:134. doi: 10.3389/fncel.2014.00134
20. Napoli E, Schneider A, Wang JY, Trivedi A, Carrillo NR, Tassone F, et al. Allopregnanolone treatment improves plasma metabolomic profile associated with GABA metabolism in fragile X-associated tremor/ataxia syndrome: a pilot study. Mol Neurobiol (2019) 56(5):3702–13. doi: 10.1007/s12035-018-1330-3
21. Chen S, Wang JM, Irwin RW, Yao J, Liu L, Brinton RD. Allopregnanolone promotes regeneration and reduces β-amyloid burden in a preclinical model of Alzheimer’s disease. PloS One (2011) 6(8):e24293. doi: 10.1371/journal.pone.0024293
22. Liao G, Cheung S, Galeano J, Ji AX, Qin Q, Bi X. Allopregnanolone treatment delays cholesterol accumulation and reduces autophagic/lysosomal dysfunction and inflammation in Npc1-/- mouse brain. Brain Res (2009) 1270:140–51. doi: 10.1016/j.brainres.2009.03.027
23. Balan I, Aurelian L, Schleicher R, Boero G, O’Buckley T, Morrow AL. Neurosteroid allopregnanolone (3alpha,5alpha-THP) inhibits inflammatory signals induced by activated MyD88-dependent toll-like receptors. Transl Psychiatry (2021) 11(1):145. doi: 10.1038/s41398-021-01266-1
24. Jolivel V, Brun S, Biname F, Benyounes J, Taleb O, Bagnard D, et al. Microglial cell morphology and phagocytic activity are critically regulated by the neurosteroid allopregnanolone: A possible role in neuroprotection. Cells (2021) 10(3):698. doi: 10.3390/cells10030698
25. Balan I, Beattie MC, O’Buckley TK, Aurelian L, Morrow AL. Endogenous neurosteroid (3alpha,5alpha)3-hydroxypregnan-20-one inhibits toll-like-4 receptor activation and pro-inflammatory signaling in macrophages and brain. Sci Rep (2019) 9(1):1220. doi: 10.1038/s41598-018-37409-6
26. Wang JM, Singh C, Liu L, Irwin RW, Chen S, Chung EJ, et al. Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA (2010) 107(14):6498–503. doi: 10.1073/pnas.1001422107
27. Wang JY, Trivedi AM, Carrillo NR, Yang J, Schneider A, Giulivi C, et al. Open-label allopregnanolone treatment of men with fragile X-associated tremor/ataxia syndrome. Neurotherapeutics (2017) 14(4):1073–83. doi: 10.1007/s13311-017-0555-6
28. Wang JM, Brinton RD. Allopregnanolone-induced rise in intracellular calcium in embryonic hippocampal neurons parallels their proliferative potential. BMC Neurosci (2008) 9 Suppl 2:S11. doi: 10.1186/1471-2202-9-S2-S11
29. Brinton RD. The neurosteroid 3 alpha-hydroxy-5 alpha-pregnan-20-one induces cytoarchitectural regression in cultured fetal hippocampal neurons. J Neurosci (1994) 14(5 Pt 1):2763–74. doi: 10.1523/JNEUROSCI.14-05-02763.1994
30. Belelli D, Lambert JJ. Neurosteroids: endogenous regulators of the GABA(A) receptor. Nat Rev Neurosci (2005) 6(7):565–75. doi: 10.1038/nrn1703
31. Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci (2014) 15(10):637–54. doi: 10.1038/nrn3819
32. Carver CM, Reddy DS. Neurosteroid interactions with synaptic and extrasynaptic GABA(A) receptors: regulation of subunit plasticity, phasic and tonic inhibition, and neuronal network excitability. Psychopharmacol (Berl) (2013) 230(2):151–88. doi: 10.1007/s00213-013-3276-5
33. Faroni A, Magnaghi V. The neurosteroid allopregnanolone modulates specific functions in central and peripheral glial cells. Front Endocrinol (Lausanne) (2011) 2:103. doi: 10.3389/fendo.2011.00103
34. Yan Y, Dempsey RJ, Sun D. Expression of Na(+)-K(+)-Cl(-) cotransporter in rat brain during development and its localization in mature astrocytes. Brain Res (2001) 911(1):43–55. doi: 10.1016/S0006-8993(01)02649-X
35. Liu J, Feng X, Wang Y, Xia X, Zheng JC. Astrocytes: GABAceptive and GABAergic cells in the brain. Front Cell Neurosci (2022) 16:892497. doi: 10.3389/fncel.2022.892497
36. Wu TW, Chen S, Brinton RD. Membrane estrogen receptors mediate calcium signaling and MAP kinase activation in individual hippocampal neurons. Brain Res (2011) 1379:34–43. doi: 10.1016/j.brainres.2011.01.034
37. Yao J, Chen S, Cadenas E, Brinton RD. Estrogen protection against mitochondrial toxin-induced cell death in hippocampal neurons: antagonism by progesterone. Brain Res (2011) 1379:2–10. doi: 10.1016/j.brainres.2010.11.090
38. Ashpole NM, Sanders JE, Hodges EL, Yan H, Sonntag WE. Growth hormone, insulin-like growth factor-1 and the aging brain. Exp Gerontol (2015) 68:76–81. doi: 10.1016/j.exger.2014.10.002
39. Budni J, Bellettini-Santos T, Mina F, Garcez ML, Zugno AI. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis (2015) 6(5):331–41. doi: 10.14336/AD.2015.0825
40. Urbanska M, Gozdz A, Macias M, Cymerman IA, Liszewska E, Kondratiuk I, et al. GSK3beta controls mTOR and prosurvival signaling in neurons. Mol Neurobiol (2018) 55(7):6050–62. doi: 10.1007/s12035-017-0823-9
41. Yu S, Yang S, Holsboer F, Sousa N, Almeida OF. Glucocorticoid regulation of astrocytic fate and function. PloS One (2011) 6(7):e22419. doi: 10.1371/journal.pone.0022419
42. Concas A, Mostallino MC, Porcu P, Follesa P, Barbaccia ML, Trabucchi M, et al. Role of brain allopregnanolone in the plasticity of gamma-aminobutyric acid type A receptor in rat brain during pregnancy and after delivery. Proc Natl Acad Sci USA (1998) 95(22):13284–9. doi: 10.1073/pnas.95.22.13284
43. Luisi S, Petraglia F, Benedetto C, Nappi RE, Bernardi F, Fadalti M, et al. Serum allopregnanolone levels in pregnant women: changes during pregnancy, at delivery, and in hypertensive patients. J Clin Endocrinol Metab (2000) 85(7):2429–33. doi: 10.1210/jcem.85.7.6675
44. Irwin RW, Yao J, Ahmed SS, Hamilton RT, Cadenas E, Brinton RD. Medroxyprogesterone acetate antagonizes estrogen up-regulation of brain mitochondrial function. Endocrinology (2011) 152:556–67.
45. Irwin RW, Yao J, To J, Hamilton RT, Cadenas E, Brinton RD. Selective oestrogen receptor modulators differentially potentiate brain mitochondrial function. J Neuroendocrinol (2012) 24(1):236–48.
46. Yao J, Zhao L, Mao Z, Chen S, Wong KC, To J, et al. Potentiation of brain mitochondrial function by S-equol and R/S-equol estrogen receptor beta-selective phytoSERM treatments. Brain Res (2013) 1514:128–41.
47. Peng J-Y, Lin C-C, Chen Y-J, Kao L-S, Liu Y-C, Chou C-C, et al. Automatic morphological subtyping reveals new roles of caspases in mitochondrial dynamics. PloS Comput Biol (2011) 7(10):e1002212. doi: 10.1371/journal.pcbi.1002212
48. Qi G, Mi Y, Shi X, Gu H, Brinton RD, Yin F. ApoE4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep (2021) 34(1):108572. doi: 10.1016/j.celrep.2020.108572
49. Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research (2015) 4:1521. doi: 10.12688/f1000research.7563.1
50. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol (2014) 15(12):550. doi: 10.1186/s13059-014-0550-8
51. Mayford M, Siegelbaum SA, Kandel ER. Synapses and memory storage. Cold Spring Harb Perspect Biol (2012) 4(6):a005751. doi: 10.1101/cshperspect.a005751
52. Abel ED. Mitochondrial dynamics and metabolic regulation in cardiac and skeletal muscle. Trans Am Clin Climatol Assoc (2018) 129:266–78.
53. Gonzalez-Reyes RE, Nava-Mesa MO, Vargas-Sanchez K, Ariza-Salamanca D, Mora-Munoz L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci (2017) 10:427. doi: 10.3389/fnmol.2017.00427
54. Mi Y, Qi G, Vitali F, Shang Y, Raikes AC, Wang T, et al. Loss of fatty acid degradation by astrocytic mitochondria triggers neuroinflammation and neurodegeneration. Nat Metab (2023) 5(3):445–65. doi: 10.1038/s42255-023-00756-4
55. Cekanaviciute E, Dietrich HK, Axtell RC, Williams AM, Egusquiza R, Wai KM, et al. Astrocytic TGF-beta signaling limits inflammation and reduces neuronal damage during central nervous system Toxoplasma infection. J Immunol (2014) 193(1):139–49. doi: 10.4049/jimmunol.1303284
56. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
57. Abdullah MO, Zeng RX, Margerum CL, Papadopoli D, Monnin C, Punter KB, et al. Mitochondrial hyperfusion via metabolic sensing of regulatory amino acids. Cell Rep (2022) 40(7):111198. doi: 10.1016/j.celrep.2022.111198
58. Brinton RD, Wang JM. Preclinical analyses of the therapeutic potential of allopregnanolone to promote neurogenesis in vitro and in vivo in transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res (2006) 3(1):11–7. doi: 10.2174/156720506775697160
59. Szabadkai G, Simoni AM, Bianchi K, De Stefani D, Leo S, Wieckowski MR, et al. Mitochondrial dynamics and Ca2+ signaling. Biochim Biophys Acta (2006) 1763(5-6):442–9. doi: 10.1016/j.bbamcr.2006.04.002
60. Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res (2008) 79(2):208–17. doi: 10.1093/cvr/cvn098
61. Patterson M, Yasuda R. Signalling pathways underlying structural plasticity of dendritic spines. Br J Pharmacol (2011) 163(8):1626–38. doi: 10.1111/j.1476-5381.2011.01328.x
62. Sakamoto K, Karelina K, Obrietan K. CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem (2011) 116(1):1–9. doi: 10.1111/j.1471-4159.2010.07080.x
63. Zampese E, Wokosin DL, Gonzalez-Rodriguez P, Guzman JN, Tkatch T, Kondapalli J, et al. Ca(2+) channels couple spiking to mitochondrial metabolism in substantia nigra dopaminergic neurons. Sci Adv (2022) 8(39):eabp8701. doi: 10.1126/sciadv.abp8701
64. Jow F, Chiu D, Lim HK, Novak T, Lin S. Production of GABA by cultured hippocampal glial cells. Neurochem Int (2004) 45(2-3):273–83. doi: 10.1016/j.neuint.2003.11.021
65. Irwin RW, Solinsky CM, Loya CM, Salituro FG, Rodgers KE, Bauer G, et al. Allopregnanolone preclinical acute pharmacokinetic and pharmacodynamic studies to predict tolerability and efficacy for Alzheimer’s disease. PloS One (2015) 10(6):e0128313. doi: 10.1371/journal.pone.0128313
66. Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell (2016) 61(5):654–66. doi: 10.1016/j.molcel.2016.01.028
67. Flippo KH, Strack S. Mitochondrial dynamics in neuronal injury, development and plasticity. J Cell Sci (2017) 130(4):671–81. doi: 10.1242/jcs.171017
68. Cheng A, Hou Y, Mattson MP. Mitochondria and neuroplasticity. ASN Neuro (2010) 2(5):e00045. doi: 10.1042/AN20100019
69. Zamora-Sanchez CJ, Hansberg-Pastor V, Salido-Guadarrama I, Rodriguez-Dorantes M, Camacho-Arroyo I. Allopregnanolone promotes proliferation and differential gene expression in human glioblastoma cells. Steroids (2017) 119:36–42. doi: 10.1016/j.steroids.2017.01.004
70. Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med (2017) 214(5):1351–70. doi: 10.1084/jem.20150237
71. Liu L, Chan C. The role of inflammasome in Alzheimer’s disease. Ageing Res Rev (2014) 15:6–15. doi: 10.1016/j.arr.2013.12.007
Keywords: allopregnanolone, calcium signaling, mitochondria, neuroplasticity, astrocytic function
Citation: Wang T, Chen S, Mao Z, Shang Y and Brinton RD (2023) Allopregnanolone pleiotropic action in neurons and astrocytes: calcium signaling as a unifying mechanism. Front. Endocrinol. 14:1286931. doi: 10.3389/fendo.2023.1286931
Received: 31 August 2023; Accepted: 13 November 2023;
Published: 22 December 2023.
Edited by:
Donald Stein, Emory University, United StatesReviewed by:
Richard Anthony DeFazio, University of Michigan, United StatesCopyright © 2023 Wang, Chen, Mao, Shang and Brinton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberta Diaz Brinton, cmJyaW50b25AYXJpem9uYS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.