 Sarah S. Malik
Sarah S. Malik Diksha Padmanabhan
Diksha Padmanabhan Rebecca L. Hull-Meichle
Rebecca L. Hull-Meichle- 1Department of Pharmacology, University of Washington, Seattle, WA, United States
- 2Research Service, Veterans Affairs Puget Sound Health Care System, Seattle, WA, United States
- 3Seattle Institute for Biomedical and Clinical Research, Seattle, WA, United States
- 4Division of Metabolism, Endocrinology, and Nutrition, Department of Medicine, University of Washington, Seattle, WA, United States
Cystic fibrosis (CF) is a multi-organ disease caused by loss-of-function mutations in CFTR (which encodes the CF transmembrane conductance regulator ion channel). Cystic fibrosis related diabetes (CFRD) occurs in 40-50% of adults with CF and is associated with significantly increased morbidity and mortality. CFRD arises from insufficient insulin release from β cells in the pancreatic islet, but the mechanisms underlying the loss of β cell function remain understudied. Widespread pathological changes in the CF pancreas provide clues to these mechanisms. The exocrine pancreas is the epicenter of pancreas pathology in CF, with ductal pathology being the initiating event. Loss of CFTR function results in ductal plugging and subsequent obliteration. This in turn leads to destruction of acinar cells, fibrosis and fatty replacement. Despite this adverse environment, islets remain relatively well preserved. However, islet composition and arrangement are abnormal, including a modest decrease in β cells and an increase in α, δ and γ cell abundance. The small amount of available data suggest that substantial loss of pancreatic/islet microvasculature, autonomic nerve fibers and intra-islet macrophages occur. Conversely, T-cell infiltration is increased and, in CFRD, islet amyloid deposition is a frequent occurrence. Together, these pathological changes clearly demonstrate that CF is a disease of the pancreas/islet microenvironment. Any or all of these changes are likely to have a dramatic effect on the β cell, which relies on positive signals from all of these neighboring cell types for its normal function and survival. A thorough characterization of the CF pancreas microenvironment is needed to develop better therapies to treat, and ultimately prevent CFRD.
1 Introduction
Cystic fibrosis (CF) is the most prevalent life-limiting autosomal recessive disease affecting populations of Northern European descent (1), although it is important to recognize that CF affects all racial/ethnic groups (2, 3). Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) leads to multi-organ disease, with progressive lung disease representing the leading cause of premature death (1, 4). With substantial advances in therapies, life expectancy of people with CF in the United States has progressively increased, and is now ~45 years (3). With this increase in survival, however, other features of CF have emerged, including CF-related diabetes (CFRD). CFRD affects 20% of adolescents and ~40-50% of adults with CF (3, 5) and, importantly, is an important risk factor for worsened CF lung disease. CFRD results in a 4-6 fold greater mortality rate compared to people with CF who do not have diabetes (6–9). The inadequate release of insulin from the islet β cell underlies the development of CFRD (10–14), manifest as a characteristic blunted and delayed insulin response to nutrient stimulation. One potential contributor is an impairment in the incretin axis (i.e. action of the hormones glucagon like peptide 1 and gastric inhibitory polypeptide to enhance insulin release in response to glucose) (15, 16). However, much remains unknown about the etiology of insulin deficiency in CF, limiting the development of improved treatments for CFRD. The focus of this article is the islet/pancreas microenvironment whose constituent cell types, under normal conditions, provide critical signals to maintain β cell function, identity and survival (Figure 1A). In CF, profound and widespread changes occur in this microenvironment (Figure 1B), providing clues to novel mechanisms that may underlie loss of insulin release in this disease (Figure 1C).
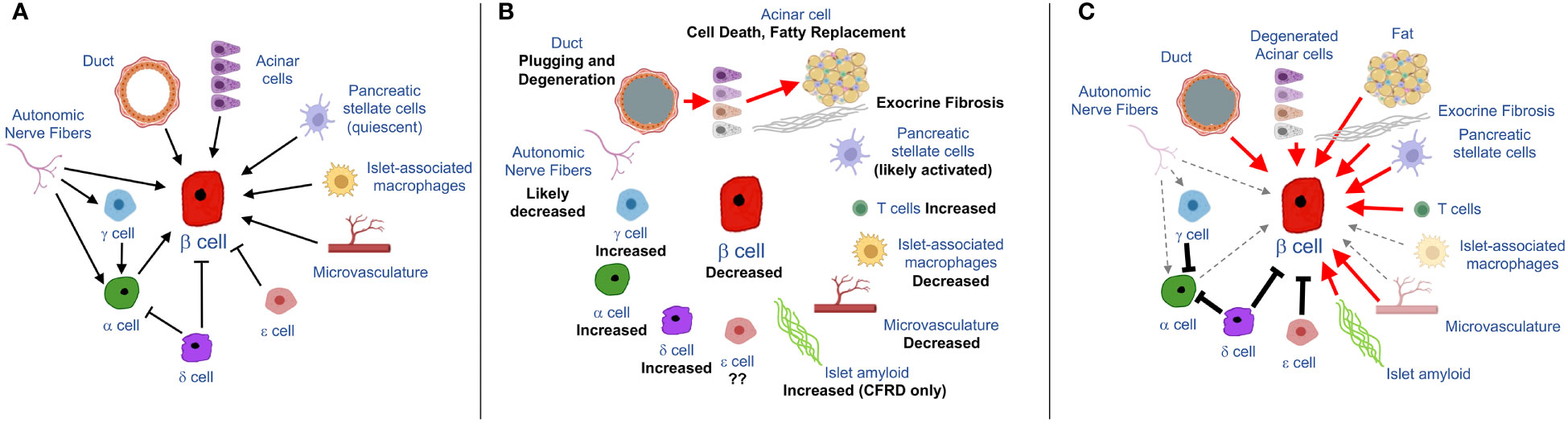
Figure 1 Schematics showing input from cells of the pancreas/islet to the β cell under normal (A) and CF (B, C) conditions. (A) Summary of physiological/homeostatic input to β cell from exocrine-, endocrine or islet-associated cell types. (B) Pathological features of the exocrine pancreas in CF, emanating from ductal pathology, as indicated by red arrows, along with changes in abundance of islet endocrine cells and support cells (nerve, vascular and immune cells). (C) hypothesized implications for β cell function and/or survival based on CF pancreatic pathology. Red arrows denote detrimental (chiefly inflammatory) signals derived from diseased cell types. Dashed grey arrows denote decreased input to the β cell due to loss of nerve fibers, macrophages or vasculature. Solid black lines denote increased inhibitory input to β and α cells (due to increased abundance of δ, γ and potentially also ϵ cells) some elements in the figure were made using BioRender.
2 Pathology and dysfunction of the exocrine and endocrine pancreas in CF
2.1 Exocrine pancreas pathology and dysfunction in CF
Within the pancreas, CFTR is predominantly expressed, at very high levels, in pancreatic ductal epithelial cells (PDECs) (17). There, its regulation of Cl- and HCO3- transport is critical for maintaining luminal pH and water balance and maintaining acinar-derived digestive enzymes in a dilute, inactive form (18, 19). Loss of CFTR function results in increased protein concentration and subsequent plugging of the ductal lumen (18). This results in duct obliteration, which in turn leads to destruction of acinar cells and fatty replacement of the exocrine pancreas. This pathology is strikingly similar to that seen in other diseases of the exocrine pancreas, including chronic pancreatitis (20). Fibrosis is also a common feature of CF pancreas disease. Its etiology remains understudied, but likely involves activation of pancreatic stellate cells (PSCs), as has been demonstrated in other exocrine pancreas diseases (21). Under normal conditions, PSCs contribute to normal tissue structure and homeostasis (21). In disease, however, activation of PSCs results in their proliferation and trans-differentiation to myofibroblasts, leading to synthesis and deposition of a fibrotic, proinflammatory extracellular matrix (21, 22). A role for PSCs in CF pancreas fibrosis is therefore likely, but remains to be clarified.
In CF, exocrine pancreas pathology is initiated very early in life [even in utero (23)], and while pancreatic morphology appears relatively preserved in infants (<1 year of age), substantial abnormalities are seen by 5 years of age (24). Consequently, exocrine pancreas insufficiency occurs in the vast majority (~85%) of people living with CF (18). Moreover, in the minority that retain exocrine pancreatic function, there is still evidence of damage to their pancreas (18) with reports of pancreatitis occurring in individuals upon treatment with highly effective CFTR modulator therapies (HEMT) (25). Therefore, exocrine pancreas pathology is itself a major, debilitating feature of the CF disease process. Moreover, as described below in section 3, emerging evidence suggests it may also be a key contributor to the pathogenesis of CFRD.
2.2 Endocrine pancreas pathology and dysfunction
Given the extensive disruption of the exocrine pancreas in CF, it is surprising that islets remain relatively intact. However, the number and/or arrangement of endocrine cell types that make up the islet are significantly altered in CF.
2.2.1 Islet β cells
The islet β cell is the most abundant islet endocrine cell and is the source of the hormone insulin, which is required for maintenance of glucose homeostasis. Impaired insulin release is common among people living with CF (10, 11, 13, 26), and is the key contributing factor to the onset of CFRD (as it is for all forms of diabetes). Defects in processing of insulin from its precursor, proinsulin have been described in CF models (27), a further indication of β cell dysfunction.
In human autopsy pancreas specimens, decreases in β cell area in adult CF subjects without diabetes when compared to non-CF controls have been reported in some (28–31) but not all studies (24, 32); data from these studies are summarized in Table 1. β cell loss does not appear to differ based on the major form of exocrine pathology (i.e. fatty vs. fibrotic) (31) although it appears that a greater degree of β cell loss is observed in younger CF subjects (24). Similarly, most studies report a modest but not severe loss of β cell mass in CFRD (24, 32–34).
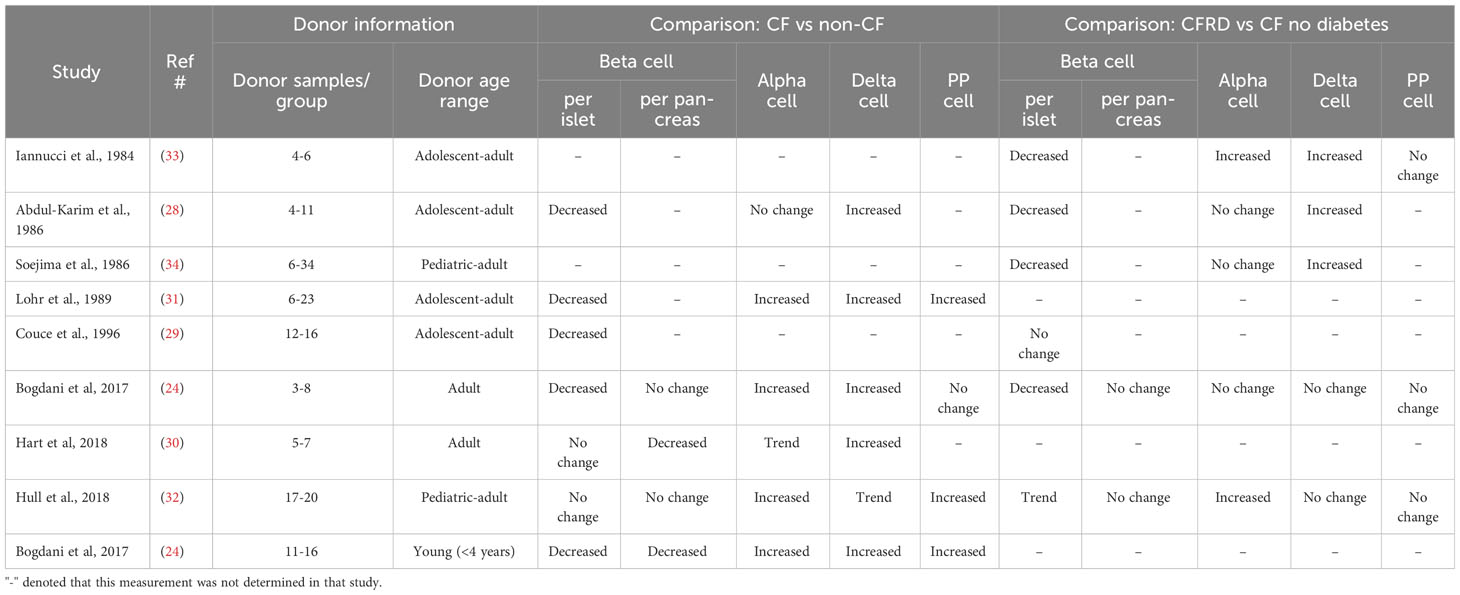
Table 1 Summary of findings from morphometric studies examining islet endocrine cell types in donors with CF (no diabetes) and/or CFRD.
The mechanisms underlying loss of β cells in CF are not well understood. β cell apoptosis may be a contributor, as has been described in one study (24). However, in T2D, it has been noted that the observed decrease in β cell mass is much greater than can be accounted for by the rate of β cell apoptosis (35). This, and other work, led to the concept that loss of β cell identity and not solely β cell death may contribute to the decreased number of functional β cells that are seen in diabetic conditions (35, 36). In β cells, markers of differentiated identity include the transcription factors, MAFA, NKX6.1, PDX1, PAX6, NKX2-2, ISL1, NEUROD1, FOXO1 and FOXA2 (37) as well as key components of the β cell secretory pathway (including GCK, SLC2A2, SLC2A1 and INS itself). In CF, islet cell de-differentiation has been suggested based on work identifying islet cells that are positive for chromogranin A but negative for islet hormones, and the presence of polyhormonal cells (38). However, which cell type(s) may lose identity markers remains unclear. Only one study to date has investigated β cell identity markers in CF. Bulk RNAseq in islets from CF vs. non-CF donors revealed no significant changes in expression of key β cell transcription factors (MAFA, NKX6.1, PDX1, PAX6, ISL1) or components of the β cell secretory pathway (INS, SLC2A2, SLC2A1, GCK, ABCC8, GLP1R) (30).
This suggests that both β cell mass and identity remain largely unperturbed in CF, despite the extensive destruction of the surrounding exocrine pancreas, and that strategies to improve their function could be a viable means to treat and even prevent CFRD.
2.2.2 Islet α cells
The islet α cell is the second most abundant islet endocrine cell type, whose main function is the secretion of glucagon, a hormone which has a range of metabolic effects, including stimulation of hepatic glucose production (39, 40). In contrast to the decrease seen in β cell abundance, α cells have been shown to be increased in autopsy pancreas specimens from donors with CF both with and without diabetes, using immunohistochemical and electron microscopy approaches (24, 30–32). However, despite the increased abundance of α cells, glucagon secretion (like insulin secretion) is impaired in CF, in response to a range of stimuli including arginine and hypoglycemia (10, 41).
A possible contributor to α cell dysfunction in CF is loss of cell identity. In islets from CF donors, a significant decrease in ARX expression (30), one of the transcription factors critical for α cell identity (37), was observed. While more research is needed, an imbalance between the abundance vs. function/identity of α cells seems to be a key feature of islet pathology in CF.
2.2.3 Islet δ, γ and ϵ cells
The population of islet δ cells is variable and much less abundant than α or β cells. δ cells secrete somatostatin and act as intra-islet paracrine negative regulators of α and β cells (42). Plasma levels of somatostatin have been reported to be increased in CF (43). However, the half-life of islet-derived somatostatin is very short and as such peripheral levels are likely not reflective of those within the islet. Therefore, the status of δ cell function in CF remains unknown. Morphologically, similar to α cells, the abundance of δ cells is significantly increased or shows an upward trend in CF islets based on multiple studies (24, 28, 30–32, 34), suggesting that local concentrations of somatostatin in the CF islet may be elevated.
γ cells make up only about 5% of islet area and are found more commonly in the head of the pancreas. These cells release pancreatic polypeptide (PP), and PP levels have been shown to be indicative of vagal input to the pancreas. In general, the abundance of γ cells, like α and δ cells, is increased in CF (24, 32), although this has not been a universal finding (33), and γ cells do not seem to be consistently increased in islets from CF donors with and without diabetes (32). Despite some inconsistencies in reports of the abundance of γ cells, there is a profound defect in PP release in CF, revealing another example of a disconnect between the abundance and function of islet endocrine cells in this disease.
ϵ cells are the fifth and final type of endocrine islet cell type in the pancreas. ϵ cells produce ghrelin, which is principally known as a gut hormone that activates the growth hormone secretagogue receptor (44). Circulating levels of ghrelin (specifically the active, acylated form) are increased in CF (45). However, a more accurate picture of islet ϵ cell function in CF required direct examination of ghrelin levels within the pancreatic islet; this has not been determined to date.
Regarding cell identity markers, less is known about the nature of these in δ, ϵ, and γ cells, although there is overlap between transcription factors expressed in ϵ and γ cells, with similarity to those expressed in α cells (37). Furthermore, HHEX has been shown to contribute to δ cell differentiation (46). Whether these identity markers are compromised in the CF islet and could thereby contribute to impaired function of δ, γ and ϵ cells remains to be determined.
2.3 Pathological changes in non-endocrine islet cells
2.3.1 Microvasculature
The pancreatic islet has an extensive microvascular capillary network which acts as a conduit for transportation of nutrients to islet endocrine cells and delivery of islet hormones to peripheral tissues (47–50). In addition, the islet microvasculature has emerged as a key source of supportive signals (e.g. growth factors, extracellular matrix components) that are necessary to maintain islet β-cell function and survival. Both of the main constituent cell types, islet endothelial cells which form the capillary wall and the constrictive pericytes which surround them, have been shown to play this positive role in modulating β cell function and survival (48, 49, 51–57).
In both T1D and T2D structural defects in the islet vasculature have been observed, including thickening and fragmentation of capillaries and an apparent increase in vascular density (58, 59). Moreover, in models of T2D, endothelial cells attain an inflamed, pro-adhesive phenotype (49, 55, 60, 61) that renders them incapable of supporting insulin release (55). In CF, changes in islet microvascular density appear to differ profoundly from what is seen in T1D and T2D. Specifically, preliminary data from our lab demonstrate a substantial decrease in capillary density in islets and exocrine pancreas (62). Moreover, islet bulk RNAseq data from human donors with CF exhibit increases in inflammatory markers, including key indicators of endothelial inflammation/activation including SELE and IL6 (30). Together, these data suggest that while there is a decrease in islet endothelial cell abundance within CF islets, those remaining endothelial cells are highly inflamed.
While some data exist regarding islet endothelial cells in CF, the other main constituent of the islet microvasculature, the pericyte, remains entirely unstudied. Pericytes surround microvascular endothelial cells and are responsible for control of blood flow (63, 64) but are also critical for microvessel stability and the prevention of vessel leakage and inflammation. Loss of pericyte attachment and/or cell death has been defined as a critical pathological event in multiple vascular diseases including tumor growth, diabetic retinopathy and kidney fibrosis, and the close interaction between endothelial cells and pericytes has been shown to be disrupted in islets in T2D and models thereof (61, 63, 65). These data suggest that loss of microvascular stability could be a common pathological feature of the diabetic islet. Whether the same is true in CF/CFRD remains an important unanswered question.
2.3.2 Innervation
It has long been appreciated that pancreatic islets are innervated by sympathetic, parasympathetic and sensory neurons (66, 67). Parasympathetic input stimulates, while sympathetic input inhibits insulin release (66, 67). Moreover, sympathetic input to the α cell is required for the glucagon response to hypoglycemia (39). Islet innervation in CF has not been well-studied, although a decrease in PGP9.5+ nerve fiber density has been reported in the CF pig pancreas (68). The profound defect in PP release in people living with CF is suggestive of a loss of vagal input to the pancreas, and the loss of islet (and exocrine) capillary density would also be expected to be reflective of impaired innervation to both pancreas compartments, given the known close association of the vasculature and autonomic nerve fibers (67). However, more data are clearly needed to delineate the state of pancreatic innervation in human CF.
2.3.3 Immune cells
While (increased) macrophages within the islet have been reported to have detrimental effects under diabetic conditions (69), it is now well established that resident intra-islet macrophages are critical for β-cell growth, regeneration following injury and function/glucose homeostasis (70–75). We previously reported that increased IL-1β positivity is a common and early feature of the islet in CF (32). Based on data from T2D, whereby increased IL-1β production is indicative of an increase in the number and/or activation state of macrophages, this observation prompted an investigation of the abundance of islet macrophages in CF. Surprisingly, we and others found an almost complete absence of intra-islet macrophages in adolescents and adults with CF (24, 32) (regardless of diabetes status), despite the continued presence of macrophages in exocrine pancreas.
Islets also contain other resident leukocyte populations, including relatively rare T-lymphocytes. Increased T cell infiltration is a hallmark of autoimmune T1D and has also been described in islets in CF pancreas sections taken from children and adults (24, 30). Flow cytometry analysis of CF donor islets leukocyte populations revealed a relatively abundant CD3+ T cell population, which largely (~60%) consisted of CD8+ T cells (30).
2.3.4 Islet amyloid
Amyloids are aggregates of misfolded proteins and have been linked to the development of numerous diseases. In the islet, amyloid deposits contain as their unique component islet amyloid polypeptide (IAPP), which is a normal secretory product of the β cell (76). Islet amyloid deposition has been recognized as a feature of islets in CF for several decades (29) and, unlike many of the other features described above is pathognomonic of CFRD, occurring in 60-70% of cases compared to only 0-20% of individuals with CF without diabetes (24, 29, 32). Moreover, islet amyloid does not appear to be a pathological feature in children with CF (24), although, deposition in CFRD appears to be markedly accelerated compared to the classically-described islet amyloid seen in subjects with T2D, where amyloid is associated with established disease in individuals around the 6th-7th decade of life (76, 77).
3 Potential mechanisms underlying CFRD
3.1 Investigation of intrinsic effects of CFTR in the β cell
Perhaps the most straightforward explanation for β cell dysfunction in CF would be that mutated CFTR has an intrinsic effect to impair insulin secretion in β cells. Indeed, some studies provide evidence for β-cell CFTR expression/activity and an effect on insulin release (27, 78–80). Conversely, studies using RNAseq, in situ hybridization and immunohistochemistry show that β cell expression of CFTR is very low and/or occurs in a small proportion of β cells (30, 81–85). Moreover, patch-clamp electrophysiology failed to detect a forskolin-activated chloride currents in human β cells (which would be consistent with CFTR activity) (30), and CFTR modulators were unable to alter insulin release in isolated human islets in response to glucose and cAMP-mediated activation (30). These latter data are consistent with no impact on insulin release or glucose tolerance in mouse models with β cell deletion of Cftr (30). Therefore, while intrinsic effects of CFTR in β cells may occur, it seems likely that extrinsic effects of the profoundly altered microenvironment in the CF pancreas are also important in dysregulation of insulin release.
3.2 Impact of pancreatic ductal pathology on β cell function
There is clear support from the literature for a role of exocrine pancreas pathology/dysfunction on insulin release, best illustrated by the fact that CF patients with pancreatic insufficiency have a greater deficit in insulin and glucagon release vs. those with residual exocrine pancreatic function (10, 13, 14). As mentioned, exocrine pathology is initiated in young children, although it remains relatively normal until around 1 year of age, suggesting there may be a window of opportunity for prevention. Therefore, understanding mechanisms whereby early stages of exocrine pancreas pathology may exert detrimental effects on the β cell is important and timely.
Some evidence exists to suggest that ductal pathology may contribute directly to β-cell dysfunction. First, small intercalating ducts are located in close proximity to islets, making them appropriately positioned to exert effects on β cells (86–88). Second, the limited available data examining an effect of pancreatic ductal epithelial cells (PDECs) on β cell function under normal circumstances generally show a positive effect. Specifically, co-incubation of human islets with PDECs ameliorated the decline in β-cell function which is observed with long term culture (89, 90). It has also been reported that co-transplantation of islet with PDECs improved islet transplantation outcomes (91–93) In contrast, one study showed that PDEC-secreted factors resulted in increased basal insulin release (with no effect on GSIS), and worsened islet transplantation outcomes (94). The reason for the discrepant data is not clear, but the authors of the latter study suggest that “inflammation may mediate the deleterious effects of ductal cells on islet cells”.
Several studies suggest that loss of CFTR expression/activity from PDECs can impair insulin release (82, 85, 95). Knockdown of CFTR in isolated islets (from ferrets or human donors) was sufficient to impair insulin release (82, 85). This interpretation is based on the authors’ demonstration that CFTR expression in isolated islets was restricted to ductal epithelial cells [the presence of ductal cells within isolated islet preparations is well supported in the literature (82, 96–99)]. A separate study developed a model whereby islets were cultured in close proximity to human PDECs (95). Here, acute CFTR inhibition in those PDECs resulted in decreased insulin release from islets. While this study had the advantage of a PDEC-selective intervention, it had some limitations, such as use of the pharmacological inhibitor CFTR (inh)-172, which has been shown to have non-specific effects (82, 100–102).
Further, PDECs with defective CFTR function likely release proinflammatory mediators that could impair insulin release from β cells. Human pluripotent stem cell (hPSC)-derived PDECs from CF donors express a proinflammatory transcriptome (103). Moreover, PDECs from CF ferrets release a pro-inflammatory secretome (104). Several of the differentially regulated proteins identified in the latter study have been reported to impact pathways known to affect β cell/glucose metabolism (105–109). For example, IGFBP7 was found to be downregulated in PDECs, although it was increased in pancreatic stellate cells (PSCs) from CFTR-/- ferrets, and exogenous treatment of wild-type whole islets from ferrets, IGFBP7 resulted in altered insulin secretion (104). Together, these data strongly suggest that PDEC dysfunction, especially in the early stages of CF pancreas disease may contribute to impaired β cell function/survival.
3.3 Impact of acinar pathology on β cell function
It is well-established that pancreatic ductal pathology is also the key initiator of pancreatic acinar destruction in CF (18, 23, 24, 110), and in turn the profound destruction and remodeling of acinar cells is a likely source of signals that may negatively impact the β cell.
There is evidence in the literature to support the existence of an acinar-islet axis: insulin is known to modulate pancreatic acinar cell function (111, 112), while pancreatic acinar-derived products affect islet cell function and proliferation (113–116). This suggests that normal acinar function is important in maintaining β cell function and, conversely, that acinar pathology or loss in CF could contribute to impaired insulin release.
Potential mediators of negative effects of inflamed acinar cells on the islet come from in vitro studies of chronic pancreatitis (which also manifests acinar dysfunction/destruction) (117). These studies show increased production of proinflammatory cytokines such as TNF-α and IL-1β from pancreatic acinar cells, which are known to mediate impaired insulin release and/or β cell death. Recent work focused on MODY8 (which occurs due to mutations in the acinar lipase CEL) showed that defective acinar cells were able to induce ER stress and β cell secretory dysfunction (118). Additionally, the extensive fibrosis and infiltrating fat that characterize the CF pancreas may themselves release factors that could impact β cell survival and function, although the nature of such factors remain unknown.
3.4 Endocrine cell cross-talk
Disruptions to the islet microenvironment likely also contribute to the insulin deficiency that characterizes CF. In α cells, several studies suggest that CFTR may have intrinsic effects (119, 120), although whether CFTR is expressed/active in α cells remains controversial and understudied. It is now well recognized that paracrine effects of α cell products (predominantly glucagon and GLP-1) are required for optimal β cell function. However, the increased α cell abundance in the face of impaired insulin release in CF suggests an impairment in that paracrine axis. Indeed, new data suggests that gene expression related to cell identity and hormone secretion is altered in α cells from CF donors (30, 121), and that β cell GLP-1R expression is also decreased (121), both of which would be consistent with impaired α-to-β cell communication within the CF islet.
For other islet endocrine cells, fewer data exist. However, the increased abundance of δ cells suggests that local islet somatostatin levels may be elevated, which would be expected to suppress both insulin and glucagon release. For PP cells, these may also be increased in CF, despite a suppression of circulating PP levels, suggesting again that local islet levels may be increased. Given that PP has been shown to inhibit glucagon secretion through the PPYR-1 receptors in α cells (122), an increase in islet PP could be another means by which glucagon (and thereby insulin) release could be impaired in CF. For ϵ cells, as mentioned, the status of islet ghrelin production in CF is unknown. However, circulating levels are increased (45) which, given the known effect of ghrelin to suppress insulin release (but enhance glucagon release) could also impact islet function. Taken together, dysregulation of islet endocrine cell composition and/or function likely play a critical role in perturbing insulin release in CF.
3.5 Changes in the pancreas/islet microenvironment
It is highly likely that the decrease in islet vascular density reported in our preliminary study has a profound effect on insulin secretion into the peripheral circulation. In fact, the profile of insulin release classically seen in CF, namely a blunted and delayed insulin response (10, 13, 14), could be explained almost entirely by the impaired delivery of nutrients to the islet along with inefficient delivery of insulin to the peripheral circulation that would be expected to occur in the face of decreased islet vascularization. The concept that impaired insulin release may be at least partly dependent on this loss of vascularity, also fits with data showing that β cell mass (24, 28–32) and identity (30) are relatively preserved in CF, suggesting again that “indirect” mechanisms likely explain the deficient insulin release. And, moreover, this is also consistent with data showing that when isolated islets from CF donors are studied ex vivo (i.e. under conditions where they are no longer dependent on vascularization for nutrient delivery and hormone release), insulin release is relatively intact (30). Of course, additional data will be required to fully test this hypothesis.
Similarly, the anticipated loss in autonomic innervation to the CF pancreas is also likely to have a major impact on islet function. This decreased innervation is likely to be a generalized effect, affecting sympathetic, parasympathetic and sensory input, as it is anticipated to occur secondary to exocrine pancreas destruction (and loss of vasculature). Therefore, CF islets may be essentially denervated, which would certainly be consistent with known defects such as impaired insulin release (e.g. due to reduced cholinergic tone), loss of the glucagon response to hypoglycemia (due to loss of sympathetic input) and essentially absent PP secretion (due to loss of vagal input).
While the activation of PSCs during fibrosis has been most widely studied in the context of pancreatic cancer and pancreatitis (21, 22) there is also some evidence to suggest the activated PSCs can be detrimental to β cells. Several studies demonstrate that activated PSCs can impair β cell development (123) induce β cell death (124–126), with some (127) but not all studies (128) also suggesting a negative impact on β cell function. Analysis of secreted factors from PSCs has been published by several groups (129–134); investigation of these data may inform our understanding of potential mechanisms underlying β cell dysfunction in CF.
Several lines of evidence suggest that the islet microenvironment in CF is highly inflamed. As described above, a diverse array of proinflammatory molecules are produced by both CFTR-defective PDECs and inflamed acinar cells in the surrounding exocrine pancreas, as well as CD8+ T-cells, which are known to produce proinflammatory cytokines, and inflamed/activated endothelial cells within the islet itself. Deposition of islet amyloid, in addition to its well-known toxic effects on β cells, has proinflammatory effects on both leukocytes and endothelial cells (61, 135, 136). Moreover, the proinflammatory cytokines IL-1β and IL-6 have both been shown to be produced by human or ferret islets, respectively, in CF (32, 82); these may derive from endocrine cells themselves, although the cellular source has not been definitively determined. Finally, the loss of intra-islet macrophages would further compromise the islet’s ability to clear cell debris or pathological material such as amyloid deposits, which are known to stimulate pathways such as inflammasome and toll like receptor signaling, further exacerbating the inflammatory milieu of the CF islet. Overall, this inflammatory assault on the islet is a prime target for therapeutic intervention, with approaches ideally targeting multiple cell types and/or inflammatory mediators.
4 Considerations for treatment of CFRD
The ideal treatment for CFRD would be correction of CFTR mutations to reverse or even prevent exocrine and endocrine pancreas disease. Highly effective CFTR modulator therapies (HEMT) have had major positive benefit in treating CF lung disease and improving nutritional status/body mass index (137–140). However, only limited data are available demonstrating their impact on insulin release and glucose tolerance [reviewed in (141)].
Since exocrine and endocrine pancreatic defects begin very early in life (12, 110, 142), CFTR modulators may need to be initiated in very young children to see substantial benefit. Encouragingly, in young children (between 4 months and 5 years old), the CFTR modulator ivacaftor shows improvements in exocrine pancreas function (143–145), with small studies (including children as young as 5 years old) also showing improved insulin release (146, 147). However, it is important to recognize that many people with CF may not be able to fully benefit from these interventions (e.g. those with already-established pancreas disease or whose CFTR mutations do not have available modulator therapies). Moreover, concerningly, the improved nutritional status with the administration of HEMTs have resulted in increased prevalence of overweight and obesity in people with CF as well as the emergence of features of metabolic syndrome (148), both of which may further increase the prevalence and/or severity of CFRD.
It seems that early adoption of HEMT may be possible in some people with CF. However, even in those people, combination of HEMT with therapies aimed at reversing or compensating for aspects of the disrupted pancreas/islet microenvironment (illustrated in Figures 1B, C) may well be necessary to improve β cell function. These include three major areas of intervention. First, the replacement of factors that are lost or whose release is impaired in CF may be beneficial. Incretin-based therapies fit into this category and have been shown to be efficacious in small studies of people with CF (16, 149) but their broad applicability remain to be demonstrated. Other islet-specific mediators may include glucagon or factors elicited from the islet microvasculature which are required for optimal insulin release. Second, correcting aberrant restraint on the β cell due to the upregulation of paracrine inhibitors such as somatostatin or ghrelin may be effective in restoring insulin release. Finally, a key component will likely be blockade of inflammatory mediators, which are released by multiple islet/pancreatic cell types (e.g. exocrine, vascular and adaptive immune cells; see Figure 1C) and which are well known to impair β cell function and identity. To develop such approaches will require efforts to generate a more comprehensive picture of the underlying pathways and mediators which result in a compromised pancreatic/islet microenvironment in CF.
Author contributions
SM: Writing – original draft, Writing – review & editing. DP: Writing – original draft, Writing – review & editing. RH-M: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Department of Veterans Affairs, VA Puget Sound Health Care System (Seattle, WA) and VA Merit Review I01-BX004063 (RLH), by the Seattle Institute for Biomedical and Clinical Research and awards from the Cystic Fibrosis Foundation (HULL20I0, HULL21P0 and HULL22G0-CFRD). SM was supported by T32 GM007750 and by a fellowship funded by the University of Washington CFF Research and Development Program (SINGH19R0).
Acknowledgments
We thank Drs. Sakeneh Zraika and Al Aplin both in the Research Service at VA Puget Sound Health Care System, Seattle, WA, USA, for valuable discussions related to the writing of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Bobadilla JL, Macek M Jr., Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum Mutat (2002) 19(6):575–606. doi: 10.1002/humu.10041
3. Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry Annual Data Report (2021). Available at: https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf.
4. Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell (1993) 73(7):1251–4. doi: 10.1016/0092-8674(93)90353-R
5. Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care (2009) 32(9):1626–31. doi: 10.2337/dc09-0586
6. Kwong E, Desai S, Chong L, Lee K, Zheng J, Wilcox PG, et al. The impact of cystic fibrosis-related diabetes on health-related quality of life. J Cyst Fibros (2019) 18(5):734–6. doi: 10.1016/j.jcf.2019.03.007
7. Marshall BC, Butler SM, Stoddard M, Moran AM, Liou TG, Morgan WJ. Epidemiology of cystic fibrosis-related diabetes. J Pediatr (2005) 146(5):681–7. doi: 10.1016/j.jpeds.2004.12.039
8. Lewis C, Blackman SM, Nelson A, Oberdorfer E, Wells D, Dunitz J, et al. Diabetes-related mortality in adults with cystic fibrosis. Role genotype Sex Am J Respir Crit Care Med (2015) 191(2):194–200. doi: 10.1164/rccm.201403-0576OC
9. Olesen HV, Drevinek P, Gulmans VA, Hatziagorou E, Jung A, Mei-Zahav M, et al. Cystic fibrosis related diabetes in Europe: Prevalence, risk factors and outcome. J Cyst Fibros (2020) 19(2):321–7. doi: 10.1016/j.jcf.2019.10.009
10. Moran A, Diem P, Klein DJ, Levitt MD, Robertson RP. Pancreatic endocrine function in cystic fibrosis. J Pediatr (1991) 118:715–23. doi: 10.1016/S0022-3476(05)80032-0
11. Merjaneh L, He Q, Long Q, Phillips LS, Stecenko AA. Disposition index identifies defective beta-cell function in cystic fibrosis subjects with normal glucose tolerance. J Cyst Fibros (2015) 14(1):135–41. doi: 10.1016/j.jcf.2014.06.004
12. Yi Y, Norris AW, Wang K, Sun X, Uc A, Moran A, et al. Abnormal glucose tolerance in infants and young children with cystic fibrosis. Am J Respir Crit Care Med (2016) 194(8):974–80. doi: 10.1164/rccm.201512-2518OC
13. Sheikh S, Gudipaty L, De Leon DD, Hadjiliadis D, Kubrak C, Rosenfeld NK, et al. Reduced beta-cell secretory capacity in pancreatic-insufficient, but not pancreatic-sufficient, cystic fibrosis despite normal glucose tolerance. Diabetes (2017) 66(1):134–44. doi: 10.2337/db16-0394
14. Nyirjesy SC, Sheikh S, Hadjiliadis D, De Leon DD, Peleckis AJ, Eiel JN, et al. Beta-cell secretory defects are present in pancreatic insufficient cystic fibrosis with 1-hour oral glucose tolerance test glucose ≥155 mg/dL. Pediatr Diabetes (2018) 19(7):1173–82. doi: 10.1111/pedi.12700
15. Frost F, Jones GH, Dyce P, Jackson V, Nazareth D, Walshaw MJ. Loss of incretin effect contributes to postprandial hyperglycaemia in cystic fibrosis-related diabetes. Diabetes Med (2019) 36(11):1367–74. doi: 10.1111/dme.14121
16. Nyirjesy SC, Peleckis AJ, Eiel JN, Gallagher K, Doliba A, Tami A, et al. Effects of GLP-1 and GIP on islet function in glucose-intolerant, pancreatic-insufficient cystic fibrosis. Diabetes (2022) 71(10):2153–65. doi: 10.2337/db22-0399
17. Marino CR, Matovcik LM, Gorelick FS, Cohn JA. Localization of the cystic fibrosis transmembrane conductance regulator in pancreas. J Clin Invest (1991) 88(2):712–6. doi: 10.1172/JCI115358
18. Wilschanski M, Novak I. The cystic fibrosis of exocrine pancreas. Cold Spring Harb Perspect Med (2013) 3(5):a009746. doi: 10.1101/cshperspect.a009746
19. McKay IR, Ooi CY. The exocrine pancreas in cystic fibrosis in the era of CFTR modulation: A mini review. Front Pediatr (2022) 10:914790. doi: 10.3389/fped.2022.914790
20. Kloppel G, Zamboni G. Acute and chronic alcoholic pancreatitis, including paraduodenal pancreatitis. Arch Pathol Lab Med (2023) 147(3):294–303. doi: 10.5858/arpa.2022-0202-RA
21. Apte MV, Pirola RC, Wilson JS. Pancreatic stellate cells: a starring role in normal and diseased pancreas. Front Physiol (2012) 3:344. doi: 10.3389/fphys.2012.00344
22. Masamune A, Watanabe T, Kikuta K, Shimosegawa T. Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin Gastroenterol Hepatol (2009) 7(11 Suppl):S48–54. doi: 10.1016/j.cgh.2009.07.038
23. Imrie JR, Fagan DG, Sturgess JM. Quantitative evaluation of the development of the exocrine pancreas in cystic fibrosis and control infants. Am J Pathol (1979) 95(3):697–708.
24. Bogdani M, Blackman SM, Ridaura C, Bellocq JP, Powers AC, Aguilar-Bryan L. Structural abnormalities in islets from very young children with cystic fibrosis may contribute to cystic fibrosis-related diabetes. Sci Rep (2017) 7(1):17231. doi: 10.1038/s41598-017-17404-z
25. Redman AW, Yoo M, Freswick P, Thompson K. Acute pancreatitis in a previously exocrine pancreatic insufficient cystic fibrosis patient who had improved pancreatic function after being treated with lumacaftor/ivacaftor. JPGN Rep (2021) 2(3):e096. doi: 10.1097/PG9.0000000000000096
26. Mohan K, Miller H, Dyce P, Grainger R, Hughes R, Vora J, et al. Mechanisms of glucose intolerance in cystic fibrosis. Diabetes Med (2009) 26(6):582–8. doi: 10.1111/j.1464-5491.2009.02738.x
27. Edlund A, Barghouth M, Huhn M, Abels M, Esguerra J, Mollet I, et al. Defective exocytosis and processing of insulin in a cystic fibrosis mouse model. J Endocrinol (2019) 241(1):45–57. doi: 10.1530/JOE-18-0570
28. Abdul-Karim FW, Dahms BB, Velasco ME, Rodman HM. Islets of Langerhans in adolescents and adults with cystic fibrosis. A quantitative study. Arch Pathol Lab Med (1986) 110(7):602–6.
29. Couce M, O'Brien TD, Moran A, Roche PC, Butler PC. Diabetes mellitus in cystic fibrosis is characterized by islet amyloidosis. J Clin Endocrinol Metab (1996) 81(3):1267–72. doi: 10.1210/jcem.81.3.8772610
30. Hart NJ, Aramandla R, Poffenberger G, Fayolle C, Thames AH, Bautista A, et al. Cystic fibrosis-related diabetes is caused by islet loss and inflammation. JCI Insight (2018) 3(8). doi: 10.1172/jci.insight.98240
31. Lohr M, Goertchen P, Nizze H, Gould NS, Gould VE, Oberholzer M, et al. Cystic fibrosis associated islet changes may provide a basis for diabetes. An immunocytochemical and morphometrical study. Virchows Arch A Pathol Anat Histopathol (1989) 414(2):179–85. doi: 10.1007/BF00718598
32. Hull RL, Gibson RL, McNamara S, Deutsch GH, Fligner CL, Frevert CW, et al. Islet interleukin-1beta immunoreactivity is an early feature of cystic fibrosis that may contribute to beta-cell failure. Diabetes Care (2018) 41(4):823–30. doi: 10.2337/dc17-1387
33. Iannucci A, Mukai K, Johnson D, Burke B. Endocrine pancreas in cystic fibrosis: an immunohistochemical study. Hum Pathol (1984) 15(3):278–84. doi: 10.1016/S0046-8177(84)80191-4
34. Soejima K, Landing BH. Pancreatic islets in older patients with cystic fibrosis with and without diabetes mellitus: morphometric and immunocytologic studies. Pediatr Pathol (1986) 6(1):25–46. doi: 10.3109/15513818609025923
35. Remedi MS, Emfinger C. Pancreatic beta-cell identity in diabetes. Diabetes Obes Metab (2016) 18 Suppl 1(Suppl 1):110–6. doi: 10.1111/dom.12727
36. Jonas JC, Sharma A, Hasenkamp W, Ilkova H, Patane G, Laybutt R, et al. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J Biol Chem (1999) 274(20):14112–21. doi: 10.1074/jbc.274.20.14112
37. Walker JT, Saunders DC, Brissova M, Powers AC. The human islet: mini-organ with mega-impact. Endocr Rev (2021) 42(5):605–57. doi: 10.1210/endrev/bnab010
38. Cory M, Moin ASM, Moran A, Rizza RA, Butler PC, Dhawan S, et al. An increase in chromogranin A-positive, hormone-negative endocrine cells in pancreas in cystic fibrosis. J Endocr Soc (2018) 2(9):1058–66. doi: 10.1210/js.2018-00143
39. Taborsky GJ Jr. The physiology of glucagon. J Diabetes Sci Technol (2010) 4(6):1338–44. doi: 10.1177/193229681000400607
40. Wewer Albrechtsen NJ, Holst JJ, Cherrington AD, Finan B, Gluud LL, Dean ED, et al. 100 years of glucagon and 100 more. Diabetologia (2023) 66(8):1378–94. doi: 10.1007/s00125-023-05947-y
41. Aitken ML, Szkudlinska MA, Boyko EJ, Ng D, Utzschneider KM, Kahn SE. Impaired counterregulatory responses to hypoglycaemia following oral glucose in adults with cystic fibrosis. Diabetologia (2020) 63(5):1055–65. doi: 10.1007/s00125-020-05096-6
42. Gao R, Yang T, Zhang Q. delta-cells: the neighborhood watch in the islet community. Biol (Basel) (2021) 10(2). doi: 10.3390/biology10020074
43. Meacham LR, Caplan DB, McKean LP, Buchanan CN, Parks JS, Culler FL. Preservation of somatostatin secretion in cystic fibrosis patients with diabetes. Arch Dis Child (1993) 68(1):123–5. doi: 10.1136/adc.68.1.123
44. Pradhan G, Samson SL, Sun Y. Ghrelin: much more than a hunger hormone. Curr Opin Clin Nutr Metab Care (2013) 16(6):619–24. doi: 10.1097/MCO.0b013e328365b9be
45. Monajemzadeh M, Mokhtari S, Motamed F, Shams S, Ashtiani MT, Abbasi A, et al. Plasma ghrelin levels in children with cystic fibrosis and healthy children. Arch Med Sci (2013) 9(1):93–7. doi: 10.5114/aoms.2012.28599
46. Zhang J, McKenna LB, Bogue CW, Kaestner KH. The diabetes gene Hhex maintains delta-cell differentiation and islet function. Genes Dev (2014) 28(8):829–34. doi: 10.1101/gad.235499.113
47. Richards OC, Raines SM, Attie AD. The role of blood vessels, endothelial cells, and vascular pericytes in insulin secretion and peripheral insulin action. Endocr Rev (2010) 31(3):343–63. doi: 10.1210/er.2009-0035
48. Peiris H, Bonder CS, Coates PT, Keating DJ, Jessup CF. The beta-cell/EC axis: how do islet cells talk to each other? Diabetes (2014) 63(1):3–11. doi: 10.2337/db13-0617
49. Hogan MF, Hull RL. The islet endothelial cell: a novel contributor to beta cell secretory dysfunction in diabetes. Diabetologia (2017) 60(6):952–9. doi: 10.1007/s00125-017-4272-9
50. Jansson L, Carlsson PO. Pancreatic blood flow with special emphasis on blood perfusion of the islets of Langerhans. Compr Physiol (2019) 9(2):799–837. doi: 10.1002/cphy.c160050
51. Johansson M, Mattsson G, Andersson A, Jansson L, Carlsson PO. Islet endothelial cells and pancreatic beta-cell proliferation: studies in vitro and during pregnancy in adult rats. Endocrinology (2006) 147(5):2315–24. doi: 10.1210/en.2005-0997
52. Nikolova G, Jabs N, Konstantinova I, Domogatskaya A, Tryggvason K, Sorokin L, et al. The vascular basement membrane: a niche for insulin gene expression and Beta cell proliferation. Dev Cell (2006) 10(3):397–405. doi: 10.1016/j.devcel.2006.01.015
53. Johansson A, Lau J, Sandberg M, Borg LA, Magnusson PU, Carlsson PO. Endothelial cell signalling supports pancreatic beta cell function in the rat. Diabetologia (2009) 52(11):2385–94. doi: 10.1007/s00125-009-1485-6
54. Houtz J, Borden P, Ceasrine A, Minichiello L, Kuruvilla R. Neurotrophin signaling Is required for glucose-induced insulin secretion. Dev Cell (2016) 39(3):329–45. doi: 10.1016/j.devcel.2016.10.003
55. Hogan MF, Liu AW, Peters MJ, Willard JR, Rabbani Z, Bartholomew EC, et al. Markers of islet endothelial dysfunction occur in male B6.BKS(D)-Leprdb/J mice and may contribute to reduced insulin release. Endocrinology (2017) 158(2):293–303. doi: 10.1210/en.2016-1393
56. Sakhneny L, Rachi E, Epshtein A, Guez HC, Wald-Altman S, Lisnyansky M, et al. Pancreatic pericytes support beta-cell function in a Tcf7l2-dependent manner. Diabetes (2018) 67(3):437–47. doi: 10.2337/db17-0697
57. Sakhneny L, Epshtein A, Landsman L. Pericytes contribute to the islet basement membranes to promote beta-cell gene expression. Sci Rep (2021) 11(1):2378. doi: 10.1038/s41598-021-81774-8
58. Brissova M, Shostak A, Fligner CL, Revetta FL, Washington MK, Powers AC, et al. Human islets have fewer blood vessels than mouse islets and the density of islet vascular structures is increased in type 2 diabetes. J Histochem Cytochem (2015) 63(8):637–45. doi: 10.1369/0022155415573324
59. Canzano JS, Nasif LH, Butterworth EA, Fu DA, Atkinson MA, Campbell-Thompson M. Islet microvasculature alterations with loss of beta-cells in patients with type 1 diabetes. J Histochem Cytochem (2018) 22155418778546:41–52. doi: 10.1369/0022155418778546
60. Lacraz G, Giroix MH, Kassis N, Coulaud J, Galinier A, Noll C, et al. Islet endothelial activation and oxidative stress gene expression is reduced by IL-1Ra treatment in the type 2 diabetic GK rat. PloS One (2009) 4(9):e6963. doi: 10.1371/journal.pone.0006963
61. Castillo JJ, Aplin AC, Hackney DJ, Hogan MF, Esser N, Templin AT, et al. Islet amyloid polypeptide aggregation exerts cytotoxic and proinflammatory effects on the islet vasculature in mice. Diabetologia (2022) 65(10):1687–700. doi: 10.1007/s00125-022-05756-9
62. Castillo JJ, Aplin AC, Akter R, Hull-Meichle RL. 13-OR: Cystic fibrosis donors exhibit striking decrease in pancreatic islet and exocrine capillaries. Diabetes (2022) 71(S1):13-OR. doi: 10.2337/db22-13-OR
63. Almaca J, Weitz J, Rodriguez-Diaz R, Pereira E, Caicedo A. The pericyte of the pancreatic islet regulates capillary diameter and local blood flow. Cell Metab (2018) 27(3):630–44 e4. doi: 10.1016/j.cmet.2018.02.016
64. Landsman L. Pancreatic pericytes in glucose homeostasis and diabetes. Adv Exp Med Biol (2019) 1122:27–40. doi: 10.1007/978-3-030-11093-2_2
65. Mateus Goncalves L, Pereira E, Werneck de Castro JP, Bernal-Mizrachi E, Almaca J. Islet pericytes convert into profibrotic myofibroblasts in a mouse model of islet vascular fibrosis. Diabetologia (2020) 63(8):1564–75. doi: 10.1007/s00125-020-05168-7
66. Ahren B. Autonomic regulation of islet hormone secretion–implications for health and disease. Diabetologia (2000) 43(4):393–410. doi: 10.1007/s001250051322
67. Hampton RF, Jimenez-Gonzalez M, Stanley SA. Unravelling innervation of pancreatic islets. Diabetologia (2022) 65(7):1069–84. doi: 10.1007/s00125-022-05691-9
68. Uc A, Olivier AK, Griffin MA, Meyerholz DK, Yao J, Abu-El-Haija M, et al. Glycaemic regulation and insulin secretion are abnormal in cystic fibrosis pigs despite sparing of islet cell mass. Clin Sci (Lond) (2015) 128(2):131–42. doi: 10.1042/CS20140059
69. Eguchi K, Nagai R. Islet inflammation in type 2 diabetes and physiology. J Clin Invest (2017) 127(1):14–23. doi: 10.1172/JCI88877
70. Tessem JS, Jensen JN, Pelli H, Dai XM, Zong XH, Stanley ER, et al. Critical roles for macrophages in islet angiogenesis and maintenance during pancreatic degeneration. Diabetes (2008) 57(6):1605–17. doi: 10.2337/db07-1577
71. Xiao X, Gaffar I, Guo P, Wiersch J, Fischbach S, Peirish L, et al. M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc Natl Acad Sci USA (2014) 111(13):E1211–20. doi: 10.1073/pnas.1321347111
72. Brissova M, Aamodt K, Brahmachary P, Prasad N, Hong JY, Dai C, et al. Islet microenvironment, modulated by vascular endothelial growth factor-A signaling, promotes beta cell regeneration. Cell Metab (2014) 19(3):498–511. doi: 10.1016/j.cmet.2014.02.001
73. Mussar K, Pardike S, Hohl TM, Hardiman G, Cirulli V, Crisa L. A CCR2+ myeloid cell niche required for pancreatic beta cell growth. JCI Insight (2017) 2(15). doi: 10.1172/jci.insight.93834
74. Chittezhath M, Gunaseelan D, Zheng X, Hasan R, Tay VSY, Lim ST, et al. Islet macrophages are associated with islet vascular remodeling and compensatory hyperinsulinemia during diabetes. Am J Physiol Endocrinol Metab (2019) 317(6):E1108–E20. doi: 10.1152/ajpendo.00248.2019
75. Nackiewicz D, Dan M, Speck M, Chow SZ, Chen YC, Pospisilik JA, et al. Islet macrophages shift to a reparative state following pancreatic beta-cell death and are a major source of islet insulin-like growth factor-1. iScience (2020) 23(1):100775. doi: 10.1016/j.isci.2019.100775
76. Hull RL, Westermark GT, Westermark P, Kahn SE. Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab (2004) 89(8):3629–43. doi: 10.1210/jc.2004-0405
77. Bell ET. Hyalinization of the islets of Langerhans in nondiabetic individuals. Am J Pathol (1959) 35(4):801–5.
78. Edlund A, Esguerra JL, Wendt A, Flodstrom-Tullberg M, Eliasson L. CFTR and Anoctamin 1 (ANO1) contribute to cAMP amplified exocytosis and insulin secretion in human and murine pancreatic beta-cells. BMC Med (2014) 12:87. doi: 10.1186/1741-7015-12-87
79. Guo JH, Chen H, Ruan YC, Zhang XL, Zhang XH, Fok KL, et al. Glucose-induced electrical activities and insulin secretion in pancreatic islet beta-cells are modulated by CFTR. Nat Commun (2014) 5:4420. doi: 10.1038/ncomms5420
80. Ntimbane T, Mailhot G, Spahis S, Rabasa-Lhoret R, Kleme ML, Melloul D, et al. CFTR silencing in pancreatic beta-cells reveals a functional impact on glucose-stimulated insulin secretion and oxidative stress response. Am J Physiol Endocrinol Metab (2016) 310(3):E200–12. doi: 10.1152/ajpendo.00333.2015
81. Boom A, Lybaert P, Pollet JF, Jacobs P, Jijakli H, Golstein PE, et al. Expression and localization of cystic fibrosis transmembrane conductance regulator in the rat endocrine pancreas. Endocrine (2007) 32(2):197–205. doi: 10.1007/s12020-007-9026-x
82. Sun X, Yi Y, Xie W, Liang B, Winter MC, He N, et al. CFTR influences beta cell function and insulin secretion through non-cell autonomous exocrine-derived factors. Endocrinology (2017) 158(10):3325–38. doi: 10.1210/en.2017-00187
83. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science (2015) 347(6220):1260419.
84. White MG, Maheshwari RR, Anderson SJ, Berlinguer-Palmini R, Jones C, Richardson SJ, et al. In situ analysis reveals that CFTR is expressed in only a small minority of beta-cells in normal adult human pancreas. J Clin Endocrinol Metab (2019) 105(5):1366–74. doi: 10.1210/clinem/dgz209
85. Norris AW, Ode KL, Merjaneh L, Sanda S, Yi Y, Sun X, et al. Survival in a bad neighborhood: pancreatic islets in cystic fibrosis. J Endocrinol (2019) 241(1):R35–50. doi: 10.1530/JOE-18-0468
86. Bertelli E, Bendayan M. Association between endocrine pancreas and ductal system. More than an epiphenomenon of endocrine differentiation and development? J Histochem Cytochem (2005) 53(9):1071–86. doi: 10.1369/jhc.5R6640.2005
87. El-Gohary Y, Wiersch J, Tulachan S, Xiao X, Guo P, Rymer C, et al. Intraislet pancreatic ducts can give rise to insulin-positive cells. Endocrinology (2016) 157(1):166–75. doi: 10.1210/en.2015-1175
88. Fowler JL, Lee SS, Wesner ZC, Olehnik SK, Kron SJ, Hara M. Three-dimensional analysis of the human pancreas. Endocrinology (2018) 159(3):1393–400. doi: 10.1210/en.2017-03076
89. Gatto C, Callegari M, Folin M, Conconi M, Paolin A, Di Falco G, et al. Effects of cryopreservation and coculture with pancreatic ductal epithelial cells on insulin secretion from human pancreatic islets. Int J Mol Med (2003) 12(6):851–4. doi: 10.3892/ijmm.12.6.851
90. Murray HE, Paget MB, Bailey CJ, Downing R. Sustained insulin secretory response in human islets co-cultured with pancreatic duct-derived epithelial cells within a rotational cell culture system. Diabetologia (2009) 52(3):477–85. doi: 10.1007/s00125-008-1247-x
91. Street CN, Lakey JR, Shapiro AM, Imes S, Rajotte RV, Ryan EA, et al. Islet graft assessment in the Edmonton Protocol: implications for predicting long-term clinical outcome. Diabetes (2004) 53(12):3107–14. doi: 10.2337/diabetes.53.12.3107
92. Webb MA, Dennison AR, James RF. The potential benefit of non-purified islets preparations for islet transplantation. Biotechnol Genet Eng Rev (2012) 28:101–14. doi: 10.5661/bger-28-101
93. Benomar K, Chetboun M, Espiard S, Jannin A, Le Mapihan K, Gmyr V, et al. Purity of islet preparations and 5-year metabolic outcome of allogenic islet transplantation. Am J Transplant (2018) 18(4):945–51. doi: 10.1111/ajt.14514
94. Marin-Canas S, Estil Les E, Llado L, San Jose P, Nacher M, Tellez N, et al. Pancreatic ductal cells may have a negative effect on human islet transplantation. PloS One (2019) 14(7):e0220064. doi: 10.1371/journal.pone.0220064
95. Shik Mun K, Arora K, Huang Y, Yang F, Yarlagadda S, Ramananda Y, et al. Patient-derived pancreas-on-a-chip to model cystic fibrosis-related disorders. Nat Commun (2019) 10(1):3124. doi: 10.1038/s41467-019-11178-w
96. Ichii H, Miki A, Yamamoto T, Molano RD, Barker S, Mita A, et al. Characterization of pancreatic ductal cells in human islet preparations. Lab Invest (2008) 88(11):1167–77. doi: 10.1038/labinvest.2008.87
97. Warnock GL, Meloche RM, Thompson D, Shapiro RJ, Fung M, Ao Z, et al. Improved human pancreatic islet isolation for a prospective cohort study of islet transplantation vs best medical therapy in type 1 diabetes mellitus. Arch Surg (2005) 140(8):735–44. doi: 10.1001/archsurg.140.8.735
98. Segerstolpe A, Palasantza A, Eliasson P, Andersson EM, Andreasson AC, Sun X, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab (2016) 24(4):593–607. doi: 10.1016/j.cmet.2016.08.020
99. Keymeulen B, Gillard P, Mathieu C, Movahedi B, Maleux G, Delvaux G, et al. Correlation between beta cell mass and glycemic control in type 1 diabetic recipients of islet cell graft. Proc Natl Acad Sci USA (2006) 103(46):17444–9. doi: 10.1073/pnas.0608141103
100. Kelly M, Trudel S, Brouillard F, Bouillaud F, Colas J, Nguyen-Khoa T, et al. Cystic fibrosis transmembrane regulator inhibitors CFTR(inh)-172 and GlyH-101 target mitochondrial functions, independently of chloride channel inhibition. J Pharmacol Exp Ther (2010) 333(1):60–9. doi: 10.1124/jpet.109.162032
101. Melis N, Tauc M, Cougnon M, Bendahhou S, Giuliano S, Rubera I, et al. Revisiting CFTR inhibition: a comparative study of CFTRinh -172 and GlyH-101 inhibitors. Br J Pharmacol (2014) 171(15):3716–27. doi: 10.1111/bph.12726
102. Friard J, Tauc M, Cougnon M, Compan V, Duranton C, Rubera I. Comparative effects of chloride channel inhibitors on LRRC8/VRAC-mediated chloride conductance. Front Pharmacol (2017) 8:328. doi: 10.3389/fphar.2017.00328
103. Hohwieler M, Illing A, Hermann PC, Mayer T, Stockmann M, Perkhofer L, et al. Human pluripotent stem cell-derived acinar/ductal organoids generate human pancreas upon orthotopic transplantation and allow disease modelling. Gut (2017) 66(3):473–86. doi: 10.1136/gutjnl-2016-312423
104. Rotti PG, Evans IA, Zhang Y, Liang B, Cunicelli N, O'Malley Y, et al. Lack of CFTR alters the ferret pancreatic ductal epithelial secretome and cellular proteome: Implications for exocrine/endocrine signaling. J Cyst Fibros (2022) 21(1):172–80. doi: 10.1016/j.jcf.2021.04.010
105. Garay-Malpartida HM, Mourao RF, Mantovani M, Santos IA, Sogayar MC, Goldberg AC. Toll-like receptor 4 (TLR4) expression in human and murine pancreatic beta-cells affects cell viability and insulin homeostasis. BMC Immunol (2011) 12:18. doi: 10.1186/1471-2172-12-18
106. Amyot J, Semache M, Ferdaoussi M, Fontes G, Poitout V. Lipopolysaccharides impair insulin gene expression in isolated islets of Langerhans via Toll-Like Receptor-4 and NF-kappaB signalling. PloS One (2012) 7(4):e36200. doi: 10.1371/journal.pone.0036200
107. Assayag-Asherie N, Sever D, Bogdani M, Johnson P, Weiss T, Ginzberg A, et al. Can CD44 be a mediator of cell destruction? The challenge of type 1 diabetes. PloS One (2015) 10(12):e0143589. doi: 10.1371/journal.pone.0143589
108. Gurlo T, Costes S, Hoang JD, Rivera JF, Butler AE, Butler PC. Beta cell-specific increased expression of calpastatin prevents diabetes induced by islet amyloid polypeptide toxicity. JCI Insight (2016) 1(18):e89590. doi: 10.1172/jci.insight.89590
109. Arima N, Sasaki Y, Lee LH, Zhang H, Figueiredo JL, Mlynarchik AK, et al. Multiorgan systems study reveals Igfbp7 as a suppressor of gluconeogenesis after gastric bypass surgery. J Proteome Res (2020) 19(1):129–43. doi: 10.1021/acs.jproteome.9b00441
110. Andersen DH. Cystic fibrosis of the pancreas. J Chronic Dis (1958) 7(1):58–90. doi: 10.1016/0021-9681(58)90185-1
111. Söling HD, Unger KO. The role of insulin in the regulation of alpha-amylase synthesis in the rat pancreas. Eur J Clin Invest (1972) 2(4):199–212. doi: 10.1111/j.1365-2362.1972.tb00645.x
112. Adler G, Kern HF. Regulation of exocrine pancreatic secretory process by insulin in vivo. Horm Metab Res (1975) 7(4):290–6. doi: 10.1055/s-0028-1093717
113. Henderson JR. Why are the islets of langerhans? Lancet (1969) 2(7618):469–70. doi: 10.1016/S0140-6736(69)90171-8
114. Aida K, Saitoh S, Nishida Y, Yokota S, Ohno S, Mao X, et al. Distinct cell clusters touching islet cells induce islet cell replication in association with over-expression of Regenerating Gene (REG) protein in fulminant type 1 diabetes. PloS One (2014) 9(4):e95110. doi: 10.1371/journal.pone.0095110
115. Aida K, Kobayashi T, Takeshita A, Jimbo E, Nishida Y, Yagihashi S, et al. Crucial role of Reg I from acinar-like cell cluster touching with islets (ATLANTIS) on mitogenesis of beta cells in EMC virus-induced diabetic mice. Biochem Biophys Res Commun (2018) 503(2):963–9. doi: 10.1016/j.bbrc.2018.06.103
116. Egozi A, Bahar Halpern K, Farack L, Rotem H, Itzkovitz S. Zonation of pancreatic acinar cells in diabetic mice. Cell Rep (2020) 32(7):108043. doi: 10.1016/j.celrep.2020.108043
117. Lugea A, Waldron RT, Mareninova OA, Shalbueva N, Deng N, Su HY, et al. Human pancreatic acinar cells: proteomic characterization, physiologic responses, and organellar disorders in ex vivo pancreatitis. Am J Pathol (2017) 187(12):2726–43. doi: 10.1016/j.ajpath.2017.08.017
118. Kahraman S, Dirice E, Basile G, Diegisser D, Alam J, Johansson BB, et al. Abnormal exocrine-endocrine cell cross-talk promotes beta-cell dysfunction and loss in MODY8. Nat Metab (2022) 4(1):76–89. doi: 10.1038/s42255-021-00516-2
119. Edlund A, Pedersen MG, Lindqvist A, Wierup N, Flodstrom-Tullberg M, Eliasson L. CFTR is involved in the regulation of glucagon secretion in human and rodent alpha cells. Sci Rep (2017) 7(1):90. doi: 10.1038/s41598-017-00098-8
120. Huang WQ, Guo JH, Yuan C, Cui YG, Diao FY, Yu MK, et al. Abnormal CFTR affects glucagon production by islet alpha cells in cystic fibrosis and polycystic ovarian syndrome. Front Physiol (2017) 8:835. doi: 10.3389/fphys.2017.00835
121. Vemireddy R, Castillo JJ, Fountaine BS, Bammler TK, Macdonald J, Hull-Meichle RL, et al. Islet GLP-1R protein expression is reduced in humans with cystic fibrosis–related diabetes. Diabetes (2023) 72:1753–P. doi: 10.2337/db23-1753-P
122. Aragon F, Karaca M, Novials A, Maldonado R, Maechler P, Rubi B. Pancreatic polypeptide regulates glucagon release through PPYR1 receptors expressed in mouse and human alpha-cells. Biochim Biophys Acta (2015) 1850(2):343–51. doi: 10.1016/j.bbagen.2014.11.005
123. Li J, Chen B, Fellows GF, Goodyer CG, Wang R. Activation of pancreatic stellate cells is beneficial for exocrine but not endocrine cell differentiation in the developing human pancreas. Front Cell Dev Biol (2021) 9:694276. doi: 10.3389/fcell.2021.694276
124. Kikuta K, Masamune A, Hamada S, Takikawa T, Nakano E, Shimosegawa T. Pancreatic stellate cells reduce insulin expression and induce apoptosis in pancreatic beta-cells. Biochem Biophys Res Commun (2013) 433(3):292–7. doi: 10.1016/j.bbrc.2013.02.095
125. Zang G, Sandberg M, Carlsson PO, Welsh N, Jansson L, Barbu A. Activated pancreatic stellate cells can impair pancreatic islet function in mice. Ups J Med Sci (2015) 120(3):169–80. doi: 10.3109/03009734.2015.1032453
126. Kim JJ, Lee E, Ryu GR, Ko SH, Ahn YB, Song KH. Hypoxia Increases beta-Cell Death by Activating Pancreatic Stellate Cells within the Islet. Diabetes Metab J (2020) 44(6):919–27. doi: 10.4093/dmj.2019.0181
127. Zha M, Xu W, Zhai Q, Li F, Chen B, Sun Z. High glucose aggravates the detrimental effects of pancreatic stellate cells on Beta-cell function. Int J Endocrinol (2014) 2014:165612. doi: 10.1155/2014/165612
128. Lee E, Ryu GR, Ko SH, Ahn YB, Song KH. A role of pancreatic stellate cells in islet fibrosis and beta-cell dysfunction in type 2 diabetes mellitus. Biochem Biophys Res Commun (2017) 485(2):328–34. doi: 10.1016/j.bbrc.2017.02.082
129. Wehr AY, Furth EE, Sangar V, Blair IA, Yu KH. Analysis of the human pancreatic stellate cell secreted proteome. Pancreas (2011) 40(4):557–66. doi: 10.1097/MPA.0b013e318214efaf
130. Amrutkar M, Aasrum M, Verbeke CS, Gladhaug IP. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer (2019) 19(1):596. doi: 10.1186/s12885-019-5803-1
131. Lenggenhager D, Amrutkar M, Santha P, Aasrum M, Lohr JM, Gladhaug IP, et al. Commonly used pancreatic stellate cell cultures differ phenotypically and in their interactions with pancreatic cancer cells. Cells (2019) 8(1). doi: 10.3390/cells8010023
132. Marzoq AJ, Mustafa SA, Heidrich L, Hoheisel JD, Alhamdani MSS. Impact of the secretome of activated pancreatic stellate cells on growth and differentiation of pancreatic tumour cells. Sci Rep (2019) 9(1):5303. doi: 10.1038/s41598-019-41740-x
133. Amrutkar M, Larsen EK, Aasrum M, Finstadsveen AV, Andresen PA, Verbeke CS, et al. Establishment and characterization of paired primary cultures of human pancreatic cancer cells and stellate cells derived from the same tumor. Cells (2020) 9(1). doi: 10.3390/cells9010227
134. Chakraborty A, Halder B, Mondal S, Barrett A, Zhi W, Csanyi G, et al. NADPH oxidase 1 in chronic pancreatitis-activated pancreatic stellate cells facilitates the progression of pancreatic cancer. Am J Cancer Res (2023) 13(1):118–42.
135. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol (2010) 11(10):897–904. doi: 10.1038/ni.1935
136. Westwell-Roper C, Dai DL, Soukhatcheva G, Potter KJ, van RN, Ehses JA, et al. IL-1 blockade attenuates islet amyloid polypeptide-induced proinflammatory cytokine release and pancreatic islet graft dysfunction. J Immunol (2011) 187(5):2755–65. doi: 10.4049/jimmunol.1002854
137. Volkova N, Moy K, Evans J, Campbell D, Tian S, Simard C, et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J Cyst Fibros (2020) 19(1):68–79. doi: 10.1016/j.jcf.2019.05.015
138. Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med (2010) 363(21):1991–2003. doi: 10.1056/NEJMoa0909825
139. Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med (2019) 381(19):1809–19. doi: 10.1056/NEJMoa1908639
140. Burgel PR, Munck A, Durieu I, Chiron R, Mely L, Prevotat A, et al. Real-life safety and effectiveness of lumacaftor-ivacaftor in patients with cystic fibrosis. Am J Respir Crit Care Med (2020) 201(2):188–97. doi: 10.1164/rccm.201906-1227OC
141. Merjaneh L, Hasan S, Kasim N, Ode KL. The role of modulators in cystic fibrosis related diabetes. J Clin Transl Endocrinol (2022) 27:100286. doi: 10.1016/j.jcte.2021.100286
142. Ode KL, Frohnert B, Laguna T, Phillips J, Holme B, Regelmann W, et al. Oral glucose tolerance testing in children with cystic fibrosis. Pediatr Diabetes (2010) 11(7):487–92. doi: 10.1111/j.1399-5448.2009.00632.x
143. Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med (2016) 4(2):107–15. doi: 10.1016/S2213-2600(15)00545-7
144. Rosenfeld M, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2-5years (KLIMB). J Cyst Fibros (2019) 18(6):838–43. doi: 10.1016/j.jcf.2019.03.009
145. Davies JC, Wainwright CE, Sawicki GS, Higgins MN, Campbell D, Harris C, et al. Ivacaftor in infants aged 4 to <12 months with cystic fibrosis and a gating mutation. Results of a two-part phase 3 clinical trial. Am J Respir Crit Care Med (2021) 203(5):585–93. doi: 10.1164/rccm.202008-3177OC
146. Bellin MD, Laguna T, Leschyshyn J, Regelmann W, Dunitz J, Billings J, et al. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediatr Diabetes (2013) 14(6):417–21. doi: 10.1111/pedi.12026
147. Kelly A, De Leon DD, Sheikh S, Camburn D, Kubrak C, Peleckis AJ, et al. Islet hormone and incretin secretion in cystic fibrosis after four months of ivacaftor therapy. Am J Respir Crit Care Med (2019) 199(3):342–51. doi: 10.1164/rccm.201806-1018OC
148. Kutney KA, Sandouk Z, Desimone M, Moheet A. Obesity in cystic fibrosis. J Clin Transl Endocrinol (2021) 26:100276. doi: 10.1016/j.jcte.2021.100276
Keywords: cystic fibrosis, cystic fibrosis-related diabetes, exocrine pancreas, islet, duct, beta cell, microenvironment
Citation: Malik SS, Padmanabhan D and Hull-Meichle RL (2023) Pancreas and islet morphology in cystic fibrosis: clues to the etiology of cystic fibrosis-related diabetes. Front. Endocrinol. 14:1269139. doi: 10.3389/fendo.2023.1269139
Received: 29 July 2023; Accepted: 03 October 2023;
Published: 23 November 2023.
Edited by:
James Shaw, Newcastle University, United KingdomReviewed by:
Günter Klöppel, Technical University of Munich, GermanyValeria Grancini, IRCCS Ca ‘Granda Foundation Maggiore Policlinico Hospital, Italy
Nicole Kattner, Newcastle University, United Kingdom
Copyright © 2023 Malik, Padmanabhan and Hull-Meichle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebecca L. Hull-Meichle, cmh1bGxAdXcuZWR1