 Hubert Kolb1,2
Hubert Kolb1,2 Kerstin Kempf
Kerstin Kempf- 1Faculty of Medicine, Heinrich-Heine-University Düsseldorf, Düsseldorf, Germany
- 2West-German Centre of Diabetes and Health, Düsseldorf Catholic Hospital Group, Düsseldorf, Germany
Experimental studies in animal models of aging such as nematodes, fruit flies or mice have observed that decreased levels of insulin or insulin signaling promotes longevity. In humans, hyperinsulinemia and concomitant insulin resistance are associated with an elevated risk of age-related diseases suggestive of a shortened healthspan. Age-related disorders include neurodegenerative diseases, hypertension, cardiovascular disease, and type 2 diabetes. High ambient insulin concentrations promote increased lipogenesis and fat storage, heightened protein synthesis and accumulation of non-functional polypeptides due to limited turnover capacity. Moreover, there is impaired autophagy activity, and less endothelial NO synthase activity. These changes are associated with mitochondrial dysfunction and oxidative stress. The cellular stress induced by anabolic activity of insulin initiates an adaptive response aiming at maintaining homeostasis, characterized by activation of the transcription factor Nrf2, of AMP activated kinase, and an unfolded protein response. This protective response is more potent in the long-lived human species than in short-lived models of aging research resulting in a stronger pro-aging impact of insulin in nematodes and fruit flies. In humans, resistance to insulin-induced cell stress decreases with age, because of an increase of insulin and insulin resistance levels but less Nrf2 activation. These detrimental changes might be contained by adopting a lifestyle that promotes low insulin/insulin resistance levels and enhances an adaptive response to cellular stress, as observed with dietary restriction or exercise.
Introduction
Humans and most animal species exhibit the phenomenon of aging prior to dying a natural death. There is an age-dependent increase of physical damage to cellular constituents and changes in cellular and organ function.
At the cellular level, age associated damage includes the accumulation of defective macromolecules such as oxidized lipids, proteins and deoxyribonucleic acid (DNA), as well as the formation of protein aggregates. There is increased production of free radicals and less adenosine triphosphate (ATP) from dysfunctional mitochondria in the context of lower availability of nicotinamide adenine dinucleotide (NAD+) and altered nutrient sensing. Cell repair and turnover mechanisms are impaired as evident from impaired proteostasis, decreased autophagy and lower stem cell activity (1–4). In most cell types, cell division is accompanied by shortening of telomeres which may prohibit proper replication of chromosomes. Further, aging is associated with modifications of DNA and histones, and there is a strong correlation between methylation patterns of DNA and chronological or biological age (5). Several of these defects initiate cellular senescence, a functional state with replicative arrest, resistance to apoptosis, often associated with secretion of a variable combination of soluble factors and exosomes which promote low-grade inflammation, fibrosis and senescence of additional cells (4, 6). There is impairment of immune functions, termed immunosenescence. Probably all organs exhibit altered or deficient functions, including the microbiome (1–11).
Can a natural course of aging be defined? Is there a primary lesion which kicks off a cascade of defects, and what is the role of insulin in this process? First of all, there is a genetic basis to the duration of life, otherwise the strikingly different lifespans between species such as between mice and humans or frogs and turtles could not be explained (12, 13). However, follow-up studies of the aging process have as yet failed to identify a primary cause and a standard sequence of events leading to functional decline of cells, organs and the organism. It has been suggested that DNA damage is an early lesion preceding other defects such as increased levels of oxygen radicals (14). However, it cannot be excluded that intracellular free radicals contribute to the accumulation of damaged DNA. It could also be argued that the primary lesion is a defective DNA repair response which would also promote the accumulation of DNA lesions. Alternatively, an impaired ability to scavenge radicals might precede increased levels of oxygen radicals (15, 16). Because of the interdependence between DNA damage, mitochondrial dysfunction, increased levels of free radicals, deficient autophagy, telomere attrition, loss of proteostasis, enhanced pro-inflammatory gene expression and cell regenerative activities, these different processes probably are part of a functional network. Aging could then be viewed as deterioration of a physiological network active within and between cells rather than being due to one primary damage initiating a linear chain of molecular events (8).
In support of the network concept is the experience from anti-aging trials. The DrugAge database of aging-related drugs lists several hundred compounds for which significant extension of the lifespan in at least one model has been reported. Drug targets include many different cell functions ranging from glutathione metabolism to synaptic transmission which argues against a dominant role of defects in only one cellular compartment (17). Similarly, genes associated with increased longevity code for many different cellular functions rather than for one critical process. Aging-associated genes are more likely to participate in the crosstalk between different pathways or biological processes, and there seems to be a network of “aging genes” directly interacting with each other (18).
We conclude that cell, organ and organismal physiology has several “weak spots” with low resistance towards metabolic, inflammatory, toxic or other types of stress. For instance, depending on genetic background, environment, lifestyle or developmental stage, there may be accumulation of DNA damage in excess of DNA repair capacity, protein aggregation during periods of high peptide synthesis overburdening protein turnover or disaggregation mechanism, high levels of oxygen radical formation in the context of intense mitochondrial activity and failing radical scavenging responses, or accumulation of oxidized lipids because of deficient autophagy (Figure 1). Such conditions may arise as consequence of high anabolic cell activity such as in response to excess concentrations of anabolic hormones like growth hormone, insulin-like growth factor (IGF) or insulin. The growth hormone – IGF – insulin signaling axis is a major modulator of the aging process (19, 20). In the present review we focus on the role of insulin which differs from that of growth hormone and the IGF system in that it is strongly linked to nutrient sensing. We suggest here that the age-associated decrease of resistance towards cellular stress may explain the unfavorable effects of insulin during aging. Thus, the actions of insulin may promote aging because of insufficient ability to cope with the cellular stress incurred by the hormone’s anabolic function.
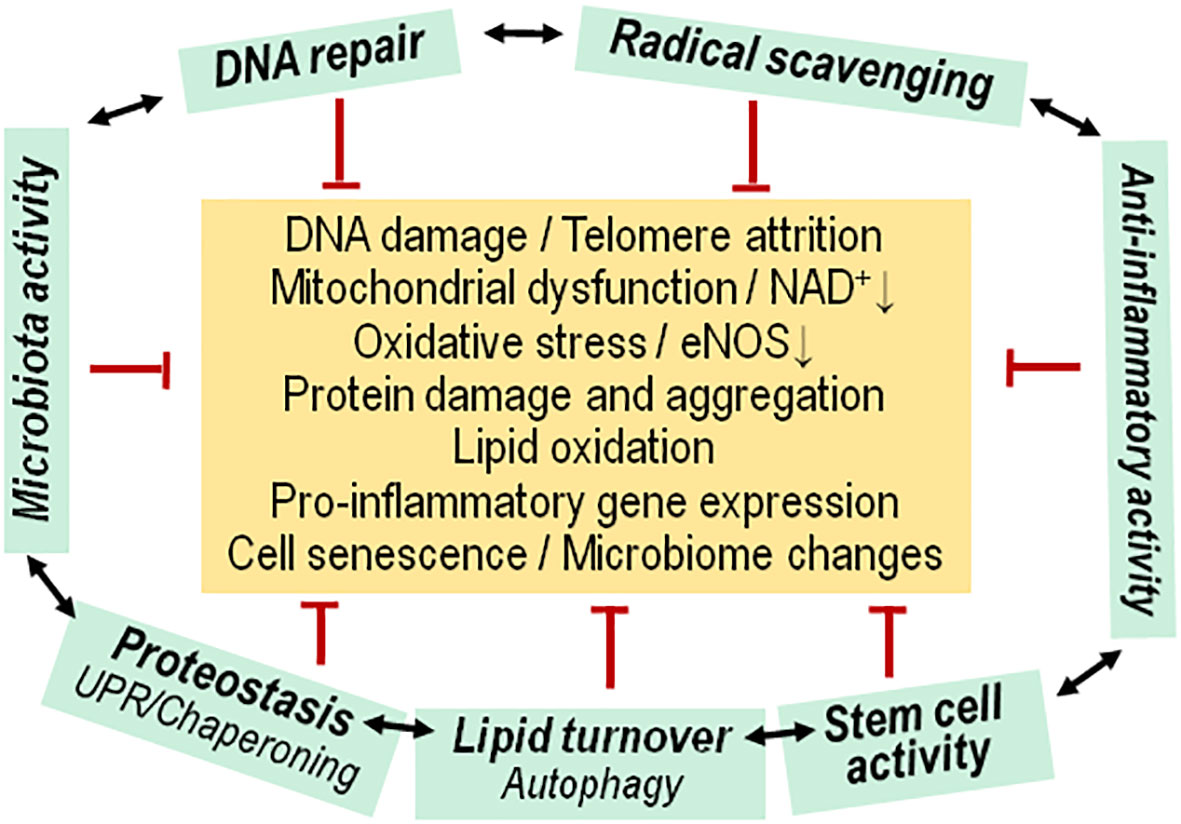
Figure 1 Functional network of cytoprotective pathways versus aging associated insults. Living cells experience a insults that usually initiate (green arrows) an adaptive, protective/repair response (red arrows) for maintaining cell functions, or there is replacement by newly differentiated cells. During the aging process, the adaptive response fails to maintain a normal physiological state of cells, Progenitor/stem cell activity is diminished, and there is concomitant dysfunction of the microbiota. Mitoch., mitochondrial.
Insulin and aging: genetics
There is a strong genetic basis to an aging-promoting effect of insulin or the insulin/IGF-1 signaling pathway. A single mutation with impact on the insulin/IGF-1 signal transduction pathway, either affecting the sole insulin/insulin-like growth factor receptor or the phosphatidyl-inositol-3-OH kinase (PI3K), more than doubles the natural lifespan of the nematode Caenorhabditis elegans (21–23). Lifespan regulation by the insulin/IGF-1 signaling pathway is similar in the fruit fly Drosophila melanogaster. Genetic interference with proper signal transduction by various approaches shares as outcome an extension of lifespan (24).
In mice or humans, the regulation of the insulin/IGF-1 signal transduction pathway is more complex because of an additional upstream anabolic hormone, growth hormone. This pituitary hormone promotes IGF-1 production from the liver and other tissues, but the two hormones have partly opposite effects. For instance, growth hormone induces insulin resistance but promotes insulin production whereas IGF-1 promotes insulin sensitivity and reduces insulin secretion (25). Therefore, outcomes of genetic disturbance of the regulatory balance between growth hormone, IGF-1 and insulin are difficult to interpret. In mice, disruption of the insulin receptor in adipose tissue was sufficient to increase median and maximum lifespan by 18% (26). A body-wide knockout of the insulin receptor leads to early postnatal lethality whereas mice heterozygous for mutant and wildtype receptors did not show an altered lifespan despite some functional impairment of insulin signaling (27). In another study, mice heterozygous for a knockout of the insulin receptor showed no differences in lifespan to wildtype littermates in females but an increase in maximum lifespan in males (28).
Many studies have observed an extended lifespan in mice if growth hormone expression, or binding to its receptor are impaired. Longevity is increased in both sexes of Ames or other dwarf mice with deficient production of growth hormone together with prolactin and thyroid stimulating hormone or with isolated growth hormone deficiency (29, 30). Mice with disruption of the growth hormone receptor gene express a similar phenotype (31). The longevity mechanism of mice with deficient growth hormone activity has not been fully elucidated, but it is of interest that there is a strong association with enhanced insulin sensitivity (32). Similar analyses of IGF-1 are hampered by the fact that lack of functional IGF-1 receptors severely impairs development. Therefore, mice heterozygous for a receptor gene knockout were analyzed. Prolongation of lifespan was modest and seen in female mice only (33–36). IGF-1 receptor function can also be affected by deletion of insulin receptor substrate genes. This approach also impairs insulin signaling. Mice lacking insulin receptor substrate 1 exhibit increased longevity (37). For the insulin receptor substrate 2 gene, deletion in all tissues of mice was not found to increase lifespan while deletion in brain tissue only promoted longevity (38) (Table 1).

Table 1 Genetic manipulation of anabolic hormone signaling versus lifespan.
The opposing effects of growth hormone and IGF-1 on insulin sensitivity and production leads to the question whether insulin action itself is more closely related to longevity than the two other anabolic hormones. In mice, modulation of circulating insulin levels and insulin sensitivity often but not always were reported to affect the lifespan which supports a role of insulin actions in the aging process. In one study, mice with reduced insulin sensitivity because of impaired insulin receptor function exhibited an increased lifespan in males but not in females. Increased insulin sensitivity because of deficiency of protein tyrosine phosphatase 1B or overexpressed peroxisome proliferator activated receptor gamma coactivator-1α was associated with a shortened lifespan (28). Another strain of mice with impaired insulin receptor function also exhibited insulin resistance and hyperinsulinemia, but without an impact on lifespan (27). Modest lowering of circulating insulin levels by 25 – 34% but not of IGF-1 via knocking out the Ins1 gene and one of two Ins2 alleles in female mice appeared to increase maximum lifespan (p < 0.059) (40).
In humans the contribution of single genes coding for components of the insulin/IGF-1 signaling pathway to longevity appears to be low with the exception of FOXO3A (41, 42) and possibly AKT1 (43, 44). However, the genetic association of single nucleotide polymorphisms with human longevity became significant when polymorphisms of 68 genes of the insulin/IGF-1 signaling pathway were analyzed together. The significance of the association was carried by alleles of nine genes, AKT1, AKT3, FOXO4, IGF2, INS, PIK3CA, SGK, SGK2, and YWHAG (45). This study did not observe the well documented association of FOXO3A with longevity, possibly because nonagenarians rather than centenarians were analyzed.
Taken together, a low activity state of the insulin/IGF-1 signaling pathway promotes longevity, effects are stronger in nematodes and fruit flies than in mice or humans, possibly due to the more complex regulatory network in mammals which has as additional player, growth hormone, which is not present invertebrates.
Another additional factor determining the outcome of insulin actions on longevity might be the overall metabolic rate. A high metabolic rate is associated with increased production of reactive oxygen species (ROS). For instance, small-breed domestic dogs exhibit a higher mass-specific metabolic and growth rate than large dogs, and therefore oxidative damage of lipids is seen. Nevertheless, small-breed dogs live significantly longer (46, 47). In mice, heavier body weight is associated with increased epigenetic aging and earlier death (48, 49). Similar findings have been reported for humans. In Southern Chinese adults, the basal metabolic rate was inversely correlated with all-cause mortality in males, but not in females (50). Within a local population, people of smaller size have a higher life expectancy, in different regions of the world (51). It may be concluded that within a species a higher growth rate is associated with shorter lifespan, but this is not explained by a higher metabolic rate.
Insulin and aging: epidemiological findings
In humans, epidemiological studies suggest a pro-aging effect of insulin. Insulin resistance increases with aging, but centenarians usually preserve normal glucose tolerance, low levels of fasting insulin and higher insulin sensitivity, when compared with adults > 75 years of age (52–54). The higher longevity in shorter men is also associated with lower fasting insulin concentrations (55).
In adults with normal glucose tolerance, there is a parallel increase of fasting insulin levels and insulin resistance with aging, and this is associated with central obesity (56, 57). Hyperinsulinemia and insulin resistance are important risk factors for type 2 diabetes as well as hypertension and cardiovascular disease (58–60). Age-related disorders associated with insulin resistance also include neurodegenerative diseases such as Alzheimer’s or Parkinson’s disease (61, 62).
Another approach of studying the health impact of hyperinsulinemia is to determine the insulinemic potential of the diet as assessed by food frequency questionnaires evaluated by measuring circulating C-peptide concentrations. Analyses of the prospective Nurses’ Health Study and the Health Professionals Follow-up Study (total of about 2,800,000 person-years) showed that a higher insulinemic potential of diet was associated with increased risk of all-cause, cardiovascular and cancer mortality (63). Of note, these associations were independent of BMI.
Insulin and aging: (patho)physiological aspects
Insulin is a potent anabolic hormone. Just doubling fasting insulin levels is enough for suppression of lipolysis by approximately 50% and promotion of lipogenesis in adipocytes while hepatic gluconeogenesis is not yet inhibited (reviewed in (64)). A Mendelian randomization analysis found that genetic variants which code for a higher insulin response to glucose challenge are strongly associated with increased BMI which is considered as proof of a causal relationship between increased insulin secretion and body weight gain (65). This fits with the observation that insulin therapy favors weight gain (66). Conversely, pharmacological lowering of circulating insulin concentrations in obese people by diazoxide caused greater weight loss than diet alone (67). Treatment of obese persons with the somatostatin analogue octreotide led to weight loss in conjunction with a decrease of insulin levels (68, 69). Lifestyle changes or other interventions known to improve risk factors of age-associated disease and cardiovascular mortality cause lower insulin levels, as reported for calorie-restricted diets, intermittent fasting or bariatric surgery (70–73). Vegetarian diets are also associated with lower insulin resistance and lower fasting insulin levels, even in comparison with matched lean controls, and appear to improve healthspan and possibly also lifespan (74, 75). Another lifestyle parameter associated with better healthspan is physical exercise, which causes lower fasting and post-challenge insulin levels as well as improved insulin sensitivity (76–78).
Although insulin is an essential hormone for growth and maintenance of complex organisms (79), the above findings suggest that elevated insulin levels promote age-associated diseases. One cellular response to permanently elevated insulin levels is partial downregulation of insulin signaling via the insulin receptor, causing the phenomenon of insulin resistance. This may involve decreased insulin receptor expression, but the major reason is impaired signal transduction because of diminished tyrosine autophosphorylation of the receptor, removal of bound phosphate residues by phosphatases and suboptimal downstream signaling along the insulin receptor substrate (IRS) – (PI3K) – protein kinase B (PKB/AKT) pathway (80–83). A higher amount of alternatively spliced type A insulin receptor lacking exon 11 also may contribute to insulin resistance by directing insulin signaling towards the mitogen activated kinase pathway which promotes cell proliferation and tumor development (84).
Signaling via the PI3K-AKT pathway is not only affected by modulation of insulin receptor function but also enzyme activities downstream. The diversity of proteins involved in the PI3K-AKT signaling pathway allows for varying outcomes of signaling, and this complexity is only partially resolved. It therefore is not surprising that “insulin resistance” does not mean full suppression of hormonal activity but only downregulation of some insulin functions such as induction of glucose transporter translocation to the cell membrane (85, 86). In addition to impaired glucose transport, insulin resistance suppresses the stimulatory effect of insulin on nitric oxide production from endothelial nitric oxide (NO) synthase because of deficient posttranslational modification of the enzyme via PI3K/AKT activity (87, 88). The resulting decreased arterial smooth muscle relaxation is aggravated by the non-suppressed insulin-dependent influx of calcium ions which enhances vascular contractility, resulting in upregulated vascular tone which increases the risk of vascular events (89, 90).
Other hormonal actions that are less or not affected by insulin resistance and may even be upregulated with the concomitant hyperinsulinemia include upregulation of PI3K-AKT dependent lipogenesis in hepatocytes and of the mechanistic target of rapamycin complex 1 (mTORC1) activity, the latter resulting in increased protein synthesis and impaired autophagy (91–95). Increased systemic insulin levels and concomitant insulin resistance during the progression to type 2 diabetes is associated with chronic overactivation of the mTORC1 signaling pathway and cell stress in the context of a high protein synthesis rate (96). During insulin resistance states (and concomitant hyperinsulinemia) there is, varying between tissues, phosphorylation of several Forkhead Box O (FOXO) transcription factors and their retention in the cytoplasm. resulting in suppression of muscle autophagy and protein degradation, among other effects (86, 97–99). The impact of elevated insulin levels on protein synthesis and autophagy is accompanied by the accumulation of proteins with multiple posttranslational modifications because of insufficient degradation which leads to endoplasmic reticulum stress (95, 100). Insulin signaling via phosphorylation of the Src homology 2 domain-containing transforming proteins (SHC) and subsequent activation of the mitogen-activated kinase protein kinase kinase (MEK) - extracellular signal-regulated kinase (ERK) is not affected by insulin resistance and contributes to these effects of hyperinsulinemia (Figure 2) (101, 102).
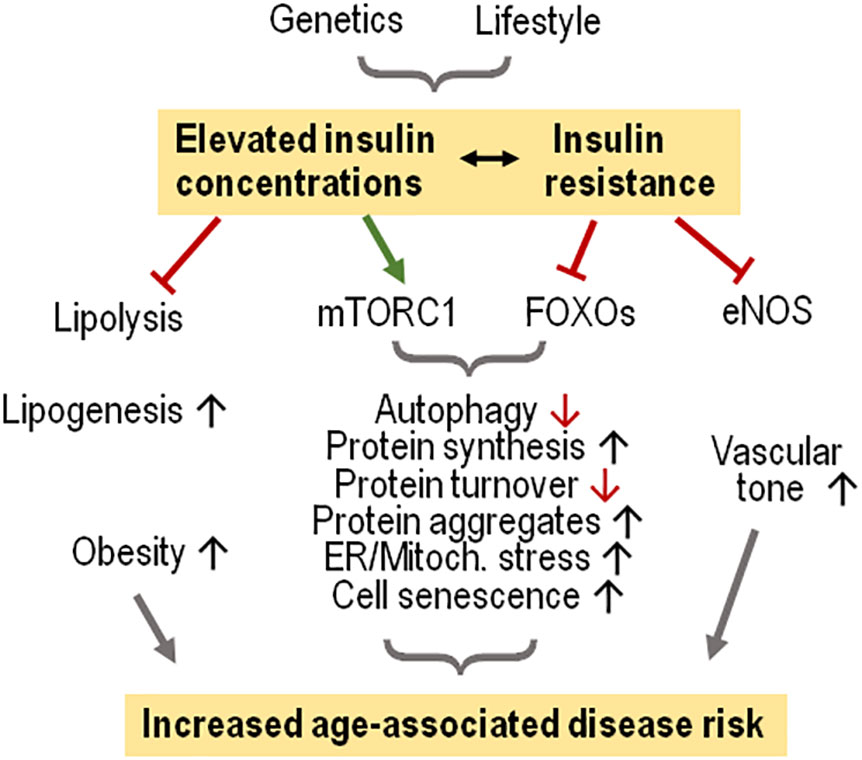
Figure 2 Elevated insulin levels and insulin resistance favor age-associated diseases in humans. Modest increases of insulin concentrations suffice to suppress lipolysis and support lipogenesis, promoting obesity. Hyperinsulinemia combined with insulin resistance cause activation of mTORC1 which in the context of less FOXO activation favors cell stress because of increased protein synthesis, eventually causing cell senescence. Insulin resistance impairs endothelial NO synthase (eNOS) activity, limiting vascular relaxation.
These findings suggest that increased insulin signaling because of elevated ambient levels causes cell stress, and there is a potentiating effect of insulin resistance. It therefore is not surprising that chronic exposure of human hepatocytes to high insulin levels (20 nmol/l) in vitro elicits a senescent cell phenotype, characterized by cell cycle arrest and adoption of a senescence-associated secretory phenotype which includes the secretion of proinflammatory mediators, microRNAs and vesicles (103). The promotion of hepatocyte senescence by hyperinsulinemia is absent in mice with a liver-specific knockout of the insulin receptor whereas enhanced senescence was still occurring in white adipose tissue. In obese persons undergoing bariatric surgery, insulin levels were closely associated with markers of senescence in liver tissue (104). Increased levels of insulin were also observed to promote senescence of human adipocytes in vitro as well as in vivo (105). High ambient insulin concentrations also drive mouse neurons into a senescence-like state, in vitro and in vivo (106).
Another age-associated marker is DNA damage. Prolonged incubation of animal or human cells with 0.5 nmol/l insulin caused DNA damage in the context of increased radical oxygen species production from nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondria (107). Whether insulin resistance or the concomitant hyperinsulinemia promotes enhances telomere attrition in peripheral blood leukocytes in addition to cell stress has not been studied in detail. Cross-sectional studies suggest that that insulin resistance is associated with increased telomere shortening in some groups but not in others (108–112). A positive association was also noted in the follow-up of cohorts (113–115) with one exception (116). These observational studies also found an association between telomere attrition and other parameters such as adiposity, hypertension or circulating sirtuin-1 concentrations. Therefore, the association between telomere length and insulin levels may also be indirect.
Insulin and aging: failure of adaptive response
As reviewed above, high insulin concentrations cause cell stress because of excess anabolic activity which include (i), increased lipogenesis and fat storage also in non-adipocytes, (ii), increased protein synthesis and accumulation of non-functional polypeptides because of limited turnover capacity, (iii), impaired autophagy activity, (iv) increased progression of stressed cells towards a senescent stage. These changes are associated with mitochondrial dysfunction and increased levels of radical oxygen species (117, 118). Hyperinsulinemia usually is accompanied by insulin resistance, but there is only partial suppression of insulin signaling, favoring lipogenesis as well as mTORC1 activation for protein synthesis and autophagy inhibition. The relevance of enhanced mTORC1 activation for the aging process has been demonstrated by treating mice with the mTORC1 inhibitor rapamycin which resulted in less proliferative and protein synthesis activity concomitant with improved autophagy and increased longevity. These changes resemble effects of dietary restriction. However, pharmacological inhibition of mTOR may reach a degree where detrimental consequences to the physiological balance are noted such as impaired immune cell activation, insulin resistance and beta islet cell damage (95, 119, 120). Insulin resistance in the presence of hyperinsulinemia helps maintain glucose homeostasis and decreasing metabolic and oxidative stress by depressing excess glucose influx (121, 122). However, the concomitant suppression of NO production from endothelial NO synthase favors a pro-oxidant and inflammatory vascular milieu as well as vasoconstriction potentially favoring vascular damage (Figure 2) (87–89).
Taken together, hyperinsulinemia in the context of insulin resistance appears to exhibit a pro-aging role. Whether these effects become clinically relevant probably depends on the body’s ability to mount an appropriate defense response for containing the detrimental consequences of hyperinsulinemia and insulin resistance. One well documented health risk associated with increased insulin levels is type 2 diabetes. We have previously argued that the progression to overt type 2 diabetes is prevented if there is a persistent protective/adaptive response which includes an anti-inflammatory defense response to nutrient-induced inflammation, increased neutralization of free radicals and improved mitochondrial function for the reduction of oxidative stress, and an upregulated ability to lessen endoplasmic reticulum stress by an unfolded protein response and autophagy (123). We suggest here that this adaptive (hormetic) response also controls the pro-aging effect of insulin. The stress signals involved in inducing a hormetic response include oxygen radicals, misfolded proteins and decreased levels of ATP (Figure 3).
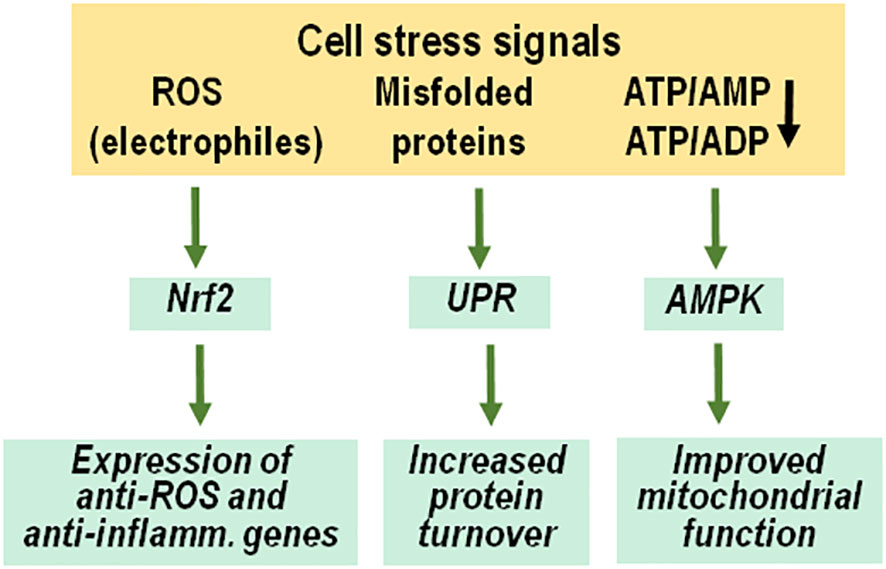
Figure 3 Adaptive response to anabolic cell stress. Molecular signals of cell stress include radical oxygen species and other electrophiles, misfolded proteins and decreased ATP/AMP and ATP/ADP ratios because of enhanced ATP consumption. These signals initiate an adaptive response to increase cellular resistance and restore proper physiological functions, including activation and nuclear transfer of Nrf2, an unfolded protein response and stimulation of AMP-activated protein kinases. ROS, radical oxygen species; UPR, unfolded protein response; AMPK, AMP-activated protein kinases, inflamm., inflammatory.
Oxygen radicals initiate a cell protective response by activation of nuclear factor erythroid 2 – related factor 2 (Nrf2), a key transcriptional factor for the expression of more than 250 genes involved in cytoprotective processes such as redox regulation, xenobiotic metabolism, DNA repair, and protein homeostasis including the unfolded protein response (124–126). There is impairment of pro-inflammatory gene expression, including the suppression of nuclear factor kappa B (NFkB) and pro-inflammatory cytokines (127, 128). Another effect of Nrf2 activation is the support of endothelial NO synthase expression and NO production (129). Thus, activation of Nrf2 is an appropriate adaptive cellular response to the oxidative, inflammatory and vascular stress caused by hyperinsulinemia and concomitant insulin resistance, with an impact on aging (130).
Loss of proteostasis because of excessive protein synthesis is a major consequence of an acute rise of insulin levels, but this is apparently contained by the unfolded protein response of the endoplasmic reticulum (100). Misfolded proteins signal the loss of proteostasis by binding to chaperone sensors which initiates a transcriptional program leading to a general increase of mechanisms involved in protein synthesis and turnover, the unfolded protein response (131). This protective cell response is impaired in the presence of experimentally induced or diabetes-associated insulin resistance (132). Low chaperone activity causes cell senescence (133).
A third important signal of cell stress is a decrease of ATP levels versus adenosine diphosphate (ADP) and adenosine monophosphate (AMP) concentrations, which results from increased consumption and deficient production of ATP. Low ATP levels lead to the activation of AMP-activated protein kinases. This group of kinases modulates the activity of many metabolic enzymes, histones and transcription factors by phosphorylation and by promoting their acetylation. One important consequence is the restoration of mitochondrial homeostasis (134–136).
As mentioned, several lifestyle factors have been observed to lower levels of fasting and postprandial insulin as well as of insulin resistance. These factors include dietary restriction and exercise (72, 137). Interestingly, dietary restriction or exercise cause an initial increase of oxidative or electrophile stress. The resulting activation of the Nrf2 system appears to mediate much of the health effects observed (138–141). Many dietary phytochemicals such as polyphenols also cause the activation of Nrf2, in part with an involvement of the hydrocarbon receptor (102, 142, 143). Another pathway of improving insulin resistance and concomitant hyperinsulinemia by lifestyle changes involves the gut, possibly by modulation of gut microbiota composition and activity may decrease gut leakage. The resulting lower levels of bacterial compounds in circulation is associated with decreased production of pro-inflammatory immune mediators and increased insulin sensitivity (144).
Discussion
The anabolic hormone insulin induces cell stress because of increased biosynthetic activity and reduced clearance/repair of damaged cellular components. Insulin resistance is a potentiating factor because of increased signaling via the mitogen-activated kinase pathway and less production of NO by endothelial NO synthase. These potentially aging-promoting effects are contained by an adaptive cellular activity characterized by anti-oxidative, anti-inflammatory, protein chaperone, DNA repair and overall turnover process which is more potent in the long-lived human species than in short-lived models of aging research (145–147). Therefore, the pro-aging impact of insulin is less controlled in short-lived animal models such as nematodes and fruit flies. The balance between insulin/insulin resistance induced cell stress and the cytoprotective response determines detrimental effects of hyperinsulinemia and insulin resistance. Controlling factors are, on the one side, levels of insulin and insulin resistance, and, on the other side, the quality of cellular resistance to anabolic stress. This fits with the observation that centenarians exhibit low circulating insulin concentrations as well as high insulin sensitivity.
Of note, lifestyle factors that are considered to improve healthspan and possibly lifespan in humans modify both sides of the balance. Dietary restriction and exercise have been found to lower levels of insulin and insulin resistance. Concomitantly, dietary restriction, dietary phytochemicals and exercise activate the Nrf2-dependent cellular stress response and modify microbiota composition and function in a favorable way. During aging, the cell stress response via Nrf2 becomes less potent but possibly not in centenarians (148–151), and there is an age-dependent increase of circulating insulin and insulin resistance (52, 152). Both processes are supporting the pro-aging effects of insulin, and both may be targeted by dietary restriction and exercise.
Author contributions
HK: Writing – original draft. KK: Writing – review & editing. SM: Writing – review & editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. The work was supported by Gesellschaft von Freunden und Förderern der Heinrich-Heine-Universität Düsseldorf e.V.
Acknowledgments
We thank Fraser W. Scott, the Ottawa Hospital Research Institute and University of Ottawa, Canada, for reviewing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jones DL, Rando TA. Emerging models and paradigms for stem cell ageing. Nat Cell Biol (2011) 13:506–12. doi: 10.1038/ncb0511-506
2. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell (2023) 186:243–78. doi: 10.1016/j.cell.2022.11.001
3. Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E. From discoveries in ageing research to therapeutics for healthy ageing. Nature (2019) 571:183–92. doi: 10.1038/s41586-019-1365-2
4. da Silva PFL, Schumacher B. Principles of the molecular and cellular mechanisms of aging. J Invest Dermatol (2021) 141:951–60. doi: 10.1016/j.jid.2020.11.018
5. Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet (2018) 19:371–84. doi: 10.1038/s41576-018-0004-3
6. Wiley CD, Campisi J. The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat Metab (2021) 3:1290–301. doi: 10.1038/s42255-021-00483-8
7. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol (2018) 14:576–90. doi: 10.1038/s41574-018-0059-4
8. Cohen AA, Ferrucci L, Fulop T, Gravel D, Hao N, Kriete A, et al. A complex systems approach to aging biology. Nat Aging (2022) 2:580–91. doi: 10.1038/s43587-022-00252-6
9. Kim M, Benayoun BA. The microbiome: an emerging key player in aging and longevity. Transl Med Aging (2020) 4:103–16. doi: 10.1016/j.tma.2020.07.004
10. Tripathi U, Misra A, Tchkonia T, Kirkland JL. Impact of senescent cell subtypes on tissue dysfunction and repair: importance and research questions. Mech Ageing Dev (2021) 198:111548. doi: 10.1016/j.mad.2021.111548
11. Palmer AK, Jensen MD. Metabolic changes in aging humans: current evidence and therapeutic strategies. J Clin Invest (2022) 132: e158451. doi: 10.1172/JCI158451
12. Jones OR, Scheuerlein A, Salguero-Gomez R, Camarda CG, Schaible R, Casper BB, et al. Diversity of ageing across the tree of life. Nature (2014) 505:169–73. doi: 10.1038/nature12789
13. Milholland B, Vijg J. Why Gilgamesh failed: the mechanistic basis of the limits to human lifespan. Nat Aging (2022) 2:878–84. doi: 10.1038/s43587-022-00291-z
14. Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ. The central role of DNA damage in the ageing process. Nature (2021) 592:695–703. doi: 10.1038/s41586-021-03307-7
15. Rattan SI, Clark BF. Understanding and modulating ageing. IUBMB Life (2005) 57:297–304. doi: 10.1080/15216540500092195
16. Leyane TS, Jere SW, Houreld NN. Oxidative stress in ageing and chronic degenerative pathologies: molecular mechanisms involved in counteracting oxidative stress and chronic inflammation. Int J Mol Sci (2022) 23:7273. doi: 10.3390/ijms23137273
17. Barardo D, Thornton D, Thoppil H, Walsh M, Sharifi S, Ferreira S, et al. The DrugAge database of aging-related drugs. Aging Cell (2017) 16:594–7. doi: 10.1111/acel.12585
18. Zhang Q, Nogales-Cadenas R, Lin JR, Zhang W, Cai Y, Vijg J, et al. Systems-level analysis of human aging genes shed new light on mechanisms of aging. Hum Mol Genet (2016) 25:2934–47. doi: 10.1093/hmg/ddw145
19. Junnila RK, List EO, Berryman DE, Murrey JW, Kopchick JJ. The GH/IGF-1 axis in ageing and longevity. Nat Rev Endocrinol (2013) 9:366–76. doi: 10.1038/nrendo.2013.67
20. Bartke A. Growth hormone and aging. Rev Endocr Metab Disord (2021) 22:71–80. doi: 10.1007/s11154-020-09593-2
21. Friedman DB, Johnson TE. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics (1988) 118:75–86. doi: 10.1093/genetics/118.1.75
22. Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature (1993) 366:461–4. doi: 10.1038/366461a0
23. Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science (1997) 277:942–6. doi: 10.1126/science.277.5328.942
24. Altintas O, Park S, Lee SJ. The role of insulin/IGF-1 signaling in the longevity of model invertebrates, C. elegans and D. melanogaster. BMB Rep (2016) 49:81–92. doi: 10.5483/BMBRep.2016.49.2.261
25. Bartke A, Brown-Borg H. Mutations affecting mamMalian aging: GH and GHR vs IGF-1 and insulin. Front Genet (2021) 12:667355. doi: 10.3389/fgene.2021.667355
26. Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science (2003) 299:572–4. doi: 10.1126/science.1078223
27. Shimizu T, Baba T, Ogawara M, Shirasawa T. Lifespan and glucose metabolism in insulin receptor mutant mice. J Aging Res (2011) 2011:315640. doi: 10.4061/2011/315640
28. Nelson JF, Strong R, Bokov A, Diaz V, Ward W. Probing the relationship between insulin sensitivity and longevity using genetically modified mice. J Gerontol A Biol Sci Med Sci (2012) 67:1332–8. doi: 10.1093/gerona/gls199
29. Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature (1996) 384:33. doi: 10.1038/384033a0
30. Alba M, Salvatori R. A mouse with targeted ablation of the growth hormone-releasing hormone gene: a new model of isolated growth hormone deficiency. Endocrinology (2004) 145:4134–43. doi: 10.1210/en.2004-0119
31. Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, et al. A mamMalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc Natl Acad Sci U.S.A. (1997) 94:13215–20. doi: 10.1073/pnas.94.24.13215
32. Masternak MM, Panici JA, Bonkowski MS, Hughes LF, Bartke A. Insulin sensitivity as a key mediator of growth hormone actions on longevity. J Gerontol A Biol Sci Med Sci (2009) 64:516–21. doi: 10.1093/gerona/glp024
33. Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature (2003) 421:182–7. doi: 10.1038/nature01298
34. Bokov AF, Garg N, Ikeno Y, Thakur S, Musi N, DeFronzo RA, et al. Does reduced IGF-1R signaling in Igf1r+/- mice alter aging? PloS One (2011) 6:e26891. doi: 10.1371/journal.pone.0026891
35. Xu J, Gontier G, Chaker Z, Lacube P, Dupont J, Holzenberger M. Longevity effect of IGF-1R(+/-) mutation depends on genetic background-specific receptor activation. Aging Cell (2014) 13:19–28. doi: 10.1111/acel.12145
36. Garratt M, Nakagawa S, Simons MJP. Life-span extension with reduced somatotrophic signaling: moderation of aging effect by signal type, sex, and experimental cohort. J Gerontol A Biol Sci Med Sci (2017) 72:1620–6. doi: 10.1093/gerona/glx010
37. Selman C, Partridge L, Withers DJ. Replication of extended lifespan phenotype in mice with deletion of insulin receptor substrate 1. PloS One (2011) 6:e16144. doi: 10.1371/journal.pone.0016144
38. Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science (2007) 317:369–72. doi: 10.1126/science.1142179
39. Coschigano KT, Holland AN, Riders ME, List EO, Flyvbjerg A, Kopchick JJ. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology (2003) 144:3799–810. doi: 10.1210/en.2003-0374
40. Templeman NM, Flibotte S, Chik JHL, Sinha S, Lim GE, Foster LJ, et al. Reduced circulating insulin enhances insulin sensitivity in old mice and extends lifespan. Cell Rep (2017) 20:451–63. doi: 10.1016/j.celrep.2017.06.048
41. Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, et al. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U.S.A. (2008) 105:13987–92. doi: 10.1073/pnas.0801030105
42. Zhao Y, Liu YS. Longevity factor FOXO3: A key regulator in aging-related vascular diseases. Front Cardiovasc Med (2021) 8:778674. doi: 10.3389/fcvm.2021.778674
43. Pawlikowska L, Hu D, Huntsman S, Sung A, Chu C, Chen J, et al. Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity. Aging Cell (2009) 8:460–72. doi: 10.1111/j.1474-9726.2009.00493.x
44. Caruso C, Ligotti ME, Accardi G, Aiello A, Duro G, Galimberti D, et al. How important are genes to achieve longevity? Int J Mol Sci (2022) 23(10):5635. doi: 10.3390/ijms23105635
45. Deelen J, Uh HW, Monajemi R, van HD, Thijssen PE, Bohringer S, et al. Gene set analysis of GWAS data for human longevity highlights the relevance of the insulin/IGF-1 signaling and telomere maintenance pathways. Age (Dordr ) (2013) 35:235–49. doi: 10.1007/s11357-011-9340-3
46. Greer KA, Canterberry SC, Murphy KE. Statistical analysis regarding the effects of height and weight on life span of the domestic dog. Res Vet Sci (2007) 82:208–14. doi: 10.1016/j.rvsc.2006.06.005
47. Jimenez AG, Downs CJ. Untangling life span and body mass discrepancies in canids: phylogenetic comparison of oxidative stress in blood from domestic dogs and wild canids. Am J Physiol Regul Integr Comp Physiol (2020) 319:R203–10. doi: 10.1152/ajpregu.00067.2020
48. Sandoval-Sierra JV, Helbing AHB, Williams EG, Ashbrook DG, Roy S, Williams RW, et al. Body weight and high-fat diet are associated with epigenetic aging in female members of the BXD murine family. Aging Cell (2020) 19:e13207. doi: 10.1111/acel.13207
49. Miller RA, Harper JM, Galecki A, Burke DT. Big mice die young: early life body weight predicts longevity in genetically heterogeneous mice. Aging Cell (2002) 1:22–9. doi: 10.1046/j.1474-9728.2002.00006.x
50. Han F, Hu F, Wang T, Zhou W, Zhu L, Huang X, et al. Association between basal metabolic rate and all-cause mortality in a prospective cohort of southern chinese adults. Front Physiol (2021) 12:790347. doi: 10.3389/fphys.2021.790347
51. Samaras TT. Should we be concerned over increasing body height and weight? Exp Gerontol (2009) 44:83–92. doi: 10.1016/j.exger.2008.02.002
52. Barbieri M, Gambardella A, Paolisso G, Varricchio M. Metabolic aspects of the extreme longevity. Exp Gerontol (2008) 43:74–8. doi: 10.1016/j.exger.2007.06.003
53. Arai Y, Kojima T, Takayama M, Hirose N. The metabolic syndrome, IGF-1, and insulin action. Mol Cell Endocrinol (2009) 299:124–8. doi: 10.1016/j.mce.2008.07.002
54. Vitale G, Barbieri M, Kamenetskaya M, Paolisso G. GH/IGF-I/insulin system in centenarians. Mech Ageing Dev (2017) 165:107–14. doi: 10.1016/j.mad.2016.12.001
55. He Q, Morris BJ, Grove JS, Petrovitch H, Ross W, Masaki KH, et al. Shorter men live longer: association of height with longevity and FOXO3 genotype in American men of Japanese ancestry. PloS One (2014) 9:e94385. doi: 10.1371/journal.pone.0094385
56. Cefalu WT, Wang ZQ, Werbel S, Bell-Farrow A, Crouse JR III, Hinson WH, Terry JG, et al. Contribution of visceral fat mass to the insulin resistance of aging. Metabolism (1995) 44:954–9. doi: 10.1016/0026-0495(95)90251-1
57. Iozzo P, Beck-Nielsen H, Laakso M, Smith U, Yki-Jarvinen H, Ferrannini E. Independent influence of age on basal insulin secretion in nondiabetic humans. European Group for the Study of Insulin Resistance. J Clin Endocrinol Metab (1999) 84:863–8. doi: 10.1210/jcem.84.3.5542
58. Marott SCW, Nordestgaard BG, Tybjaerg-Hansen A, Benn M. Causal associations in type 2 diabetes development. J Clin Endocrinol Metab (2019) 104:1313–24. doi: 10.1210/jc.2018-01648
59. Wang F, Han L, Hu D. Fasting insulin, insulin resistance and risk of hypertension in the general population: A meta-analysis. Clin Chim Acta (2017) 464:57–63. doi: 10.1016/j.cca.2016.11.009
60. Reaven G. Insulin resistance and coronary heart disease in nondiabetic individuals. Arterioscler Thromb Vasc Biol (2012) 32:1754–9. doi: 10.1161/ATVBAHA.111.241885
61. Kellar D, Craft S. Brain insulin resistance in Alzheimer's disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol (2020) 19:758–66. doi: 10.1016/S1474-4422(20)30231-3
62. Athauda D, Foltynie T. Insulin resistance and Parkinson's disease: A new target for disease modification? Prog Neurobiol (2016) 145-146:98–120. doi: 10.1016/j.pneurobio.2016.10.001
63. Wan Y, Tabung FK, Lee DH, Fung TT, Willett WC, Giovannucci EL. Dietary insulinemic potential and risk of total and cause-specific mortality in the nurses' Health study and the health professionals follow-up study. Diabetes Care (2022) 45:451–9. doi: 10.2337/dc21-1530
64. Kolb H, Stumvoll M, Kramer W, Kempf K, Martin S. Insulin translates unfavourable lifestyle into obesity. BMC Med (2018) 16:232. doi: 10.1186/s12916-018-1225-1
65. Astley CM, Todd JN, Salem RM, Vedantam S, Ebbeling CB, Huang PL, et al. Genetic evidence that carbohydrate-stimulated insulin secretion leads to obesity. Clin Chem (2018) 64:192–200. doi: 10.1373/clinchem.2017.280727
66. Koivisto VA. Insulin therapy in type II diabetes. Diabetes Care (1993) 16 Suppl 3:29–39. doi: 10.2337/diacare.16.3.29
67. Alemzadeh R, Langley G, Upchurch L, Smith P, Slonim AE. Beneficial effect of diazoxide in obese hyperinsulinemic adults. J Clin Endocrinol Metab (1998) 83:1911–5. doi: 10.1210/jc.83.6.1911
68. Doyle ME, Egan JM. Pharmacological agents that directly modulate insulin secretion. Pharmacol Rev (2003) 55:105–31. doi: 10.1124/pr.55.1.7
69. Lustig RH, Greenway F, Velasquez-Mieyer P, Heimburger D, Schumacher D, Smith D, et al. A multicenter, randomized, double-blind, placebo-controlled, dose-finding trial of a long-acting formulation of octreotide in promoting weight loss in obese adults with insulin hypersecretion. Int J Obes (Lond) (2006) 30:331–41. doi: 10.1038/sj.ijo.0803074
70. Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: norMalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia (2011) 54:2506–14. doi: 10.1007/s00125-011-2204-7
71. Kraus WE, Bhapkar M, Huffman KM, Pieper CF, Krupa DS, Redman LM, et al. 2 years of calorie restriction and cardiometabolic risk (CALERIE): exploratory outcomes of a multicentre, phase 2, randomised controlled trial. Lancet Diabetes Endocrinol (2019) 7:673–83. doi: 10.1016/S2213-8587(19)30151-2
72. Mattson MP, Longo VD, Harvie M. Impact of intermittent fasting on health and disease processes. Ageing Res Rev (2017) 39:46–58. doi: 10.1016/j.arr.2016.10.005
73. Pories WJ, Dohm GL. Diabetes: have we got it all wrong? Hyperinsulinism as the culprit: surgery provides the evidence. Diabetes Care (2012) 35:2438–42. doi: 10.2337/dc12-0684
74. Valachovicova M, Krajcovicova-Kudlackova M, Blazicek P, Babinska K. No evidence of insulin resistance in normal weight vegetarians. A Case control study. Eur J Nutr (2006) 45:52–4. doi: 10.1007/s00394-005-0563-x
75. Norman K, Klaus S. Veganism, aging and longevity: new insight into old concepts. Curr Opin Clin Nutr Metab Care (2020) 23:145–50. doi: 10.1097/MCO.0000000000000625
76. Jelleyman C, Yates T, O'Donovan G, Gray LJ, King JA, Khunti K, et al. The effects of high-intensity interval training on glucose regulation and insulin resistance: a meta-analysis. Obes Rev (2015) 16:942–61. doi: 10.1111/obr.12317
77. Short KR, Vittone JL, Bigelow ML, Proctor DN, Rizza RA, Coenen-Schimke JM, et al. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes (2003) 52:1888–96. doi: 10.2337/diabetes.52.8.1888
78. Timmerman KL, Ballard KD, Volk GA, Deal MA, Meisler AJ, Karrow JM, et al. Altering physical activity influences insulin responses to glucose ingestion in healthy adults. Int J Sports Med (2018) 39:972–7. doi: 10.1055/a-0735-9641
79. Accili D, Drago J, Lee EJ, Johnson MD, Cool MH, Salvatore P, et al. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet (1996) 12:106–9. doi: 10.1038/ng0196-106
80. Bertacca A, Ciccarone A, Cecchetti P, Vianello B, Laurenza I, Maffei M, et al. Continually high insulin levels impair Akt phosphorylation and glucose transport in human myoblasts. Metabolism (2005) 54:1687–93. doi: 10.1016/j.metabol.2005.06.019
81. Catalano KJ, Maddux BA, Szary J, Youngren JF, Goldfine ID, Schaufele F. Insulin resistance induced by hyperinsulinemia coincides with a persistent alteration at the insulin receptor tyrosine kinase domain. PloS One (2014) 9:e108693. doi: 10.1371/journal.pone.0108693
82. Chen Y, Huang L, Qi X, Chen C. Insulin receptor trafficking: consequences for insulin sensitivity and diabetes. Int J Mol Sci (2019) 20:5007. doi: 10.3390/ijms20205007
83. Pei J, Wang B, Wang D. Current studies on molecular mechanisms of insulin resistance. J Diabetes Res (2022) 2022:1863429. doi: 10.1155/2022/1863429
84. Renna LV, Bose F, Brigonzi E, Fossati B, Meola G, Cardani R. Aberrant insulin receptor expression is associated with insulin resistance and skeletal muscle atrophy in myotonic dystrophies. PloS One (2019) 14:e0214254. doi: 10.1371/journal.pone.0214254
85. Madsen RR, Vanhaesebroeck B. Cracking the context-specific PI3K signaling code. Sci Signal (2020) 13:eaay2940. doi: 10.1126/scisignal.aay2940
86. Batista TM, Haider N, Kahn CR. Defining the underlying defect in insulin action in type 2 diabetes. Diabetologia (2021) 64:994–1006. doi: 10.1007/s00125-021-05415-5
87. Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). J Biol Chem (2001) 276:30392–8. doi: 10.1074/jbc.M103702200
88. King GL, Park K, Li Q. Selective insulin resistance and the development of cardiovascular diseases in diabetes: the 2015 edwin bierman award lecture. Diabetes (2016) 65:1462–71. doi: 10.2337/db16-0152
89. Gutierrez A, Contreras C, Sanchez A, Prieto D. Role of phosphatidylinositol 3-kinase (PI3K), mitogen-activated protein kinase (MAPK), and protein kinase C (PKC) in calcium signaling pathways linked to the alpha(1)-adrenoceptor in resistance arteries. Front Physiol (2019) 10:55. doi: 10.3389/fphys.2019.00055
90. Pinho JF, Medeiros MA, Capettini LS, Rezende BA, Campos PP, Andrade SP, et al. Phosphatidylinositol 3-kinase-delta up-regulates L-type Ca2+ currents and increases vascular contractility in a mouse model of type 1 diabetes. Br J Pharmacol (2010) 161:1458–71. doi: 10.1111/j.1476-5381.2010.00955.x
91. Semple RK, Sleigh A, Murgatroyd PR, Adams CA, Bluck L, Jackson S, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest (2009) 119:315–22. doi: 10.1172/JCI37432
92. Wu X, Chen K, Williams KJ. The role of pathway-selective insulin resistance and responsiveness in diabetic dyslipoproteinemia. Curr Opin Lipidol (2012) 23:334–44. doi: 10.1097/MOL.0b013e3283544424
93. Ter Horst KW, Vatner DF, Zhang D, Cline GW, Ackermans MT, Nederveen AJ, et al. Hepatic insulin resistance is not pathway selective in humans with nonalcoholic fatty liver disease. Diabetes Care (2021) 44:489–98. doi: 10.2337/dc20-1644
94. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol (2011) 12:21–35. doi: 10.1038/nrm3025
95. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol (2020) 21:183–203. doi: 10.1038/s41580-019-0199-y
96. Guillen C, Benito M. mTORC1 overactivation as a key aging factor in the progression to type 2 diabetes mellitus. Front Endocrinol (Lausanne) (2018) 9:621. doi: 10.3389/fendo.2018.00621
97. O'Neill BT, Lee KY, Klaus K, Softic S, Krumpoch MT, Fentz J, et al. Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J Clin Invest (2016) 126:3433–46. doi: 10.1172/JCI86522
98. Sajan M, Hansen B, Ivey R III, Sajan J, Ari C, Song S, et al. brain insulin signaling is increased in insulin-resistant states and decreases in FOXOs and PGC-1alpha and increases in abeta1-40/42 and phospho-tau may abet alzheimer development. Diabetes (2016) 65:1892–903. doi: 10.2337/db15-1428
99. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell (2004) 117:399–412. doi: 10.1016/S0092-8674(04)00400-3
100. Boden G, Cheung P, Salehi S, Homko C, Loveland-Jones C, Jayarajan S, et al. Insulin regulates the unfolded protein response in human adipose tissue. Diabetes (2014) 63:912–22. doi: 10.2337/db13-0906
101. Williams KJ, Wu X. Imbalanced insulin action in chronic over nutrition: Clinical harm, molecular mechanisms, and a way forward. Atherosclerosis (2016) 247:225–82. doi: 10.1016/j.atherosclerosis.2016.02.004
102. Kolb H, Kempf K, Röhling M, Martin S. Insulin: too much of a good thing is bad. BMC Med (2020) 18:224. doi: 10.1186/s12916-020-01688-6
103. Baboota RK, Spinelli R, Erlandsson MC, Brandao BB, Lino M, Yang H, et al. Chronic hyperinsulinemia promotes human hepatocyte senescence. Mol Metab (2022) 64:101558. doi: 10.1016/j.molmet.2022.101558
104. Meijnikman AS, van Olden CC, Aydin O, Herrema H, Kaminska D, Lappa D, et al. Hyperinsulinemia is highly associated with markers of hepatocytic senescence in two independent cohorts. Diabetes (2022) 71:1929–36. doi: 10.2337/db21-1076
105. Li Q, Hagberg CE, Silva CH, Lang S, Hyvonen MT, Salehzadeh F, et al. Obesity and hyperinsulinemia drive adipocytes to activate a cell cycle program and senesce. Nat Med (2021) 27:1941–53. doi: 10.1038/s41591-021-01501-8
106. Chow HM, Shi M, Cheng A, Gao Y, Chen G, Song X, et al. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat Neurosci (2019) 22:1806–19. doi: 10.1038/s41593-019-0505-1
107. Othman EM, Leyh A, Stopper H. Insulin mediated DNA damage in mamMalian colon cells and human lymphocytes in vitro. Mutat Res (2013) 745-746:34–9. doi: 10.1016/j.mrfmmm.2013.03.006
108. Aviv A, Valdes A, Gardner JP, Swaminathan R, Kimura M, Spector TD. Menopause modifies the association of leukocyte telomere length with insulin resistance and inflammation. J Clin Endocrinol Metab (2006) 91:635–40. doi: 10.1210/jc.2005-1814
109. Demissie S, Levy D, Benjamin EJ, Cupples LA, Gardner JP, Herbert A, et al. Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the Framingham Heart Study. Aging Cell (2006) 5:325–30. doi: 10.1111/j.1474-9726.2006.00224.x
110. Al-Attas OS, Al-Daghri NM, Alokail MS, Alfadda A, Bamakhramah A, Sabico S, et al. Adiposity and insulin resistance correlate with telomere length in middle-aged Arabs: the influence of circulating adiponectin. Eur J Endocrinol (2010) 163:601–7. doi: 10.1530/EJE-10-0241
111. Yang M, Jiang P, Jin C, Wang J. Longer telomere length and its association with lower levels of C-peptide. Front Endocrinol (Lausanne) (2017) 8:244. doi: 10.3389/fendo.2017.00244
112. Aguiar SS, Rosa TS, Neves RVP, Leite PLA, Maciel LA, Gutierrez SD, et al. Telomere length, SIRT1, and insulin in male master athletes: the path to healthy longevity? Int J Sports Med (2022) 43:29–33. doi: 10.1055/a-1510-9259
113. Gardner JP, Li S, Srinivasan SR, Chen W, Kimura M, Lu X, et al. Rise in insulin resistance is associated with escalated telomere attrition. Circulation (2005) 111:2171–7. doi: 10.1161/01.CIR.0000163550.70487.0B
114. Mangge H, Herrmann M, Almer G, Zelzer S, Moeller R, Horejsi R, et al. Telomere shortening associates with elevated insulin and nuchal fat accumulation. Sci Rep (2020) 10:6863. doi: 10.1038/s41598-020-63916-6
115. Ghoussaini R, Tamim H, Elbejjani M, Makki M, Nasreddine L, Ismaeel H, et al. C-peptide is a predictor of telomere shortening: A five-year longitudinal study. Front Endocrinol (Lausanne) (2022) 13:978747. doi: 10.3389/fendo.2022.978747
116. Verhulst S, Dalgard C, Labat C, Kark JD, Kimura M, Christensen K, et al. A short leucocyte telomere length is associated with development of insulin resistance. Diabetologia (2016) 59:1258–65. doi: 10.1007/s00125-016-3915-6
117. Prasun P. Mitochondrial dysfunction in metabolic syndrome. Biochim Biophys Acta Mol Basis Dis (2020) 1866:165838. doi: 10.1016/j.bbadis.2020.165838
118. Miwa S, Kashyap S, Chini E, von ZT. Mitochondrial dysfunction in cell senescence and aging. J Clin Invest (2022) 132:e158447. doi: 10.1172/JCI158447
119. Barlow AD, Nicholson ML, Herbert TP. Evidence for rapamycin toxicity in pancreatic beta-cells and a review of the underlying molecular mechanisms. Diabetes (2013) 62:2674–82. doi: 10.2337/db13-0106
120. Blagosklonny MV. Rapamycin for longevity: opinion article. Aging (Albany NY) (2019) 11:8048–67. doi: 10.18632/aging.102355
121. Hoehn KL, Salmon AB, Hohnen-Behrens C, Turner N, Hoy AJ, Maghzal GJ, et al. Insulin resistance is a cellular antioxidant defense mechanism. Proc Natl Acad Sci U.S.A. (2009) 106:17787–92. doi: 10.1073/pnas.0902380106
122. Nolan CJ, Ruderman NB, Kahn SE, Pedersen O, Prentki M. Insulin resistance as a physiological defense against metabolic stress: implications for the management of subsets of type 2 diabetes. Diabetes (2015) 64:673–86. doi: 10.2337/db14-0694
123. Kolb H, Eizirik DL. Resistance to type 2 diabetes mellitus: a matter of hormesis? Nat Rev Endocrinol (2011) 8:183–92. doi: 10.1038/nrendo.2011.158
124. Silva-Palacios A, Ostolga-Chavarria M, Zazueta C, Konigsberg M. Nrf2: Molecular and epigenetic regulation during aging. Ageing Res Rev (2018) 47:31–40. doi: 10.1016/j.arr.2018.06.003
125. Dodson M, de la Vega MR, Cholanians AB, Schmidlin CJ, Chapman E, Zhang DD. Modulating NRF2 in disease: timing is everything. Annu Rev Pharmacol Toxicol (2019) 59:555–75. doi: 10.1146/annurev-pharmtox-010818-021856
126. He F, Ru X, Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci (2020) 21:4777. doi: 10.3390/ijms21134777
127. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun (2016) 7:11624. doi: 10.1038/ncomms11624
128. van der Horst D, Carter-Timofte ME, van GJ, Laguette N, Dinkova-Kostova AT, Olagnier D. Regulation of innate immunity by Nrf2. Curr Opin Immunol (2022) 78:102247. doi: 10.1016/j.coi.2022.102247
129. Heiss EH, Schachner D, Werner ER, Dirsch VM. Active NF-E2-related factor (Nrf2) contributes to keep endothelial NO synthase (eNOS) in the coupled state: role of reactive oxygen species (ROS), eNOS, and heme oxygenase (HO-1) levels. J Biol Chem (2009) 284:31579–86. doi: 10.1074/jbc.M109.009175
130. Trougakos IP. Nrf2, stress and aging. Aging (Albany NY) (2019) 11:5289–91. doi: 10.18632/aging.102143
131. Klaips CL, Jayaraj GG, Hartl FU. Pathways of cellular proteostasis in aging and disease. J Cell Biol (2018) 217:51–63. doi: 10.1083/jcb.201709072
132. Boden G, Cheung P, Kresge K, Homko C, Powers B, Ferrer L. Insulin resistance is associated with diminished endoplasmic reticulum stress responses in adipose tissue of healthy and diabetic subjects. Diabetes (2014) 63:2977–83. doi: 10.2337/db14-0055
133. Hebishy M, Shintouo CM, Dufait I, Debacq-Chainiaux F, Bautmans I, Njemini R. Heat shock proteins and cellular senescence in humans: A systematic review. Arch Gerontol Geriatr (2023) 113:105057. doi: 10.1016/j.archger.2023.105057
134. Grahame HD. AMP-activated protein kinase: a key regulator of energy balance with many roles in human disease. J Intern Med (2014) 276:543–59. doi: 10.1111/joim.12268
135. Salminen A, Kauppinen A, KaarnIranta K. AMPK/Snf1 signaling regulates histone acetylation: Impact on gene expression and epigenetic functions. Cell Signal (2016) 28:887–95. doi: 10.1016/j.cellsig.2016.03.009
136. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol (2018) 19:121–35. doi: 10.1038/nrm.2017.95
137. Gradinariu V, Ard J, van Dam RM. Effects of dietary quality, physical activity and weight loss on glucose homeostasis in persons with and without prediabetes in the PREMIER trial. Diabetes Obes Metab (2023) 2023:2714–22. doi: 10.1111/dom.15160
138. Calabrese EJ, Kozumbo WJ. The hormetic dose-response mechanism: Nrf2 activation. Pharmacol Res (2021) 167:105526. doi: 10.1016/j.phrs.2021.105526
139. Lettieri-Barbato D, Minopoli G, Caggiano R, Izzo R, Santillo M, Aquilano K, et al. Fasting drives nrf2-related antioxidant response in skeletal muscle. Int J Mol Sci (2020) 21:7780. doi: 10.3390/ijms21207780
140. Done AJ, Traustadottir T. Nrf2 mediates redox adaptations to exercise. Redox Biol (2016) 10:191–9. doi: 10.1016/j.redox.2016.10.003
141. Mallard AR, Spathis JG, Coombes JS. Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) and exercise. Free Radic Biol Med (2020) 160:471–9. doi: 10.1016/j.freeradbiomed.2020.08.024
142. Qin S, Hou DX. Multiple regulations of Keap1/Nrf2 system by dietary phytochemicals. Mol Nutr Food Res (2016) 60:1731–55. doi: 10.1002/mnfr.201501017
143. Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol (2019) 19:184–97. doi: 10.1038/s41577-019-0125-8
144. Bodogai M, O'Connell J, Kim K, Kim Y, Moritoh K, Chen C, et al. Commensal bacteria contribute to insulin resistance in aging by activating innate B1a cells. Sci Transl Med (2018) 10:eaat4271. doi: 10.1126/scitranslmed.aat4271
145. Burkart V, Liu H, Bellmann K, Wissing D, Jaattela M, Cavallo MG, et al. Natural resistance of human beta cells toward nitric oxide is mediated by heat shock protein 70. J Biol Chem (2000) 275:19521–8. doi: 10.1074/jbc.M002265200
146. Soo SK, Rudich ZD, Ko B, Moldakozhayev A, AlOkda A, Van Raamsdonk JM. Biological resilience and aging: Activation of stress response pathways contributes to lifespan extension. Ageing Res Rev (2023) 88:101941. doi: 10.1016/j.arr.2023.101941
147. Gomez J, Mota-Martorell N, Jove M, Pamplona R, Barja G. Mitochondrial ROS production, oxidative stress and aging within and between species: Evidences and recent advances on this aging effector. Exp Gerontol (2023) 174:112134. doi: 10.1016/j.exger.2023.112134
148. Smith EJ, Shay KP, Thomas NO, Butler JA, Finlay LF, Hagen TM. Age-related loss of hepatic Nrf2 protein homeostasis: Potential role for heightened expression of miR-146a. Free Radic Biol Med (2015) 89:1184–91. doi: 10.1016/j.freeradbiomed.2015.11.003
149. Tang M, Ji C, Pallo S, Rahman I, Johnson GVW. Nrf2 mediates the expression of BAG3 and autophagy cargo adaptor proteins and tau clearance in an age-dependent manner. Neurobiol Aging (2018) 63:128–39. doi: 10.1016/j.neurobiolaging.2017.12.001
150. Pomatto LCD, Sun PY, Yu K, Gullapalli S, Bwiza CP, Sisliyan C, et al. Limitations to adaptive homeostasis in an hyperoxia-induced model of accelerated ageing. Redox Biol (2019) 24:101194. doi: 10.1016/j.redox.2019.101194
151. Davinelli S, Willcox DC, Scapagnini G. Extending healthy ageing: nutrient sensitive pathway and centenarian population. Immun Ageing (2012) 9:9. doi: 10.1186/1742-4933-9-9
Keywords: insulin, insulin resistance, aging, longevity, senescence, oxidative stress, proteostasis, Nrf2
Citation: Kolb H, Kempf K and Martin S (2023) Insulin and aging – a disappointing relationship. Front. Endocrinol. 14:1261298. doi: 10.3389/fendo.2023.1261298
Received: 25 July 2023; Accepted: 25 August 2023;
Published: 03 October 2023.
Edited by:
James Harper, Sam Houston State University, United StatesReviewed by:
Paolina Crocco, University of Calabria, ItalyAlessandro Bitto, University of Washington, Seattle, United States
Norma Edith Lopez, Universidad Autónoma Metropolitana Iztapalapa, Mexico
Copyright © 2023 Kolb, Kempf and Martin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kerstin Kempf, a2Vyc3Rpbi5rZW1wZkB3ZGd6LmRl