
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 20 December 2023
Sec. Renal Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1281107
 Emma S. Speedtsberg1,2,3
Emma S. Speedtsberg1,2,3 Martin Tepel1,2,3*†
Martin Tepel1,2,3*†Background and aims: Outcome trials using sodium glucose cotransporter type 2 inhibitors have consistently shown their potential to preserve kidney function in diabetic and nondiabetic patients. Several mechanisms have been introduced which may explain the nephroprotective effect of sodium glucose cotransporter type 2 inhibitors beyond lowering blood glucose. This current narrative review has the objective to describe main underlying mechanisms causing a nephroprotective effect and to show similarities as well as differences between proposed mechanisms which can be observed in patients with diabetic and nondiabetic chronic kidney disease.
Methods: We performed a narrative review of the literature on Pubmed and Embase. The research string comprised various combinations of items including “chronic kidney disease”, “sodium glucose cotransporter 2 inhibitor” and “mechanisms”. We searched for original research and review articles published until march, 2022. The databases were searched independently and the agreements by two authors were jointly obtained.
Results: Sodium glucose cotransporter type 2 inhibitors show systemic, hemodynamic, and metabolic effects. Systemic effects include reduction of blood pressure without compensatory activation of the sympathetic nervous system. Hemodynamic effects include restoration of tubuloglomerular feedback which may improve pathologic hyperfiltration observed in most cases with chronic kidney disease. Current literature indicates that SGLT2i may not improve cortical oxygenation and may reduce medullar oxygenation.
Conclusion: Sodium glucose cotransporter type 2 inhibitors cause nephroprotective effects by several mechanisms. However, several mediators which are involved in the underlying pathophysiology may be different between diabetic and nondiabetic patients.
Chronic kidney disease (CKD) affects around one in ten people worldwide and is largely a contributor to mortality and reduced quality of life (1–4). Because the number of people living with risk factors for kidney disease increases, the number of patients suffering from CKD and the number of patients dying from the disease, continues to rise (1–4). The most common causes of CKD include diabetes mellitus (DM), hypertension, and glomerulonephritis (GN) (4). The definition of CKD includes structural or functional changes with persistence of at least 3 months. The diagnosis is based on either the presence of kidney damage markers, for example, albuminuria, or a decreased glomerular filtration rate (GFR) (4). Until recently, blockade of the renin angiotensin aldosterone system (RAAS) with angiotensin converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARB) have been cornerstones to slow progressive decline of kidney function (5). Several large outcome trials have consistently shown the nephroprotective potential of sodium glucose cotransporter type 2 inhibitors (SGLT2i) in diabetic patients with CKD (6–9).
The recent KDIGO clinical practice guideline for diabetes management in chronic kidney disease recommends treatment of patients with type 2 diabetes mellitus, chronic kidney disease, and an estimated glomerular filtration rate more the 20mL per 1.73m2 with SGLT2i (10). The authors noticed that start of SGLT2i treatment may cause a reversible decrease in the eGFR and was not an indication for discontinuation (10). Due to lacking evidence administration of SGLT2i does not apply to kidney transplant recipients (10). The EMPA-Kidney trial showed that administration of SGLT2i, empagliflozin, to patients with kidney disease for 2 years significantly reduced the composite endpoint, progression of kidney disease or death from cardiovascular causes. The outcome was observed in 13.1% (432 of 3304 patients) in the empagliflozin group and in 16.9% (558 of 3305 patients) in the placebo group (hazard ratio, 0.72, 95% CI 0.64 to 0.82) (11). It is important to mention that the results were consistent among patients with or without diabetes (11). The authors also indicated that SGLT2i, empagliflozin, was beneficial in patients with an eGFR less than 30 ml per minute or a low urinary albumin-to-creatinine ratio (11).
A recent publication presented the American Diabetes Association (ADA) and Kidney Disease Improving Global Outcomes (KDIGO) consensus statement that SGLT2i is recommended for patients with diabetes mellitus type 2, chronic kidney disease, and an eGFR more than 20 ml/min/1.73m2 (12). This recommendation was based on strong evidence from large outcome trials that SGLT2i may reduce the progression of chronic kidney disease, heart failure and atherosclerotic cardiovascular disease in these patients (12). The authors indicate that SGLT2i reduce intra-glomerular pressure (12).
As shown in Figure 1, the reabsorption of filtered glucose from the tubular lumen is caused by two transporters located apically on the proximal tubule cells (13). SGLT2 is situated in the S1 segment, whereas sodium glucose cotransporter type 1 (SGLT1) is situated in the distal S3 segments of the proximal tubule located at the corticomedullary junction (13). These transporters work in conjunction with the basolateral sodium potassium pumps, which uses energy in the form of adenosine triphosphate (ATP) to create an electrochemical gradient. The transport of glucose to the bloodstream is facilitated passively by glucose transporter type 2 (GLUT2). Since SGLT2 transporters are responsible for the uptake of filtered glucose in cotransport with sodium in a 1:1 ratio, inhibition of SGLT2 causes glucosuria and increases natriuresis (14). Which mechanisms may link the increased loss of glucose and sodium with kidney protection and lower mortality? Several mechanisms regarding the nephroprotective effect in diabetic as well as non-diabetic patients with CKD have been proposed. The objective of this review is to describe the main mechanisms underlying the nephroprotective effect of SGLT2i in patients with diabetic nephropathy. The second aim is to demonstrate similarities and differences in patients with diabetic and nondiabetic CKD regarding these mechanisms. We present the following article in accordance with the Narrative Review reporting checklist.
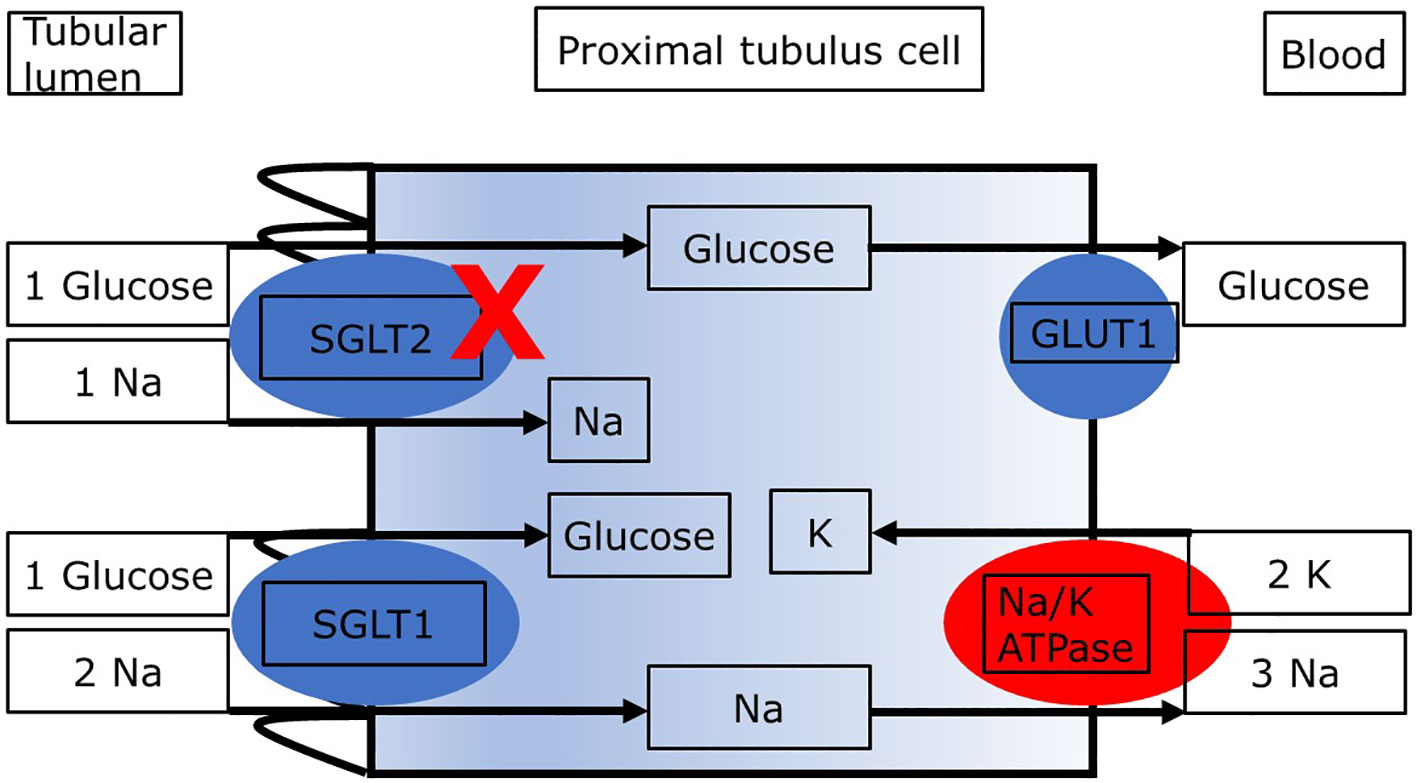
Figure 1 Proximal tubule cell with transporters relevant to the reabsorption of glucose. Blockade of SGLT2 results in increased delivery of glucose and sodium to downstream segments and loss in the urine. ATP, adenosine triphosphate; GLUT2, glucose transporter 2; SGLT1, sodium glucose cotransporter type 2. The sodium:glucose coupling ratio is 1:1 for SGLT2 and it is 2:1 for SGLT1. The different sodium:glucose coupling ratios may impact renal energy expenditure with SGLT2i.
The research strategy summary is given in Table 1. The databases Pubmed and Embase were searched for original research and review articles from start until March, 2022. Based on the objectives, relevant blocks were constructed and terms and synonymous for each block were identified (Table 2). The main words were “chronic kidney disease, sodium glucose transporter 2 inhibitor, and mechanism”. Synonyms were found from articles by the initial unstructured research, and by selecting “show index” for each word in the search function in Pubmed. Quotes were used to ensure that the words were not searched for individually and truncation was used to allow the word to have multiple endings. The individual words within each block were combined with OR, while each block was combined with AND. From this, a search string for each database was constructed, including both free text terms and keywords. The studies identified from the two databases, were imported into the reference system Endnote (Clarivate Analytics, Philadelphia, USA) and duplicates were removed automatically. Then they were exported to the screening tool Rayyan (Rayyan Systems Inc, Cambridge, England) and additional duplicates were removed manually. The studies were screened for title and abstract, and exclusion was done by the authors if words from the three blocks were not included, or if the study was made in animals only. The remaining full text articles were screened and included if they did not meet the following exclusion criteria: animal studies only, the population did not have kidney disease, the article was a comment, the language was not Danish or English (Figure 2). Additional studies were identified through included references.
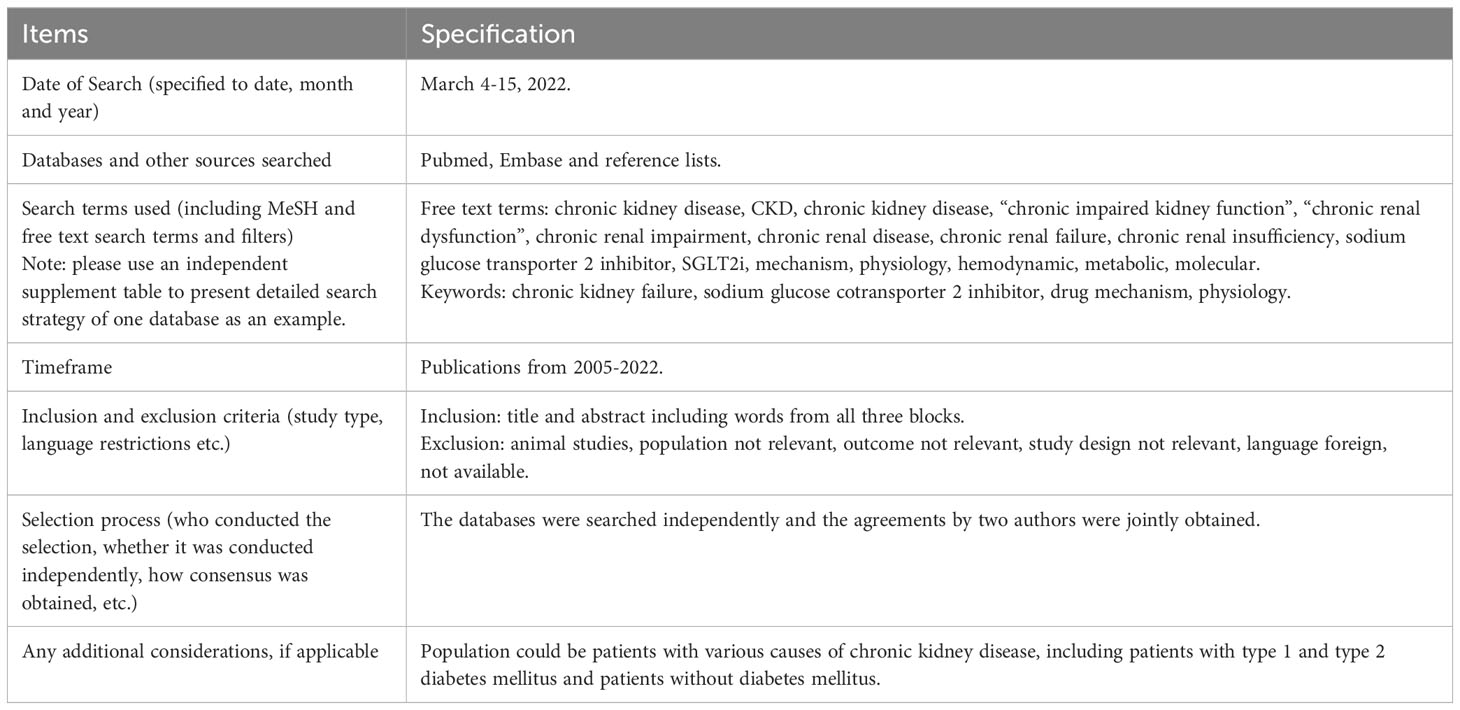
Table 1 Research strategy summary.
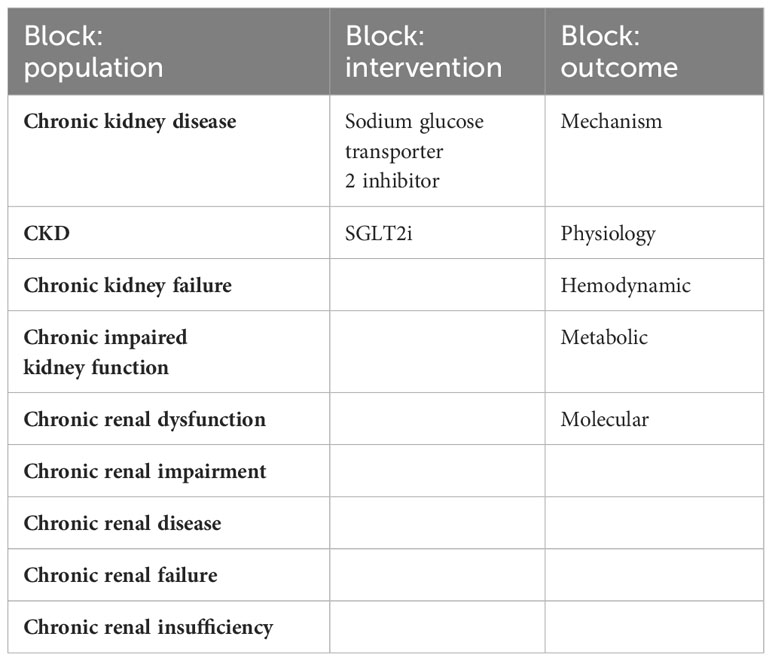
Table 2 The table shows the tree blocks which were constructed based on the objectives, with terms and synonymous relevant to each block.
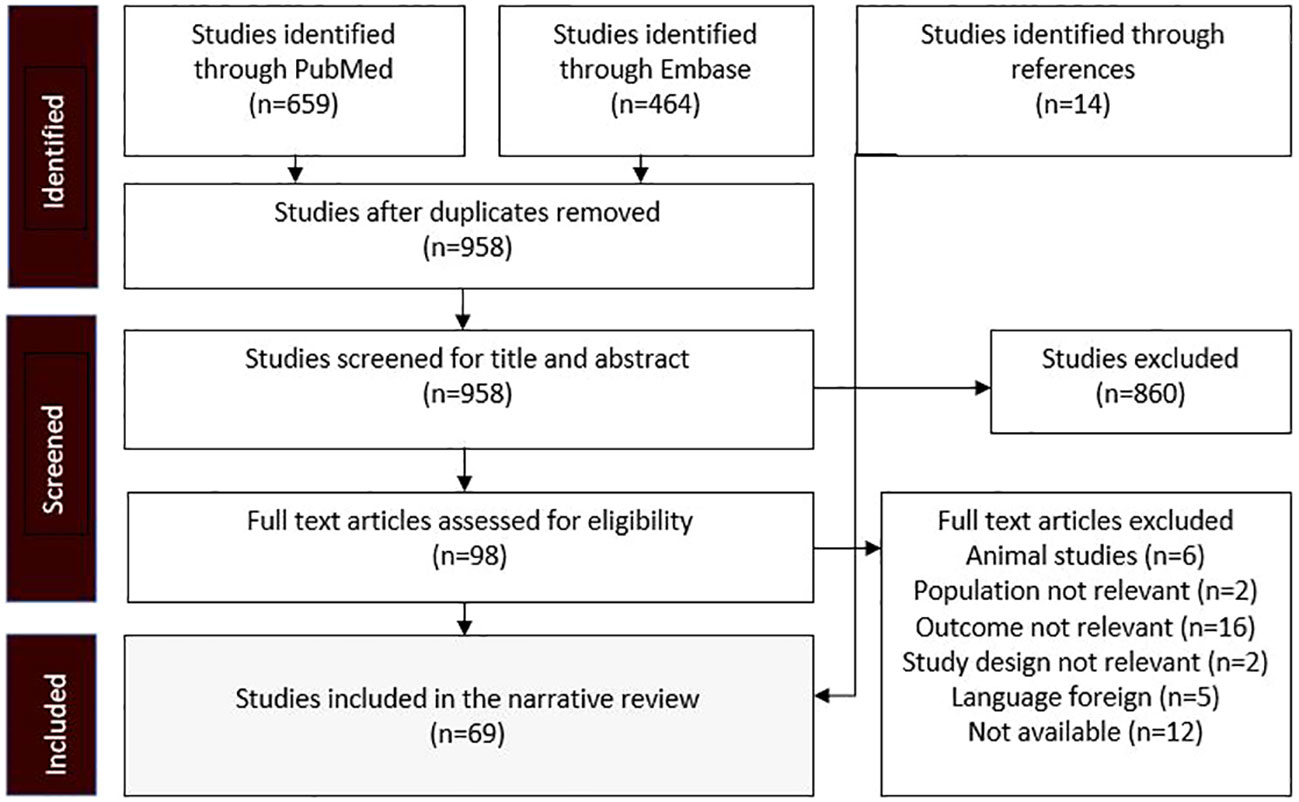
Figure 2 Flow diagram showing the process whereby studies were identified, screened, and included. The exclusion criteria were predetermined before the research.
The nephroprotective effects of SGLT2i are summarized in Table 3, and have been attributed to systemic, hemodynamic, and metabolic mechanisms. The details extracted from references included in this review are given in Table 4. Some of the mechanisms may affect risk factors which are observed more often in diabetic patients, whereas others may target features common to all patients with CKD.
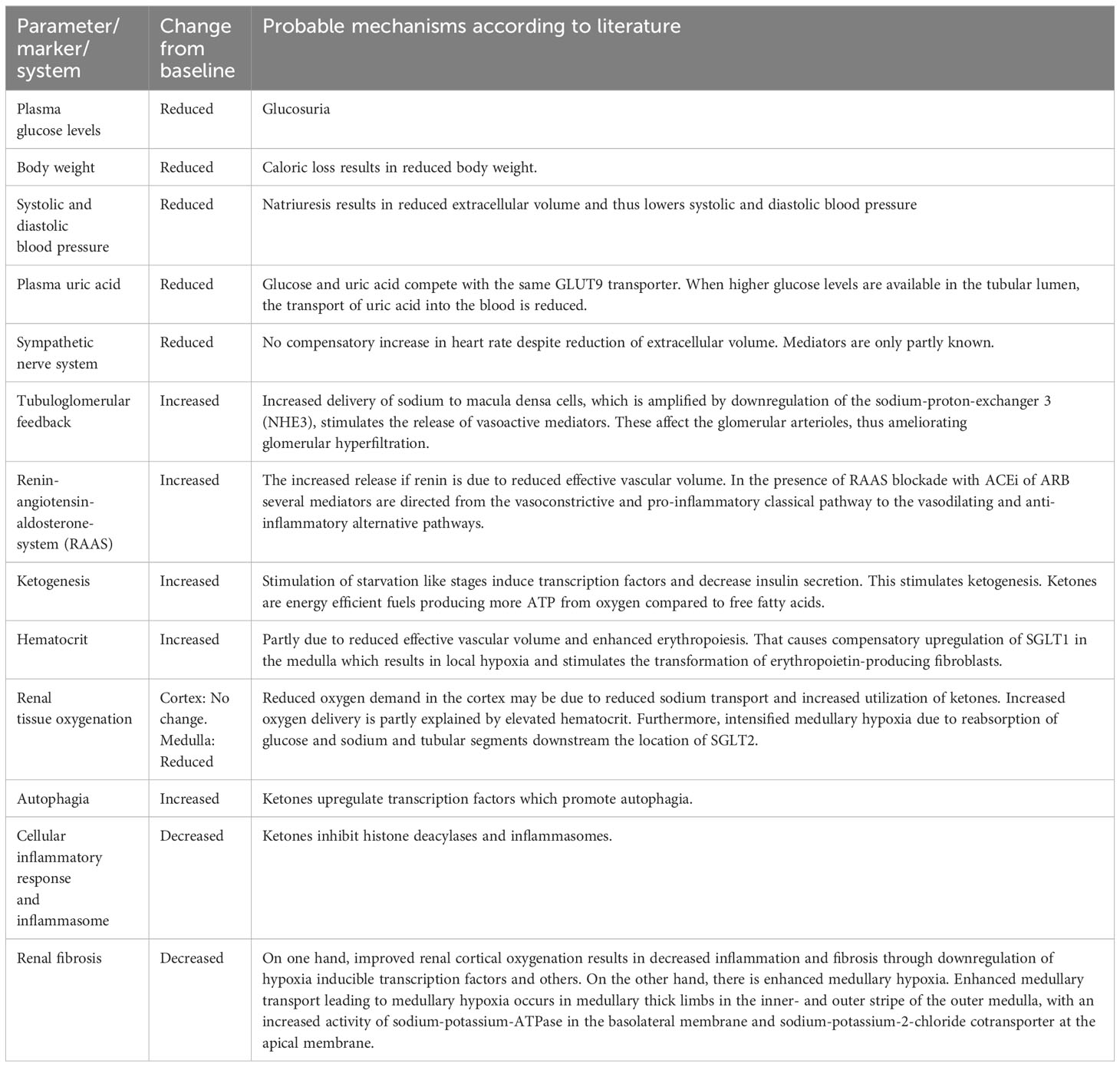
Table 3 Parameters/markers/systems affected, changes from baseline, and the probable mechanisms of SGLT2 inhibitors.
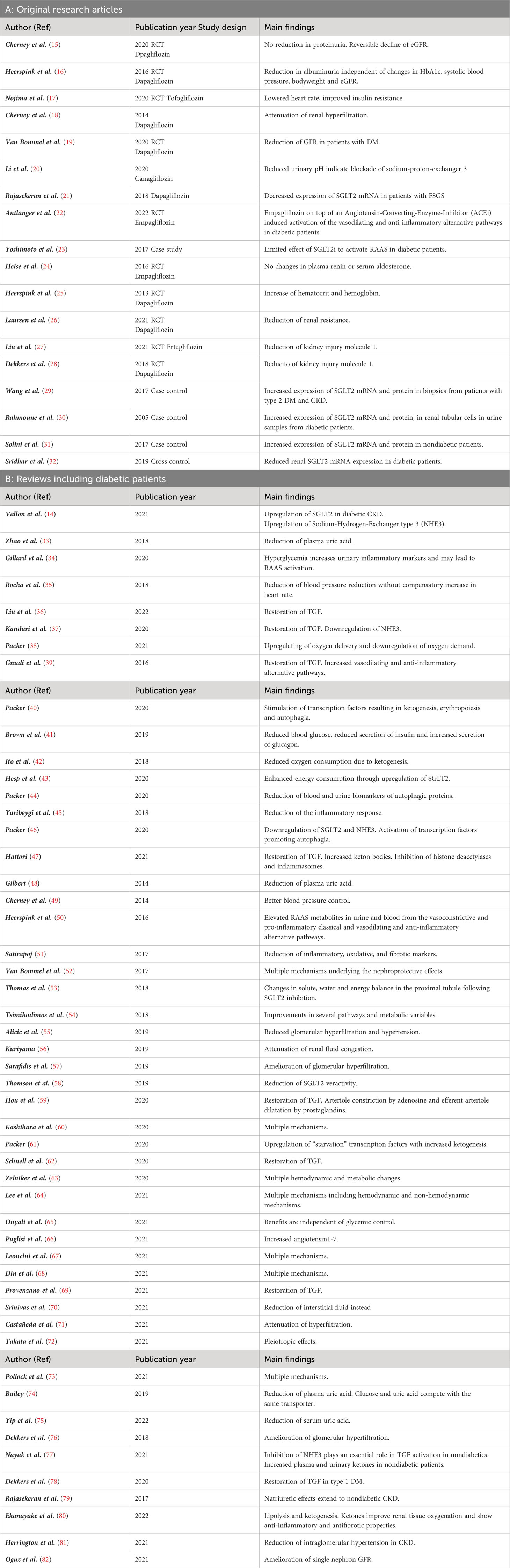
Table 4 Studies included in this narrative review.
Due to glucosuria and natriuresis, a modest reduction in hemoglobin A1c (HbA1c), bodyweight, as well as systolic and diastolic blood pressure have been observed in several studies in diabetic patients with CKD. In nondiabetic patients with CKD, Cherney et al. showed a reduction in bodyweight, but no significant changes in HbA1c or blood pressure (15). Since overweight as well as increased blood pressure are known risk factors for progression of CKD, these mechanisms likely confer nephroprotection by SGLT2i. Heerspink et al. found, that a reduction in albuminuria in patients with type 2 DM treated with a SGLT2i appeared to be independent of changes in HbA1c, bodyweight and systolic blood pressure (16). It should be noted that the effect on glucosuria is attenuated in patients with reduced kidney function, while blood pressure lowering is quite consistent across different levels of kidney function (73). Large outcome studies showed that sodium glucose cotransporter type 2 inhibitors (SGLT2i) reduce systolic/diastolic blood pressure by approximately 4 mmHg/2 mmHg (83). Compared to placebo mean reduction of 24-hour diastolic blood pressure were 1.0 mmHg, 1.3 mmHg, and 4.8 mmHg in patients with eGFR more than 90 ml/min per 1.73 m2, eGFR between 90 and 60 ml/min per 1.73 m2, and eGFR between 60 and 30 ml/min per 1.73 m2, respectively (84). SGLT2i do not act as osmotic diuretics. But low-level ketoacidosis which is observed after administration of SGLT2i can reduce blood pressure (80). In the Dahl salt-sensitive rat model of hypertension, the co-administration of β-hydroxybutyrate reduced elevated blood pressure in a salt-rich diet (85).
Reduced plasma uric acid is a consequence of increased glucose in the tubular lumen, which is taken up by the same glucose transporter type 9, at the expense of uric acid. Results have been conflicting, as to whether lowering of plasma uric acid provides nephroprotection. Some studies indicate that hyperuricemia is associated with increased risk of kidney disease in patients with type 1 DM (86, 87) and in patients with type 2 DM (74). Other studies showed that interventions to reduce plasma uric acid could retard the progression of CKD (88, 89). In contrast, other studies indicated that uric acid may not be directly involved in the development of CKD in diabetic patients, but is a downstream marker of kidney damage (90), hence trials using febuxostat and allopurinol failed to show a large nephroprotective effect (91, 92). Zhao et al. found that the reduction of plasma uric acid is attenuated with lower estimated glomerular filtration rate (eGFR) (33). These studies concluded that uric acid lowering alone may not provide nephroprotection, but they do not exclude that in a combination with other mechanisms offered by SGLT2i, the observed lowering of plasma uric acid may have an additional beneficial effect (34).
Elevated concentrations of plasma uric acid have been associated with increased risk of development and progression of CKD in nondiabetic patients, and interventions to reduce uric acid, may contribute to the nephroprotective effect in these patients (74). A recent meta-analysis of 43 randomized controlled trials indicated that SGLT2i reduced plasma uric acid levels in both diabetic and nondiabetic patients (75). However, the effect may be smaller in nondiabetic patients, because the concentration of plasma uric acid is generally lower, and the uricosuric effect may be smaller due to the lower filtered glucose, capable of competing with the glucose transporter type 9 (74). The beneficial effects of SGLT2i may be due to the fact that fractional uric acid excretion was strongly correlated to fractional glucose excretion (93).
The sympathetic nervous system (SNS) is not activated upon SGLT2 inhibition, which is proved by the ability of SGLT2i to reduce systolic and diastolic blood pressure without a compensatory increase in heart rate (35). That may be due to lower adipose tissue insulin resistance (17). Furthermore, SGLT2i, dapagliflozin, may directly attenuate the sympathetic response (94).
The restoration of the tubuloglomerular feedback (TGF) mechanism has been considered to be an outstanding explanation why SGLT2i offers nephroprotection, because it targets common steps in the pathogenesis of CKD, in particular the glomerular hyperfiltration (76). Vasodilation of the afferent arteriole which can be observed in patients with diabetes mellitus or in patients with high protein intake causes glomerular hyperfiltration (95). Activation of renin-angiotensin-aldosterone system leads to efferent arteriolar vasoconstriction which causes glomerular hypertension (95). Furthermore, glomerular hyperfiltration is a consequence of reduced number of nephrons in CKD, resulting in a compensatory increase in glomerular filtration in the remaining nephrons (4).
Large outcome trials consistently showed a significant initial decline of eGFR following administration of SGLT2i (5–9). By blocking the reabsorption of glucose and sodium, an increased amount of sodium can be observed at the macula densa cells, which leads to the release of nucleosides finally affecting the tone of the afferent arteriole. Different mediators may contribute and different effects on glomerular vascular tone have been proposed, depending on the cause which resulted in hyperfiltration.
In patients with type 1 DM, Cherney et al. measured eGFR using inulin clearance and renal plasma flow using paraaminohippurate clearance together with circulating levels of RAAS and nitric oxide. These values were measured under clamped euglycemic and hyperglycemic conditions at baseline and at the end of treatment with SGLT2i. Cherney et al. observed that attenuation of hyperfiltration was accompanied by decreased renal plasma flow, increased renal vascular resistance and no changes of vasodilators, including urinary prostaglandins and nitric oxide (18). They suggested that SGLT2i affect TGF and afferent arteriole constriction, and mentioned adenosine as a major vasoconstrictor involved. In patients with type 2 DM, van Bommel et al. showed that attenuation of hyperfiltration was associated with increased levels of urinary adenosine and prostaglandins but no increase in renal vascular resistance (19). That may point to high baseline RAAS inhibition and high afferent arteriole constriction at baseline, which may limit further vasoconstriction. Adenosine has several receptors, and binding to adenosine A1 receptors on the efferent arteriole likely causes vasodilation, which may be reinforced by increased production of vasodilating prostaglandins (36).
In nondiabetic patients, Cherney et al. demonstrated the ability of SGLT2i to attenuate hyperfiltration, as indicated by the initial decline of eGFR during treatment (15). They did not detect a reduction in albuminuria which has been shown in previous studies in diabetic patients (15). One reason could be the short duration of treatment. Another reason could be differences in underlying disease pathologies because some sources of proteinuria are less responsive to changes in eGFR (15). Finally, an explanation could be a weaker activation of macula densa cells in comparison to diabetic patients, because the amount of filtered glucose is already lower in nondiabetic patients, and the initial decline of eGFR leads to further lowering of glucose and sodium delivery to macula densa cells. Hence TGF might not be activated sufficiently to reduce proteinuria in nondiabetic patients who already have a low GFR (15). They did not observe an association between the initial decline of eGFR and changes in adenosine or prostaglandins. Therefore, other vasoactive mediators, i.e., endothelin and nitric oxide could be involved in nondiabetic patients (15).
Downregulation of sodium hydrogen exchanger type 3 and thus reduced reabsorption of sodium may also contribute to restoration of TGF (37). Downregulation of sodium hydrogen exchanger type 3 may occur because the activities of sodium hydrogen exchanger type 3 and SGLT2 are closely linked, i.e., SGLT2 presumably increases the activity of sodium hydrogen exchanger type 3 (96). This was demonstrated clinically by reduced urine pH following SGLT2i, due to the urinary loss of hydrogen (20). By using a mathematical model of renal function and volume homeostasis in combination with clinical data, it has been predicted that inhibition of apical proximal tubule sodium hydrogen exchanger type 3 is required for the natriuretic effect induced by SGLT2i in humans (97). Since sodium and chloride remain the sole solute sensor for macula densa cells, downregulation of sodium hydrogen exchanger type 3 may also play an essential role in TGF activation in nondiabetic patients (77).
Given the TGF mechanism, it is possible that several kidney diseases might benefit from SGLT2i (78). Diseases like obesity induced nephropathy, hypertensive nephropathy as well as several types of GN are characterized by renal hemodynamic changes including glomerular hypertension and hyperfiltration (79). They therefore share a common step in the pathogenesis leading to CKD, namely damage and loss of nephrons and thus hyperfiltration in remaining nephrons, creating a vicious cycle. Kidney diseases characterized by glomerular hyperfiltration will likely benefit from SGLT2 inhibition via restoration of TGF, but the mediators involved likely are different in various diseases.
Despite being an important and well-studied mechanism, restoration of TGF may only partly explain the large nephroprotective effect of SGLT2i. Rajasekeran et al. questions whether this mechanism is central in all subtypes of CKD, as they failed to observe any favorable renal hemodynamic alterations or attenuation in albuminuria in patients with focal segmental glomerulosclerosis (FSGS) after administration of an SGLT2i (21). This could be due to the loss of transporters in these patients. In addition, Heerspink et al. showed that in patients with type 2 DM, a reduction in albuminuria was maintained even after adjustment for changes in eGFR, suggesting that other mechanisms may be involved (16). To support the contribution from other mechanisms, Packer claims that patients with very low filtration rates still benefit from SGLT2i treatment, despite the fact that amelioration of hyperfiltration by SGLT2i is probably limited in these patients (38).
Blood pressure is affected by both natriuresis as well as the Renin-Angiotensin-Aldosterone-System.
An increased sodium delivery to the macula densa activates the tubuloglomerular feedback increasing the resistance in the afferent arteriole (76). In contrast, the Renin-Angiotensin-Aldosterone-System mainly affects the resistance in the efferent arteriole. Furthermore, SGLT2i-induced activation of the tubuloglomerular feedback may reduce glomerular filtration by affecting the afferent arteriole thereby reducing excreted sodium.
Reduced effective circulating volume, which may be a consequence of SGLT2i, results in increased release of renin and thus production of angiotensin I from angiotensinogen. In the presence of RAAS blockade, by an ACEi or an ARB, angiotensin I is converted to angiotensin 1-7 by angiotensin converting enzyme 2. These are mediators of the vasodilating and anti-inflammatory alternative pathways, and the importance of these mediators as key opposing effectors to angiotensin II has been well established (98). Antlanger et al. reported elevated plasma angiotensin I and angiotensin 1-7 after administration of an SGLT2i on top of an ACEi in patients with type 2 DM (22). They propose that ACEi cannot fully reverse CKD progression due to normalization of angiotensin II levels after long-term therapy, a phenomenon termed “ACEi escape” (22). They conclude that suppression of angiotensin II with RAAS blockade in combination with stimulation of the vasodilating and anti-inflammatory alternative pathways by SGLT2i could therefore be of importance (22). The presumed theory is that angiotensin II induces vasoconstriction and inflammation while angiotensin 1-7 promote vasodilatation and have anti-inflammatory properties (39). In contrast, Yoshimoto et al. conclude that the ability of SGLT2i to activate RAAS in patients with type 2 DM is limited (23). They found no increase in urinary angiotensinogen during treatment with different SGLT2i. In addition, Heise et al. did not observe any changes in plasma renin or serum aldosterone during SGLT2i (24).
Continuous glucosuria, and thereby loss of calories through the urine, simulates a starvation like state resulting in a metabolic shift from glycolysis to lipolysis and ketogenesis (80). This is presumably due to upregulation of several transcription factors normally induced in the fasting state (40). In addition, the decline in blood glucose leads to reduced secretion of insulin and an increased secretion of glucagon (41). In addition, ketogenesis is associated to direct upregulation of energy deprivation sensors like AMPK (adenosine monophosphate-activated kinase) and SIRT1 (Sirtuin 1) (99).
Ketone bodies are an efficient fuel substrate because they generate more ATP for the same amount of oxygen compared to free fatty acids (FFA) (80). Ketogenesis could therefore probably contribute to improved renal tissue oxygenation, by reducing renal oxygen consumption (42). Packer claims, that it is unlikely that the ability of SGLT2i to increase ketone bodies, is responsible for the nephroprotective effect in diabetic nephropathy, since circulating levels of ketone bodies are already increased in diabetic patients in the absence of treatment (38). During treatment with SGLT2i, a doubling of ketone bodies in plasma has been observed in nondiabetic patients similar to what is found in diabetic patients (100). This is accompanied by increased levels of ketone bodies and metabolites from ketogenesis in the urine (77). Because impaired tissue oxygenation plays an equally crucial role in progression of CKD of various subtypes, its reversal may be important (43). Due to this, the beneficial effects from ketone bodies through reduced consumption of oxygen, likely extends to nondiabetic patients (80).
SGLT2i elevates hematocrit through several pathways. First, because of an increased delivery of glucose to the transporters downstream in the medullary segments, a compensatory upregulation of SGLT1 likely occurs, resulting in increased oxygen demand, and thus a risk of hypoxia in this area (35). It should be noted that that the sodium reabsorption independent of glucose by transporters in the inner stripe of the outer medullar may contribute to outer medullary hypoxia. SGLT2i may induce erythropoietin due to increased hypoxia at the corticomedullary junction, related to the translocation of tubular transport from cortical segments to medullary thick ascending limbs (101). Studies showed that this mechanism may stimulate erythropoietin (EPO) producing fibroblasts (25, 77). Animal studies support the described mechanisms for example determination of the intrarenal distribution of tissue oxygenation following SGLTi with the use of oxygen microelectrodes (102). Gullaksen et al. used Blood Oxygenation Level Dependent Magnetic Resonance Imaging (BOLD-MRI) for calculating an apparent relaxation rate in patients with diabetes mellitus type 2. Administration of empagliflocin for 32 weeks changed cortical oxygenation from 23.6 Hz (95%CI, 23.1-24.1) to 23.3 Hz (965% CI, 2.5-24.0; p=0.231) (103). Administration of empagliflozin for 32 weeks reduced medullary oxygenation from 24.5 Hz (95%CI, 23.9-24.9) to 25.4 Hz (95%CI, 24.7-26.2; p= 0.003; where higher apparent relaxation rate corresponds to a lower oxygenation) (103). They indicated that apparent relaxation rate is not a direct measure of oxygenation and is dependent on deoxyhemoglobin concentrations (103). Furthermore, they observed that compared to baseline values the estimated marginal means of both hematocrit and plasma erythropoietin increased after administration of empagliflozin. They concluded that on the contrary to their initial hypothesis empagliflozin reduced medullary kidney oxygenation and hypothesized that the hypoxia generated by empagliflozin stimulates erythropoietin synthesis which may mediate kidney protection (103). The complex association linking the impact of SGLT2i to proteinuria at the glomerular and tubular level, to renal oxygenation, and on the progression to chronic kidney disease has recently been reviewed in-depth by Heyman et al. (104). Increased transglomerular hydraulic pressure induces hyperfiltration and increases the albumin leak across the filtration barrier (104). The reduction of transglomerular hydraulic pressure and hyperfiltration by Angiotensin Receptor Blockers or SGLT2i may attenuate or prevent albuminuria for the long-term (104).
Second, SGLT2 inhibition likely reduces the effective circulating volume. Heerspink et al. showed that administration of a SGLT2i increased hematocrit, hemoglobin, and transiently elevated reticulocyte count and erythropoietin concentrations (25). They suggest that both volume constriction and increased red blood cell mass may contribute to that effect.
Increased ketogenesis as well as elevated hematocrit may improve renal tissue oxygenation, by compensating imbalances between oxygen consumption and oxygen delivery (76). Because tubular sodium reabsorption largely contributes to energy utilization and thus oxygen consumption, reduced proximal tubule transporter activity and thus workload may be important (43). Laursen et al. were able to demonstrate that a single high dose of the SGLT2i dapagliflozin improved renal cortical oxygenation within six hours in patients with type 1 DM and albuminuria. They did not observe changes in renal blood flow or blood oxygen saturation. Therefore, they suggested that the improvement was due to a reduction in tubular workload (26). Liu et al. showed that treatment with the SGLT2i ertugliflozin was associated with sustained lowering of kidney injury molecule 1, a biomarker specific to proximal tubules in patients with type 2 DM (27). This biomarker is sensitive and specific to kidney injury, with increased secretion from tubular cells to the urine under hypoxic conditions, and it correlates well with the onset and progression of CKD (27, 105).
Fibrosis is likely the results of dysfunctional autophagia in combination with inflammation, and it is characterized by fewer number of functional nephrons. Autophagia is typically suppressed in states of nutrient overabundance (44). The beneficial effect of SGLT2i on fibrosis may be secondary to oxidative and organellar stress (99). In diabetic patients, autophagic proteins are decreased, and the levels correlate with the stage of CKD (106, 107). Autophagia is important for the clearance of damaged proteins and organelles, and thus the prevention of inflammation (106). Inflammation has been proposed as being a prominent feature of CKD (45, 108, 109). Treatment with SGLT2 inhibitors may contribute to increased autophagia, decreased inflammation and thereby prevent fibrosis through their actions regarding ketone bodies. Ketone bodies upregulates transcription factors of the starvation like stage which likely promote autophagia (46). Because ketone bodies are not fully utilized, they may also work as inhibitors of histone deacetylases and inflammasomes (47). The improved tissue oxygenation may also contribute to reduced inflammation and fibrosis, which may be enhanced by hypoxia inducible transcription factors (38). Bessho reported that the SGLT2i luseoglifozin inhibited hypoxia-induced hypoxia inducible factor-1α protein expression in human renal proximal tubular epithelial cells (110). Dekkers et al. showed a reduction in urinary markers of inflammation, including kidney injury molecule, in diabetic patients upon SGLT2 inhibition (28). The reduction correlated positively with the reduction in albuminuria and eGFR (28). Due to several beneficial effects of SGLT2i this therapy has been introduced together with other agents to maximally slow CKD progression.
Most studies in humans reported an increased expression of SGLT2 in diabetic patients compared to healthy controls. Wang et al. found an increased expression of SGLT2 mRNA and protein in biopsies from patients with type 2 DM and CKD compared to healthy controls (29). Rahmoune et al. also found increased expression of SGLT2 mRNA and protein in proximal tubular cells from urine samples from patients with type 2 DM compared to healthy controls (30). In contrast, Solini et al. observed reduced expression of SGLT2 mRNA and protein in tissue from nephrectomies obtained from patients with type 2 DM and renal carcinoma (31). These conflicting results may be due to methodological differences including the type of tissue obtained, methods of measurement or sample bias. It could also be a consequence of differences regarding the diseases. Furthermore, the CKD stage may also be a crucial parameter since expression of mRNA from tubular cells have been reported to correlate with GFR (32).
Regarding the expression of SGLT2 in nondiabetic patients, results have been conflicting. On one hand, Raisekeran et al. reported decreased expression of SGLT2 mRNA in biopsies from patients with obesity related FSGS compared to control kidney donors (21). This reduction may reflect proximal tubule cell injury and the absence of stimulatory hyperglycemic milieu. On the other hand, Sridhar et al. detected increased expression of SGLT2 mRNA in biopsies from control kidney donors and patients with nondiabetic nephropathy involving different subtypes of GN, compared to patients with diabetic nephropathy (32). They observed no differences across GN subtypes. Renal biopsies are infrequent in diabetic patients with CKD and reserved for advanced proteinuria or severe insufficiency, thus decreased SGLT2 mRNA could reflect the more advanced stages of CKD in these patients.
Until recently, RAAS blockade using ACEi and ARB have been the cornerstones for the treatment of diabetic and nondiabetic patients with CKD. Several trials have demonstrated the efficiency of these treatments compared to placebo. It should be noted that the event rates were much higher in these trials, which yielded a number needed to treat ranging from 4 to 23 (111–115). In most trials investigating SGLT2i, the patients received SGLT2i on top of RAAS blocking agents. In the placebo-controlled trials using SGLT2i, the number needed to treat ranged from 9 to 93 (5, 7, 9, 116–118). Thus, comparing the different trial designs may indicate that blocking both, RAAS and SGLT2, may be necessary to prevent progression of kidney disease (Table 5). Different mechanisms observed in patients with diabetes mellitus type 1, diabetes mellitus type 2, and nondiabetic chronic kidney disease are summarized in Table 6.
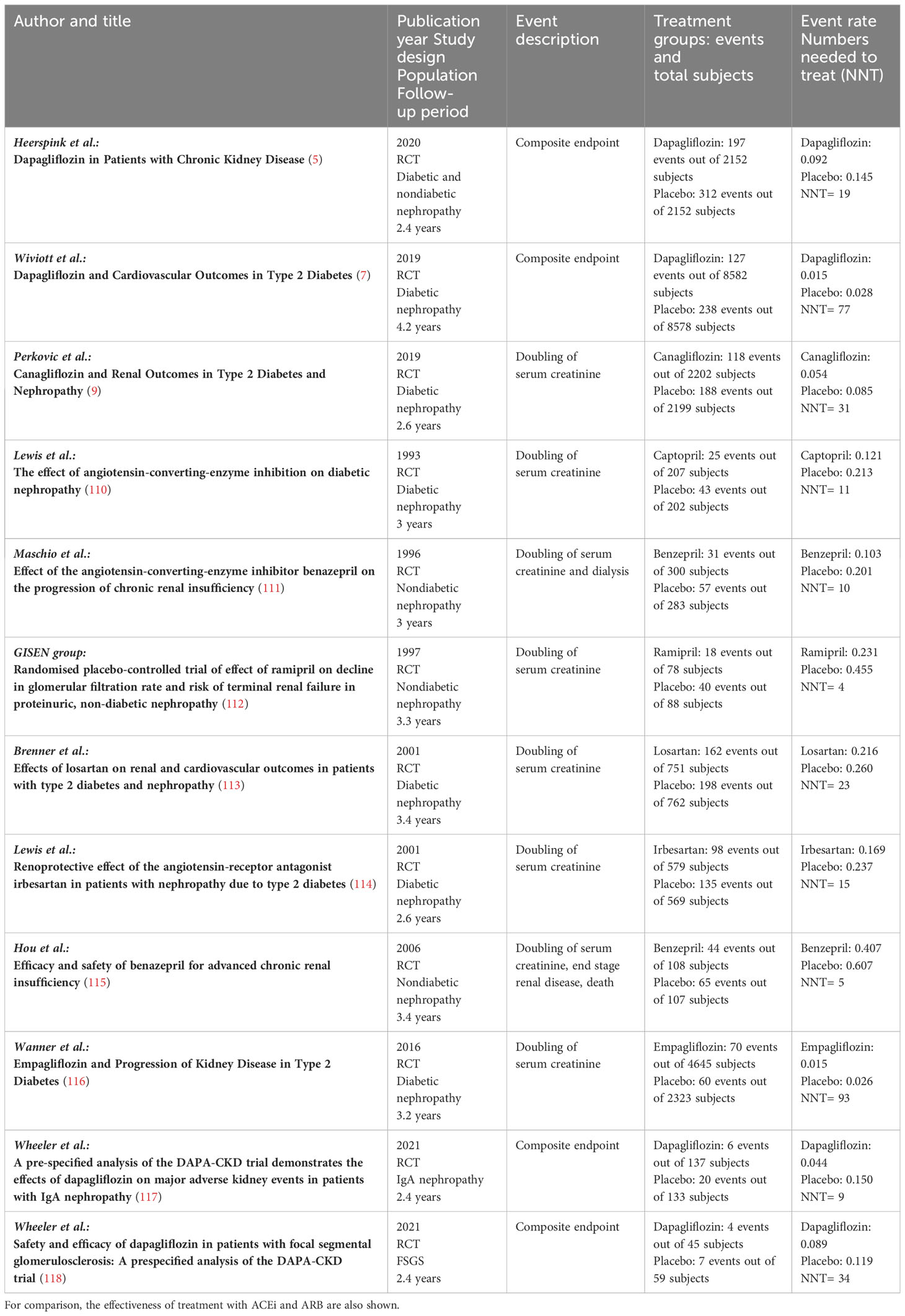
Table 5 Effectiveness of treatment with SGLT2 inhibitors compared to placebo as observed in major clinical outcome studies in diabetic and nondiabetic patients.
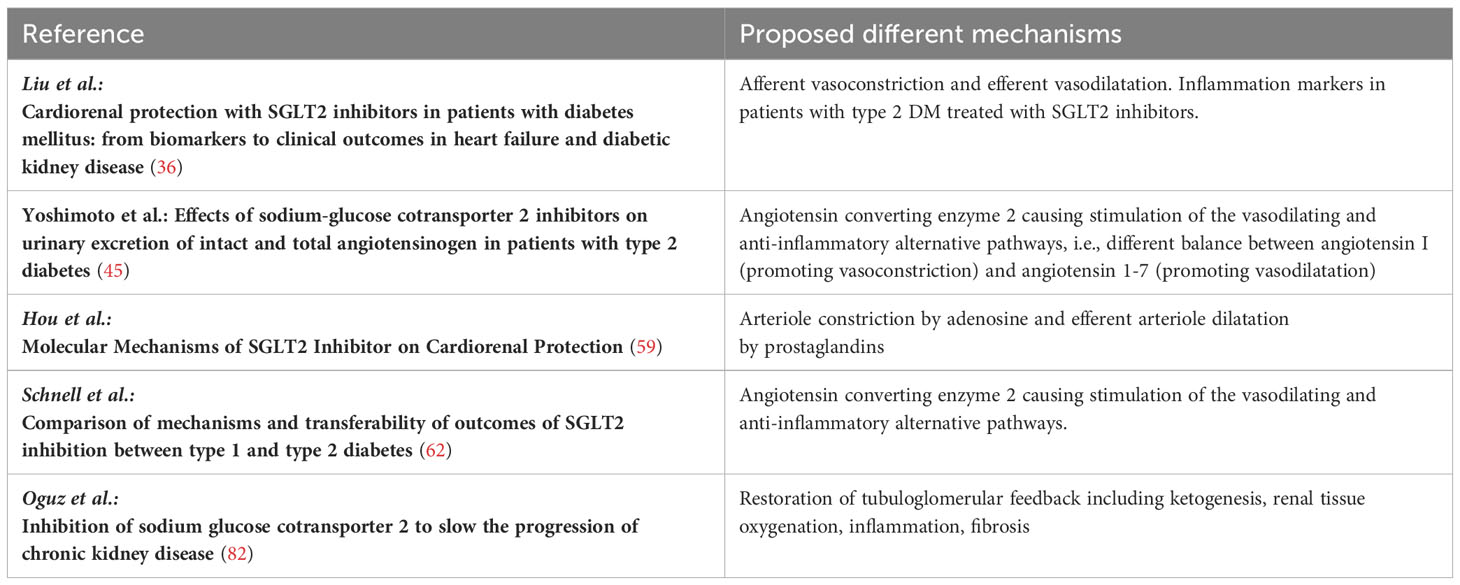
Table 6 Different mechanisms observed in patients with diabetic mellitus type 1, diabetes mellitus type 2, and nondiabetic chronic kidney disease.
This narrative review is written in accordance to the principles stated by Green et al. (119). This review highlights the beneficial effects of sodium glucose cotransporter type 2 inhibitors in patines with kidney diseases. The effects can be attributed to systemic, hemodynamic, and metabolic effects. SGLT2i show beneficial effects on blood pressure and restoration of tubuloglomerular feedback.
Nephroprotection offered by SGLT2i can be attributed to systemic, hemodynamic, and metabolic mechanisms, with restoration of tubuloglomerular feedback likely being most important. Diabetic and nondiabetic patients with CKD share common features which are targeted by SGLT2i. These include similar steps in the pathogenesis, namely glomerular hyperfiltration, as well as final common pathways involving imbalances in tissue oxygenation, inflammation, and fibrosis. The main mechanisms underlying the nephroprotective effects in diabetic patients seem transferable to nondiabetic patients. According to current literature, the underlying mediators may be different. The underlying disease may affect the cellular expression of SGLT2 and may therefore determine the benefit from the SGLT2i treatment.
ES: Methodology, Validation, Writing – original draft, Writing – review & editing, Data curation, Formal analysis, Investigation. MT: Methodology, Validation, Writing – original draft, Writing – review & editing, Conceptualization, Supervision, Visualization.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ACEi, angiotensin converting enzyme inhibitor; ARB, angiotensin receptor blocker; ATP, adenosine triphosphate; CKD, chronic kidney disease; DM, diabetes mellitus; eGFR, estimated glomerular filtration rate; FSGS, focal segmental glomerulosclerosis; GLUT2, glucose transporter 2; GN, glomerulonephritis; HbA1c, glycated hemoglobin A1c; RCT, randomized controlled trial; NNT, number needed to treat; RAAS, renin angiotensin aldosterone system; SGLT2i, sodium glucose cotransporter type 2 inhibitor; SNS, sympathetic nervous system; TGF, tubuloglomerular feedback.
1. Zhao L, Zou Y, Wu Y, Cai L, Zhao Y, Wang Y, et al. Metabolic phenotypes and risk of end-stage kidney disease in patients with type 2 diabetes. Front Endocrinol (Lausanne) (2023) 14:1103251. doi: 10.3389/fendo.2023.1103251
2. Scholze A, Tepel M. Role of leptin in reverse epidemiology in chronic kidney disease. Semin Dial (2007) 20:534–8. doi: 10.1111/j.1525-139X.2007.00334.x
3. Deng Y, Li N, Wu Y, Wang M, Yang S, Zheng Y, et al. Global, regional, and national burden of diabetes-related chronic kidney disease from 1990 to 2019. Front Endocrinol (Lausanne) (2021) 12:672350. doi: 10.3389/fendo.2021.672350
4. Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet (2017) 389(10075):1238–52. doi: 10.1016/S0140-6736(16)32064-5
5. Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, et al. Dapagliflozin in patients with chronic kidney disease. N Engl J Med (2020) 383:1436–46. doi: 10.1056/NEJMoa2024816
6. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
7. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med (2019) 380:347–57. doi: 10.1056/NEJMoa1812389
8. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
9. Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med (2019) 380:2295–306. doi: 10.1056/NEJMoa1811744
10. Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work Group. KDIGO 2022 Clinical practice guideline for diabetes management in chronic kidney disease. Kidney Int (2022) 102:S1–S127. doi: 10.1016/j.kint.2022.06.008
11. The EMPA-KIDNEY Collaborative Group, Herrington WG, Staplin N, Wanner C, Green JB, Hauske SJ, et al. Empagliflozin in patients with chronic kidney disease. N Engl J Med (2023) 388:117–27. doi: 10.1056/NEJMoa2204233
12. de Boer IH, Khunti K, Sadusky T, Tuttle KR, Neumiller JJ, Rhee CM, et al. Diabetes management in chronic kidney disease: a consensus report by the American Diabetes Association (ADA) and Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int (2022) 102:974–89. doi: 10.1016/j.kint.2022.08.012
13. Sano R, Shinozaki Y, Ohta T. Sodium-glucose cotransporters: Functional properties and pharmaceutical potential. J Diabetes Investig (2020) 11:770–82. doi: 10.1111/jdi.13255
14. Vallon V, Verma S. Effects of SGLT2 inhibitors on kidney and cardiovascular function. Annu Rev Physiol (2021) 83:503–28. doi: 10.1146/annurev-physiol-031620-095920
15. Cherney DZI, Dekkers CCJ, Barbour SJ, Cattran D, Abdul Gafor AH, Greasley PJ, et al. Effects of the SGLT2 inhibitor dapagliflozin on proteinuria in non-diabetic patients with chronic kidney disease (DIAMOND): a randomised, double-blind, crossover trial. Lancet Diabetes Endocrinol (2020) 8:582–93. doi: 10.1016/S2213-8587(20)30162-5
16. Heerspink HJ, Johnsson E, Gause-Nilsson I, Cain VA, Sjostrom CD. Dapagliflozin reduces albuminuria in patients with diabetes and hypertension receiving renin-angiotensin blockers. Diabetes Obes Metab (2016) 18:590–7.
17. Nojima T, Matsubayashi Y, Yoshida A, Suganami H, Abe T, Ishizawa M, et al. Influence of an SGLT2 inhibitor, tofogliflozin, on the resting heart rate in relation to adipose tissue insulin resistance. Diabetes Med (2020) 37:1316–25.
18. Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation (2014) 129:587–97.
19. van Bommel EJM, Muskiet MHA, van Baar MJB, Tonneijck L, Smits MM, Emanuel AL, et al. The renal hemodynamic effects of the SGLT2 inhibitor dapagliflozin are caused by post-glomerular vasodilatation rather than pre-glomerular vasoconstriction in metformin-treated patients with type 2 diabetes in the randomized, double-blind RED trial. Kidney Int (2020) 97:202–12.
20. Li J, Neal B, Perkovic V, de Zeeuw D, Neuen BL, Arnott C, et al. Mediators of the effects of canagliflozin on kidney protection in patients with type 2 diabetes. Kidney Int (2020) 98:769–77.
21. Rajasekeran H, Reich HN, Hladunewich MA, Cattran D, Lovshin JA, Lytvyn Y, et al. Dapagliflozin in focal segmental glomerulosclerosis: a combined human-rodent pilot study. Am J Physiol Renal Physiol (2018) 314:F412–22. doi: 10.1152/ajprenal.00445.2017
22. Antlanger M, Domenig O, Kaltenecker CC, Kovarik JJ, Rathkolb V, Müller MM, et al. Combined sodium glucose co-transporter-2 inhibitor and angiotensin-converting enzyme inhibition upregulates the renin-angiotensin system in chronic kidney disease with type 2 diabetes: Results of a randomized, double-blind, placebo-controlled exploratory trial. Diabetes Obes Metab (2022) 24:816–26. doi: 10.1111/dom.14639
23. Yoshimoto T, Furuki T, Kobori H, Miyakawa M, Imachi H, Murao K, et al. Effects of sodium-glucose cotransporter 2 inhibitors on urinary excretion of intact and total angiotensinogen in patients with type 2 diabetes. J Investig Med (2017) 65:1057–61. doi: 10.1136/jim-2017-000445
24. Heise T, Jordan J, Wanner C, Heer M, Macha S, Mattheus M, et al. Pharmacodynamic effects of single and multiple doses of empagliflozin in patients with type 2 diabetes. Clin Ther (2016) 38:2265–76. doi: 10.1016/j.clinthera.2016.09.001
25. Lambers Heerspink HJ, de Zeeuw D, Wie L, Leslie B, List J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes Metab (2013) 15:853–62. doi: 10.1111/dom.12127
26. Laursen JC, Søndergaard-Heinrich N, de Melo JML, Haddock B, Rasmussen IKB, Safavimanesh F, et al. Acute effects of dapagliflozin on renal oxygenation and perfusion in type 1 diabetes with albuminuria: A randomised, double-blind, placebo-controlled crossover trial. EClinicalMedicine (2021) 37:100895. doi: 10.1016/j.eclinm.2021.100895
27. Liu H, Sridhar VS, Lovblom LE, Lytvyn Y, Burger D, Burns K, et al. Markers of kidney injury, inflammation, and fibrosis associated with ertugliflozin in patients with CKD and diabetes. Kidney Int Rep (2021) 6:2095–104. doi: 10.1016/j.ekir.2021.05.022
28. Dekkers CCJ, Petrykiv S, Laverman GD, Cherney DZ, Gansevoort RT, Heerspink HJL. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes Metab (2018) 20:1988–93. doi: 10.1111/dom.13301
29. Wang XX, Levi J, Luo Y, Myakala K, Herman-Edelstein M, Qiu L, et al. SGLT2 Protein Expression Is Increased in Human Diabetic Nephropathy: SGLT2 protein inhibition decreases renal lipid accumulation, inflammation, and the development of nephropathy in diabetic mice. J Biol Chem (2017) 292:5335–48. doi: 10.1074/jbc.M117.779520
30. Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes (2005) 54:3427–34. doi: 10.2337/diabetes.54.12.3427
31. Solini A, Rossi C, Mazzanti CM, Proietti A, Koepsell H, Ferrannini E. Sodium-glucose co-transporter (SGLT)2 and SGLT1 renal expression in patients with type 2 diabetes. Diabetes Obes Metab (2017) 19:1289–94. doi: 10.1111/dom.12970
32. Srinivasan V Sridhar, Ambinathan JPN, Kretzler M, Pyle LL, Bjornstad P, Eddy S, et al. Renal SGLT mRNA expression in human health and disease: a study in two cohorts. Am J Physiol Renal Physiol (2019) 317:F1224–30. doi: 10.1152/ajprenal.00370.2019
33. Zhao Y, Xu L, Tian D, Xia P, Zheng H, Wang L, et al. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes Obes Metab (2018) 20:458–62. doi: 10.1111/dom.13101
34. Gillard P, Schnell O, Groop PH. The nephrological perspective on SGLT-2 inhibitors in type 1 diabetes. Diabetes Res Clin Pract (2020) 170:108462. doi: 10.1016/j.diabres.2020.108462
35. de Albuquerque Rocha N, Neeland IJ, McCullough PA, Toto RD, McGuire DK. Effects of sodium glucose co-transporter 2 inhibitors on the kidney. Diabetes Vasc Dis Res (2018) 15:375–86. doi: 10.1177/1479164118783756
36. Liu H, Sridhar VS, Boulet J, Dharia A, Khan A, Lawler PR, et al. Cardiorenal protection with SGLT2 inhibitors in patients with diabetes mellitus: from biomarkers to clinical outcomes in heart failure and diabetic kidney disease. Metabolism (2022) 126:154918. doi: 10.1016/j.metabol.2021.154918
37. Kanduri SR, Kovvuru K, Hansrivijit P, Thongprayoon C, Vallabhajosyula S, Pivovarova AI, et al. SGLT2 inhibitors and kidney outcomes in patients with chronic kidney disease. J Clin Med (2020) 9:2723. doi: 10.3390/jcm9092723
38. Packer M. Mechanisms leading to differential hypoxia-inducible factor signaling in the diabetic kidney: modulation by SGLT2 inhibitors and hypoxia mimetics. Am J Kidney Dis (2021) 77:280–6. doi: 10.1053/j.ajkd.2020.04.016
39. Gnudi L, Karalliedde J. Beat it early: putative renoprotective haemodynamic effects of oral hypoglycaemic agents. Nephrol Dial Transplant (2016) 31:1036–43. doi: 10.1093/ndt/gfv093
40. Packer M. Role of deranged energy deprivation signaling in the pathogenesis of cardiac and renal disease in states of perceived nutrient overabundance. Circulation (2020) 141:2095–105. doi: 10.1161/CIRCULATIONAHA.119.045561
41. Brown E, Rajeev SP, Cuthbertson DJ, Wilding JPH. A review of the mechanism of action, metabolic profile and haemodynamic effects of sodium-glucose co-transporter-2 inhibitors. Diabetes Obes Metab (2019) 21 Suppl 2:9–18. doi: 10.1111/dom.13650
42. Ito M, Tanaka T. The anticipated renoprotective effects of sodium-glucose cotransporter 2 inhibitors. Intern Med (2018) 57:2105–14. doi: 10.2169/internalmedicine.9842-17
43. Hesp AC, Schaub JA, Prasad PV, Vallon V, Laverman GD, Bjornstad P, et al. The role of renal hypoxia in the pathogenesis of diabetic kidney disease: a promising target for newer renoprotective agents including SGLT2 inhibitors? Kidney Int (2020) 98:579–89. doi: 10.1016/j.kint.2020.02.041
44. Packer M. Role of impaired nutrient and oxygen deprivation signaling and deficient autophagic flux in diabetic CKD development: implications for understanding the effects of sodium-glucose cotransporter 2-inhibitors. J Am Soc Nephrol (2020) 31:907–19. doi: 10.1681/ASN.2020010010
45. Yaribeygi H, Butler AE, Atkin SL, Katsiki N, Sahebkar A. Sodium-glucose cotransporter 2 inhibitors and inflammation in chronic kidney disease: Possible molecular pathways. J Cell Physiol (2018) 234:223–30. doi: 10.1002/jcp.26851
46. Packer M. Mitigation of the adverse consequences of nutrient excess on the kidney: A unified hypothesis to explain the renoprotective effects of sodium-glucose cotransporter 2 inhibitors. Am J Nephrol (2020) 51:289–93. doi: 10.1159/000506534
47. Hattori Y. Beneficial effects on kidney during treatment with sodium-glucose cotransporter 2 inhibitors: proposed role of ketone utilization. Heart Fail Rev (2021) 26:947–52. doi: 10.1007/s10741-020-10065-7
48. Gilbert RE. Sodium-glucose linked transporter-2 inhibitors: potential for renoprotection beyond blood glucose lowering? Kidney Int (2014) 86:693–700. doi: 10.1038/ki.2013.451
49. Cherney DZ, Perkins BA. Sodium-glucose cotransporter 2 inhibition in type 1 diabetes: simultaneous glucose lowering and renal protection? Can J Diabetes (2014) 38:356–63. doi: 10.1016/j.jcjd.2014.05.006
50. Heerspink HJL, Perkins BA, Fitchett DH, Husain M, Cherney DZI. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation (2016) 134:752–72. doi: 10.1161/CIRCULATIONAHA.116.021887
51. Satirapoj B. Sodium-glucose cotransporter 2 inhibitors with renoprotective effects. Kidney Dis (Basel) (2017) 3:24–32. doi: 10.1159/000471765
52. van Bommel EJ, Muskiet MH, Tonneijck L, Kramer MH, Nieuwdorp M, van Raalte DH. SGLT2 inhibition in the diabetic kidney-from mechanisms to clinical outcome. Clin J Am Soc Nephrol (2017) 12:700–10. doi: 10.2215/CJN.06080616
53. Thomas MC, Cherney DZI. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia (2018) 61:2098–107. doi: 10.1007/s00125-018-4669-0
54. Tsimihodimos V, Filippatos TD, Elisaf MS. SGLT2 inhibitors and the kidney: Effects and mechanisms. Diabetes Metab Syndr (2018) 12:1117–23. doi: 10.1016/j.dsx.2018.06.003
55. Alicic RZ, Neumiller JJ, Johnson EJ, Dieter B, Tuttle KR. Sodium-glucose cotransporter 2 inhibition and diabetic kidney disease. Diabetes (2019) 68:248–57. doi: 10.2337/dbi18-0007
56. Kuriyama S. A potential mechanism of cardio-renal protection with sodium-glucose cotransporter 2 inhibitors: amelioration of renal congestion. Kidney Blood Pressure Res (2019) 44:449–56. doi: 10.1159/000501081
57. Sarafidis P, Ferro CJ, Morales E, Ortiz A, Malyszko J, Hojs R, et al. SGLT-2 inhibitors and GLP-1 receptor agonists for nephroprotection and cardioprotection in patients with diabetes mellitus and chronic kidney disease. A consensus statement by the EURECA and the diabetes working groups of the ERA-EDTA. Nephrol Dial Transplant (2019) 34:208–30. doi: 10.1093/ndt/gfy407
58. Thomson SC, Vallon V. Renal effects of sodium-glucose co-transporter inhibitors. Am J Cardiol (2019) 124(Suppl 1):S28–35. doi: 10.1016/j.amjcard.2019.10.027
59. Hou YC, Zheng CM, Yen TH, Lu KC. Molecular mechanisms of SGLT2 inhibitor on cardiorenal protection. Int J Mol Sci (2020) 21:7833. doi: 10.3390/ijms21217833
60. Kashihara N, Kidokoro K, Kanda E. Renoprotective effects of sodium-glucose cotransporter-2 inhibitors and underlying mechanisms. Curr Opin Nephrol Hypertens (2020) 29:112–8. doi: 10.1097/MNH.0000000000000561
61. Packer M. Role of ketogenic starvation sensors in mediating the renal protective effects of SGLT2 inhibitors in type 2 diabetes. J Diabetes Complications (2020) 34:107647. doi: 10.1016/j.jdiacomp.2020.107647
62. Schnell O, Valensi P, Standl E, Ceriello A. Comparison of mechanisms and transferability of outcomes of SGLT2 inhibition between type 1 and type 2 diabetes. Endocrinol Diabetes Metab (2020) 3:e00129. doi: 10.1002/edm2.129
63. Zelniker TA, Braunwald E. Mechanisms of cardiorenal effects of sodium-glucose cotransporter 2 inhibitors: JACC state-of-the-art review. J Am Coll Cardiol (2020) 75:422–34. doi: 10.1016/j.jacc.2019.11.031
64. Lee JF, Berzan E, Sridhar VS, Odutayo A, Cherney DZI. Cardiorenal protection in diabetic kidney disease. Endocrinol Metab (Seoul) (2021) 36:256–69. doi: 10.3803/EnM.2021.987
65. Onyali CB, Anim-Koranteng C, Shah HE, Bhawnani N, Ethirajulu A, Alkasabera A, et al. Role of selective sodium-glucose co-transporter-2 inhibitors in managing cardio-renal complications in type 2 diabetes mellitus: beyond glycemic control. Cureus (2021) 13:e17452. doi: 10.7759/cureus.17452
66. Puglisi S, Rossini A, Poli R, Dughera F, Pia A, Terzolo M, et al. Effects of SGLT2 inhibitors and GLP-1 receptor agonists on renin-angiotensin-aldosterone system. Front Endocrinol (Lausanne) (2021) 12:738848. doi: 10.3389/fendo.2021.738848
67. Leoncini G, Russo E, Bussalino E, Barnini C, Viazzi F, Pontremoli R. SGLT2is and renal protection: from biological mechanisms to real-world clinical benefits. Int J Mol Sci (2021) 22:4441. doi: 10.3390/ijms22094441
68. Sharaf El Din UAA, Salem MM, Abdulazim DO. Sodium-glucose cotransporter 2 inhibitors as the first universal treatment of chronic kidney disease. Nefrol (Engl Ed) (2022) 42:390–403. doi: 10.1016/j.nefroe.2022.08.001
69. Provenzano M, Pelle MC, Zaffina I, Tassone B, Pujia R, Ricchio M, et al. Sodium-glucose co-transporter-2 inhibitors and nephroprotection in diabetic patients: more than a challenge. Front Med (Lausanne) (2021) 8:654557. doi: 10.3389/fmed.2021.654557
70. Srinivas N, Sarnaik MK, Modi S, Pisipati Y, Vaidya S, Syed Gaggatur N, et al. Sodium-glucose cotransporter 2 (SGLT-2) inhibitors: delving into the potential benefits of cardiorenal protection beyond the treatment of type-2 diabetes mellitus. Cureus (2021) 13:e16868. doi: 10.7759/cureus.16868
71. Castaneda AM, Dutra-Rufato A, Juarez MJ, Grosembacher L, Gonzalez-Torres H, Musso CG. Sodium-glucose cotransporter 2 inhibitors (SGLT2i): renal implications. Int Urol Nephrol (2021) 53:291–9. doi: 10.1007/s11255-020-02585-w
72. Takata T, Isomoto H. Pleiotropic effects of sodium-glucose cotransporter-2 inhibitors: renoprotective mechanisms beyond glycemic control. Int J Mol Sci (2021) 22:4374. doi: 10.3390/ijms22094374
73. Pollock C, Neuen BL. Sodium-glucose cotransporter 2 inhibition: rationale and mechanisms for kidney and cardiovascular protection in people with and without diabetes. Adv Chronic Kidney Dis (2021) 28:298–308. doi: 10.1053/j.ackd.2021.02.006
74. Bailey CJ. Uric acid and the cardio-renal effects of SGLT2 inhibitors. Diabetes Obes Metab (2019) 21:1291–8. doi: 10.1111/dom.13670
75. Yip ASY, Leong S, Teo YH, Teo YN, Syn NLX, See RM, et al. Effect of sodium-glucose cotransporter-2 (SGLT2) inhibitors on serum urate levels in patients with and without diabetes: a systematic review and meta-regression of 43 randomized controlled trials. Ther Adv Chronic Dis (2022) 13:20406223221083509. doi: 10.1177/20406223221083509
76. Dekkers CCJ, Gansevoort RT, Heerspink HJL. New diabetes therapies and diabetic kidney disease progression: the role of SGLT-2 inhibitors. Curr Diabetes Rep (2018) 18:27. doi: 10.1007/s11892-018-0992-6
77. Nayak S, Rathore V, Bharati J, Sahu KK. Extending the ambit of SGLT2 inhibitors beyond diabetes: a review of clinical and preclinical studies on non-diabetic kidney disease. Expert Rev Clin Pharmacol (2021) 14:1513–26. doi: 10.1080/17512433.2021.2028620
78. Dekkers CCJ, Gansevoort RT. Sodium-glucose cotransporter 2 inhibitors: extending the indication to non-diabetic kidney disease? Nephrol Dial Transplant (2020) 35(Suppl 1):i33–42. doi: 10.1093/ndt/gfz264
79. Rajasekeran H, Cherney DZ, Lovshin JA. Do effects of sodium-glucose cotransporter-2 inhibitors in patients with diabetes give insight into potential use in non-diabetic kidney disease? Curr Opin Nephrol Hypertens (2017) 26:358–67. doi: 10.1097/MNH.0000000000000343
80. Ekanayake P, Mudaliar S. A novel hypothesis linking low-grade ketonaemia to cardio-renal benefits with sodium-glucose cotransporter-2 inhibitors. Diabetes Obes Metab (2022) 24:3–11. doi: 10.1111/dom.14562
81. Herrington WG, Savarese G, Haynes R, Marx N, Mellbin L, Lund LH, et al. Cardiac, renal, and metabolic effects of sodium-glucose co-transporter 2 inhibitors: a position paper from the European Society of Cardiology ad-hoc task force on sodium-glucose co-transporter 2 inhibitors. Eur J Heart Fail (2021) 23:1260–75. doi: 10.1002/ejhf.2286
82. Oguz F, Demoulin N, Thissen JP, Jadoul M, Morelle J. Inhibition of sodium-glucose cotransporter 2 to slow the progression of chronic kidney disease. Acta Clin Belg (2021) 77:805–14. doi: 10.1080/17843286.2021.1966583
83. Sternlicht H, Bakris GL. Blood pressure lowering and sodium-glucose co-transporter 2 inhibitors (SGLT2is): more than osmotic diuresis. Curr Hypertens Rep (2019) 21:12. doi: 10.1007/s11906-019-0920-4
84. Cherney DZI, Cooper ME, Tikkanen I, Pfarr E, Johansen OE, Woerle HJ, et al. Pooled analysis of phase III trials indicate contrasting influences of renal function on blood pressure, body weight, and HbA1c reductions with empagliflozin. Kidney Int (2018) 93:231–44. doi: 10.1016/j.kint.2017.06.017
85. Chakraborty S, Galla S, Cheng X, Yeo JY, Mell B, Singh V, et al. Saltresponsive metabolite, β-hydroxybutyrate, attenuates hypertension. Cell Rep (2018) 25:677–89.e4. doi: 10.1016/j.celrep.2018.09.058
86. Ficociello LH, Rosolowsky ET, Niewczas MA, Maselli NJ, Weinberg JM, Aschengrau A, et al. High-normal serum uric acid increases risk of early progressive renal function loss in type 1 diabetes: results of a 6-year follow-up. Diabetes Care (2010) 33:1337–43. doi: 10.2337/dc10-0227
87. Hovind P, Rossing P, Tarnow L, Johnson RJ, Parving HH. Serum uric acid as a predictor for development of diabetic nephropathy in type 1 diabetes: an inception cohort study. Diabetes (2009) 58:1668–71. doi: 10.2337/db09-0014
88. Bose B, Badve SV, Hiremath SS, Boudville N, Brown FG, Cass A, et al. Effects of uric acid-lowering therapy on renal outcomes: a systematic review and meta-analysis. Nephrol Dial Transplant (2014) 29:406–13. doi: 10.1093/ndt/gft378
89. Goicoechea M, Garcia de Vinuesa S, Verdalles U, Verde E, Macias N, Santos A, et al. Allopurinol and progression of CKD and cardiovascular events: long-term follow-up of a randomized clinical trial. Am J Kidney Dis (2015) 65:543–9. doi: 10.1053/j.ajkd.2014.11.016
90. Ahola AJ, Sandholm N, Forsblom C, Harjutsalo V, Dahlstrom E, Groop PH, et al. The serum uric acid concentration is not causally linked to diabetic nephropathy in type 1 diabetes. Kidney Int (2017) 91:1178–85. doi: 10.1016/j.kint.2016.11.025
91. Kimura K, Hosoya T, Uchida S, Inaba M, Makino H, Maruyama S, et al. Febuxostat therapy for patients with stage 3 CKD and asymptomatic hyperuricemia: A randomized trial. Am J Kidney Dis (2018) 72:798–810. doi: 10.1053/j.ajkd.2018.06.028
92. Badve SV, Pascoe EM, Tiku A, Boudville N, Brown FG, Cass A, et al. Effects of allopurinol on the progression of chronic kidney disease. N Engl J Med (2020) 382:2504–13. doi: 10.1056/NEJMoa1915833
93. Suijk DLS, van Baar MJB, van Bommel EJM, Iqbal Z, Krebber MM, Vallon V, et al. SGLT2 inhibition and uric acid excretion in patients with type 2 diabetes and normal kidney function. Clin J Am Soc Nephrol (2022) 17:663–71. doi: 10.2215/CJN.11480821
94. Kim HK, Ishizawa R, Fukazawa A, Wang Z, Bezan Petric U, Hu MC, et al. Dapagliflozin attenuates sympathetic and pressor responses to stress in young prehypertensive spontaneously hypertensive rats. Hypertension (2022) 79:1824–34. doi: 10.1161/HYPERTENSIONAHA.122.19177
95. Helal I, Fick-Brosnahan GM, Reed-Gitomer B, Schrier RW. Glomerular hyperfiltration: definitions, mechanisms and clinical implications. Nat Rev Nephrol (2012) 8:293–300. doi: 10.1038/nrneph.2012.19
96. Coady MJ, El Tarazi A, Santer R, Bissonnette P, Sasseville LJ, Calado J, et al. MAP17 is a necessary activator of renal na+/glucose cotransporter SGLT2. J Am Soc Nephrol (2017) 28:85–93. doi: 10.1681/ASN.2015111282
97. Hallow KM, Greasley PJ, Helmlinger G, Chu L, Heerspink HJ, Boulton DW. Evaluation of renal and cardiovascular protection mechanisms of SGLT2 inhibitors: model-based analysis of clinical data. Am J Physiol Renal Physiol (2018) 315:F1295–F306. doi: 10.1152/ajprenal.00202.2018
98. Jiang F, Yang J, Zhang Y, Dong M, Wang S, Zhang Q, et al. Angiotensin-converting enzyme 2 and angiotensin 1-7: novel therapeutic targets. Nat Rev Cardiol (2014) 11:413–26. doi: 10.1038/nrcardio.2014.59
99. Packer M. Autophagy-dependent and -independent modulation of oxidative and organellar stress in the diabetic heart by glucose-lowering drugs. Cardiovasc Diabetol (2020) 19:62. doi: 10.1186/s12933-020-01041-4
100. Ekanayake P, Hupfeld C, Mudaliar S. Sodium-glucose cotransporter type 2 (SGLT-2) inhibitors and ketogenesis: the good and the bad. Curr Diabetes Rep (2020) 20:74. doi: 10.1007/s11892-020-01359-z
101. Heyman SN, Abassi Z. Gliflozins, erythropoietin, and erythrocytosis: is it renal normoxia- or hypoxia-driven? J Clin Med (2023) 12:4871. doi: 10.3390/jcm12144871
102. O’Neill J, Fasching A, Pihl L, Patinha D, Franzen S, Palm F. Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am J Physiol Renal Physiol (2015) 309:F227–34. doi: 10.1152/ajprenal.00689.2014
103. Gullaksen S, Vernstrøm L, Sørensen SS, Ringgaard S, Laustsen C, Funck KL, et al. Separate and combined effects of semaglutide and empagliflozin on kidney oxygenation and perfusion in people with type 2 diabetes: a randomised trial. Diabetologia (2023) 66:813–25. doi: 10.1007/s00125-023-05876-w
104. Heyman SN, Raz I, Dwyer JP, Weinberg Sibony R, Lewis JB, Abassi Z. Diabetic proteinuria revisited: updated physiologic perspectives. Cells (2022) 11:2917. doi: 10.3390/cells11182917
105. Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol (2006) 17:17–25. doi: 10.1681/ASN.2005070757
106. Naguib M, Rashed LA. Serum level of the autophagy biomarker Beclin-1 in patients with diabetic kidney disease. Diabetes Res Clin Pract (2018) 143:56–61. doi: 10.1016/j.diabres.2018.06.022
107. Matboli M, Azazy AEM, Adel S, Bekhet MM, Eissa S. Evaluation of urinary autophagy transcripts expression in diabetic kidney disease. J Diabetes Complications (2017) 31:1491–8. doi: 10.1016/j.jdiacomp.2017.06.009
108. Aranda-Rivera AK, Cruz-Gregorio A, Pedraza-Chaverri J, Scholze A. Nrf2 activation in chronic kidney disease: promises and pitfalls. Antioxid (Basel) (2022) 11:1112. doi: 10.3390/antiox11061112
109. Rasmussen M, Hansen KH, Scholze A. Nrf2 protein serum concentration in human CKD shows a biphasic behavior. Antioxid (Basel) (2023) 12:932. doi: 10.3390/antiox12040932
110. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med (1993) 329:1456–62. doi: 10.1056/NEJM199311113292004
111. Maschio G, Alberti D, Janin G, Locatelli F, Mann JF, Motolese M, et al. Effect of the angiotensin-converting-enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting-Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med (1996) 334:939–45. doi: 10.1056/NEJM199604113341502
112. Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Lancet (1997) 349(9069):1857–63.
113. Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med (2001) 345:861–9. doi: 10.1056/NEJMoa011161
114. Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med (2001) 345:851–60. doi: 10.1056/NEJMoa011303
115. Hou FF, Zhang X, Zhang GH, Xie D, Chen PY, Zhang WR, et al. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N Engl J Med (2006) 354:131–40. doi: 10.1056/NEJMoa053107
116. Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med (2016) 375:323–34. doi: 10.1056/NEJMoa1515920
117. Wheeler DC, Toto RD, Stefánsson BV, Jongs N, Chertow GM, Greene T, et al. A pre-specified analysis of the DAPA-CKD trial demonstrates the effects of dapagliflozin on major adverse kidney events in patients with IgA nephropathy. Kidney Int (2021) 100:215–24. doi: 10.1016/j.kint.2021.03.033
118. Wheeler DC, Jongs N, Stefansson BV, Chertow GM, Greene T, Hou FF, et al. Safety and efficacy of dapagliflozin in patients with focal segmental glomerulosclerosis: A prespecified analysis of the DAPA-CKD trial. Nephrol Dial Transplant (2022) 37:1647–56. doi: 10.1093/ndt/gfab335
Keywords: diabetic chronic kidney disease, nondiabetic chronic kidney disease, sodium glucose cotransporter type 2 inhibitors, nephroprotection, pathophysiologic mechanisms
Citation: Speedtsberg ES and Tepel M (2023) Narrative review investigating the nephroprotective mechanisms of sodium glucose cotransporter type 2 inhibitors in diabetic and nondiabetic patients with chronic kidney disease. Front. Endocrinol. 14:1281107. doi: 10.3389/fendo.2023.1281107
Received: 21 August 2023; Accepted: 16 November 2023;
Published: 20 December 2023.
Edited by:
Giuseppe Remuzzi, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, ItalyReviewed by:
George Bakris, University of Chicago Medical Center, United StatesCopyright © 2023 Speedtsberg and Tepel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Tepel, bXRlcGVsQGhlYWx0aC5zZHUuZGs=
†ORCID: Martin Tepel, orcid.org/0000-0002-0086-0997
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.