
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 29 August 2023
Sec. Pituitary Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1242588
This article is part of the Research TopicInsights in HyperprolactinemiaView all 6 articles
 Laurent Vroonen1
Laurent Vroonen1 Albert Beckers1Severine Camby2
Albert Beckers1Severine Camby2 Thomas Cuny3Pablo Beckers4
Thomas Cuny3Pablo Beckers4 Marie-Lise Jaffrain-Rea5,6Muriel Cogne7
Marie-Lise Jaffrain-Rea5,6Muriel Cogne7 Luciana Naves8
Luciana Naves8 Amandine Ferriere9
Amandine Ferriere9 Pauline Romanet10
Pauline Romanet10 Atanaska Elenkova11Auli Karhu12,13Thierry Brue3,10
Atanaska Elenkova11Auli Karhu12,13Thierry Brue3,10 Anne Barlier10
Anne Barlier10 Patrick Pétrossians1
Patrick Pétrossians1 Adrian F. Daly1*
Adrian F. Daly1*Introduction: Prolactinomas are the most frequent type of pituitary adenoma encountered in clinical practice. Dopamine agonists (DA) like cabergoline typically provide sign/ symptom control, normalize prolactin levels and decrease tumor size in most patients. DA-resistant prolactinomas are infrequent and can occur in association with some genetic causes like MEN1 and pathogenic germline variants in the AIP gene (AIPvar).
Methods: We compared the clinical, radiological, and therapeutic characteristics of AIPvar-related prolactinomas (n=13) with unselected hospital-treated prolactinomas (“unselected”, n=41) and genetically-negative, DA-resistant prolactinomas (DA-resistant, n=39).
Results: AIPvar-related prolactinomas occurred at a significantly younger age than the unselected or DA-resistant prolactinomas (p<0.01). Males were more common in the AIPvar (75.0%) and DA- resistant (49.7%) versus unselected prolactinomas (9.8%; p<0.001). AIPvar prolactinomas exhibited significantly more frequent invasion than the other groups (p<0.001) and exhibited a trend to larger tumor diameter. The DA-resistant group had significantly higher prolactin levels at diagnosis than the AIPvar group (p<0.001). Maximum DA doses were significantly higher in the AIPvar and DA-resistant groups versus unselected. DA-induced macroadenoma shrinkage (>50%) occurred in 58.3% in the AIPvar group versus 4.2% in the DA-resistant group (p<0.01). Surgery was more frequent in the AIPvar and DA- resistant groups (43.8% and 61.5%, respectively) versus unselected (19.5%: p<0.01). Radiotherapy was used only in AIPvar (18.8%) and DA-resistant (25.6%) groups.
Discussion: AIPvar confer an aggressive phenotype in prolactinomas, with invasive tumors occurring at a younger age. These characteristics can help differentiate rare AIPvar related prolactinomas from DA-resistant, genetically-negative tumors.
Prolactinomas are the most frequent (53%) clinically relevant pituitary adenomas, occurring with a prevalence of about 1 case in every 2000 of the general population (1). Typically, prolactinomas are diagnosed in premenopausal women, most often as microadenomas that present with menstrual disturbance and galactorrhea (2). With increasing age, the epidemiology of prolactinomas changes, with males accounting for an increasing proportion of patients (3, 4). In general, dopamine agonists (DA), such as, cabergoline are the first-line treatment for prolactinomas, as these agents are effective in normalizing prolactin secretion and controlling signs and symptoms in the majority of cases. Prolactinomas that are resistant to DA at labelled doses may require careful dose escalation, or referral for neurosurgical resection (5, 6).
Despite being the predominant form of pituitary adenomas, relatively little is known about the pathophysiology of prolactinomas. Most pituitary adenomas are sporadic and are believed to arise from somatic genetic changes in a single cell that later undergoes clonal expansion (7). Few consistent molecular genetic abnormalities have been identified to date in studies of prolactinoma tissue. Prolactinomas can occur as part of an inherited multiorgan endocrine tumor syndrome, such as multiple endocrine neoplasia (MEN) 1, MEN4, and pheochromocytoma/paraganglioma/pituitary adenoma (3P) Association (8). In MEN1, like in sporadic tumors, prolactinomas are the most frequent pituitary adenoma seen (9–11). Prolactinomas also occur in the setting of familial isolated pituitary adenoma (FIPA) kindreds, in which two or more related members in a family have isolated pituitary adenomas in the absence of MEN1 or other multiple endocrine neoplasias (12). In FIPA, prolactinomas tend to be larger and occur at a younger age than sporadic tumors (12). About 15-25% of FIPA kindreds have an underlying pathogenic AIP gene variant (AIPvar), which typically leads to acromegaly-gigantism, although many adenomas are mixed growth hormone and prolactin-secreting, but prolactinomas are seen occasionally (13–15). Apparently sporadic prolactinomas with an aggressive phenotype occurring in young individuals can also be due to AIPvar (16, 17). In acromegaly, AIPvar leads to a clinical phenotype of resistance to the first-generation somatostatin analogs, octreotide and lanreotide (16). Due to their comparative rarity, less is known about the characteristics of prolactinomas due to AIPvar, although DA resistance has been described (16). To better characterize the profile of patients with prolactinomas due to AIPvar, we performed a retrospective study in which the demographic, tumoral and therapeutic characteristics of AIPvar patients were compared with two control groups: an unselected group of prolactinomas from a real-world clinical setting and AIP-negative, DA-resistant prolactinoma patients.
This was an international, retrospective study performed in subjects with prolactinoma and an accompanying pathogenic or probably pathogenic germline variant in the AIP gene. The study population consisted of three groups: the AIPvar group, a dopamine agonist (DA)-resistant group, and an unselected prolactinoma group. The international, DA-resistant comparator group consisted of a series of subjects referred for genetic testing due to prolactinomas that were resistant to DA treatment at the maximum labelled dose and in which no germline variants or deletions in AIP, MEN1, or CDKN1B were found on genetic testing. In practice, DA resistance was taken as a lack of normalization of serum prolactin following treatment with the maximum labelled dose of 2 mg/week of cabergoline for at least 6 months (18). The second comparator group were subjects with prolactinomas that were not filtered or pre-selected in terms of clinical characteristics or genetic screening results. This group represented a “real-world” population studied at the Department of Endocrinology, Centre Hospitalier Universitaire de Liège, Belgium.
To be eligible, all subjects from all groups had to have proven radiological evidence of a pituitary adenoma and to have demonstrated elevated levels of prolactin that was associated with clinical symptoms. Individuals with non-tumoral, drug-induced or unexplained hyperprolactinemia were not included. The demographic and clinical characteristics of all subjects were collected and included the following criteria: age, sex, age at first symptoms and diagnosis, tumoral maximum diameter (mm) on magnetic resonance imaging (MRI), invasion (yes/no; unilateral, bilateral), extrasellar extension, optic chiasma compression, treatments used, surgery (number of interventions), radiotherapy, prolactin level at diagnosis and nadir levels under treatments.
Genetic testing for sequence variants in AIP, MEN1 and CDKN1B was performed as previously described (13, 19). Only pathogenic and probably pathogenic variants were selected. In addition, multiplex ligation-specific probe amplification (MLPA) kits were used to identify deletions of whole or part of these three genes (SALSA® MLPA® Probemix P244, MRC-Holland, The Netherlands). Previously we reported the clinical characteristics of some of these pathogenic AIPvar patients but without comparisons with control groups (16). The study was approved by the Ethics Committee of the University of Liège and all patients provided informed consent.
Data on discrete variables were expressed as medians and interquartile ranges (Q1-Q3) and analyzed using Wilcoxon’s test. Categorical variables in the different groups were assessed using Pearson’s chi-squared test with Yates’ continuity correction. Statistical analyses were performed with the R software package (R Core Team 2015; http://www.R-project.org). Graphics were plotted using the ggplot2 library (https://ggplot2.tidyverse.org). Ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York. ISBN 978-3-319-24277-4)
The study population consisted of 13 subjects with AIPvar, 39 subjects with genetically-negative, DA-resistant prolactinomas and 41 unselected subjects with prolactinomas. There were 11 different germline AIPvar in the AIPvar group, all of which were previously described. Three variants in five subjects led to protein truncation: p.Gln14X (n=2), p.Tyr268X, and p.Gly117Alafs*39 (n=2). The remaining AIPvar were missense (p.Arg56Cys, p.Leu58Asn, p.Leu70Met, pVal195Ala, p.Lys241Glu, p.Tyr268Cys, and p.Arg271Trp) or a splice site change (c.100-18 C>T (IVS1)) in one subject each. Another three subjects with the variants p.Arg16His (n=1) and p.Arg304Gln (n=2), for which pathogenicity is debatable, were not included in the statistical analyses. Ten subjects came from heterogeneous acromegaly-prolactinoma FIPA kindreds (all subjects presented spontaneously and none had been identified on family screening) and three were apparently sporadic (without full available studies of parents’ AIP status).
The sex distribution was significantly different between the unselected controls (female: 90.2%) and the AIPvar (female: 30.8%; p<0.001) and DA-resistant groups (female: 51.3%; p<0.001). There was no statistically significant difference between the AIPvar and DA-resistant groups in terms of sex distribution (p=0.14). The AIPvar group had a significantly younger median age at first symptoms (18.0 years; Q1-Q3: 15.0-23.0) than either the unselected prolactinoma (34.0 years; Q1-Q3: 25.0-43.0; p<0.001) or DA-resistant groups (33.0 years; 19.0-43.0; p<0.01, Figure 1A). Similarly, the median age at diagnosis was significantly younger in the AIPvar prolactinoma group (19.0 years; Q1-Q3:15.0-24.0) versus the unselected (35.0 years; Q1-Q3: 25.0-43.0; p<0.001) and DA-resistant groups (35.0 years; Q1-Q3: 19.0-44.0: p<0.01, Figure 1B).
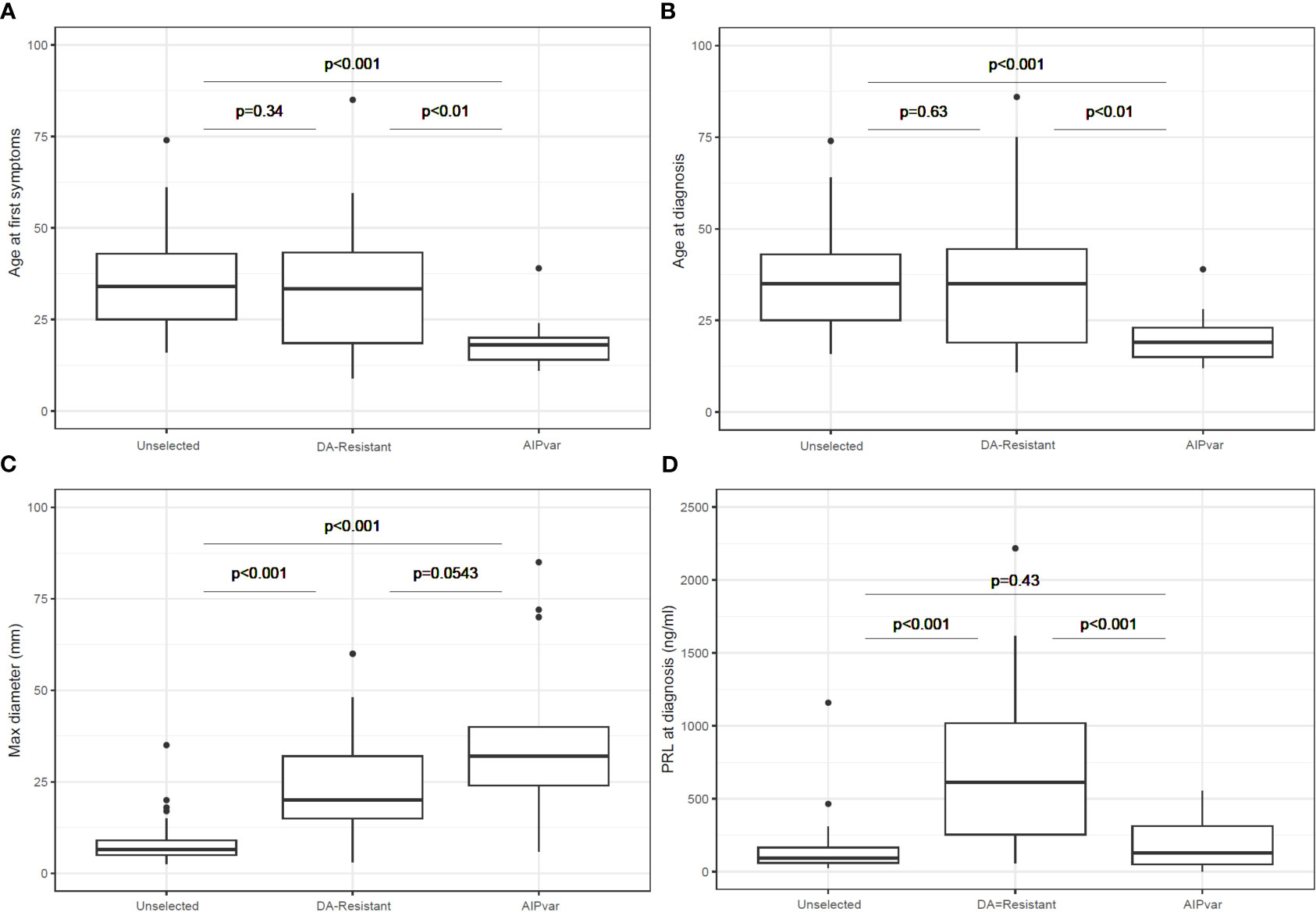
Figure 1 Comparisons between groups with AIPvar-related, unselected and DA-resistant prolactinomas in terms of age at first symptoms (A), age at diagnosis (B), maximum tumor diameter at diagnosis (C) and prolactin secretion at diagnosis (D). Box and whisker plots show medians as horizontal lines and the lower and upper limits of the box correspond to the first and third quartiles; the whiskers extend the box to 1.5 times the inter-quartile range (IQR) or to the most extreme values if they lie within this range.
AIPvar-associated prolactinomas had a significantly larger median maximum diameter (32.0 mm; Q1-Q3: 26-41) than the unselected prolactinoma group (6.5 mm; Q1-Q3: 5.0-9.0; p<0.001); there was a trend towards a larger tumor size in the AIPvar versus the DA-resistant groups (20.0 mm; Q1-Q3: 15.0-32.0; p=0.054: Figure 1C). At diagnosis, macroadenomas predominated in the AIPvar (92.3%) and DA-resistant groups (89.7%), whereas they constituted only 24.4% of the unselected prolactinomas. Giant adenomas (≥40 mm) were present in 5/13 (38.5%) of the AIPvar group, 7/39 (17.9%) of the DA-resistant and in none of the unselected patients (p<0.01 unselected vs AIPvar). Eleven of 13 (84.6%) AIPvar prolactinomas had suprasellar extension, versus 28.2% of the DA-resistant and 7.3% of the unselected groups (p ≤ 0.01 AIPvar vs DA-resistant or unselected). Also, chiasmal compression was >4 times more frequent at diagnosis in the AIPvar group (61.5%) as compared to the DA-resistant prolactinomas (12.8%) and >8-fold higher than in the unselected prolactinoma controls (p ≤ 0.01 AIPvar vs DA-resistant or unselected). Fully 76.9% of AIPvar-related prolactinomas had invaded the cavernous sinuses (30.8% bilaterally) at the time of diagnosis, as compared with 28.2% of DA-resistant prolactinomas (0% bilateral) and only 14.6% (0% bilateral) of the unselected group (p<0.001 AIPvar vs DA-resistant or unselected).
In contrast to the above differences in tumor characteristics, the DA-resistant prolactinoma group had a significantly higher prolactin level at diagnosis as compared with both the unselected controls and the AIPvar group (Figure 1D; p<0.001). There was no significant difference in prolactin levels at diagnosis between the AIPvar and unselected controls (p=0.43). There was no evidence of interference by elevated macroprolactin forms in the biochemical analyses of the AIPvar group. The median maximal dose of cabergoline in the unselected prolactinoma group was significantly lower (0.5: Q1-Q3: 0.25-0.75 mg) as compared with the AIPvar (3.5: Q1-Q3: 2.0-5.0 mg; p<0.001) and the DA-resistant groups (3.5: Q1-Q3: 3.0-4.0 mg; p<0.001, Figure 2A); AIPvar and DA-resistant groups did not differ significantly (p=0.41). The median decrease in prolactin from baseline at last follow-up was approximately the same across all three study groups (90.0-95.4%; Figure 2B). As shown in the waterfall plot in Figure 2C, however, the high prolactin at baseline in the DA-resistant group meant that these >90% decreases were insufficient to normalise prolactin levels in many cases. Hyperprolactinemia remained at the maximum dose of DA in 4/13 (30.7%) in the AIPvar group, 5/41 (12.2%) in the unselected group and in 26/39 (66.7%) of the DA-resistant group. We also examined the relationship between prolactin secretion at diagnosis and tumor diameter. As shown in Figure 2D, most tumors in the unselected group were small and the prolactin level steadily increased with rising tumor size ( ± 23.5 mg/dL per each mm of tumor diameter, p<0.001). In the DA-resistant group, tumors were larger and secreted more prolactin; this secretion rose significantly at a rate of ± 154.0 mg/dL for each mm increase in diameter (p<0.001). Prolactinomas in the AIPvar group, which were larger than in the unselected group, had a different secretory pattern, with the prolactin levels barely increasing by 5.8 mg/dL for each mm of tumor diameter (p=0.028).
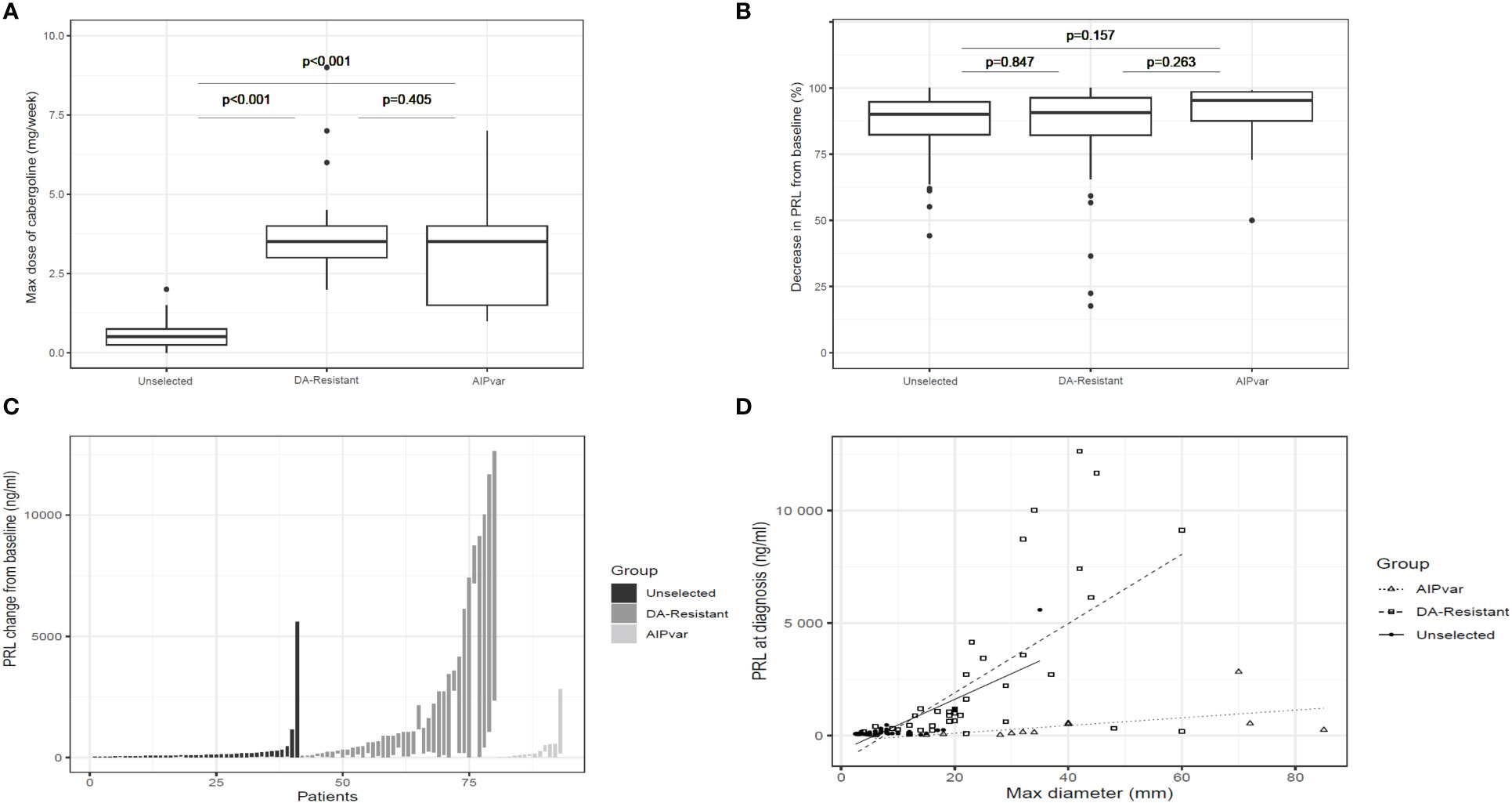
Figure 2 Dopamine agonist treatment of prolactinomas. Comparisons between groups with AIPvar-related, unselected and DA-resistant prolactinomas according to median weekly dose of cabergoline (A); median percentage reduction from baseline at maximum cabergoline dose in the three study groups (B); a waterfall plot depicts the individual reductions in prolactin from baseline to nadir levels under maximum cabergoline doses in the three groups (C). The relationship of prolactinoma maximum diameter and prolactin secretion at baseline across the three study groups was compared in panel (D) Box and whisker plots show medians as horizontal lines and the lower and upper limits of the box correspond to the first and third quartiles; the whiskers extend the box to 1.5 times the inter-quartile range (IQR) or to the most extreme values if they lie within this range.
Shrinkage of >50% following DA treatment occurred in 7/12 with evaluable MRI data in the AIPvar group, as compared with only 1/24 in the DA-resistant group (p<0.01). Surgery was more frequent in the AIPvar and DA-resistant groups (53.8% and 61.5%, respectively) as compared with the unselected prolactinomas (19.5%: p<0.01 for AIPvar and DA-resistant versus unselected patients). No patient in the unselected prolactinoma group received radiotherapy, whereas it was used in 23.1% and 25.6% of the AIPvar and DA-resistant groups, respectively (p<0.03 for AIPvar and DA-resistant versus unselected prolactinomas).
Prolactinomas are the most frequently encountered pituitary adenoma in clinical practice (1, 20). In children, adolescents and young adults, prolactinomas are also the most common pituitary adenoma (21). Most present as microadenomas in fertile women that are responsive to labelled doses of cabergoline (2). Prolactinomas that are resistant or refractory to these DA doses are rarer, and some can have aggressive clinical characteristics (22, 23). The pathophysiology of DA resistance in prolactinomas is largely unknown, but factors like altered expression of dopamine D2 receptor levels have been suggested (24). Although AIP has been implicated in resistance to somatostatin analogs in AIPvar somatotropinomas (16, 25), this has not been explored in details in AIPvar prolactinomas. As AIPvar have been rarely reported in prolactinomas occurring in FIPA kindreds, sporadic or resistant cases, in the current study we wanted to further characterize AIPvar associated prolactinomas as compared to relevant controls. To this end, we chose two study groups: 1) an unselected population of prolactinoma patients which represented a real-world population typical of those encountered in specialist (hospital) endocrine practice, in whom genetic testing is not routinely employed; 2) a population of DA-resistant prolactinomas with negative genetic testing.
From an epidemiological point of view, AIPvar prolactinomas had a median age at symptom onset and diagnosis that was >10 years younger than unselected and DA-resistant control; 75% were males. In addition, AIPvar-associated prolactinomas presented as large macroadenomas (median diameter 33.0 mm), with suprasellar extension, and frequent chiasmal compression and invasion. They differed from the unselected control group that was comprised predominantly (approx. 90%) of female microprolactinoma patients with a median age of 35 at diagnosis. The unselected control group responded well overall to DA treatment, with 82% having normal prolactin at a weekly cabergoline dose of ≤2.0 mg. As most of these patients had small microprolactinomas, it was not readily feasible to capture data on our shrinkage criteria (>50% from baseline) on MRI across this group. There were a few unselected prolactinoma patients that were either uncontrolled on DA or that had surgery (n=8), which reflects a hospital-based population (not community managed) and emphasizes that DA-led management is usually successful. With the other control group, we defined a population in which none of the known genetic causes of pituitary adenomas, including MEN1 and AIPvar was present. That comparison shows that AIPvar-related prolactinomas have certain differences from DA-resistant tumors, and these characteristics might be relevant when considering which prolactinoma patients for genetic testing. Young males with large or giant, invasive prolactinomas were typical of the AIPvar group. In contrast, sex distribution (predominantly male), percentage of macroadenomas, and higher prolactin secretion at diagnosis did not discriminate AIPvar-related tumors from genetically-negative, DA-resistant controls. Indeed, unlike the DA-resistant group, in which a sub-set of patients are characterized by very high prolactin and large tumor size, in the AIPvar group prolactin levels increased little with rising tumor volume, which is unusual for prolactinomas in general (26). Furthermore, the AIPvar group had a lower rate of surgery, radiotherapy and a lower maximal cabergoline dose than the DA-resistant group. Control of hyperprolactinemia was achieved at the maximum DA dose in almost twice as many AIPvar patients as compared with the DA-resistant patients, indicating that while large and invasive prolactinomas are characteristic of AIPvar-related tumors, DA resistance is not absolute and control with DA or multimodal therapy is achievable. As noted by others, even large, complex prolactinomas related to AIPvar can have clinically-relevant hormonal and tumoral responses to chronic DA therapy (27). Unlike in the AIPvar group, the DA-resistant group had a sub-set of patients that are characterized by very high prolactin and large tumor size. The molecular pathways that explain why large prolactinomas can differ in terms of their secretory abilities remain unclear. Differing therapeutic responses to DA may be related to dopamine D2 receptor levels regulation, although this remains to be studied in AIPvar-related prolactinomas.
Overall, these results suggest that the main consequence of genetic disturbance of AIP favors earlier and more aggressive tumoral growth in prolactinomas. This echoes some, but not all, aspects of the more typical clinical presentation of AIPvar-related pituitary tumors, namely acromegaly and pituitary gigantism. In patients with AIPvar-related acromegaly, an earlier age at onset/diagnosis, male predominance, larger tumor size and relative resistance to medical treatment was seen as compared to AIP wild-type acromegaly controls (16). In AIPvar-related prolactinomas, we found additionally that invasion at baseline was a clinical characteristic that differentiated that group from DA-resistant and unselected subgroups. There was also a higher percentage of giant adenomas in the AIPvar group as compared with the DA-resistant group, which presumably contributed to higher rates of chiasmal compression in the AIPvar group. We did not observe cases of clinical or radiological apoplexy in any of the treatment groups in the study.
Our results suggest that when considering prolactinoma patients for AIP testing, DA-resistance should not be the only factor and should be accompanied by large and invasive tumoral growth in young patients. This echoes the negative results with using isolated somatostatin analog resistance as a factor for AIP testing in otherwise unselected acromegaly populations (28). Aggressive growth beginning at a young age was also a defining characteristic of AIPvar-related prolactinomas in other studies. In 90 pediatric patients prolactinomas, Kumar et al. reported on 18 cases of giant prolactinomas; of these, 18.8% of patients tested had an AIP pathogenic variant and AIP status was a predictor of requirement for other therapies in addition to DA (29). In a large study of 77 patients aged <20 with macroprolactinomas, Salenave et al. found that 5/55 (9%) of subjects had an AIP pathogenic variant, and 3/59 (5%) had a MEN1 pathogenic variant. Interestingly, there was a relationship between DA-resistance and MEN1 variant status, but not AIPvar status (30).
Numerous screening studies in series of patients with unselected pituitary adenomas, pediatric-onset pituitary adenomas, pituitary macroadenomas, and FIPA have been performed (13, 15, 17, 28, 30–42). About two-thirds of individuals with AIPvar-related pituitary adenomas present with acromegaly, usually in the setting of FIPA, and among this group, pituitary gigantism occurs in 32% (16). Prolactinomas are the second most common presentation (14.5%) and tumors with mixed growth hormone and prolactin secretion also occur (9.5%) (14). Among AIPvar patients, Hernandez Ramirez et al. reported that 7/31 variants occurred in patients with prolactinomas and of a total of 175 AIPvar positive subjects in the UK series, 19 (10.9%) had prolactinomas (27, 43). Although prolactinomas form an integral part of the AIPvar related spectrum of pituitary adenomas, they usually occur along with acromegaly in heterogeneous FIPA kindreds. In contrast, prolactinoma-only families that are a frequent presentation of FIPA are almost always AIPvar negative (14, 44). To date, only one FIPA family with homogeneous prolactinoma and a pathogenic AIPvar has been reported (27). In that Scottish family with a p.Arg304Ter pathogenic variant, there were four subjects with prolactinomas. The presentation ranged from a male in his 40’s with a giant partially cystic adenoma, to his sisters in their mid-30’s with microadenomas and amenorrhea (one sister had two pituitary lesions) and the son of one of the sisters with an asymptomatic but biologically active macroprolactinoma who was diagnosed on family AIP screening. Responses to cabergoline in that family were relatively good, although doses were limited by tolerability or were being up-titrated in three of the four reported patients.
How to adequately define DA resistance in terms of dose and response criteria remains unclear and has been the subject of significant debate (5). For this and previous work, we used the dose definition proposed by Molitch, which relates to the upper limit of the package insert/labelled dose of cabergoline, at 2.0 mg per week (18, 45). This cut-off has a logical basis, being derived from the highest dose at which safety and efficacy was established for hyperprolactinemia treatment from registration studies. As noted by Maiter, all cut-off dose levels of cabergoline or other DAs are essentially arbitrary, given that the ideal and safe dose to which patients can be up-titrated is unknown and depends on the strictness of the criteria for an acceptable hormonal and symptomatic response (5). Cabergoline is generally safe within its prescribed dose range and the cumulative risk of adverse events like cardiac valvulopathy has been shown to be low (46–48). Tolerability of DA treatment and its relationship with psychological disorders should always be assessed on a patient-by-patient basis (49, 50). The degree of shrinkage that is deemed clinically relevant is also arbitrary and a wide range of cut-offs have been suggested from 30-80% (5). The pragmatic approach is to be guided by the clinical status of the patient; for tumor shrinkage, the main goal should be alleviation of potential mass effects like optic chiasmal impingement. In the case of the mildly hyperprolactinemic patient on DA with sign/symptom resolution and whose tumor size is controlled, it is unclear if there is a tangible medical benefit to be gained by further up-titrating DA to achieve strictly numerically normal prolactin levels.
This study is a retrospective series in which the patient population was referred for genetic testing due to factors relating to an aggressive disease profile, including FIPA, prolactinomas occurring at a young age and an aggressive disease history. For this reason, we included a DA resistant control group that was genetically negative, and this allowed us to demonstrate that AIPvar-related prolactinomas have specific characteristics. The other unselected control group came from a single center, whereas the AIPvar and DA-resistant populations were multi-center, international cohorts. Local management decisions in the unselected controls could influence the comparisons, but in general therapeutic choices followed standard guidelines, so any center effect would be marginal. The comparisons on tumor shrinkage on DA were limited by the high frequency of very small micro-prolactinomas in the unselected controls. In the AIPvar group, the genetic variants seen were rare, but there remains uncertainty about their definitive pathogenic nature. This is an ongoing limitation in the field of AIP testing, as the functional data from in vitro models give varying interpretations of pathogenicity (25, 51–53). Altering testing recommendations based on this rare AIP-related presentation of prolactinomas should be done with caution. Like in acromegaly, there is no apparent value in performing AIP (or other) germline genetic testing in unselected prolactinomas at this time.
In conclusion, we confirm that prolactinomas occurring in sporadic and FIPA patients with germline AIPvar have aggressive clinical characteristics. In general, AIPvar confer a growth and aggression phenotype in prolactinomas, with larger, invasive tumors occurring at a younger age than in controls, but extremely elevated prolactin levels are not characteristic. These features differentiate AIPvar related prolactinomas from DA-resistant, genetically negative tumors. AIPvar and DA-resistant, genetically-negative groups were similar in terms of sex (predominantly male), and prolactin secretion at diagnosis. AIPvar related tumors were, however, not more challenging to treat than the wild-type DA resistant prolactinoma group, with the latter undergoing more frequent surgery, radiotherapy and using a higher maximal cabergoline dose.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Hospitalo-Facultaire Ethics Committee, University of Liege. The patients/participants provided their written informed consent to participate in this study.
LV, ABe and AD conceived of and designed the study. LV, SC, TC, M-LJ-R, MC, LN, AF, AE, AK, TB, ABa and AD identified subjects and collected clinical information on active and control groups. PB and PR conducted genetic analyses. LV, AD, PP designed the study database. PP conducted statistical analyses. LV, PP and AD wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
The study received support from FIRS grants from the Centre Hospitalier Universitaire de Liege 2018-2020.
The authors would like to acknowledge Dr. Lucien Marchand and Prof. Antoine Tabarin for discussions on potential patients; Dr. Emilie Castermans and Ms. Nathalie Sacre for discussions on AIP and other sequence analyses.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Daly AF, Beckers A. The epidemiology of pituitary adenomas. Endocrinol Metab Clin North Am (2020) 49:347–55. doi: 10.1016/j.ecl.2020.04.002
2. Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab (2011) 96:273–88. doi: 10.1210/jc.2010-1692
3. Vasilev V, Daly AF, Vroonen L, et al. Resistant prolactinomas. J Endocrinol Invest (2011) 34:312–316. doi: 10.1007/BF03347092
4. Chanson P, Maiter D. The epidemiology, diagnosis and treatment of Prolactinomas: The old and the new. Best Pract Res Clin Endocrinol Metab (2019) 33:101290. doi: 10.1016/j.beem.2019.101290
5. Maiter D. Management of dopamine agonist-resistant prolactinoma. Neuroendocrinology (2019) 109:42–50. doi: 10.1159/000495775
6. Souteiro P, Karavitaki N. Dopamine agonist resistant prolactinomas: any alternative medical treatment? Pituitary (2020) 23:27–37. doi: 10.1007/s11102-019-00987-3
7. Herman V, Fagin J, Gonsky R, Kovacs K, Melmed S. Clonal origin of pituitary adenomas. J Clin Endocrinol Metab (1990) 71:1427–33. doi: 10.1210/jcem-71-6-1427
8. Vandeva S, Daly AF, Petrossians P, Zacharieva S, Beckers A. Somatic and germline mutations in the pathogenesis of pituitary adenomas. Eur J Endocrinol (2019) 181:R235–54. doi: 10.1530/EJE-19-0602
9. Vergès B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, et al. Pituitary disease in MEN type 1 (MEN1): Data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab (2002) 87:457–65. doi: 10.1210/jcem.87.2.8145
10. de Laat JM, Dekkers OM, Pieterman CRC, Kluijfhout WP, Hermus AR, Pereira AM, et al. Long-term natural course of pituitary tumors in patients with MEN1: results from the dutchMEN1 study group (DMSG). J Clin Endocrinol Metab (2015) 100:3288–96. doi: 10.1210/JC.2015-2015
11. Le Bras M, Leclerc H, Rousseau O, Goudet P, Cuny T, Castinetti F, et al. Pituitary adenoma in patients with multiple endocrine neoplasia type 1: a cohort study. Eur J Endocrinol (2021) 185:863–73. doi: 10.1530/EJE-21-0630
12. Daly AF, Jaffrain-Rea M-L, Ciccarelli A, Valdes-Socin H, Rohmer V, Tamburrano G, et al. Clinical characterization of familial isolated pituitary adenomas. J Clin Endocrinol Metab (2006) 91:3316–23. doi: 10.1210/jc.2005-2671
13. Daly AF, Vanbellinghen J-F, Sok KK, Jaffrain-Rea M-L, Naves LA, Guitelman MA, et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: Analysis in 73 families. J Clin Endocrinol Metab (2007) 92:1891–6. doi: 10.1210/jc.2006-2513
14. Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev (2013) 34:239–77. doi: 10.1210/er.2012-1013
15. Leontiou CA, Gueorguiev M, van der Spuy J, Quinton R, Lolli F, Hassan S, et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab (2008) 93:2390–401. doi: 10.1210/jc.2007-2611
16. Daly AF, Tichomirowa MA, Petrossians P, Heliövaara E, Jaffrain-Rea M-L, Barlier A, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab (2010) 95:E373–83. doi: 10.1210/jc.2009-2556
17. Tichomirowa MA, Barlier A, Daly AF, Jaffrain-Rea M-L, Ronchi C, Yaneva M, et al. High prevalence of AIP gene mutations following focused screening in young patients with sporadic pituitary macroadenomas. Eur J Endocrinol (2011) 165:509–15. doi: 10.1530/EJE-11-0304
18. Molitch ME. Management of medically refractory prolactinoma. J Neurooncol (2014) 117:421–8. doi: 10.1007/s11060-013-1270-8
19. Tichomirowa MA, Lee M, Barlier A, Daly AF, Marinoni I, Jaffrain-Rea M-L, et al. Cyclin-dependent kinase inhibitor 1B (CDKN1B) gene variants in AIP mutation-negative familial isolated pituitary adenoma kindreds. Endocr Relat Cancer (2012) 19:233–41. doi: 10.1530/ERC-11-0362
20. Chanson P, Maiter D. Chapter 16 – prolactinoma. In: The pituitary (2017). (London, UK: Academic Press) 467–514. doi: 10.1016/B978-0-12-804169-7.00016-7
21. Arya VB, Aylwin SJB, Hulse T, Ajzensztejn M, Kalitsi J, Kalogirou N, et al. Prolactinoma in childhood and adolescence-Tumour size at presentation predicts management strategy: Single centre series and a systematic review and meta-analysis. Clin Endocrinol (Oxf) (2021) 94:413–23. doi: 10.1111/cen.14394
22. Vroonen L, Jaffrain-Rea M-L, Petrossians P, Tamagno G, Chanson P, Vilar L, et al. Prolactinomas resistant to standard doses of cabergoline: a multicenter study of 92 patients. Eur J Endocrinol (2012) 167:651–62. doi: 10.1530/EJE-12-0236
23. Vroonen L, Daly AF, Beckers A. Epidemiology and management challenges in prolactinomas. Neuroendocrinology (2019) 109:20–7. doi: 10.1159/000497746
24. Pivonello C, Patalano R, Negri M, Pirchio R, Colao A, Pivonello R, et al. Resistance to dopamine agonists in pituitary tumors: molecular mechanisms. Front Endocrinol (Lausanne) (2021) 12:791633. doi: 10.3389/fendo.2021.791633
25. Bogner EM, Daly AF, Gulde S, Karhu A, Irmler M, Beckers J, et al. miR-34a is upregulated in AIP-mutated somatotropinomas and promotes octreotide resistance. Int J Cancer (2020) 147:3523–38. doi: 10.1002/ijc.33268
26. Delgrange E, Trouillas J, Maiter D, Donckier J, Tourniaire J. Sex-related difference in the growth of prolactinomas: A clinical and proliferation marker study1. J Clin Endocrinol Metab (1997) 82:2102–7. doi: 10.1210/jcem.82.7.4088
27. Carty DM, Harte R, Drummond RS, Ward R, Magid K, Collier D, et al. AIP variant causing familial prolactinoma. Pituitary (2021) 24:48–52. doi: 10.1007/s11102-020-01085-5
28. Daly AF, Cano DA, Venegas-Moreno E, Petrossians P, Dios E, Castermans E, et al. AIP and MEN1 mutations and AIP immunohistochemistry in pituitary adenomas in a tertiary referral center. Endocr Connect (2019) 8:338–48. doi: 10.1530/EC-19-0027
29. Kumar S, Sarathi V, Lila AR, Sehemby M, Memon SS, Karlekar M, et al. Giant prolactinoma in children and adolescents: a single-center experience and systematic review. Pituitary (2022) 25:819–30. doi: 10.1007/s11102-022-01250-y
30. Salenave S, Ancelle D, Bahougne T, Raverot G, Kamenický P, Bouligand J, et al. Macroprolactinomas in children and adolescents: factors associated with the response to treatment in 77 patients. J Clin Endocrinol Metab (2015) 100:1177–86. doi: 10.1210/jc.2014-3670
31. Tuncer FN, Çiftçi Doğanşen S, Serbest E, Tanrıkulu S, Ekici Y, Bilgiç B, et al. Screening of AIP gene variations in a cohort of turkish patients with young-onset sporadic hormone-secreting pituitary adenomas. Genet Test Mol Biomarkers (2018) 22:702–8. doi: 10.1089/gtmb.2018.0133
32. Martínez de LaPiscina I, Portillo Najera N, Rica I, Gaztambide S, Webb SM, Santos A, et al. Clinical and genetic characteristics in patients under 30 years with sporadic pituitary adenomas. Eur J Endocrinol (2021) 185:485–96. doi: 10.1530/EJE-21-0075
33. Georgitsi M, Raitila A, Karhu A, Tuppurainen K, Makinen MJ, Vierimaa O, et al. Molecular diagnosis of pituitary adenoma predisposition caused by aryl hydrocarbon receptor-interacting protein gene mutations. Proc Natl Acad Sci U.S.A. (2007) 104:4101–5. doi: 10.1073/pnas.0700004104
34. Barlier A, Vanbellinghen J-F, Daly AF, Silvy M, Jaffrain-Rea M-L, Trouillas J, et al. Mutations in the aryl hydrocarbon receptor interacting protein gene are not highly prevalent among subjects with sporadic pituitary adenomas. J Clin Endocrinol Metab (2007) 92:1952–5. doi: 10.1210/jc.2006-2702
35. Cazabat L, Guillaud-Bataille M, Bertherat J, Raffin-Sanson ML. Mutations of the gene for the aryl hydrocarbon receptor-interacting protein in pituitary adenomas. Horm Res (2009) 71:132–41. doi: 10.1159/000197869
36. Cazabat L, Bouligand J, Salenave S, Bernier M, Gaillard S, Parker F, et al. Germline AIP mutations in apparently sporadic pituitary adenomas: prevalence in a prospective single-center cohort of 443 patients. J Clin Endocrinol Metab (2012) 97:E663–70. doi: 10.1210/jc.2011-2291
37. Cuny T, Pertuit M, Sahnoun-Fathallah M, Daly A, Occhi G, Odou MF, et al. Genetic analysis in young patients with sporadic pituitary macroadenomas: besides AIP don’t forget MEN1 genetic analysis. Eur J Endocrinol / Eur Fed Endocrine Societies (2013) 168:533–41. doi: 10.1530/EJE-12-0763
38. Occhi G, Trivellin G, Ceccato F, De Lazzari P, Giorgi G, Dematte S, et al. Prevalence of AIP mutations in a large series of sporadic Italian acromegalic patients and evaluation of CDKN1B status in acromegalic patients with multiple endocrine neoplasia. Eur J Endocrinol (2010) 163:369–76. doi: 10.1530/EJE-10-0327
39. Araujo PB, Kasuki L, de Azeredo Lima CH, Ogino L, Camacho AHS, Chimelli L, et al. AIP mutations in Brazilian patients with sporadic pituitary adenomas: a single-center evaluation. Endocr Connect (2017) 6:914–25. doi: 10.1530/EC-17-0237
40. Cai F, Zhang YD, Zhao X, Yang YK, Ma SH, Dai CX, et al. Screening for AIP gene mutations in a Han Chinese pituitary adenoma cohort followed by LOH analysis. Eur J Endocrinol (2013) 169:867–84. doi: 10.1530/EJE-13-0442
41. Preda V, Korbonits M, Cudlip S, Karavitaki N, Grossman AB. Low rate of germline AIP mutations in patients with apparently sporadic pituitary adenomas before the age of 40: a single-centre adult cohort. Eur J Endocrinol (2014) 171:659–66. doi: 10.1530/EJE-14-0426
42. Stratakis CA, Tichomirowa MA, Boikos S, Azevedo MF, Lodish M, Martari M, et al. The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin Genet (2010) 78:457–63. doi: 10.1111/j.1399-0004.2010.01406.x
43. Hernández-Ramírez LC, Gabrovska P, Dénes J, Stals K, Trivellin G, Tilley D, et al. Landscape of familial isolated and young-onset pituitary adenomas: prospective diagnosis in AIP mutation carriers. J Clin Endocrinol Metab (2015) 100:E1242–54. doi: 10.1210/jc.2015-1869
44. Vacchiano V, Seleme S, Daly AF, Beckers A, Valdés-Socin H. Clinical and genetic studies of a three-member familial isolated pituitary adenoma with homogeneous prolactinomas. Medicina (B Aires) (2020) 80:181–4.
45. Vroonen L, Jaffrain-Rea M-L, Petrossians P, Tamagno G, Chanson P, Vilar L, et al. Prolactinomas resistant to standard doses of cabergoline: a multicenter study of 92 patients. Eur J Endocrinol (2012) 167(5):651–62. doi: 10.1530/EJE-12-0236
46. Steeds R, Stiles C, Sharma V, Chambers J, Lloyd G, Drake W. Echocardiography and monitoring patients receiving dopamine agonist therapy for hyperprolactinaemia: A joint position statement of the British Society of Echocardiography, the British Heart Valve Society and the Society for Endocrinology. Clin Endocrinol (Oxf) (2019) 90:662–9. doi: 10.1111/cen.13940
47. Daly AF, Beckers A. A hard look at cardiac safety with dopamine agonists in endocrinology. J Clin Endocrinol Metab (2021) 106(6):e2452–e2454. doi: 10.1210/clinem/dgab073
48. Caputo C, Prior D, Inder WJ. The need for annual echocardiography to detect cabergoline-associated valvulopathy in patients with prolactinoma: A systematic review and additional clinical data. Lancet Diabetes Endocrinol (2015) 3:906–13. doi: 10.1016/S2213-8587(14)70212-8
49. Hamidianjahromi A, Tritos NA. Impulse control disorders in hyperprolactinemic patients on dopamine agonist therapy. Rev Endocr Metab Disord (2022) 23:1089–99. doi: 10.1007/s11154-022-09753-6
50. De Sousa SMC, Baranoff J, Rushworth RL, Butler J, Sorbello J, Vorster J, et al. Impulse control disorders in dopamine agonist-treated hyperprolactinemia: prevalence and risk factors. J Clin Endocrinol Metab (2020) 105:e108–18. doi: 10.1210/clinem/dgz076
51. Aflorei ED, Klapholz B, Chen C, Radian S, Dragu AN, Moderau N, et al. In vivo bioassay to test the pathogenicity of missense human AIP variants. J Med Genet (2018) 55:522. doi: 10.1136/jmedgenet-2017-105191
52. Hernández-Ramírez LC, Martucci F, Morgan RML, Trivellin G, Tilley D, Ramos-Guajardo N, et al. Rapid proteasomal degradation of mutant proteins is the primary mechanism leading to tumorigenesis in patients with missense AIP mutations. J Clin Endocrinol Metab (2016) 101:3144–54. doi: 10.1210/jc.2016-1307
Keywords: prolactinoma, genetic, resistance, dopamine agonist, cabergoline, AIP, MEN1
Citation: Vroonen L, Beckers A, Camby S, Cuny T, Beckers P, Jaffrain-Rea M-L, Cogne M, Naves L, Ferriere A, Romanet P, Elenkova A, Karhu A, Brue T, Barlier A, Pétrossians P and Daly AF (2023) The clinical and therapeutic profiles of prolactinomas associated with germline pathogenic variants in the aryl hydrocarbon receptor interacting protein (AIP) gene. Front. Endocrinol. 14:1242588. doi: 10.3389/fendo.2023.1242588
Received: 19 June 2023; Accepted: 17 July 2023;
Published: 29 August 2023.
Edited by:
Maria Mercedes Pineyro, Universidad de la República, UruguayReviewed by:
Liza Das, Post Graduate Institute of Medical Education and Research (PGIMER), IndiaCopyright © 2023 Vroonen, Beckers, Camby, Cuny, Beckers, Jaffrain-Rea, Cogne, Naves, Ferriere, Romanet, Elenkova, Karhu, Brue, Barlier, Pétrossians and Daly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrian F. Daly, YWRyaWFuLmRhbHlAY2h1bGllZ2UuYmU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.