 Xiaodan Zhang1
Xiaodan Zhang1 Zirui Luo
Zirui Luo Yu Li
Yu Li Wangen Li
Wangen Li- 1Department of Endocrinology, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
- 2The Second Clinical Medicine School, Guangzhou Medical University, Guangzhou, China
Diabetes is a global health problem which is accompanied with multi-systemic complications. It is of great significance to elucidate the pathogenesis and to identify novel therapies of diabetes and diabetic complications. Sestrin2, a stress-inducible protein, is primarily involved in cellular responses to various stresses. It plays critical roles in regulating a series of cellular events, such as oxidative stress, mitochondrial function and endoplasmic reticulum stress. Researches investigating the correlations between Sestrin2, diabetes and diabetic complications are increasing in recent years. This review incorporates recent findings, demonstrates the diverse functions and regulating mechanisms of Sestrin2, and discusses the potential roles of Sestrin2 in the pathogenesis of diabetes and diabetic complications, hoping to highlight a promising therapeutic direction.
1 Introduction
Diabetic mellitus (DM) is a metabolic disease characterized by hyperglycemia. It is a type of disease in which defects in insulin production and activity lead to abnormal glucose metabolism. Continuous hyperglycemia leads to impaired cellular autophagy and oxidative stress response, which further induces inflammatory response and the stimulation of coagulation, and finally gives rise to the occurrence of complications in multiple organs and systems (1). In recent years, the number of patients with diabetes has increased dramatically globally due to an aging population, changes in the lifestyle, and the increase prevalence of obesity. Among various types of DM, type 2 DM (T2DM) is the major type and accounts for nearly 90% of all DM cases. Diabetes and diabetic chronic complications have become important causes of disability and death for individuals, and have posed huge economic burdens worldwide (2).
As an important member of the Sestrins (Sesns) protein family, Sestrin2 is a newly discovered stress-inducible protein widely distributed in animals. Sestrin2 gene was originally identified in human neuroblastoma cells as a hypoxia-activated gene (3). Sestrin2 accumulates in mammalian cells in various pathophysiological states such as hypoxia, starvation, radiation, oxidative stress and endoplasmic reticulum (ER) stress (4). Previous studies on Sestrin2 mostly focused on metabolic disease such as obesity, age-related diseases and malignant tumors. Recent researches indicated that Sestrin2 also plays critical roles in the pathogenesis of cardiovascular diseases, kidney diseases, liver diseases, respiratory diseases, and diseases of the nervous system and exerts protective effects on several organs (5). Complicated mechanism is involved, including regulation of oxidative stress, mitochondrial function, ER stress, autophagy, metabolism and inflammatory response (5, 6). In recent years, increasing numbers of studies report about correlations between Sestrin2 and diabetes, indicating that Sestrin2 might become a novel therapeutic target for diabetes. In this review, we summarize the recent findings and discuss the potential role and underlying mechanism of Sestrin2 in diabetes and diabetic complications.
2 Sestrin2 pathways and modulating mechanisms
As a sensitive stress receptor, Sestrin2 is activated in stress conditions. A variety of adverse stresses could promote Sestrin2 expression, such as oxidative stress, ER stress, hypoxia, energetic stress, and age- and obesity-associated metabolic pathological conditions (7–10). Up-regulated Sestrin2 exerts pleiotropic biological effects in diverse physiological and pathological states, through attenuating oxidative stress, and modulating a series of cellular events such as autophagy, ER stress, mitochondrial biogenesis, protein synthesis, cell energy homeostasis and apoptosis, while many of these responsive pathways are interconnected (5, 11–14).
2.1 Upstream factors of Sestrin2 signaling
In response to stress, Sestrin2 could be regulated by various transcription factors, including tumor-suppressor protein p53, hypoxia inducible factor-1α (HIF-1α), nuclear factor erythroid 2-related factor-2 (Nrf2), nuclear factor-κB (NF-κB), activated transcription factor 4 (ATF4), c-Jun NH(2)- terminal kinase (JNK)/c-Jun, Foxhead box O3 (FoxO), activated protein 1 (AP-1), and CCAAT. A series of stress conditions, such as oxidative stress, ER stress, DNA damage, hypoxia and mitochondrial dysfunction, provoke the release of these upstream factors, and result in altered expression and activity of Sestrin2 (15, 16). The mediating effects of Sestrin2 in stress conditions will be further discussed below.
2.2 Downstream pathways of Sestrin2 signaling
After being induced, Sestrin2 thereafter mediates several signaling pathways, including adenosine monophosphate-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) pathway (17), Kelch-like ECH-associated protein1/Nrf2 (Keap1/Nrf2) pathway (18), the mitogen-activated protein kinase8/JNK1 (MAPK8/JNK1) pathway (19), AMPK/peroxisome proliferator-activated receptor γ coactivator-1α (AMPK/PGC-1α) pathway (20), extracellular regulated protein kinases (ERK1/2) pathway (21), thrombospondin-1/transforming growth factor-β1/Smad3 (TSP-1/TGF-β1/Smad3) pathway (22), and TGF-β/NADPH Oxidase 4 (NOX4)/ROS signaling pathway (23). Among these signaling pathways, AMPK/mTOR and Nrf2/Keap1 are the principal ones that Sestrin2 is suggested to be involved in the pathogenesis of diabetes and diabetic complications.
2.2.1 The AMPK/mTOR pathway
Comprising of two distinct protein complexes (mTORC1 and mTORC2), mTOR functions as a crucial sensor for energy, nutrient, and redox states, and thereafter regulates protein synthesis and autophagy (24). Persistent mTOR stimulation is linked with a wide range of diseases such as diabetes, obesity, cardiovascular diseases, cancer, autoimmune diseases and metabolic disorders (25). mTORC1 can phosphorylate and suppress autophagy-initiating protein kinases unc-51-like kinase 1 (ULK1), so as to inhibit cellular autophagic catabolism (26). mTORC2 is insensitive to nutrients but is sensitive to growth factors via phosphatidylinositol 3 kinase (PI3K) signaling, and thereby regulates metabolism and cytoskeletal tissue (27) and functions as a crucial controller of lipid metabolism (28). mTORC1 could negatively regulate mTORC2 activity. mTOR is strongly associated with T2DM and many of its chronic complications. Both mTORC1 and mTORC2 play significant roles in the regulation of insulin signaling. mTORC1/ribosomal protein S6 kinase 1 (S6K1) and mTORC2/protein kinase B (AKT), is critical for the maintenance of insulin sensitivity and that their dysfunction contributes to the development of T2DM (29). mTORC1 in pancreatic β-cells controls cell size, proliferation, survival, maturation, protein translation, insulin processing and secretion, and autophagy. mTORC1/S6K1 pathway regulates the apoptosis and autophagy of β-cell, while mTORC1/4E-BP2-eIF4E pathway regulates the proliferation of β-cell. Loss of β-cell-specific mTORC1 leads to diabetes and β-cell failure (30). mTORC2 is essential for maintaining a balance between the proliferation and the cell size of β-cells (31). Recently, it is reported that mTORC2 regulates glucose-stimulated insulin secretion in β-cell by enhancing actin filament remodeling (32). Besides the direct regulating effects on β-cell, mTORC2 also modulates glucose uptake in peripheral tissues including adipose, skeletal muscle and brown adipose tissues (33–35). Moreover, mTORC2 participates in the regulation of hepatic insulin sensitivity, glycolysis, and lipogenesis (35, 36).
Sestrin2 exerts antioxidant and apoptosis-associated effects in a variety of diseases through the inhibition of mammalian target of rapamycin complex 1 (mTORC1) and/or the activation of AMPK (21–23). Sestrin2 can suppress mTORC1 through AMPK-dependent or -independent pathways. It was presumed that Sestrin2 regulates AMPK activation by orchestrating the recruitment of liver kinase B1 (LKB1), independent of calmodulin-dependent protein kinase 2 (CAMKK2), as well as promoting LKB1/AMPKα1β1γ1 complex expression (37, 38). Except the AMPK signaling pathway, Sestrin2 can also directly bind to RagA/B regulatory protein complex 2 (GATOR2), mediate the discharge of GATOR1, stimulate GATOR1 to inhibit RagA/B, and suppress mTORC1 inimitably (39). Sestrin2 promotes mTORC2 activity through its interaction with mTORC2, as well as the inhibition of feedback loop (40). GATOR2-mTORC2 axis is essential for Sestrin2-induced AKT activation (41), which exerts various glucose- and lipid-regulating effects (42).
2.2.2 The Keap1/Nrf2 pathway
As a member of a family of basic leucine transcription factors, Nrf2 is involved in a serious of important cellular events including redox regulation, DNA repair, metabolic homeostasis, and apoptosis prevention (43). Nrf2 acts as a crucial transcription factor that can modulate antioxidant gene expression through its interaction with the antioxidant response elements (AREs). Keap1 acts as a sensor of oxidative stress, as well as a inhibitor of Nrf2 (44). Under physiological circumstance, Keap1 binds to Nrf2 in the cytoplasm and is inactivated (45). Keap1/Nrf2 signaling plays a key role in diverse diseases, including diabetes, cancer, neurodegenerative diseases, airway diseases, inflammatory diseases, cardiovascular diseases, and aging (44, 46, 47). A growing body of evidence suggest Nrf2 as a key regulator in the development and progression of diabetes and its complications (43). Nrf2 contributes to the suppression of inflammation of pancreatic β-cell, the maintenance of autophagy in pancreatic β-cells under ROS stimulation, and the regulation of the ER-associated degradation (48, 49). Besides regulating β-cell function, Keap1/Nrf2 pathway also demonstrates protective effects in diabetic complications, i.e. diabetic kidney disease (50), diabetic cardiomyopathy (51) and diabetic neuropathy (52), which is further elucidated below. In fact, Nrf2 has been indicated to be involved in mediating all aspects of diabetic complications across every diabetes-relevant organ (43).
The Nrf2 activators up-regulate the expression of Sestrin2 in a time- and dose-dependent manner and the Nfr2-ARE pathway activation seems to be necessary for Sestrin2 induction. In turn, Sestrin2 can function as a positive regulator of Nrf2 signaling, activate the Nrf2 pathway by augmenting autophagy-directed degradation of Keap1, which targets and breaks down Nrf2 (6, 53). Sestrin2 overexpression suppresses oxidative stress and cell apoptosis by activating Nrf2-ARE signaling (54, 55). Also, Sestrin2/Nrf2 signaling may be important for the mediation of ER stress as a downstream regulator of the protein kinase R-like endoplasmic reticulum kinase (PERK) pathway (56), which is illustrated below.
2.3 Sestrin2 and autophagy
Autophagy is a distinct type of programmed cellular death. Autophagy helps maintain cell survival and tissue stabilization by degrading misfolded and aged intracellular proteins and dysfunctional organelles during stress. The process of autophagy is regulated by various pathways and involves diverse organelles such as mitochondria, ER, ribosomes, peroxisomes, and lysosomes. The dysfunction of autophagy is related to a myriad of diseases, such as diabetes, cardiovascular diseases, cancer, neurodegenerative diseases, liver diseases, and inflammatory diseases (57–59). Autophagy takes part in the regulation of pancreatic β-cells and protection of insulin target tissues. Dysfunctional autophagy is detrimental for the maintenance of β-cell function and reduces insulin secretion. Furthermore, inhibition of autophagy leads to chronic ER stress and β-cell apoptosis. The disruption of autophagy also contributes to diverse diabetic complications (1, 60).
Autophagy activation is required for the antioxidant effects of Sestrin2 (61). After activated by the JNK pathways (62), Sestrin2 is involved in modulation of autophagy through AMPK/mTORC1, Keap1/Nrf2, p53/Sestrin2 and PI3K/AKT/mTOR pathways (63–65). Furthermore, Sestrin2 has been indicated to be interacted with BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), which is also a promoter of autophagy (66).
2.4 Sestrin2 and oxidative stress
Oxidative stress is considered to be an imbalance in redox properties in certain cellular environments, and plays a crucial role in the development of numerous human diseases, such as diabetes, obesity, and myocardial injury (67, 68). Oxidative stress has been proved to play key roles in the pathogenesis of diabetes and diabetic complications. The high glucose activates various molecular and biochemical pathways, causing increased ROS production, which thereby leads to insulin resistance, β-cell dysfunction and diabetic complications (69).
Sestrin2 is essential for the maintenance of cellular homeostasis under oxidative stress. In various types of diseases, Sestrin2 is up-regulated and is important for the resistance to oxidative stress injury. Under oxidative stress, Sestrin2 is activated by various transcription factors, including NF-κB, activator protein-1 (AP-1), CCAAT-enhancer-binding protein beta (C/ERPβ), forkhead box O3 (FOXO3), and p53 (70). Sestrin2 helps to maintain the balance of oxidative metabolism by exerting two major biological functions. First, as an antioxidant enzyme, Sestrin2 is able to directly reduce the accumulation of ROS (71). Second, Sestrin2 can exert antioxidant effects through several signaling pathways, such as Keap1/Nrf2 pathway (6) and AMPK/mTORC1 pathway (72), which have been described above.
2.5 Sestrin2 and ER stress
ER stress is provoked when unfolded or misfolded proteins accumulate in the endoplasmic network lumen in pathophysiological conditions (73). Many physiological and pathological factors, such as inflammation, hypoxia and oxidative stress, disturb the homeostasis of ER and lead to ER malfunction, which thereby causes ER stress and promotes the unfolded protein response (UPR). Three ER transmembrane receptors inositol-requiring enzyme 1 (IRE1, also known as ERN1), PERK and activating transcription factor 6 (ATF6) mediate ER state by regulating UPR (74). The activation of UPR impairs cellular survival by improving protein folding ability, inhibiting protein production and accumulation, inducing ER stress-related gene transcription, and strengthening the self-repair ability of ER. But if ER stress persists or continues for prolong periods, UPR is not enough to maintain ER homeostasis, and apoptosis ultimately occurs (75). ER stress has significant impact on maintaining cellular homeostasis (76). ER stress plays mediating roles in the pathogenesis of a series of diseases, such as diabetes, obesity, inflammation, neurodegenerative diseases, cancer, and autoimmune diseases (77). Numerous studies have proved the role of ER stress in diabetes. Disturbed ER homeostasis and unmitigated ER stress trigger or exacerbate β-cell dysfunction, and contribute to insulin resistance in diabetes (60, 78). Diabetic complications are closely associated with dysregulation of UPR signaling pathways (79).
Increasing evidence has shown that Sestrin2 is activated under ER stress (8, 80). How Sestrin2 expression is induced by ER stress is not fully revealed. The PERK and IRE1/X-box binding protein 1 (XBP1) arms of the UPR appear to be required (8). Also, the activation of transcription molecules, such as HIF-1, activating transcription factor 4 (ATF4) and Nrf2, is suggested to be necessary for ER stress-induced expression of Sestrin2 (72). Once induced, Sestrin2 in turn prevents protein synthesis by inhibiting mTORC1 (81). Sestrin2 inhibits the phosphorylation of JNK and p38 as well as poly ADP-ribose polymerase (PARP) cleavage, and prevents the adverse effect of excessive ER stress (80). The AMPK/mTORC1 pathway, Keap1/Nrf2 pathway, CCAAT-enhancer-binding protein homologous protein, phosphorylation of both p38 and JNK, and Sestrin2-mediated UPR is involved in the protective effects of Sestrin2 against ER stress-associated diseases (4, 82, 83).
2.6 Sestrin2 and mitochondrial function
Mitochondria are the prime organelle which not only offers energy substrates to cells but also controls the fate of cells via mediating diverse cellular processes such as autophagy, apoptosis, cellular mobilization and metabolism (84, 85). Mitochondria possess a quality control system, including mitochondrial dynamics (fusion and fission), mitophagy and mitochondrial biogenesis, which is critical for maintaining a well-functioning mitochondrial network (86, 87). Altered mitochondrial functionality is involved in a variety of diseases, such as diabetes, obesity, neurological disorders, and cardiovascular diseases (88–91). A myriad of evidence has revealed crucial roles of mitochondrial dynamics, mitophagy, and mitochondrial biogenesis in the pathogenesis of diabetes. Dysregulations of mitochondrial functions and dynamics could result in β-cell dysfunction and insulin resistance (92, 93).
Recent studies have showed that Sestrin2 may play an important role in maintaining cellular homeostasis by restoring mitochondrial function and metabolism (7, 94). Mitochondrial superoxide mediates Sestrin2 activation in the process of mitochondrial quality control (95). Sestrin2 can thereby secure the mitochondria from oxidative lesion, both in vivo and in vitro (96, 97). Mitophagy is a subtype of autophagy, which helps to remove dysfunctional mitochondria as well as is crucial for maintenance of the functionality and integrity of the mitochondrial network. Several studies indicated that Sestrin2 is involved in regulating the pace of mitophagy (95, 98). Sestrin2 stimulates ULK1- mediated phosphorylation of Beclin1 and strengthens the interaction between Beclin1 and Parkin. Then mitophagy is provoked as Parkin’s shift on the surface of mitochondria (95, 99). Sestrin2-mediated autophagy and mitophagy can ameliorate mitochondrial dysfunction and prevent cell apoptosis (100).
Several signaling pathways participate the regulating mechanisms of Sestrin2 in mitochondrial function and metabolism. Kim et al. reported that Sestrin2 suppresses the overactivation of the NLRP3 inflammasome and alleviates mitochondrial injury. Sestrin2 promotes perinuclear clustering-damaged mitochondria through regulating the aggregation of SQSTM1 and its binding to Lys63-linked ubiquitins on the surface of damaged mitochondria (98). Sestrin2 overexpression suppresses inflammation by inducing AMPK/PGC-1α-mediated mitochondrial biogenesis (101). Sestrin2/LKB1/AMPK pathway is also indicated to function in mitochondrial quality control enhancement, including mitochondrial biogenesis and mitophagy (38).
2.7 Sestrin2 and apoptosis
Apoptosis is an active programmed cell death process, characterized by specific biochemical and morphological alterations such as cellular shrinkage, nuclear condensation and chromatin condensation along the nuclear membrane (102). There are three major signaling pathways that modulate apoptosis, namely the mitochondrial, death receptor and ER pathways (103). Pancreatic β-cell apoptosis is the determining factor for the decline of β-cell function and impaired insulin secretion in diabetes (104). Also, apoptosis of organ-specific cells has been identified and characterized in the development of diabetic complications (105, 106).
In different cell types and under different pathophysiological conditions, Sestrin2 may exert opposite effects on apoptosis. In most studies targeting non-tumor cells, Sestrin2 is involved in anti-apoptotic signaling pathways. However, in most tumor studies, Sestrin2 elicits proapototic effects in cancer cells (107). Ding B et al. reported that Sestrin2 is protective for overall cell energy metabolism and mitochondrial function. Sestrin2 overexpression can reduce cell apoptosis by reducing ROS aggregation, maintaining mitochondrial membrane potential, reducing ATP consumption and restoring mitochondrial DNA level (7). However, as shown by the study of Seo K et al. (108) and Budanov AV et al. (109), Sestrin2 overexpression can promote cell apoptosis. Bidirectional regulating roles of Sestrin2 in apoptosis are indicated and require further validation (110).
3 The roles of Sestrin2 in diabetes
A summary of researches which investigated the roles of Sestrin2 in diabetes, diabetic complications and diabetes-associated conditions is presented in Table 1. Figure 1 summarizes the effects of Sestrin2 on diabetes-associated signaling pathways. As mentioned before, diabetes is characterized by changes in AMPK and mTOR, the principle nutrient level sensing mechanisms (129). The chronic continuous activation of mTORC1 is accompanied by continuous inhibition of hepatocyte autophagy, leading to insulin resistance and T2DM mainly by suppressing the phosphorylation of insulin receptor substrates (130). Continuous cellular mTORC1 activation under overnutrition promotes protein and lipid synthesis and inhibits autophagy catabolism (111). One of the major negative feedback mechanisms that prevent the harmful effects of chronic mTORC1 continuous activation is the transcriptional activation of Sestrin2. Chronic mTORC1 activation mediated by stress responses such as overnutrition eventually results in Sestrin2 overactivation (111, 131). After activation, Sestrin2 stimulates AMPK signaling, which in turn impairs mTORC1 activation and, therefore, triggers autophagy (111, 132). The major target organs and tissues of insulin resistance include liver, muscle and adipose tissue. Sestrin2 is found to be highly accumulated in muscle, liver, and adipose tissues in models of T2DM and obesity (41, 111). As reported by Lee et al., Sestrin2 can activate AMPK, attenuate mTORC1-S6K activity in the liver, thereby lowering blood glucose level in obese mice. Sestrin2 ablation activates hepatic mTORC1-S6K signaling, and enhances insulin resistance, hepatic steatosis and diabetic progression, indicating a key role of Sestrin2 in cell metabolic homeostasis (111). Insulin up-regulates Sestrin2 expression in mouse primary hepatic cells and the upregulation of Sestrin2 by insulin was shown to be regulated via PI3K/PKB/mTOR signaling pathway, indicating a feedback mediation of Sestrin2 on insulin signaling transduction (112). Also, Sestrin2 is identified to induce autophagy, maintain insulin sensitivity and glucose metabolism by regulating AMPK/mTORC1 signaling pathway (64, 113). Sestrin2/AMPK/mTORC1 signaling pathway is indicated to contribute to the maintenance of β-cell function and resistance to pathological stresses associated with diabetes (114). Therefore, based on these evidences, Sestrin2 is a potential insulin sensitizer and one of the key factors for β-cell homeostasis. Deficiency and/or dysfunction of Sestrin2 may result in insulin resistance and the development of diabetes (70).
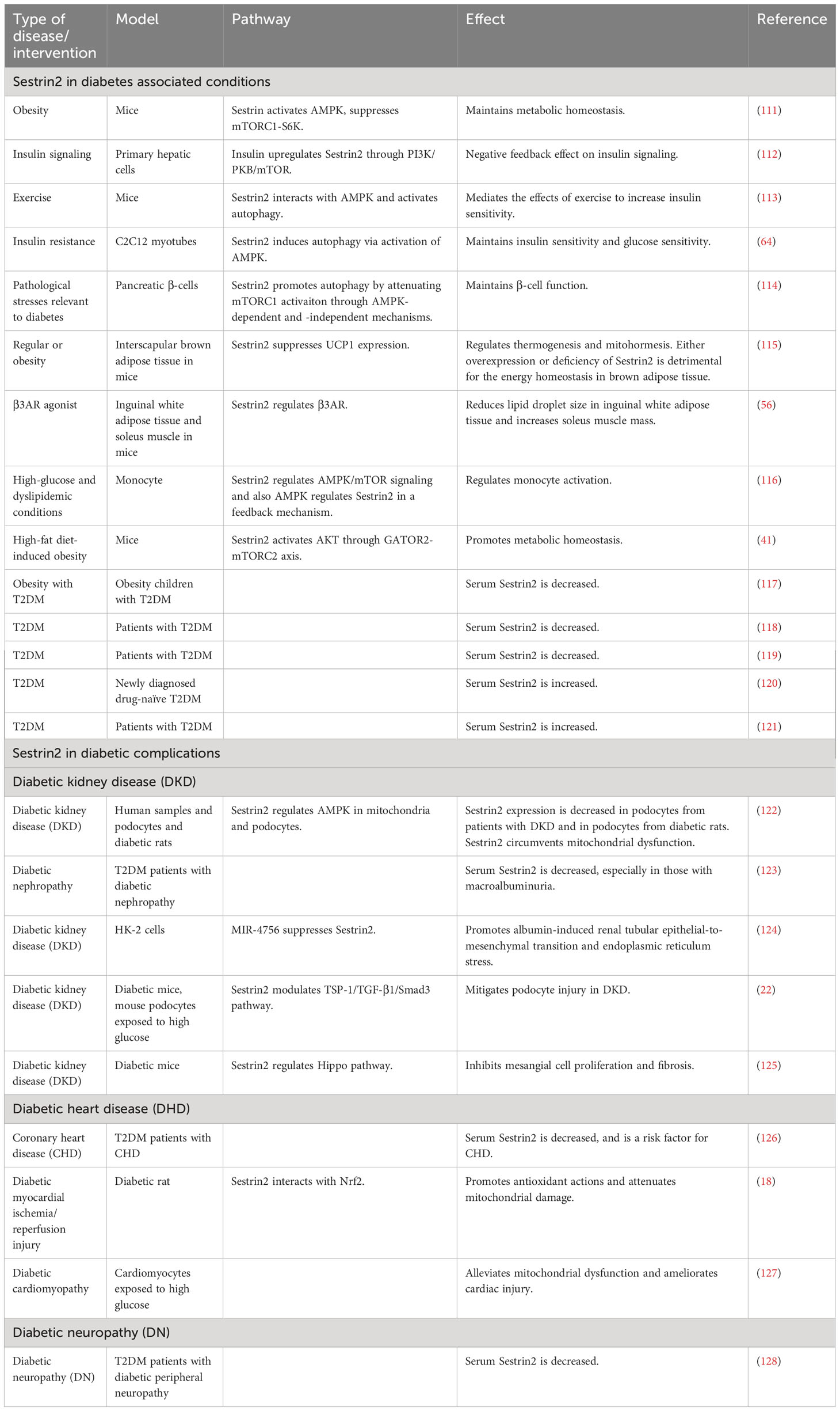
Table 1 The roles of Sestrin2 in diabetes and diabetic complications.
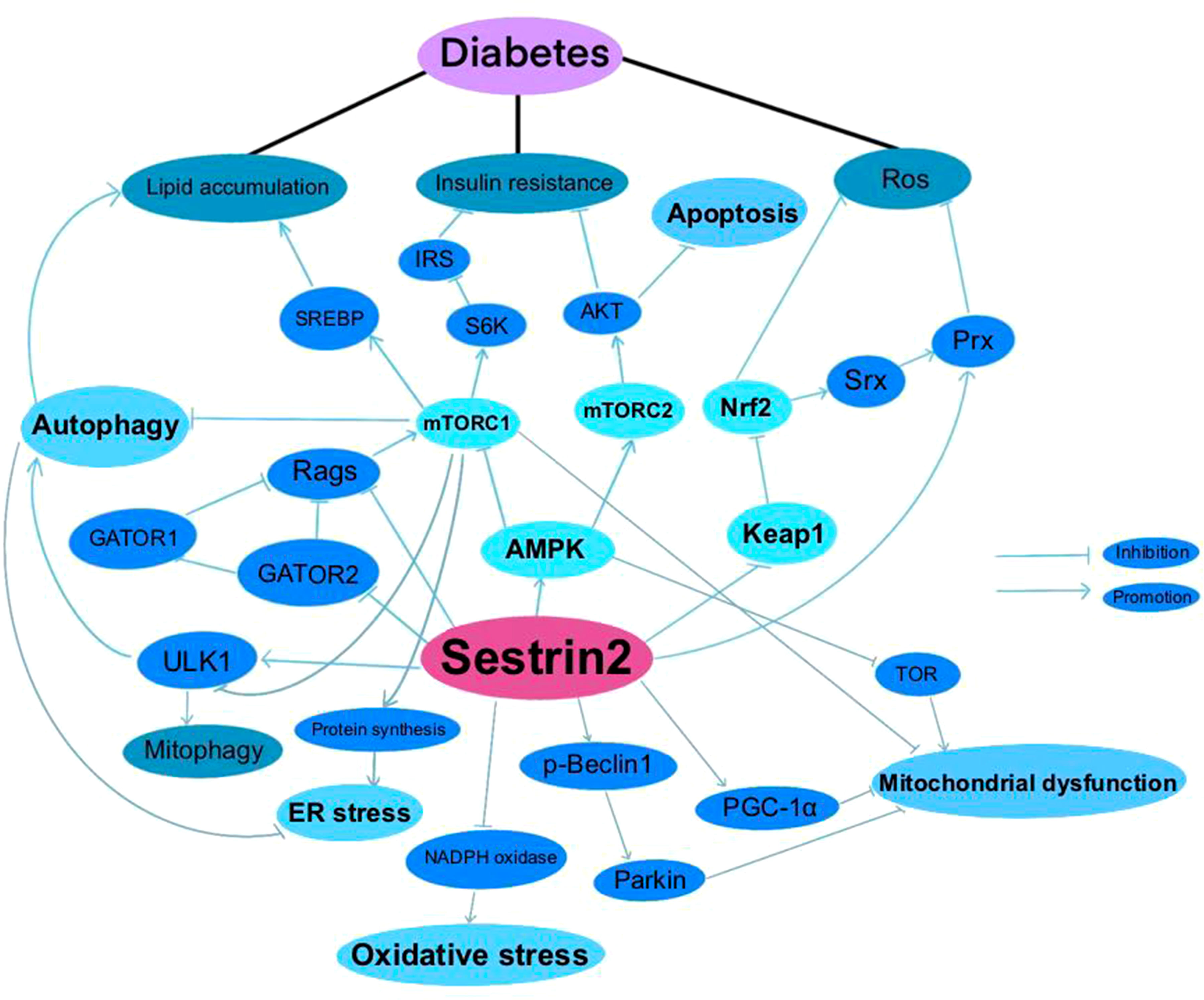
Figure 1 Sestrin2 and Diabetes.
Besides the role in the modulation of insulin signaling, there are studies referring to the regulating effects of Sestrin2 on peripheral tissues which play key roles in the pathogenesis of diabetes, such as adipose tissues and skeletal muscle. Growing evidence displays adipose tissue as an endocrine organ which produces multiple adipokines regulating diverse aspects of β-cell function and viability. Adipose tissue malfunction is crucially involved in the development of diabetes (133, 134). Recent finding suggested Sestrin2 as a regulator of motohormesis in brown adipose tissue (135). Also, Sestrin2 was found to be related to beneficial body composition changes, including the decrease of lipid droplet size in inguinal white adipose tissue and the increase of soleus muscle mass (56). Monocytes and macrophages are critically involved in atherosclerosis and participate in the atherosclerotic lesion progression associated with diabetes (136). Sestrin2 was shown to play a principal role in regulating monocyte activation through the AMPK/mTOR pathway in diabetes and also AMPK mediates Sestrin2 in a feedback way (116).
As described before, except for the classical AMPK/mTORC1 pathway, Sestrin2 can exert downregulating effects on mTORC1 through other mechanisms such as GATOR2-GATOR1-mTORC1 signaling pathway (39). On the other hand, Sestrin2 could increase mTORC2 activity through its ability to interact with mTORC2 via GATOR2-mTORC2 signaling pathway during high-fat diet-induced obesity (41). However, these mediating pathways of Sestrin2 on mTORC have not been investigated and confirmed in diabetic models.
Several clinical studies have investigated the changes of circulatory levels of Sestrin2 in patients with obesity, T2DM, and metabolic syndrome. However, no consensus was reached. It was indicated that circulatory Sestrin2 is lowered in diabetes and negatively correlates with glycemic levels (117, 118, 123). Also, as shown by the study of Golpour et al., plasma Sestrin2 level presents a trend of decrease in obesity and T2DM (119). On the contrary, some studies reported significant high serum levels of Sestrin2 in patients with T2DM, obesity, and metabolic syndrome (120, 121). Sestrin2 concentration significantly correlates with insulin resistance and percentage body fat (120).
4 The roles of Sestrin2 in diabetic complications
Rather than a disease of mere hyperglycemia, diabetes brings real harm and devastating effects by leading to a series of complications in peripheral systems, organs and tissues such as kidney, cardiovascular system, retina, and the nervous system (137). Diabetic complications are often irreversible, causing severe injury and increasing mortality in patients with diabetes. Recent researches have suggested the contribution of alterations of Sestrin2 and the related pathways in the pathogenesis of diabetic complications.
4.1 Diabetic kidney disease
Diabetic kidney disease (DKD) is a typical chronic microvascular diabetic complication and is a major cause of chronic kidney disease and end-stage renal disease worldwide (138). About 30%-50% of patients with T1DM or T2DM eventually develop DKD, resulting a significant increase of mortality in these patients. Clinically, patients with DKD often exhibit proteinuria, hypertension and edema, while laboratory tests present increased urinary albumin excretion and decreased estimated glomerular filtration rate (eGFR). A series of signaling pathways contributes to the pathogenesis of DKD, including AMPK/mTOR pathway, MAPKs/Erk1/2 pathway, PI3K/AKT pathway and the advanced glycation end products (AGEs) pathway (139–141). As shown by Puelles et al., hyperglycemia can induce oxidative stress and other pathophysiological processes through AMPK/mTOR signaling, leading to podocyte injury and proteinuria, therefore leading to the loss of renal function (139). Activated mTORC1 signaling is a feature of DKD, which causes podocyte and tubular damage by suppressing autophagy and in turn promotes progressive kidney dysfunction (142, 143).
The activation of Sestrin2 could inhibit AMPK/mTOR signaling, promote autophagy and reduce the susceptibility of renal cells to diabetes-related damage. The potential therapeutic role of Sestrin2 in DKD was initially found in a human proximal tubule cell line (HK-2) model, illustrating that overexpression of Sestrin2 represses DKD-induced renal epithelial tubular cell epithelial-to-mesenchymal transition and ER stress, but its mechanism is still unclear. The researchers further found that administration of microRNA-4756 regulates DKD-induced renal tubular epithelial cell damage by the interaction with Sestrin2 (124). Later, Lin et al. reported that Sestrin2 activation increases the level of AMPK phosphorylation, and thereby ameliorates mitochondrial dysfunction of podocytes under high glucose conditions (122). However, it is worth noting that though overactivation of mTORC1 in diabetes aggravates kidney lesions, mTOR activity is necessary to maintain podocyte homeostasis. Genetic deletion of mTOR in mouse podocytes induces proteinuria and progressive glomerulosclerosis. A tightly balanced mTOR activity is essential to maintain normal renal function in diabetes (142). Clinically, serum Sestrin2 levels were found to significantly decrease in T2DM patients with diabetic nephropathy, especially in the ones with macroalbuminuria (123). In recent years, sodium-glucose co-transporter 2 (SGLT2) inhibitors have been well-documented to protect the renal function in patients with and without T2DM and slow down the progression towards end-stage kidney disease (144). It has been shown that the Sestrin2/AMPK pathway plays a critical role in the protective actions of SGLT2 inhibitors on metabolism, fibrosis, and organ damage in obese mice (145). Specially, studies have demonstrated that activation of AMPK by inhibiting SGLT2 is a main protective mechanism in diabetic nephropathy (146). Nevertheless, in another study investigating the working mechanisms of empagliflozin, Sestrin2/AMPK pathway was not activated in nondiabetic rats and did not participate in the renal protective effects of empagliflozin (147).
TGF-β1 is a decisive regulator of renal fibrosis and overactivation of TGF-β1 could cause progressive renal injury (148). Hyperglycemia and insulin resistance enhance the expression of Angiotensin II, which increases ROS production and activates TGF-β1 signaling (149). Smad2/3 complex, PI3K/AKT/mTOR, protein kinase C (PKC), MAPK, interleukin like kinase (ILK) and Wnt/beta-catenin signaling are among the downstream targets that modulate profibrogenic effects of TGF-β1 (150–152). Thrombospondin-1 (TSP-1) is an extracellular matrix protein that mediates a wide range of biological processes. TSP-1 is vital to maintain normal glucose metabolism. Also, TSP-1 is involved in the pathophysiology of multiple diabetic complications, including diabetic cardiomyopathy, neuropathy and nephropathy (153). TSP-1 mediates the activation of latent TGF-β1, which is indispensable for maintaining the normal function of islet. Nevertheless, during chronic hyperglycemia, TGF-β1 exacerbates diabetic nephropathy by inducing renal fibrosis (154). Both TGF-β and TSP-1 have been indicated to play causal roles in insulin resistance and obesity-related renal fibrosis, except for TGF-β-dependent and independent roles of TSP-1 (155, 156). Recently, Song et al. reported that Sestrin2 remedies podocyte injury in DKD through the coordination with TSP-1/TGF-β1/Smad3 pathway, suggesting that Sestrin2/TSP-1/TGF-β1 signaling is critically involved in renal protection (22).
The Hippo pathway, a kinase cascade that regulates cellular proliferation, differentiation, and tissue homeostasis, is inhibited in diabetic conditions. The Hippo pathway has been indicated to be involved in the development and progression of DKD (157, 158). PI3K/AKT signaling is related to the Hippo pathway and both of these pathways take part in the pathogenesis of DKD (158). Sestrin2 overexpression was found to alleviate renal damage via regulating Hippo pathway in DKD mice (125). Considering the interactions among Sestrin2 and multiple signaling pathways, Sestrin2 may be critically in involved in the development of DKD and thus may perform as a latent therapeutic target for DKD. Figure 2 summarizes Sestrin2 signaling pathways in diabetic kidney disease.
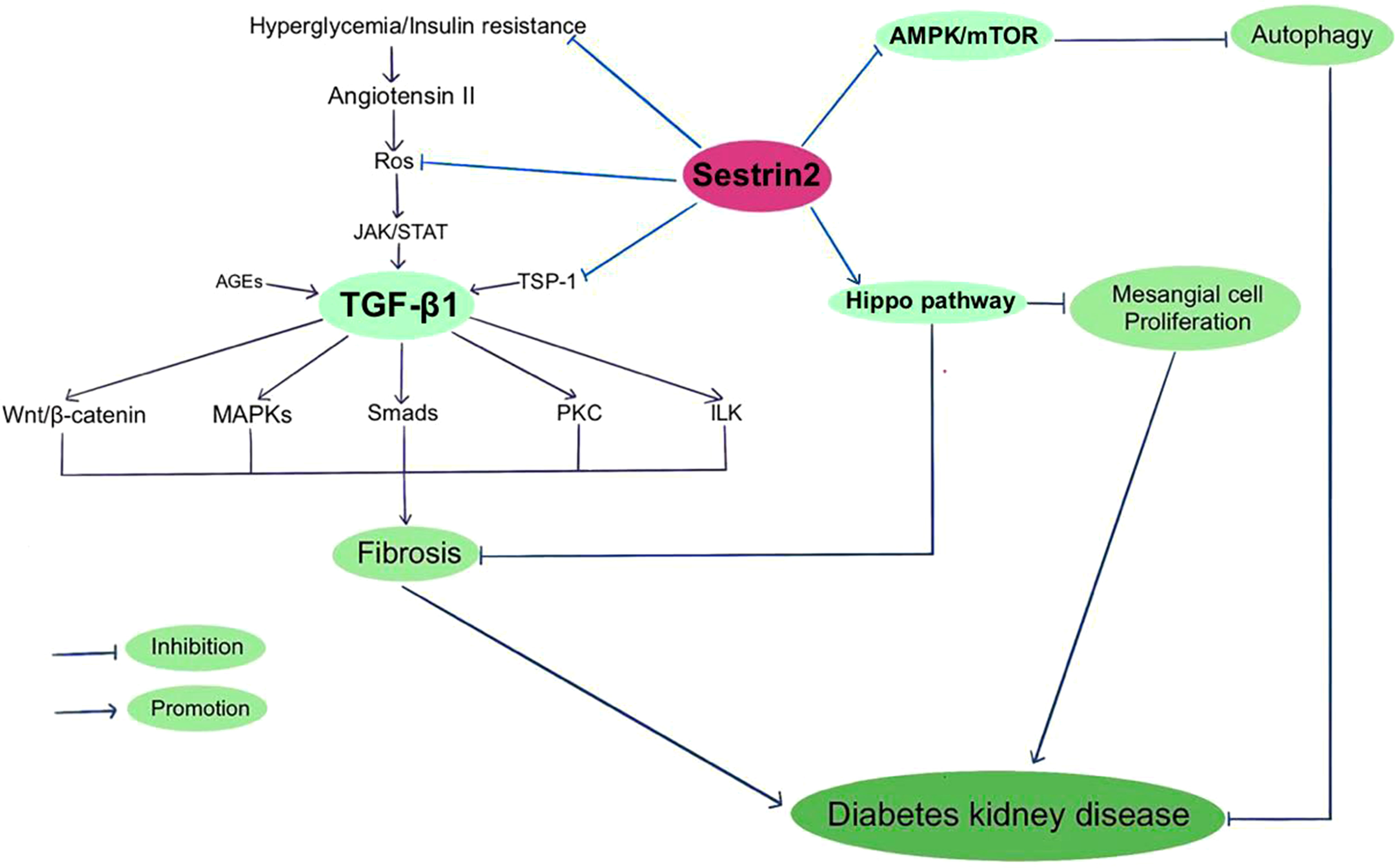
Figure 2 Sestrin2 and Diabetic kidney disease.
4.2 Diabetic cardiovascular complications/diabetic heart disease
Diabetic heart disease (DHD) is a major cause of death in patients with diabetes. It refers to abnormal heart structure and manifestations in patients with diabetes in the absence of other cardiac risk factors. DHD is a conglomeration of coronary artery disease, heart failure, diabetic cardiomyopathy (DCM) and diabetic cardiac autonomic neuropathy, and is characterized by molecular, structural, and functional changes in the myocardium (159, 160). The pathogenesis includes the macrovascular and microvascular lesions and cardiac autonomic neuropathy caused by oxidative stress, inflammatory response, mitochondrial dysfunction, AGEs, alterations at the level of insulin signaling, gene regulation, ER stress, hypoxia, neurohumoral activation, apoptosis, and exosome dysregulation (160, 161).
mTOR signaling is found to play a key role in the development of DHD. Activation of mTORC1 either by strengthening PI3K/AKT signaling or disruption of tuberous sclerosis complex 1 (TSC1) drives cardiac hypertrophy (162, 163). Also, mTORC1 inhibition exerts cardioprotective effect against myocardial ischemia and DCM by activating autophagy (164). Inhibition of mTOR signaling by application of melatonin reduces myocardial damages and protects against DCM (165). mTORC2 seems to exert reverse effect on cardiac remodeling. Suppression of mTORC1 and activation of mTORC2 exert beneficial effects on myocardial ischemia and adverse cardiac remodeling (29). TGF-βs are central effectors of myocardial fibrosis (166). TGF-β-driven fibrosis is regulated by canonical or noncanonical pathways and is mediated by coreceptors and by interacting networks. The activation of canonical or Smad-dependent signaling pathways causes phosphorylation and activation of SMAD proteins. The activation of noncanonical pathways include PI3K/AKT, ERK, JNK, RhoA and MAPK pathways (167, 168). In the dilated cardiomyopathy model, the increase of myocardial expression of TGF-β and activation of downstream Smad 2 and Smad 3 signal cascades have been unanimously confirmed. Overexpression and activation of TGF-β1 in DCM induces cardiac fibrosis, which can be alleviated by administration of telmisartan, empagliflozin, dapagliflozin, epigallocatechin gallate, or cannabidiol, possibly due to the inhibitory effects on TGF-β signaling (169–174). TSP-1 is suggested to play a significant role in DCM. TSP-1 upregulation in the diabetic heart stabilizes the cardiac matrix and promotes vascular rarefaction in obese diabetic mice. TSP-1 enhancement in the myocardium may be a crucial regulator in diabetes-associated impaired angiogenesis (175). The effect of TSP-1 are mediated regulated by activation of TGF-β, angiostatic actions, matrix metalloproteinase inhibition and direct stimulation of CD36 signaling (176). Downregulating TSP-1 and TGF-β1 improves the heart function and ameliorates vascular fibrosis in diabetic rats (177, 178).
As previously described, Sestrin2 participates in the modulation of oxidative stress, mitochondrial biogenesis, ER stress and apoptosis. Also, the AMPK/mTOR pathway and TSP-1/TGF-β1 pathway are constitute parts of the regulating mechanism of Sestrin2. It is reasonable to postulate that Sestrin2 may take part in the pathogenesis of DHD. But the researches investigating the role of Sestrin2 in DHD are relatively few (rare). Previously, Sestrin2 is considered to be cardioprotective in several models of cardiovascular diseases, including myocardial infarction and cardiac dysfunction induced by ER stress or lipopolysaccharide, via AMPK/mTOR signaling cascade (97, 179). Besides, increasing evidence indicate a protective role for Sestrin2 against the development and progression of cardiomyopathy and heart failure in model of pressure-overload cardiac remodeling, via Nrf2/Keap1 pathway (180). Also, Sestrin2 is indicated to modulate cardiac inflammatory response through maintaining oxidative stress through MAPK/JNK pathway during ischemia and reperfusion (181). Sestrin2 suppression aggravates ER stress-induced oxidative stress and apoptosis in endothelial cells (182). Clinically, several studies have investigated the variations in plasma Sestrin2 protein levels in patients with cardiomyopathy and/or heart failure, and displayed conflicting results (183–185). Wang et al. reported that plasma Sestrin2 level was increased in patients with chronic heart failure (CHD) and was positively related to the severity of CHD. Increment of Sestrin2 concentrations prominently increased the occurrence of major adverse cardiac events and suggested poor prognosis (183). Also, plasma Sestrin2 levels were found to be increased in patients with coronary heart disease (CAD) and positively related to the severity of CAD (184, 186). However, lower serum Setrin2 levels were indicated in patients with septic cardiomyopathy and in T2DM patients with CHD (126, 185). Low Sestrin2 level was a risk factor for CHD in T2DM patients (126). The contradictory results concerning the beneficial or harmful effects of sestrin2 in cardiomyopathy and heart failure need to be further clarified. In recent years, a few studies investigated the role of Sestrin2 in DHD. Zhou et al. reported that Sestrin2 may enhance antioxidative actions and alleviate mitochondrial lesion by interacting with Nrf2 to prevent the diabetic rat heart from ischemia/reperfusion injury (18). Our previous research showed that inhibition of enhanced Sestrin2 expression attenuates cardiac injury in DCM, which may be mainly attributed to the restoration of mitochondrial function (127). Some antiglycemic agents, such as metformin and empagliflozin, were found to be cardioprotective through Sestrin2-associated mechanism. As shown by Yang et al., metformin can activate AMPK, thereby promoting autophagy by suppressing the mTOR pathway in DCM (187). Sestrin2 was suggested to participate in cardioprotective effects of metformin in a model of acute kidney injury (188). Sun et al. found that empagliflozin improves obesity-related cardiac dysfunction via regulating Sestrin2-mediated AMPK/mTOR signaling and maintaining redox homeostasis (145). Figure 3 summarizes Sestrin2 signaling pathways in diabetic heart disease.
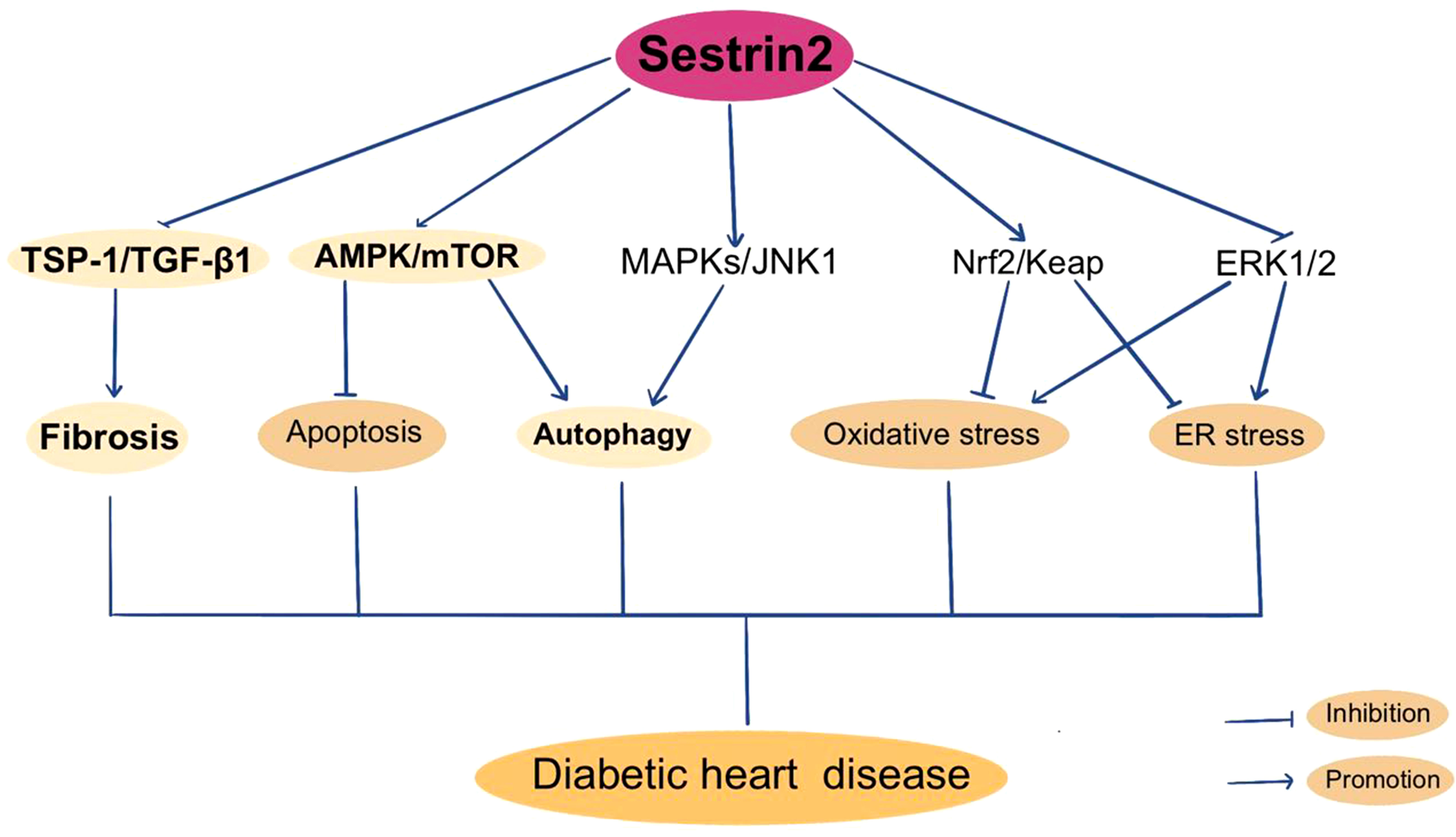
Figure 3 Sestrin2 and Diabetic heart disease.
4.3 Diabetic ocular complications
Diabetes can cause various ocular complications, such as diabetic retinopathy (DR), cataract, diabetic papillopathy, glaucoma, and ocular surface diseases (189). DR is a major diabetic complication characterized by retinal microvascular lesion and is a major cause of vision loss in working middle-aged adults. Complicated mechanism is included in the pathogenesis of DR, including increased free radical production, activated AMPK/mTOR signaling, renin-angiotensin pathway, TGF-β/Smad signaling, and the kallikrein-kinin system, the formation of AGEs, and increased inflammatory factors and vascular endothelial growth factor (VEGF) (190–192). mTOR signaling is considered to play multiple roles in the pathogenesis of DR. mTORC1 is indispensable for the hypoxic-induced expression of VEGF, which is an important pathogenic event in DR (193). Also, mTORC1 affects DR development by negatively regulating autophagy (194). PI3K/AKT/mTOR signaling pathway is associated with the early pathogenesis of DR (195). Promoting autophagy and enhancing AMPK/mTOR signaling pathway can protect retinal Muller cells from apoptosis caused by high glucose (196). Aberrant TGF-β signaling pathway is involved in the pathogenesis of DR (155). TGF-β1 protects retinal ganglion cells from hyperglycemia-induced oxidative damage through promoting cell antioxidation and neuroprotection pathways, including Nrf2/Keap1 signaling (197). Increased TGF-β signaling induced by diabetes protects retinal vessels in diabetic rats and may prevent rapid retinopathy progression (198).
Based on its pleiotropic modulating effects, Sestrin2 may have an impact on the pathogenesis of DR. So far there are several studies exploring the role of Sestrin2 in models of ocular lesions, but still no reports in diabetic ocular complications have been found. Previously, Hanus et al. demonstrated that upregulation of Sestrin2 protects retinal pigment epithelial cells from oxidative stress-induced necrosis (199). Sestrin2 could also secure retinal ganglion cells from oxidative stress-induced apoptosis through Keap1/Nrf2 pathway, which suggests a significant role of Sestrin2 in retinal degeneration in glaucoma (200). But on the other hand, Sestrin2 is indicated to be a negative modulator of corneal epithelial cell proliferation. The downregulation of Sestrin2 leads to the synergistic activation of mTORC1 and Hippo signaling, thus promoting reepithelialization of the corneal wound (201).
4.4 Diabetic neuropathy
Diabetic neuropathy (DN) is another frequent chronic complication of diabetes, consisting of four major types including peripheral neuropathy, autonomic neuropathy, proximal neuropathy, and mononeuropathy (202). The pathogenesis of DN is complicated, including changes of various metabolic pathways and vascular pathways. Three main pathological events contribute to the progression of DN, including chronic low-grade inflammation, endothelial dysfunction and oxidative stress (203). Consistent hyperglycemia in diabetes induces activation/inhibition of diverse pathways, including polyol, hexosamine, AGEs, PARP, MAPK, mTOR, NF-κB and tumor necrosis factor-α (TNF-α) pathway, which contribute to the pathogenesis and progression of DN (204, 205). Among them, overactive mTORC1 interferes with synaptic plasticity and is one of the main factors contribute to chronic neuropathy. Activation of mTOR exacerbates the hyperalgesia in diabetic rats, while suppression of mTORC1 activity is indicated to lead to an anti-injury effect in experimental model of diabetic small fiber neuropathy (206, 207). With the inhibition of PI3K/AKT/mTOR pathway, autophagy is enhanced and hyperalgesia is alleviated in diabetes rats (208). So far, no research exploring the role of Sestrin2 in DN can be found. But there are a few studies in other disease models. In denervated atrophy, Sestrin2 has been proved to prevent the change of muscle fiber type from slow contraction to fast contraction through AMPK/PGC-1α signaling, and protect muscle quality (209). Regulation of UPR and mitophagy is also included in the mechanism by which Sestrin2 protects against denervated muscle atrophy (210). Zhang et al. demonstrated that overexpressing Sestrin2 significantly reduces oxidative stress of neurons in model of cerebral ischemia-reperfusion injury through modulating the activity of Nrf2 (12). Mao et al. conducted a clinical study and documented that serum Sestrin2 is lowered in T2DM patients with diabetic peripheral neuropathy (128).
5 Possible pharmacological mediators of Sestrin2
The exploration of Therapeutic strategies through mediation of Sestrin2 is now underway. Several natural products and medications in diabetes have been shown to alter the expression levels of Sestrin2, which lead to the possibility of novel treatments in diabetes and diabetic complications targeting Sestrin2 and the associated pathways (211). Initially, a few studies investigated the potential mediator of Sestrin2 in the field of tumor therapies, including several small molecules (212). Recently, natural-derived mediator of Sestrin2, such as Gallic acid (213) and eupatilin (Unpublished data of our research), are also indicated to be potential therapeutic agents of obesity and diabetes. Some antidiabetic medications are indicated to be involved in the regulation of Sestrin2 signaling in diabetes-associated conditions, some of which have been illustrated in the previous sections of this review. Sestrin2 can be targeted by empagliflozin in the treatment of obesity-related nonalcoholic fatty liver disease (214). Another antidiabetic agent, liraglutide, is shown to alleviate obesity-related fatty liver disease via modulating the Sestrin2-mediated Nrf2-HO-1 pathway (215). Intervention through gene editing of Sestrin2 also presents beneficial effects in organ and tissue protection (216), but further investigation is needed in the context of diabetes.
6 Conclusions and perspectives
Diabetes is a condition causing multi-organ injuries and is a major global threaten for human health. As a stress-induced protein, Sestrin2 can be activated by diverse stresses and can exert pleiotropic effects. Sestrin2 can interact with various signaling perspectives involved in the development of diabetes. Increasing numbers of studies indicate a prominent role of Sestrin2 in the pathogenesis of diabetes and diabetic complications. Sestrin2 may functions in a multitude of ways and offer exciting prospects for the treatment of diabetes and diabetic complications, though currently the strong supporting evidence is limited. Despite of the protective roles of Sestrn2 in various conditions, the pros and cons of excessive activation or inhibition of Sestrin2 is yet to be confirmed. How to exert accurate mediation under different conditions also remains elusive. The modulation of Sestrin2 activity to effectively achieve homeostasis might be more appropriate. Further researches are needed to thoroughly reveal the relationship between Sestrin2 and diabetes which includes multi-organ injuries, to disclose associated signaling pathways and to explore potential treatment protocols.
Author contributions
XZ: Writing – review & editing, Conceptualization, Writing – original draft. ZL: Writing – original draft. JL: Writing – original draft. YLin: Writing – original draft. YLi: Writing – original draft, Project administration, Resources, Software. WL: Writing – review & editing, Supervision, Validation.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (grant no. 81800726), Plan on enhancing scientific research in Guangzhou Medical University (grant no. 2023-184), and The Student innovation ability improvement program of Guangzhou Medical University (grant no. 2022-85).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ge X, Wang L, Fei A, Ye S, Zhang Q. Research progress on the relationship between autophagy and chronic complications of diabetes. Front Physiol (2022) 13:956344. doi: 10.3389/fphys.2022.956344
2. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
3. Budanov AV, Lee JH, Karin M. Stressin' Sestrins take an aging fight. EMBO Mol Med (2010) 2:388–400. doi: 10.1002/emmm.201000097
4. Park HJ, Yang SG, Koo DB. SESN2/NRF2 signaling activates as a direct downstream regulator of the PERK pathway against endoplasmic reticulum stress to improve the in vitro maturation of porcine oocytes. Free Radic Biol Med (2022) 178:413–27. doi: 10.1016/j.freeradbiomed.2021.12.258
5. Ala M, Eftekhar SP. Target sestrin2 to rescue the damaged organ: mechanistic insight into its function. Oxid Med Cell Longev (2021) 2021:8790369. doi: 10.1155/2021/8790369
6. Pasha M, Eid AH, Eid AA, Gorin Y, Munusamy S. Sestrin2 as a novel biomarker and therapeutic target for various diseases. Oxid Med Cell Longev (2017) 2017:3296294. doi: 10.1155/2017/3296294
7. Ding B, Parmigiani A, Divakaruni AS, Archer K, Murphy AN, Budanov AV. Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death. Sci Rep (2016) 6:22538. doi: 10.1038/srep22538
8. Saveljeva S, Cleary P, Mnich K, Ayo A, Pakos-Zebrucka K, Patterson JB, et al. Endoplasmic reticulum stress-mediated induction of SESTRIN 2 potentiates cell survival. Oncotarget (2016) 7:12254–66. doi: 10.18632/oncotarget.7601
9. Lee JH, Bodmer R, Bier E, Karin M. Sestrins at the crossroad between stress and aging. Aging (Albany NY) (2010) 2:369–74. doi: 10.18632/aging.100157
10. Che X, Chai J, Fang Y, Zhang X, Zu A, Li L, et al. Sestrin2 in hypoxia and hypoxia-related diseases. Redox Rep (2021) 26:111–6. doi: 10.1080/13510002.2021.1948774
11. Chen T, Li T, Wang J. p53 mediates PEDF−induced autophagy in human umbilical vein endothelial cells through sestrin2 signaling. Mol Med Rep (2019) 20:1443–50. doi: 10.3892/mmr.2019.10319
12. Zhang LL, Zhang ZJ. Sestrin2 aggravates oxidative stress of neurons by decreasing the expression of Nrf2. Eur Rev Med Pharmacol Sci (2018) 22:3493–501. doi: 10.26355/eurrev_201806_15176
13. Wu CL, Chen SD, Yin JH, Hwang CS, Yang DI. Nuclear factor-kappaB-dependent sestrin2 induction mediates the antioxidant effects of BDNF against mitochondrial inhibition in rat cortical neurons. Mol Neurobiol (2016) 53:4126–42. doi: 10.1007/s12035-015-9357-1
14. Han D, Kim H, Kim S, Le QA, Han SY, Bae J, et al. Sestrin2 protects against cholestatic liver injury by inhibiting endoplasmic reticulum stress and NLRP3 inflammasome-mediated pyroptosis. Exp Mol Med (2022) 54:239–51. doi: 10.1038/s12276-022-00737-9
15. Wang BJ, Wang S, Xiao M, Zhang J, Wang AJ, Guo Y, et al. Regulatory mechanisms of Sesn2 and its role in multi-organ diseases. Pharmacol Res (2021) 164:105331. doi: 10.1016/j.phrs.2020.105331
16. Pan C, Chen Z, Li C, Han T, Liu H, Wang X. Sestrin2 as a gatekeeper of cellular homeostasis: Physiological effects for the regulation of hypoxia-related diseases. J Cell Mol Med (2021) 25:5341–50. doi: 10.1111/jcmm.16540
17. Li Y, Zhang J, Zhou K, Xie L, Xiang G, Fang M, et al. Elevating sestrin2 attenuates endoplasmic reticulum stress and improves functional recovery through autophagy activation after spinal cord injury. Cell Biol Toxicol (2021) 37:401–19. doi: 10.1007/s10565-020-09550-4
18. Zhou XR, Ru XC, Xiao C, Pan J, Lou YY, Tang LH, et al. Sestrin2 is involved in the Nrf2-regulated antioxidative signaling pathway in luteolin-induced prevention of the diabetic rat heart from ischemia/reperfusion injury. Food Funct (2021) 12:3562–71. doi: 10.1039/d0fo02942d
19. Lanna A, Gomes DC, Muller-Durovic B, McDonnell T, Escors D, Gilroy DW, et al. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat Immunol (2017) 18:354–63. doi: 10.1038/ni.3665
20. Li L, Xiao L, Hou Y, He Q, Zhu J, Li Y, et al. Sestrin2 silencing exacerbates cerebral ischemia/reperfusion injury by decreasing mitochondrial biogenesis through the AMPK/PGC-1α Pathway in rats. Sci Rep (2016) 6:30272. doi: 10.1038/srep30272
21. Dong B, Xue R, Sun Y, Dong Y, Liu C. Sestrin 2 attenuates neonatal rat cardiomyocyte hypertrophy induced by phenylephrine via inhibiting ERK1/2. Mol Cell Biochem (2017) 433:113–23. doi: 10.1007/s11010-017-3020-2
22. Song S, Shi C, Bian Y, Yang Z, Mu L, Wu H, et al. Sestrin2 remedies podocyte injury via orchestrating TSP-1/TGF-β1/Smad3 axis in diabetic kidney disease. Cell Death Dis (2022) 13:663. doi: 10.1038/s41419-022-05120-0
23. Hwang CY, Han YH, Lee SM, Cho SM, Yu DY, Kwon KS. Sestrin2 attenuates cellular senescence by inhibiting NADPH oxidase 4 expression. Ann Geriatr Med Res (2020) 24:297–304. doi: 10.4235/agmr.20.0051
24. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell (2002) 110:163–75. doi: 10.1016/s0092-8674(02)00808-5
25. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol (2011) 12:21–35. doi: 10.1038/nrm3025
26. Zhu Z, Yang C, Iyaswamy A, Krishnamoorthi S, Sreenivasmurthy SG, Liu J, et al. Balancing mTOR signaling and autophagy in the treatment of parkinson's disease. Int J Mol Sci (2019) 20:728. doi: 10.3390/ijms20030728
27. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol (2018) 19:121–35. doi: 10.1038/nrm.2017.95
28. Lamming DW, Sabatini DM. A Central role for mTOR in lipid homeostasis. Cell Metab (2013) 18:465–9. doi: 10.1016/j.cmet.2013.08.002
29. Yang L, Zhang Z, Wang D, Jiang Y, Liu Y. Targeting mTOR signaling in type 2 diabetes mellitus and diabetes complications. Curr Drug Targets (2022) 23:692–710. doi: 10.2174/1389450123666220111115528
30. Blandino-Rosano M, Barbaresso R, Jimenez-Palomares M, Bozadjieva N, Werneck-de-Castro JP, Hatanaka M, et al. Loss of mTORC1 signalling impairs β-cell homeostasis and insulin processing. Nat Commun (2017) 8:16014. doi: 10.1038/ncomms16014
31. Gu Y, Lindner J, Kumar A, Yuan W, Magnuson MA. Rictor/mTORC2 is essential for maintaining a balance between beta-cell proliferation and cell size. Diabetes (2011) 60:827–37. doi: 10.2337/db10-1194
32. Blandino-Rosano M, Scheys JO, Werneck-de-Castro JP, Louzada RA, Almaça J, Leibowitz G, et al. Novel roles of mTORC2 in regulation of insulin secretion by actin filament remodeling. Am J Physiol Endocrinol Metab (2022) 323:E133–e144. doi: 10.1152/ajpendo.00076.2022
33. Yu D, Tomasiewicz JL, Yang SE, Miller BR, Wakai MH, Sherman DS, et al. Calorie-restriction-induced insulin sensitivity is mediated by adipose mTORC2 and not required for lifespan extension. Cell Rep (2019) 29:236–248.e3. doi: 10.1016/j.celrep.2019.08.084
34. Jung SM, Hung CM, Hildebrand SR, Sanchez-Gurmaches J, Martinez-Pastor B, Gengatharan JM, et al. Non-canonical mTORC2 Signaling Regulates Brown Adipocyte Lipid Catabolism through SIRT6-FoxO1. Mol Cell (2019) 75:807–822.e8. doi: 10.1016/j.molcel.2019.07.023
35. Hung CM, Calejman CM, Sanchez-Gurmaches J, Li H, Clish CB, Hettmer S, et al. Rictor/mTORC2 loss in the Myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Rep (2014) 8:256–71. doi: 10.1016/j.celrep.2014.06.007
36. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science (2012) 335:1638–43. doi: 10.1126/science.1215135
37. Sanli T, Linher-Melville K, Tsakiridis T, Singh G. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PloS One (2012) 7:e32035. doi: 10.1371/journal.pone.0032035
38. Yan M, Jin S, Liu Y, Wang L, Wang Z, Xia T, et al. Cajaninstilbene acid ameliorates acetaminophen-induced liver injury through enhancing sestrin2/AMPK-mediated mitochondrial quality control. Front Pharmacol (2022) 13:824138. doi: 10.3389/fphar.2022.824138
39. Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, et al. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep (2014) 9:1–8. doi: 10.1016/j.celrep.2014.09.014
40. Byun JK, Choi YK, Kim JH, Jeong JY, Jeon HJ, Kim MK, et al. A positive feedback loop between sestrin2 and mTORC2 is required for the survival of glutamine-depleted lung cancer cells. Cell Rep (2017) 20:586–99. doi: 10.1016/j.celrep.2017.06.066
41. Kowalsky AH, Namkoong S, MettEtal E, Park HW, Kazyken D, Fingar DC, et al. The GATOR2-mTORC2 axis mediates Sestrin2-induced AKT Ser/Thr kinase activation. J Biol Chem (2020) 295:1769–80. doi: 10.1074/jbc.RA119.010857
42. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell (2017) 169:381–405. doi: 10.1016/j.cell.2017.04.001
43. Dodson M, Shakya A, Anandhan A, Chen J, Garcia JGN, Zhang DD. NRF2 and diabetes: the good, the bad, and the complex. Diabetes (2022) 71:2463–76. doi: 10.2337/db22-0623
44. Lee S, Hu L. Nrf2 activation through the inhibition of Keap1-Nrf2 protein-protein interaction. Med Chem Res (2020) 29:846–67. doi: 10.1007/s00044-020-02539-y
45. Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci (2013) 34:340–6. doi: 10.1016/j.tips.2013.04.005
46. Jiménez-Osorio AS, Picazo A, González-Reyes S, Barrera-Oviedo D, Rodríguez-Arellano ME, Pedraza-Chaverri J. Nrf2 and redox status in prediabetic and diabetic patients. Int J Mol Sci (2014) 15:20290–305. doi: 10.3390/ijms151120290
47. Abed DA, Goldstein M, Albanyan H, Jin H. Discovery of direct inhibitors of Keap1-Nrf2 protein-protein interaction as potential therapeutic and preventive agents. Acta Pharm Sin B (2015) 5:285–99. doi: 10.1016/j.apsb.2015.05.008
48. Choudhury S, Ghosh S, Gupta P, Mukherjee S, Chattopadhyay S. Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2/NF-κB and SAPK/JNK pathway. Free Radic Res (2015) 49:1371–83. doi: 10.3109/10715762.2015.1075016
49. Zhang L, Li J, Ma J, Chen X, Chen K, Jiang Z, et al. The relevance of nrf2 pathway and autophagy in pancreatic cancer cells upon stimulation of reactive oxygen species. Oxid Med Cell Longev (2016) 2016:3897250. doi: 10.1155/2016/3897250
50. Shopit A, Niu M, Wang H, Tang Z, Li X, Tesfaldet T, et al. Protection of diabetes-induced kidney injury by phosphocreatine via the regulation of ERK/Nrf2/HO-1 signaling pathway. Life Sci (2020) 242:117248. doi: 10.1016/j.lfs.2019.117248
51. Ge ZD, Lian Q, Mao X, Xia Z. Current status and challenges of NRF2 as a potential therapeutic target for diabetic cardiomyopathy. Int Heart J (2019) 60:512–20. doi: 10.1536/ihj.18-476
52. Kumar A, Mittal R. Nrf2: a potential therapeutic target for diabetic neuropathy. Inflammopharmacology (2017) 25:393–402. doi: 10.1007/s10787-017-0339-y
53. Li Y, Wu J, Yu S, Zhu J, Zhou Y, Wang P, et al. Sestrin2 promotes angiogenesis to alleviate brain injury by activating Nrf2 through regulating the interaction between p62 and Keap1 following photothrombotic stroke in rats. Brain Res (2020) 1745:146948. doi: 10.1016/j.brainres.2020.146948
54. Liu W, Xu C, Zou Z, Weng Q, Xiao Y. Sestrin2 suppresses ferroptosis to alleviate septic intestinal inflammation and barrier dysfunction. Immunopharmacol Immunotoxicol (2023) 45:123–32. doi: 10.1080/08923973.2022.2121927
55. Wang H, Xi J, Zhang Z, Li J, Guo L, Li N, et al. Sestrin2 is increased in calcific aortic disease and inhibits osteoblastic differentiation in valvular interstitial cells via the nuclear factor E2-related factor 2 pathway. J Cardiovasc Pharmacol (2022) 80:609–15. doi: 10.1097/fjc.0000000000001314
56. Park MJ, Kim JW, Roh E, Choi KM, Baik SH, Hwang HJ, et al. Sestrin2 regulates beneficial β3-adrenergic receptor-mediated effects observed in inguinal white adipose tissue and soleus muscle. Endocrinol Metab (Seoul) (2022) 37:552–7. doi: 10.3803/EnM.2022.1421
57. Oh SJ, Lee MS. Role of autophagy in the pathogenesis of diabetes and therapeutic potential of autophagy modulators in the treatment of diabetes and metabolic syndrome. J Korean Med Sci (2022) 37:e276. doi: 10.3346/jkms.2022.37.e276
58. Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy (2016) 12:245–60. doi: 10.1080/15548627.2015.1071759
59. Kim KH, Lee MS. Autophagy–a key player in cellular and body metabolism. Nat Rev Endocrinol (2014) 10:322–37. doi: 10.1038/nrendo.2014.35
60. Kulkarni A, Muralidharan C, May SC, Tersey SA, Mirmira RG. Inside the β Cell: molecular stress response pathways in diabetes pathogenesis. Endocrinology (2022) 164:bqac184. doi: 10.1210/endocr/bqac184
61. Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab (2013) 17:73–84. doi: 10.1016/j.cmet.2012.12.002
62. Liang Y, Zhu J, Huang H, Xiang D, Li Y, Zhang D, et al. SESN2/sestrin 2 induction-mediated autophagy and inhibitory effect of isorhapontigenin (ISO) on human bladder cancers. Autophagy (2016) 12:1229–39. doi: 10.1080/15548627.2016.1179403
63. Jin HR, Du CH, Wang CZ, Yuan CS, Du W. Ginseng metabolite Protopanaxadiol induces Sestrin2 expression and AMPK activation through GCN2 and PERK. Cell Death Dis (2019) 10:311. doi: 10.1038/s41419-019-1548-7
64. Li H, Liu S, Yuan H, Niu Y, Fu L. Sestrin 2 induces autophagy and attenuates insulin resistance by regulating AMPK signaling in C2C12 myotubes. Exp Cell Res (2017) 354:18–24. doi: 10.1016/j.yexcr.2017.03.023
65. Shen T, Alvarez-Garcia O, Li Y, Olmer M, Lotz MK. Suppression of Sestrins in aging and osteoarthritic cartilage: dysfunction of an important stress defense mechanism. Osteoarthritis Cartilage (2017) 25:287–96. doi: 10.1016/j.joca.2016.09.017
66. Gao A, Jiang J, Xie F, Chen L. Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin Chim Acta (2020) 506:72–83. doi: 10.1016/j.cca.2020.02.024
67. Yaribeygi H, Sathyapalan T, Atkin SL, Sahebkar A. Molecular mechanisms linking oxidative stress and diabetes mellitus. Oxid Med Cell Longev (2020) 2020:8609213. doi: 10.1155/2020/8609213
68. Münzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC. Impact of oxidative stress on the heart and vasculature: part 2 of a 3-part series. J Am Coll Cardiol (2017) 70:212–29. doi: 10.1016/j.jacc.2017.05.035
69. Panigrahy SK, Bhatt R, Kumar A. Reactive oxygen species: sources, consequences and targeted therapy in type 2 diabetes. J Drug Targeting (2017) 25:93–101. doi: 10.1080/1061186x.2016.1207650
70. Gong L, Wang Z, Wang Z, Zhang Z. Sestrin2 as a potential target for regulating metabolic-related diseases. Front Endocrinol (Lausanne) (2021) 12:751020. doi: 10.3389/fendo.2021.751020
71. Liu Y, Du X, Huang Z, Zheng Y, Quan N. Sestrin 2 controls the cardiovascular aging process via an integrated network of signaling pathways. Ageing Res Rev (2020) 62:101096. doi: 10.1016/j.arr.2020.101096
72. Gao A, Li F, Zhou Q, Chen L. Sestrin2 as a potential therapeutic target for cardiovascular diseases. Pharmacol Res (2020) 159:104990. doi: 10.1016/j.phrs.2020.104990
73. Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol (2017) 13:477–91. doi: 10.1038/nrneurol.2017.99
74. Gorman AM, Healy SJ, Jäger R, Samali A. Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol Ther (2012) 134:306–16. doi: 10.1016/j.pharmthera.2012.02.003
75. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep (2006) 7:880–5. doi: 10.1038/sj.embor.7400779
76. Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell (2000) 101:249–58. doi: 10.1016/s0092-8674(00)80835-1
77. Zhao H, Liu H, Yang Y, Lan T, Wang H, Wu D. Hydrogen sulfide plays an important role by regulating endoplasmic reticulum stress in diabetes-related diseases. Int J Mol Sci (2022) 23:7170. doi: 10.3390/ijms23137170
78. Moon S, Jung HS. Endoplasmic reticulum stress and dysregulated autophagy in human pancreatic beta cells. Diabetes Metab J (2022) 46:533–42. doi: 10.4093/dmj.2022.0070
79. Prasad MK, Mohandas S, Ramkumar KM. Role of ER stress inhibitors in the management of diabetes. Eur J Pharmacol (2022) 922:174893. doi: 10.1016/j.ejphar.2022.174893
80. Jegal KH, Park SM, Cho SS, Byun SH, Ku SK, Kim SC, et al. Activating transcription factor 6-dependent sestrin 2 induction ameliorates ER stress-mediated liver injury. Biochim Biophys Acta Mol Cell Res (2017) 1864:1295–307. doi: 10.1016/j.bbamcr.2017.04.010
81. Park HW, Park H, Ro SH, Jang I, Semple IA, Kim DN, et al. Hepatoprotective role of Sestrin2 against chronic ER stress. Nat Commun (2014) 5:4233. doi: 10.1038/ncomms5233
82. Hwang HJ, Jung TW, Choi JH, Lee HJ, Chung HS, Seo JA, et al. Knockdown of sestrin2 increases pro-inflammatory reactions and ER stress in the endothelium via an AMPK dependent mechanism. Biochim Biophys Acta Mol Basis Dis (2017) 1863:1436–44. doi: 10.1016/j.bbadis.2017.02.018
83. Yang Y, Guo G, Zhou W, Ge Y, Fan Z, Liu Q, et al. Sestrin2 protects against bavachin induced ER stress through AMPK/mTORC1 signaling pathway in HepG2 cells. J Pharmacol Sci (2021) 145:175–86. doi: 10.1016/j.jphs.2020.11.012
84. Okamoto K, Kondo-Okamoto N. Mitochondria and autophagy: critical interplay between the two homeostats. Biochim Biophys Acta (2012) 1820:595–600. doi: 10.1016/j.bbagen.2011.08.001
85. Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annu Rev Genet (2009) 43:95–118. doi: 10.1146/annurev-genet-102108-134850
86. Sygitowicz G, Sitkiewicz D. Mitochondrial quality control: the role in cardiac injury. Front Biosci (Landmark Ed) (2022) 27:96. doi: 10.31083/j.fbl2703096
87. Yan X, Wang B, Hu Y, Wang S, Zhang X. Abnormal mitochondrial quality control in neurodegenerative diseases. Front Cell Neurosci (2020) 14:138. doi: 10.3389/fncel.2020.00138
88. Georgiev A, Granata C, Roden M. The role of mitochondria in the pathophysiology and treatment of common metabolic diseases in humans. Am J Physiol Cell Physiol (2022) 322:C1248–c1259. doi: 10.1152/ajpcell.00035.2022
89. Wang L, Yang Z, He X, Pu S, Yang C, Wu Q, et al. Mitochondrial protein dysfunction in pathogenesis of neurological diseases. Front Mol Neurosci (2022) 15:974480. doi: 10.3389/fnmol.2022.974480
90. Chang X, Li Y, Cai C, Wu F, He J, Zhang Y, et al. Mitochondrial quality control mechanisms as molecular targets in diabetic heart. Metabolism (2022) 137:155313. doi: 10.1016/j.metabol.2022.155313
91. Li Y, Ma Y, Dang QY, Fan XR, Han CT, Xu SZ, et al. Assessment of mitochondrial dysfunction and implications in cardiovascular disorders. Life Sci (2022) 306:120834. doi: 10.1016/j.lfs.2022.120834
92. Weksler-Zangen S. Is type 2 diabetes a primary mitochondrial disorder? Cells (2022) 11:1617. doi: 10.3390/cells11101617
93. Shan Z, Fa WH, Tian CR, Yuan CS, Jie N. Mitophagy and mitochondrial dynamics in type 2 diabetes mellitus treatment. Aging (Albany NY) (2022) 14:2902–19. doi: 10.18632/aging.203969
94. Kumar A, Dhiman D, Shaha C. Sestrins: Darkhorse in the regulation of mitochondrial health and metabolism. Mol Biol Rep (2020) 47:8049–60. doi: 10.1007/s11033-020-05769-w
95. Kumar A, Shaha C. SESN2 facilitates mitophagy by helping Parkin translocation through ULK1 mediated Beclin1 phosphorylation. Sci Rep (2018) 8:615. doi: 10.1038/s41598-017-19102-2
96. Quan N, Wang L, Chen X, Luckett C, Cates C, Rousselle T, et al. Sestrin2 prevents age-related intolerance to post myocardial infarction via AMPK/PGC-1α pathway. J Mol Cell Cardiol (2018) 115:170–8. doi: 10.1016/j.yjmcc.2018.01.005
97. Hwang HJ, Kim JW, Chung HS, Seo JA, Kim SG, Kim NH, et al. Knockdown of sestrin2 increases lipopolysaccharide-induced oxidative stress, apoptosis, and fibrotic reactions in H9c2 cells and heart tissues of mice via an AMPK-dependent mechanism. Mediators Inflamm (2018) 2018:6209140. doi: 10.1155/2018/6209140
98. Kim MJ, Bae SH, Ryu JC, Kwon Y, Oh JH, Kwon J, et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy (2016) 12:1272–91. doi: 10.1080/15548627.2016.1183081
99. Wang P, Wang L, Lu J, Hu Y, Wang Q, Li Z, et al. SESN2 protects against doxorubicin-induced cardiomyopathy via rescuing mitophagy and improving mitochondrial function. J Mol Cell Cardiol (2019) 133:125–37. doi: 10.1016/j.yjmcc.2019.06.005
100. Jegal KH, Ko HL, Park SM, Byun SH, Kang KW, Cho IJ, et al. Eupatilin induces Sestrin2-dependent autophagy to prevent oxidative stress. Apoptosis (2016) 21:642–56. doi: 10.1007/s10495-016-1233-6
101. Sun J, Song FH, Wu JY, Zhang LQ, Li DY, Gao SJ, et al. Sestrin2 overexpression attenuates osteoarthritis pain via induction of AMPK/PGC-1α-mediated mitochondrial biogenesis and suppression of neuroinflammation. Brain Behav Immun (2022) 102:53–70. doi: 10.1016/j.bbi.2022.02.015
102. Fleisher TA. Apoptosis. Ann Allergy Asthma Immunol (1997) 78:245–9. doi: 10.1016/s1081-1206(10)63176-6
103. Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta (2016) 1863:2977–92. doi: 10.1016/j.bbamcr.2016.09.012
104. You S, Zheng J, Chen Y, Huang H. Research progress on the mechanism of beta-cell apoptosis in type 2 diabetes mellitus. Front Endocrinol (Lausanne) (2022) 13:976465. doi: 10.3389/fendo.2022.976465
105. Simó R, Simó-Servat O, Bogdanov P, Hernández C. Diabetic retinopathy: role of neurodegeneration and therapeutic perspectives. Asia Pac J Ophthalmol (Phila) (2022) 11:160–7. doi: 10.1097/apo.0000000000000510
106. Wei J, Zhao Y, Liang H, Du W, Wang L. Preliminary evidence for the presence of multiple forms of cell death in diabetes cardiomyopathy. Acta Pharm Sin B (2022) 12:1–17. doi: 10.1016/j.apsb.2021.08.026
107. Chen Y, Huang T, Yu Z, Yu Q, Wang Y, Hu J, et al. The functions and roles of sestrins in regulating human diseases. Cell Mol Biol Lett (2022) 27:2. doi: 10.1186/s11658-021-00302-8
108. Seo K, Seo S, Ki SH, Shin SM. Sestrin2 inhibits hypoxia-inducible factor-1α accumulation via AMPK-mediated prolyl hydroxylase regulation. Free Radic Biol Med (2016) 101:511–23. doi: 10.1016/j.freeradbiomed.2016.11.014
109. Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene (2002) 21:6017–31. doi: 10.1038/sj.onc.1205877
110. Qu J, Luo M, Zhang J, Han F, Hou N, Pan R, et al. A paradoxical role for sestrin 2 protein in tumor suppression and tumorigenesis. Cancer Cell Int (2021) 21:606. doi: 10.1186/s12935-021-02317-9
111. Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab (2012) 16:311–21. doi: 10.1016/j.cmet.2012.08.004
112. Chai D, Wang G, Zhou Z, Yang H, Yu Z. Insulin increases sestrin 2 content by reducing its degradation through the PI 3 K/mTOR signaling pathway. Int J Endocrinol (2015) 2015:505849. doi: 10.1155/2015/505849
113. Liu X, Niu Y, Yuan H, Huang J, Fu L. AMPK binds to Sestrins and mediates the effect of exercise to increase insulin-sensitivity through autophagy. Metabolism (2015) 64:658–65. doi: 10.1016/j.metabol.2015.01.015
114. Yamani L, Li B, Larose L. Nck1 deficiency improves pancreatic β cell survival to diabetes-relevant stresses by modulating PERK activation and signaling. Cell Signal (2015) 27:2555–67. doi: 10.1016/j.cellsig.2015.09.016
115. Ro SH, Nam M, Jang I, Park HW, Park H, Semple IA, et al. Sestrin2 inhibits uncoupling protein 1 expression through suppressing reactive oxygen species. Proc Natl Acad Sci U S A (2014) 111:7849–54. doi: 10.1073/pnas.1401787111
116. Sundararajan S, Jayachandran I, Balasubramanyam M, Mohan V, Venkatesan B, Manickam N. Sestrin2 regulates monocyte activation through AMPK-mTOR nexus under high-glucose and dyslipidemic conditions. J Cell Biochem (2018) 120:8201–13. doi: 10.1002/jcb.28102
117. Mohany KM, Al Rugaie O, Al-Wutayd O, Al-Nafeesah A. Investigation of the levels of circulating miR-29a, miR-122, sestrin 2 and inflammatory markers in obese children with/without type 2 diabetes: a case control study. BMC Endocr Disord (2021) 21:152. doi: 10.1186/s12902-021-00829-z
118. Sundararajan S, Jayachandran I, Subramanian SC, Anjana RM, Balasubramanyam M, Mohan V, et al. Decreased Sestrin levels in patients with type 2 diabetes and dyslipidemia and their association with the severity of atherogenic index. J Endocrinol Invest (2021) 44:1395–405. doi: 10.1007/s40618-020-01429-9
119. Golpour P, Nourbakhsh M, Mazaherioun M, Janani L, Nourbakhsh M, Yaghmaei P. Improvement of NRF2 gene expression and antioxidant status in patients with type 2 diabetes mellitus after supplementation with omega-3 polyunsaturated fatty acids: A double-blind randomised placebo-controlled clinical trial. Diabetes Res Clin Pract (2020) 162:108120. doi: 10.1016/j.diabres.2020.108120
120. Chung HS, Hwang HJ, Hwang SY, Kim NH, Seo JA, Kim SG, et al. Association of serum Sestrin2 level with metabolic risk factors in newly diagnosed drug-naïve type 2 diabetes. Diabetes Res Clin Pract (2018) 144:34–41. doi: 10.1016/j.diabres.2018.07.024
121. Ashimawy HM AA. Association of serum Sestrin-2 level with insulin resistance, metabolic syndrome, and diabetic nephropathy in patients with type 2 diabetes. Egyptian J Internal Med (2019) 31:107–14. doi: 10.4103/ejim.ejim_85_18
122. Lin Q, Ma Y, Chen Z, Hu J, Chen C, Fan Y, et al. Sestrin−2 regulates podocyte mitochondrial dysfunction and apoptosis under high−glucose conditions via AMPK. Int J Mol Med (2020) 45:1361–72. doi: 10.3892/ijmm.2020.4508
123. Mohany KM, Rugaie O. Association of serum sestrin 2 and betatrophin with serum neutrophil gelatinase associated lipocalin levels in type 2 diabetic patients with diabetic nephropathy. J Diabetes Metab Disord (2020) 19:249–56. doi: 10.1007/s40200-020-00498-0
124. Jia Y, Zheng Z, Yang Y, Zou M, Li J, Wang L, et al. MiR-4756 promotes albumin-induced renal tubular epithelial cell epithelial-to-mesenchymal transition and endoplasmic reticulum stress via targeting Sestrin2. J Cell Physiol (2019) 234:2905–15. doi: 10.1002/jcp.27107
125. Bian Y, Shi C, Song S, Mu L, Wu M, Qiu D, et al. Sestrin2 attenuates renal damage by regulating Hippo pathway in diabetic nephropathy. Cell Tissue Res (2022) 390:93–112. doi: 10.1007/s00441-022-03668-z
126. Tian X, Gao Y, Zhong M, Kong M, Zhao L, Feng Z, et al. The association between serum Sestrin2 and the risk of coronary heart disease in patients with type 2 diabetes mellitus. BMC Cardiovasc Disord (2022) 22:281. doi: 10.1186/s12872-022-02727-1
127. Zhang X, Deng X, Ye H, Chen Z, Li W. Inhibition of Sestrin2 overexpression in diabetic cardiomyopathy ameliorates cardiac injury via restoration of mitochondrial function. Exp Ther Med (2022) 23:265. doi: 10.3892/etm.2022.11191
128. Mao EW, Cheng XB, Li WC, Kan CX, Huang N, Wang HS, et al. Association between serum Sestrin2 level and diabetic peripheral neuropathy in type 2 diabetic patients. World J Clin Cases (2021) 9:11156–64. doi: 10.12998/wjcc.v9.i36.11156
129. Eid AA, Lee DY, Roman LJ, Khazim K, Gorin Y. Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol Cell Biol (2013) 33:3439–60. doi: 10.1128/mcb.00217-13
130. Howell JJ, Ricoult SJ, Ben-Sahra I, Manning BD. A growing role for mTOR in promoting anabolic metabolism. Biochem Soc Trans (2013) 41:906–12. doi: 10.1042/bst20130041
131. Kimball SR, Gordon BS, Moyer JE, Dennis MD, Jefferson LS. Leucine induced dephosphorylation of Sestrin2 promotes mTORC1 activation. Cell Signal (2016) 28:896–906. doi: 10.1016/j.cellsig.2016.03.008
132. Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell (2008) 134:451–60. doi: 10.1016/j.cell.2008.06.028
133. Cianci R, Franza L, Massaro MG, Borriello R, Tota A, Pallozzi M, et al. The crosstalk between gut microbiota, intestinal immunological niche and visceral adipose tissue as a new model for the pathogenesis of metabolic and inflammatory diseases: the paradigm of type 2 diabetes mellitus. Curr Med Chem (2022) 29:3189–201. doi: 10.2174/0929867329666220105121124
134. Biondi G, Marrano N, Borrelli A, Rella M, Palma G, Calderoni I, et al. Adipose tissue secretion pattern influences β-cell wellness in the transition from obesity to type 2 diabetes. Int J Mol Sci (2022) 23:5522. doi: 10.3390/ijms23105522
135. Ro SH, Semple I, Ho A, Park HW, Lee JH. Sestrin2, a regulator of thermogenesis and mitohormesis in brown adipose tissue. Front Endocrinol (Lausanne) (2015) 6:114. doi: 10.3389/fendo.2015.00114
136. Kanter JE, Hsu CC, Bornfeldt KE. Monocytes and macrophages as protagonists in vascular complications of diabetes. Front Cardiovasc Med (2020) 7:10. doi: 10.3389/fcvm.2020.00010
137. Demir S, Nawroth PP, Herzig S, Üstünel B. Emerging targets in type 2 diabetes and diabetic complications. Adv Sci (Weinh) (2021) 8:e2100275. doi: 10.1002/advs.202100275
138. Sagoo MK, Gnudi L. Diabetic nephropathy: an overview. Methods Mol Biol (2020) 2067:3–7. doi: 10.1007/978-1-4939-9841-8_1
139. Puelles VG, van der Wolde JW, Wanner N, Scheppach MW, Cullen-McEwen LA, Bork T, et al. mTOR-mediated podocyte hypertrophy regulates glomerular integrity in mice and humans. JCI Insight (2019) 4:e99271. doi: 10.1172/jci.insight.99271
140. Gajjala PR, Sanati M, Jankowski J. Cellular and molecular mechanisms of chronic kidney disease with diabetes mellitus and cardiovascular diseases as its comorbidities. Front Immunol (2015) 6:340. doi: 10.3389/fimmu.2015.00340
141. Sedeek M, Gutsol A, Montezano AC, Burger D, Nguyen Dinh Cat A, Kennedy CR, et al. Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of Type 2 diabetes. Clin Sci (Lond) (2013) 124:191–202. doi: 10.1042/cs20120330
142. Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest (2011) 121:2197–209. doi: 10.1172/jci44774
143. Kogot-Levin A, Hinden L, Riahi Y, Israeli T, Tirosh B, Cerasi E, et al. Proximal tubule mTORC1 is a central player in the pathophysiology of diabetic nephropathy and its correction by SGLT2 inhibitors. Cell Rep (2020) 32:107954. doi: 10.1016/j.celrep.2020.107954
144. Skrabic R, Kumric M, Vrdoljak J, Rusic D, Skrabic I, Vilovic M, et al. SGLT2 inhibitors in chronic kidney disease: from mechanisms to clinical practice. Biomedicines (2022) 10:2458. doi: 10.3390/biomedicines10102458
145. Sun X, Han F, Lu Q, Li X, Ren D, Zhang J, et al. Empagliflozin ameliorates obesity-related cardiac dysfunction by regulating sestrin2-mediated AMPK-mTOR signaling and redox homeostasis in high-fat diet-induced obese mice. Diabetes (2020) 69:1292–305. doi: 10.2337/db19-0991
146. Guo Y, Ran Z, Zhang Y, Song Z, Wang L, Yao L, et al. Marein ameliorates diabetic nephropathy by inhibiting renal sodium glucose transporter 2 and activating the AMPK signaling pathway in db/db mice and high glucose-treated HK-2 cells. BioMed Pharmacother (2020) 131:110684. doi: 10.1016/j.biopha.2020.110684
147. Ala M, Khoshdel MRF, Dehpour AR. Empagliflozin enhances autophagy, mitochondrial biogenesis, and antioxidant defense and ameliorates renal ischemia/reperfusion in nondiabetic rats. Oxid Med Cell Longev (2022) 2022:1197061. doi: 10.1155/2022/1197061
148. Ma TT, Meng XM. TGF-β/smad and renal fibrosis. Adv Exp Med Biol (2019) 1165:347–64. doi: 10.1007/978-981-13-8871-2_16
149. Chang AS, Hathaway CK, Smithies O, Kakoki M. Transforming growth factor-β1 and diabetic nephropathy. Am J Physiol Renal Physiol (2016) 310:F689–f696. doi: 10.1152/ajprenal.00502.2015
150. Zhao L, Zou Y, Liu F. Transforming growth factor-beta1 in diabetic kidney disease. Front Cell Dev Biol (2020) 8:187. doi: 10.3389/fcell.2020.00187
151. Lu Q, Wang WW, Zhang MZ, Ma ZX, Qiu XR, Shen M, et al. ROS induces epithelial-mesenchymal transition via the TGF-β1/PI3K/Akt/mTOR pathway in diabetic nephropathy. Exp Ther Med (2019) 17:835–46. doi: 10.3892/etm.2018.7014
152. Lv W, Booz GW, Wang Y, Fan F, Roman RJ. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur J Pharmacol (2018) 820:65–76. doi: 10.1016/j.ejphar.2017.12.016
153. Gutierrez LS, Gutierrez J. Thrombospondin 1 in metabolic diseases. Front Endocrinol (Lausanne) (2021) 12:638536. doi: 10.3389/fendo.2021.638536
154. Lu A, Miao M, Schoeb TR, Agarwal A, Murphy-Ullrich JE. Blockade of TSP1-dependent TGF-β activity reduces renal injury and proteinuria in a murine model of diabetic nephropathy. Am J Pathol (2011) 178:2573–86. doi: 10.1016/j.ajpath.2011.02.039
155. Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab (2011) 14:67–79. doi: 10.1016/j.cmet.2011.04.013
156. Maimaitiyiming H, Clemons K, Zhou Q, Norman H, Wang S. Thrombospondin1 deficiency attenuates obesity-associated microvascular complications in ApoE-/- mice. PloS One (2015) 10:e0121403. doi: 10.1371/journal.pone.0121403
157. Lei D, Chengcheng L, Xuan Q, Yibing C, Lei W, Hao Y, et al. Quercetin inhibited mesangial cell proliferation of early diabetic nephropathy through the Hippo pathway. Pharmacol Res (2019) 146:104320. doi: 10.1016/j.phrs.2019.104320
158. Qian X, He L, Hao M, Li Y, Li X, Liu Y, et al. YAP mediates the interaction between the Hippo and PI3K/Akt pathways in mesangial cell proliferation in diabetic nephropathy. Acta Diabetol (2021) 58:47–62. doi: 10.1007/s00592-020-01582-w
159. Rawal S, Manning P, Katare R. Cardiovascular microRNAs: as modulators and diagnostic biomarkers of diabetic heart disease. Cardiovasc Diabetol (2014) 13:44. doi: 10.1186/1475-2840-13-44
160. Rajbhandari J, Fernandez CJ, Agarwal M, Yeap BXY, Pappachan JM. Diabetic heart disease: A clinical update. World J Diabetes (2021) 12:383–406. doi: 10.4239/wjd.v12.i4.383
161. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res (2020) 126:1501–25. doi: 10.1161/circresaha.120.315913
162. Davogustto GE, Salazar RL, Vasquez HG, Karlstaedt A, Dillon WP, Guthrie PH, et al. Metabolic remodeling precedes mTORC1-mediated cardiac hypertrophy. J Mol Cell Cardiol (2021) 158:115–27. doi: 10.1016/j.yjmcc.2021.05.016
163. Proud CG. Ras, PI3-kinase and mTOR signaling in cardiac hypertrophy. Cardiovasc Res (2004) 63:403–13. doi: 10.1016/j.cardiores.2004.02.003
164. Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy (2015) 11:1146–60. doi: 10.1080/15548627.2015.1051295
165. Kandemir YB, Tosun V, Güntekin Ü. Melatonin protects against streptozotocin-induced diabetic cardiomyopathy through the mammalian target of rapamycin (mTOR) signaling pathway. Adv Clin Exp Med (2019) 28:1171–7. doi: 10.17219/acem/103799
166. Frangogiannis N. Transforming growth factor-β in tissue fibrosis. J Exp Med (2020) 217:e20190103. doi: 10.1084/jem.20190103
167. Peng ML, Fu Y, Wu CW, Zhang Y, Ren H, Zhou SS. Signaling pathways related to oxidative stress in diabetic cardiomyopathy. Front Endocrinol (Lausanne) (2022) 13:907757. doi: 10.3389/fendo.2022.907757
168. Hachana S, Larrivée B. TGF-β Superfamily signaling in the eye: implications for ocular pathologies. Cells (2022) 11:2336. doi: 10.3390/cells11152336
169. Rajesh M, Mukhopadhyay P, Bátkai S, Patel V, Saito K, Matsumoto S, et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J Am Coll Cardiol (2010) 56:2115–25. doi: 10.1016/j.jacc.2010.07.033
170. Shen N, Li X, Zhou T, Bilal MU, Du N, Hu Y, et al. Shensong Yangxin Capsule prevents diabetic myocardial fibrosis by inhibiting TGF-β1/Smad signaling. J Ethnopharmacol (2014) 157:161–70. doi: 10.1016/j.jep.2014.09.035
171. Li C, Zhang J, Xue M, Li X, Han F, Liu X, et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol (2019) 18:15. doi: 10.1186/s12933-019-0816-2
172. Tian J, Zhang M, Suo M, Liu D, Wang X, Liu M, et al. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J Cell Mol Med (2021) 25:7642–59. doi: 10.1111/jcmm.16601
173. Wu W, Liu W, Kong D. Telmisartan alleviates inflammatory response and myocardial apoptosis in diabetic cardiomyopathy rats through TGF-β1/Smad signaling pathway. Minerva Med (2021) 112:411–2. doi: 10.23736/s0026-4806.19.06201-3
174. Gui L, Wang F, Hu X, Liu X, Yang H, Cai Z, et al. Epigallocatechin gallate protects diabetes mellitus rats complicated with cardiomyopathy through TGF-β1/JNK signaling pathway. Curr Pharm Des (2022) 28:2758–70. doi: 10.2174/1381612828666220902115437
175. Gonzalez-Quesada C, Cavalera M, Biernacka A, Kong P, Lee DW, Saxena A, et al. Thrombospondin-1 induction in the diabetic myocardium stabilizes the cardiac matrix in addition to promoting vascular rarefaction through angiopoietin-2 upregulation. Circ Res (2013) 113:1331–44. doi: 10.1161/circresaha.113.302593
176. Kong P, Cavalera M, Frangogiannis NG. The role of thrombospondin (TSP)-1 in obesity and diabetes. Adipocyte (2014) 3:81–4. doi: 10.4161/adip.26990
177. Sun H, Zhao Y, Bi X, Li S, Su G, Miao Y, et al. Valsartan blocks thrombospondin/transforming growth factor/Smads to inhibit aortic remodeling in diabetic rats. Diagn Pathol (2015) 10:18. doi: 10.1186/s13000-015-0246-8
178. Yu J, Fei J, Azad J, Gong M, Lan Y, Chen G. Myocardial protection by Salvia miltiorrhiza Injection in streptozotocin-induced diabetic rats through attenuation of expression of thrombospondin-1 and transforming growth factor-β1. J Int Med Res (2012) 40:1016–24. doi: 10.1177/147323001204000320
179. Zhang J, Yao L, Li S, Ferdous M, Zhao P. ER stress induces myocardial dysfunction and cardiac autophagy in Sestrin2 knockout mice. Am J Transl Res (2022) 14:5800–11.
180. Zhang N, Liao HH, Feng H, Mou SQ, Li WJ, Aiyasiding X, et al. Knockout of AMPKα2 blocked the protection of sestrin2 overexpression against cardiac hypertrophy induced by pressure overload. Front Pharmacol (2021) 12:716884. doi: 10.3389/fphar.2021.716884
181. Ren D, Quan N, Fedorova J, Zhang J, He Z, Li J. Sestrin2 modulates cardiac inflammatory response through maintaining redox homeostasis during ischemia and reperfusion. Redox Biol (2020) 34:101556. doi: 10.1016/j.redox.2020.101556
182. Fatima MT, Hasan M, Abdelsalam SS, Sivaraman SK, El-Gamal H, Zahid MA, et al. Sestrin2 suppression aggravates oxidative stress and apoptosis in endothelial cells subjected to pharmacologically induced endoplasmic reticulum stress. Eur J Pharmacol (2021) 907:174247. doi: 10.1016/j.ejphar.2021.174247
183. Wang H, Li N, Shao X, Li J, Guo L, Yu X, et al. Increased plasma sestrin2 concentrations in patients with chronic heart failure and predicted the occurrence of major adverse cardiac events: A 36-month follow-up cohort study. Clin Chim Acta (2019) 495:338–44. doi: 10.1016/j.cca.2019.04.084
184. Kishimoto Y, Aoyama M, Saita E, Ikegami Y, Ohmori R, Kondo K, et al. Association between plasma sestrin2 levels and the presence and severity of coronary artery disease. Dis Markers (2020) 2020:7439574. doi: 10.1155/2020/7439574
185. Huang R, Chen F, Zeng A, Ke J, Lin S. Serum sestrin2 was lower in septic shock patients with cardiomyopathy. Dis Markers (2022) 2022:1390373. doi: 10.1155/2022/1390373
186. Ye J, Wang M, Xu Y, Liu J, Jiang H, Wang Z, et al. Sestrins increase in patients with coronary artery disease and associate with the severity of coronary stenosis. Clin Chim Acta (2017) 472:51–7. doi: 10.1016/j.cca.2017.07.020
187. Yang F, Qin Y, Wang Y, Meng S, Xian H, Che H, et al. Metformin inhibits the NLRP3 inflammasome via AMPK/mTOR-dependent effects in diabetic cardiomyopathy. Int J Biol Sci (2019) 15:1010–9. doi: 10.7150/ijbs.29680
188. Iglesias M, Wang H, Krause-Hauch M, Ren D, Zoungrana LI, Li Z, et al. Sestrin2 mediates metformin rescued the age-related cardiac dysfunctions of cardiorenal syndrome type 3. Cells (2023) 12:845. doi: 10.3390/cells12060845
189. Sayin N, Kara N, Pekel G. Ocular complications of diabetes mellitus. World J Diabetes (2015) 6:92–108. doi: 10.4239/wjd.v6.i1.92
190. Heng LZ, Comyn O, Peto T, Tadros C, Ng E, Sivaprasad S, et al. Diabetic retinopathy: pathogenesis, clinical grading, management and future developments. Diabetes Med (2013) 30:640–50. doi: 10.1111/dme.12089
191. Wong TY, Cheung CM, Larsen M, Sharma S, Simó R. Diabetic retinopathy. Nat Rev Dis Primers (2016) 2:16012. doi: 10.1038/nrdp.2016.12
192. Saika S, Yamanaka O, Okada Y, Tanaka S, Miyamoto T, Sumioka T, et al. TGF beta in fibroproliferative diseases in the eye. Front Biosci (Schol Ed) (2009) 1:376–90. doi: 10.2741/s32
193. Wei J, Jiang H, Gao H, Wang G. Blocking mammalian target of rapamycin (mTOR) attenuates HIF-1α Pathways engaged-vascular endothelial growth factor (VEGF) in diabetic retinopathy. Cell Physiol Biochem (2016) 40:1570–7. doi: 10.1159/000453207
194. Lopes de Faria JM, Duarte DA, Montemurro C, Papadimitriou A, Consonni SR, Lopes de Faria JB. Defective autophagy in diabetic retinopathy. Invest Ophthalmol Vis Sci (2016) 57:4356–66. doi: 10.1167/iovs.16-19197
195. Zeng J, Zhao H, Chen B. DJ-1/PARK7 inhibits high glucose-induced oxidative stress to prevent retinal pericyte apoptosis via the PI3K/AKT/mTOR signaling pathway. Exp Eye Res (2019) 189:107830. doi: 10.1016/j.exer.2019.107830
196. Chen H, Ji Y, Yan X, Su G, Chen L, Xiao J. Berberine attenuates apoptosis in rat retinal Müller cells stimulated with high glucose via enhancing autophagy and the AMPK/mTOR signaling. BioMed Pharmacother (2018) 108:1201–7. doi: 10.1016/j.biopha.2018.09.140
197. Chen HY, Ho YJ, Chou HC, Liao EC, Tsai YT, Wei YS, et al. The role of transforming growth factor-beta in retinal ganglion cells with hyperglycemia and oxidative stress. Int J Mol Sci (2020) 21:6482. doi: 10.3390/ijms21186482
198. Dagher Z, Gerhardinger C, Vaz J, Goodridge M, Tecilazich F, Lorenzi M. The increased transforming growth factor-β Signaling induced by diabetes protects retinal vessels. Am J Pathol (2017) 187:627–38. doi: 10.1016/j.ajpath.2016.11.007
199. Hanus J, Zhang H, Chen DH, Zhou Q, Jin P, Liu Q, et al. Gossypol acetic acid prevents oxidative stress-induced retinal pigment epithelial necrosis by regulating the foxO3/sestrin2 pathway. Mol Cell Biol (2015) 35:1952–63. doi: 10.1128/mcb.00178-15
200. Fan Y, Xing Y, Xiong L, Wang J. Sestrin2 overexpression alleviates hydrogen peroxide-induced apoptosis and oxidative stress in retinal ganglion cells by enhancing Nrf2 activation via Keap1 downregulation. Chem Biol Interact (2020) 324:109086. doi: 10.1016/j.cbi.2020.109086
201. Lee JS, Park HW, Cho KJ, Lyu J. Sestrin2 inhibits YAP activation and negatively regulates corneal epithelial cell proliferation. Exp Mol Med (2020) 52:951–62. doi: 10.1038/s12276-020-0446-5
202. Sivakumar PM, Prabhakar PK, Cetinel S,RN, Prabhawathi V. Molecular insights on the therapeutic effect of selected flavonoids on diabetic neuropathy. Mini Rev Med Chem (2022) 22:1828–46. doi: 10.2174/1389557522666220309140855
203. Méndez-Morales ST, Pérez-De Marcos JC, Rodríguez-Cortés O, Flores-Mejía R, Martínez-Venegas M, Sánchez-Vera Y, et al. Diabetic neuropathy: Molecular approach a treatment opportunity. Vascul Pharmacol (2022) 143:106954. doi: 10.1016/j.vph.2022.106954
204. Sanaye MM, Kavishwar SA. Diabetic neuropathy: review on molecular mechanisms. Curr Mol Med (2023) 23:97–110. doi: 10.2174/1566524021666210816093111
205. Dewanjee S, Das S, Das AK, Bhattacharjee N, Dihingia A, Dua TK, et al. Molecular mechanism of diabetic neuropathy and its pharmacotherapeutic targets. Eur J Pharmacol (2018) 833:472–523. doi: 10.1016/j.ejphar.2018.06.034
206. Wu LY, Li M, Qu ML, Li X, Pi LH, Chen Z, et al. High glucose up-regulates Semaphorin 3A expression via the mTOR signaling pathway in keratinocytes: A potential mechanism and therapeutic target for diabetic small fiber neuropathy. Mol Cell Endocrinol (2018) 472:107–16. doi: 10.1016/j.mce.2017.11.025
207. He WY, Zhang B, Zhao WC, He J, Wang Y, Zhang L, et al. mTOR activation due to APPL1 deficiency exacerbates hyperalgesia via Rab5/Akt and AMPK signaling pathway in streptozocin-induced diabetic rats. Mol Pain (2019) 15:1744806919880643. doi: 10.1177/1744806919880643
208. Liu K, Yang Y, Zhou F, Xiao Y, Shi L. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy and relieves hyperalgesia in diabetic rats. Neuroreport (2020) 31:644–9. doi: 10.1097/wnr.0000000000001461
209. Yang X, Xue P, Liu Z, Li W, Li C, Chen Z. SESN2 prevents the slow-to-fast myofiber shift in denervated atrophy via AMPK/PGC-1α pathway. Cell Mol Biol Lett (2022) 27:66. doi: 10.1186/s11658-022-00367-z
210. Yang X, Xue P, Yuan M, Xu X, Wang C, Li W, et al. SESN2 protects against denervated muscle atrophy through unfolded protein response and mitophagy. Cell Death Dis (2021) 12:805. doi: 10.1038/s41419-021-04094-9
211. Chen SD, Yang JL, Hsieh YH, Lin TK, Lin YC, Chao AC, et al. Potential roles of sestrin2 in alzheimer's disease: antioxidation, autophagy promotion, and beyond. Biomedicines (2021) 9:1308. doi: 10.3390/biomedicines9101308
212. Qu N, Qu J, Huang N, Zhang K, Ye T, Shi J, et al. Calycosin induces autophagy and apoptosis via Sestrin2/AMPK/mTOR in human papillary thyroid cancer cells. Front Pharmacol (2022) 13:1056687. doi: 10.3389/fphar.2022.1056687
213. Sousa JN, Queiroz L, de Paula AMB, Guimarães ALS, Lescano CH, Aguilar CM, et al. Gallic acid as a Sestrin (SESN2) activator and potential obesity therapeutic agent: A molecular docking study. Gene (2023) 883:147683. doi: 10.1016/j.gene.2023.147683
214. Ma Y, Zhang G, Kuang Z, Xu Q, Ye T, Li X, et al. Empagliflozin activates Sestrin2-mediated AMPK/mTOR pathway and ameliorates lipid accumulation in obesity-related nonalcoholic fatty liver disease. Front Pharmacol (2022) 13:944886. doi: 10.3389/fphar.2022.944886
215. Han X, Ding C, Zhang G, Pan R, Liu Y, Huang N, et al. Liraglutide ameliorates obesity-related nonalcoholic fatty liver disease by regulating Sestrin2-mediated Nrf2/HO-1 pathway. Biochem Biophys Res Commun (2020) 525:895–901. doi: 10.1016/j.bbrc.2020.03.032
Keywords: diabetes, diabetic complications, Sestrin2, oxidative stress, mitochondrial function
Citation: Zhang X, Luo Z, Li J, Lin Y, Li Y and Li W (2023) Sestrin2 in diabetes and diabetic complications. Front. Endocrinol. 14:1274686. doi: 10.3389/fendo.2023.1274686
Received: 08 August 2023; Accepted: 03 October 2023;
Published: 18 October 2023.
Edited by:
Jian Ma, Harbin Medical University, ChinaReviewed by:
Yan Yao, Columbia University, United StatesYiqiong Liu, Harvard Medical School, United States
Copyright © 2023 Zhang, Luo, Li, Lin, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wangen Li, bGl3ZzY2MEAxMjYuY29t