 Jayashree Jena
Jayashree Jena Luis Miguel García-Peña
Luis Miguel García-Peña Renata O. Pereira
Renata O. Pereira
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 25 September 2023
Sec. Cellular Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1264530
This article is part of the Research TopicRole of Mitochondrial Stress Response in Metabolic HealthView all 6 articles
Various models of mitochondrial stress result in induction of the stress-responsive cytokines fibroblast growth factor 21 (FGF21) and growth differentiation factor 15 (GDF15). This is an adaptive mechanism downstream of the mitochondrial integrated stress response frequently associated with improvements in systemic metabolic health. Both FGF21 and GDF15 have been shown to modulate energy balance and glucose homeostasis, and their pharmacological administration leads to promising beneficial effects against obesity and associated metabolic diseases in pre-clinical models. Furthermore, endogenous upregulation of FGF21 and GDF15 is associated with resistance to diet-induced obesity (DIO), improved glucose homeostasis and increased insulin sensitivity. In this review, we highlight several studies on transgenic mouse models of mitochondrial stress and will compare the specific roles played by FGF21 and GDF15 on the systemic metabolic adaptations reported in these models.
Chronic mitochondrial stress is associated with the activation of stress response pathways such as the mitochondrial unfolded protein response (UPRmt) (1) and the integrated stress response (ISR) (2) in various tissues such as liver, adipose tissue and skeletal muscle. This is believed to be an adaptive mechanism to mitigate stress at the cellular level, but is also associated with cell non-autonomous mechanisms via the secretion of mitokines (3).
Metabolic stress and changes in mitochondrial membrane potential can result in mitochondrial dysfunction, thereby inducing the mitochondrial stress response (1). The UPRmt mediates retrograde mitochondria to nucleus signaling to alleviate mitochondrial stress (4). In mammals, it has been shown that the UPRmt is strongly coupled with the ISR (5). The ISR can be induced by four different eukaryotic initiation factor 2 alpha (eIF2α) kinases that act as early responders to disturbances in cellular homeostasis (2). Once activated, the ISR leads to phosphorylation of eIF2α and induction of its main effector, the activating transcription factor 4 (ATF4), which coordinates activation of transcriptional programs aiming at restoring cellular homeostasis. The ISR can promote UPRmt activation, thereby reducing the stress on mitochondrial chaperones and proteases, and attenuating mitochondrial dysfunction (5). ATF4 can also mediate the transcription of fibroblast growth factor 21 (FGF21) and growth differentiation factor 15 (GDF15) (6–9), mitokines that can act distally to regulate systemic metabolic homeostasis (10, 11).
FGF21 and GDF15 can also be induced under physiological stress conditions, such as in response to an obesogenic diet (12, 13), exercise (14, 15) and cold exposure (16, 17). Furthermore, pharmacological administration of FGF21 or GDF15 ameliorates obesity and related metabolic complications by improving energy and glucose homeostasis (18, 19). Nonetheless, there are several yet unresolved questions regarding the endogenous roles played by these cytokines in response to physiological versus mitochondrial stress.
In this review, we will focus on the systemic metabolic adaptations mediated by endogenous FGF21 and GDF15 induction in response to mitochondrial stress and activation of the mitochondrial stress response. We will summarize various transgenic mouse models harboring mitochondrial defects of different etiologies and in different tissues but resulting in similar improvements in metabolic health. Finally, we will discuss downstream mechanisms that are predominantly regulated by either FGF21 or GDF15 and their possible synergistic effects on systemic metabolism.
FGF21 is a member of the fibroblast growth factor (FGF) family that can be released into the circulation to act as a hormone. High FGF21 levels have been reported in various metabolic diseases, including obesity, fatty liver disease, and diabetes (20, 21). In both, rodents (22) and humans (23), circulating levels of FGF21 are derived primarily from the liver under physiological conditions, however Fgf21 mRNA can be detected in numerous tissues including the pancreas, muscle, and adipose tissues (24, 25). Circulating levels of FGF21 are low under baseline conditions and are markedly induced by numerous cellular and nutritional stress signals (21). Depending on the metabolic state, induction of FGF21 instructs the system to reestablish homeostasis through actions on multiple tissues (21). In the context of metabolism, FGF21 has been proposed to function in an endocrine manner as a ‘master sensitizer’ of specific hormonal signals that maintain energy balance via the regulation of glucose and lipid metabolism. Furthermore, previous studies have established a role for FGF21 in enhancing insulin sensitivity, increasing energy homeostasis, decreasing hepatic triglycerides, and regulating macronutrient preferences (21, 26). The multiple metabolic effects of FGF21 are mediated by both its central and peripheral actions, and by its fine-tuning of inter-organ metabolic crosstalk (26).
Besides its endogenous roles, pharmacological administration of FGF21 can decrease plasma glucose levels by more than 50% in animal models with genetically induced and diet-induced obesity (DIO) mainly through peripheral glucose disposal (21). Furthermore, prolonged administration of FGF21 or FGF21 analogs has important metabolic effects, including a marked decrease in body weight in rodents and non-human primates, and more modest effects on body weight in humans (10, 27, 28). In the context of obesity, pharmacological administration of FGF21 to obese rodents reverses diabetes and obesity through increasing energy expenditure (28, 29). Moreover, prolonged FGF21 administration to DIO mice significantly decreases body weight, adiposity, and hepatic triglycerides and cholesterol. FGF21 also reverses plasma hyperglycemia and hypertriglyceridemia and increases insulin sensitivity (28). In individuals with obesity and type 2 diabetes mellitus, FGF21 analogs alleviate dyslipidemia and increase adiponectin levels, but have minimal effects on glycemic control, thereby highlighting interspecies differences in the actions of FGF21 (26). Similarly, in patients with non-alcoholic steatohepatitis, FGF21 analogues ameliorate hepatic steatosis, liver stiffness and biomarkers of liver fibrosis (26), however the long-term effects of FGF21 in clinical outcomes remain unknown.
Growth differentiation factor 15 (GDF15), also known as macrophage inhibitory cytokine-1 [MIC-1] (12), is a stress-induced cytokine originally classified as a divergent member of the transforming growth factor beta (TGF-β) superfamily (30). GDF15 is synthesized as pro-GDF15 dimer in the cytoplasm and is subsequently cleaved and secreted as mature GDF15 (25kDa) into the blood stream (31). GDF15 is expressed in several tissues including liver (12, 32), lungs (33), kidney (34), and adipose tissue (12, 17). Levels of mature circulating GDF15 can be measured and are normally very low in humans under normal physiological conditions (35, 36). However, GDF15 levels are markedly elevated in various pathological conditions including cancer (37), inflammatory diseases (38), cardiovascular disease (39, 40), obesity (12) and mitochondrial dysfunction (41).
Glial cell–derived neurotrophic factor (GDNF) family receptor α-like protein (GFRAL) has been recently identified as GDF15’s receptor (18, 42). GFRAL’s expression is restricted to neurons of the area postrema and solitary tract nucleus in the brain (43). Mature GDF15 binds to its receptor GFRAL in the brain inducing phosphorylation of RET (tyrosine kinase co-receptor) and other intracellular signaling molecules, such as AKT and ERK1/2, thereby controlling appetite (12). Furthermore, studies also suggest mature GDF15 may affect several metabolic pathways independently of the GFRAL/RET receptor pathway and can modulate metabolism independently of changes in food intake. Indeed, systemic overexpression of GDF15 prevents obesity and insulin resistance by modulating metabolic activity and enhancing the expression of thermogenic and lipolytic genes in brown adipose tissue (BAT) and white adipose tissue (WAT) (44–46). Some of these effects were independent of changes in food intake (43).
Obesity is also associated with increased serum concentrations of GDF15. An increase of up to 10-fold in serum GDF15 levels was observed in obese mice (12). Several studies have reported the role of GDF15 towards reducing food intake, body weight, and adiposity and improving glucose tolerance under normal and obesogenic diets (12, 45, 47). Indeed, GDF15 and GFRAL knockout mice fed a high-fat diet (HFD) display a slight increase in fat depots and body weight when compared to their wild-type counterparts (43, 48). Conversely, GDF15 overexpression is associated with leanness and other improved metabolic parameters in mice (44–46).
Besides its endogenous roles, pharmacological administration of GDF15 has gained significant interest owing to its role as an appetite suppressor and a regulator of energy homeostasis (18, 49). Pre-clinical studies administering recombinant GDF15 in rodents and non-human primates revealed the potential of GDF15 for the treatment of metabolic disorders such as obesity and diabetes. GDF15 treatment potently reduces food intake in ob/ob mice (50), and in non-human primates with spontaneous obesity (18). Several studies have shown that treatment with recombinant-GDF15 attenuates obesity and improves glycemic control through GFRAL-dependent suppression of food intake (18, 42). Furthermore, administration of a long-acting GDF15 molecule to non-human primates maintained the decrease in appetite and body weight for 4 weeks, suggesting this molecule might be used for chronic treatment of obesity (51). In addition to changes in food intake and body weight, recombinant GDF15 also reduces liver steatosis and improves glycemic control via GFRAL in genetically induced and DIO mouse models (45, 46). Noteworthy, besides reducing food intake, treatment with recombinant GDF15 reduces adiposity and corrects metabolic dysfunction in obese mice by increasing thermogenesis in adipocytes (35), and by increasing energy expenditure via increased adrenergic stimulation and futile calcium cycling in skeletal muscle through GFRAL signaling (52). Recent studies also uncovered roles for GDF15 in mediating the beneficial effects of compounds with anti-obesity properties, including metformin (53), capsaicin (54), resveratrol (55) and conjugated linoleic acid (56). Furthermore, GDF15 may also interact with other appetite regulators such as leptin to regulate body weight and adiposity (57). Lastly, a GLP-1/GDF15 dual agonist effectively lowers body weight and caloric intake in various animal models, including obese non-human primates (58).
These promising pre-clinical studies have now been extended to clinical trials. Indeed, results from a recently completed clinical trial using a new long-acting GDF15 receptor agonist show significant reduction in body weight in rodents and in non-human primates. However, despite displaying an acceptable safety profile and reducing food intake in humans, body weight was only modestly affected (49). These interesting results highlight potential differences in GDF15 signaling and mechanisms of action between rodents, non-human primates and human subjects. Furthermore, this suggests that additional GDF15-mediated mechanisms that might be independent of GFRAL signaling, or food intake reduction are likely required to promote weight loss in humans.
FGF21 and GDF15 can be induced in response to mitochondrial stress, and as such are recognized as mitokines (59, 60). Mitokines are signaling molecules that enable communication between local mitochondrial stress and distant cells and tissues, that can be strongly induced in mouse models of metabolic dysregulation and mitochondrial dysfunction (59). Mitochondrial dysfunction of various etiologies, such as defective mitochondrial oxidative phosphorylation (OxPhos) and coupling efficiency (59, 61), impaired quality control (46) or dynamics (6, 62, 63) activate the UPRmt and the ISR, leading to increased phosphorylation of eIF2α (2). eIF2α phosphorylation inhibits overall translation, while selectively allowing for the translation of stress responsive proteins such as the activating transcription factor 4 (ATF4). Downstream of eIF2α, ATF4 directly induces Fgf21 transcription and via induction of C/EBP homologous protein (CHOP) it indirectly induces Gdf15 expression (59, 64) (Figure 1).
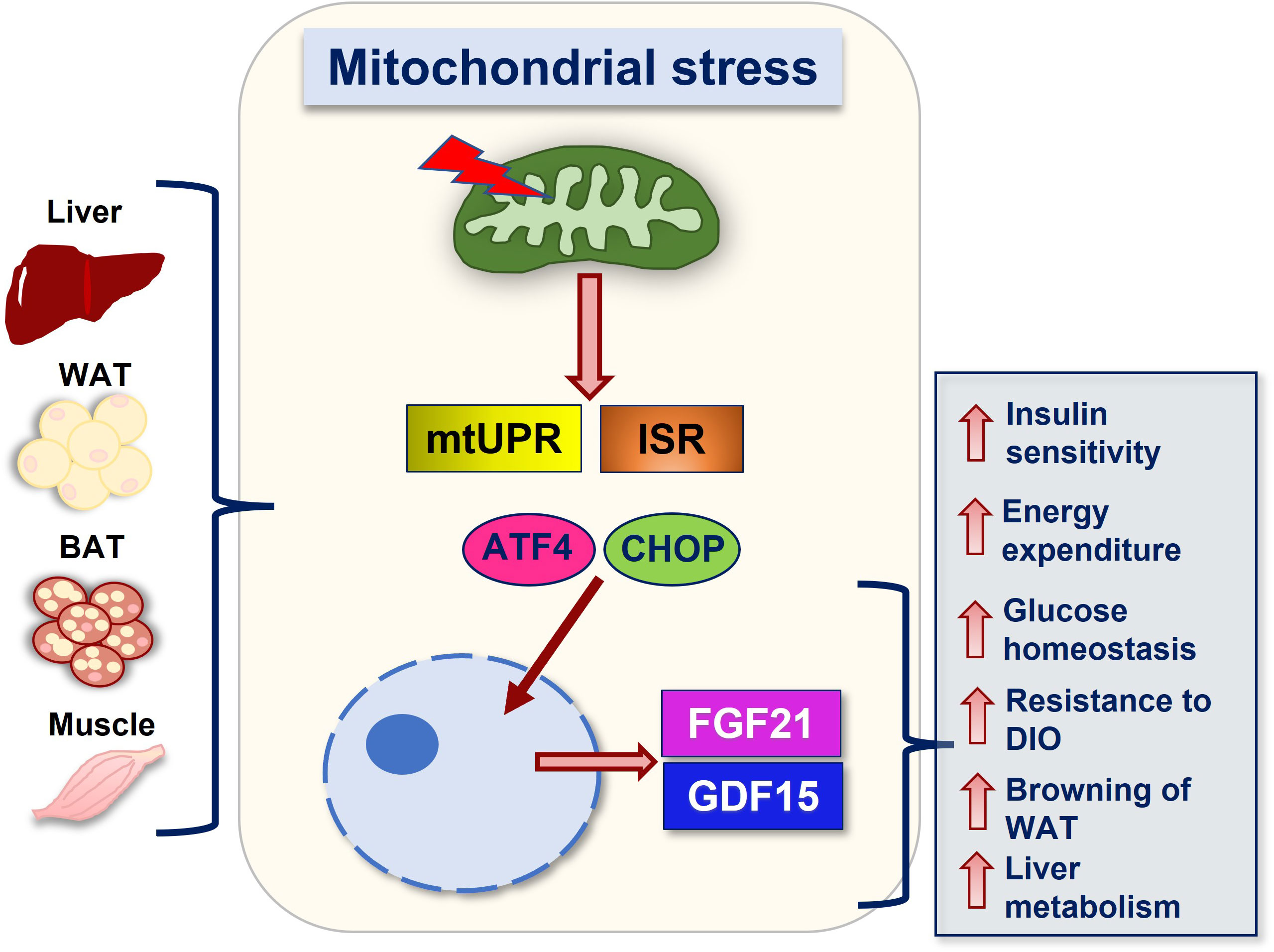
Figure 1 Summary of FGF21 and GDF15 mediated actions downstream of mitochondrial stress. Mitochondrial stress of various etiologies and in various tissues lead to the activation of stress response pathways such as the mtUPR and the ISR, resulting in the translation of transcription factors such as ATF4 and its downstream target CHOP, which promote induction of FGF21 and GDF15, respectively. Secretion of these factors into the circulation mediates metabolic adaptations in response to mitochondrial stress. Together, these adaptations promote improvements in systemic metabolic health, including increases in energy expenditure, resistance to diet-induced obesity (DIO), improved insulin sensitivity and glucose homeostasis, attenuated hepatic steatosis, and increased browning. Abbreviations: WAT (white adipose tissue), BAT (brown adipose tissue), UPRmt (mitochondrial unfolded protein response), ISR (integrated stress response), ATF4 (activating transcription factor 4), CHOP (C/EBP homologous protein), FGF21 (fibroblast growth factor 21) and GDF15 (growth differentiation factor 15).
Several mouse models of mitochondrial stress have shown an upregulation of FGF21 as a protective mechanism against mitochondrial injury and metabolic dysregulation. Ectopic expression of the uncoupling protein 1 (UCP1) in skeletal muscle in vivo (UCP1-Tg) leads to induction of FGF21, which is required to increase browning of white adipose tissue (WAT), with only mild effects on the metabolic phenotype of these mice (9, 59). In mice lacking UCP1 in brown adipose tissue (BAT), the main effector in canonical BAT thermogenesis, induction of FGF21 promotes resistance to DIO (61). In models of disrupted mitochondrial dynamics in skeletal muscle (62), BAT (6) and liver (63), secretion of FGF21 resulted in protection against DIO and improved metabolic phenotypes. Genetic impairment of adipocytes OxPhos function in vivo also protects mice from DIO and insulin resistance, with ablation of FGF21 in this model leading to increased body weight and adiposity, and hepatic steatosis after 8 weeks on high-fat diet (HFD) (41). ATG7 deletion selectively in skeletal muscle inhibits mitophagy and induces FGF21 as a mitokine, leading to protection from obesity and insulin resistance (65). Another model of impaired mitophagy also showed upregulated Fgf21 expression, and a role for ISR-mediated secretion of FGF21 in promoting thermogenic remodeling of adipose tissue (66). Furthermore, several studies of gain- and loss-of-function indicate that FGF21 is a key metabolic mediator to improve compromised mitochondrial function and reduce inflammation and apoptosis in skeletal muscle (67).
Various transgenic mouse models of mitochondrial dysfunction also result in GDF15 induction in a tissue-specific manner, leading to improved systemic metabolic homeostasis (60, 68). Elevated GDF15 expression in and secretion from skeletal muscle has been described in human subjects with mitochondrial myopathies (69–71). Furthermore, mouse models of selective mitochondrial dysfunction in muscle have higher levels of circulating GDF15, which is associated with improved aspects of systemic metabolism. Indeed, mice with muscle-specific deletion of a component of mtDNA ribosomes, Crif-1, have impaired oxidative phosphorylation and upregulation of Gdf15 expression in and secretion from muscle via activation of a UPRmt-ATF4-CHOP axis. This increase in GDF15 levels was required to promote resistance to DIO and insulin resistance, due to an increase in lipolysis in adipocytes and hepatocytes (60). In another model of mitochondrial dysfunction caused by Ant1 deficiency in muscle, Gdf15 levels were highly up regulated (72). Moreover, UCP1-tg mice also leads to increased GDF15 circulating levels. Deletion of either Fgf21 or Gdf15 in the UCP1-tg background showed that FGF21 was of minor importance for the metabolic adaptations of this particular mouse model (9), whereas ablation of Gdf15 led to a progressive body mass increase due to an accumulation of body fat, and abolished the increased insulin sensitivity reported in this mouse model. These effects were also associated with day-time restricted anorexia, which was dependent on GDF15 (7).
Mice with liver-specific mitochondrial stress induced by a loss of function of Crif-1 have aberrant OxPhos and activated UPRmt, increasing the secretion of GDF15 and FGF21 as mitokines. By using whole body GDF15 or FGF21 knock out mouse models in conjunction with Crif-1 deletion in liver, the authors concluded that GDF15 is necessary to regulate body mass and fat mass and to prevent diet-induced hepatic steatosis, whereas FGF21 is requited to improve insulin sensitivity, energy expenditure and thermogenesis in WAT (32).
Furthermore, adipocyte-specific deletion of Crif-1 in mice decreases adipocyte OxPhos function and induces FGF21 and GDF15 as mitokines, thereby promoting resistance to DIO, and improving glucose homeostasis. Long-term induction of GDF15 in this model was required to attenuate the progression of obesity by increasing energy expenditure, while FGF21 did not affect energy expenditure, but remarkably ameliorated DIO and insulin resistance (41). Finally, a report by Miyake et al. shows that induction of GDF15 secretion mediated by activation of the integrated stress response (ISR) in adipocytes reduces food intake via GFRAL signaling and decreases body weight in mice fed HFD (64).
It is important to note that, although chronic mild mitochondrial stress can lead to adaptive responses that result in improved metabolic health, unresolved mitochondrial stress is central to the pathogenesis of many major metabolic disorders such as obesity, insulin resistance and type 2 diabetes (4, 73). During OxPhos, in addition to generating ATP, mitochondria also generate reactive oxygen species (ROS) as toxic by-products. In excess, ROS may damage mitochondrial and cellular DNA, proteins, lipids and other molecules, contributing to mitochondrial dysfunction (74).
Mitochondrial dysfunction has been shown to be associated with insulin resistance in various tissues including skeletal muscle (75), liver (76) and adipose tissue (77). In skeletal muscle, impaired mitochondrial oxidative capacity, reduced ATP production rates and increased ROS levels have all been reported as contributing factors in the pathogenesis of insulin resistance (4). Impaired mitochondrial β-oxidation is found in patients with nonalcoholic fatty liver disease (NAFLD), contributing to hepatic steatosis, liver injury (78, 79) and fibrosis (80). Mitochondria are also crucial for adipose tissue function (73), therefore, unsurprisingly, impaired mitochondrial function and dynamics disrupts adipocyte homeostasis (77, 81) and is a central driver for obesity, inflammation and associated metabolic disorders (82).
Both GDF15 and FGF21 are central endocrine regulators of systemic energy metabolism and glucose homeostasis with robust therapeutical potential for the treatment of obesity and associated metabolic diseases (10, 18). Pre-clinical studies suggest that GDF15 acts predominantly on reducing energy intake (42, 51), whereas FGF21 might play a primary role on increasing energy expenditure (10, 21). Nonetheless, recent clinical trials were unable to show a robust effect on weight loss, even though other metabolic parameters seem to be ameliorated by FGF21 and GDF15 treatment (49, 83, 84). Therefore, further studies will be required to determine the primary targets and fundamental mechanisms of actions of both factors in humans so that more effective therapies can be developed.
In mouse models of mild mitochondrial stress, chronically elevated FGF21 and GDF15 provide resistance to DIO and insulin resistance and ameliorate diet-induced hepatic steatosis and glucose intolerance (6, 32, 62). These effects are likely caused by the lifelong induction of a catabolic state in these models. However, induction of FGF21 levels in adult obese mice, by inducible deletion of the mitochondrial fusion protein optic atrophy 1 (OPA1) in skeletal muscle, was able to reverse DIO, suggesting that induction of this pathway after the onset of obesity may still exert beneficial metabolic effects (62). Noteworthy, GDF15 and FGF21 play divergent roles in regulating energy metabolism and glucose homeostasis as part of the mitochondrial stress response. These different roles are dictated by the type of mitochondrial stress, the tissue directly affected by the stress and the metabolic state of the animal. Together, studies support a predominant role for GDF15 rather than FGF21 in promoting resistance to DIO in response to mitochondrial stress and suggests a role for GDF15 in promoting browning during DIO (32). Conversely, FGF21 is required to increase insulin sensitivity, energy expenditure and UCP1-mediated thermogenesis in inguinal white adipose tissue (iWAT) of regular chow-fed mice (6, 32). However, whether FGF21 and GDF15 interact to modulate energy balance and glucose homeostasis in response to mitochondrial stress remains to be investigated.
Finally, while our understanding of the role of endogenous FGF21 and GDF15 in the regulation of metabolic homeostasis in response to mitochondrial stress has advanced in the recent years with the characterization of various mouse models, several outstanding questions remain to be addressed. Although studies have shown that the pharmacological actions of FGF21 are primarily mediated by its receptor in the brain and in adipose tissues, to date, the only GDF15 receptor identified is strictly expressed in discrete areas of the brain. Nonetheless, studies suggest GDF15 may exert some of its effects independently of GFRAL via activation of yet non-identified receptors. Moreover, the role of these cytokines in response to physiological stressors such as exercise and cold exposure remain to be assessed. Better understanding of the roles of endogenous FGF21 and GDF15 in health and disease might lead to the development of new therapies to treat metabolic disorders.
JJ: Writing – review & editing, Writing – original draft. LG-P: Writing – original draft, Writing – review & editing. RP: Writing – review & editing, Conceptualization, Funding acquisition, Project administration, Resources, Supervision.
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by NIH DK125405 to RP; by the Diabetes Research Training Program funded by the NIH (T32DK112751-01) to JJ; and by the NIH 1R25GM116686 to LG-P.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Zhou Z, Fan Y, Zong R, Tan K. The mitochondrial unfolded protein response: A multitasking giant in the fight against human diseases. Ageing Res Rev (2022) 81:101702. doi: 10.1016/j.arr.2022.101702
2. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep (2016) 17:1374–95. doi: 10.15252/embr.201642195
3. Mick E, Titov DV, Skinner OS, Sharma R, Jourdain AA, Mootha VK. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. Elife (2020) 9:554–64. doi: 10.7554/eLife.49178
4. Hu F, Liu F. Mitochondrial stress: a bridge between mitochondrial dysfunction and metabolic diseases? Cell Signal (2011) 23:1528–33. doi: 10.1016/j.cellsig.2011.05.008
5. Lu H, Wang X, Li M, Ji D, Liang D, Liang C, et al. Mitochondrial unfolded protein response and integrated stress response as promising therapeutic targets for mitochondrial diseases. Cells (2022) 12. doi: 10.3390/cells12010020
6. Pereira RO, Marti A, Olvera AC, Tadinada SM, Bjorkman SH, Weatherford ET, et al. OPA1 deletion in brown adipose tissue improves thermoregulation and systemic metabolism via FGF21. Elife (2021) 10. doi: 10.7554/eLife.66519.sa2
7. Ost M, Igual Gil C, Coleman V, Keipert S, Efstathiou S, Vidic V, et al. Muscle-derived GDF15 drives diurnal anorexia and systemic metabolic remodeling during mitochondrial stress. EMBO Rep (2020) 21:e48804. doi: 10.15252/embr.201948804
8. Forsstrom S, Jackson CB, Carroll CJ, Kuronen M, Pirinen E, Pradhan S, et al. Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab (2019) 30:1040–1054.e7. doi: 10.1016/j.cmet.2019.08.019
9. Ost M, Coleman V, Voigt A, van Schothorst EM, Keipert S, van der Stelt I, et al. Muscle mitochondrial stress adaptation operates independently of endogenous FGF21 action. Mol Metab (2016) 5:79–90. doi: 10.1016/j.molmet.2015.11.002
10. BonDurant LD, Ameka M, Naber MC, Markan KR, Idiga SO, Acevedo MR, et al. FGF21 regulates metabolism through adipose-dependent and -independent mechanisms. Cell Metab (2017) 25:935–944.e4. doi: 10.1016/j.cmet.2017.03.005
11. Breit SN, Brown DA, Tsai VW. The GDF15-GFRAL pathway in health and metabolic disease: friend or foe? Annu Rev Physiol (2021) 83:127–51. doi: 10.1146/annurev-physiol-022020-045449
12. Patel S, Alvarez-Guaita A, Melvin A, Rimmington D, Dattilo A, Miedzybrodzka EL, et al. GDF15 provides an endocrine signal of nutritional stress in mice and humans. Cell Metab (2019) 29:707–718 e8. doi: 10.1016/j.cmet.2018.12.016
13. Zhang X, Yeung DC, Karpisek M, Stejskal D, Zhou ZG, Liu F, et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes (2008) 57:1246–53. doi: 10.2337/db07-1476
14. Klein AB, Kleinert M, Richter EA, Clemmensen C. GDF15 in appetite and exercise: essential player or coincidental bystander? Endocrinology (2022) 163:(2022). doi: 10.1210/endocr/bqab242
15. Morville T, Sahl RE, Trammell SA, Svenningsen JS, Gillum MP, Helge JW, et al. Divergent effects of resistance and endurance exercise on plasma bile acids, FGF19, and FGF21 in humans. JCI Insight (2018) 3. doi: 10.1172/jci.insight.122737
16. Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev (2012) 26:271–81. doi: 10.1101/gad.177857.111
17. Campderros L, Moure R, Cairo M, Gavalda-Navarro A, Quesada-Lopez T, Cereijo R, et al. Brown adipocytes secrete GDF15 in response to thermogenic activation. Obes (Silver Spring) (2019) 27(10):1606–16. doi: 10.1002/oby.22584
18. Mullican SE, Lin-Schmidt X, Chin CN, Chavez JA, Furman JL, Armstrong AA, et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat Med (2017) 23:1150–7. doi: 10.1038/nm.4392
19. Claflin KE, Sullivan AI, Naber MC, Flippo KH, Morgan DA, Neff TJ, et al. Pharmacological FGF21 signals to glutamatergic neurons to enhance leptin action and lower body weight during obesity. Mol Metab (2022) 64:101564. doi: 10.1016/j.molmet.2022.101564
20. Kliewer SA, Mangelsdorf DJ. A dozen years of discovery: insights into the physiology and pharmacology of FGF21. Cell Metab (2019) 29:246–53. doi: 10.1016/j.cmet.2019.01.004
21. Flippo KH, Potthoff MJ. Metabolic messengers: FGF21. Nat Metab (2021) 3:309–17. doi: 10.1038/s42255-021-00354-2
22. Markan KR, Naber MC, Ameka MK, Anderegg MD, Mangelsdorf DJ, Kliewer SA, et al. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes (2014) 63:4057–63. doi: 10.2337/db14-0595
23. Hansen JS, Clemmesen JO, Secher NH, Hoene M, Drescher A, Weigert C, et al. Glucagon-to-insulin ratio is pivotal for splanchnic regulation of FGF-21 in humans. Mol Metab (2015) 4:551–60. doi: 10.1016/j.molmet.2015.06.001
24. Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta (2000) 1492:203–6. doi: 10.1016/S0167-4781(00)00067-1
25. Giralt M, Gavalda-Navarro A, Villarroya F. Fibroblast growth factor-21, energy balance and obesity. Mol Cell Endocrinol (2015) 418(Pt 1):66–73. doi: 10.1016/j.mce.2015.09.018
26. Geng L, Lam KSL, Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrinol (2020) 16:654–67. doi: 10.1038/s41574-020-0386-0
27. Moyers JS, Shiyanova TL, Mehrbod F, Dunbar JD, Noblitt TW, Otto KA, et al. Molecular determinants of FGF-21 activity-synergy and cross-talk with PPARgamma signaling. J Cell Physiol (2007) 210:1–6. doi: 10.1002/jcp.20847
28. Xu J, Stanislaus S, Chinookoswong N, Lau YY, Hager T, Patel J, et al. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models–association with liver and adipose tissue effects. Am J Physiol Endocrinol Metab (2009) 297:E1105–14. doi: 10.1152/ajpendo.00348.2009
29. Adams AC, Yang C, Coskun T, Cheng CC, Gimeno RE, Luo Y, et al. The breadth of FGF21's metabolic actions are governed by FGFR1 in adipose tissue. Mol Metab (2012) 2:31–7. doi: 10.1016/j.molmet.2012.08.007
30. Tsai VW, Lin S, Brown DA, Salis A, Breit SN. Anorexia-cachexia and obesity treatment may be two sides of the same coin: role of the TGF-b superfamily cytokine MIC-1/GDF15. Int J Obes (Lond) (2016) 40:193–7. doi: 10.1038/ijo.2015.242
31. Baek SJ, Eling T. Growth differentiation factor 15 (GDF15): A survival protein with therapeutic potential in metabolic diseases. Pharmacol Ther (2019) 198:46–58. doi: 10.1016/j.pharmthera.2019.02.008
32. Kang SG, Choi MJ, Jung SB, Chung HK, Chang JY, Kim JT, et al. Differential roles of GDF15 and FGF21 in systemic metabolic adaptation to the mitochondrial integrated stress response. iScience (2021) 24:102181. doi: 10.1016/j.isci.2021.102181
33. Coll AP, Chen M, Taskar P, Rimmington D, Patel S, Tadross JA, et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature (2020) 578:444–8. doi: 10.1038/s41586-019-1911-y
34. Paralkar VM, Vail AL, Grasser WA, Brown TA, Xu H, Vukicevic S, et al. Cloning and characterization of a novel member of the transforming growth factor-beta/bone morphogenetic protein family. J Biol Chem (1998) 273:13760–7. doi: 10.1074/jbc.273.22.13760
35. Tsai VW, Zhang HP, Manandhar R, Lee-Ng KKM, Lebhar H, Marquis CP, et al. Treatment with the TGF-b superfamily cytokine MIC-1/GDF15 reduces the adiposity and corrects the metabolic dysfunction of mice with diet-induced obesity. Int J Obes (Lond) (2018) 42:561–71. doi: 10.1038/ijo.2017.258
36. Tsai VW, Husaini Y, Manandhar R, Lee-Ng KK, Zhang HP, Harriott K, et al. Anorexia/cachexia of chronic diseases: a role for the TGF-beta family cytokine MIC-1/GDF15. J Cachexia Sarcopenia Muscle (2012) 3:239–43. doi: 10.1007/s13539-012-0082-6
37. Modi A, Dwivedi S, Roy D, Khokhar M, Purohit P, Vishnoi J, et al. Growth differentiation factor 15 and its role in carcinogenesis: an update. Growth Factors (2019) 37:190–207. doi: 10.1080/08977194.2019.1685988
38. Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, et al. GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell (2019) 178:1231–1244.e11. doi: 10.1016/j.cell.2019.07.033
39. Xu J, Kimball TR, Lorenz JN, Brown DA, Bauskin AR, Klevitsky R, et al. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res (2006) 98:342–50. doi: 10.1161/01.RES.0000202804.84885.d0
40. Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J, et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med (2011) 17:581–8. doi: 10.1038/nm.2354
41. Choi MJ, Jung SB, Lee SE, Kang SG, Lee JH, Ryu MJ, et al. An adipocyte-specific defect in oxidative phosphorylation increases systemic energy expenditure and protects against diet-induced obesity in mouse models. Diabetologia (2020) 63:837–52. doi: 10.1007/s00125-019-05082-7
42. Yang L, Chang CC, Sun Z, Madsen D, Zhu H, Padkjaer SB, et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat Med (2017) 23:1158–66. doi: 10.1038/nm.4394
43. Aguilar-Recarte D, Barroso E, Palomer X, Wahli W, Vazquez-Carrera M. Knocking on GDF15's door for the treatment of type 2 diabetes mellitus. Trends Endocrinol Metab (2022) 33:741–54. doi: 10.1016/j.tem.2022.08.004
44. Macia L, Tsai VW, Nguyen AD, Johnen H, Kuffner T, Shi YC, et al. Macrophage inhibitory cytokine 1 (MIC-1/GDF15) decreases food intake, body weight and improves glucose tolerance in mice on normal & obesogenic diets. PloS One (2012) 7:e34868. doi: 10.1371/journal.pone.0034868
45. Chrysovergis K, Wang X, Kosak J, Lee SH, Kim JS, Foley JF, et al. NAG-1/GDF-15 prevents obesity by increasing thermogenesis, lipolysis and oxidative metabolism. Int J Obes (Lond) (2014) 38:1555–64. doi: 10.1038/ijo.2014.27
46. Kim JM, Kosak JP, Kim JK, Kissling G, Germolec DR, Zeldin DC, et al. NAG-1/GDF15 transgenic mouse has less white adipose tissue and a reduced inflammatory response. Mediators Inflamm (2013) 2013:641851. doi: 10.1155/2013/641851
47. Tran T, Yang J, Gardner J, Xiong Y. GDF15 deficiency promotes high fat diet-induced obesity in mice. PloS One (2018) 13:e0201584. doi: 10.1371/journal.pone.0201584
48. Patel S, Haider A, Alvarez-Guaita A, Bidault G, El-Sayed Moustafa JS, Guiu-Jurado E, et al. Combined genetic deletion of GDF15 and FGF21 has modest effects on body weight, hepatic steatosis and insulin resistance in high fat fed mice. Mol Metab (2022) 65:101589. doi: 10.1016/j.molmet.2022.101589
49. Benichou O, Coskun T, Gonciarz MD, Garhyan P, Adams AC, Du Y, et al. Discovery, development, and clinical proof of mechanism of LY3463251, a long-acting GDF15 receptor agonist. Cell Metab (2023) 35:274–286.e10. doi: 10.1016/j.cmet.2022.12.011
50. Xiong Y, Walker K, Min X, Hale C, Tran T, Komorowski R, et al. Long-acting MIC-1/GDF15 molecules to treat obesity: Evidence from mice to monkeys. Sci Transl Med (2017) 9. doi: 10.1126/scitranslmed.aan8732
51. Mullican SE, Rangwala SM. Uniting GDF15 and GFRAL: therapeutic opportunities in obesity and beyond. Trends Endocrinol Metab (2018) 29:560–70. doi: 10.1016/j.tem.2018.05.002
52. Wang D, Townsend LK, DesOrmeaux GJ, Frangos SM, Batchuluun B, Dumont L, et al. GDF15 promotes weight loss by enhancing energy expenditure in muscle. Nature (2023) 619:143–50. doi: 10.1038/s41586-023-06249-4
53. Gerstein HC, Pare G, Hess S, Ford RJ, Sjaarda J, Raman K, et al. Growth differentiation factor 15 as a novel biomarker for metformin. Diabetes Care (2017) 40:280–3. doi: 10.2337/dc16-1682
54. Lee SH, Richardson RL, Dashwood RH, Baek SJ. Capsaicin represses transcriptional activity of beta-catenin in human colorectal cancer cells. J Nutr Biochem (2012) 23:646–55. doi: 10.1016/j.jnutbio.2011.03.009
55. Baek SJ, Wilson LC, Lee CH, Eling TE. Dual function of nonsteroidal anti-inflammatory drugs (NSAIDs): inhibition of cyclooxygenase and induction of NSAID-activated gene. J Pharmacol Exp Ther (2002) 301:1126–31. doi: 10.1124/jpet.301.3.1126
56. Lee SH, Yamaguchi K, Kim JS, Eling TE, Safe S, Park Y, et al. Conjugated linoleic acid stimulates an anti-tumorigenic protein NAG-1 in an isomer specific manner. Carcinogenesis (2006) 27:972–81. doi: 10.1093/carcin/bgi268
57. Breit SN, Manandhar R, Zhang HP, Lee-Ng M, Brown DA, Tsai VW. GDF15 enhances body weight and adiposity reduction in obese mice by leveraging the leptin pathway. Cell Metab (2023) 35(8):1341-1355.e3. doi: 10.1016/j.cmet.2023.06.009
58. Zhang Y, Zhao X, Dong X, Zhang Y, Zou H, Jin Y, et al. Activity-balanced GLP-1/GDF15 dual agonist reduces body weight and metabolic disorder in mice and non-human primates. Cell Metab (2023) 35:287–298 e4. doi: 10.1016/j.cmet.2023.01.001
59. Keipert S, Ost M. Stress-induced FGF21 and GDF15 in obesity and obesity resistance. Trends Endocrinol Metab (2021) 32:904–15. doi: 10.1016/j.tem.2021.08.008
60. Chung HK, Kim JT, Kim HW, Kwon M, Kim SY, Shong M, et al. GDF15 deficiency exacerbates chronic alcohol- and carbon tetrachloride-induced liver injury. Sci Rep (2017) 7:17238. doi: 10.1038/s41598-017-17574-w
61. Keipert S, Lutter D, Schroeder BO, Brandt D, Stahlman M, Schwarzmayr T, et al. Endogenous FGF21-signaling controls paradoxical obesity resistance of UCP1-deficient mice. Nat Commun (2020) 11:624. doi: 10.1038/s41467-019-14069-2
62. Pereira RO, Tadinada SM, Zasadny FM, Oliveira KJ, Pires KMP, Olvera A, et al. OPA1 deficiency promotes secretion of FGF21 from muscle that prevents obesity and insulin resistance. EMBO J (2017) 36:2126–45. doi: 10.15252/embj.201696179
63. Wang L, Ishihara T, Ibayashi Y, Tatsushima K, Setoyama D, Hanada Y, et al. Disruption of mitochondrial fission in the liver protects mice from diet-induced obesity and metabolic deterioration. Diabetologia (2015) 58:2371–80. doi: 10.1007/s00125-015-3704-7
64. Miyake M, Zhang J, Yasue A, Hisanaga S, Tsugawa K, Sakaue H, et al. Integrated stress response regulates GDF15 secretion from adipocytes, preferentially suppresses appetite for a high-fat diet and improves obesity. iScience (2021) 24:103448. doi: 10.1016/j.isci.2021.103448
65. Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med (2013) 19:83–92. doi: 10.1038/nm.3014
66. Fu T, Xu Z, Liu L, Guo Q, Wu H, Liang X, et al. Mitophagy directs muscle-adipose crosstalk to alleviate dietary obesity. Cell Rep (2018) 23:1357–72. doi: 10.1016/j.celrep.2018.03.127
67. Ji K, Zheng J, Lv J, Xu J, Ji X, Luo YB, et al. Skeletal muscle increases FGF21 expression in mitochondrial disorders to compensate for energy metabolic insufficiency by activating the mTOR-YY1-PGC1alpha pathway. Free Radic Biol Med (2015) 84:161–70. doi: 10.1016/j.freeradbiomed.2015.03.020
68. Aguilar-Recarte D, Barroso E, Guma A, Pizarro-Delgado J, Pena L, Ruart M, et al. GDF15 mediates the metabolic effects of PPARbeta/delta by activating AMPK. Cell Rep (2021) 36:109501. doi: 10.1016/j.celrep.2021.109501
69. Fujita Y, Ito M, Kojima T, Yatsuga S, Koga Y, Tanaka M. GDF15 is a novel biomarker to evaluate efficacy of pyruvate therapy for mitochondrial diseases. Mitochondrion (2015) 20:34–42. doi: 10.1016/j.mito.2014.10.006
70. Montero R, Yubero D, Villarroya J, Henares D, Jou C, Rodriguez MA, et al. GDF-15 is elevated in children with mitochondrial diseases and is induced by mitochondrial dysfunction. PloS One (2016) 11:e0148709. doi: 10.1371/journal.pone.0148709
71. Kalko SG, Paco S, Jou C, Rodriguez MA, Meznaric M, Rogac M, et al. Transcriptomic profiling of TK2 deficient human skeletal muscle suggests a role for the p53 signalling pathway and identifies growth and differentiation factor-15 as a potential novel biomarker for mitochondrial myopathies. BMC Genomics (2014) 15:91. doi: 10.1186/1471-2164-15-91
72. Morrow RM, Picard M, Derbeneva O, Leipzig J, McManus MJ, Gouspillou G, et al. Mitochondrial energy deficiency leads to hyperproliferation of skeletal muscle mitochondria and enhanced insulin sensitivity. Proc Natl Acad Sci U.S.A. (2017) 114:2705–10. doi: 10.1073/pnas.1700997114
73. Moos WH, Faller DV, Glavas IP, Harpp DN, Kamperi N, Kanara I, et al. Pathogenic mitochondrial dysfunction and metabolic abnormalities. Biochem Pharmacol (2021) 193:114809. doi: 10.1016/j.bcp.2021.114809
74. Bar-Ziv R, Bolas T, Dillin A. Systemic effects of mitochondrial stress. EMBO Rep (2020) 21:e50094. doi: 10.15252/embr.202050094
75. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes (2002) 51:2944–50. doi: 10.2337/diabetes.51.10.2944
76. Mansouri A, Gattolliat CH, Asselah T. Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterology (2018) 155:629–47. doi: 10.1053/j.gastro.2018.06.083
77. AlZaim I, Eid AH, Abd-Elrahman KS, El-Yazbi AF. Adipose tissue mitochondrial dysfunction and cardiometabolic diseases: On the search for novel molecular targets. Biochem Pharmacol (2022) 206:115337. doi: 10.1016/j.bcp.2022.115337
78. Perez-Carreras M, Del Hoyo P, Martin MA, Rubio JC, Martin A, Castellano G, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology (2003) 38:999–1007. doi: 10.1002/hep.1840380426
79. Pessayre D, Fromenty B. NASH: a mitochondrial disease. J Hepatol (2005) 42:928–40. doi: 10.1016/j.jhep.2005.03.004
80. Li X, Zhang W, Cao Q, Wang Z, Zhao M, Xu L, et al. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov (2020) 6:80. doi: 10.1038/s41420-020-00316-9
81. Pereira RO, Olvera AC, Marti A, Fang S, White JR, Westphal M, et al. OPA1 regulates lipid metabolism and cold-induced browning of white adipose tissue in mice. Diabetes (2022) 71:2572–83. doi: 10.2337/db22-0450
82. Heinonen S, Jokinen R, Rissanen A, Pietilainen KH. White adipose tissue mitochondrial metabolism in health and in obesity. Obes Rev (2020) 21:e12958. doi: 10.1111/obr.12958
83. Bhatt DL, Bays HE, Miller M, Cain JE 3rd, Wasilewska K, Andrawis NS, et al. Investigators, The FGF21 analog pegozafermin in severe hypertriglyceridemia: a randomized phase 2 trial. Nat Med (2023) 29:1782–92. doi: 10.1038/s41591-023-02427-z
Keywords: mitochondrial stress, FGF21, GDF15, integrated stress response, metabolic health, energy balance
Citation: Jena J, García-Peña LM and Pereira RO (2023) The roles of FGF21 and GDF15 in mediating the mitochondrial integrated stress response. Front. Endocrinol. 14:1264530. doi: 10.3389/fendo.2023.1264530
Received: 20 July 2023; Accepted: 11 September 2023;
Published: 25 September 2023.
Edited by:
Joanna K. Filipowska, City of Hope National Medical Center, United StatesReviewed by:
Kai Wang, Shanghai Children’s Medical Center, ChinaCopyright © 2023 Jena, García-Peña and Pereira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renata O. Pereira, cmVuYXRhLXBlcmVpcmFAdWlvd2EuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.