 Anasuya Guha1*
Anasuya Guha1* Ales Vicha2
Ales Vicha2 Tomas Zelinka3Martin Kana4
Tomas Zelinka3Martin Kana4 Zdenek Musil5
Zdenek Musil5 Karel Pacak6Jan Betka4
Karel Pacak6Jan Betka4 Martin Chovanec1
Martin Chovanec1 Jan Plzak4Jan Boucek4
Jan Plzak4Jan Boucek4- 1Department of Otorhinolaryngology, Charles University, 3rd Faculty of Medicine and University Hospital Kralovske Vinohrady, Prague, Czechia
- 2Department of Pediatric Hematology and Oncology, Charles University, 2nd Faculty of Medicine and University Hospital Motol, Prague, Czechia
- 33rd Department of Medicine, Department of Endocrinology and Metabolsim of the 1st Faculty of Medicine and General University Hospital in Prague, Prague, Czechia
- 4Department of Otorhinolaryngology and Head and Neck Surgery, Charles University, 1st Faculty of Medicine and University Hospital Motol, Prague, Czechia
- 5Institute of Biology and Medical Genetics of the 1st Faculty of Medicine and General University Hospital in Prague, Prague, Czechia
- 6Section of Medical Neuroendocrinology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, United States
Introduction: Head and neck paragangliomas (HNPGLs) are rare neuroendocrine tumors, which are mostly benign in nature. Amongst all genes, Succinate Dehydrogenase Subunit D (SDHD) is the most commonly mutated in familial HNPGLs. In about 30% of HNPGLs, germline mutations in SDHD can also occur in the absence of positive family history, thus giving rise to “occult familial” cases. Our aim was to evaluate the pattern of SDHD germline mutations in Czech patients with HNPGLs.
Materials and methods: We analyzed a total of 105 patients with HNPGLs from the Otorhinolaryngology departments of 2 tertiary centers between 2006 – 2021. All underwent complex diagnostic work-up and were also consented for genetic analysis.
Results: Eighty patients aged 13-76 years were included; around 60% with multiple PGLs were males. Carotid body tumor was the most frequently diagnosed tumor. Germline SDHD mutation was found in only 12% of the Czech patients; approximately 78% of those harboring the mutation had negative family history. The mutation traits had higher affiliation for multiple tumors with nearly 70% patients of ≤ 40 years of age.
Conclusion: An SDHD mutation variant was shared amongst unrelated patients but no founder-effect was established. Our findings confirmed that the pattern of SDHD mutation distribution amongst HNPGLs in Czech Republic differs from most studies worldwide.
1 Introduction
Head and neck paragangliomas (HNPGLs) are classified as tumors originating from extra-adrenal paraganglias (1, 2), with an overall incidence of 0.3 to 1 in 100 000 (3, 4). Gender distribution shows a higher female predominance of 3-4:1; most patients become symptomatic between their fourth and seventh decade of life (5, 6). Carotid body paragangliomas (CBPGLs) represent 60% of all types of HNPGLs, with bilateral presentation in about 10% of patients (5, 7, 8). Other frequently detected HNPGLs include jugulotympanic (<35-40%) (9) and vagal (<5%) (10). These neuroendocrine tumors are mostly benign and non-secretory in nature. Patients can remain asymptomatic for long periods, however, the tumors can be found incidentally either during ultrasound of the neck or due to symptoms arising from cranial nerve dysfunction (6). A multidisciplinary approach is required for the management of such tumors.
This disease can occur in a sporadic or hereditary form, hence genetic analysis plays a pivotal role in differentiating the forms. This form of investigation has been popularized for the early detection, management and prediction of tumors in familial cases. It is also now known that nearly 40-50% of all HNPGLs are hereditary (11), including a significant subset without known family history.
Although many genes have been linked to pheochromocytomas and paragangliomas, the mitochondrial complex II genes, subunits of Succinate Dehydrogenase (SDHx) genes, have been most often identified as susceptible to the development of HNPGLs (5, 6). The discovery of Succinate Dehydrogenase Subunit D (SDHD) gene in families with Paraganglioma syndrome type 1 (PGL1) in 2000, helped in understanding the molecular mechanism of paraganglioma inheritance (4, 6, 12). Subsequently, it was shown, that mutations in other subunits (A – C) of SDHx along with the SDH Assembly Factor (SDHAF2) genes lead to Paraganglioma syndrome types 2 to 5 (PGL2-5), which are all inherited in an autosomal dominant manner (3–7). In hereditary syndromes, jugular, vagal and carotid PGLs are observed in 26%, 31%, and 39% of cases respectively (3, 13). Young age (≤ 40 years) with multiple tumors, positive family history, presence of carotid body tumor as well as bilateral presentation have a higher predilection for familial forms of the disease (6, 9, 13, 14). PGL1, related to the SDHD gene, has the highest affinity for HNPGLs (13). Germline mutations in SDHx genes occurring in suspected sporadic HNPGLs, due to the absence of positive family history, suggests the possibility of “occult familial” cases. This concept is mainly seen with SDHD (14, 15). Similarly, the risk of occult paragangliomas amongst SDHD carriers is also relatively high (16). Therefore, the evidence supports the fact, that, patients with head and neck paragangliomas should undergo genetic testing (4, 6, 11, 17).
We decided to study the distribution pattern of SDHD mutations amongst our cohort of patients with HNPGLs. From a clinical point of view, these findings will also have an impact on the early management including screening of family members of such patients in standard clinical practice.
2 Materials and methods
2.1 Patients
Between 2006 and 2021, 105 patients with HNPGLs were referred for consultation to the departments of Otorhinolaryngology across 2 tertiary centers. A multidisciplinary approach was adopted in all patients. Patients underwent standard examination including clinical, biochemical and radiological (anatomical and functional imaging) investigations. The HNPGLs were classified by focality and localization [carotid body (CBPGLs), jugular (JPGLs), tympanic (TPGLs) and vagal (VPGLs)] based on clinical and radiological findings. Furthermore, other forms of paragangliomas and sites of metastases were also identified. Plasma metanephrine, normetanepherine and chromogranin A were used to assess secretory activity of tumors and risk of malignity. The importance and possibility of genetic analysis were discussed with all the patients and referred accordingly. A treatment plan was advocated in each case, with the decision on interventional therapy or ‘wait and scan’ approach.
2.2 Protocol for Genetic investigations
Genetic examination was recommended for all our patients and consent was obtained accordingly. Those who did not consent or failed to attend their tests were excluded from this study. Peripheral blood samples were collected to initiate the process. Genomic DNA was extracted from 10mL of EDTA-anticoagulated blood using standardized methods. In our genetic center, we use Sanger Sequencing for single gene mutation analysis to exclude SDHD first followed by Next Generation Sequencing (NGS). However, in cases with multiple HNPGLs or if requested by the referring physician, NGS was done first. In the context of sharing similar research interests for pheochromocytomas and paragangliomas (PPGLs), we also performed Whole Exome Sequencing (WES) for 13 patients to compare results with NGS examination in Czech Republic. This was done in collaboration with the National Institutes of Health and National Cancer Institute, Bethesda, USA.
On identification of an index patient with positive germline mutation, they were advised to contact their first degree relatives at risk to undergo genetic counselling and if necessary preventive scanning in order to evaluate carrier status.
2.2.1 Sanger sequencing
The extracted DNA from peripheral blood samples were checked for quality control. These were analyzed using specific primers for SDHD exons 1-4 (primer sequence available on request). DNA fragments were sequenced in both forward and backward directions using an automatic fluorescent ABI Prism™ 3130 Genetic Analyzer (PE Applied Biosystems). DNA sequence analysis was then done using the Mutation Surveyor (Carolina Biosystems®, USA) software.
2.2.2 Next generation sequencing
Capture-based next-generation DNA sequencing was performed on a NextSeq 500 instrument (Illumina®, San Diego, California, USA). A custom Pheochromocytoma/Paraganglioma panel was used. This covers the entire coding as well as selected intronic and promoter regions of 123 genes, which are of particular relevance in these tumors. Agilent capture system was used (SSEL XT HS Reagent Kit, Agilent). Reads were aligned against the reference genome (GRch38). GENOVESA (BIOXSYS®, Czech Republic) software was used for analysis.
2.2.3 Whole exome sequencing
This technique of sequencing consisted two main processes, namely target-enrichment and sequencing. Sample preparation included purification and quality control of DNA samples. The next step was target-enrichment (DNA fragmentation and exome capture). This was performed to select and capture exome from DNA samples. Seventy Exome samples were pooled and sequenced on NovaSeq 6000 S2 (Illumina®, USA) run using Agilent® SureSelect Human All Exon V7 and paired-end sequencing mode. The samples have 100M to 189M pass filter reads, with Q30 above 89%. The samples were mapped and variants were called using Dynamic Read Analysis for GENomics (Dragen; Illumina®, USA).
3 Results
3.1 Patient demographics and characteristics of tumors
A total of 105 patients were referred with HNPGLs; however 80 (25 males; 55 females) patients of 13-76 years completed genetic testing. Approximately 40% of patients were ≤ 40 years of age. Seventy-six patients were of Czech origin, the other four were from Poland, Hungary, Slovakia and Syria. Only 7 patients had a positive family history of HNPGLs; six were females. A total of 102 head and neck tumors were found amongst 80 patients; 94% had benign tumors (Table 1). Four out of five patients with metastatic disease, had solitary tumors. CBPGLs were the most commonly diagnosed tumors, followed by JPGLs, TPGLs and VPGLs. All patients with positive family history had CBPGLs. Bilateral carotid body tumors were seen in 8 patients; approximately 63% being males. Tympanic paragangliomas were almost exclusively found in females. Amongst 15 patients with multiple HNPGLs, 93% were of Czech origin. Representative data from previously published results on patients with multiple tumors have been included here (18). About 67% of patients with multiple tumors were of young age (≤ 40 years) and had higher male predominance. Five patients had paragangliomas located below the neck; three patients had mediastinal PGLs whilst retroperitoneal tumors were detected in 2 others. Pheochromocytomas were not seen in our cohort. Raised plasma metanephrine and normetanephrine levels were detected in 2 related patients, diagnosed with solitary CBPGL and mediastinal PGL. Amongst those with multiple HNPGLs, Chromogranin A was elevated in two patients (one had retroperitoneal and the other had mediastinal PGL) and Normetanephrine was higher in another patient (18). Amidst those with benign tumors, 64 patients had intervention (89% surgery; 5% radiotherapy; 6% combination therapy), 10 were allocated to ‘wait and scan’ and one patient died from respiratory complications of advanced disease. Those with metastases had combination therapy.
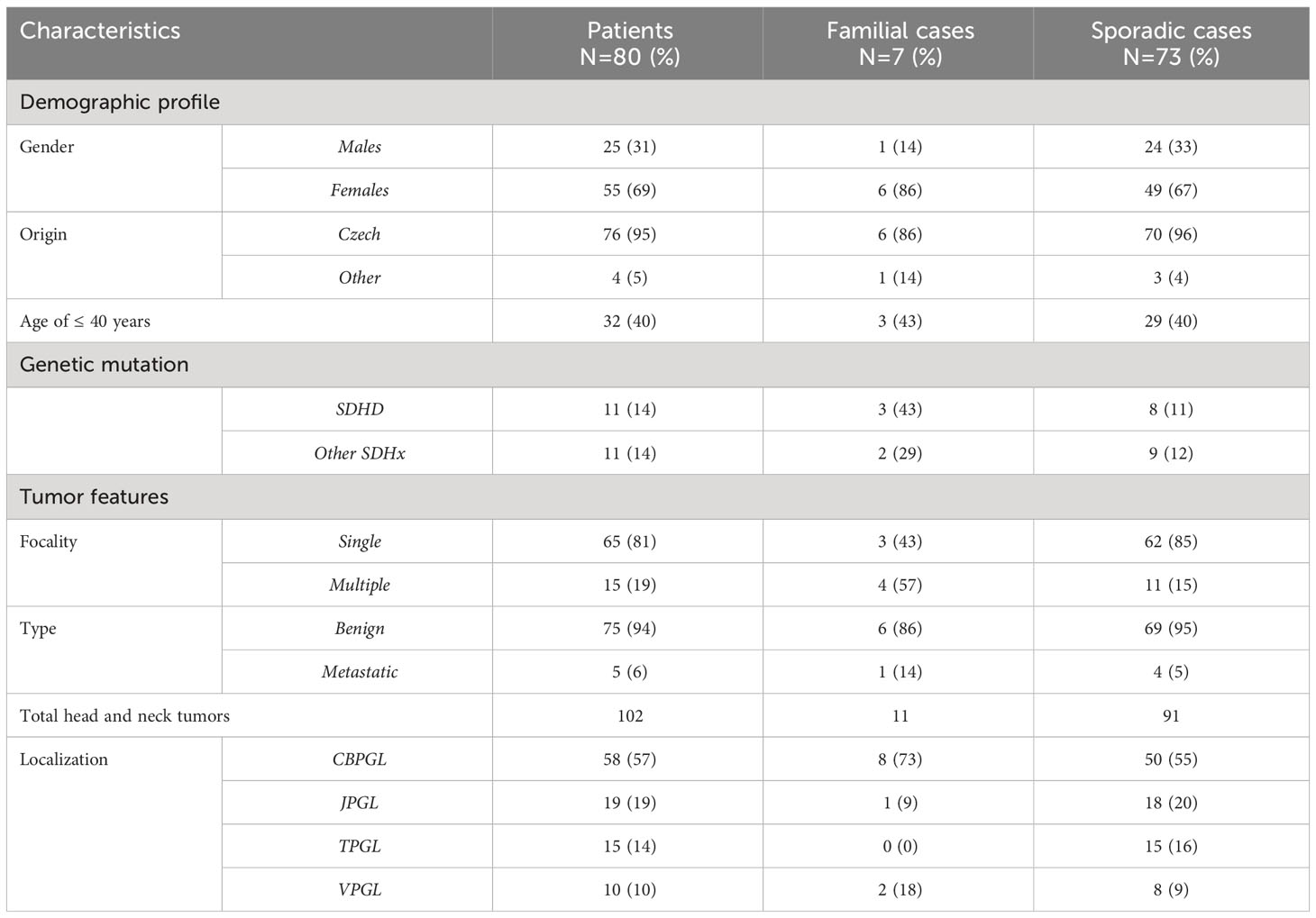
Table 1 Characteristics of patients with HNPGLs.
3.2 Germline mutation analysis in HNPGLs
On completion of all genetic analysis, which included the NGS panel genes for PPGLs and in certain cases, the use of the extended genetic library for WES, interestlingly, only SDHx pathogenic germline mutations were found in the entire cohort. Germline mutations were detected in 22 patients; eight were found with SDHB gene mutations, whilst 3 had SDHC and only 11 had SDHD mutation (Table 2). We only reported pathogenic and likely pathogenic variants according to the American College of Medical Genetics and Genomics (ACMG) classification and Clinical Variants Database of germline mutation (ClinVar) database. No novel mutation was found. Nine patients were of Czech origin, one was from Poland and the other was of Slovakian origin. Three patients had positive family history including a patient from Poland (18). The variants of SDHD mutation showed a higher affiliation for patients ≤ 40 years old with multiple tumors. Approximately 64% with SDHD mutation were females. For the purposes of our study and to accurately assess the frequency of SDHD mutation amongst Czech patients, we excluded the 4 patients with other nationalities.
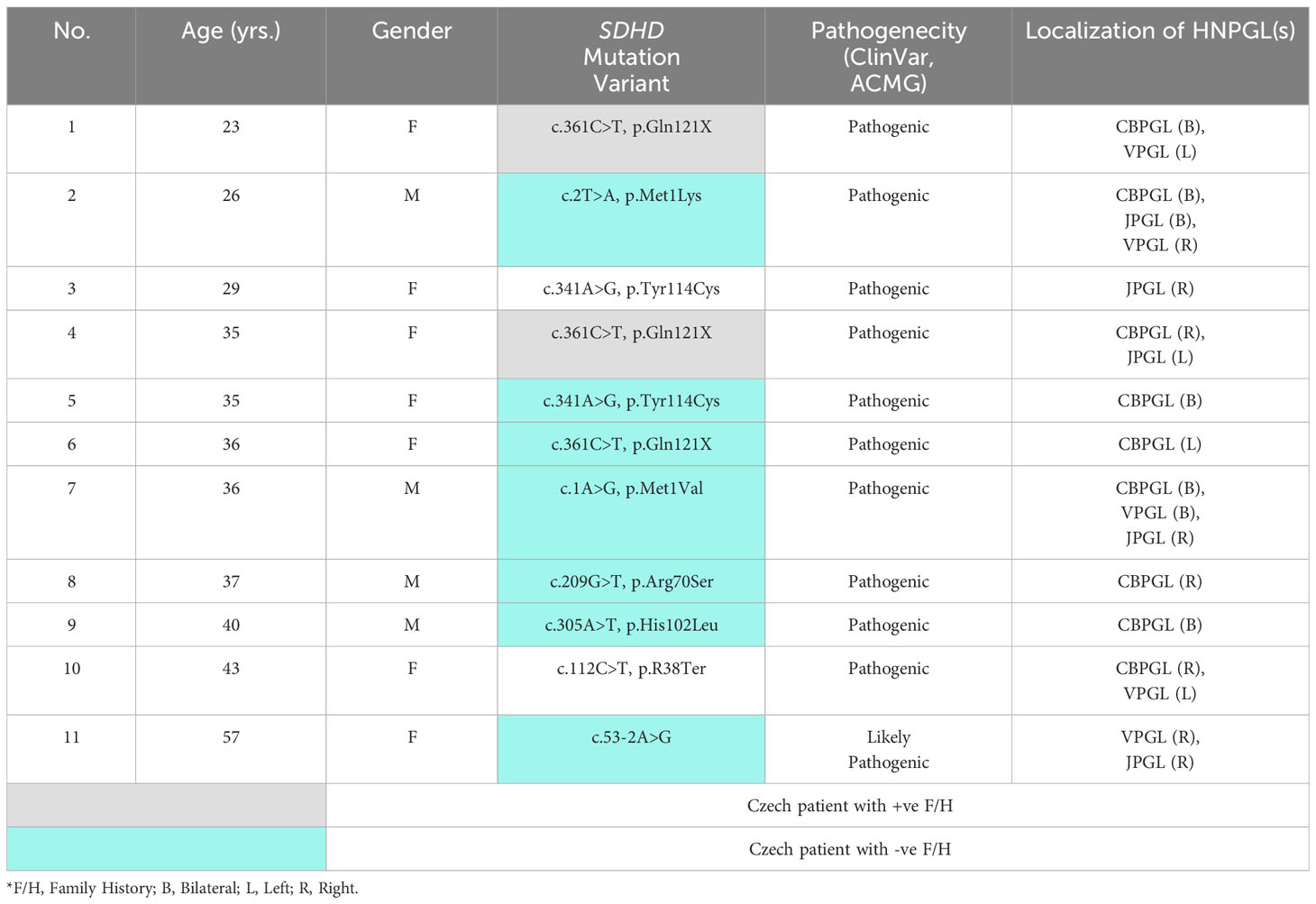
Table 2 SDHD germline mutation analysis in patients with HNPGLs.
The SDHD mutation was found only in 12% of Czech patients, where 78% were occult familial cases (Table 2). CBPGLs were seen in 8 out of 9 patients, bilateral tumors being present in 56% cases. The c.361C>T, p.Gln121X variant was reported in 2 familial as well as in a suspected sporadic case. These patients were unrelated. All of them were females below 40 years of age and had CBPGLs. The youngest patient with a positive family history and multiple HNPGLs was diagnosed with metastatic disease. Lymph node, bone and liver metastases were detected on 68Ga-DOTA-TOC PET-CT whole body imaging. The 35-year old female patient with multiple benign HNPGLs also has a sister with multiple paragangliomas. The last patient with the same variant had a single tumor and negative family history. Both children of this 36-year old female patient also tested positive for this mutation, however, they are clinically silent due to the probability of maternal imprinting.
The variant c.341A>G, p.Tyr114Cys causing occult PGL1 in patient no. 5 with benign bilateral CBPGLs was also found in the Slovakian patient with single JPGL. Highest number of tumors including a mediastinal PGL was diagnosed in the 36-year old male patient, who developed very advanced disease that led to his death from severe lower cranial nerve dysfunction (18). The other 4 SDHD mutation variants were also occult familial cases with benign tumors. The last patient in this series was diagnosed with the second tumor after 3 years follow-up (18). As already mentioned, eleven patients had SDHx gene mutations other than SDHD. In comparison to total SDHx gene mutations, SDHD gene mutation was seen in 47%.
4 Discussion
During the period of our study, 80 out of 105 patients diagnosed with HNPGLs were included. Amongst these patients, 95% were of Czech origin. We demonstrated higher female predominance (F:M = 2.2:1), typically seen in HNPGLs, but less than expected for females (5). Fifteen patients had multiple tumors including 5 patients with PGLs below the neck. Only 3 (one with SDHB and two family members with SDHC germline mutation) patients had elevated catecholamines; Chromogranin A levels were raised in 2 unrelated patients with SDHD germline mutation. Approximately 6% of all patients had metastatic disease.
Carotid body tumors (57%) were the most commonly found HNPGLs, followed by JPGLs (19%), TPGLs (14%) and VPGLs (10%). This pattern is in accordance with most reported studies (5, 7, 9, 17). Eight out of fifty patients with CBPGLs, had bilateral presentation. All 7 patients with positive family history including 1 of Polish origin had carotid body tumors (43% solitary; 57% bilateral). These tumors are usually of non-hereditary form in about 60% of the cases (3, 9), a finding confirmed by our study too. It should be mentioned that up to 72% patients diagnosed with SDHx germline mutation including nine out of the eleven with SDHD gene mutation had an affinity for CBPGLs. Therefore, the presence of carotid body tumor, whether as solitary or bilateral should also be considered a risk for germline disease (19, 20). Jugulotympanic tumors were almost exclusively seen in female patients, an observation made in a large muticentric study too (17). Bilateral jugular PGLs were seen in 2 males only.
Researchers worldwide reported that head and neck paragangliomas (solitary or multiple) represent a strong predictor for SDHD mutation even in small cohort of patients (15, 21–32) (Table 3). The SDHD mutation was surprisingly found in only 9 out of 76 Czech patients.
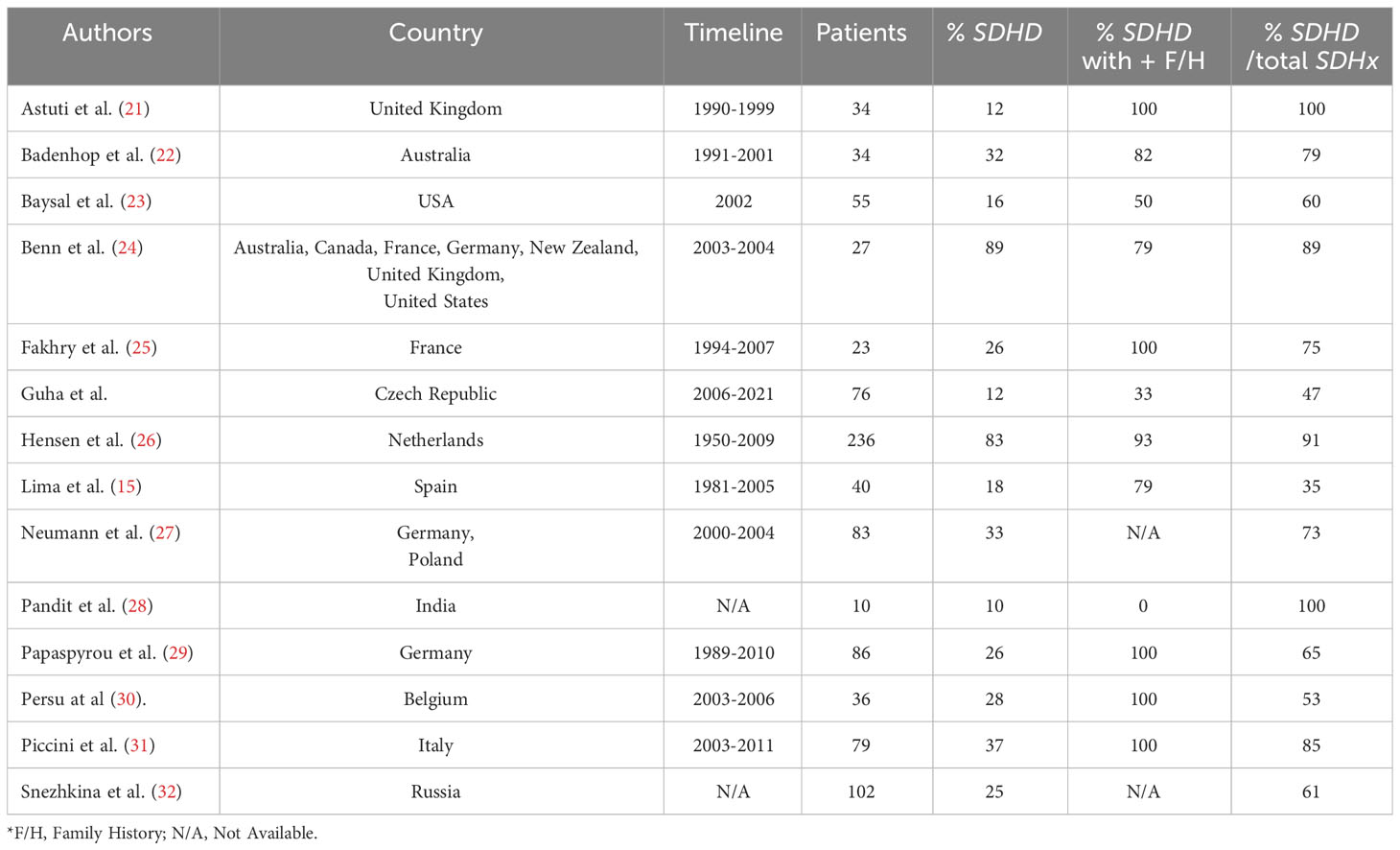
Table 3 Worldwide distribution of SDHD germline mutations in patients with HNPGLs.
It is already well established, that those with the familial form of the disease, usually present at a younger age (less than 40 years) and with multiple tumors (9, 11, 17, 19, 20). Similarly, the SDHD variants discovered in our study exhibited a strong association with young age and multiple tumors.
Furthermore, a large number of studies showed that the percentage of germline SDHD mutations in positive family history could be as high as 80% to 100% (Table 3). This occurrence is most typical for Netherlands and is supported by consistent findings (14, 26, 33). In contrast, only one-third of Czech patients with known family history showed germline pathogenic SDHD mutation. Interestingly ‘occult familial cases’ were observed in almost 78% of patients diagnosed with PGL1. This may be partially explained by the pattern of transmission. Theoretically, this autosomal dominant syndrome can be inherited both via the paternal and maternal lines, but in maternal transmission, PGLs almost never develop. There is still a 50% chance of maternally derived carriers transmitting the mutation to their offspring, hence PGL1 can seem to skip generations (3).
The most frequently reported variant amongst Czech patients was the SDHD c.361C>T, p.Gln121X, a pathogenic point mutation. This was seen in 2 unrelated familial as well as in a suspected sporadic case; all of them were below the age of 40 years (Table 2). This mutation was also observed in another unrelated young male patient from Czech Republic with negative family history and retroperitoneal PGL (34). The youngest patient amongst the three with c.361C>T, p.Gln121X in this study, had a positive family history presented with multiple tumors and metastatic liver disease, which is an unusual feature. This sort of uncommon presentation was also seen in a family in Brazil; all diagnosed members were of young age. Apart from HNPGLs, pheochromocytomas were also seen. Here, the youngest of three members, an 11-year old boy was also diagnosed with metastatic PPGL affecting the lung (35). Despite our findings, a founder effect in Czech Republic related to c.361C>T, p.Gln121X could not be established.
The variants diagnosed in patients no. 7, 10 and 11 have been discussed in detail in a previous study (18). The 36-year old male patient with c.1A>G, p.Met1Val, initially diagnosed with jugular tumor, had rapid progression over a span of 10 years leading to the development of 5 more PGLs. No signs of metastasis was detected. The oldest patient with c.53-2A>G also primarily presented with a solitary VPGL.
The c.341A>G, p.Tyr114Cys protein variant was found in 2 unrelated young female patients of different origins, but of close geographical locations. The Czech patient had bilateral CBPGLs, whilst the Slovakian patient had solitary JPGL. This is a missense mutation of pathogenic variant. This was reported in a large study from Italy, showing the endemic nature of PGL1 in Trentino natives, thus accounting for one of the oldest and largest SDHD founder effect ever seen (36). The other missense mutation c.209G>T, p.Arg70Ser, a variant of the p.R70M, was related to patient no. 8 with solitary CBPGL. This has also been reported in several studies (19, 30, 37, 38). On a retrospective study done from the Mayo Clinic, the c.305A>T, p.His102.Leu was identified in a 30-year old with a single CBPGL (39), findings being similar to our 40-year old patient with bilateral carotid body tumors.
Metastasis with SDHD mutation was detected in 1 Czech patient with positive family history. This is rarely seen in patients with HNPGLs, and is an even more unexpected finding in relation to PGL1. As such predisposition to malignancy with hereditary background is highest amongst those with SDHB mutation, and about 1-3% with SDHD (18, 40), which is synchronous with our findings.
Lastly, comparative analysis indicated that most studies have a high rate of SDHD mutations in comparison to other SDHx gene mutations in the pathogenesis of HNPGLs; at least 8 out of 13 studies showed a ratio of above 70% (21, 22, 24–28, 31) (Table 3). This proportion was less than 50% amongst our patients, as seen in the Spanish cohort (15).
We could contemplate that patients may be unaware of their family history or their family members remained asymptomatic and therefore undiagnosed. The disease being mostly of benign nature and slow progression, most patients may find it difficult to understand the risk associated with transmitting the mutation. The one major limitation of our study was the inability to test most of the at-risk first-degree relatives, despite index patients receiving genetic counselling. It has been proposed that the probability of ascertaining a mutation decreases to 40% in patients without a family history (33). Here, another factor to consider would be the migration trend of Czech inhabitants to other countries, which might have an impact on research associated with such disease.
The importance of the genetic mutation profile we carried out amongst our patients not only demonstrated a low absolute frequency of SDHD gene mutation amongst the Czech population, but also showed inconsistency in patients with known family history. In comparison to most studies, there is a significant discrepancy that arose between the expected and actual outcome in terms of observed frequency of SDHD mutation. The ratio of SDHD gene in comparison to total SDHx gene mutations was also lower. More importantly, we also determined a high incidence of ‘occult familial cases’, which is not a common phenomenon for HNPGLs. It should be considered, that there is a high prevalence of occult paragangliomas in asymptomatic carriers of SDHD and SDHB gene mutations. As such, one clinical surveillance disclosed that up to 59.6% of asymptomatic SDHD carriers can have occult HNPGLs (16). Absence of family history does not rule out the presence of germline mutations in SDHx genes, especially SDHD. Patients with undetected germline mutations are not only at risk of developing multiple tumors but may also transmit the mutation to the next generation (33, 41). Despite a number of recommendations being suggested regarding early detection of HNPGLs and determination of genetic profile, the uncommonness of these tumors and delayed presentation will always present a certain risk of underestimated cases reported in any cohort.
5 Conclusion
Germline SDHD gene mutation was found in only 12% of all Czech patients and 78% could be described as ‘occult familial cases’. We were able to establish the relationship between germline SDHD and the presence of multiple tumors in younger patients, but with known family history, the affinity was lower. Our results showed a different pattern in comparison to other studies worldwide. The SDHD c.361C>T, p.Gln121X variant was the most frequently detected mutation in Czech patients, however a founder effect was not established. Therefore, the key to prediction and early management of HNPGLs should include reiterating the importance of genetic testing to patients and ascertaining a comprehensive guidance protocol for all physicians involved in the care of such patients.
Data availability statement
All relevant data is contained within the article: The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
All procedures performed in studies involving human subjects were in compliance with the Helsinki declaration and further in accordance with local ethical guidelines of the institutional ethical committees of Charles University, Prague, Czech Republic. Informed consent was obtained for all patients undergoing intervention according to the individual hospital regulations, institutional guidelines of Charles University and those defined by the practice codes of the Ministry of Health of the Czech Republic. Additional informed consent was obtained for genetic testing of patients. No identifying information of the patients have been included in this manuscript.
Author contributions
AG: Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft. AV: Data curation, Funding acquisition, Investigation, Methodology, Resources, Software, Validation, Writing – review & editing. TZ: Funding acquisition, Investigation, Methodology, Resources, Validation, Writing – review & editing. MK: Data curation, Methodology, Resources, Writing – review & editing. ZM: Investigation, Resources, Software, Validation, Writing – review & editing. KP: Formal Analysis, Investigation, Methodology, Resources, Writing – review & editing. JBe: Funding acquisition, Methodology, Resources, Writing – review & editing. MC: Data curation, Investigation, Methodology, Resources, Writing – review & editing. JP: Data curation, Funding acquisition, Methodology, Resources, Writing – review & editing. JBo: Data curation, Funding acquisition, Methodology, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors declare that this study received funding from the Ministry of Health of the Czech Republic (grant no. NU21-08-00280 and NU23-01-00323). The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
We appreciate the support given to our study by the Ministry of Health of the Czech Republic (grant no. NU21-03-00273), the COOPERATIO program (Surgical Disciplines; Oncology and Hematology), Charles University, the Grant Agency of the Czech Republic (grant no. 22-07091S) and The EXCELES project, The National Institute for Cancer Research Programme (Project No. LX22NPO5102) - Funded by the European Union - Next Generation EU, Prague, Czech Republic. We would like to kindly thank all our clinical and laboratory staff who supported us in our research. We are also very grateful to all the patients who have participated in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mete O, Asa SL, Gill AJ, Kimura N, de Krijger RR, Tischler A. Overview of the 2022 WHO classification of paragangliomas and pheochromocytomas. Endocr Pathol (2022) 33(1):90–114. doi: 10.1007/s12022-022-09704-6
2. Mete O, Wenig BM. Update from the 5th edition of the World Health Organization Classification of head and neck tumors: overview of the 2022 WHO classification of head and neck neuroendocrine neoplasms. Head Neck Pathol (2022) 16(1):123–42. doi: 10.1007/s12105-022-01435-8
3. Taïeb D, Kaliski A, Boedeker CC, Martucci V, Fojo T, Adler JR, et al. Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev (2014) 35(5):795–819. doi: 10.1210/er.2014-1026
4. Sandow L, Thawani R, Kim MS, Heinrich MC. Paraganglioma of the head and neck: A review. Endocrine Practice (2023) 29(2):141–7. doi: 10.1016/j.eprac.2022.10.002
5. Boedeker CC. Paragangliomas and paraganglioma syndromes. GMS Curr Top Otorhinolaryngol Head Neck Surg (2011) 10:Doc03. doi: 10.3205/cto000076
6. Guha A, Musil Z, Vicha A, Zelinka T, Pacak K, Astl J, et al. A systematic review on the genetic analysis of paragangliomas: Primarily focused on head and neck paragangliomas. Neoplasma (2019) 66:671–80. doi: 10.4149/neo_2018_181208N933
7. Erickson D, Kudva YC, Ebersold MJ, Thompson GB, Grant CS, van Heerden JA, et al. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab (2001) 86(11):5210–6. doi: 10.1210/jcem.86.11.8034
8. Robertson V, Poli F, Hobson B, Saratzis A, Ross Naylor A. A systematic review and meta-analysis of the presentation and surgical management of patients with carotid body tumours. Eur J Vasc Endovascular Surgery (2019) 57(4):477–86. doi: 10.1016/j.ejvs.2018.10.038
9. Smith JD, Harvey RN, Darr OA, Prince ME, Bradford CR, Wolf GT, et al. Head and neck paragangliomas: A two-decade institutional experience and algorithm for management. Laryngoscope Investig Otolaryngol (2017) 2(6):380–9. doi: 10.1002/lio2.122
10. Biller HF, Som P, Lawson W, Rosenfeld R. Glomus vagale tumors. Ann Otol Rhinol Laryngol (1989) 98(1):21–6. doi: 10.1177/000348948909800105
11. Taïeb D, Wanna GB, Ahmad M, Lussey-Lepoutre C, Perrier ND, Nölting S, et al. Clinical consensus guideline on the management of phaeochromocytoma and paraganglioma in patients harbouring germline SDHD pathogenic variants. Lancet Diabetes Endocrinol (2023) 11(5):345–61. doi: 10.1016/S2213-8587(23)00038-4
12. Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science (1979) 287(5454):848–51. doi: 10.1126/science.287.5454.848
13. Gimenez-Roqueplo A-P, Dahia P, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Hormone Metab Res (2012) 44(5):328–33. doi: 10.1055/s-0031-1301302
14. Dannenberg H, Dinjens WNM, Abbou M, Van Urk H, Pauw BKH, Mouwen D, et al. Frequent germ-line succinate dehydrogenase subunit D gene mutations in patients with apparently sporadic parasympathetic paraganglioma. Clin Cancer Res (2002) 8(7):2061–6.
15. Lima J, Feijão T, Ferreira da Silva A, Pereira-Castro I, Fernandez-Ballester G, Máximo V, et al. High frequency of germline succinate dehydrogenase mutations in sporadic cervical paragangliomas in Northern Spain: mitochondrial succinate dehydrogenase structure-function relationships and clinical-pathological correlations. J Clin Endocrinol Metab (2007) 92(12):4853–64. doi: 10.1210/jc.2007-0640
16. Heesterman BL, Bayley JP, Tops CM, Hes FJ, van Brussel BTJ, Corssmit EPM, et al. High prevalence of occult paragangliomas in asymptomatic carriers of SDHD and SDHB gene mutations. Eur J Hum Genet (2013) 21(4):469–70. doi: 10.1038/ejhg.2012.203
17. Richter S, Qiu B, Ghering M, Kunath C, Constantinescu G, Luths C, et al. Head/neck paragangliomas: focus on tumor location, mutational status and plasma methoxytyramine. Endocr Relat Cancer (2022) 29(4):213–24. doi: 10.1530/ERC-21-0359
18. Guha A, Vicha A, Zelinka T, Musil Z, Chovanec M. Genetic variants in patients with multiple head and neck paragangliomas: dilemma in management. Biomedicines (2021) 9(6):626. doi: 10.3390/biomedicines9060626
19. Burnichon N, Rohmer V, Amar L, Herman P, Leboulleux S, Darrouzet V, et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab (2009) 94(8):2817–27. doi: 10.1210/jc.2008-2504
20. Neumann HPH, Erlic Z, Boedeker CC, Rybicki LA, Robledo M, Hermsen M, et al. Clinical predictors for germline mutations in head and neck paraganglioma patients: cost reduction strategy in genetic diagnostic process as fall-out. Cancer Res (2009) 69(8):3650–6. doi: 10.1158/0008-5472.CAN-08-4057
21. Astuti D, Hart-Holden N, Latif F, Lalloo F, Black GC, Lim C, et al. Genetic analysis of mitochondrial complex II subunits SDHD, SDHB and SDHC in paraganglioma and phaeochromocytoma susceptibility. Clin Endocrinol (Oxf) (2003) 59(6):728–33. doi: 10.1046/j.1365-2265.2003.01914.x
22. Badenhop RF. The prevalence of SDHB, SDHC, and SDHD mutations in patients with head and neck paraganglioma and association of mutations with clinical features. J Med Genet (2004) 41(7):e99–9. doi: 10.1136/jmg.2003.011551
23. Baysal BE, Willett-Brozick JE, Lawrence EC, Drovdlic CM, Savul SA, McLeod DR, et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet (2002) 39(3):178–83. doi: 10.1136/jmg.39.3.178
24. Benn DE, Gimenez-Roqueplo A-P, Reilly JR, Bertherat J, Burgess J, Byth K, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab (2006) 91(3):827–36. doi: 10.1210/jc.2005-1862
25. Fakhry N, Niccoli-Sire P, Barlier-Seti A, Giorgi R, Giovanni A, Zanaret M. Cervical paragangliomas: is SDH genetic analysis systematically required? Eur Arch Oto-Rhino-Laryngol (2008) 265(5):557–63. doi: 10.1007/s00405-007-0517-4
26. Hensen EF, Siemers MD, Jansen JC, Corssmit EPM, Romijn JA, Tops CMJ, et al. Mutations in SDHD are the major determinants of the clinical characteristics of Dutch head and neck paraganglioma patients. Clin Endocrinol (Oxf) (2011) 75(5):650–5. doi: 10.1111/j.1365-2265.2011.04097.x
27. Neumann HPH, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA (2004) 292(8):943–51. doi: 10.1001/jama.292.8.943
28. Pandit R, Khadilkar K, Sarathi V, Kasaliwal R, Goroshi M, Khare S, et al. Germline mutations and genotype–phenotype correlation in Asian Indian patients with pheochromocytoma and paraganglioma. Eur J Endocrinol (2016) 175(4):311–23. doi: 10.1530/EJE-16-0126
29. Papaspyrou K, Mewes T, Rossmann H, Fottner C, Schneider-Raetzke B, Bartsch O, et al. Head and neck paragangliomas: Report of 175 patients (1989-2010). Head Neck (2012) 34(5):632–7. doi: 10.1002/hed.21790
30. Persu A, Hamoir M, Grégoire V, Garin P, Duvivier E, Reychler H, et al. High prevalence of SDHB mutations in head and neck paraganglioma in Belgium. J Hypertens (2008) 26(7):1395–401. doi: 10.1097/HJH.0b013e3282ffdc54
31. Piccini V, Rapizzi E, Bacca A, Di Trapani G, Pulli R, Giachè V, et al. Head and neck paragangliomas: genetic spectrum and clinical variability in 79 consecutive patients. Endocr Relat Cancer (2012) 19(2):149–55. doi: 10.1530/ERC-11-0369
32. Snezhkina AV, Fedorova MS, Pavlov VS, Kalinin DV, Golovyuk AL, Pudova EA, et al. Mutation frequency in main susceptibility genes among patients with head and neck paragangliomas. Front Genet (2020) 18:614908. doi: 10.3389/fgene.2020.614908
33. Taschner PEM, Jansen JC, Baysal BE, Bosch A, Rosenberg EH, Bröcker-Vriends AHJT, et al. Nearly all hereditary paragangliomas in The Netherlands are caused by two founder mutations in the SDHD gene. Genes Chromosomes Cancer (2001) 31(3):274–81. doi: 10.1002/gcc.1144
34. Vosecka T, Vicha A, Zelinka T, Jencova P, Pacak K, Duskova J, et al. Absence of BRAF mutation in pheochromocytoma and paraganglioma. Neoplasma (2017) 64(2):278–82. doi: 10.4149/neo_2017_215
35. Gómez AM, Soares DC, Costa AAB, Pereira DP, Achatz MI, Formiga MN. Pheochromocytoma and paraganglioma: implications of germline mutation investigation for treatment, screening, and surveillance. Arch Endocrinol Metab (2019) 11:369–75. doi: 10.20945/2359-3997000000145
36. Schiavi F, Demattè S, Cecchini ME, Taschin E, Bobisse S, Del Piano A, et al. The endemic paraganglioma syndrome type 1: origin, spread, and clinical expression. J Clin Endocrinol Metab (2012) 97(4):E637–41. doi: 10.1210/jc.2011-2597
37. van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol (2009) 10(8):764–71. doi: 10.1016/S1470-2045(09)70164-0
38. Haller F, Moskalev EA, Faucz FR, Barthelmeß S, Wiemann S, Bieg M, et al. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocr Relat Cancer (2014) 21(4):567–77. doi: 10.1530/ERC-14-0254
39. Sen I, Young WF, Kasperbauer JL, Polonis K, Harmsen WS, Colglazier JJ, et al. Tumor-specific prognosis of mutation-positive patients with head and neck paragangliomas. J Vasc Surg (2020) 71(5):1602–1612.e2. doi: 10.1016/j.jvs.2019.08.232
40. Fliedner SMJ, Lehnert H, Pacak K. Metastatic paraganglioma. Semin Oncol (2010) 37(6):627–37. doi: 10.1053/j.seminoncol.2010.10.017
Keywords: HNPGL, CBPGL, paraganglioma syndrome, germline mutation, SDHD gene
Citation: Guha A, Vicha A, Zelinka T, Kana M, Musil Z, Pacak K, Betka J, Chovanec M, Plzak J and Boucek J (2023) High incidence of occult familial SDHD cases amongst Czech patients with head and neck paragangliomas. Front. Endocrinol. 14:1278175. doi: 10.3389/fendo.2023.1278175
Received: 15 August 2023; Accepted: 06 November 2023;
Published: 08 December 2023.
Edited by:
Ben Nephew, Worcester Polytechnic Institute, United StatesReviewed by:
Karim Abid, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandKatarzyna Ziemnicka, Poznan University of Medical Sciences, Poland
Copyright © 2023 Guha, Vicha, Zelinka, Kana, Musil, Pacak, Betka, Chovanec, Plzak and Boucek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anasuya Guha, YW5hc3V5YS5ndWhhQG91dGxvb2suY29t