 Yanzhong Liu
Yanzhong Liu Dan Wang
Dan Wang Yi-Ping Liu*
Yi-Ping Liu*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 31 October 2023
Sec. Clinical Diabetes
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1237934
This article is part of the Research Topic The Underlying Mechanisms and Novel Approaches for Diabetes and its Related Complications View all 36 articles
Type 2 diabetes mellitus (T2DM) has become a major health problem, threatening the quality of life of nearly 500 million patients worldwide. As a typical multifactorial metabolic disease, T2DM involves the changes and interactions of various metabolic pathways such as carbohydrates, amino acid, and lipids. It has been suggested that metabolites are not only the endpoints of upstream biochemical processes, but also play a critical role as regulators of disease progression. For example, excess free fatty acids can lead to reduced glucose utilization in skeletal muscle and induce insulin resistance; metabolism disorder of branched-chain amino acids contributes to the accumulation of toxic metabolic intermediates, and promotes the dysfunction of β-cell mitochondria, stress signal transduction, and apoptosis. In this paper, we discuss the role of metabolites in the pathogenesis of T2DM and their potential as biomarkers. Finally, we list the effects of anti-hyperglycemic drugs on serum/plasma metabolic profiles.
According to the latest statistics from the International Diabetes Federation (IDF), as of 2021, about 537 million adults worldwide had diabetes, and this number is expected to increase to 783 million by 2045, causing 6.7 million deaths and high health costs every year (1). T2DM is the most common type of diabetes mellitus, accounting for about 90% of the total number of diabetes mellitus. It is of great significance to explore the pathogenesis of T2DM and develop precise and reliable prevention and treatment strategies. T2DM is a typical metabolic disease, usually accompanied by the disorder of systemic metabolic networks including carbohydrates, lipids, and amino acids, which is very suitable for metabolomics and lipidomics studies. Metabolites are not only ending products of genome regulation and cellular energy transfer, reflecting biological situations that have occurred or are occurring in the body, but also have multiple functions such as signaling molecules, immune regulation, and environmental sensors. Thus, the exploration of metabolite changes can reflect the metabolic phenotype of T2DM in a relatively comprehensive way. In contrast to biopsies, blood sample collection is a minimally invasive method with the advantages of rapid, economical and high availability, and is essential for facilitating mapping of disease metabolic profiles and prognostic diagnosis.
Metabolites are commonly recognized as end products of a wide range of gene transcription and biochemical reactions, and there is growing evidence that metabolites can be involved in disease development as biomarkers (Figure 1) and regulators. When acting as regulators, metabolites have an impact on the pathogenesis of T2DM in at least the following aspects: (1) Metabolites can regulate the downstream signaling pathway of insulin and directly affect insulin sensitivity; (2) Accumulation of harmful metabolic intermediates; (3) Cause organelle dysfunction; (4) Directly or indirectly mediates the inflammatory response of target tissues. A comprehensive understanding of metabolic pathways may be a novel direction for the prevention and treatment of T2DM.
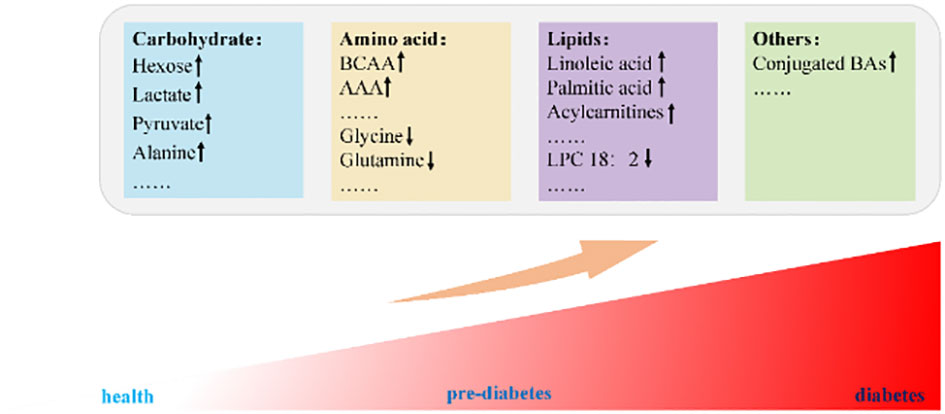
Figure 1 BCAA, branched-chain amino acids; AAA, aromatic amino acids; LPC, lysophosphatidylcholine; BAs, bile acids.
Chronic hyperglycemia is not only a major feature of diabetes, but also a key factor in accelerating its progression and inducing complications. High glucose can damage the mitochondrial aerobic metabolic flux of pancreatic β-cells and reduce insulin content, which may be the pathological basis of the progressive decline of β-cell function in patients with T2DM. Excess fructose can cause a dramatic increase in hepatocyte carbohydrate response element binding protein (ChREBP) activity, which not only mediates changes in circulating triglycerides and high density lipoprotein (HDL) levels, but also is an important upstream regulator of a key enzyme in BCAA metabolism, branch chain ketoate dehydrogenase kinase (BCKDK)/metal ion-dependent protein phosphatase (PPM1K), integration affects BCAA oxidation and lipid metabolism (2). Evidence suggests that overproduction of the subtype ChREBP-β mediates glucose toxicity and subsequent cell death in β-cells. Overexpression of the different subtypes of ChREBP-α enhances glucose-stimulated β-cell proliferation and antagonizes Chrebp-β-cell death mediated by Nuclear factor erythroid 2-related factor 2 (Nrf2) antioxidant pathway (3).
Single-cell sequencing results showed that genes related to oxidative phosphorylation and ATP synthesis were significantly downregulated in the islets of T2DM patients (4), and this phenomenon was verified in animal models. Nearly all glycolytic enzymes were significantly upregulated in the diabetic mouse islets. In contrast, genes, proteins, and BCAA metabolic pathways associated with mitochondrial oxidative phosphorylation were significantly reduced (5, 6). Haythorne et al. have found that the impairment of β-cell function by high glucose is not glucose per se, but mediated by metabolic intermediates associated with increased glycolysis flux, one or more metabolites located between phosphofructokinase (PFK) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). By stimulating mammalian rapamycin complex 1 (mTORC1) and inhibiting AMPK activity, preventing pyruvate from entering the TCA cycle, and the imbalance of NADH/NAD+ in mitochondria and cytoplasm exacerbates the accumulation of upstream metabolites of GAPDH, creating a vicious cycle. This may partly explain the impaired oxidative phosphorylation of mitochondria. More importantly, glucose stimulation at 8mM appears to be sufficient to initiate this cycle, suggesting that cumulative impairment of pancreatic function may have already begun in patients with early impaired glucose tolerance (7).
Impaired glucose oxidative phosphorylation metabolic pathways and increased glycolysis flux are determinants of increased lactate levels. In patients at high risk of CVD, plasma lactate, pyruvate, glycerol-3 phosphate, and isocitrate were significantly positively correlated with the risk of T2DM (23%-44% higher for every 1 SD increase) (8, 9), and was associated with various pathological phenomena of T2DM: (1) Increased blood lactate concentration reflects decreased mitochondrial oxidation capacity and is strongly positively correlated with IR index (10); (2) Blood lactate level may reflect liver dysfunction in T2DM patients (9); (3) Blood lactate levels may be indicative of susceptibility to T2DM to some extent (11). However, the effect of lactate as a signaling molecule on disease is complex and may depend on exposure duration and specificity of tissue and organ (Figure 2). Recent evidence suggests that lactate signaling is involved in inflammatory response (12–14), cells proliferation and migration (15), appetite regulation (16), redox homeostasis regulation (17), histone modification (18), and vascular cells damage (19–21). Diabetes is commonly accompanied by oxidation and systemic chronic inflammation. Elevated lactate levels can lead to an increase in NADH/NAD+ ratio, and mitochondria actively oxidize lactate to produce additional ROS accordingly. When antioxidants are out of balance, oxidative damage may be caused. Lactate is one of the main fuels of TCA cycle. Acute lactate exposure can stimulate mitochondrial coupling efficiency and promote bioenergetics of mitochondria in heart, skeletal muscle, and liver (22), while chronic hyper-lactate exposure may negatively affect mitochondrial respiration rate, reduced metabolic flexibility (23).

Figure 2 Lactate is involved in physiological and pathological processes of various tissues, including cardiovascular disease, immune response, fatty acid metabolism, cognitive function, and tumor cell angiogenesis. OXPHOS, oxidative phosphorylation; VSMC, vascular smooth muscle cell.
Chronic hyperglycemia can also trigger the activation of collateral glucose metabolic pathways, such as polyol, hexosamine, and AGE, resulting in oxidative stress, promoting the transcription of pro-inflammatory factors and apoptosis, and thus contribute to the occurrence and development of diabetes mellitus and its complications. AGE, cytokines and ROS can promote the production of triose phosphate, an intermediate of glucose metabolism, and increase de novo synthesis of diacylglycerol (DAG), the activator of protein kinase C (PKC) (24). In the polyol pathway, aldose reductase activated by hyperglycemia reduces glucose to sorbitol, which is further metabolized to fructose by sorbitol dehydrogenase. An increase in plasma fructose concentration is positively correlated with the development of T2DM and can lead to liver insulin resistance (IR) and non-alcoholic fatty liver disease development (25–28). In tissues such as nerves, sorbitol cannot easily cross the cell membrane, so the accumulation of sorbitol and fructose can increase the osmotic pressure in the cytoplasm and cause the leakage of myoinositol, a deficiency of which can lead to the production of DAG. On the other hand, the activation of the polyol pathway leads to the consumption of NADPH and promotes the conversion of NAD+ to NADH, which may not only reduce the production of the antioxidant glutathione, resulting in oxidative stress (29), but also competitively inhibit GAPDH, down-regulate glycolytic flux, and promote the transfer of more glucose to non-traditional metabolic pathways (such as hexosamine), thus aggravating glucotoxicity (30, 31). As the hexosamine biosynthesis pathway is also activated in diabetes, fructose-6-phosphate is detached from glycolysis and subsequently catalyzed to glucosamine 6-phosphate by fructose 6-phosphate aminotransferase. Glucosamine 6-phosphate forms the end product uridine diphosphate-N-acetylglucosamine through acetylation and isomerization. It then serves as the basic substrates for the formation of glycosyl side chains in post-translational modifications of proteins and lipids (32). This post-translational modification can aggravate glucotoxicity by regulating target protein stability, activity and subcellular localization, which can promote liver gluconeogenesis, lead to impaired insulin signaling and pancreatic β-cell function (33), and is directly involved in the pathogenesis of several diabetic complications, especially in cardiovascular disease and kidney dysfunction (34).
Since Felig et al. found in the 1970s that the increased concentration of circulating amino acids in obese people is associated with decreased insulin sensitivity (35), a large number of studies have confirmed the value of amino acids in the early identification and risk stratification of diabetes and its complications (36–40). Among the known and relatively clear amino acid biomarkers, plasma branched-chain amino acids (BCAA) and aromatic amino acids (AAA) increased significantly (Table 1), while glycine and glutamine decreased in diabetes and prediabetes (25, 41–43, 52–56).
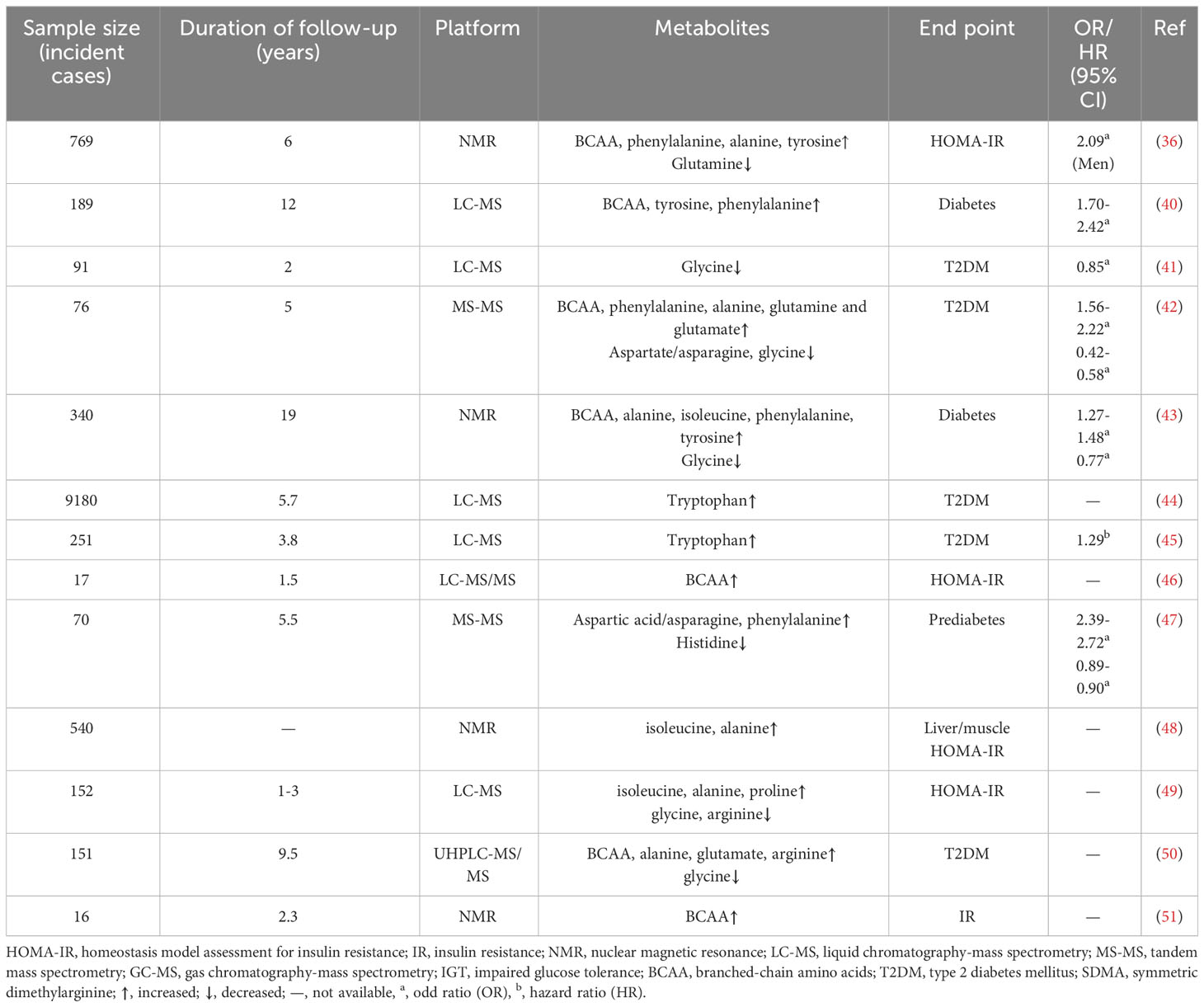
Table 1 Association of amino acids profiles with T2DM in cohort studies.
BCAA is most closely related to homeostasis model assessment for insulin resistance (HOMA-IR) and blood glucose (46, 57, 58). The results of large sample size genome-wide association studies (GWAS) show that BCAA contributes to the increase of the incidence of IR and T2DM (59). Under physiological conditions, BCAA promotes protein synthesis or inhibits its breakdown by activating the mTOR signaling pathway, a catalytic subunit of two distinct structural and functional complexes mTORC1 and mTORC2. mTORC1 promotes protein synthesis and regulates autophagy, and mTORC2 is a classic insulin/PI3K signaling pathway effector (60). BCAA supplementation alone did not significantly affect skeletal muscle mass and glycemic control in patients with T2DM (61), nor worsen diet-induced insulin resistance and glucose intolerance in obese mice (62). Whereas HFD combined with BCAA supplementation caused chronic activation of mTORC1, p70-S6 kinase (p70S6K), and phosphorylation of insulin receptor substrate 1 (IRS1) serine, promoting the accumulation of multiple acylcarnitines in muscle, decreased insulin sensitivity (37), which can be reversed by the mTOR inhibitor rapamycin (63). Reducing dietary BCAA intake rapidly reduced diet-induced obesity, improved glucose tolerance, reversed fatty acyl-coA accumulation in skeletal muscle, normalized glycine content, and improved skeletal muscle insulin sensitivity (64, 65). In leucine-incubated skeletal muscle, AMPK activity decreased by more than 50%, phosphorylation of mTOR and p70S6K was concentration-dependent, phosphorylation of insulin-stimulated Akt was impaired, and AMPK agonist was used to inhibit these changes (66).
Paradoxically, Leucine has also been suggested to increase GLUT4-mediated glucose uptake, stimulate insulin-dependent PI3K and protein kinase C (PKC) signaling cascades, and increase mitochondrial biogenesis and substrate oxidation capacity (67, 68). Leucine supplementation has been shown to reduce body weight by 32% and improve insulin sensitivity, plasma total cholesterol, and low-density lipoprotein cholesterol (LDL) levels in mice (69). This may be related to the insulinotropic properties of BCAA (especially Leu), short-term (4 weeks) BCAA-restricted diet decreased postprandial insulin secretion, increased postprandial insulin sensitivity and mitochondrial metabolism efficiency in adipose tissue (70). On the other hand, Long-term (60 weeks) supplementation with amino acids has also been shown to improve glycemic control and insulin sensitivity in older non-obese (BMI within 19∼23) T2DM patients (71). Since protein degradation is commonly increased in populations with poor IR and T2DM control (72), and the provision of additional BCAAs in the diet can mitigate protein degradation (73), it is necessary to tailor nutritional programs to different populations. In addition, valine and isoleucine are major contributors to the production of cyclic odd-chain FA (74), among which valine not only promotes α oxidation by activating PPARα, but also promotes odd-chain FA production by providing PrCoA as a substrate (75).
Different organs also show differences in amino acid profiles, liver but not muscle IR was associated with increased levels of leucine and tyrosine, leucine deprivation enhances insulin sensitivity by increasing AMPK phosphorylation and inhibiting the mTOR/S6K pathway in the mouse liver (76), while both showed higher levels of isoleucine and alanine and lower levels of glycine (48). In the liver, BCAA supplementation activates mTORC1 and suppresses mTORC2, blocks insulin-mediated Akt2 phosphorylation, and promotes its ubiquitination and degradation, negative regulation of Akt2 increases FoxO1-mediated gluconeogenesis and inhibits liver lipogenesis mediated by the sterol-regulatory element binding protein (SREBP)1/INSIG2a signaling pathway (77). It is believed that when FA are excessive, the accumulation of metabolic intermediates of BCAA (rather than BCAA itself) can competitively “block” FA β oxidation flux, resulting in the accumulation of BCAA and incomplete oxidation products of FA, leading to a corresponding decrease in glucose utilization (37). All of these suggest that the increase of BCAA in T2DM is likely to be a downstream effect caused by obesity and IR, and then plays a further mediating role in disease development (37).
Studies have suggested that increased IR leads to increased levels of circulating fasting BCAA and inflammation (78, 79). Obesity and T2DM reduce the activity of metabolic enzymes involved in BCAA catabolism, leading to BCAA accumulation (80). BCAA catabolism involves the first transamination of BCAA aminotransferase (BCAT) to branch alpha-ketoic acid (BCKA), followed by decarboxylation of BCKA by BCKA dehydrogenase complex (BCKDC), which is activated by dephosphorylation of PPM1K phosphatase and deactivated by phosphorylation of BCKD kinase. The expression of BCAT and BCKDC is relatively low in the liver, where adipose tissue and skeletal muscle are major sites of BCAA oxidative metabolism (81, 82). In human and animal models of metabolic syndrome, hypoxia, inflammation, and ER stress in adipose tissue can lead to a significant decrease in the level of BCAA catabolic enzyme (83, 84), and the accumulation of BCAA directly inhibits the activity of pyruvate dehydrogenase (PDH) and reduces the oxidative metabolism of glucose and FA (85). Another study suggested that reduced oxygenation in adipose tissue inhibited BCAA catabolism (86). Oxygen partial pressure in subcutaneous adipose tissue was negatively correlated with the expression of markers of inflammation and fibrosis. Meanwhile, hypoxia inhibited the catabolism and oxidation of BCAA, resulting in increased plasma BCAA concentration, thus promoting IR. Surgical weight loss interventions can reverse the increase in plasma concentrations by improving BCAA metabolism in adipose tissue, suggesting that changes in plasma BCAA reflect IR or relative insulin deficiency in obesity (87). Muscle biopsies in patients with T2DM also showed decreased expression of two enzymes necessary for valine and isoleucine metabolism (88). In contrast, increased BCAA catabolism effectively reduced plasma BCAA levels in T2DM patients, significantly improved peripheral glucose utilization, and increased pyruvate mitochondrial oxidation flux by 10% in muscle (89).
In addition to BCAA and AAA, multiple additional amino acids and derivatives are associated with diabetes progression (49, 90, 91). Studies have shown that glutamate is significantly increased in people with T2DM, while glutamine and glycine are associated with a 15% and 11% reduction in diabetes risk, respectively (52). After adjusting for BMI, concentrations of aspartic acid, asparagine, and histidine were strongly correlated with the incidence of prediabetes. For every 1 standard deviation increase in baseline aspartic acid and asparagine levels, the risk of prediabetes increased by 2.72 times, while for every 1 standard deviation increase in baseline histidin level, the risk of prediabetes decreased by 10% (47). This may be related to histidine’s role in regulating gluconeogenesis and anti-inflammatory (92).
Studies have shown that alanine, tryptophan, and trytophan-related metabolic intermediates are associated with a higher risk of T2DM and prediabetes (40, 43, 44, 93), and that kynurenine is the main metabolic intermediate of tryptophan, chronic inflammatory can induce activation of the tryptophan/kynurenine metabolic pathway (94), and may mediate the increased mortality associated with inflammation in T2DM (95). Circulating kynurenine levels are also affected by dietary tryptophan intake. As a result, it has been suggested that the ratio of kynurenine/tryptophan can reflect the metabolic status of tryptophan further than that of tryptophan or kynurenine concentration alone (96). This partly explains why the level of tryptophan increases at the beginning of T2DM and reverses as the disease progresses (45), and why there is no significant association between the plasma kynurenine/tryptophan ratio and T2DM risk, but the urine kynurenine/tryptophan rate is strongly associated with T2DM risk (96). In addition, indolepropionate, a tryptophan-breaking metabolite derived from the gut microbiome, was negatively associated with T2DM risk, while increased indolelactate was associated with higher T2DM risk (44, 53).
Elevated levels of lysine and its metabolic intermediate 2-aminoadipic acid were associated with an increased risk of T2DM. 2-aminoadipic acid metabolism occurs primarily in mitochondria and is broken down into acetyl-CoA before entering the (tricarboxylic acid, TCA) cycle. Plasma 2-Aminoadipic acid level increases by 47% in obesity and is positively correlated with IR (97). As a novel biomarker to predict the risk of T2DM, 2-aminoadipic acid, independent of common BCAA and AAA, has been shown to increase in concentration 12 years before the onset of diabetes symptoms (98), and as a biomarker to predict childhood obesity and related metabolic disorders 2 years later (99). In addition, the level of circulating 2-Aminoadipic acid is significantly negatively correlated with HDL, which is closely associated with cardiovascular complications such as atherosclerosis and coronary artery calcification (100, 101).
Elevated blood levels of triacylglycerols (TAGs) are traditional risk indicators for T2DM (48, 102, 103). Free fatty acids (FFA) are non-esterified fatty acids in the serum that comes primarily from the breakdown of TAG. When caloric intake exceeds the normal storage and consumption capacity of lipids, fatty acids “spillover” will result in increased FFA (104). Elevated fasting FFA is associated with a three-fold increased risk of impaired glucose tolerance or T2DM over the next 5∼8 years (105). After clinical intervention, the level of FFA can also serve as an effective prognostic evaluation index (106). FFA can be divided into saturated fatty acids (SFA), monounsaturated fatty acids (MUFA), and polyunsaturated fatty acids (PUFA) according to the difference of hydrocarbon saturation. Serum FFA variations of different types are generally suggestive of hyperglycemia or T2DM (Table 2). Specifically, increased levels of partial n-6, n-7 and n-9 were significantly positively correlated with elevated blood glucose, while some n-3 PUFA was significantly inversely correlated with T2DM (107, 108, 113, 116). In addition, different chain lengths of SFA have particular metabolic and biological effects. Increased circulating concentrations of C15:0, C17:0 and C24:0 and very long chain of SFA are associated with lower risk of T2DM (114, 117), while C14:0, C16:0, C16:1, and C18:0 are positively correlated with T2DM risk (109, 111).
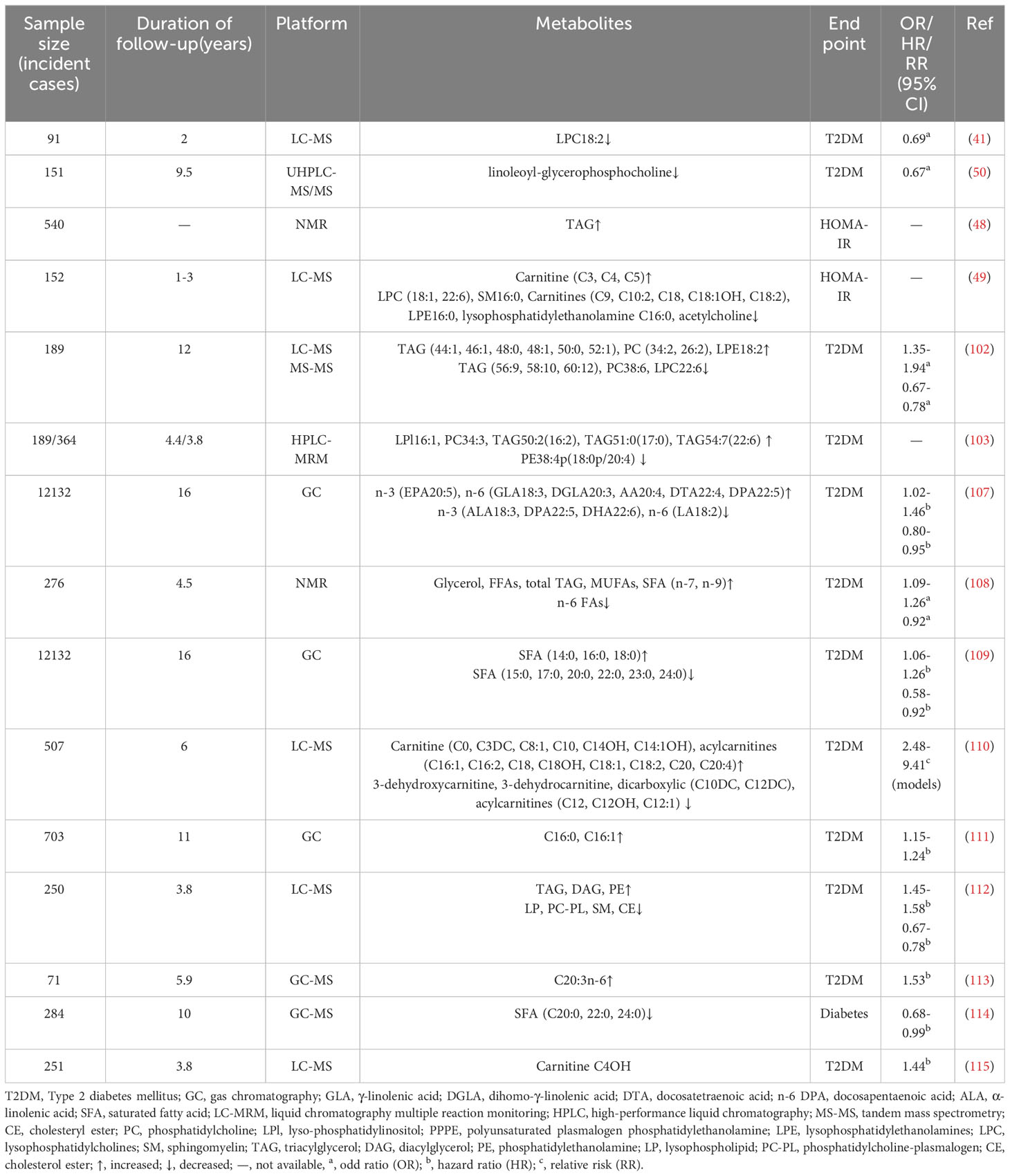
Table 2 Association of lipids profiles with T2DM in cohort studies.
Increased FFA release and oxidation rate can antagonize glucose oxidation, resulting in the disturbance of pyruvate metabolism and impaired insulin sensitivity (118, 119), and this damage to glucose homeostasis by FFA is commonly referred to as lipotoxicity. Extensive evidence has demonstrated the role of lipotoxicity in IR and pancreatic β-cell injury (120, 121). Elevated plasma FFA levels are also known to cause TAG and DAG deposition in a variety of tissues and organs, gradually accumulating DAG enhances NADPH oxidase activity through the PKC pathway, exacerbates oxidative stress and cytokine transcription, and promotes cell differentiation, proliferation, and apoptosis. Activation of the PKC pathway is a critical mechanism leading to diabetes cardiovascular disease (122). Thus, lipid management is absolutely recommended for the prevention and treatment of vascular complications of T2DM.
The inflammatory response mediated by SFA is an essential cause of IR and β-cell damage (Figure 3). SFA and lipopolysaccharide synergistically amplifies the effects of decreased β-cell viability, increased apoptosis, and decreased basal insulin secretion, and significantly alleviates lipid-induced β-cell damage by blocking toll-like receptor 4 (TLR4) or overexpressing neutral ceramidase (NCDase) activity (123, 124). Palmitate (C16:00) is the most abundant SFA in dietary and plasma, palmitate can enhance the interaction between TLR and myeloid differentiation primary response protein MyD88, mediating β-cell death (125). On the one hand, it directly inhibits insulin signal transduction by activating the phosphorylation of Jun N-terminal kinase (JNK) and the inhibitor of nuclear factor-κB (NF-κB) kinas (IKKβ). On the other hand, degradation of inhibitor of NF-κB (IκB) leads to nuclear translocation of NF-κB, this increases cytokine transcription and exacerbates inflammation. TLR4, IKKβ, or JNK knockout inhibited the expression of inflammatory cytokines in adipocytes and macrophages and protected mice against lipid-induced IR (126–128). In contrast, some PUFA such as docosahexaenoic acid (DHA) inhibit the production of TLR4-induced inflammatory cytokines (129), improve insulin sensitivity and insulin secretion capacity to some extent, and reduce the risk of T2DM (128, 130).
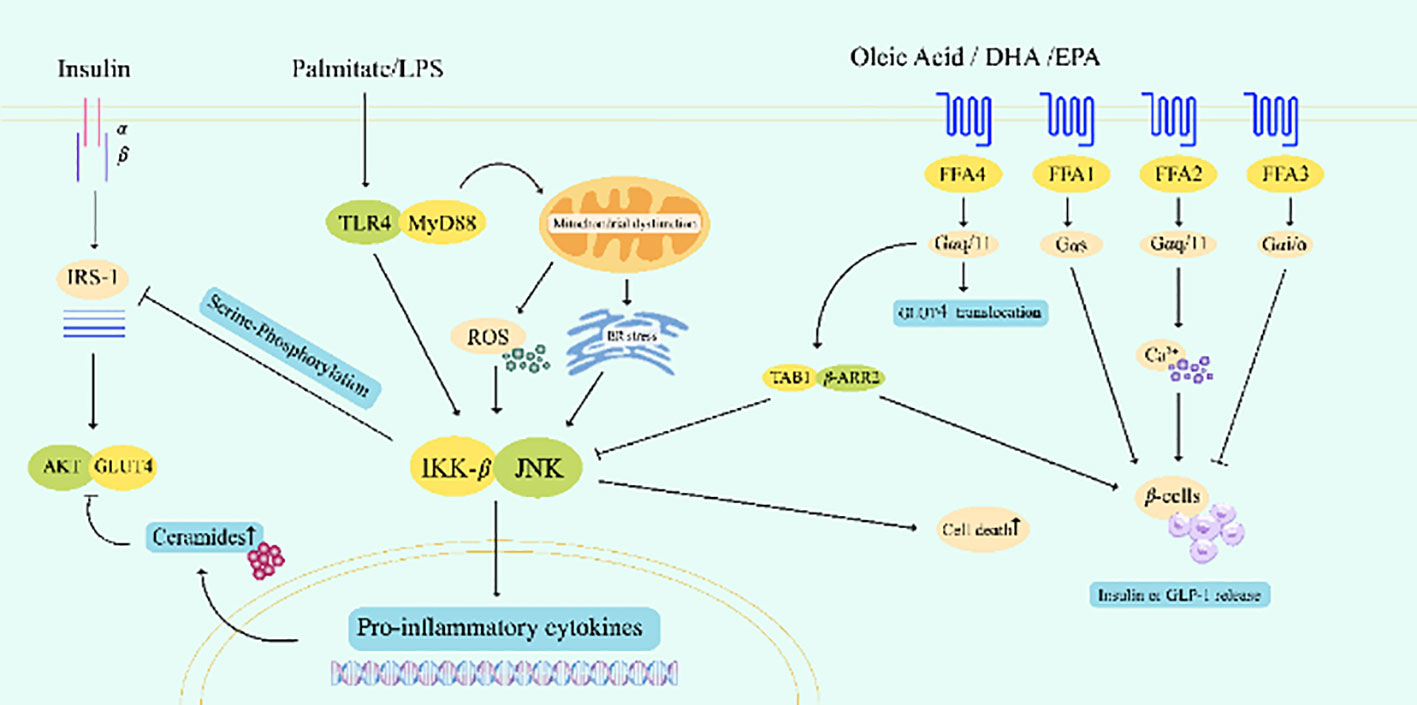
Figure 3 Palmitate cooperated with LPS to amplify TLR4-related signaling pathways, directly or indirectly inhibited insulin signaling, and caused β-cell apoptosis in islets. The activation of FFA4 by certain PUFAs such as oleic acid, DHA and EPA can competitively bind TAB1 through the recruitment of β-arrestin-2, inhibit the phosphorylation and activation of TAK1, inhibit the pro-inflammatory response, and promote the release of insulin and GLP-1. LPS, lipopolysaccharide; IRS-1, insulin receptor substrate 1; TLR4, toll-like receptor 4; AKT, protein kinase B; GLUT, glucose transporter; ROS, reactive oxygen species; IKK-β, inhibitor kappa B kinase-β; JNK, Jun N-terminal kinase; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; FFA, free fatty acid receptor; TAB1, TAK1 binding protein 1; β-ARR2, β-arrestin-2; GLP-1, glucagon-like peptide-1.
In specific lipids and derivatives, baseline lysophosphatidylcholine (LPC), phosphatidylcholine (PC), sphingomyelin, and cholesterol esters were inversely associated with T2DM risk (112). LPC were strongly associated with IR and β-cell dysfunction (50). Decreased concentrations of LPC18:2 and 16:0 are associated with the onset of T2DM (41, 131), and increased levels of PC O-16:1/0:0, (O-18:1/0:0)/(P-18:0/0:0) and LPC 20:2 can increase the 10-year risk of T2DM by 29% (132). Diacylphosphatidylcholine C32:1, C36:1, C38:3 and C40:5 were positively associated with T2DM risk. PC O-20:0/O-20:0, 22:6/20:4, LPC18:0, sphingomyelin C16:1 and acyl-alkyl-phosphatidylcholine C34:3, C40:6, C42:5, C44:4 and C44:5 were negatively correlated with T2DM risk (133–135). In the early stages of dysglycemia and IR, fasting concentration of linoleoylglycerophosphocholine is decreased, independent of classical predictors, as an indicator of worsening glucose tolerance (50).
Ceramide is a relatively minor component of the total cellular lipidome with a particularly low abundance, and increased ceramide content has been shown to be positively correlated with HOMA-IR, fasting glucose, and cardiovascular diseases (136, 137). Elevated ceramides are key lipotoxic species in skeletal muscle, liver, adipose tissue, and vascular cells, and contribute to disease progression by interfering with insulin signaling, stimulating lipid uptake, and enhancing inflammatory cytokines (138–142). Of the different types of sphingolipids, C16 ceramides and C18 ceramides are more damaging to adipose and liver function. C16:0 ceramides can impair mitochondrial oxidative phosphorylation by inhibiting mitochondrial complex II and promoting mitochondrial fission, reduce mitochondrial respiration, and promote the release of cytochrome c to induce apoptosis by increasing the permeability of mitochondrial outer membrane (139, 143). Inhibition of ceramide synthesis can improve insulin sensitivity and prevent obesity-induced diabetes (144). It also increases brown adipocyte numbers, mitochondrial activity, and promotes the polarization of adipose tissue macrophages towards the M2 anti-inflammatory phenotype (145).
Acylcarnitines, metabolites of FA, play crucial roles in cellular energy metabolism and are gradually considered as influential biomarkers of metabolic disorders in metabolic syndrome, diabetes, cardiovascular diseases and other diseases. For example, C2, C3DC-CH3, C4, C5, C7 and C26 have been observed to be associated with HOMA-IR (49). In cross-sectional studies, acylcarnitines were elevated in IGT and diabetic individuals (146), reflecting incomplete fatty acid beta oxidation in the organism, but acetylcarnitine C2 did not predict IGT or T2DM years before onset, so it is more likely to be a quick-acting event (41).
Recently, FFA ligand-specific G-protein-coupled receptor (GPR) including GPR40 (also known as FFA1), GPR43 (FFA2), GPR41 (FFA3), GPR120 (FFA4) has been extensively studied. In HFD-fed mice, FFA2 function is more mediated by Gi/o and activated by short-chain FA, which inhibit insulin signaling in adipose tissue, increasing energy expenditure and improving insulin sensitivity in different tissues, including liver and muscle (147, 148). Propionic acid (C3) and valeric acid (C5) can increase basal glucose uptake in adipocytes and muscle cells by activating FFA3, while this effect is decreased after FFA3 inhibition (149).
FFA1 and FFA4, as long chain FFA receptors, are expressed in a variety of tissues and cells, such as adipocytes, macrophages, and pancreatic β-cells. FFA1 activation alone or synergistically amplifies glucose-dependent insulin secretion by affecting cellular Ca2+ signaling and increasing intracellular Ca2+ concentration (150, 151), which is critical for maintaining the homeostasis of glucose and lipid metabolism in IR individuals, making it an attractive research target for the regulation of glucose and lipid metabolism (152, 153). However, the exact mechanism of FFA1 in β-cells is still under debate. Although FFA1 mediates insulin secretion in response to acute FFA exposure, long-term activation of FFA1 is also involved in lipotoxicity to β-cells. The diversity of FFAs may partly explain the difference in efficacy, for example, palmitate increases endoplasmic reticulum stress and mitochondrial dysfunction through FFA1 activation (154–157), resulting in increased apoptosis and decreased insulin secretion. In addition, FFAR1 also responds to trans isomers of conjugated linoleic acid or arachidonic acid (158), regulating the crosstalk between Akt/mTOR and IRS-1 signaling in β-cells under lipotoxicity conditions, promoting the progression of IR and T2DM (159). Oleic acid can inhibit the activation of JNK and NF-κB, inhibit inflammatory cytokine secretion, and improve insulin sensitivity (160), while palmitoleic acid can reverse the HFD-induced proinflammatory polarization of macrophages by activating AMPK and FFA4 (161, 162), independently of the PPAR-α mechanism (163). n-3 PUFA activates FFA4, which inhibits inflammation and increases insulin sensitivity (162). This is at least in part through the regulation of NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome and macrophage conversion to the M2 anti-inflammatory standard (164), and FFA4 also mediates a variety of effects such as glucagon-like peptide-1 (GLP-1) secretion, islet function, and appetite control (165, 166).
α-hydroxybutyrate (α-HB) is an organic acid derived from α-ketobutyrate, a by-product of amino acids such as methionine and threonine catabolism and glutathione synthesis (167). Increasing evidence has shown that α-HB is an early predictor of IR and impaired glucose tolerance (168, 169), the combination of α-HB and L-glycerophosphate choline showed similar accuracy to glucose in OGTT assay, and the plasma level of α-HB was negatively correlated with insulin sensitivity (Table 3). It has also been shown to be associated with β-cell dysfunction in statistical and mechanism studies (50, 51, 170). Since glutathione is a vital antioxidant, which can inhibit oxidative damage caused by an imbalance of lipid peroxides and free radicals in cells, the potential mechanism of increased α-HB may be a compensatory increase in liver glutathione synthesis flux after body REDOX dysplasia (169).
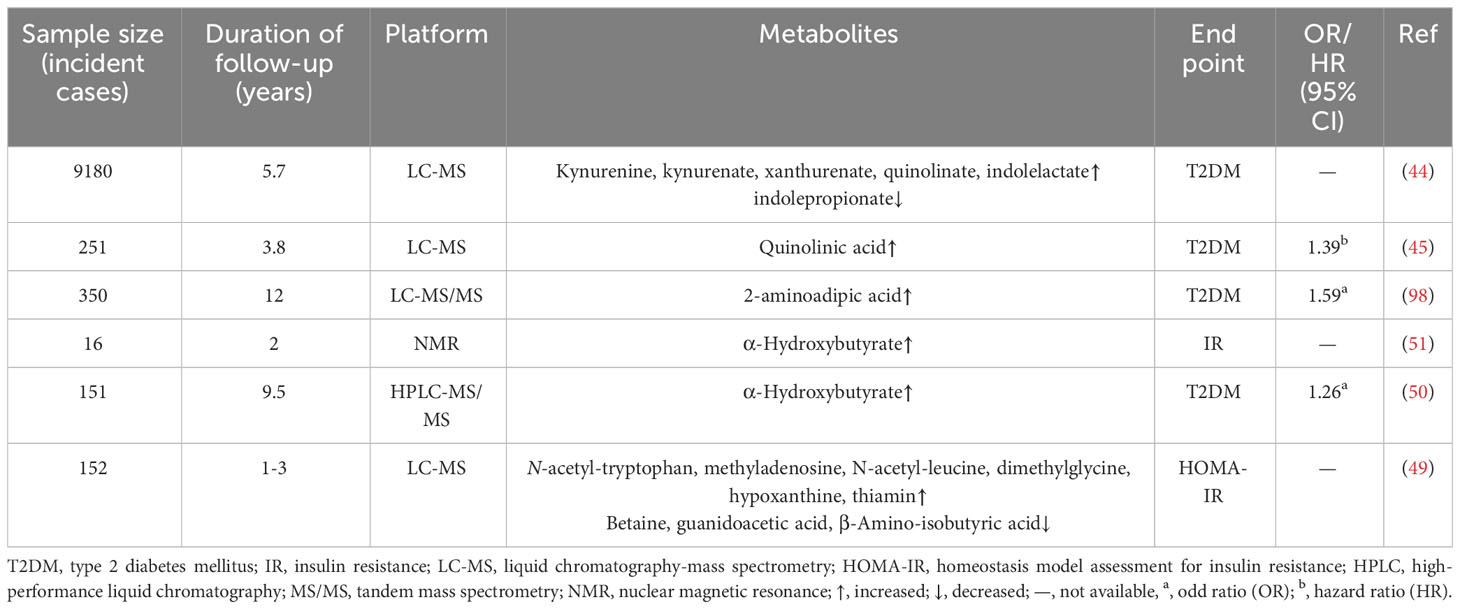
Table 3 Association of metabolic intermediate profiles with T2DM in cohort studies.
3-Hydroxyisobutyrate (3-HIB), an intermediate product of valine decomposition, is considered a higher sensitive biomarker of T2DM than valine. Impaired valine catabolism increases 3-HIB production, leading to increased lipid oxidation and acylcarnitine accumulation. Currently, 3-HIB is believed to mediate the occurrence of IR by affecting FA uptake in endothelial cells, and 3-HIB treatment has a regulatory effect on mitochondrial metabolism in white and brown adipocytes (171). Recent studies have shown that changes in the gut microbiome are involved in the metabolic disorders of T2DM, where imidazole propionate (ImP) is a product of histidine microbial metabolism. Serum ImP expression is upregulated in T2DM patients due to changes in microbial metabolism rather than histidine intake per se (172). It affects insulin receptor substrates and inflammatory signals by activating p38γ/MAPK/p62/mTORC1 signals, leading to impaired glucose metabolism (173). In addition, ImP (but not the precursor histidine) also showed a significant association with diastolic blood pressure in the overweight/obesity population, showing a possible role in CVD complications (174).
Serum concentrations of 3-carboxy-4-methyl-5-propyl-2-furan propionic acid (CMPF), the main endogenous metabolite of furan FA, are elevated in patients with impaired glucose tolerance and T2DM, and can directly act on pancreatic β-cells to lead to impaired insulin secretion (175–177). However, one study showed the opposite result: compared with the control group, the concentration of serum CMPF in the T2DM group was lower and was negatively correlated with the changes in serum TAG (178), although this study was limited by the small sample size, it still suggested that the metabolism of CMPF might be affected by race, diet and other factors. Supplementation of n-3 FA can increase the level of CMPF, and there is a positive correlation between docosahexaenoic acid (DPA) and DHA levels and CMPF, but no significant relationship between eicosapentaenoic acid (EPA) (179). It should be noted that although exogenous n-3 FA intake (e.g. fish) can significantly increase circulating CMPF concentration, it is still far below the level of T2DM and low doses of CMPF do not have a significant effect on glucose metabolism (180).
Bile acids (BAs) can act as signaling regulators for lipids and glucose metabolism, and the concentration of BAs changed has been linked to metabolic disorders such as IR. Studies have shown that after adjusting for age, sex, BMI, waist circumference, and fasting blood glucose, increased circulating 12α-hydroxylated BAs concentration is significantly associated with increased HOMA-IR and fasting blood glucose (132, 181), but it cannot be used as an effective predictor of diabetes (182, 183). This may be because decreased insulin sensitivity and impaired glucose tolerance occur before the rise in BAs. The increase in circulating BAs is not the factor that causes the change in glucose metabolism, but its downstream effect (184). Recent study has also confirmed that the increase of circulating BAs in T2DM individuals is positively correlated with fasting blood glucose, HbA1c, and HOMA-IR, which may be due to insulin signaling dysfunction. However, insulin treatment did not significantly affect the total level of BAs. Therefore, more studies are needed on the composition of BAs and its role as a regulator in metabolic disorders (185).
Many of commonly used anti-hyperglycemic medications can also have pleiotropic effects on the metabolite profile, which may positively affect T2DM and complications. In the following, we summarize the therapeutic effects of current mainstream hypoglycemic drugs on metabolites associated with different diabetes risks.
Metformin is a common drug for T2DM management. In randomized controlled trials, taking metformin was associated with increased levels of betaine, alanine, histidine, leucine/isoleucine and decreased levels of carnitine, phenylalanine, tyrosine and valine (186, 187). In terms of blood lipids, metformin was associated with an increase in TAG of higher carbon numbers (188), and elevated levels of the latter seemed to predict a reduced risk of T2DM (102), and very-low-density lipoprotein (VLDL)-triglyceride levels were significantly reduced (189). In another small sample size study, T2DM patients treated with metformin had increased serum trimethylamine-N-oxide, α-HB, and tryptophan, while acetoacetic acid, phenylalanine, and LPC (16:0, 18:0, and 18:2) were decreased (190).
Compared with metformin, pioglitazone increased myocardial glucose uptake and decreased hepatic TAG content (191, 192), but did not show any effect on subcutaneous fat volume. Compared with rosiglitazone, pioglitazone has a smaller increase in LDL and a larger increase in HDL, and promotes the transformation of VLDL to larger LDL by reducing asymmetric dimethylarginine levels (193, 194), which has a protective effect on cardiovascular diseases.
Treatment with liraglutide can significantly reduce serum tyrosine, valine and isoleucine levels in obese people, but has no significant effect on T2DM patients (195, 196). After liraglutide treatment, ceramides, phospholipids, hexocyl-ceramides, LPC, sphingolipids, and TAG were significantly deregulated in T2DM patients, demonstrating the cardiovascular system benefits of liraglutide (197–200). Exenatide treatment for 6 months was effective in reducing body weight, cysteine, and FFA concentration, while levels of aminoisobutyric acid, anandamide, and sarcosine tended to increase (201, 202). The efficacy of duraglutide was also associated with a significant reduction in 2-hydroxybutyric acid and a significant upregulation of threonine compared to placebo (203). In addition, high doses of trusted downregulated BCAA, glutamate, 3-hydroxyisobutyrate, branched-chain ketoacids, and 2-hydroxybutyrate (204).
Studies have shown that 6 months of vildagliptin treatment can reduce the level of asymmetric dimethylarginine in T2DM patients (205), but has no significant effects on FFA, glycerol, lactic acid and pyruvate (206, 207).
The present investigation shows that 3 months of metformin plus pioglitazone can significantly reduce the levels of phenylalanine/tyrosine, citrulline/arginine, and lysine/α-aminoadipic acid in T2DM and obese adults (208). Compared with treatment alone, the combination of pioglitazone and exenatide reduced hepatic fat and plasma TAG more significantly (209).
At present, the effects of hypoglycemic drugs on serum metabolites in patients with T2DM are more focused on the effects of lipids and lipoproteins, but the number of studies on amino acids and metabolic derivatives are limited. GLP-1 agonists have shown relatively better effects on lipid and amino acid metabolites, and improvements in metabolites associated with cardiovascular risk have been observed in short-term trials, but long-term follow-up evidence is still lacking. An anti-hyperglycemic drug’s effect on blood metabolites needs more prospective, intervention and randomized clinical trial studies to confirm the molecular mechanism of further metabolites.
As a typical metabolic disease, exploring changes in metabolites and their regulatory mechanisms is closer to the essence of T2DM. Among the promising metabolites, blood concentrations of hexose, BCAA, AAA, TAG, phospholipids and sphingomyelins were significantly and positively associated with T2DM incidence, while glycine and glutamine were negatively associated with T2DM risk. However, using only one metabolite type as a biomarker has many limitations in terms of disease duration, race, or diet, so a comprehensive judgment of multiple metabolite prediction models is necessary. Understanding the metabolism of metabolites in specific tissues and the influence of the regulation of corresponding receptors on immune response and biological efficacy, as well as verifying causality through mechanism studies, is key to metabolite research. Finally, while we have an initial understanding of the functions of metabolites as regulators, the results of dietary interventions do not completely match our expectations. How dietary nutrients cause changes in metabolic pathways and certain protein signaling pathways, as well as the role of gut flora in metabolite synthesis and downstream regulation, will be attractive topics.
YL mainly wrote the manuscript. DW ccollects data, corrects and provides fund support. Y-PL reviews, editing and provides fund support. All authors have read and agreed to the published version of the manuscript.
This research was funded by Natural Science Foundation of Fujian Province (2017J01627), and China Postdoctoral Science Foundation (2021M700782).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Magliano DJ, Boyko EJ, committee IDFDAtes. IDF Diabetes Atlas. Idf diabetes atlas. Brussels: International Diabetes Federation (2021).
2. White PJ, McGarrah RW, Grimsrud PA, Tso SC, Yang WH, Haldeman JM, et al. The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab (2018) 27(6):1281–93.e7. doi: 10.1016/j.cmet.2018.04.015
3. Katz LS, Brill G, Zhang P, Kumar A, Baumel-Alterzon S, Honig LB, et al. Maladaptive positive feedback production of ChREBPβ underlies glucotoxic β-cell failure. Nat Commun (2022) 13(1):4423. doi: 10.1038/s41467-022-32162-x
4. Segerstolpe Å, Palasantza A, Eliasson P, Andersson EM, Andréasson AC, Sun X, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab (2016) 24(4):593–607. doi: 10.1016/j.cmet.2016.08.020
5. Hou J, Li Z, Zhong W, Hao Q, Lei L, Wang L, et al. Temporal transcriptomic and proteomic landscapes of deteriorating pancreatic islets in type 2 diabetic rats. Diabetes (2017) 66(8):2188–200. doi: 10.2337/db16-1305
6. Haythorne E, Rohm M, van de Bunt M, Brereton MF, Tarasov AI, Blacker TS, et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat Commun (2019) 10(1):2474. doi: 10.1038/s41467-019-10189-x
7. Haythorne E, Lloyd M, Walsby-Tickle J, Tarasov AI, Sandbrink J, Portillo I, et al. Altered glycolysis triggers impaired mitochondrial metabolism and mTORC1 activation in diabetic β-cells. Nat Commun (2022) 13(1):6754. doi: 10.1038/s41467-022-34095-x
8. Guasch-Ferré M, Santos JL, Martínez-González MA, Clish CB, Razquin C, Wang D, et al. Glycolysis/gluconeogenesis- and tricarboxylic acid cycle-related metabolites, Mediterranean diet, and type 2 diabetes. Am J Clin Nutr (2020) 111(4):835–44. doi: 10.1093/ajcn/nqaa016
9. Ishitobi M, Hosaka T, Morita N, Kondo K, Murashima T, Kitahara A, et al. Serum lactate levels are associated with serum alanine aminotransferase and total bilirubin levels in patients with type 2 diabetes mellitus: A cross-sectional study. Diabetes Res Clin practice (2019) 149:1–8. doi: 10.1016/j.diabres.2019.01.028
10. Santos JL, Cataldo LR, Cortés-Rivera C, Bravo C, Díaz-Casanova L, Martínez JA, et al. Plasma lactate and leukocyte mitochondrial DNA copy number as biomarkers of insulin sensitivity in non-diabetic women. J Physiol Biochem (2019) 75(3):285–97. doi: 10.1007/s13105-019-00672-w
11. Memon B, Elsayed AK, Bettahi I, Suleiman N, Younis I, Wehedy E, et al. iPSCs derived from insulin resistant offspring of type 2 diabetic patients show increased oxidative stress and lactate secretion. Stem Cell Res Ther (2022) 13(1):428. doi: 10.1186/s13287-022-03123-4
12. Russo S, Kwiatkowski M, Govorukhina N, Bischoff R, Melgert BN. Meta-inflammation and metabolic reprogramming of macrophages in diabetes and obesity: the importance of metabolites. Front Immunol (2021) 12:746151. doi: 10.3389/fimmu.2021.746151
13. Ranganathan P, Shanmugam A, Swafford D, Suryawanshi A, Bhattacharjee P, Hussein MS, et al. GPR81, a cell-surface receptor for lactate, regulates intestinal homeostasis and protects mice from experimental colitis. J Immunol (Baltimore Md: 1950) (2018) 200(5):1781–9. doi: 10.4049/jimmunol.1700604
14. Li X, Yang Y, Zhang B, Lin X, Fu X, An Y, et al. Lactate metabolism in human health and disease. Signal transduction targeted Ther (2022) 7(1):305. doi: 10.1038/s41392-022-01206-5
15. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res (2011) 71(22):6921–5. doi: 10.1158/0008-5472.CAN-11-1457
16. McCarthy SF, Islam H, Hazell TJ. The emerging role of lactate as a mediator of exercise-induced appetite suppression. Am J Physiol Endocrinol Metab (2020) 319(4):E814–e9. doi: 10.1152/ajpendo.00256.2020
17. Carrière A, Lagarde D, Jeanson Y, Portais JC, Galinier A, Ader I, et al. The emerging roles of lactate as a redox substrate and signaling molecule in adipose tissues. J Physiol Biochem (2020) 76(2):241–50. doi: 10.1007/s13105-019-00723-2
18. Chen AN, Luo Y, Yang YH, Fu JT, Geng XM, Shi JP, et al. Lactylation, a novel metabolic reprogramming code: current status and prospects. Front Immunol (2021) 12:688910. doi: 10.3389/fimmu.2021.688910
19. Yang K, Holt M, Fan M, Lam V, Yang Y, Ha T, et al. Cardiovascular dysfunction in COVID-19: association between endothelial cell injury and lactate. Front Immunol (2022) 13:868679. doi: 10.3389/fimmu.2022.868679
20. Bae KH, Choi YK, Bae KH, Bae KH, Bae KH, et al. Role of lactate dehydrogenase a (LDH-A) in diabetic vascular complication. J Diabetes. doi: 10.1530/endoabs.49.EP383
21. Zhu Y, Han XQ, Sun XJ, Yang R, Ma WQ, Liu NF. Lactate accelerates vascular calcification through NR4A1-regulated mitochondrial fission and BNIP3-related mitophagy. Apoptosis. doi: 10.1007/s10495-020-01592-7
22. Young A, Oldford C, Mailloux RJ. Lactate dehydrogenase supports lactate oxidation in mitochondria isolated from different mouse tissues. Redox Biol (2020) 28:101339. doi: 10.1016/j.redox.2019.101339
23. San-Millan I, Sparagna GC, Chapman HL, Warkins VL, Chatfield KC, Shuff SR, et al. Chronic lactate exposure decreases mitochondrial function by inhibition of fatty acid uptake and cardiolipin alterations in neonatal rat cardiomyocytes. Front Nutr (2022) 9:809485. doi: 10.3389/fnut.2022.809485
24. Mizukami H, Osonoi S. Pathogenesis and molecular treatment strategies of diabetic neuropathy collateral glucose-utilizing pathways in diabetic polyneuropathy. Int J Mol Sci (2020) 22(1):94. doi: 10.3390/ijms22010094
25. Allalou A, Nalla A, Prentice KJ, Liu Y, Zhang M, Dai FF, et al. A predictive metabolic signature for the transition from gestational diabetes mellitus to type 2 diabetes. Diabetes (2016) 65(9):2529–39. doi: 10.2337/db15-1720
26. Fiehn O, Garvey WT, Newman JW, Lok KH, Hoppel CL, Adams SH. Plasma metabolomic profiles reflective of glucose homeostasis in non-diabetic and type 2 diabetic obese African-American women. PloS One (2010) 5(12):e15234. doi: 10.1371/journal.pone.0015234
27. Bian C, Wang Y, Li J, Gao J, Luan Z, Cui X, et al. Endogenous fructose is correlated with urinary albumin creatinine ratios and uric acid in type 2 diabetes mellitus. Diabetes Res Clin practice (2021) 179:109034. doi: 10.1016/j.diabres.2021.109034
28. Basaranoglu M, Basaranoglu G, Sabuncu T, Sentürk H. Fructose as a key player in the development of fatty liver disease. World J gastroenterol (2013) 19(8):1166–72. doi: 10.3748/wjg.v19.i8.1166
29. Tang WH, Martin KA, Hwa J. Aldose reductase, oxidative stress, and diabetic mellitus. Front Pharmacol (2012) 3:87. doi: 10.3389/fphar.2012.00087
30. Tang WH, Wu S, Wong TM, Chung SK, Chung SS. Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radical Biol Med (2008) 45(5):602–10. doi: 10.1016/j.freeradbiomed.2008.05.003
31. Yang Y, Hayden MR, Sowers S, Bagree SV, Sowers JR. Retinal redox stress and remodeling in cardiometabolic syndrome and diabetes. Oxid Med Cell longevity (2010) 3(6):392–403. doi: 10.4161/oxim.3.6.14786
32. Kang Q, Yang C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol (2020) 37:101799. doi: 10.1016/j.redox.2020.101799
33. Issad T, Masson E, Pagesy P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab (2010) 36(6 Pt 1):423–35. doi: 10.1016/j.diabet.2010.09.001
34. Issad T, Kuo M. O-GlcNAc modification of transcription factors, glucose sensing and glucotoxicity. Trends Endocrinol metabolism: TEM (2008) 19(10):380–9. doi: 10.1016/j.tem.2008.09.001
35. Felig P, Marliss E, Cahill GF Jr. Plasma amino acid levels and insulin secretion in obesity. New Engl J Med (1969) 281(15):811–6. doi: 10.1056/NEJM196910092811503
36. Würtz P, Soininen P, Kangas AJ, Rönnemaa T, Lehtimäki T, Kähönen M, et al. Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care (2013) 36(3):648–55. doi: 10.2337/dc12-0895
37. Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab (2012) 15(5):606–14. doi: 10.1016/j.cmet.2012.01.024
38. Ramzan I, Ardavani A, Vanweert F, Mellett A, Atherton PJ, Idris I. The association between circulating branched chain amino acids and the temporal risk of developing type 2 diabetes mellitus: A systematic review & Meta-analysis. Nutrients (2022) 14(20):4411. doi: 10.3390/nu14204411
39. Cosentino RG, Churilla JR, Josephson S, Molle-Rios Z, Hossain MJ, Prado WL, et al. Branched-chain amino acids and relationship with inflammation in youth with obesity: A randomized controlled intervention study. J Clin Endocrinol Metab (2021) 106(11):3129–39. doi: 10.1210/clinem/dgab538
40. Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med (2011) 17(4):448–53. doi: 10.1038/nm.2307
41. Wang-Sattler R, Yu Z, Herder C, Messias AC, Floegel A, He Y, et al. Novel biomarkers for pre-diabetes identified by metabolomics. Mol Syst Biol (2012) 8:615. doi: 10.1038/msb.2012.43
42. Palmer ND, Stevens RD, Antinozzi PA, Anderson A, Bergman RN, Wagenknecht LE, et al. Metabolomic profile associated with insulin resistance and conversion to diabetes in the Insulin Resistance Atherosclerosis Study. J Clin Endocrinol Metab (2015) 100(3):E463–8. doi: 10.1210/jc.2014-2357
43. Tillin T, Hughes AD, Wang Q, Würtz P, Ala-Korpela M, Sattar N, et al. Diabetes risk and amino acid profiles: cross-sectional and prospective analyses of ethnicity, amino acids and diabetes in a South Asian and European cohort from the SABRE (Southall And Brent REvisited) Study. Diabetologia (2015) 58(5):968–79. doi: 10.1007/s00125-015-3517-8
44. Qi Q, Li J, Yu B, Moon JY, Chai JC, Merino J, et al. Host and gut microbial tryptophan metabolism and type 2 diabetes: an integrative analysis of host genetics, diet, gut microbiome and circulating metabolites in cohort studies. Gut (2022) 71(6):1095–105. doi: 10.1136/gutjnl-2021-324053
45. Yu E, Papandreou C, Ruiz-Canela M, Guasch-Ferre M, Clish CB, Dennis C, et al. Association of tryptophan metabolites with incident type 2 diabetes in the PREDIMED trial: A case-cohort study. Clin Chem (2018) 64(8):1211–20. doi: 10.1373/clinchem.2018.288720
46. McCormack SE, Shaham O, McCarthy MA, Deik AA, Wang TJ, Gerszten RE, et al. Circulating branched-chain amino acid concentrations are associated with obesity and future insulin resistance in children and adolescents. Pediatr Obes (2013) 8(1):52–61. doi: 10.1111/j.2047-6310.2012.00087.x
47. Owei I, Umekwe N, Stentz F, Wan J, Dagogo-Jack S. Amino acid signature predictive of incident prediabetes: A case-control study nested within the longitudinal pathobiology of prediabetes in a biracial cohort. Metabolism: Clin experimental (2019) 98:76–83. doi: 10.1016/j.metabol.2019.06.011
48. Vogelzangs N, van der Kallen CJH, van Greevenbroek MMJ, van der Kolk BW, Jocken JWE, Goossens GH, et al. Metabolic profiling of tissue-specific insulin resistance in human obesity: results from the Diogenes study and the Maastricht Study. Int J Obes (2005) (2020) 44(6):1376–86. doi: 10.1038/s41366-020-0565-z
49. Papandreou C, Bulló M, Ruiz-Canela M, Dennis C, Deik A, Wang D, et al. Plasma metabolites predict both insulin resistance and incident type 2 diabetes: a metabolomics approach within the Prevención con Dieta Mediterránea (PREDIMED) study. Am J Clin Nutr (2019) 109(3):626–34. doi: 10.1093/ajcn/nqy262
50. Ferrannini E, Natali A, Camastra S, Nannipieri M, Mari A, Adam KP, et al. Early metabolic markers of the development of dysglycemia and type 2 diabetes and their physiological significance. Diabetes (2013) 62(5):1730–7. doi: 10.2337/db12-0707
51. Tricò D, Prinsen H, Giannini C, de Graaf R, Juchem C, Li F, et al. Elevated α-hydroxybutyrate and branched-chain amino acid levels predict deterioration of glycemic control in adolescents. J Clin Endocrinol Metab (2017) 102(7):2473–81. doi: 10.1210/jc.2017-00475
52. Guasch-Ferré M, Hruby A, Toledo E, Clish CB, Martínez-González MA, Salas-Salvadó J, et al. Metabolomics in prediabetes and diabetes: A systematic review and meta-analysis. Diabetes Care (2016) 39(5):833–46. doi: 10.2337/dc15-2251
53. Morze J, Wittenbecher C, Schwingshackl L, Danielewicz A, Rynkiewicz A, Hu FB, et al. Metabolomics and type 2 diabetes risk: an updated systematic review and meta-analysis of prospective cohort studies. Diabetes Care (2022) 45(4):1013–24. doi: 10.2337/dc21-1705
54. Xu F, Tavintharan S, Sum CF, Woon K, Lim SC, Ong CN. Metabolic signature shift in type 2 diabetes mellitus revealed by mass spectrometry-based metabolomics. J Clin Endocrinol Metab (2013) 98(6):E1060–5. doi: 10.1210/jc.2012-4132
55. Tai ES, Tan ML, Stevens RD, Low YL, Muehlbauer MJ, Goh DL, et al. Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia (2010) 53(4):757–67. doi: 10.1007/s00125-009-1637-8
56. Zhang X, Wang Y, Hao F, Zhou X, Han X, Tang H, et al. Human serum metabonomic analysis reveals progression axes for glucose intolerance and insulin resistance statuses. J Proteome Res (2009) 8(11):5188–95. doi: 10.1021/pr900524z
57. Menni C, Fauman E, Erte I, Perry JR, Kastenmüller G, Shin SY, et al. Biomarkers for type 2 diabetes and impaired fasting glucose using a nontargeted metabolomics approach. Diabetes (2013) 62(12):4270–6. doi: 10.2337/db13-0570
58. Würtz P, Wang Q, Kangas AJ, Richmond RC, Skarp J, Tiainen M, et al. Metabolic signatures of adiposity in young adults: Mendelian randomization analysis and effects of weight change. PloS Med (2014) 11(12):e1001765. doi: 10.1371/journal.pmed.1001765
59. Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T, et al. Genetic predisposition to an impaired metabolism of the branched-chain amino acids and risk of type 2 diabetes: A mendelian randomization analysis. PloS Med (2016) 13(11):e1002179. doi: 10.1371/journal.pmed.1002179
60. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.03.035
61. Matsuda T, Suzuki H, Sugano Y, Suzuki Y, Yamanaka D, Araki R, et al. Effects of branched-chain amino acids on skeletal muscle, glycemic control, and neuropsychological performance in elderly persons with type 2 diabetes mellitus: an exploratory randomized controlled trial. Nutrients (2022) 14(19):3917. doi: 10.3390/nu14193917
62. Lee J, Vijayakumar A, White PJ, Xu Y, Ilkayeva O, Lynch CJ, et al. BCAA supplementation in mice with diet-induced obesity alters the metabolome without impairing glucose homeostasis. Endocrinology (2021) 162(7):bqab062. doi: 10.1210/endocr/bqab062
63. Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab (2009) 9(4):311–26. doi: 10.1016/j.cmet.2009.02.002
64. White PJ, Lapworth AL, An J, Wang L, McGarrah RW, Stevens RD, et al. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol Metab (2016) 5(7):538–51. doi: 10.1016/j.molmet.2016.04.006
65. Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J Physiol (2018) 596(4):623–45. doi: 10.1113/JP275075
66. Saha AK, Xu XJ, Lawson E, Deoliveira R, Brandon AE, Kraegen EW, et al. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes (2010) 59(10):2426–34. doi: 10.2337/db09-1870
67. Schnuck JK, Sunderland KL, Gannon NP, Kuennen MR, Vaughan RA. Leucine stimulates PPARβ/δ-dependent mitochondrial biogenesis and oxidative metabolism with enhanced GLUT4 content and glucose uptake in myotubes. Biochimie (2016) 128-129:1–7. doi: 10.1016/j.biochi.2016.06.009
68. Nishitani S, Matsumura T, Fujitani S, Sonaka I, Miura Y, Yagasaki K. Leucine promotes glucose uptake in skeletal muscles of rats. Biochem Biophys Res Commun (2002) 299(5):693–6. doi: 10.1016/s0006-291x(02)02717-1
69. Zhang Y, Guo K, LeBlanc RE, Loh D, Schwartz GJ, Yu YH. Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes (2007) 56(6):1647–54. doi: 10.2337/db07-0123
70. Karusheva Y, Koessler T, Strassburger K, Markgraf D, Mastrototaro L, Jelenik T, et al. Short-term dietary reduction of branched-chain amino acids reduces meal-induced insulin secretion and modifies microbiome composition in type 2 diabetes: a randomized controlled crossover trial. Am J Clin Nutr (2019) 110(5):1098–107. doi: 10.1093/ajcn/nqz191
71. Solerte SB, Fioravanti M, Locatelli E, Bonacasa R, Zamboni M, Basso C, et al. Improvement of blood glucose control and insulin sensitivity during a long-term (60 weeks) randomized study with amino acid dietary supplements in elderly subjects with type 2 diabetes mellitus. Am J Cardiol (2008) 101(11a):82e–8e. doi: 10.1016/j.amjcard.2008.03.006
72. Nair KS, Garrow JS, Ford C, Mahler RF, Halliday D. Effect of poor diabetic control and obesity on whole body protein metabolism in man. Diabetologia (1983) 25(5):400–3. doi: 10.1007/BF00282518
73. Louard RJ, Barrett EJ, Gelfand RA. Overnight branched-chain amino acid infusion causes sustained suppression of muscle proteolysis. Metabolism: Clin experimental (1995) 44(4):424–9. doi: 10.1016/0026-0495(95)90047-0
74. Crown SB, Marze N, Antoniewicz MR. Catabolism of branched chain amino acids contributes significantly to synthesis of odd-chain and even-chain fatty acids in 3T3-L1 adipocytes. PloS One (2015) 10(12):e0145850. doi: 10.1371/journal.pone.0145850
75. Bishop CA, Schulze MB, Klaus S, Weitkunat K. The branched-chain amino acids valine and leucine have differential effects on hepatic lipid metabolism. FASEB J (2020) 34(7):9727–39. doi: 10.1096/fj.202000195R
76. Xiao F, Huang Z, Li H, Yu J, Wang C, Chen S, et al. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes (2011) 60(3):746–56. doi: 10.2337/db10-1246
77. Zhao H, Zhang F, Sun D, Wang X, Zhang X, Zhang J, et al. Branched-chain amino acids exacerbate obesity-related hepatic glucose and lipid metabolic disorders via attenuating akt2 signaling. Diabetes (2020) 69(6):1164–77. doi: 10.2337/db19-0920
78. Mahendran Y, Jonsson A, Have CT, Allin KH, Witte DR, Jørgensen ME, et al. Genetic evidence of a causal effect of insulin resistance on branched-chain amino acid levels. Diabetologia (2017) 60(5):873–8. doi: 10.1007/s00125-017-4222-6
79. Wang Q, Holmes MV, Davey Smith G, Ala-Korpela M. Genetic support for a causal role of insulin resistance on circulating branched-chain amino acids and inflammation. Diabetes Care (2017) 40(12):1779–86. doi: 10.2337/dc17-1642
80. Vanweert F, de Ligt M, Hoeks J, Hesselink MKC, Schrauwen P, Phielix E. Elevated plasma branched-chain amino acid levels correlate with type 2 diabetes-related metabolic disturbances. J Clin Endocrinol Metab (2021) 106(4):e1827–e36. doi: 10.1210/clinem/dgaa751
81. Herman MA, She P, Peroni OD, Lynch CJ, Kahn BB. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem (2010) 285(15):11348–56. doi: 10.1074/jbc.M109.075184
82. Suryawan A, Hawes JW, Harris RA, Shimomura Y, Jenkins AE, Hutson SM. A molecular model of human branched-chain amino acid metabolism. Am J Clin Nutr (1998) 68(1):72–81. doi: 10.1093/ajcn/68.1.72
83. Lackey DE, Lynch CJ, Olson KC, Mostaedi R, Ali M, Smith WH, et al. Regulation of adipose branched-chain amino acid catabolism enzyme expression and cross-adipose amino acid flux in human obesity. Am J Physiol Endocrinol Metab (2013) 304(11):E1175–87. doi: 10.1152/ajpendo.00630.2012
84. Burrill JS, Long EK, Reilly B, Deng Y, Armitage IM, Scherer PE, et al. Inflammation and ER stress regulate branched-chain amino acid uptake and metabolism in adipocytes. Mol Endocrinol (Baltimore Md) (2015) 29(3):411–20. doi: 10.1210/me.2014-1275
85. Li T, Zhang Z, Kolwicz SC Jr., Abell L, Roe ND, Kim M, et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab (2017) 25(2):374–85. doi: 10.1016/j.cmet.2016.11.005
86. Cifarelli V, Beeman SC, Smith GI, Yoshino J, Morozov D, Beals JW, et al. Decreased adipose tissue oxygenation associates with insulin resistance in individuals with obesity. J Clin Invest (2020) 130(12):6688–99. doi: 10.1172/JCI141828
87. She P, Van Horn C, Reid T, Hutson SM, Cooney RN, Lynch CJ. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Metab (2007) 293(6):E1552–63. doi: 10.1152/ajpendo.00134.2007
88. Lefort N, Glancy B, Bowen B, Willis WT, Bailowitz Z, De Filippis EA, et al. Increased reactive oxygen species production and lower abundance of complex I subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes (2010) 59(10):2444–52. doi: 10.2337/db10-0174
89. Vanweert F, Neinast M, Tapia EE, van de Weijer T, Hoeks J, Schrauwen-Hinderling VB, et al. A randomized placebo-controlled clinical trial for pharmacological activation of BCAA catabolism in patients with type 2 diabetes. Nat Commun (2022) 13(1):3508. doi: 10.1038/s41467-022-31249-9
90. Zsuga J, Török J, Magyar MT, Valikovics A, Gesztelyi R, Lenkei A, et al. Dimethylarginines at the crossroad of insulin resistance and atherosclerosis. Metabolism: Clin experimental (2007) 56(3):394–9. doi: 10.1016/j.metabol.2006.10.023
91. Schutte AE, Schutte R, Huisman HW, van Rooyen JM, Fourie CM, Malan L, et al. Dimethylarginines: their vascular and metabolic roles in Africans and Caucasians. Eur J endocrinol (2010) 162(3):525–33. doi: 10.1530/EJE-09-0865
92. DiNicolantonio JJ, McCarty MF, OKeefe JH. Role of dietary histidine in the prevention of obesity and metabolic syndrome. Open heart (2018) 5(2):e000676. doi: 10.1136/openhrt-2017-000676
93. Fikri AM, Smyth R, Kumar V, Al-Abadla Z, Abusnana S, Munday MR. Pre-diagnostic biomarkers of type 2 diabetes identified in the UAE’s obese national population using targeted metabolomics. Sci Rep (2020) 10(1):17616. doi: 10.1038/s41598-020-73384-7
94. Oxenkrug GF. Metabolic syndrome, age-associated neuroendocrine disorders, and dysregulation of tryptophan-kynurenine metabolism. Ann New York Acad Sci (2010) 1199:1–14. doi: 10.1111/j.1749-6632.2009.05356.x
95. Scarale MG, Mastroianno M, Prehn C, Copetti M, Salvemini L, Adamski J, et al. Circulating metabolites associate with and improve the prediction of all-cause mortality in type 2 diabetes. Diabetes (2022) 71(6):1363–70. doi: 10.2337/db22-0095
96. Rebnord EW, Strand E, Midttun Ø, Svingen GFT, Christensen MHE, Ueland PM, et al. The kynurenine:tryptophan ratio as a predictor of incident type 2 diabetes mellitus in individuals with coronary artery disease. Diabetologia (2017) 60(9):1712–21. doi: 10.1007/s00125-017-4329-9
97. Short KR, Chadwick JQ, Teague AM, Tullier MA, Wolbert L, Coleman C, et al. Effect of obesity and exercise training on plasma amino acids and amino metabolites in american Indian adolescents. J Clin Endocrinol Metab (2019) 104(8):3249–61. doi: 10.1210/jc.2018-02698
98. Wang TJ, Ngo D, Psychogios N, Dejam A, Larson MG, Vasan RS, et al. 2-Aminoadipic acid is a biomarker for diabetes risk. J Clin Invest (2013) 123(10):4309–17. doi: 10.1172/JCI64801
99. Lee HJ, Jang HB, Kim WH, Park KJ, Kim KY, Park SI, et al. 2-Aminoadipic acid (2-AAA) as a potential biomarker for insulin resistance in childhood obesity. Sci Rep (2019) 9(1):13610. doi: 10.1038/s41598-019-49578-z
100. Shi M, Wang C, Mei H, Temprosa M, Florez JC, Tripputi M, et al. Genetic architecture of plasma alpha-aminoadipic acid reveals a relationship with high-density lipoprotein cholesterol. J Am Heart Assoc (2022) 11(11):e024388. doi: 10.1161/JAHA.121.024388
101. Saremi A, Howell S, Schwenke DC, Bahn G, Beisswenger PJ, Reaven PD. Advanced glycation end products, oxidation products, and the extent of atherosclerosis during the VA diabetes trial and follow-up study. Diabetes Care (2017) 40(4):591–8. doi: 10.2337/dc16-1875
102. Rhee EP, Cheng S, Larson MG, Walford GA, Lewis GD, McCabe E, et al. Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. J Clin Invest (2011) 121(4):1402–11. doi: 10.1172/JCI44442
103. Lu J, Lam SM, Wan Q, Shi L, Huo Y, Chen L, et al. High-coverage targeted lipidomics reveals novel serum lipid predictors and lipid pathway dysregulation antecedent to type 2 diabetes onset in normoglycemic chinese adults. Diabetes Care (2019) 42(11):2117–26. doi: 10.2337/dc19-0100
104. Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatol (Baltimore Md) (2010) 51(2):679–89. doi: 10.1002/hep.23280
105. Salgin B, Ong KK, Thankamony A, Emmett P, Wareham NJ, Dunger DB. Higher fasting plasma free fatty acid levels are associated with lower insulin secretion in children and adults and a higher incidence of type 2 diabetes. J Clin Endocrinol Metab (2012) 97(9):3302–9. doi: 10.1210/jc.2012-1428
106. Ni Y, Zhao L, Yu H, Ma X, Bao Y, Rajani C, et al. Circulating unsaturated fatty acids delineate the metabolic status of obese individuals. EBioMedicine (2015) 2(10):1513–22. doi: 10.1016/j.ebiom.2015.09.004
107. Forouhi NG, Imamura F, Sharp SJ, Koulman A, Schulze MB, Zheng J, et al. Association of Plasma Phospholipid n-3 and n-6 Polyunsaturated Fatty Acids with Type 2 Diabetes: The EPIC-InterAct Case-Cohort Study. PloS Med (2016) 13(7):e1002094. doi: 10.1371/journal.pmed.1002094
108. Mahendran Y, Cederberg H, Vangipurapu J, Kangas AJ, Soininen P, Kuusisto J, et al. Glycerol and fatty acids in serum predict the development of hyperglycemia and type 2 diabetes in Finnish men. Diabetes Care (2013) 36(11):3732–8. doi: 10.2337/dc13-0800
109. Forouhi NG, Koulman A, Sharp SJ, Imamura F, Kröger J, Schulze MB, et al. Differences in the prospective association between individual plasma phospholipid saturated fatty acids and incident type 2 diabetes: the EPIC-InterAct case-cohort study. Lancet Diabetes endocrinol (2014) 2(10):810–8. doi: 10.1016/S2213-8587(14)70146-9
110. Sun L, Liang L, Gao X, Zhang H, Yao P, Hu Y, et al. Early prediction of developing type 2 diabetes by plasma acylcarnitines: A population-based study. Diabetes Care (2016) 39(9):1563–70. doi: 10.2337/dc16-0232
111. Harris WS, Luo J, Pottala JV, Margolis KL, Espeland MA, Robinson JG. Red blood cell fatty acids and incident diabetes mellitus in the women’s health initiative memory study. PloS One (2016) 11(2):e0147894. doi: 10.1371/journal.pone.0147894
112. Razquin C, Toledo E, Clish CB, Ruiz-Canela M, Dennis C, Corella D, et al. Plasma lipidomic profiling and risk of type 2 diabetes in the PREDIMED trial. Diabetes Care (2018) 41(12):2617–24. doi: 10.2337/dc18-0840
113. Lankinen MA, Stančáková A, Uusitupa M, Ågren J, Pihlajamäki J, Kuusisto J, et al. Plasma fatty acids as predictors of glycaemia and type 2 diabetes. Diabetologia (2015) 58(11):2533–44. doi: 10.1007/s00125-015-3730-5
114. Lemaitre RN, Fretts AM, Sitlani CM, Biggs ML, Mukamal K, King IB, et al. Plasma phospholipid very-long-chain saturated fatty acids and incident diabetes in older adults: the Cardiovascular Health Study. Am J Clin Nutr (2015) 101(5):1047–54. doi: 10.3945/ajcn.114.101857
115. Guasch-Ferré M, Ruiz-Canela M, Li J, Zheng Y, Bulló M, Wang DD, et al. Plasma acylcarnitines and risk of type 2 diabetes in a mediterranean population at high cardiovascular risk. J Clin Endocrinol Metab (2019) 104(5):1508–19. doi: 10.1210/jc.2018-01000
116. Huang T, Bhulaidok S, Cai Z, Xu T, Xu F, Wahlqvist ML, et al. Plasma phospholipids n-3 polyunsaturated fatty acid is associated with metabolic syndrome. Mol Nutr Food Res (2010) 54(11):1628–35. doi: 10.1002/mnfr.201000025
117. Fretts AM, Imamura F, Marklund M, Micha R, Wu JHY, Murphy RA, et al. Associations of circulating very-long-chain saturated fatty acids and incident type 2 diabetes: a pooled analysis of prospective cohort studies. Am J Clin Nutr (2019) 109(4):1216–23. doi: 10.1093/ajcn/nqz005
118. Fryk E, Olausson J, Mossberg K, Strindberg L, Schmelz M, Brogren H, et al. Hyperinsulinemia and insulin resistance in the obese may develop as part of a homeostatic response to elevated free fatty acids: A mechanistic case-control and a population-based cohort study. EBioMedicine (2021) 65:103264. doi: 10.1016/j.ebiom.2021.103264
119. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet (London England) (1963) 1(7285):785–9. doi: 10.1016/s0140-6736(63)91500-9
120. Wanner C, Tonelli M. KDIGO Clinical Practice Guideline for Lipid Management in CKD: summary of recommendation statements and clinical approach to the patient. Kidney Int (2014) 85(6):1303–9. doi: 10.1038/ki.2014.31
121. Grant PJ, Cosentino F. The 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: New features and the ‘Ten Commandments’ of the 2019 Guidelines are discussed by Professor Peter J. Grant and Professor Francesco Cosentino, the Task Force chairmen. Eur Heart J (2019) 40(39):3215–7. doi: 10.1093/eurheartj/ehz687
122. Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes (2000) 49(11):1939–45. doi: 10.2337/diabetes.49.11.1939
123. Xu YN, Wang Z, Zhang SK, Xu JR, Pan ZX, Wei X, et al. Low-grade elevation of palmitate and lipopolysaccharide synergistically induced β-cell damage via inhibition of neutral ceramidase. Mol Cell endocrinol (2022) 539:111473. doi: 10.1016/j.mce.2021.111473
124. Luo F, Feng Y, Ma H, Liu C, Chen G, Wei X, et al. Neutral ceramidase activity inhibition is involved in palmitate-induced apoptosis in INS-1 cells. Endocrine J (2017) 64(8):767–76. doi: 10.1507/endocrj.EJ16-0512
125. Lee SM, Choi SE, Lee JH, Lee JJ, Jung IR, Lee SJ, et al. Involvement of the TLR4 (Toll-like receptor4) signaling pathway in palmitate-induced INS-1 beta cell death. Mol Cell Biochem (2011) 354(1-2):207–17. doi: 10.1007/s11010-011-0820-7
126. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest (2006) 116(11):3015–25. doi: 10.1172/JCI28898
127. Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature (2002) 420(6913):333–6. doi: 10.1038/nature01137
128. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Sci (New York NY) (2001) 293(5535):1673–7. doi: 10.1126/science.1061620
129. Weatherill AR, Lee JY, Zhao L, Lemay DG, Youn HS, Hwang DH. Saturated and polyunsaturated fatty acids reciprocally modulate dendritic cell functions mediated through TLR4. J Immunol (Baltimore Md: 1950) (2005) 174(9):5390–7. doi: 10.4049/jimmunol.174.9.5390
130. Imamura F, Micha R, Wu JH, de Oliveira Otto MC, Otite FO, Abioye AI, et al. Effects of saturated fat, polyunsaturated fat, monounsaturated fat, and carbohydrate on glucose-insulin homeostasis: A systematic review and meta-analysis of randomized controlled feeding trials. PloS Med (2016) 13(7):e1002087. doi: 10.1371/journal.pmed.1002087
131. Drogan D, Dunn WB, Lin W, Buijsse B, Schulze MB, Langenberg C, et al. Untargeted metabolic profiling identifies altered serum metabolites of type 2 diabetes mellitus in a prospective, nested case control study. Clin Chem (2015) 61(3):487–97. doi: 10.1373/clinchem.2014.228965
132. Fall T, Salihovic S, Brandmaier S, Nowak C, Ganna A, Gustafsson S, et al. Non-targeted metabolomics combined with genetic analyses identifies bile acid synthesis and phospholipid metabolism as being associated with incident type 2 diabetes. Diabetologia (2016) 59(10):2114–24. doi: 10.1007/s00125-016-4041-1
133. Floegel A, Stefan N, Yu Z, Mühlenbruch K, Drogan D, Joost HG, et al. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes (2013) 62(2):639–48. doi: 10.2337/db12-0495
134. Zhao J, Zhu Y, Hyun N, Zeng D, Uppal K, Tran VT, et al. Novel metabolic markers for the risk of diabetes development in American Indians. Diabetes Care (2015) 38(2):220–7. doi: 10.2337/dc14-2033
135. Park JE, Lim HR, Kim JW, Shin KH. Metabolite changes in risk of type 2 diabetes mellitus in cohort studies: A systematic review and meta-analysis. Diabetes Res Clin practice (2018) 140:216–27. doi: 10.1016/j.diabres.2018.03.045
136. Huynh K, Barlow CK, Jayawardana KS, Weir JM, Mellett NA, Cinel M, et al. High-throughput plasma lipidomics: detailed mapping of the associations with cardiometabolic risk factors. Cell Chem Biol (2019) 26(1):71–84.e4. doi: 10.1016/j.chembiol.2018.10.008
137. Lemaitre RN, Yu C, Hoofnagle A, Hari N, Jensen PN, Fretts AM, et al. Circulating sphingolipids, insulin, HOMA-IR, and HOMA-B: the strong heart family study. Diabetes (2018) 67(8):1663–72. doi: 10.2337/db17-1449
138. Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, et al. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metab (2015) 22(2):266–78. doi: 10.1016/j.cmet.2015.06.007
139. Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, et al. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab (2014) 20(4):687–95. doi: 10.1016/j.cmet.2014.09.015
140. Kolak M, Westerbacka J, Velagapudi VR, Wågsäter D, Yetukuri L, Makkonen J, et al. Adipose tissue inflammation and increased ceramide content characterize subjects with high liver fat content independent of obesity. Diabetes (2007) 56(8):1960–8. doi: 10.2337/db07-0111
141. Coen PM, Hames KC, Leachman EM, DeLany JP, Ritov VB, Menshikova EV, et al. Reduced skeletal muscle oxidative capacity and elevated ceramide but not diacylglycerol content in severe obesity. Obes (Silver Spring Md) (2013) 21(11):2362–71. doi: 10.1002/oby.20381
142. Adams JM 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, et al. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes (2004) 53(1):25–31. doi: 10.2337/diabetes.53.1.25
143. Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Sci (New York NY) (1993) 259(5102):1769–71. doi: 10.1126/science.8456305
144. Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab (2007) 5(3):167–79. doi: 10.1016/j.cmet.2007.01.002
145. Chaurasia B, Kaddai VA, Lancaster GI, Henstridge DC, Sriram S, Galam DL, et al. Adipocyte ceramides regulate subcutaneous adipose browning, inflammation, and metabolism. Cell Metab (2016) 24(6):820–34. doi: 10.1016/j.cmet.2016.10.002
146. Adams SH, Hoppel CL, Lok KH, Zhao L, Wong SW, Minkler PE, et al. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr (2009) 139(6):1073–81. doi: 10.3945/jn.108.103754
147. Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun (2013) 4:1829. doi: 10.1038/ncomms2852
148. Bjursell M, Admyre T, Göransson M, Marley AE, Smith DM, Oscarsson J, et al. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am J Physiol Endocrinol Metab (2011) 300(1):E211–220. doi: 10.1152/ajpendo.00229.2010
149. Han JH, Kim IS, Jung SH, Lee SG, Son HY, Myung CS. The effects of propionate and valerate on insulin responsiveness for glucose uptake in 3T3-L1 adipocytes and C2C12 myotubes via G protein-coupled receptor 41. PloS One (2014) 9(4):e95268. doi: 10.1371/journal.pone.0095268
150. Fujiwara K, Maekawa F, Yada T. Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet beta-cells: mediation by PLC and L-type Ca2+ channel and link to insulin release. Am J Physiol Endocrinol Metab (2005) 289(4):E670–677. doi: 10.1152/ajpendo.00035.2005
151. Nolan CJ, Madiraju MS, Delghingaro-Augusto V, Peyot ML, Prentki M. Fatty acid signaling in the beta-cell and insulin secretion. Diabetes (2006) 55 Suppl 2:S16–23. doi: 10.2337/db06-s003
152. Chen Y, Ren Q, Zhou Z, Deng L, Hu L, Zhang L, et al. HWL-088, a new potent free fatty acid receptor 1 (FFAR1) agonist, improves glucolipid metabolism and acts additively with metformin in ob/ob diabetic mice. Br J Pharmacol (2020) 177(10):2286–302. doi: 10.1111/bph.14980
153. Li Z, Liu C, Zhou Z, Hu L, Deng L, Ren Q, et al. A novel FFA1 agonist, CPU025, improves glucose-lipid metabolism and alleviates fatty liver in obese-diabetic (ob/ob) mice. Pharmacol Res (2020) 153:104679. doi: 10.1016/j.phrs.2020.104679
154. Akerfeldt MC, Howes J, Chan JY, Stevens VA, Boubenna N, McGuire HM, et al. Cytokine-induced beta-cell death is independent of endoplasmic reticulum stress signaling. Diabetes (2008) 57(11):3034–44. doi: 10.2337/db07-1802
155. Lin N, Chen H, Zhang H, Wan X, Su Q. Mitochondrial reactive oxygen species (ROS) inhibition ameliorates palmitate-induced INS-1 beta cell death. Endocrine (2012) 42(1):107–17. doi: 10.1007/s12020-012-9633-z
156. Wu J, Sun P, Zhang X, Liu H, Jiang H, Zhu W, et al. Inhibition of GPR40 protects MIN6 β cells from palmitate-induced ER stress and apoptosis. J Cell Biochem (2012) 113(4):1152–8. doi: 10.1002/jcb.23450
157. Kristinsson H, Bergsten P, Sargsyan E. Free fatty acid receptor 1 (FFAR1/GPR40) signaling affects insulin secretion by enhancing mitochondrial respiration during palmitate exposure. Biochim Biophys Acta (2015) 1853(12):3248–57. doi: 10.1016/j.bbamcr.2015.09.022
158. Yamashima T. Dual effects of the non-esterified fatty acid receptor ‘GPR40’ for human health. Prog Lipid Res (2015) 58:40–50. doi: 10.1016/j.plipres.2015.01.002
159. Marafie SK, Al-Shawaf EM, Abubaker J, Arefanian H. Palmitic acid-induced lipotoxicity promotes a novel interplay between Akt-mTOR, IRS-1, and FFAR1 signaling in pancreatic β-cells. Biol Res (2019) 52(1):44. doi: 10.1186/s40659-019-0253-4
160. Finucane OM, Lyons CL, Murphy AM, Reynolds CM, Klinger R, Healy NP, et al. Monounsaturated fatty acid-enriched high-fat diets impede adipose NLRP3 inflammasome-mediated IL-1β secretion and insulin resistance despite obesity. Diabetes (2015) 64(6):2116–28. doi: 10.2337/db14-1098
161. Chan KL, Pillon NJ, Sivaloganathan DM, Costford SR, Liu Z, Théret M, et al. Palmitoleate reverses high fat-induced proinflammatory macrophage polarization via AMP-activated protein kinase (AMPK). J Biol Chem (2015) 290(27):16979–88. doi: 10.1074/jbc.M115.646992
162. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell (2010) 142(5):687–98. doi: 10.1016/j.cell.2010.07.041
163. Souza CO, Teixeira AA, Lima EA, Batatinha HA, Gomes LM, Carvalho-Silva M, et al. Palmitoleic acid (n-7) attenuates the immunometabolic disturbances caused by a high-fat diet independently of PPARα. Mediators inflammation (2014) 2014:582197. doi: 10.1155/2014/582197
164. Im DS. Functions of omega-3 fatty acids and FFA4 (GPR120) in macrophages. Eur J Pharmacol (2016) 785:36–43. doi: 10.1016/j.ejphar.2015.03.094
165. Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med (2005) 11(1):90–4. doi: 10.1038/nm1168
166. Freitas RDS, Campos MM. Understanding the appetite modulation pathways: The role of the FFA1 and FFA4 receptors. Biochem Pharmacol (2021) 186:114503. doi: 10.1016/j.bcp.2021.114503
167. Landaas S. The formation of 2-hydroxybutyric acid in experimental animals. Clinica chimica acta; Int J Clin Chem (1975) 58(1):23–32. doi: 10.1016/0009-8981(75)90481-7
168. Sarswat PK, Mishra YK, Free ML. Fabrication and response of alpha-hydroxybutyrate sensors for rapid assessment of cardiometabolic disease risk. Biosensors bioelectronics (2017) 89(Pt 1):334–42. doi: 10.1016/j.bios.2016.07.019
169. Gall WE, Beebe K, Lawton KA, Adam KP, Mitchell MW, Nakhle PJ, et al. alpha-hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population. PloS One (2010) 5(5):e10883. doi: 10.1371/journal.pone.0010883
170. Varvel SA, Pottala JV, Thiselton DL, Caffrey R, Dall T, Sasinowski M, et al. Serum α-hydroxybutyrate (α-HB) predicts elevated 1 h glucose levels and early-phase β-cell dysfunction during OGTT. BMJ Open Diabetes Res Care (2014) 2(1):e000038. doi: 10.1136/bmjdrc-2014-000038
171. Nilsen MS, Jersin R, Ulvik A, Madsen A, McCann A, Svensson PA, et al. 3-hydroxyisobutyrate, A strong marker of insulin resistance in type 2 diabetes and obesity that modulates white and brown adipocyte metabolism. Diabetes (2020) 69(9):1903–16. doi: 10.2337/db19-1174
172. Molinaro A, Bel Lassen P, Henricsson M, Wu H, Adriouch S, Belda E, et al. Imidazole propionate is increased in diabetes and associated with dietary patterns and altered microbial ecology. Nat Commun (2020) 11(1):5881. doi: 10.1038/s41467-020-19589-w
173. Koh A, Molinaro A, Ståhlman M, Khan MT, Schmidt C, Mannerås-Holm L, et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell (2018) 175(4):947–61.e17. doi: 10.1016/j.cell.2018.09.055
174. van Son J, Serlie MJ, Ståhlman M, Bäckhed F, Nieuwdorp M, Aron-Wisnewsky J. Plasma imidazole propionate is positively correlated with blood pressure in overweight and obese humans. Nutrients (2021) 13(8):2706. doi: 10.3390/nu13082706
175. Prentice KJ, Luu L, Allister EM, Liu Y, Jun LS, Sloop KW, et al. The furan fatty acid metabolite CMPF is elevated in diabetes and induces β cell dysfunction. Cell Metab (2014) 19(4):653–66. doi: 10.1016/j.cmet.2014.03.008
176. Yi J, Jin H, Zhang R, Zhang S, Chen P, Yu X, et al. Increased serum 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid (CMPF) levels are associated with glucose metabolism in Chinese pregnant women. J endocrinological Invest (2018) 41(6):663–70. doi: 10.1007/s40618-017-0789-5
177. Zhang S, Chen P, Jin H, Yi J, Xie X, Yang M, et al. Circulating 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid (CMPF) levels are associated with hyperglycemia and β cell dysfunction in a Chinese population. Sci Rep (2017) 7(1):3114. doi: 10.1038/s41598-017-03271-1
178. Zheng JS, Lin M, Imamura F, Cai W, Wang L, Feng JP, et al. Serum metabolomics profiles in response to n-3 fatty acids in Chinese patients with type 2 diabetes: a double-blind randomized controlled trial. Sci Rep (2016) 6:29522. doi: 10.1038/srep29522
179. Liu G, Gibson RA, Callahan D, Guo XF, Li D, Sinclair AJ. Pure omega 3 polyunsaturated fatty acids (EPA, DPA or DHA) are associated with increased plasma levels of 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid (CMPF) in a short-term study in women. Food Funct (2020) 11(3):2058–66. doi: 10.1039/c9fo02440a
180. Lankinen MA, Hanhineva K, Kolehmainen M, Lehtonen M, Auriola S, Mykkänen H, et al. CMPF does not associate with impaired glucose metabolism in individuals with features of metabolic syndrome. PloS One (2015) 10(4):e0124379. doi: 10.1371/journal.pone.0124379
181. Haeusler RA, Astiarraga B, Camastra S, Accili D, Ferrannini E. Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes (2013) 62(12):4184–91. doi: 10.2337/db13-0639
182. Chávez-Talavera O, Wargny M, Pichelin M, Descat A, Vallez E, Kouach M, et al. Bile acids associate with glucose metabolism, but do not predict conversion from impaired fasting glucose to diabetes. Metabolism: Clin experimental (2020) 103:154042. doi: 10.1016/j.metabol.2019.154042
183. Lee SG, Lee YH, Choi E, Cho Y, Kim JH. Fasting serum bile acids concentration is associated with insulin resistance independently of diabetes status. Clin Chem Lab Med (2019) 57(8):1218–28. doi: 10.1515/cclm-2018-0741
184. Jørgensen NB, Dirksen C, Bojsen-Møller KN, Kristiansen VB, Wulff BS, Rainteau D, et al. Improvements in glucose metabolism early after gastric bypass surgery are not explained by increases in total bile acids and fibroblast growth factor 19 concentrations. J Clin Endocrinol Metab (2015) 100(3):E396–406. doi: 10.1210/jc.2014-1658
185. Bishay RH, Tonks KT, George J, Samocha-Bonet D, Meyerowitz-Katz G, Chisholm DJ, et al. Plasma bile acids more closely align with insulin resistance, visceral and hepatic adiposity than total adiposity. J Clin Endocrinol Metab (2021) 106(3):e1131–e9. doi: 10.1210/clinem/dgaa940
186. Safai N, Suvitaival T, Ali A, Spégel P, Al-Majdoub M, Carstensen B, et al. Effect of metformin on plasma metabolite profile in the Copenhagen Insulin and Metformin Therapy (CIMT) trial. Diabetic medicine: J Br Diabetic Assoc (2018) 35(7):944–53. doi: 10.1111/dme.13636
187. Preiss D, Rankin N, Welsh P, Holman RR, Kangas AJ, Soininen P, et al. Effect of metformin therapy on circulating amino acids in a randomized trial: the CAMERA study. Diabetic medicine: J Br Diabetic Assoc (2016) 33(11):1569–74. doi: 10.1111/dme.13097
188. Zhang Y, Hu C, Hong J, Zeng J, Lai S, Lv A, et al. Lipid profiling reveals different therapeutic effects of metformin and glipizide in patients with type 2 diabetes and coronary artery disease. Diabetes Care (2014) 37(10):2804–12. doi: 10.2337/dc14-0090
189. Green CJ, Marjot T, Walsby-Tickle J, Charlton C, Cornfield T, Westcott F, et al. Metformin maintains intrahepatic triglyceride content through increased hepatic de novo lipogenesis. Eur J endocrinol (2022) 186(3):367–77. doi: 10.1530/EJE-21-0850
190. Huo T, Cai S, Lu X, Sha Y, Yu M, Li F. Metabonomic study of biochemical changes in the serum of type 2 diabetes mellitus patients after the treatment of metformin hydrochloride. J Pharm Biomed analysis (2009) 49(4):976–82. doi: 10.1016/j.jpba.2009.01.008
191. van der Meer RW, Rijzewijk LJ, de Jong HW, Lamb HJ, Lubberink M, Romijn JA, et al. Pioglitazone improves cardiac function and alters myocardial substrate metabolism without affecting cardiac triglyceride accumulation and high-energy phosphate metabolism in patients with well-controlled type 2 diabetes mellitus. Circulation (2009) 119(15):2069–77. doi: 10.1161/CIRCULATIONAHA.108.803916
192. Jonker JT, Wang Y, de Haan W, Diamant M, Rijzewijk LJ, van der Meer RW, et al. Pioglitazone decreases plasma cholesteryl ester transfer protein mass, associated with a decrease in hepatic triglyceride content, in patients with type 2 diabetes. Diabetes Care (2010) 33(7):1625–8. doi: 10.2337/dc09-1935
193. Goldberg RB, Kendall DM, Deeg MA, Buse JB, Zagar AJ, Pinaire JA, et al. A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care (2005) 28(7):1547–54. doi: 10.2337/diacare.28.7.1547
194. Tahara N, Yamagishi S-I, Mizoguchi M, Tahara A, Imaizumi T. Pioglitazone decreases asymmetric dimethylarginine levels in patients with impaired glucose tolerance or type 2 diabetes. Rejuvenation Res (2013) (No.5):344–51. doi: 10.1089/rej.2013.1434
195. Angelidi AM, Kokkinos A, Sanoudou D, Connelly MA, Alexandrou A, Mingrone G, et al. Early metabolomic, lipid and lipoprotein changes in response to medical and surgical therapeutic approaches to obesity. Metabolism: Clin experimental (2023) 138:155346. doi: 10.1016/j.metabol.2022.155346
196. Peradze N, Farr OM, Perakakis N, Lázaro I, Sala-Vila A, Mantzoros CS. Short-term treatment with high dose liraglutide improves lipid and lipoprotein profile and changes hormonal mediators of lipid metabolism in obese patients with no overt type 2 diabetes mellitus: a randomized, placebo-controlled, cross-over, double-blind clinical trial. Cardiovasc diabetol (2019) 18(1):141. doi: 10.1186/s12933-019-0945-7
197. Koska J, Lopez L, D’Souza K, Osredkar T, Deer J, Kurtz J, et al. Effect of liraglutide on dietary lipid-induced insulin resistance in humans. Diabetes Obes Metab (2018) 20(1):69–76. doi: 10.1111/dom.13037
198. Zobel EH, Wretlind A, Ripa RS, Rotbain Curovic V, von Scholten BJ, Suvitaival T, et al. Ceramides and phospholipids are downregulated with liraglutide treatment: results from the LiraFlame randomized controlled trial. BMJ Open Diabetes Res Care (2021) 9(1):e002395. doi: 10.1136/bmjdrc-2021-002395
199. Rizzo M, Chandalia M, Patti AM, Di Bartolo V, Rizvi AA, Montalto G, et al. Liraglutide decreases carotid intima-media thickness in patients with type 2 diabetes: 8-month prospective pilot study. Cardiovasc Diabetol (2014) 13:49. doi: 10.1186/1475-2840-13-49
200. Jendle J, Hyötyläinen T, Orešič M, Nyström T. Pharmacometabolomic profiles in type 2 diabetic subjects treated with liraglutide or glimepiride. Cardiovasc diabetol (2021) 20(1):237. doi: 10.1186/s12933-021-01431-2
201. Rodgers M, Migdal AL, Rodríguez TG, Chen ZZ, Nath AK, Gerszten RE, et al. Weight loss outcomes among early high responders to exenatide treatment: A randomized, placebo controlled study in overweight and obese women. Front endocrinol (2021) 12:742873. doi: 10.3389/fendo.2021.742873
202. Chaudhuri A, Ghanim H, Vora M, Sia CL, Korzeniewski K, Dhindsa S, et al. Exenatide exerts a potent antiinflammatory effect. J Clin Endocrinol Metab (2012) 97(1):198–207. doi: 10.1210/jc.2011-1508
203. Gerstein HC, Lee SF, Paré G, Bethel MA, Colhoun HM, Hoover A, et al. Biomarker changes associated with both dulaglutide and cardiovascular events in the REWIND randomized controlled trial: A nested case-control post hoc analysis. Diabetes Care (2023) 46(5):1046–51. doi: 10.2337/dc22-2397
204. Pirro V, Roth KD, Lin Y, Willency JA, Milligan PL, Wilson JM, et al. Effects of tirzepatide, a dual GIP and GLP-1 RA, on lipid and metabolite profiles in subjects with type 2 diabetes. J Clin Endocrinol Metab (2022) 107(2):363–78. doi: 10.1210/clinem/dgab722
205. Cakirca M, Karatoprak C, Zorlu M, Kiskac M, Kanat M, Cikrikcioglu MA, et al. Effect of vildagliptin add-on treatment to metformin on plasma asymmetric dimethylarginine in type 2 diabetes mellitus patients. Drug design Dev Ther (2014) 8:239–43. doi: 10.2147/DDDT.S52545
206. Boschmann M, Engeli S, Dobberstein K, Budziarek P, Strauss A, Boehnke J, et al. Dipeptidyl-peptidase-IV inhibition augments postprandial lipid mobilization and oxidation in type 2 diabetic patients. J Clin Endocrinol Metab (2009) 94(3):846–52. doi: 10.1210/jc.2008-1400
207. Monami M, Lamanna C, Desideri CM, Mannucci E. DPP-4 inhibitors and lipids: systematic review and meta-analysis. Adv Ther (2012) 29(1):14–25. doi: 10.1007/s12325-011-0088-z
208. Irving BA, Carter RE, Soop M, Weymiller A, Syed H, Karakelides H, et al. Effect of insulin sensitizer therapy on amino acids and their metabolites. Metabolism: Clin experimental (2015) 64(6):720–8. doi: 10.1016/j.metabol.2015.01.008
Keywords: diabetes mellitus, metabolites, amino acids, lipids, carbohydrates
Citation: Liu Y, Wang D and Liu Y-P (2023) Metabolite profiles of diabetes mellitus and response to intervention in anti-hyperglycemic drugs. Front. Endocrinol. 14:1237934. doi: 10.3389/fendo.2023.1237934
Received: 10 June 2023; Accepted: 16 October 2023;
Published: 31 October 2023.
Edited by:
Åke Sjöholm, Gävle Hospital, SwedenReviewed by:
Athanasia K. Papazafiropoulou, Tzaneio Hospital, GreeceCopyright © 2023 Liu, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi-Ping Liu, eXBsaXUxOTY2QGZqbnUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.