
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 21 September 2023
Sec. Bone Research
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1244361
 Isabel Mazarico-Altisent1*†
Isabel Mazarico-Altisent1*† Ismael Capel1
Ismael Capel1 Neus Baena2Maria Rosa Bella-Cueto3Santi Barcons4Xavier Guirao4Rocío Pareja1Andreea Muntean1Valeria Arsentales1Assumpta Caixàs1Mercedes Rigla1
Neus Baena2Maria Rosa Bella-Cueto3Santi Barcons4Xavier Guirao4Rocío Pareja1Andreea Muntean1Valeria Arsentales1Assumpta Caixàs1Mercedes Rigla1Background: Approximately 10% of primary hyperparathyroidism cases are hereditary, due to germline mutations in certain genes. Although clinically relevant, a systematized genetic diagnosis is missing due to a lack of firm evidence regarding individuals to test and which genes to evaluate.
Methods: A customized gene panel (AIP, AP2S1, CASR, CDC73, CDKN1A, CDKN1B, CDKN2B, CDKN2C, GCM2, GNA11, MEN1, PTH, RET, and TRPV6) was performed in 40 patients from the Mediterranean area with suspected familial hyperparathyroidism (≤45 years of age, family history, high-risk histology, associated tumour, multiglandular disease, or recurrent hyperparathyroidism). We aimed to determine the prevalence of germline variants in these patients, to clinically characterize the probands and their relatives, and to compare disease severity in carriers versus those with a negative genetic test.
Results: Germline variants were observed in 9/40 patients (22.5%): 2 previously unknown pathogenic/likely pathogenic variants of CDKN1B (related to MEN4), 1 novel variant of uncertain significance of CDKN2C, 4 variants of CASR (3 pathogenic/likely pathogenic variants and 1 variant of uncertain significance), and 2 novel variants of uncertain significance of TRPV6. Familial segregation studies allowed diagnosis and early treatment of PHPT in first-degree relatives of probands.
Conclusion: The observed prevalence of germline variants in the Mediterranean cohort under study was remarkable and slightly higher than that seen in other populations. Genetic screening for suspected familial hyperparathyroidism allows the early diagnosis and treatment of PHPT and other related comorbidities. We recommend genetic testing for patients with primary hyperparathyroidism who present with high-risk features.
Primary hyperparathyroidism (PHPT) is a common endocrine disorder, with an overall incidence of 13-78 per 100,000 person-years in the world (1). Owing to more widespread screening for hypercalcemia, this frequency has risen in recent years (1–3). The prevalence rises with age and typically exhibits a female predominance (1). If left untreated, PHPT can cause hypercalcaemic syndrome, with increased mortality, muscle weakness, nephrocalcinosis, renal stones, osteoporosis, bone fractures, and heart failure among others (1). Up to 10% of all PHPT cases are due to germline variants in recurrent genes (4, 5), a situation known as familial primary hyperparathyroidism (FHPT) (6). This prevalence increases to 15-26% in patients with risk features (7). However, it appears that FHPT is underdiagnosed and underreported in the literature, likely due to the limited number of genetic studies performed in clinical practice and an insufficient understanding of these conditions.
FHPT may occur either alone (without other related tumours) or as part of a syndrome, such as multiple endocrine neoplasia (MEN) types 1, 2A, and 4, and hyperparathyroidism-jaw tumour syndrome. MEN1 is caused by inactivating germline variants of the MEN1 tumour suppressor gene that produce PHPT, gastro-entero-pancreatic neuroendocrine tumours, pituitary adenomas, and other tumours, such as bronchial/thymic carcinoids or lipomas (8). MEN2A is caused by activating variants in the proto-oncogene RET and typically presents as medullary thyroid carcinoma, pheochromocytoma, and parathyroid tumours (20-30%) with a lower penetrance (9). Patients with MEN4 express MEN1-like milder phenotypes due to CDKN1B germline variants and, less frequently, other non-endocrine neoplasms such as breast cancer, prostate cancer, colon cancer, papillary thyroid carcinoma, angiomyolipoma, and meningioma, although only approximately 100 cases have been reported to date (10, 11). Variants in other cyclin-dependent kinase inhibitors (CDKIs), such as CDKN1A, CDKN2B, and CDKN2C, have been described in patients with the MEN1 phenotype or suspected FHPT, despite limited data (11, 12). Recently, germline variants of the MAX tumour suppressor gene have been associated with a new entity, MEN5. It is thought to cause pheochromocytomas and paragangliomas and, possibly, parathyroid adenomas and functional pituitary adenomas (13, 14). Hyperparathyroidism-jaw tumour syndrome is caused by germline inactivating variants of the CDC73 (also called HRPT2) tumour suppressor gene. These mutations can result in aggressive forms of PHPT (parathyroid carcinoma, atypical parathyroid adenoma), fibroosseous jaw tumours, and renal and uterine tumours (15, 16).
FHPT may also present as non-syndromic PHPT, which includes familial hypocalciuric hypercalcemia (FHH) forms (caused by variants of CASR, GNA11, or AP2S1), neonatal severe hyperparathyroidism, and other forms caused by several genes classified as familial isolated hyperparathyroidism (FIHP). FIHP is diagnosed by exclusion because the genetic cause is unknown in most cases (17–19). GCM2 proto-oncogene variants are present in about 18% of these cases, while about 30% involve mutations in MEN1 or CDC73 with incomplete expression or loss-of-function variants of the CASR gene (1, 4, 20–22). Other genes related to familial hyperparathyroidism are AIP (23) and TRPV6 (24). In recent years, the number of genes potentially involved in parathyroid tumorigenesis is constantly expanding (25).
Most authors propose performing genetic testing in patients with PHPT who present some risk features suspicious of FHPT: diagnosis under 30-45 years, high-risk parathyroid histology (multiglandular disease (MGD), parathyroid carcinoma, atypical adenoma, or cystic adenoma), carrying tumours related to syndromic FHPT, family history of PHPT, or a calcium creatinine clearance ratio (CCCR) <0,02 (7). However, consensual systematized genetic screening for FHPT has not been widely implemented because of the lack of firm evidence on which individuals should be tested and which genes should be evaluated. Few studies on the systematic search for germline variants in patients with suspected FHPT have been conducted, and those that have been done have shown mixed results. In addition, these studies were performed in various clinical scenarios, with heterogeneous genetic testing methodologies and various genes assessed (7, 26, 27). To our knowledge, none of these were carried out among the Spanish population, and only a few involved people living in the Mediterranean area.
Genetic testing is of considerable interest to patients with PHPT of a presumed familial origin. This allows for: 1) Predicting which patients are at risk of developing other neoplasms (ex. neuroendocrine tumours in MEN1 or MEN4). 2) Planning the best management of PHPT for each patient and aid in surgical necessity and planning. 3) Guiding surveillance for other clinical features associated with a given genetic anomaly. 4) Carrying out cascade testing of asymptomatic relatives, which allows for early diagnosis of potentially severe syndromes and reduces healthcare burden and psychological stress in mutation-negative relatives. 5) Offering optimal genetic counselling if needed via prenatal/preimplantation diagnosis (1, 3, 7, 11, 15).
The aim of this study was to perform genetic screening, at our hospital, of patients with PHPT who presented some risk features, in order to: 1) Determine the prevalence of germline mutations in the analysed genes in our cohort. 2) Clinically characterize carriers of variants and their first-degree relatives. 3) Compare disease severity in carriers of variants with respect to those without a genetic alteration found and compare the prevalence of germline variants in our population with those in other cohorts.
An ambispective observational study was conducted. We collected data from patients diagnosed with PHPT who also exhibited some risk factors for FHPT. The diagnosis of PHPT was established by inappropriately non-suppressed or high parathyroid hormone levels (PTH) (reference value [RV] 12.32-41.99 pmol/L) with normal or elevated albumin-corrected serum calcium levels (RV 2.1-2.55 mmol/L). Patients were included in the study when at least one inclusion criterion was met: 1) Age at onset of PHPT ≤ 45 years. 2) High-risk parathyroid histology (parathyroid carcinoma, atypical parathyroid adenoma, cystic parathyroid adenoma, or biopsy-documented MGD defined as having two or more glands affected by either adenomas or hyperplasia). 3) Recurrent or persistent PHPT (established by presenting corrected serum calcium levels ≥ 2.62 mmol/L three months after parathyroid surgery). 4) Family history of PHPT in at least one first-degree relative. 5) Other tumour/s related to syndromic FHPT (gastro-entero-pancreatic neuroendocrine tumours, pituitary adenoma, jaw tumour, renal or uterine tumour, medullary thyroid carcinoma, breast tumour, malignant hemopathy, or others). Having a known mutation in any of the genes under study, as well as having a first-degree relative with a known mutation in any of them, were the only exclusion criteria.
We retrospectively reviewed the medical records, from pathologic data base, of all patients who underwent surgery for hyperparathyroidism between 1989-2019 at the Parc Taulí University Hospital. In total, 103/485 patients met the histological inclusion criteria. We also examined all patients referred to the Endocrinology and Nutrition Department of our hospital for PHPT from January 2017 to December 2019 to include patients with PHPT who had not yet undergone surgery. Of them, 23/197 met inclusion criteria. In total, 126 patients met at least one of the inclusion criteria. Seventy-three patients were deceased or unreachable and 13 declined to participate. In the end, 40 index cases were included. The study design is illustrated in Figure 1.
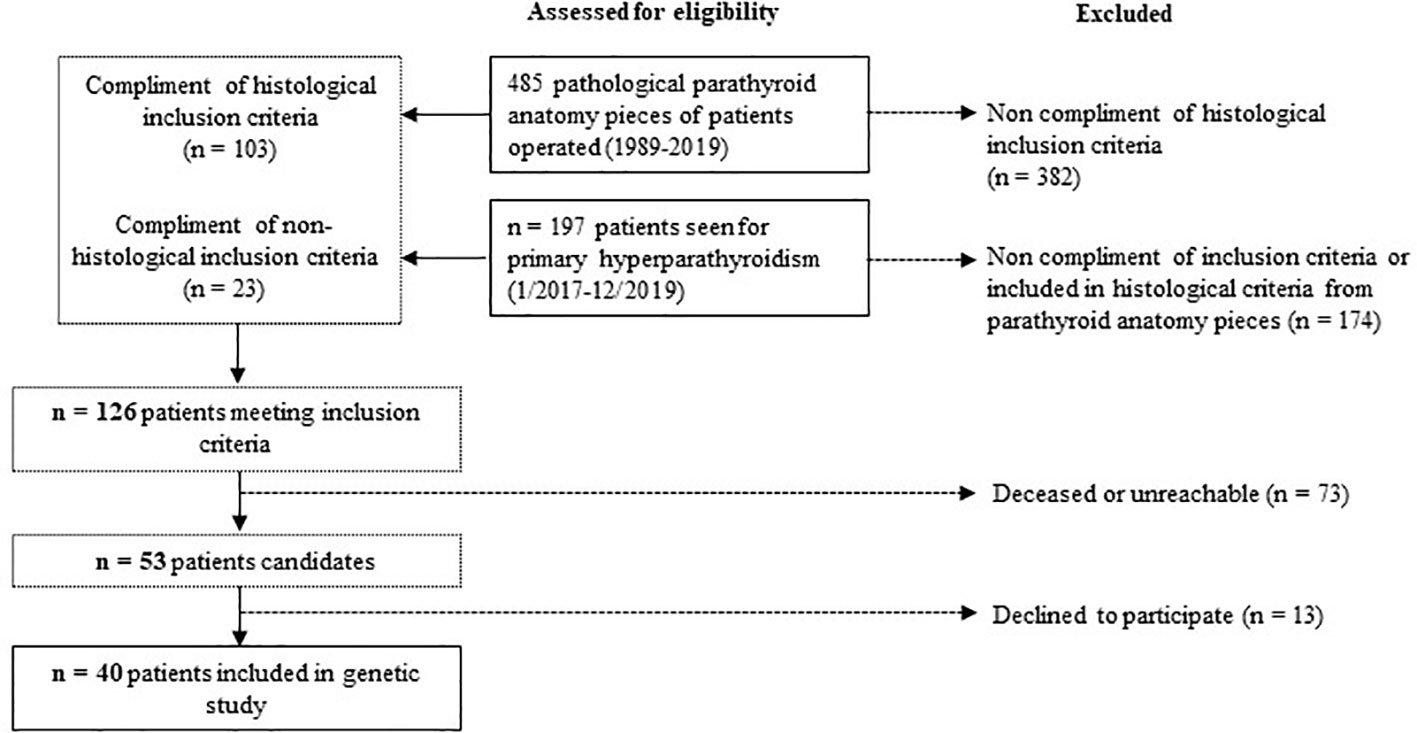
Figure 1 Study design.
Patients who met the inclusion criteria and agreed to participate were interviewed to obtain detailed medical and family histories between February and July 2020. Baseline data were recorded for each patient, including age at presentation, sex, initial clinical presentation, preoperative and postoperative biochemical and hormonal findings (PTH, calcium, phosphate, and 25-hydroxy vitamin D levels), and calcium creatinine clearance ratio (CCCR, calculated using a 24-hour urinary calcium collection and one blood sample) at diagnosis when available. We reviewed the preoperative imaging results of the parathyroid glands, type of surgery performed, surgical findings, histopathology, postoperative data, and follow-up outcomes.
Written informed consent was obtained from all patients, and the study was approved by the institutional Ethics Committee of Institut d’Investigació i Innovació Parc Taulí I3PT.
DNA was extracted from blood and other tissue samples using the Gentra Puregene DNA reagent (Qiagen, Valencia, CA). Genetic screening for FHPT was performed using a customized gene panel that included AIP, AP2S1, CASR, CDC73, CDKN1A, CDKN1B, CDKN2B, CDKN2C, GCM2, GNA11, MEN1, PTH, RET, and TRPV6, through both sequence analysis and copy number variation (CNV) analysis. This gene panel was selected based on recent data and previous studies (15, 28, 29).
In the analysis, the mean sequencing depth was > 150 times, and > 99% of the target nucleotides were covered with > 20× sequencing depth for all assays. The target nucleotides included all protein-coding exons of the genes in the panels in addition to 20 base pairs inside each intron/exon boundary. The panel was also customized by adding oligonucleotides that targeted deep intronic variants (≥ 20 base pairs from the intron/exon boundary) and noncoding variants (promoter region and 5′ or 3′ untranslated regions), which were reported to cause disease in association with PHPT. The sequence reads of each sample were mapped to the human reference genome (GRCh37/hg19). The Burrows–Wheeler Aligner software was used for reading alignment. Duplicate read marking, local realignment around indels, base quality score recalibration, and variant calling were performed using the genomic analysis toolkit algorithms (Sentieon) for nDNA. Variant data were annotated using a collection of tools (VcfAnno and VEP) with a variety of public variant databases, including, but not limited to, the Genome Aggregation Database control population cohorts (gnomAD), ClinVar, and the Human Gene Mutation Database (30).
The sequence variant analysis pipeline was validated in the Clinical Laboratory Improvement Amendments (CLIA) and College of American Pathologists (CAP)-accredited Blueprint Genetics diagnostic laboratory. The series of selected quality criteria included the variant call quality score, variant genomic location, sequence content, and integrative genomics viewer visual analysis. This algorithm was established based on the outcome of internal validation performed in the CLIA and CAP-accredited Blueprint Genetics diagnostic laboratory.
CNV analysis was performed bioinformatically from next-generation sequencing data using a bioinformatics pipeline; one component was a CNV kit and the other involved in-house developed proprietary technology. CNVs were confirmed by digital polymerase chain reaction. The CNV analysis pipeline was validated in the CLIA and CAP-accredited Blueprint Genetics diagnostic laboratories. Sanger sequencing of the candidate variants was performed on the patient and family member samples to confirm the presence of the variant and the pattern of inheritance.
Variants were classified into five categories according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines (31): benign, likely benign, variant of uncertain significance (VUS), likely pathogenic (LP), and pathogenic (P). We reported all the variants classified as P, LP, or VUS. If any variant was found, genetic counselling of the proband and cascade genetic testing were offered to all at-risk family members. If required, loss of heterozygosity and CNV in tumours and immunohistochemistry studies were performed.
Results corresponding to quantitative variables were presented as the mean ± standard deviation. Data associated with qualitative characteristics were reported as percentages. The normality of quantitative variables was assessed using the Shapiro-Wilk normality test. The significance of the difference between two means was assessed using Student’s T-test for normal variables and the Wilcoxon rank sum test for non-normal variables. The significance of the difference between more than two means was assessed using the Kruskal-Wallis test for non-normal variables. The significance of the difference between percentages was assessed using Pearson’s chi-square test. Differences were considered statistically significant at P < 0.05. Calculations were performed using R Commander version 4.0.5.
The clinical characteristics of the 40 participants (27 females and 13 males) are displayed in Table 1. The mean age at diagnosis of primary hyperparathyroidism was 51 ± 15.8 years. The circumstances leading to diagnosis of PHPT were diverse: 30% incidental findings of hypercalcemia, 20% symptomatic renal lithiasis, 17.5% palpable cervical nodes, 17.5% neuromuscular symptoms, and other clinical features in the remaining patients. Thirty-two patients (80%) underwent surgery, with mean highest PTH levels of 50.28 (12.32-41.99) pmol/L (RV 1.06-6.89), mean albumin adjusted serum calcium of 3.03 ± 0.5 mmol/L, and mean nadir phosphate levels of 0.84 ± 0.39 mmol/L. The inclusion criteria were as follows: 15 subjects were under 45 years of age at diagnosis of PHPT; 28 cases met the histology criteria (one parathyroid carcinoma, 11 atypical parathyroid adenomas, six cystic parathyroid adenomas, and five MGD); four patients presented with recurrence of PHPT after surgery; two cases were included because of family history of PHPT; and seven patients presented tumours that could be related to familial syndrome. 32% of the patients met two or more inclusion criteria.
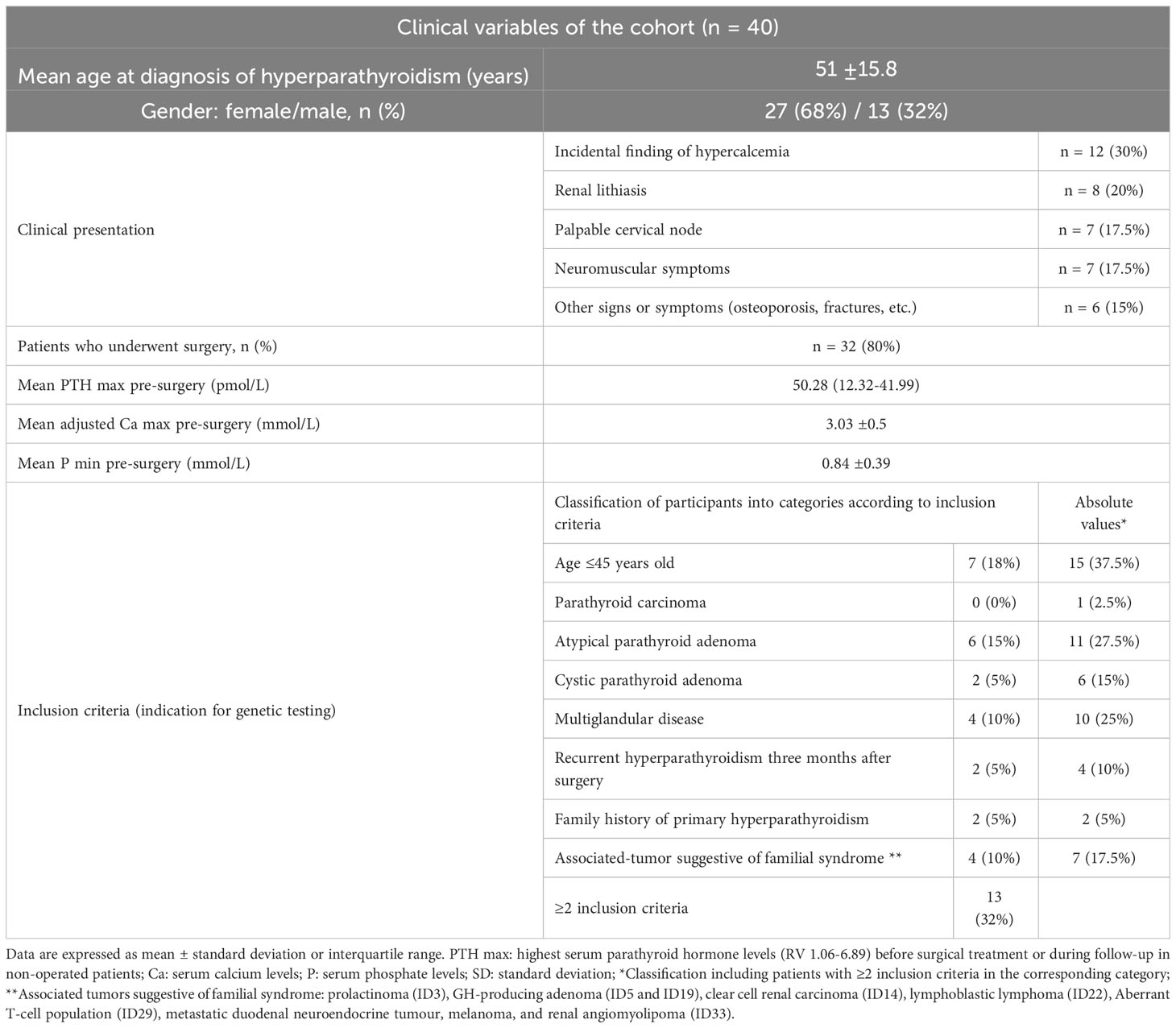
Table 1 Clinical variables of the 40 participants.
Genetic sequencing of the targeted panel identified five pathogenic/likely pathogenic germline variants in five of the 40 participants (three in CASR and two in CDKN1B) (12.5%) and four variants of uncertain significance (one in CDKN2C, one in CASR, and two in TRPV6) (10%). There was one frameshift variant, one nonsense variant, and seven missense variants. Table 2 summarizes the genetic and clinical characteristics of patients with germline variants.
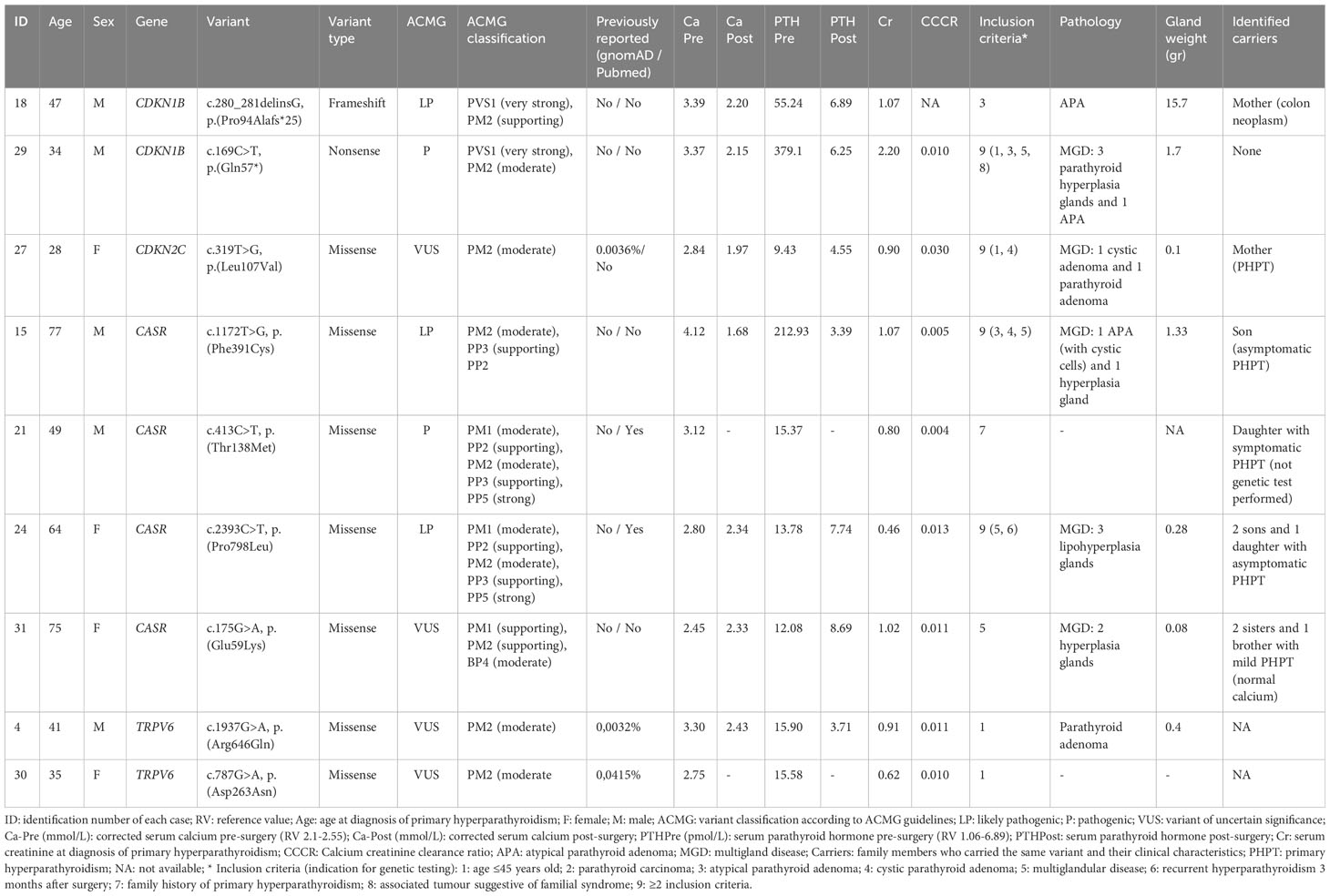
Table 2 Genetic and clinical characteristics of patients displaying germline variants in any of the genes included in the panel.
Four patients carried heterozygous missense variants of the CASR gene (ID15, ID21, ID24, and ID31): two likely pathogenic, one pathogenic, and one VUS. Two of the CASR variant carriers had CCCR < 0.01, and the others were between 0.01-0.02. Of the four patients, three underwent surgical treatment.
Proband ID15 presented with severe symptomatic hypercalcemia at the time of PHPT diagnosis that required hospital admission. Computed tomography of the neck revealed a partially cystic lesion posterior to the thyroid gland, suggesting cystic parathyroid adenoma. Surgery was performed, and the pathological diagnosis was MGD due to one atypical parathyroid adenoma with cystic cells and one hyperplastic parathyroid gland. After surgery, the patient remained normocalcemic and asymptomatic for four years of follow-up until now. A genetic test revealed c.1172T>G, p.(Phe391Cys) variant of CASR, classified as LP, according to ACMG. Biochemical tests detected hypercalcemia plus CCCR 0.01 in one of the two family members who carried the identical germline variant as the proband; conversely, no alterations in calcium or PTH serum levels were found on non-mutation carrier relatives (see family pedigree in Figure 2).
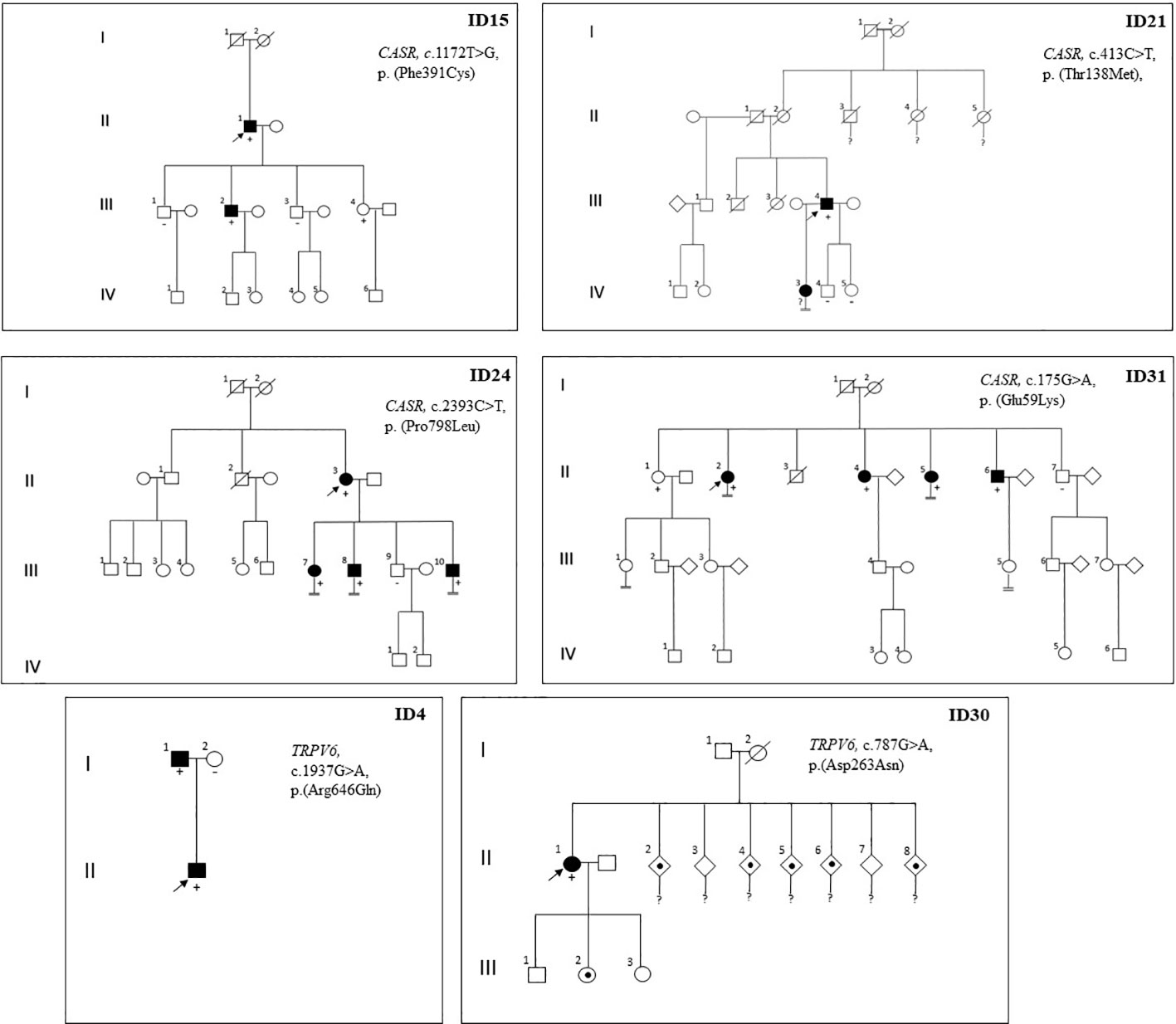
Figure 2 Pedigree of probands with detection of germline variant of CASR or TRPV6 genes and their relatives. Family members are indicated by generation (Roman numerals) and individuals (Arabic numerals). Arrows indicate index case; Circle, female; Square, male; Double line below descent line, no offspring; Empty symbol, unaffected family member; Filled symbol, affected family member (inappropriately non-suppressed or high parathyroid hormone levels with normal or elevated serum calcium levels); Filled spot, history of nephrolithiasis or severe vitamin D deficiency; Slashed symbol, deceased; +, mutation carrier; -, non mutation carrier;?, no genetic test performed.
Cases ID24 and ID31 were diagnosed with PHPT in an osteoporosis study (defined by a T score below -2.5 SD). In both cases, the Sestamibi scan findings were consistent with a single parathyroid lesion. Even so, bilateral surgery was performed in both cases due to the observation of multiple enlarged glands during the procedure. Pathology study showed multiglandular disease in both cases (three lipohyperplasia glands in case ID24 and two hyperplasia glands in case ID31) (See Table 2). Densitometric osteoporosis improved after parathyroid excision in both cases, although patient ID24 showed persistent biochemical PHPT after surgery. Cascade genetic testing and blood tests were offered to all first-degree relatives. Biochemical PHPT (characterized by elevated or inadequately normal PTH levels with varying degrees of hypercalcemia) was detected in multiple family members who share the same variants as the probands (Figure 2). They were offered clinical and analytical follow-up, although to date none of them required parathyroid surgery. In the ID31 family, segregation was observed in all but one sibling. Yet, variant c.175G>A, p.(Glu59Lys) was classified as VUS for the time being, according to the ACMG criteria (PM1 (supporting), PM2 (supporting), and BP4 (moderate)). Case ID21 was included in the study because he had one daughter with PHPT. His imaging localization studies were negative, so surgery was not performed, but treatment with cinacalcet was initiated (30 mg once daily) and calcium plasma levels decreased from 3.12 to 2.74 mmol/L (RV 2.1-2.55). The proband’s daughter declined to undergo a genetic test.
As our group recently reported, three novel CDKIs germline variants were identified in three unrelated cases: two likely pathogenic variants of CDKN1B and one VUS variant of CDKN2C (see Table 2) (11). Patients harbouring CDKN1B variants were included in the study because of atypical parathyroid adenomas, plus age under 45 years at diagnosis in case ID29. Case ID27, who carried the CDKN2C variant, was included due to having a young age at diagnosis and cystic parathyroid adenoma. Neoplasm screening showed other non-endocrine tumours in the three cases: single colon adenoma with dysplasia and atypical lipomas (defined as lipomatous neoplasms with focal cellular atypia) in case ID18, an aberrant T-cell population in case ID29, and a non-functional pituitary adenoma in case ID27. Familial segregation analysis of these variants identified the same variant of the CDKN2C gene in ID27’s mother, who was diagnosed with PHPT with moderate hypercalcemia and osteoporosis due to parathyroid adenoma. She underwent surgery and subsequently achieved normal calcium and PTH levels. Genetic analysis of tumour samples showed uniparental disomy of chromosome 1, including CDKN2C, in case ID27. Neither loss of heterozygosity nor CNV for the CDKN1B gene was found in cases ID18 and ID29, respectively. Clinical data are comprehensively described in the mentioned paper (11).
We detected two variants of the TRPV6 gene, classified as VUS, in two unrelated cases that were included in the study by virtue of having a young age at PHPT diagnosis (Table 2). Both probands had a CCCR of 0.01. Case ID4, who had a clinical history of nephrolithiasis, was diagnosed with PHPT at 41 years old. He underwent surgery for a single parathyroid adenoma; after surgery, his PTH and calcium plasma levels remained normal. Familial segregation analysis was performed on his parents (his only living first-degree relatives). His father carried the same genetic variant and was diagnosed with asymptomatic normocalcemic primary hyperparathyroidism through analytical tests. Case ID30 was diagnosed with PHPT and severe vitamin D deficiency after her newborn developed severe hypocalcaemia due to rickets. Vitamin D treatment was started for both the patient and the newborn. After normalizing vitamin D serum levels, PTH and calcium levels remained elevated in case ID30. Neck ultrasound and Sestamibi scan revealed a nodule suggestive of a parathyroid lesion within the thymus. The proband was advised to undergo surgery, but she failed to follow up, and the family segregation research could not be completed. The only information available about the first-degree relatives is that five of the seven siblings of the index case had a history of nephrolithiasis (see family pedigree in Figure 2).
We analysed the clinical and biochemical features of the study participants according to their genetic test results (see Table 3). The median age at PHPT diagnosis of subjects with a positive genetic test result was not significantly different from that of patients with negative genetic results. Mean PTH and calcium serum levels pre-surgery of patients carrying genetic variants were higher than those with a negative genetic study, but the difference was not statistically significant. Serum phosphate levels were statistically similar between both groups. The only statistically significant difference found between the two groups was the CCCR, which was lower in the group of patients with a positive genetic test (0.01 vs 0.02, P = 0.0038).
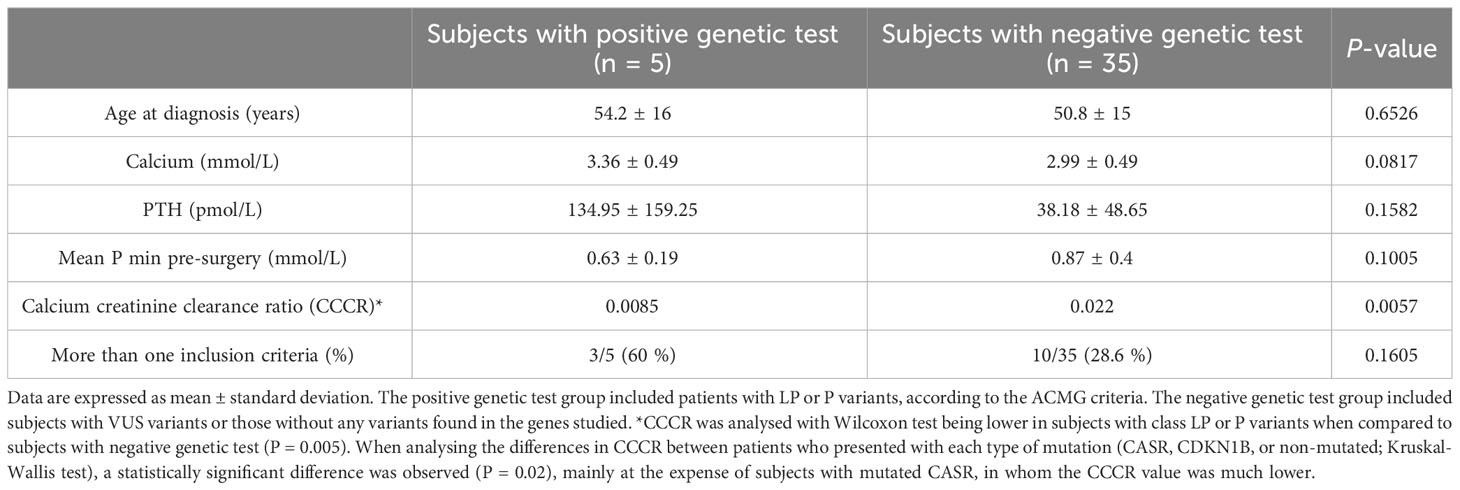
Table 3 Characteristics of patients based on genetic test outcomes.
In this study, we provide a well-defined cohort of 40 patients who presented features indicative of hereditary hyperparathyroidism. As far as we know, this is the largest cohort of patients with suspected FHPT reported in Spain where an expanded panel of genes related to FHPT was systematically studied. We identified nine germline variants (two of CDKN1B, one of CDKN2C, four of CASR, and two of TRPV6) in nine participants (22.5%): five pathogenic or likely pathogenic variants (12.5%) and four VUSs (10%). The most common confirmed genetic disorder in our sample was FHH type 1 (due to CASR variants), followed by MEN4 (due to CDKN1B variants). Notably, seven of these variants have not been previously reported (Table 2).
The prevalence of germline variants found in our study is consistent with some previous studies conducted in individuals with clinical diagnoses of FHPT and is slightly higher than other cohorts. In the Japanese population, CDC73, MEN1, and CASR were studied in 13 families with suspected FHPT due to variable risk phenotypes, resulting in four variants (30%) (32). Conversely, only one MEN1 mutation was found in all the genes studied in a cohort of 29 Finnish patients with suspected FHPT (3%) (28). Other studies in different susceptible populations were conducted, resulting in a variable prevalence of germline variants: 8/86 (9.3%) in the American cohort (29), 9/62 (15%) in South Australia (7), and 18/68 (26%) in New Zealand (7). One of the largest studies to date identified 19 pathogenic germline variants (11 CASR, 6 MEN1, 1 CDC73, and 1 AP2S1) in 121 British patients referred for genetic testing for suspected FHPT (16%) (33). However, all these results are hardly comparable, as the authors used different risk criteria for FHPT and the genes assessed differed between the studies (7, 21, 22, 27, 33, 34).
In our study, the inclusion criteria were based on the established features of inherited tumour susceptibility. The histological diagnosis of the surgical pathology specimens included in this study was based on the outdated WHO criteria. However, it is important to note that the nomenclature has since been updated. The concept parathyroid hyperplasia is no longer generally supported in the context of PHPT as it consists of multiple “clonal” neoplastic proliferations. The 2022 WHO classification approves PHPT-related multiglandular parathyroid disease as germline susceptibility-driven neoplasm. Also, previously referred to as atypical parathyroid adenomas are now recognized as atypical parathyroid tumours (35). As shown in Table 3, in a comparison of different characteristics of the participants according to the genetic results, the only statistically significant difference was in CCCR. There were no statistically significant differences in the mean age of the group carrying LP or P variants compared to that of subjects with a negative genetic study (54.2 vs. 50.8 years, respectively). Calcium and PTH concentrations were somewhat higher in the group of patients with a positive genetic test but without statistically significant differences from the group with a negative test.
Interestingly, subjects carrying LP or P variants had a lower CCCR than those with a negative genetic study (Wilcoxon test, P = 0.057). When we analysed the differences in CCCR between patients who presented with each type of mutation (CASR, CDKN1B, or non-mutated; Kruskal-Wallis test), a statistically significant difference was observed (P = 0.02) but mainly at the expense of subjects with mutated CASR, in whom the CCCR value was much lower (see Table 3). Of note, a CCCR of <0.01 is generally considered a well-planned screening method of FHH. However, this cut-off is of limited clinical value due to reduced diagnostic sensitivity (only captures ~65% of FHH type 1 patients) and reduced specificity (~18% of surgically confirmed PHPT cases have CCCR <0.01). Also, CCCR is not validated for diagnosing FHH in patients with renal impairment, vitamin D insufficiency, or pregnancy (36).
Apart from CCCR, we did not find a trait that was clearly more frequent in patients in whom we found germline variants. Although the differences were not statistically significant, the group of patients carrying a genetic variant had a greater tendency to present more than one inclusion criterion, i.e., various risk factors (60% vs. 28.6% in patients with a negative genetic study). This suggests that having more than one risk characteristic may increase the possibility of positive genetic screening. However, it should be noted that our sample was small and that this is a descriptive study, so it is difficult to draw firm conclusions. Studies with a larger number of patients would be necessary to corroborate this hypothesis.
As mentioned above, four subjects were diagnosed with FHH type 1 due to CASR variants. Variants c.413C>T, p.(Thr138Met) and c.2393C>T, p.(Pro798Leu) were earlier reported in patients with a diagnosis of FHH (37, 38). Instead, variants c.1172T>G, p.(Phe391Cys) and c.175G>A, p.(Glu59Lys) were not previously described. Among the three patients who underwent surgery, one achieved a cure, while the remaining two did not, which is typically observed in cases of FHH. However, both of these cases showed improvement in osteoporosis following the surgery. This supports the notion that parathyroid surgery might be beneficial and even curative in some cases of FHH. Of note, the c.175G>A, p.(Glu59Lys) variant was not documented in gnomID, a reference population database. Therefore, this variant was classified as a VUS according to ACMG criteria (Table 2). Even so, we believed it was appropriate to report this variant since the clinical features of the index case and most of the carrier relatives were consistent with FHH type 1. As noted in previous publications, FHH is a rare autosomal dominant disorder with low or variable penetrance, which could be the reason for the incomplete segregation of the disease in the ID31 family (39).
Historically, FHH and PHPT have been classified as distinct conditions owing to some differences, such as lower urinary calcium excretion (typically CCCR ≤0.01), generally a more benign clinical course, and the controversial role of parathyroid surgery in FHH patients, as parathyroid hyperplasia of more than one gland is present in the majority of cases. However, recent research has shown that FHH can be classified as a subtype of FHPT because they share a similar pathophysiology. The CASR gene is known to play a role in parathyroid growth; therefore, we suppose that some variants may promote both hyperplasia and parathyroid adenoma formation (33). There are several reported cases of parathyroid adenoma in patients with FHH (33, 40, 41). To our knowledge, we report for the first time a case of atypical parathyroid tumour in a patient with FHH type 1 (ID15). In FHH type 1, CASR hypoactivity leads to increased renal calcium reabsorption and increased PTH production, which can result in hypercalcemia with inappropriately normal or elevated PTH. Although traditionally considered rare, 40% of FHH cases could present with atypical features like renal and bone damage, hypercalciuria, severe hypercalcemia, CCCR>0.02, or parathyroid adenoma (42). In agreement with this, of the four CASR variant carriers we reported, one had severe hypercalcemia requiring hospitalization (ID15) and three had bone damage (osteoporosis). The presence of osteoporosis more strongly suggests the acquisition of autonomy of one or more parathyroid glands.
A prevalence of 7.5% (3/40) of variants in CDKIs was found in our group. Two novel likely pathogenic CDKN1B variants were found in patients with atypical parathyroid adenomas (Table 2). CDKN1B mutations are responsible for MEN4 syndrome, which causes a MEN1-like phenotype with other related tumours (11, 43). Additionally, a novel variant of the CDKN2C gene with uniparental disomy was classified as VUS in a patient with PHPT and a non-functional pituitary adenoma. To our knowledge, CDKIs variants have not been reported in patients with atypical or cystic adenomas. As variants in CDKIs have been shown to be a potential cause of FHPT, which may also give rise to other tumours, we suggest that these genes should be added to the FHPT study gene panels. Despite the potential significance of germline variants of these genes, the reported incidence among affected patients is limited (11, 43). Further research is required to determine the actual prevalence of these mutations and the extent of their phenotypic impact.
We report two variants of uncertain significance in TRPV6 in two patients with PHPT diagnosed before the age of 45 years (ID4 and ID30). TRPV6 encodes the TRPV6 channel, which plays a vital role in intestinal and renal calcium absorption. TRPV6 protein is expressed in many epithelial tissues, including the intestine, kidney, placenta, epididymis, and exocrine glands such as the pancreas, prostate, salivary glands, sweat, and mammary glands. Transient severe neonatal hyperparathyroidism has been reported in patients with missense variants of TRPV6, inherited in an autosomal recessive manner. The disease is characterized by transplacental maternal-foetal calcium transport interference, causing a foetal calcium deficit, resulting in neonatal hyperparathyroidism and insufficient bone mineralization (44, 45).
Variants in the TRPV6 gene have also been associated with nephrolithiasis and chronic pancreatitis when inherited in an autosomal recessive manner. However, most studies investigating the genotypic and phenotypic effects of TRPV6 were conducted in mice (45, 46). In the present study, two cases (ID4 and ID30) were diagnosed with primary hyperparathyroidism in young adulthood and carried a single TRPV6 variant. To our knowledge, there have been no reports of adults with dominant inheritance of TRPV6 involvement that presented with PHPT. Thus, we report these variants to expand the understanding of this gene. However, their clinical significance remains unclear and should be interpreted with caution. Further research is necessary to determine the clinical implications of TRPV6 variants inherited in a dominant manner. A potential strategy could be to perform whole exome sequencing (WES) or whole genome sequencing (WGS) to study patients with inconclusive genetic test results. This approach may help to uncover a second variant that was not detected by a gene panel and may reveal novel genes associated with FHPT.
We did not find any variants in the remaining genes included in the panel. One point of note is the absence of MEN1 in the studied cohort. The average percentage of MEN1 pathogenic variants in families with clinical FIHP is around 18% (26). This absence in our cohort could be attributed to the exclusion of individuals previously known to carry MEN1 mutations. In our hospital, MEN1 gene analysis is frequently performed in clinical practice rather than in other genes (47). It should be noted that previous studies associated atypical parathyroid tumours with mutations in CDC73 (48). In contrast, variants in CDC73 were not found in 11 patients with these tumours included in our cohort.
This study has several limitations. The exclusion of cases of FHPT with known mutations in any of the studied genes might have distorted the actual prevalence of germline variants in the at-risk population, which could be higher than reported. Also, there may be a selection bias as: some patients may not express risk features because a variable clinical expressivity and/or different penetrance of the characteristics associated with each of the various syndromic forms of PHPT at the time of PHPT diagnosis, etc (49). Another limitation was the small number of participants, which made it difficult to extract statistically significant results applicable to the population. It should be noted that four of the variants found in our cohort were classified as VUS, so their clinical relevance should be interpreted with caution. However, this classification system is not absolute and is dependent on the accuracy of the available evidence at the time of assessment (50). In addition, we did not conduct functional analysis; therefore, we could not confirm whether the variants caused the characteristics observed in the patients, particularly in the case of those classified as VUS. On the other hand, the genetic testing we conducted was limited to gene panel sequencing.
In summary, we found a remarkable prevalence of genetic variants among the genes studied in the at-risk population. This is consistent with the hypothesis that FPHP is probably an underdiagnosed entity owing to non-standardized genetic screening. In our cohort, genetic testing allowed the early diagnosis and treatment of PHPT and other related comorbidities of probands and their first-degree relatives. We suggest genetic testing in patients with PHPT who present any of the risk characteristics considered in the inclusion criteria of our study. WES strategy is replacing gene panel studies in clinical laboratories. Thus, we propose a filtering procedure using the aforementioned customized gene panel updated as a first-tier virtual panel for patients with suspected FHPT.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Written informed consent was obtained from all patients, and the study was approved by the institutional Ethics Committee of Institut d’Investigació i Innovació Parc Taulí I3PT. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
IC and IM-A designed the study, analysed and interpreted the data, and wrote, reviewed, and edited the manuscript. NB performed and interpreted genetic data and wrote and edited the manuscript. MB-C, SB, XG, and RP participated in editing the manuscript. AC and MR provided scientific guidance. All authors approved the final version of the manuscript to be published.
The study was financed with research funds from the Endocrinology and Nutrition Department, Parc Taulí University Hospital, Institut d’Investigació i Innovació Parc Taulí (I3PT), Sabadell, Barcelona, Spain.
The authors thank the staff of our hospital for suggestions and guidance and the Blueprint Genetic laboratory for the clinical support. Great appreciation goes to all our study participants for the time they devoted.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Minisola S, Arnold A, Belaya Z, Brandi ML, Clarke B, Hannan FM, et al. Epidemiology, pathophysiology and genetics of primary hyperparathyroidism. J Bone Mineral Res (2022) 37(11):2315–29. doi: 10.1002/jbmr.4665
2. Bilezikian JP. Primary hyperparathyroidism. J Clin Endocrinol Metab (2018) 103(11):3993–4004. doi: 10.1210/jc.2018-01225
3. Capel I, Mazarico-Altisent I, Baena N. Genetic study in primary hyperparathyroidism: Which patients and which genes to study? Endocrinol Diabetes Nutr (2022) 69(4):237–9. doi: 10.1016/j.endien.2022.02.020
4. Blau JE, Simonds WF. Familial hyperparathyroidism. Front Endocrinol (Lausanne) (2021) 12(February):1–14. doi: 10.3389/fendo.2021.623667
5. Bilezikian JP, Brandi ML, Eastell R, Silverberg SJ, Udelsman R, Marcocci C, et al. Guidelines for the management of asymptomatic primary hyperparathyroidism: Summary statement from the fourth international workshop. J Clin Endocrinol Metab (2014) 99(10)3561–9. doi: 10.1210/jc.2014-1413
6. Thakker RV, Bouloux P, Wooding C, Chotai K, Broad PM, Spurr NK, et al. Association of parathyroid tumors in multiple endocrine neoplasia type 1 with loss of alleles on chromosome 11. N Engl J Med (1989) 321(4):218–24. doi: 10.1056/NEJM198907273210403
7. de Sousa SMC, Carroll RW, Henderson A, Burgess J, Clifton-Bligh RJ. A contemporary clinical approach to genetic testing for heritable hyperparathyroidism syndromes. Endocrine. (2022) 75:23–32. doi: 10.1007/s12020-021-02927-3
8. Thakker R v, Newey PJ, Walls G v, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab (2012) 97(9):2990–3011. doi: 10.1210/jc.2012-1230
9. American Thyroid Association Guidelines Task Force 1, Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid (2009) 19(6):565–612. doi: 10.1089/thy.2008.0403
10. Frederiksen A, Rossing M, Hermann P, Ejersted C, Thakker R v, Frost M. Clinical features of multiple endocrine neoplasia type 4: Novel pathogenic variant and review of published cases. J Clin Endocrinol Metab (2019) 104(9):3637–46. doi: 10.1210/jc.2019-00082
11. Mazarico-Altisent I, Capel I, Baena N, Bella-Cueto MR, Barcons S, Guirao X, et al. Novel germline variants of CDKN1B and CDKN2C identified during screening for familial primary hyperparathyroidism. J Endocrinol Invest (2022) 46(4):829–40. doi: 10.1007/s40618-022-01948-7
12. Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab (2009) 94(5):1826–34. doi: 10.1210/jc.2008-2083
13. Nosé V, Gill A, Teijeiro JMC, Perren A, Erickson L. Overview of the 2022 WHO classification of familial endocrine tumor syndromes. Endocrine Pathol (2022) 33:197–227. doi: 10.1007/s12022-022-09705-5
14. Seabrook AJ, Harris JE, Velosa SB, Kim E, McInerney-Leo AM, Dwight T, et al. Multiple endocrine tumors associated with germline MAX mutations: Multiple endocrine neoplasia type 5? J Clin Endocrinol Metab (2021) 106(4):1163–82. doi: 10.1210/clinem/dgaa957
15. Thakker RV. Genetics of parathyroid tumours. J Intern Med(2016) 280(6):574–83. doi: 10.1111/joim.12523
16. Simonds WF, James-Newton LA, Agarwal SK, Yang B, Skarulis MC, Hendy GN, et al. Familial isolated hyperparathyroidism clinical and genetic characteristics of 36 kindreds. Medicine. (2002) 81(1):1–26. doi: 10.1097/00005792-200201000-00001
17. Simonds WF, Robbins CM, Agarwal SK, Hendy GN, Carpten JD, Marx SJ. Familial isolated hyperparathyroidism is rarely caused by germline mutation in HRPT2, the gene for the hyperparathyroidism-jaw tumor syndrome. J Clin Endocrinol Metab (2004) 89(1):96–102. doi: 10.1210/jc.2003-030675
18. Pontikides N, Karras S, Kaprara A, Anagnostis P, Mintziori G, Goulis DG, et al. Genetic basis of familial isolated hyperparathyroidism: A case series and a narrative review of the literature. J Bone Miner Metab (2014) 32(4):351–66. doi: 10.1007/s00774-013-0551-9
19. Guan B, Welch JM, Sapp JC, Ling H, Li Y, Johnston JJ, et al. GCM2-activating mutations in familial isolated hyperparathyroidism. Am J Hum Genet (2016) 99(5):1034–44. doi: 10.1016/j.ajhg.2016.08.018
20. Marx SJ, Goltzman D. Evolution of our understanding of the hyperparathyroid syndromes: A historical perspective. J Bone Mineral Res John Wiley Sons Inc.; (2019) 34:22–37. doi: 10.1002/jbmr.3650
21. el Lakis M, Nockel P, Gaitanidis A, Guan B, Agarwal S, Welch J, et al. Probability of positive genetic testing results in patients with family history of primary hyperparathyroidism. J Am Coll Surg (2018) 226(5):933–8. doi: 10.1016/j.jamcollsurg.2018.01.007
22. Warner J, Epstein M, Sweet A, Singh D, Burgess J, Stranks S, et al. Genetic testing in familial isolated hyperparathyroidism: Unexpected results and their implications. J Med Genet (2004) 41(3):155–60. doi: 10.1136/jmg.2003.016725
23. Pardi E, Marcocci C, Borsari S, Saponaro F, Torregrossa L, Tancredi M, et al. Aryl hydrocarbon receptor interacting protein (AIP) mutations occur rarely in sporadic parathyroid adenomas. J Clin Endocrinol Metab (2013) 98(7):2800–10. doi: 10.1210/jc.2012-4029
24. Suzuki Y, Sawada H, Tokumasu T, Suzuki S, Ninomiya S. Novel TRPV6 mutations in the spectrum of transient neonatal hyperparathyroidism. J Physiol Sci (2020) 70(1):33. doi: 10.1186/s12576-020-00761-2
25. Jha S, Simonds WF. Molecular and Clinical Spectrum of Primary Hyperparathyroidism The symptoms of PHPT can be non-specific, potentially delaying the diagnosis. Jha: Endocr Rev. 24:bnad009. doi: 10.1210/endrev/bnad009
26. Cetani F, Pardi E, Ambrogini E, Lemmi M, Borsari S, Cianferotti L, et al. Genetic analyses in familial isolated hyperparathyroidism: Implication for clinical assessment and surgical management. Clin Endocrinol (Oxf). (2006) 64(2):146–52. doi: 10.1111/j.1365-2265.2006.02438.x
27. Pardi E, Borsari S, Saponaro F, Bogazzi F, Urbani C, Mariotti S, et al. Mutational and large deletion study of genes implicated in hereditary forms of primary hyperparathyroidism and correlation with clinical features. PLoS One (2017) 12(10):e0186485. doi: 10.1371/journal.pone.0186485
28. Vierimaa O, Villablanca A, Alimov A, Georgitsi M, Raitila A, Vahteristo P, et al. Mutation analysis of MEN1, HRPT2, CASR, CDKN1B, and AIP genes in primary hyperparathyroidism patients with features of genetic predisposition. J Endocrinol Invest (2009) 32:512–8. doi: 10.3275/6107
29. Starker LF, Åkerström T, Long WD, Delgado-Verdugo A, Donovan P, Udelsman R, et al. Frequent germ-line mutations of the MEN1, CASR, and HRPT2/CDC73 genes in young patients with clinically non-familial primary hyperparathyroidism. Horm Cancer. (2012) 3(1–2):44–51. doi: 10.1007/s12672-011-0100-8
30. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr Protoc Bioinf (2013) 43(1110):1–33. doi: 10.1002/0471250953.bi1110s43
31. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
32. Mizusawa N, Uchino S, Iwata T, Tsuyuguchi M, Suzuki Y, Mizukoshi T, et al. Genetic analyses in patients with familial isolated hyperparathyroidism and hyperparathyroidism-jaw tumour syndrome. Clin Endocrinol (Oxf). (2006) 65(1):9–16. doi: 10.1111/j.1365-2265.2006.02534.x
33. Mariathasan S, Andrews K, Thompson E, Armstrong R, Park SM, Casey R. Genetic testing for hereditary hyperparathyroidism in a large UK cohort. Endocrine Abstracts (2019) 93(4):409–18. doi: 10.1530/endoabs.63.OC1.1
34. el Allali Y, Hermetet C, Bacchetta J, Amouroux C, Rothenbuhler A, Porquet-Bordes V, et al. Presenting features and molecular genetics of primary hyperparathyroidism in the paediatric population. Eur J Endocrinol (2021) 184(2):347–55. doi: 10.1530/EJE-20-1119
35. Erickson LA, Mete O, Juhlin CC, Perren A, Gill AJ. Overview of the 2022 WHO classification of parathyroid tumors. Endocrine Pathol (2022) 33:64–89. doi: 10.1007/s12022-022-09709-1
36. Eastell R, Brandi ML, Costa AG, D’Amour P, Shoback DM, Thakker RV. Diagnosis of asymptomatic primary hyperparathyroidism: Proceedings of the fourth international workshop. J Clin Endocrinol Metab (2014) 99(10):3570–9. doi: 10.1210/jc.2014-1414
37. Chou YHW, Pollak MR, Brandi ML, Toss G, Arnqvist H, Atkinson AB, et al. Mutations in the human ca2+-sensing-receptor gene that cause familial hypocalciuric hypercalcemia. Am J Hum Genet (1995) 56(5):1075–9.
38. Hannan FM, Nesbit MA, Zhang C, Cranston T, Curley AJ, Harding B, et al. Identification of 70 calcium-sensing receptor mutations in hyper- and hypo-calcaemic patients: Evidence for clustering of extracellular domain mutations at calcium-binding sites. Hum Mol Genet (2012) 21(12):2768–78. doi: 10.1093/hmg/dds105
39. Rotundo G, Turco EM, Ruotolo G, Torrente I, Candido O, Lopez G, et al. Generation of an induced pluripotent stem cell line CSSi015-A (9553), carrying a point mutation c.2915C > T in the human calcium sensing receptor (CasR) gene. Stem Cell Res (2023) 67:103023. doi: 10.1016/j.scr.2023.103023
40. Burski K, Torjussen B, Paulsen AQ, Boman H, Bollerslev J. Parathyroid adenoma in a subject with familial hypocalciuric hypercalcemia: Coincidence or causality? (2002). Available at: https://academic.oup.com/jcem/article/87/3/1015/2846641.
41. Elizabeth Forde H, Hill AD, Smith D. Parathyroid adenoma in a patient with familial hypocalciuric hypercalcaemia (2014). Available at: http://group.bmj.com/group/rights-licensing/permissions.
42. Mouly C, Vargas-Poussou R, Lienhardt A, Silve C, Hureaux M, Magdelaine C, et al. Clinical characteristics of familial hypocalciuric hypercalcaemia type 1: A multicentre study of 77 adult patients. Clin Endocrinol (Oxf). (2020) 93(3):248–60. doi: 10.1111/cen.14211
43. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinology. (2014) 386:2–15. doi: 10.1016/j.mce.2013.08.002
44. Suzuki Y, Chitayat D, Sawada H, Deardorff MA, Mclaughlin HM, Begtrup A, et al. TRPV6 variants interfere with maternal-fetal calcium transport through the placenta and cause transient neonatal hyperparathyroidism. Am J Hum Genet [Internet]. (2018) 102(6):1104–14. doi: 10.1016/j.ajhg.2018.04.006
45. Nett V, Erhardt N, Wyatt A, Wissenbach U. BBA - General Subjects Human TRPV6-pathies caused by gene mutations. Biochim Biophys Acta Gen Subj (2021) 1865(6):129873. doi: 10.1016/j.bbagen.2021.129873
46. Khattar V, Wang L, Peng JB. Calcium selective channel TRPV6: Structure, function, and implications in health and disease. Gene (2022) 817:146192. doi: 10.1016/j.gene.2022.146192
47. Lagarde A, Mougel G, Coppin L, Haissaguerre M, Le Collen L, Mohamed A, et al. Systematic detection of mosaicism by using digital NGS reveals three new MEN1 mosaicisms. Endocr Connect (2022) 11(11):e220093. doi: 10.1530/EC-22-0093
48. Cetani F, Marcocci C, Torregrossa L, Pardi E. Atypical parathyroid adenomas: Challenging lesions in the differential diagnosis of endocrine tumors. Endocrine-Related Cancer (2019) 26:R441–64. doi: 10.1530/ERC-19-0135
49. Alberto F. Genetics of parathyroids disorders: Overview. Best Pract Research: Clin Endocrinol Metab (2018) 32:781–90. doi: 10.1016/j.beem.2018.09.011
Keywords: parathyroid, primary hyperparathyroidism, multiple endocrine neoplasia type 1, multiple endocrine neoplasia type 4, cyclin-dependent kinase inhibitors, familial hyperparathyroidism, familial hypocalciuric hypercalcemia
Citation: Mazarico-Altisent I, Capel I, Baena N, Bella-Cueto MR, Barcons S, Guirao X, Pareja R, Muntean A, Arsentales V, Caixàs A and Rigla M (2023) Genetic testing for familial hyperparathyroidism: clinical-genetic profile in a Mediterranean cohort. Front. Endocrinol. 14:1244361. doi: 10.3389/fendo.2023.1244361
Received: 22 June 2023; Accepted: 04 September 2023;
Published: 21 September 2023.
Edited by:
Antonino Catalano, University of Messina, ItalyReviewed by:
Anne Barlier, Ludwig Maximilian University of Munich, GermanyCopyright © 2023 Mazarico-Altisent, Capel, Baena, Bella-Cueto, Barcons, Guirao, Pareja, Muntean, Arsentales, Caixàs and Rigla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabel Mazarico-Altisent, aXNhbWF6YXJpY29AZ21haWwuY29t
†ORCID: Isabel Mazarico-Altisent, orcid.org/0000-0001-9651-7650
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.