 Sayani Mukherjee
Sayani Mukherjee Silje Skrede
Silje Skrede Martha Haugstøyl1,2
Martha Haugstøyl1,2 Miguel López
Miguel López Johan Fernø
Johan Fernø
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 31 August 2023
Sec. Experimental Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1232171
This article is part of the Research TopicCentral and Peripheral Mechanism Interfering in Metabolic SyndromeView all 5 articles
Obesity is associated with chronic, low-grade inflammation. Excessive nutrient intake causes adipose tissue expansion, which may in turn cause cellular stress that triggers infiltration of pro-inflammatory immune cells from the circulation as well as activation of cells that are residing in the adipose tissue. In particular, the adipose tissue macrophages (ATMs) are important in the pathogenesis of obesity. A pro-inflammatory activation is also found in other organs which are important for energy metabolism, such as the liver, muscle and the pancreas, which may stimulate the development of obesity-related co-morbidities, including insulin resistance, type 2 diabetes (T2D), cardiovascular disease (CVD) and non-alcoholic fatty liver disease (NAFLD). Interestingly, it is now clear that obesity-induced pro-inflammatory signaling also occurs in the central nervous system (CNS), and that pro-inflammatory activation of immune cells in the brain may be involved in appetite dysregulation and metabolic disturbances in obesity. More recently, it has become evident that microglia, the resident macrophages of the CNS that drive neuroinflammation, may also be activated in obesity and can be relevant for regulation of hypothalamic feeding circuits. In this review, we focus on the action of peripheral and central macrophages and their potential roles in metabolic disease, and how macrophages interact with other immune cells to promote inflammation during obesity.
The increasing prevalence of obesity represents a serious health concern in both developed and developing countries. Obesity is associated with several co-morbidities, such as type 2 diabetes (T2D), atherosclerosis, non-alcoholic fatty liver disease (NAFLD) and cancer, and is estimated to be responsible for the deaths of more than 4 million people each year (1). Inflammation is believed to be important in the pathogenesis of obesity and its co-morbidities. Indeed, dysregulation of immune cells located in metabolically active tissues, in particular in the adipose tissue (AT), has been shown to contribute to disease progression. A plethora of immune cells may contribute to a pro-inflammatory state in obesity, but macrophages seem to be of special importance, representing up to 50% of the immune cells in the obese AT (2). In recent years it has become clear that obesity-induced inflammation also occurs in the central nervous system (CNS) (3), which may contribute to the metabolic phenotype as well as to CNS-specific pathologies associated with obesity, including neurodegenerative diseases like Alzheimer’s disease and Parkinson’s disease (4, 5). However, how peripheral and central inflammation is connected in obesity remains unclear. In this review we focus on macrophages in metabolically active tissues and their role in metabolic regulation, and we examine how central and peripheral immune cells interact during obesity.
Macrophages are part of the innate immune system, and together with other innate immune cells, such as neutrophils, eosinophils, and natural killer (NK) cells, they form the first line of immune defense that respond quickly to foreign invaders. The adaptive immune system, on the other hand, elicits a slower response, but has the ability to generate an immunologic memory towards specific pathogens and to trigger effective immune responses when exposed to the same pathogens in the future (6). Accumulating evidence indicates that the innate immune system may also form such a long-term memory after encountering a stimulus, a concept termed “trained immunity” that involves sustained epigenetic reprogramming that potentiates inflammatory responses to future challenges (7–9). Obesity has been shown to promote such trained immunity in innate immune cells, where for example obesity-induced changes in AT macrophages (ATMs) were maintained after weight loss in mice (10), and elevated free fatty acids induced a sustained inflammatory phenotype in myeloid cells in vitro (11).
Innate and adaptive immune cells can be categorized as either circulating or tissue-resident cells. Circulating immune cells are those who are transported in the blood stream, recognizing and killing infected/abnormal cells. Examples of circulating innate immune cells are monocytes and macrophages, whereas T cells and B cells represent circulating adaptive immune cells (12, 13). Tissue-resident immune cells play integral roles at all stages of the immune response. It should be noted that in addition to responding to infectious challenges and mediating the resolution of inflammation, these cells also have important roles in maintaining tissue homeostasis and repair (10, 14). Tissue-resident immune cells are found in AT, liver, pancreas, brain and intestine, where they express specific surface markers depending on the tissue that they reside in (15–17) and the functions they exert. Examples of major tissue-resident immune cells are ATMs, hepatic Kupffer cells (KC) and CNS microglia.
In recent years, there has been increased awareness of the importance of ATMs in obesity. Adiposity has been shown to affect their abundance, subset ratios, and phenotype diversity, which can contribute to alterations in whole body metabolism. As a response to specific microenvironmental stimuli, including metabolic stress signals such as fatty acids and glucose, levels of proinflammatory M1-like ATM subtypes are elevated, whereas alternatively activated M2-like macrophages, with anti-inflammatory properties, are reduced (18). The M1-like ATMs have been shown to impair insulin signaling in both mice and humans (19). In contrast, the M2-like ATMs are believed to mediate protection from these metabolic disturbances and to play a role in AT homeostatic functions (20, 21). Various subcategories of M2 macrophages are found, including M2a, M2b, M2c, and M2d (22). Furthermore, it is now well established that there exists a complex range of ATM phenotypes that display characteristics differing from the classical M1- and M2-like ATMs (21, 23, 24). These cells differ in their expression of cell surface markers, cytokine secretion profile, and biological characteristics, which is described in more detail below.
Although ATMs are considered the most important immune cells that mediate metabolic (dys)regulation during obesity, other cells such as T cells and NK cells are also of importance. This is partly due to their role in modulating ATM polarization, emphasizing that the interaction between immune cells plays a significant role in inflammation in obese mice and in people with obesity (25–29). In this review we aim to describe how immune cells, especially macrophages, are involved in obesity-related metabolic inflammation, and to illustrate how the balance between pro- and anti-inflammatory immune cells is important to maintain a healthy tissue homeostasis during changes in nutritional status.
Obesity is a consequence of excess nutrient intake and a positive energy balance that increases both the size of mature adipocytes (hypertrophy) and the number of adipocytes by de novo formation from preadipocytes (hyperplasia) (30). Fatty acid-binding protein (FABP4/aP2) has been shown to be important in controlling adipocyte size, differentiation and the recruitment of new adipocytes (31). Further, it regulates mitochondrial redox signaling through uncoupling protein 2 (in contrast to UCP1, which controls energy expenditure, UCP2 mainly controls ADP/ATP ratio) (32) in a process commonly termed “adipose tissue remodeling”, which is important to adapt to the increased energy load. However, during obesity, hypertrophy may not be compensated by sufficient blood vessel vascularization, which may lead to hypoxia, mitochondrial dysfunction and, subsequently, AT stress. In turn, this can lead to an increase in pro-inflammatory responses. Normally, the AT immune cells play vital roles in maintaining AT homeostasis during tissue remodeling, but pro-inflammatory activation over time may lead to chronic inflammation and the activation of reactive super oxygen species (ROS), impairing AT hormonal signaling and function (33).
In hypoxic AT, anaerobic glucose breakdown may fuel pro-inflammatory processes via activation of hypoxia-inducible factor 1-alpha (HIF-1a) (34). Moreover, the hypoxic signal has been shown to inhibit the mitochondrial electron transport chain and decrease oxidative phosphorylation (OXPHOS), leading to increased ROS production and a shift towards glycolytic processes that may further stimulate the inflammatory phenotype (35). The activation of HIF-1a also contributes to direct regulation of innate and adaptive immune cells in the AT, including epithelial cells, neutrophils, macrophages, dendritic cells (DC), T cells, B cells, NK cells and innate lymphoid cells (36). For macrophages, the elevated glycolysis leads to a shift towards M1-like ATM, as well as increased expression of genes involved in macrophage adhesion and inhibition of macrophage migration from the hypoxic AT (37, 38). It has also been reported that obesity activates the FABP4/UCP2 axis in macrophages, resulting in the activation of inflammasomes (32). Further, the activation of toll-like receptors (TLRs) by elevated fatty acids during obesity is essential for the capacity of ATMs to generate pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNFa) and interleukin (IL)-6 (39). This is mediated probably through the activation of pathways regulated by the nuclear factor kappa B/c-Jun N-terminal kinases (NFκB/JNK) and Inhibitory kappa B kinase (IκB) inhibitor (40, 41). The M2-like ATMs rely on mitochondrial OXPHOS and are thus reduced under these conditions, rendering the hypoxic environment in the expanding AT supportive of an elevated M1/M2 ratio (18, 42).
All these studies suggest that expansion of AT during obesity can trigger adipocyte hypertrophy and hypoxia, which finally may lead to chronic, low-grade inflammation where ATM activity and polarization play an important role.
In AT of both animals and people with obesity, pro-inflammatory macrophages form so-called crown-like structures (CLS), surrounding dead or dying adipocytes, in particular evident in the visceral AT (VAT) (43, 44). In recent years it has become clear that there are numerous types of ATMs that can be characterized based on the combination of surface proteins they express. Also, recent advances in single-cell sequencing have demonstrated that a variety of macrophage subsets exist that differ from the conventional M1-/M2-like macrophages, some of them exhibiting proinflammatory characteristics, while others are more metabolically active (23, 24, 45). In humans, CLS macrophages have been shown to express the surface proteins CD206 and CD11c (46), and are associated with systemic markers of metabolic dysfunction, supporting the idea that pro-inflammatory ATMs may have unfavorable effects on metabolic health (45–48). However, more recent literature has claimed that CLS are indispensable in controlling metabolic homeostasis, and that their accumulation around dying adipocytes is a mechanism to get rid of excessive adipocyte lipids in a non-harmful way (49–51). Nevertheless, the excessive inflammation associated with CLS can result in “collateral damage”, where the effect on metabolism depends on the type of pro-inflammatory and/or metabolically active ATMs that predominate in the AT. Interestingly, pro-inflammatory ATMs does not seem to express the classical surface markers of activated M1 macrophages, CD38, CD319, and CD274 (18). Instead, in addition to the combination of CD206 and CD11c as described above, ATMs have been shown to express ABCA1, CD36, and PLIN2, defining so-called metabolically activated macrophages (MMe) (18). These cells promote the clearance of dead adipocytes via lysosomal exocytosis (51). NADPH oxidase 2 (NOX2) was shown to be a major mediator of the inflammatory and adipocyte-clearing properties of MMe macrophages, and Nox2−/− mice exhibited insulin resistance, liver inflammation, and visceral lipoatrophy, characterized by deposition of dead adipocytes and dysfunctional ATM lysosomal exocytosis (51). In another study, mouse Ly6c-expressing ATMs have been shown to be predominant outside of CLS and to display adipogenic properties, while CD9-lipid associated ATMs (LAMs) were found to be present within CLS of HFD-fed mice and humans with obesity (50). The LAMs expressed “triggering receptor expressed on myeloid cell 2” (TREM2) lysosomal acid lipase, and controlled lipid metabolism and phagocytosis in mouse AT (50). Interestingly, TREM2 deletion has been shown to cause weight gain, hypercholesterolemia, and glucose intolerance in mice due to the loss of LAM function, lipid uptake, and storage (14), indicating that Trem2+ LAM cells may be key in the mitigation of metabolic disruption in the AT. Other ATMs that have been described are iron rich (MFe) macrophages, shown to have an inflammatory phenotype in obese mice (52), and antioxidant macrophages (Mox) ATMs, essential to iron and oxidative stress handling (53). In agreement with findings described above (46), a recent study from our laboratory found a positive correlation between the level of insulin resistance in 80 patients with obesity and the pro-inflammatory ATM ratio between M1-like (CD206/CD11c expressing cells) and M2-like (CD206 expressing cells) in both VAT and subcutaneous AT (SAT) (29). However, using a surface proteomics approach we also identified several new surface markers on these immune cells, indicating that a plethora of M1-like and M2-like ATM subtypes exist in the human AT (54). According to these findings, it is obvious that ATMs exist in pro- and anti-inflammatory forms, but in the light of recent findings the M1/M2 dichotomy is gradually replaced by a more differentiated view on ATM characteristics. ATMs act in response to obesity in a temporal and site-specific manner that may be both beneficial and harmful.
The details of how the different ATM subtype’s function and act in response to metabolic stimuli is further complicated by their interaction with other adipose-resident immune cells, such as T cells, invariant natural killer T (iNKT) cells, gamma delta (γδ) T cells, NK cells, DCs, eosinophils and their diverse phenotypes that may manifest in the obese AT. T cells are the second most prevalent immune cells in AT. It has been reported that in obesity there is an increased frequency of pro-inflammatory CD4+ T cells (e.g., Th1 and Th17 cells), and cytotoxic CD8+ T cells, and reduction in anti-inflammatory CD4+ T cells (e.g., Th2 cells) and FOXp3 regulatory T cells (Tregs) (55). The Th1 cells and CD8+ T cells have been shown to stimulate M1 ATM polarization in obese mice AT through IFNg production. There may, however, be several subgroups of AT-T cells with potentially different metabolic effects. An in-depth analysis of T cells in human AT depots has been conducted by our laboratory. Here, distinct subsets of T cells were shown to express markers such as CD26 and the chemokine receptor type 5 (CCR5), as well as obesity-specific genes known to activate pro-inflammatory mechanisms (56).
iNKT cells are reported to be highly expressed in AT of lean mice, and their abundance is reduced in the obese state (44). In human AT, iNKT cells are less abundant. AT-resident NKT cells have been demonstrated to modulate macrophage phenotype; IL-10-producing iNKT cells have been shown to trigger M2 polarization in steady-state AT. Further, iNKT-induced M2 polarization has been reported following acute or prolonged HFD challenges in an IL-4-dependent manner in mouse models and in human subjects with obesity (57, 58). In addition, AT-resident iNKT cells have been shown to trigger M2 macrophage polarization in the absence of HFD challenge in mice, via IL-10 (59). These results indicate that iNKT cells mitigate pro-inflammatory effects and induce metabolically favorable signaling in AT.
γδ T cells represent another subset of T cells that may act in AT (60). Usually, γδ T cells are prevalent in lean AT, but a further increase during obesity has been reported (61). Unlike the conventional CD4+ and CD8+ T cells, γδ T cells are negative for CD4 and CD8 markers, and express γδ T cell receptors (TCRs) (62). Further, the activation of these cells are mediated by TCR in a MHC-independent manner, and by receptors shared with NK cells (e.g., NKG2D and:DNAM-1) (63). The increase of infiltrating γδ T cells in AT in obesity has been shown to trigger the accumulation of ATMs, inflammation, and insulin resistance in mice, indicating that they are pro-inflammatory (61). In accordance with this finding, another study found reduced HFD-induced inflammation in AT of mice lacking γδ T cells (64). Nevertheless, further research is necessary to determine the exact role of γδ T cells is in obesity and obesity-related diseases.
NK cells and other innate lymphoid cells (ILCs) have been suggested as important mediators of the metabolic dysfunction of AT in obesity, in part through regulation of ATM activity. In AT of lean mice, NK cells were shown to exert homeostatic roles through killing M2-like ATMs, likely a mechanism to prevent them from turning pro-inflammatory (65). HFD promotes early accumulation of NK cells, especially in VAT, displaying reduced killing ability and increased IFNγ production that drive the polarization and maintenance of M1-like ATMs and development of insulin resistance (27, 66). Although less studied, AT NK cells in individuals with obesity were shown to display altered phenotype and function (67) (68), pointing to a similar role of these cells in human AT. However, the exact crosstalk between human NK cells and ATMs needs to be further elucidated.
Recently, DC has been found to play a crucial role in AT inflammation during obesity (69). DC expressing CD11c+ have been shown to be increased significantly in AT following HFD. Further, the DC increase in AT has been shown to be associated with CLS, thus confirming their association with ATMs. Importantly, a DC-null mouse model had a reduced number of AT macrophages, whereas DC replacement increased AT macrophage numbers in the DC-null mice. Finally, mice lacking DC did not gain weight or develop metabolic abnormalities when given an HFD (69). These data demonstrate that DC play an important role in systemic metabolic responses during obesity, and macrophage infiltration in AT.
Like other white blood cells, eosinophils are also involved in fighting disease and infections. In addition, eosinophils play a role in obesity that has become more evident in recent years. For example, the alternative activation of AT macrophages in obesity has been reported to be inextricably linked with eosinophils by an IL-4- or IL-13-dependent process (70). Briefly, IL-4 production by eosinophils was reported to promote thermogenesis in WAT, thereby increasing energy expenditure, limiting weight gain and improving glucose tolerance. In contrast, decreased eosinophil count was shown to be associated with increased weight gain and glucose intolerance in obese animals (70). Further, this study showed that M2-macrophages are greatly suppressed by the absence of eosinophils in WAT of obese mice. Similarly, eosinophils have been shown to control glucose homeostasis in people with obesity (71). These data suggest that eosinophils play a role in maintaining an anti-inflammatory ATM population to promote metabolic homeostasis.
Taken together, these studies confirm that in addition to ATMs, other immune cells are involved in metabolic inflammation and in the regulation of AT inflammation. These cells release proinflammatory cytokines which may directly affect metabolic pathways, but also modulate ATMs phenotypic characteristics to constitute a positive inflammatory feedback look. However, the role of macrophages and other immune cells during obesity is not limited to AT. We will now briefly discuss their role in other metabolic organs, such as the liver, pancreas, and gut.
The liver is important in a number of homeostatic functions, including detoxification, glucose metabolism, and synthesis of bile acids, proteins and lipids (72, 73). It is therefore noteworthy that obesity is a major cause of the most common chronic liver disease, non-alcoholic fatty liver disease (NAFLD), with recent data suggesting a world-wide prevalence of NAFLD of astonishing 40% (74). A sub-group of NAFLD patients develops chronic inflammation in the liver, which over time can lead to liver fibrosis, also known as non-alcoholic steatohepatitis (NASH) (1). The liver contains numerous immune cells, and accumulating evidence suggests that fatty acids and pro-inflammatory mediators released from AT may be important for liver immune cell activation and NASH progression, pointing towards the AT for a potential strategy to treat NASH (75). Several types of tissue resident immune cells (macrophages, neutrophils, B lymphocytes, T lymphocytes, and NK cells) have been reported to be associated with liver inflammation (76). The KC-liver macrophages constitute the largest population of liver immune cells and play an important role in NASH development (77, 78). They can be found within sinusoids, in contact with endothelial cells (79). Like ATMs, KCs can also be divided into pro- and anti-inflammatory M1/M2 subtypes (80), and the importance of KC polarization in the initiation and development of NAFLD and NASH is supported by several recent studies (81, 82). Additionally, KC subsets with distinct transcriptional profiles have been identified in NAFLD/NASH, reminiscent of heterogeneous ATMs in obesity. A recent study using scRNA-seq identified three distinct clusters of recruited macrophages (termed Mo-MFs) and a cluster of tissue resident KCs in HFD-fed mice (83). Another recent study, also using scRNA-seq, found distinct clusters of KCs in NASH livers. Two subsets of KC were identified by analyzing the levels of Cd5l expression and finally Trem2+ NASH-associated macrophages (NAMs) were identified by analyzing the levels of Trem2, Cd9, and Gpnmb expression in both mice and humans (84). These studies indicate that, like the AT, steatotic livers contain a heterogeneous pool of macrophages (84).
KC polarization may activate other immune cells in the liver that are relevant for obesity-related metabolic disease. One example is NK cells that normally participate in the defense against viral infections and tumor development, but that in the setting of obesity may promote fibrosis and NASH development (85). It has been shown that NK cell activity in the liver is stimulated by KCs (86). This is somewhat contrary to observations in AT, where inflammatory signals derived from NK-cells stimulate the pro-inflammatory polarization of ATMs (66). KCs have been shown to activate NK cells in the liver by generating IL-18 (87, 88), but also to suppress NK cell activity through the production of IL-10 that inhibits IFNγ expression and renders NK cells hyporesponsive in mice (86). Recent studies suggest that CD8+ T cells may also play important roles in liver immunopathology and NASH development (89, 90). However, there is still much to be discovered about the contribution of CD8+ T cells and their interaction with KCs under metabolic conditions leading to NASH. These findings indicate that KC polarization plays a major role in haptic inflammatory signaling during obesity and is thus involved in the development of NAFLD and NASH.
There has been little research on NK cells and inflammation in the liver during obesity. However, a recent study has reported that NK cells are responsible in inducing endoplasmic reticulum (ER) stress, and thus promote insulin resistance in obesity (91). In this study, HFD-fed mice display elevated production of proinflammatory cytokine osteopontin (OPN) in NK cells and this leads to ER stress and insulin resistance in the liver. At the AT level, OPN mediates macrophage infiltration, inflammation, and insulin resistance in mice during obesity. However, there is not yet a clear understanding of the role of liver NK cells derived OPN, and whether they are able to mediate inflammation in the liver by accumulating more macrophages.
Like in AT, the liver of obese (HFD) mice have been shown to be overpopulated with CD11c expressing DCs (69). This study also reported that increased liver DCs accumulated more macrophages, suggesting a macrophage-DC interaction and that DCs are involved in promoting liver inflammation during obesity (69). This is supported by human studies showing that DCs are critical in the development of liver fibrosis during obesity, thereby proving its involvement in liver inflammation (92, 93).
Overall, all these studies confirm that liver macrophages and their interaction with other liver immune cells are important mediators of metabolic dysfunction, and that targeting hepatic immune cells may represent a feasible strategy for the treatment of NASH and other metabolic disorders.
The skeletal muscle plays a significant role in metabolism. Normally, skeletal muscle is responsible for the majority of insulin-stimulated glucose disposal, suggesting that dysregulation of skeletal muscle metabolism can influence whole-body glucose homeostasis and insulin sensitivity (94). Indeed, obesity is associated with inflammation in myocytes/muscle cells, which may contribute to muscle inflammation. Nevertheless, changes in myocyte secretion of cytokines do not appear to constitute the major component of skeletal muscle inflammation in obesity. Instead, there is an increased accumulation of immune cells – in particular, macrophages and T cells - in the AT depots adjacent to the myocytes (95). This may explain why skeletal muscle immune cells in obesity resemble those identified in the AT, including the formation of CLS (96). AT resident immune cells may also help explain the fluctuations in the level of immune cell population and inflammation often observed in small-muscle biopsies, since this may be caused by variations in the ratio of muscle cells and AT (97). This may also explain why weight loss does not always seem to alter macrophage numbers in skeletal muscle (98).
Macrophages and T cells residing in skeletal muscles and their role in metabolic disorders have not been studied as extensively as in other tissues. One study showed that resveratrol alleviates obesity-induced skeletal muscle inflammation via decreasing M1 macrophage polarization and increasing the regulatory T cell population (99), suggesting that targeting these cell types may be of clinical importance. Studies exploring skeletal muscle macrophage activity are needed to explore the inflammatory pathway associated with obesity and comorbidities.
The pancreas is essential for the regulation of macronutrient digestion. Maintaining metabolism/energy homeostasis via its exocrine and endocrine components, the pancreas controls blood sugar levels through insulin secretion and production in response to glucose intake (100).. The pancreatic immune system is composed mainly of macrophages and T cells (101, 102). Generally, pancreatic macrophages are present from embryonic development and shown to play a role in pancreatic islet morphogenesis and remodeling during the fetal and neonatal stages, but they may also be involved in adult pancreatic regeneration (103). However, the phenotypic characteristics of pancreatic macrophages remain enigmatic. Unlike ATMs and liver macrophage categorization, no M2 vs. M1 polarization paradigm has been suggested for metabolic regulation and dysfunction in islet macrophages. Healthy islets often have macrophages expressing M1 markers (CD11c, MHC-II), producing IL-1β, TNF-α, and expressing the pro-inflammatory transcription factor interferon regulatory factor (IRF)-5 (104–106). However, they do not express M2 markers (CD206), as compared to exocrine pancreatic stromal macrophages (105). It has been reported that a number of factors or stimuli in obesity can change the phenotype and function of islet macrophages, resulting in both acute and chronic pancreatitis (AP and CP). For example, like other tissue resident macrophages, islet macrophages have been shown to be activated by excess free fatty acids (FFA) (107). Further, infiltration of macrophages results in inflammation and tissue destruction in obesity, shown in islets from obese rodent models (88) and in T2D patients (89). Interestingly, caloric restriction and bariatric surgery have shown to diminish obesity-induced proinflammatory macrophage infiltration in the pancreas in experimental models (108), supporting the idea that pancreatic macrophages play a crucial role during obesity and obesity-induced pancreatic steatosis. Pancreatic macrophages have also been shown to be involved in autoimmune T1D. T1D occurs when the pancreatic beta cells lose their endocrine function, and genetically susceptible individuals develop a chronic lack of insulin in the system. Here, macrophages have been shown to accumulate autoreactive T cells, which leads to inflammation of the islets (“insulitis”) or immune-mediated degeneration of insulin-producing pancreatic beta cells. Overall, all this evidence suggests that, pancreatic macrophages, and T cells are the central mediators of low-grade chronic inflammation associated with obesity and T2D (109).
Microbes that reside in the gut influence the host’s health directly through metabolic functions and immune system regulation. The innate immune system in the gut consists of a variety of cells, such as macrophages, ILC, DCs, and eosinophils, as well as epithelia. However, macrophages are the dominant innate immune cells in the gut, including a range of subtypes. At least five macrophage subpopulations were identified in mice and human intestine using flow cytometry (110). These subtypes have various functions, including maintenance of Tregs, crucial for responding to orally ingested antigens (111), and scavenging without causing inflammation. In obesity, it has been reported that there are significant changes in the gut-immune cell composition, leading to metabolic inflammation and gut dysbiosis via upregulation of inflammatory mediators like TLR4, TNF, and NFkB (112, 113). Gut dysbiosis enhances intestinal permeability by elevating the levels of gut bacterial lipopolysaccharides (LPS), which is then released into the systemic circulation to exacerbate low-grade inflammation and insulin resistance.
Among the gut-resident immune cells, increased levels of macrophages, a reduction of Tregs and increased Th1, CD8+, and γδ T cells have been reported in HFD-fed mice (114–116). In addition to these cells, ILC2s (produce proinflammatory IL-5 and IL-13) from the small intestine, have been also shown to be involved in the diet-induced obesity (117). A reduction in ILC3s (produce proinflammatory IL-17 and IL-22) levels in the lamina propria of HFD-fed mice has been shown to be correlated with reduced epithelial barrier integrity, increased serum LPS levels in gut, resulting in inflammation (115).
Overall, all these studies suggest that obesity-induced dysregulation of the gut metabolism may affect immune cell function, interaction, and composition, with some macrophage subtypes shown to play significant roles in obesity-induced metabolic diseases. These studies have revealed how microbiota affect systemic inflammation, suggesting that restoring gut microbiota could be a promising therapeutic target to treat obesity as an alternative to drugs and bariatric surgery.
Metabolic signaling between tissues occurs via neuronal connections and biochemical messengers in the circulation, such as adipokines, neuropeptides, chemokines, and cytokines. However, recent findings have highlighted that AT and other metabolic tissues also communicate via small extracellular vesicles (sEV) that are membrane particles derived from the cell (118). Because of their unique structure and physical characteristics, sEV have turned out to play a significant role in a variety of physiological processes, including immune responses, tissue repair and cell to cell communication (119). In addition, they are characterized by molecules biomarkers that are candidates for future diagnostic use in metabolic diseases like T2D (120).
sEV have a diameter of 30-200 nanometers and are constitutively produced from late endosomes and are secreted after fusion of multivesicular bodies (MVBs) with the plasma membrane. The cargo of sEV is composed of lipids, proteins, nucleic acids, organelles and other elements. There are several markers that confirm the exosome’s structural organization, including CD63, CD81, CD9, apoptosis-linked gene 2-interacting protein X (ALIX), heat-shock proteins (HSP60, HSP70, and HSP90) and tumor susceptibility gene 101 (TSG101). Commonly used non-specific sEV markers include glucose-regulated protein 94 (Grp94), calnexin (ER markers), GM130 (Golgi marker) and Cytochrome C (mitochondrial marker). These markers, which are not endogenously expressed on sEV, indicate that the sEV are released via an endocytic pathway and distinguish the sEV from necrotic bodies and other vesicles.
Recently, it has been reported that adipocyte-derived sEV exert a pivotal role in adipocyte-macrophage crosstalk during obesity (121). For example, adipocytes have been reported to release lipid-laden sEV (122) that express the lipid droplet-associated protein perilipin1 and carry phospholipids, neutral lipids, and free cholesterol that are taken up by ATMs to induce their differentiation. In another study, adipocytes from obese mice were found to release sEV with different mitochondrial composition than those derived from lean mice (123). This study also demonstrated that sEV cargo can control ATM activity and intra-organ transport of damaged mitochondria during metabolic stress.
It has been demonstrated that miRNA-containing sEV released by adipocytes influence ATMs phenotypes and function, as well as insulin sensitivity (124). For instance, it has been reported that rodents possess higher quantity of adipocyte-derived sEV-containing miR-34a during obesity (124, 125). This miRNA has been found to be interconnected with insulin resistance and metabolic inflammations by acting on ATMs. A study by Ying et al. (126) found that sEV enriched with miR-155 were mostly released from M1 macrophages isolated from obese AT. These miR-155 containing sEV have been shown to develop insulin resistance when injected into lean mice by directly targeting PPARg, leading to a reduced insulin expression and action. Besides acting as an inflammatory mediator, adipocyte-derived sEV may reduce inflammation, as demonstrated in adipocyte stem cells, and facilitate M2 macrophage polarization, thus offering a probable therapeutic intervention for obesity and metabolic disorders (127). Indeed, lean AT contained macrophages that emit sEV enriched in miR-690, were shown to improve systemic insulin sensitivity in obese mice (128).
These reports confirm that sEV are important in cellular crosstalk within the adipose tissue. Further, these data support that obesity-induced inflammation is governed by the sEV/ATM axis, which not only provides mechanistic details of AT inflammation in metabolic disease, but also opens for a new approach to treating such diseases. Moreover, recent evidence suggests that AT derived sEV can be released into the circulation and target other major metabolic organs, such as the liver, the skeletal muscle, and the heart (121, 129). Still, there are relatively few studies that have investigated whether sEV mediate the connections between AT and the brain and influence central inflammation (130, 131) (Figure 1). Currently, researchers are also studying the role of native (132), stem cell-derived (127, 133), and bioengineered sEVs (134–136)in other metabolic tissues, which may lead to improved understanding of whole-body metabolic control and therapeutic approaches to obesity.
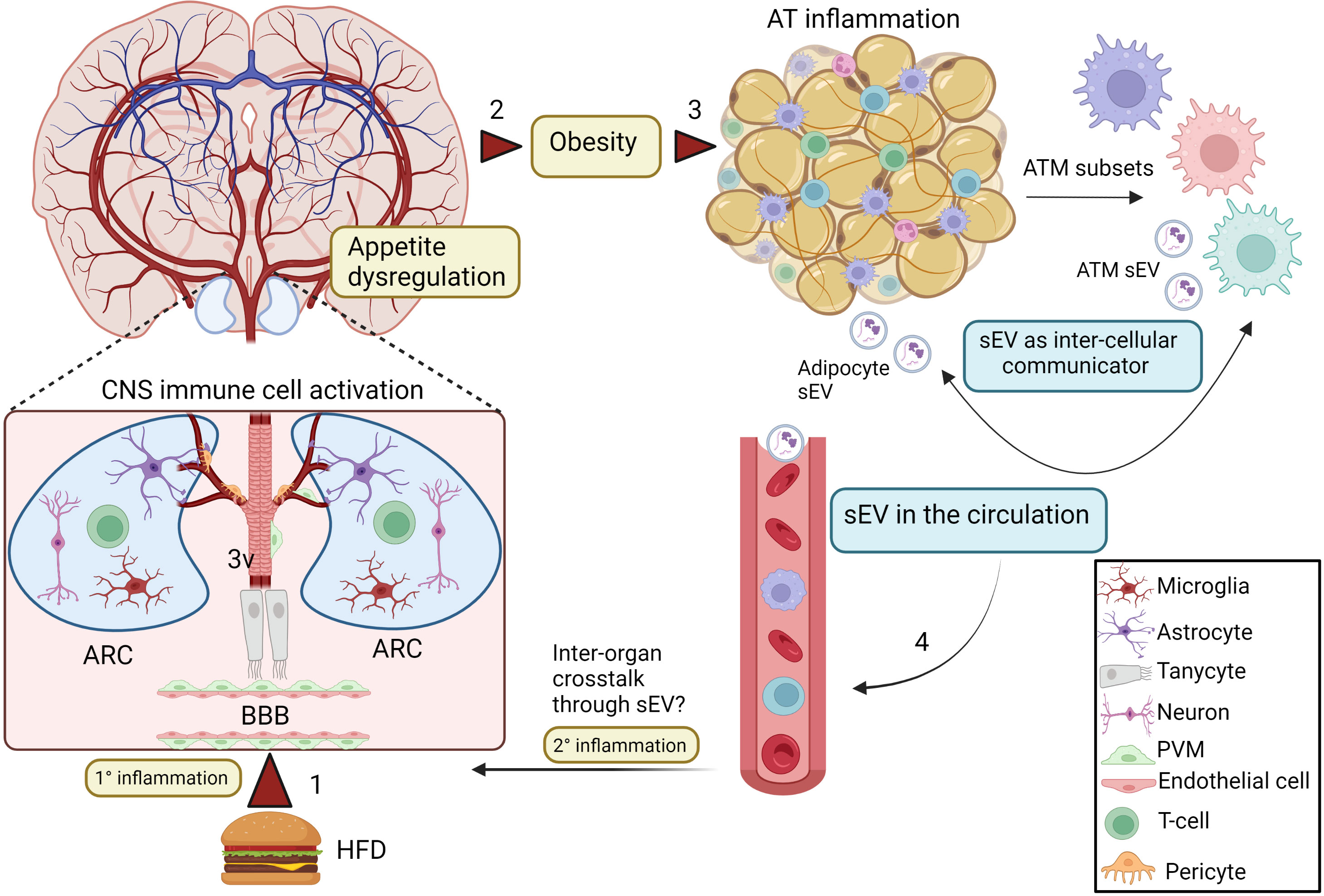
Figure 1 sEV as inflammatory mediators and cell to cell communicators in obesity. Excess nutrient intake may directly stimulate inflammation (1° inflammation) in the ARC (1), which leads to appetite dysregulation and obesity (2). This process involves activation of hypothalamic glial cells and neuronal interactions. In addition, tanycytes, that are special ependymal cells in the third ventricle of the brain (3V) are also involved in this inflammatory process in the CNS. Obesity also promotes inflammation and dysregulation of AT homeostasis (3), including increased macrophage infiltration and generation of various pro-inflammatory ATM subsets. During this altered microenvironment, both ATMs and adipocytes release sEV, which can mediate inter-cellular communication. AT-derived sEV can be released in the systemic circulation (4), but whether it can travel to the brain during obesity to participate in the secondary central inflammation from the circulation (2° inflammation) is still an open question. Created with BioRender.com. ARC, Arcuate nucleus; 3V, third ventricle; AT, adipose tissue; ATMs, adipose tissue macrophages; sEV, small extracellular vesicles; PVM, perivascular macrophage.
The autonomic nervous system (ANS) is a part of the peripheral nervous system that regulates involuntary physiological functions. ANS is further divided into the sympathetic, parasympathetic, and enteric nervous systems.
The innervation by sympathetic nervous system (SNS) of WAT was reported in the early 90s (137), and modulation mediated by SNS has been shown to influence weight changes in both rodents and humans through the release of norepinephrine (NE) (acting both as hormone and neurotransmitter) from sympathetic nerve endings (138–140). Macrophages have also been reported to release NE as well as expressing adrenergic NE receptors, and their activity can be affected by obesity-induced stress and the level of this neurotransmitter, suggesting a macrophage-SNS connection (141, 142). For instance, one study reported that anti-inflammatory ATMs release NE under cold stress (142), suggesting a role for these macrophages in the physiological response to reduced temperature. Other studies, however, reported that ATMs do not release NE (143, 144). A recent study shed new light on the connection between ATMs and NE: a cluster of sympathetic neuron–associated macrophages (SAMs) that reside in the AT was found to regulate obesity directly via the uptake and clearing of NE (145). This study demonstrated that SAMs act as a reservoir for NE and control NE uptake/elimination via the activation of the NE transporter solute carrier family 6 member 2 (SLC6A2) and the monoamine oxidase-A (MAO-A). Indeed, NE uptake by SAMs was blocked by genetic ablation of Slc6a2, which also led to increased browning of white fat, upregulation of thermogenesis, and weight loss in obese mice (145). Additionally, this study indicates that the molecular machinery encoding the NE clearance pathway is conserved across species, including humans, suggesting that obesity could be treated by targeting this pathway (145).
With regard to parasympathetic signaling, vagus nerve cholinergic signaling is an important major component. Vagus nerve activity regulates body weight and regulates inflammation in the GI tract and the liver. Ablation of vagus nerve signaling can promote hyperphagia and obesity, while vagus nerve stimulation promotes resolution of inflammation and obesity in mice through a mechanism that requires α7- acetylcholine (Ach) receptor (α7nAChR) subunit (146, 147). Intriguingly, it has been reported that macrophages, KCs, and DCs express 7nAChR, suggesting macrophage-vagus nerve crosstalk (148–151). For instance, peripheral activation of α7nAChR in mice has been shown to reduce NF-κB nuclear translocation and JAK2/STAT3 signaling, as well as lowering of proinflammatory cytokine production (152). This vagal nerve-macrophage signaling has also been reported to play a significant role in suppressing obesity-induced hepatic inflammation in mice models of NASH or NAFLD (153, 154). Similarly, in randomized clinical trials, modulation of this regulatory circuitry triggered by oral ingestion of acetylcholinesterase inhibitors has been shown to play a role in alleviating obesity-induced comorbidities in humans (155, 156), suggesting that modulating this circuitry may be an effective method of controlling obesity and obesity-related diseases. However, whether macrophages were involved in the manifestation of this effect remains unclear.
It is known that the enteric nervous system (ENS) controls intestinal motility, absorption of nutrients and whole body energy homeostasis (157). Interestingly, intestinal muscularis macrophages have been demonstrated to connect enteric neurons by releasing bone morphogenic protein 2 (BMP2, an osteogenic factor) which is required by enteric neurons for peristalsis to occur (158). As a result, the enteric neurons release the growth factor colony stimulating factor -1 (CSF-1) that supports the survival and function of enteric macrophages, confirming a symbiotic relationship. Also, it has been reported that the synapses between enteric neurons and muscularis macrophages express β2 adrenergic receptors (159), further underlining a relationship between macrophages and the peripheral nervous system. Thus, any changes in metabolism during obesity could negatively affect intestinal motility, nutrient absorption, and energy metabolism. For example, gastrointestinal alterations, mainly dysmotility, have been reported at the onset of obesity or in HFD animal models (160, 161). Further, in response to bacterial stimulation, NE was shown to be released by sympathetic neurons and to stimulate adrenergic signaling, which in turn activates a macrophage response in the intestine (159). This indicates that gut microbiota changes caused by obesity or diet changes may affect the relationship between enteric neurons and macrophages.
In view of the observations made above, macrophages can actively interact with peripheral nervous system in obesity, suggesting that modulating peripheral nervous system-macrophage axis may be useful in treating obesity in the future.
So far, we have focused on how activation of peripheral tissue immune cells, in particular macrophages, may impact organismal metabolic control. However, the obesity-induced central inflammation is more complex. Although it is becoming increasingly clear that microglia, which are specialized macrophages in the CNS, may contribute to obesity-associated metabolic changes (162), recent evidence also indicate that this central inflammation is influenced by the activity of other CNS cells, such as astrocytes and tanycytes, and their interactions with neurons (163, 164).
In particular, the microglial cell population located in the mediobasal hypothalamus (MBH), an anatomically distinct brain region that includes the arcuate nucleus (ARC) and median eminence, can be activated to a pro-inflammatory phenotype in response to nutrient excess (165, 166). For example, palmitic acid (PA)-induced neuro-inflammation has been shown to involve microglial activation (167, 168). Interestingly, the deactivation of FABP4 in microglia has been shown to decrease the PA induced inflammation by expressing more microglial UCP2 (169), indicating that overnutrition can trigger microglial activation, which may exacerbate hypothalamic inflammation.
It is well known that the hypothalamic neuronal population in the arcuate nucleus (ARC) controls metabolic feedback and regulates energy homeostasis in the body (170–172). Two functionally antagonistic neuronal populations contribute to this regulatory function; one cluster of neurons that expresses the orexigenic neuropeptides agouti-related peptide (AgRP) and neuropeptide Y (NPY), and a second cluster that expresses the anorexigenic peptides proopiomelanocortin (POMC) and cocaine and amphetamine regulated transcript (CART). Interestingly, the activity of these neurons has been reported to be altered by microglial activation during obesity (173). For instance, microglial activation increased TNFα secretion, leading to mitochondrial stress and altered firing rates in adjacent POMC neurons, thus contributing to the development of obesity in mice (174). On the contrary, increased exercise levels in mice led to reduced hypothalamic microglial activation and improvement of glucose tolerance, suggesting that microglial inflammation may be a potential therapeutic target to ameliorate metabolic dysfunction (175).
Microglia are derived from yolk sac and usually do not get replaced by circulating progenitors derived from the bone marrow (176). Nevertheless, during obesity, microglia-like myeloid cells from the circulation may infiltrate the MBH to be engaged in inflammatory crosstalk with the resident microglia (177). It may be difficult to separate the different myeloid cell populations in the brain, but cell surface markers to define distinct microglial phenotypes have been reported (178). Interestingly, it was shown that a positive energy balance in the form of high glucose treatment triggered the expression of many of the pro-inflammatory markers on microglial cells recognized from peripheral macrophages, including ionized calcium-binding adapter molecule 1 (Iba1), CD68, high mobility group box-1 protein (HMGB1), and CD11b (179), whereas the expression of anti-inflammatory microglial markers, such as CD206 and arginase-1, were decreased. Further, mimicking the environment of diabetes (characterized by hyperglycemia and hyperlipidemia) in cultured microglial cells, supplement with high glucose and free fatty acids (mixture of palmitic and oleic acids) was shown to promote Iba1 and CD11b expression, induce microglial morphological changes, raise oxidative stress levels, and stimulate IL-1 production and TNF production. This indicates that elevated levels of circulating nutrients may change microglial activity and phenotype to cause central inflammation, which may lead to dysregulation of feeding behavior in the in vivo setting.
Microglial activity can also be modulated by circulating nutrients, neuropeptides and hormones that are altered in obesity (180). The POMC-derived peptide alpha melanocyte stimulating hormone (α-MSH) and hormones like ghrelin and leptin have gained attention due to their ability to reduce or induce inflammation in the brain (181–184). Fatty acids and leptin are both elevated in the obese state and can promote cytokine secretion from microglia through activation of the TLR4/IKK/NF-κB pathway. Leptin has also been shown to induce NF-κB through the LepR/IRS1/AKT pathway (185, 186). In addition to hypothalamic inflammation, altered microglial homeostasis in the hippocampus and amygdala has been reported during HFD feeding in mice, although the activation of microglia in these areas mainly affects cognitive performance (187).
Other than parenchymal macrophages or microglia, non-parenchymal macrophages, including perivascular macrophages (PVMs), choroid plexus macrophages, and meningeal macrophages, inhabit the interface between the brain and the periphery where they function as “guardians”/”guards” against invading microorganisms and suppress harmful inflammation (150). PVMs are considered important defense immune cells, but with dual physiological functions (188). On the one hand, PVMs has been shown to mediate activation in the HPA axis by prostanoids (lipid mediators that stimulate the inflammatory response) (188, 189), whereas on the other hand PVMs have been shown to limit endothelial involvement in inflammatory processes, thereby inhibiting CNS responses to inflammatory insults. Nevertheless, during obesity PVMs seem to mostly be involved in pro-inflammatory activation. Elevated saturated fatty acid (SFA) levels and inflammatory molecules in the circulation have been shown to activate PVM signaling molecules, such as nitric oxide (NO), which may mediate increased vascular permeability and accumulation of lipids in the hypothalamus (3, 151). Vascular endothelial growth factor (VEGF) is another signaling molecule expressed by PVM. Elevated serum VEGF level is associated with obesity, poor glycemic control, and T2D (190, 191). Interestingly, the expression of VEGF in PVMs has been reported to be increased during the first four weeks of HFD feeding in mice, a period during which no significant increase in the expression of VEGF is evident in the spleen, liver, or AT (192). This suggests PVMs as a primary source of this pro-inflammatory growth factor. The PVMs may also have an effect on microglial cell function during obesity, and it was reported that PVMs, together with meningeal macrophages, may induce microgliosis in the hypothalamus (177). Indeed, the number of meningeal macrophages is elevated during HFD, indicating that these cells can trigger diet-induced microgliosis (177). The choroid plexus macrophages have also been shown to play critical roles in brain inflammation, but this seems to occur during neurodegenerative diseases such as Alzheimer’s disease, neurodevelopmental and psychiatric disorders (193–195). The role of these cells in inflammation induced by HFD and obesity is still elusive.
The astrocytes are another type of glial cell found in the CNS, and normally they are responsible for supporting differentiation and homeostasis in neurons, as well as influencing synaptic activity. However, during HFD induced obesity, astrocytes have been reported to interact with microglia and augment hypothalamic inflammations (196, 197) probably due to an increase in astrocytic NF-κB signaling (198), or due to increased chemokine signaling in microglia. For instance, conditioned medium of palmitate-treated reactive astrocytes has been shown to increase the expression of microglial chemokine CCL2, which promotes migration of microglia (196), supporting that interactions between astrocytes and microglia may contribute to obesity-induced inflammation in the CNS.
Taken together, these studies confirm that central immune cells, especially microglia, play a major role in obesity-induced central inflammation. Although the brain contains other immune cells such as T cells, B cells, and NK cells, their role and interaction with microglia and astroglia remain unexplored. Central inflammation affects appetite regulation, and thus peripheral metabolism through increased food intake and obesity Box 1. Further research on the interactions between CNS- and peripheral immune cells and between different types of brain-resident immune cells in a context of obesity is needed to better understand the how obesity-related central inflammation affect whole-body metabolism.
Tissue-resident, pro-inflammatory immune cells give rise to systemic inflammation and play a role in the development of metabolic dysregulation associated with obesity. The purpose of this review has been to shed light on peripheral and central immune cells in obesity and obesity-related disorders, with particular focus on macrophages. Macrophages are characterized by significant diversity and plasticity both within and between tissues. Their phenotypes are modulated by intricate communication with other immune cells as well as with parenchymal cells, such as adipocytes and neurons. During obesity, inflammation in peripheral tissues leads to release of pro-inflammatory cytokines into the circulation, from where they may cross the BBB to further activate brain resident macrophages and stimulate inflammation in the CNS. However, hormones, neuropeptides, and dietary fatty acids have also been shown to change macrophage function in the brain directly, independent of peripheral inflammation. Adding another layer of regulation are the sEVs, which are particularly interesting for their involvement in inter-organ communications. The adipocyte/ATM axis is important for obesity-induced inflammation and the involvement of sEV during this process seems to be of great importance. The contribution of sEVs to the CNS inflammation is also an interesting area of research that needs to be further pursued.
The development of anti-obesity therapies has proved to be extremely challenging from a methodological, technical, and societal perspective. Therefore, in-depth research into neglected areas can lead to new anti-obesity therapies that can benefit medical fields and society. For example, when it comes to the role of central macrophages in obesity and CNS inflammation, most studies have focused on microglia. However, other macrophage subsets may also be of relevance, such as those in the perivascular space, meninges, and choroid plexus that have so far been mostly neglected. Some studies have shown their connection to hypothalamic inflammation during diet-induced obesity, but their exact role in hypothalamic inflammation remains unclear, and more research is needed to understand how these non-parenchymal macrophages contribute to obesity-induced inflammation and if they affect regulation of food intake or energy expenditure.
Exploiting the properties of sEV is a promising strategy for targeted delivery of molecules to specific CNS cell types, potentially leading to more specific clinical effects and reduced side-effects. Efforts in this field should aim to understand both basic-mechanistic concepts as well as translate them into clinical practice. Due to the extremely complex nature of obesity-related inflammatory activation, involving numerous organs, cell types, signaling pathways and feedback mechanisms, the distance from “bench to bedside” appears long. However, in the face of the escalating global obesity epidemic, the pursuit of novel therapeutic agents, resting on sound fundamental research, should be continued.
BOX 1: Can peripheral and central inflammation be affected by obesity in a bidirectional manner?
Several studies have indicated that during obesity, peripheral tissues and the CNS are in constant communication with each other, and that this signaling includes inflammatory mediators (199–201). However, it has proven difficult to identify the origin of the inflammatory signal. On the one hand, inflamed AT has been shown to send pro-inflammatory, adipokine-mediated signals to the hypothalamus, which then controls energy homeostasis, feeding behavior, and metabolic rate. Also, circulating myeloid cells can be recruited to the CNS and subsequently be involved in hypothalamic inflammation that contribute to further food intake dysregulation (177). On the other hand, it was shown that inflammatory signaling in the hypothalamus occurred in response to nutrient overload before weight gain occurred and peripheral inflammation had taken place, suggesting that peripheral inflammation is rather a consequence of obesity than the source of an initial stimulating signal to the CNS (177, 202). Thus, the inflammation in the peripheral tissues that is relevant for systemic insulin sensitivity and metabolic dysfunction seems to be secondary to obesity, and that hypothalamic inflammation and dysregulation of food intake is the primary signal. Nevertheless, a positive feedback loop from the periphery to the CNS seems to be important in maintaining obesogenic signaling (Figure 2) (177, 203).
The ability of the peripheral tissues to communicate metabolic status to the CNS is of great importance. One example is the gut hormone glucagon-like peptide-1 (GLP-1) that is released into the bloodstream after feeding (204, 205). In general, it stimulates insulin secretion by potentiating the insulinotropic effects of glucose, and it also reduces appetite by acting in the hypothalamus (206). The beneficial effects of GLP-1 have been exploited pharmacologically, and there is currently a surge in GLP1R analogues and other gut hormone-based therapies that is becoming available for clinical use. Interestingly, gut hormones may also affect CNS inflammation; dysbiosis in the gut during obesity has been shown to cause the activation of hypothalamic glial cells (especially microglia) through abnormal GLP-1 receptor (GLP-1R) signaling (207). The GLP-1R signaling has also been established in Tanycytes (a special type of ependymoglial cell located near the third ventricle), and they have been reported to be involved in obesity-related CNS inflammation. Blocking GLP1R in tanycytes has been shown to prevent the transport of the anti-diabetic drug and GLP1R agonist liraglutide into the brain and its activation in the hypothalamic neurons, as well as its anti-obesity effects on food intake and body weight (208). This shows the potential of glial-like cell types in the hypothalamus to modulate GLP-1R signaling from the gut and targeting this signaling could develop new treatments for obesity. Weight loss surgery further emphasizes gut-macrophage-neuron crosstalk and the importance of bidirectional communication between the CNS and the periphery. For example, Roux-en-Y gastric bypass (RGYB) has been shown to alter the interaction between microglia and POMC neurons due to alteration in circulating humoral factors and leads to improvement in hypothalamic inflammation (irrespective of weight loss) and leptin sensitivity, suggesting a CNS-periphery crosstalk (209). In respect to humoral factors, the sEV could serve as promising candidates for a CNS-targeted strategy in the treatment of obesity. sEV can be loaded with specific cargos (e.g. lipids, proteins, DNA, and RNA) that can be transported in the circulation and targeted to specific cell types in the hypothalamus, which in turn can modulate CNS inflammation and ameliorate whole body metabolism. These possibilities emphasize the importance to understand more about how inflammation and immune cell are involved in the cross talk between the periphery and the brain, insight that will be of uttermost important in the quest for future therapies for obesity and metabolic disease.
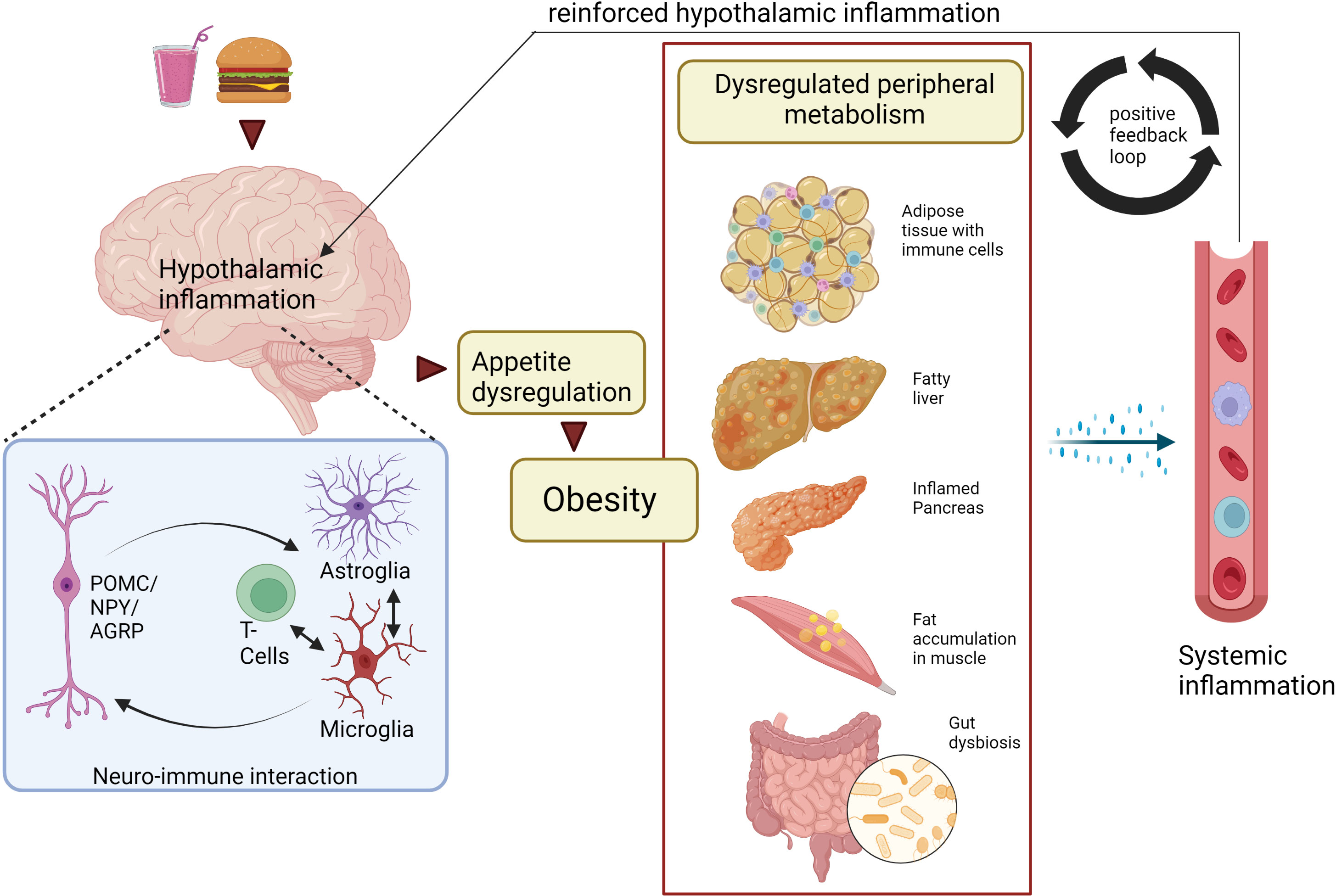
Figure 2 Peripheral and central macrophage interaction during obesity: a bidirectional mechanism? Neuro-immune interactions trigger hypothalamic inflammation and obesity. As a result, tissue-resident macrophages and other immune cells in AT, liver, pancreas, muscle, gut are activated, thereby dysregulating metabolism within these peripheral tissues. Furthermore, the tissue-resident immune cells release proinflammatory cytokines and chemokines into the circulation, some of which can enter the brain and exacerbate hypothalamic inflammation though the activation of glial cells (especially microglia) and tissue resident T-cells. Ultimately, this will result in sustained CNS inflammation and obesity, suggesting a bidirectional interaction between CNS and peripheral inflammation. Created with BioRender.com. CNS, Central Nervous system; AT, adipose tissue; POMC, Pro-opiomelanocortin neurons; NPY, Neuropeptide Y neurons; AgRP, Agouti-related protein.
SM drafted the article which was critically reviewed by all authors during the development of the article. All authors listed have made a substantial, direct and intellectual contribution to the work, and have approved it for publication.
This work was funded by EuroNanoMed III (Grant No. EURONANOMED2019050/ENAMEP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol (2018) 15(1):11–20. doi: 10.1038/nrgastro.2017.109
2. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/JCI19246
3. Lee CH, Kim HJ, Lee Y-S, Kang GM, Lim HS, Lee S-h, et al. Hypothalamic macrophage inducible nitric oxide synthase mediates obesity-associated hypothalamic inflammation. Cell Rep (2018) 25(4):934–46.e5. doi: 10.1016/j.celrep.2018.09.070
4. Forny-Germano L, De Felice FG. Vieira MNdN. Role Leptin Adiponectin Obesity-Associated Cogn Decline Alzheimer’s Disease. Front Neurosci (2019) 12:1027. doi: 10.3389/fnins.2018.01027
5. Martin-Jiménez CA, Gaitán-Vaca DM, Echeverria V, González J, Barreto GE. Relationship between obesity, alzheimer’s disease, and parkinson’s disease: an astrocentric view. Mol Neurobiol (2016) 54(9):7096–115. doi: 10.1007/s12035-016-0193-8
6. Mackay LK, Kallies A. Transcriptional regulation of tissue-resident lymphocytes. Trends Immunol (2017) 38(2):94–103. doi: 10.1016/j.it.2016.11.004
7. Ieronymaki E, Daskalaki MG, Lyroni K, Tsatsanis C. Insulin signaling and insulin resistance facilitate trained immunity in macrophages through metabolic and epigenetic changes. Front Immunol (2019) 10:1330. doi: 10.3389/fimmu.2019.01330
8. Bekkering S, Saner C, Riksen NP, Netea MG, Sabin MA, Saffery R, et al. Trained immunity: linking obesity and cardiovascular disease across the life-course? Trends Endocrinol Metab (2020) 31(5):378–89. doi: 10.1016/j.tem.2020.01.008
9. Chavakis T, Alexaki VI, Ferrante AW Jr.. Macrophage function in adipose tissue homeostasis and metabolic inflammation. Nat Immunol (2023) 24(5):757–66. doi: 10.1038/s41590-023-01479-0
10. Cottam MA, Caslin HL, Winn NC, Hasty AH. Multiomics reveals persistence of obesity-associated immune cell phenotypes in adipose tissue during weight loss and weight regain in mice. Nat Commun (2022) 13(1):2950. doi: 10.1038/s41467-022-30646-4
11. Gianfrancesco MA, Dehairs J, L'Homme L, Herinckx G, Esser N, Jansen O, et al. Saturated fatty acids induce NLRP3 activation in human macrophages through k+ efflux resulting from phospholipid saturation and Na, K-ATPase disruption. Biochim Biophys Acta (BBA) - Mol Cell Biol Lipids (2019) 1864(7):1017–30. doi: 10.1016/j.bbalip.2019.04.001
12. SantaCruz-Calvo S, Bharath L, Pugh G, SantaCruz-Calvo L, Lenin RR, Lutshumba J, et al. Adaptive immune cells shape obesity-associated type 2 diabetes mellitus and less prominent comorbidities. Nat Rev Endocrinol (2021) 18(1):23–42. doi: 10.1038/s41574-021-00575-1
13. Kapellos TS, Bonaguro L, Gemünd I, Reusch N, Saglam A, Hinkley ER, et al. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front Immunol (2019) 10:2035. doi: 10.3389/fimmu.2019.02035
14. Liu R, Nikolajczyk BS. Tissue immune cells fuel obesity-associated inflammation in adipose tissue and beyond. Front Immunol (2019) 10:1587. doi: 10.3389/fimmu.2019.01587
15. Ni Y, Ni L, Zhuge F, Xu L, Fu Z, Ota T. Adipose tissue macrophage phenotypes and characteristics: the key to insulin resistance in obesity and metabolic disorders. Obesity (2020) 28(2):225–34. doi: 10.1002/oby.22674
16. Chen K-HE, Lainez NM, Coss D. Sex differences in macrophage responses to obesity-mediated changes determine migratory and inflammatory traits. J Immunol (2021) 206(1):141–53. doi: 10.4049/jimmunol.2000490
17. Lee CH, Lam KSL. Obesity-induced insulin resistance and macrophage infiltration of the adipose tissue: a vicious cycle. J Diabetes Invest (2019) 10(1):29–31. doi: 10.1111/jdi.12918
18. Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab (2014) 20(4):614–25. doi: 10.1016/j.cmet.2014.08.010
19. Orliaguet L, Dalmas E, Drareni K, Venteclef N, Alzaid F. Mechanisms of macrophage polarization in insulin signaling and sensitivity. Front Endocrinol (2020) 11:62. doi: 10.3389/fendo.2020.00062
20. Morris DL, Singer K, Lumeng CN. Adipose tissue macrophages: phenotypic plasticity and diversity in lean and obese states. Curr Opni Clin Nutr Metab Care (2011) 14(4):341–6. doi: 10.1097/MCO.0b013e328347970b
21. Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes (2008) 57(12):3239–46. doi: 10.2337/db08-0872
22. Li P, Ma C, Li J, You S, Dang L, Wu J, et al. Proteomic characterization of four subtypes of M2 macrophages derived from human THP-1 cells. J Zhejiang University-SCIENCE B (2022) 23(5):407–22. doi: 10.1631/jzus.B2100930
23. Maniyadath B, Zhang Q, Gupta RK, Mandrup S. Adipose tissue at single-cell resolution. Cell Metab (2023) 35(3):386–413. doi: 10.1016/j.cmet.2023.02.002
24. Russo L, Lumeng CN. Properties and functions of adipose tissue macrophages in obesity. Immunology (2018) 155(4):407–17. doi: 10.1111/imm.13002
25. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med (2009) 15(8):914–20. doi: 10.1038/nm.1964
26. Morris DL, Cho KW, DelProposto JL, Oatmen KE, Geletka LM, Martinez-Santibanez G, et al. Adipose tissue macrophages function as antigen-presenting cells and regulate adipose tissue cd4+ t cells in mice. Diabetes (2013) 62(8):2762–72. doi: 10.2337/db12-1404
27. Lee B-C, Kim M-S, Pae M, Yamamoto Y, Eberlé D, Shimada T, et al. Adipose natural killer cells regulate adipose tissue macrophages to promote insulin resistance in obesity. Cell Metab (2016) 23(4):685–98. doi: 10.1016/j.cmet.2016.03.002
28. Fernø J, Strand K, Mellgren G, Stiglund N, Björkström NK. Natural killer cells as sensors of adipose tissue stress. Trends Endocrinol Metab (2020) 31(1):3–12. doi: 10.1016/j.tem.2019.08.011
29. Strand K, Stiglund N, Haugstøyl ME, Kamyab Z, Langhelle V, Lawrence-Archer L, et al. Subtype-specific surface proteins on adipose tissue macrophages and their association to obesity-induced insulin resistance. Front Endocrinol (2022) 13:856530. doi: 10.3389/fendo.2022.856530
30. Muir LA, Neeley CK, Meyer KA, Baker NA, Brosius AM, Washabaugh AR, et al. Adipose tissue fibrosis, hypertrophy, and hyperplasia: correlations with diabetes in human obesity. Obesity (2016) 24(3):597–605. doi: 10.1002/oby.21377
31. Huang Y, Jin C, Zheng Y, Li X, Zhang S, Zhang Y, et al. Knockdown of lncRNA MIR31HG inhibits adipocyte differentiation of human adipose-derived stem cells via histone modification of FABP4. Sci Rep (2017) 7(1):8080. doi: 10.1038/s41598-017-08131-6
32. Steen KA, Xu H, Bernlohr DA. FABP4/aP2 regulates macrophage redox signaling and inflammasome activation via control of UCP2. Mol Cell Biol (2023) 37(2):e00282–16. doi: 10.1128/mcb.00282-16
33. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature (2017) 542(7640):177–85. doi: 10.1038/nature21363
34. Lee Yun S, Kim J-w, Osborne O, Oh Da Y, Sasik R, Schenk S, et al. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell (2014) 157(6):1339–52. doi: 10.1016/j.cell.2014.05.012
35. Schöttl T, Kappler L, Fromme T, Klingenspor M. Limited OXPHOS capacity in white adipocytes is a hallmark of obesity in laboratory mice irrespective of the glucose tolerance status. Mol Metab (2015) 4(9):631–42. doi: 10.1016/j.molmet.2015.07.001
36. Sadiku P, Walmsley SR. Hypoxia and the regulation of myeloid cell metabolic imprinting: consequences for the inflammatory response. EMBO Rep (2019) 20(5):e47388. doi: 10.15252/embr.201847388
37. Castoldi A, Naffah de Souza C, Câmara NOS, Moraes-Vieira PM. The macrophage switch in obesity development. Front Immunol (2016) 6:637. doi: 10.3389/fimmu.2015.00637
38. Turner L, Scotton C, Negus R, Balkwill F. Hypoxia inhibits macrophage migration. Eur J Immunol (1999) 29(7):2280–7. doi: 10.1002/(sici)1521-4141(199907)29:07<2280::Aid-immu2280>3.0.Co;2-c
39. McKernan K, Varghese M, Patel R, Singer K. Role of TLR4 in the induction of inflammatory changes in adipocytes and macrophages. Adipocyte (2020) 9(1):212–22. doi: 10.1080/21623945.2020.1760674
40. Park S-H, Liu Z, Sui Y, Helsley RN, Zhu B, Powell DK, et al. IKKβ is essential for adipocyte survival and adaptive adipose remodeling in obesity. Diabetes (2016) 65(6):1616–29. doi: 10.2337/db15-1156
41. Hill AA, Anderson-Baucum EK, Kennedy AJ, Webb CD, Yull FE, Hasty AH. Activation of NF-kappaB drives the enhanced survival of adipose tissue macrophages in an obesogenic environment. Mol Metab (2015) 4(10):665–77. doi: 10.1016/j.molmet.2015.07.005
42. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25(12):677–86. doi: 10.1016/j.it.2004.09.015
43. Geng J, Zhang X, Prabhu S, Shahoei SH, Nelson ER, Swanson KS, et al. 3D microscopy and deep learning reveal the heterogeneity of crown-like structure microenvironments in intact adipose tissue. Sci Adv (2021) 7(8):eabe2480. doi: 10.1126/sciadv.abe2480
44. Lynch L, Nowak M, Varghese B, Clark J, Hogan Andrew E, Toxavidis V, et al. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity (2012) 37(3):574–87. doi: 10.1016/j.immuni.2012.06.016
45. Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci U.S.A. (2018) 115(22):E5096–E105. doi: 10.1073/pnas.1802611115
46. Wentworth JM, Naselli G, Brown WA, Doyle L, Phipson B, Smyth GK, et al. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes (2010) 59(7):1648–56. doi: 10.2337/db09-0287
47. Bigornia SJ, Farb MG, Mott MM, Hess DT, Carmine B, Fiscale A, et al. Relation of depot-specific adipose inflammation to insulin resistance in human obesity. Nutr Diabetes (2012) 2(3):e30–e. doi: 10.1038/nutd.2012.3
48. Landgraf K, Rockstroh D, Wagner IV, Weise S, Tauscher R, Schwartze JT, et al. Evidence of early alterations in adipose tissue biology and function and its association with obesity-related inflammation and insulin resistance in children. Diabetes (2015) 64(4):1249–61. doi: 10.2337/db14-0744
49. Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW, DeFuria J, Jick Z, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes (2007) 56(12):2910–8. doi: 10.2337/db07-0767
50. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell (2019) 178(3):686–98.e14. doi: 10.1016/j.cell.2019.05.054
51. Coats BR, Schoenfelt KQ, Barbosa-Lorenzi VC, Peris E, Cui C, Hoffman A, et al. Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet-induced obesity. Cell Rep (2017) 20(13):3149–61. doi: 10.1016/j.celrep.2017.08.096
52. Ameka MK, Beavers WN, Shaver CM, Ware LB, Kerchberger VE, Schoenfelt KQ, et al. An iron refractory phenotype in obese adipose tissue macrophages leads to adipocyte iron overload. Int J Mol Sci (2022) 23(13):7417. doi: 10.3390/ijms23137417
53. Marques L, Negre-Salvayre A, Costa L, Canonne-Hergaux F. Iron gene expression profile in atherogenic Mox macrophages. Biochim Biophys Acta (BBA) - Mol Basis Dis (2016) 1862(6):1137–46. doi: 10.1016/j.bbadis.2016.03.004
54. Nance SA, Muir L, Lumeng C. Adipose tissue macrophages: regulators of adipose tissue immunometabolism during obesity. Mol Metab (2022) 66:101642. doi: 10.1016/j.molmet.2022.101642
55. Cipolletta D. Adipose tissue-resident regulatory T cells: phenotypic specialization, functions and therapeutic potential. Immunology (2014) 142(4):517–25. doi: 10.1111/imm.12262
56. Haugstøyl ME, Cornillet M, Strand K, Stiglund N, Sun D, Lawrence-Archer L, et al. Distinct T cell subsets in adipose tissue are associated with obesity. Eur J Immunol (2022) 53(2):e2249990. doi: 10.1002/eji.202249990
57. Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KSL, et al. Activation of natural killer t cells promotes m2 macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (il-4)/stat6 protein signaling axis in obesity. J Biolo Chem (2012) 287(17):13561–71. doi: 10.1074/jbc.M112.350066
58. Ji Y, Sun S, Xia S, Yang L, Li X, Qi L. Short term high fat diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer t cells and interleukin-4. J Biolo Chem (2012) 287(29):24378–86. doi: 10.1074/jbc.M112.371807
59. Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of treg cells and macrophages in adipose tissue. Nat Immunol (2014) 16(1):85–95. doi: 10.1038/ni.3047
60. Goldberg EL, Shchukina I, Asher JL, Sidorov S, Artyomov MN, Dixit VD. Ketogenesis activates metabolically protective γδ T cells in visceral adipose tissue. Nat Metab (2020) 2(1):50–61. doi: 10.1038/s42255-019-0160-6
61. Mehta P, Nuotio-Antar AM, Smith CW. γδ T cells promote inflammation and insulin resistance during high fat diet-induced obesity in mice. J Leukocyte Biol (2015) 97(1):121–34. doi: 10.1189/jlb.3A0414-211RR
62. Pang DJ, Neves JF, Sumaria N, Pennington DJ. Understanding the complexity of γδ T-cell subsets in mouse and human. Immunology (2012) 136(3):283–90. doi: 10.1111/j.1365-2567.2012.03582.x
63. Correia DV, Lopes AC, Silva-Santos B. Tumor cell recognition by γδ T lymphocytes. OncoImmunology (2014) 2(1):e22892. doi: 10.4161/onci.22892
64. Le Menn G, Sibille B, Murdaca J, Rousseau A-S, Squillace R, Vergoni B, et al. Decrease in αβ/γδ T-cell ratio is accompanied by a reduction in high-fat diet-induced weight gain, insulin resistance, and inflammation. FASEB J (2018) 33(2):2553–62. doi: 10.1096/fj.201800696RR
65. Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold C, et al. Adipose type one innate lymphoid cells regulate macrophage homeostasis through targeted cytotoxicity. Immunity (2017) 46(2):273–86. doi: 10.1016/j.immuni.2017.01.008
66. Wensveen FM, Jelencic V, Valentic S, Sestan M, Wensveen TT, Theurich S, et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol (2015) 16(4):376–85. doi: 10.1038/ni.3120
67. O'Rourke RW, Gaston GD, Meyer KA, White AE, Marks DL. Adipose tissue NK cells manifest an activated phenotype in human obesity. Metabolism (2013) 62(11):1557–61. doi: 10.1016/j.metabol.2013.07.011
68. Haugstoyl ME, Cornillet M, Strand K, Stiglund N, Sun D, Lawrence-Archer L, et al. Phenotypic diversity of human adipose tissue-resident NK cells in obesity. Front Immunol (2023) 14:1130370. doi: 10.3389/fimmu.2023.1130370
69. Stefanovic-Racic M, Yang X, Turner MS, Mantell BS, Stolz DB, Sumpter TL, et al. Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes (2012) 61(9):2330–9. doi: 10.2337/db11-1523
70. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science (2011) 332(6026):243–7. doi: 10.1126/science.1201475
71. de Oliveira MC, Silveira ALM, de Oliveira ACC, Lana JP, Costa KA, Vieira ELM, et al. Eosinophils protect from metabolic alterations triggered by obesity. Metabolism (2023), 155613. doi: 10.1016/j.metabol.2023.155613
72. Rui L. Energy metabolism in the liver. Compr Physiol (2014) 4(1):177–97. doi: 10.1002/cphy.c130024
74. Riazi K, Azhari H, Charette JH, Underwood FE, King JA, Afshar EE, et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol (2022) 7(9):851–61. doi: 10.1016/S2468-1253(22)00165-0
75. Rodríguez-Rodríguez E, López-Sobaler AM, Ortega RM, Delgado-Losada ML, López-Parra AM, Aparicio A. Association between neutrophil-to-Lymphocyte ratio with abdominal obesity and healthy eating index in a representative older Spanish population. Nutrients (2020) 12(3):855. doi: 10.3390/nu12030855
76. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest (2017) 127(1):55–64. doi: 10.1172/jci88881
77. Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat Commun (2019) 10(1):1076. doi: 10.1038/s41467-019-09046-8
78. Remmerie A, Martens L, Scott CL. Macrophage subsets in obesity, aligning the liver and adipose tissue. Fronti Endocrinol (2020) 11:259. doi: 10.3389/fendo.2020.00259
79. MacPhee PJ, Schmidt EE, Groom AC. Evidence for kupffer cell migration along liver sinusoids, from high-resolution in vivo microscopy. Am J Physiol Gastroint Liver Physiol (1992) 263(1):G17–23. doi: 10.1152/ajpgi.1992.263.1.G17
80. Li W, Chang N, Li L. Heterogeneity and function of kupffer cells in liver injury. Front Immunol (2022) 13:940867. doi: 10.3389/fimmu.2022.940867
81. Ganz M, Szabo G. Immune and inflammatory pathways in NASH. Hepatol Int (2013) 7(S2):771–81. doi: 10.1007/s12072-013-9468-6
82. Santos JPMd, Maio MCd, Lemes MA, Laurindo LF, Haber JFdS, Bechara MD, et al. Non-alcoholic steatohepatitis (NASH) and organokines: what is now and what will be in the future. Int J MolSci (2022) 23(1):498. doi: 10.3390/ijms23010498
83. Krenkel O, Hundertmark J, Abdallah AT, Kohlhepp M, Puengel T, Roth T, et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut (2020) 69(3):551–63. doi: 10.1136/gutjnl-2019-318382
84. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and nash liver revealed by single-cell secretome gene analysis. Mol Cell (2019) 75(3):644–60.e5. doi: 10.1016/j.molcel.2019.07.028
85. Cepero-Donates Y, Lacraz G, Ghobadi F, Rakotoarivelo V, Orkhis S, Mayhue M, et al. Interleukin-15-mediated inflammation promotes non-alcoholic fatty liver disease. Cytokine (2016) 82:102–11. doi: 10.1016/j.cyto.2016.01.020
86. Fu HY, Bao WM, Yang CX, Lai WJ, Xu JM, Yu HY, et al. Kupffer cells regulate natural killer cells via the NK group 2, member d (nkg2d)/retinoic acid early inducible-1 (RAE-1) interaction and cytokines in a primary biliary cholangitis mouse model. Med Sci Monit (2020) 26:e923726. doi: 10.12659/MSM.923726
87. Streetz K, Fregien B, Plümpe Jr, Körber K, Kubicka S, Sass G, et al. Dissection of the intracellular pathways in hepatocytes suggests a role for jun kinase and ifn regulatory factor-1 in con a-induced liver failure. J Immunol (2001) 167(1):514–23. doi: 10.4049/jimmunol.167.1.514
88. Tsutsui H, Nakanishi K, Matsui K, Higashino K, Okamura H, Miyazawa Y, et al. IFN-gamma-inducing factor up-regulates fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J Immunol (1996) 157(9):3967–73.
89. Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M, et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature (2021) 592(7854):444–9. doi: 10.1038/s41586-021-03233-8
90. Haas JT, Vonghia L, Mogilenko DA, Verrijken A, Molendi-Coste O, Fleury S, et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat Metab (2019) 1(6):604–14. doi: 10.1038/s42255-019-0076-1
91. Wu J, Wu D, Zhang L, Lin C, Liao J, Xie R, et al. NK cells induce hepatic ER stress to promote insulin resistance in obesity through osteopontin production. J Leukocyte Biol (2020) 107(4):589–96. doi: 10.1002/jlb.3ma1119-173r
92. Barranco-Fragoso B, Pal SC, Diaz-Orozco LE, Dorantes-Heredia R, Qi X, Mendez-Sanchez N. Identification of hepatic dendritic cells in liver biopsies showing steatosis in patients with metabolic dysfunction-associated fatty liver disease (MAFLD) associated with obesity. Med Sci Monit (2022) 28:e937528. doi: 10.12659/MSM.937528
93. Ramírez-Pérez OL, Barranco-Fragoso B, Pichardo-Bahena R, Cruz-Ramón VC, Chinchilla-López P, Canizales-Quinteros S, et al. The role of dendritic cells in different stages of non-alcoholic fatty liver disease. J Hepatol (2018) 68:S363–S4. doi: 10.1016/s0168-8278(18)30953-x
94. Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest (2017) 127(1):43–54. doi: 10.1172/JCI88880
95. Fink LN, Costford SR, Lee YS, Jensen TE, Bilan PJ, Oberbach A, et al. Pro-inflammatory macrophages increase in skeletal muscle of high fat-fed mice and correlate with metabolic risk markers in humans. Obes (Silver Spring) (2014) 22(3):747–57. doi: 10.1002/oby.20615
96. Khan IM, Perrard XY, Brunner G, Lui H, Sparks LM, Smith SR, et al. Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int J Obes (Lond) (2015) 39(11):1607–18. doi: 10.1038/ijo.2015.104
97. Tam CS, Sparks LM, Johannsen DL, Covington JD, Church TS, Ravussin E. Low macrophage accumulation in skeletal muscle of obese type 2 diabetics and elderly subjects. Obes (Silver Spring) (2012) 20(7):1530–3. doi: 10.1038/oby.2012.24
98. Bruun JM, Helge JW, Richelsen B, Stallknecht B. Diet and exercise reduce low-grade inflammation and macrophage infiltration in adipose tissue but not in skeletal muscle in severely obese subjects. Am J Physiol Endocrinol Metab (2006) 290(5):E961–7. doi: 10.1152/ajpendo.00506.2005
99. Shabani M, Sadeghi A, Hosseini H, Teimouri M, Babaei Khorzoughi R, Pasalar P, et al. Resveratrol alleviates obesity-induced skeletal muscle inflammation via decreasing M1 macrophage polarization and increasing the regulatory T cell population. Sci Rep (2020) 10(1):3791. doi: 10.1038/s41598-020-60185-1
100. Campbell JE, Newgard CB. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat Rev Mol Cell Biol (2021) 22(2):142–58. doi: 10.1038/s41580-020-00317-7
101. Saffern M, Merad M. The two faces of pancreas tissue-resident macrophages. Nat Rev Immunol (2022) 22(6):336–. doi: 10.1038/s41577-022-00728-x
102. Weitz JR, Makhmutova M, Almaça J, Stertmann J, Aamodt K, Brissova M, et al. Mouse pancreatic islet macrophages use locally released ATP to monitor beta cell activity. Diabetologia (2017) 61(1):182–92. doi: 10.1007/s00125-017-4416-y
103. Criscimanna A, Coudriet GM, Gittes GK, Piganelli JD, Esni F. Activated macrophages create lineage-specific microenvironments for pancreatic acinar- and β-cell regeneration in mice. Gastroenterology (2014) 147(5):1106–18.e11. doi: 10.1053/j.gastro.2014.08.008
104. Dalmas E, Lehmann FM, Dror E, Wueest S, Thienel C, Borsigova M, et al. Interleukin-33-activated islet-resident innate lymphoid cells promote insulin secretion through myeloid cell retinoic acid production. Immunity (2017) 47(5):928–42.e7. doi: 10.1016/j.immuni.2017.10.015
105. Calderon B, Carrero JA, Ferris ST, Sojka DK, Moore L, Epelman S, et al. The pancreas anatomy conditions the origin and properties of resident macrophages. J Exp Med (2015) 212(10):1497–512. doi: 10.1084/jem.20150496
106. Carrero JA, McCarthy DP, Ferris ST, Wan X, Hu H, Zinselmeyer BH, et al. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc Natl Acad Sci (2017) 114(48):E10418–27. doi: 10.1073/pnas.1713543114
107. Xia W, Lu Z, Chen W, Zhou J, Zhao Y. Excess fatty acids induce pancreatic acinar cell pyroptosis through macrophage M1 polarization. BMC Gastroenterol (2022) 22(1):72. doi: 10.1186/s12876-022-02146-8
108. Otero A, Becerril S, Martin M, Cienfuegos JA, Valenti V, Moncada R, et al. Effect of guanylin peptides on pancreas steatosis and function in experimental diet-induced obesity and after bariatric surgery. Front Endocrinol (Lausanne) (2023) 14:1185456. doi: 10.3389/fendo.2023.1185456
109. Cucak H, Grunnet LG, Rosendahl A. Accumulation of M1-like macrophages in type 2 diabetic islets is followed by a systemic shift in macrophage polarization. J Leukocyte Biol (2014) 95(1):149–60. doi: 10.1189/jlb.0213075
110. Rohm TV, Fuchs R, Müller RL, Keller L, Baumann Z, Bosch AJT, et al. Obesity in humans is characterized by gut inflammation as shown by pro-inflammatory intestinal macrophage accumulation. Front Immunol (2021) 12:668654. doi: 10.3389/fimmu.2021.668654
111. Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory t cells in the lamina propria. Immunity (2011) 34(2):237–46. doi: 10.1016/j.immuni.2011.01.016
112. Araújo JR, Tomas J, Brenner C, Sansonetti PJ. Impact of high-fat diet on the intestinal microbiota and small intestinal physiology before and after the onset of obesity. Biochimie (2017) 141:97–106. doi: 10.1016/j.biochi.2017.05.019
113. de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastroint Liver Physiol (2010) 299(2):G440–G8. doi: 10.1152/ajpgi.00098.2010
114. Garidou L, Pomié C, Klopp P, Waget A, Charpentier J, Aloulou M, et al. The gut microbiota regulates intestinal CD4 t cells expressing rorγt and controls metabolic disease. Cell Metab (2015) 22(1):100–12. doi: 10.1016/j.cmet.2015.06.001
115. Luck H, Tsai S, Chung J, Clemente-Casares X, Ghazarian M, Revelo Xavier S, et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab (2015) 21(4):527–42. doi: 10.1016/j.cmet.2015.03.001
116. Hong C-P, Park A, Yang B-G, Yun CH, Kwak M-J, Lee G-W, et al. Gut-specific delivery of t-helper 17 cells reduces obesity and insulin resistance in mice. Gastroenterology (2017) 152(8):1998–2010. doi: 10.1053/j.gastro.2017.02.016
117. Sasaki T, Moro K, Kubota T, Kubota N, Kato T, Ohno H, et al. Innate lymphoid cells in the induction of obesity. Cell Rep (2019) 28(1):202–17.e7. doi: 10.1016/j.celrep.2019.06.016
118. Margolis L, Sadovsky Y. The biology of extracellular vesicles: the known unknowns. PloS Biol (2019) 17(7):e3000363. doi: 10.1371/journal.pbio.3000363
119. Stahl P, Raposo G, Samuelson I, Vidal-Puig Antonio J. Fed-EXosome: extracellular vesicles and cell–cell communication in metabolic regulation. Essays Biochem (2018) 62(2):165–75. doi: 10.1042/ebc20170087
120. Jafari N, Llevenes P, Denis GV. Exosomes as novel biomarkers in metabolic disease and obesity-related cancers. Nat Rev Endocrinol (2022) 18(6):327–8. doi: 10.1038/s41574-022-00666-7
121. Crewe C, Funcke J-B, Li S, Joffin N, Gliniak CM, Ghaben AL, et al. Extracellular vesicle-based interorgan transport of mitochondria from energetically stressed adipocytes. Cell Metab (2021) 33(9):1853–68.e11. doi: 10.1016/j.cmet.2021.08.002
122. Flaherty SE, Grijalva A, Xu X, Ables E, Nomani A, Ferrante AW. A lipase-independent pathway of lipid release and immune modulation by adipocytes. . Sci (2019) 363(6430):989–93. doi: 10.1126/science.aaw2586
123. Greenhill C. Mitochondria transported from adipocytes in extracellular vesicles. Nat Rev Endocrinol (2021) 17(11):637. doi: 10.1038/s41574-021-00563-5
124. Pan Y, Hui X, Hoo RLC, Ye D, Chan CYC, Feng T, et al. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J Clin Invest (2019) 129(2):834–49. doi: 10.1172/jci123069
125. Zhou Y, Tan C. miRNAs in adipocyte-derived extracellular vesicles: multiple roles in development of obesity-associated disease. Front Mol Biosci (2020) 7:171. doi: 10.3389/fmolb.2020.00171
126. Ying W, Riopel M, Bandyopadhyay G, Dong Y, Birmingham A, Seo JB, et al. Adipose tissue macrophage-derived exosomal mirnas can modulate in vivo and in vitro insulin sensitivity. Cell (2017) 171(2):372–84.e12. doi: 10.1016/j.cell.2017.08.035
127. Zhao H, Shang Q, Pan Z, Bai Y, Li Z, Zhang H, et al. Exosomes from adipose-derived stem cells attenuate adipose inflammation and obesity through polarizing m2 macrophages and beiging in white adipose tissue. Diabetes (2018) 67(2):235–47. doi: 10.2337/db17-0356
128. Ying W, Gao H, Dos Reis FCG, Bandyopadhyay G, Ofrecio JM, Luo Z, et al. MiR-690, an exosomal-derived miRNA from M2-polarized macrophages, improves insulin sensitivity in obese mice. Cell Metab (2021) 33(4):781–90.e5. doi: 10.1016/j.cmet.2020.12.019
129. Crewe C, Scherer PE. Intercellular and interorgan crosstalk through adipocyte extracellular vesicles. Rev Endocr Metab Disord (2021) 23(1):61–9. doi: 10.1007/s11154-020-09625-x
130. Gao J, Li X, Wang Y, Cao Y, Yao D, Sun L, et al. Adipocyte-derived extracellular vesicles modulate appetite and weight through mTOR signalling in the hypothalamus. Acta Physiol (2019) 228(2):e13339. doi: 10.1111/apha.13339
131. Yoshida M, Satoh A, Lin JB, Mills KF, Sasaki Y, Rensing N, et al. Extracellular vesicle-contained enampt delays aging and extends lifespan in mice. Cell Metab (2019) 30(2):329–42.e5. doi: 10.1016/j.cmet.2019.05.015
132. Michel LYM. Extracellular vesicles in adipose tissue communication with the healthy and pathological heart. Int J Mol Sci (2023) 24(9):7745. doi: 10.3390/ijms24097745
133. Hu X, Pan J, Li Y, Jiang Y, Zheng H, Shi R, et al. Extracellular vesicles from adipose-derived stem cells promote microglia M2 polarization and neurological recovery in a mouse model of transient middle cerebral artery occlusion. Stem Cell Res Ther (2022) 13(1):21. doi: 10.1186/s13287-021-02668-0
134. Mukherjee S, Diéguez C, Fernø J, López M. Obesity wars: hypothalamic sEVs a new hope. Trends Mol Med (2023) 29(8):622–34. doi: 10.1016/j.molmed.2023.04.006
135. Milbank E, Dragano N, Vidal-Gómez X, Rivas-Limeres V, Garrido-Gil P, Wertheimer M, et al. Small extracellular vesicle targeting of hypothalamic AMPKα1 promotes weight loss in leptin receptor deficient mice. Metabolism (2023) 139:155350. doi: 10.1016/j.metabol.2022.155350
136. Milbank E, Dragano NRV, González-García I, Garcia MR, Rivas-Limeres V, Perdomo L, et al. Small extracellular vesicle-mediated targeting of hypothalamic AMPKα1 corrects obesity through BAT activation. Nat Metab (2021) 3(10):1415–31. doi: 10.1038/s42255-021-00467-8
137. Youngstrom TG, Bartness TJ. Catecholaminergic innervation of white adipose tissue in Siberian hamsters. Am J Physiol Regul Integr Comp Physiol (1995) 268(3):R744–R51. doi: 10.1152/ajpregu.1995.268.3.R744
138. Spraul M, Ravussin E, Fontvieille AM, Rising R, Larson DE, Anderson EA. Reduced sympathetic nervous activity. A potential Mech predisposing to body weight gain. J Clin Invest (1993) 92(4):1730–5. doi: 10.1172/jci116760
139. Ye Y, Abu El Haija M, Morgan DA, Guo D, Song Y, Frank A, et al. Endocannabinoid receptor-1 and sympathetic nervous system mediate the beneficial metabolic effects of gastric bypass. Cell Rep (2020) 33(4):108270. doi: 10.1016/j.celrep.2020.108270
140. Dettoni JL, Consolim-Colombo FM, Drager LF, Rubira MC, Cavasin de Souza SBP, Irigoyen MC, et al. Cardiovascular effects of partial sleep deprivation in healthy volunteers. J Appl Physiol (2012) 113(2):232–6. doi: 10.1152/japplphysiol.01604.2011
141. Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature (2017) 550(7674):119–23. doi: 10.1038/nature24022
142. Nguyen KD, Qiu Y, Cui X, Goh YPS, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature (2011) 480(7375):104–8. doi: 10.1038/nature10653
143. Fischer K, Ruiz HH, Jhun K, Finan B, Oberlin DJ, van der Heide V, et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat Med (2017) 23(5):623–30. doi: 10.1038/nm.4316
144. Fischer AW, de Jong JMA, Sass F, Schlein C, Heeren J, Petrovic N. Thermoneutrality-induced macrophage accumulation in brown adipose tissue does not impair the tissue’s competence for cold-induced thermogenic recruitment. Front Endocrinol (2020) 11:568682. doi: 10.3389/fendo.2020.568682
145. Pirzgalska RM, Seixas E, Seidman JS, Link VM, Sánchez NM, Mahú I, et al. Sympathetic neuron–associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med (2017) 23(11):1309–18. doi: 10.1038/nm.4422
146. Caravaca AS, Gallina AL, Tarnawski L, Shavva VS, Colas RA, Dalli J, et al. Vagus nerve stimulation promotes resolution of inflammation by a mechanism that involves Alox15 and requires the alpha7nAChR subunit. Proc Natl Acad Sci U.S.A. (2022) 119(22):e2023285119. doi: 10.1073/pnas.2023285119
147. Yao G, Kang L, Li J, Long Y, Wei H, Ferreira CA, et al. Effective weight control via an implanted self-powered vagus nerve stimulation device. Nat Commun (2018) 9(1):5349. doi: 10.1038/s41467-018-07764-z
148. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature (2000) 405(6785):458–62. doi: 10.1038/35013070
149. Li Y, Xu Z, Yu Y, Yuan H, Xu H, Zhu Q, et al. The vagus nerve attenuates fulminant hepatitis by activating the src kinase in kuppfer cells. Scand J Immunol (2014) 79(2):105–12. doi: 10.1111/sji.12141
150. Kanauchi Y, Yamamoto T, Yoshida M, Zhang Y, Lee J, Hayashi S, et al. Cholinergic anti-inflammatory pathway ameliorates murine experimental Th2-type colitis by suppressing the migration of plasmacytoid dendritic cells. Sci Rep (2022) 12(1):54. doi: 10.1038/s41598-021-04154-2
151. Matteoli G, Gomez-Pinilla PJ, Nemethova A, Di Giovangiulio M, Cailotto C, van Bree SH, et al. A distinct vagal anti-inflammatory pathway modulates intestinal muscularis resident macrophages independent of the spleen. Gut (2014) 63(6):938–48. doi: 10.1136/gutjnl-2013-30467
152. Deng J, Wang M, Guo Y, Fischer H, Yu X, Kem D, et al. Activation of alpha7nAChR via vagus nerve prevents obesity-induced insulin resistance via suppressing endoplasmic reticulum stress-induced inflammation in kupffer cells. Med Hypotheses (2020) 140:109671. doi: 10.1016/j.mehy.2020.109671
153. Nishio T, Taura K, Iwaisako K, Koyama Y, Tanabe K, Yamamoto G, et al. Hepatic vagus nerve regulates kupffer cell activation via alpha7 nicotinic acetylcholine receptor in nonalcoholic steatohepatitis. J Gastroenterol (2017) 52(8):965–76. doi: 10.1007/s00535-016-1304-z
154. Li DJ, Liu J, Hua X, Fu H, Huang F, Fei YB, et al. Nicotinic acetylcholine receptor alpha7 subunit improves energy homeostasis and inhibits inflammation in nonalcoholic fatty liver disease. Metabolism (2018) 79:52–63. doi: 10.1016/j.metabol.2017.11.002
155. Consolim-Colombo FM, Sangaleti CT, Costa FO, Morais TL, Lopes HF, Motta JM, et al. Galantamine alleviates inflammation and insulin resistance in patients with metabolic syndrome in a randomized trial. JCI Insight (2017) 2(14):e93340. doi: 10.1172/jci.insight.93340
156. Sangaleti CT, Katayama KY, De Angelis K, Lemos de Moraes T, Araujo AA, Lopes HF, et al. The cholinergic drug galantamine alleviates oxidative stress alongside anti-inflammatory and cardio-metabolic effects in subjects with the metabolic syndrome in a randomized trial. Front Immunol (2021) 12:613979. doi: 10.3389/fimmu.2021.613979
157. Dou D, Chen Qiao Q, Zhong Z-q, Xia X-w, Ding W-J. Regulating the enteric nervous system against obesity in mice by electroacupuncture. Neuroimmunomodulation (2020) 27(1):48–57. doi: 10.1159/000506483
158. Muller Paul A, Koscsó B, Rajani Gaurav M, Stevanovic K, Berres M-L, Hashimoto D, et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell (2014) 158(2):300–13. doi: 10.1016/j.cell.2014.04.050
159. Gabanyi I, Muller Paul A, Feighery L, Oliveira Thiago Y, Costa-Pinto Frederico A, Mucida D. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell (2016) 164(3):378–91. doi: 10.1016/j.cell.2015.12.023
160. Wan X, Yin J, Chen J. Characteristics of intestinal myoelectrical and motor activities in diet-induced obese rats: obesity and motility. Digest Dis Sci (2019) 64(6):1478–85. doi: 10.1007/s10620-019-5458-4
161. Seelig E, Henning E, Keogh JM, Gillett D, Shin E, Buscombe J, et al. Obesity due to melanocortin 4 receptor (MC4R) deficiency is associated with delayed gastric emptying. Clin Endocrinol (2021) 96(2):270–5. doi: 10.1111/cen.14615
162. Valdearcos M, Robblee MM, Benjamin DI, Nomura DK, Xu AW, Koliwad SK. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep (2014) 9(6):2124–38. doi: 10.1016/j.celrep.2014.11.018
163. García-Cáceres C, Balland E, Prevot V, Luquet S, Woods SC, Koch M, et al. Role of astrocytes, microglia, and tanycytes in brain control of systemic metabolism. Nat Neurosci (2018) 22(1):7–14. doi: 10.1038/s41593-018-0286-y
164. Kälin S, Heppner FL, Bechmann I, Prinz M, Tschöp MH, Yi C-X. Hypothalamic innate immune reaction in obesity. Nat Rev Endocrinol (2015) 11(6):339–51. doi: 10.1038/nrendo.2015.48
165. Kreutzer C, Peters S, Schulte DM, Fangmann D, Turk K, Wolff S, et al. Hypothalamic inflammation in human obesity is mediated by environmental and genetic factors. Diabetes (2017) 66(9):2407–15. doi: 10.2337/db17-0067
166. Lainez NM, Jonak CR, Nair MG, Ethell IM, Wilson EH, Carson MJ, et al. Diet-induced obesity elicits macrophage infiltration and reduction in spine density in the hypothalami of male but not female mice. Front Immunol (2018) 9:1992. doi: 10.3389/fimmu.2018.01992
167. Mao L, Hochstetter D, Yao L, Zhao Y, Zhou J, Wang Y, et al. Green tea polyphenol (–)-epigallocatechin gallate (EGCG) attenuates neuroinflammation in palmitic acid-stimulated bv-2 microglia and high-fat diet-induced obese mice. Int J Mol Sci (2019) 20(20):5081. doi: 10.3390/ijms20205081
168. Howe A-M, Burke S, O’Reilly ME, McGillicuddy FC, Costello DA. Palmitic acid and oleic acid differently modulate tlr2-mediated inflammatory responses in microglia and macrophages. Mol Neurobiol (2022) 59(4):2348–62. doi: 10.1007/s12035-022-02756-z
169. Duffy CM, Xu H, Nixon JP, Bernlohr DA, Butterick TA. Identification of a fatty acid binding protein4-UCP2 axis regulating microglial mediated neuroinflammation. Mol Cell Neurosci (2017) 80:52–7. doi: 10.1016/j.mcn.2017.02.004
170. López M, Nogueiras R, Tena-Sempere M, Diéguez C. Hypothalamic AMPK: a canonical regulator of whole-body energy balance. Nat Rev Endocrinol (2016) 12(7):421–32. doi: 10.1038/nrendo.2016.67
171. Seoane-Collazo P, Fernø J, Gonzalez F, Diéguez C, Leis R, Nogueiras R, et al. Hypothalamic-autonomic control of energy homeostasis. Endocrine (2015) 50(2):276–91. doi: 10.1007/s12020-015-0658-y
172. Contreras C, Nogueiras R, Diéguez C, Rahmouni K, López M. Traveling from the hypothalamus to the adipose tissue: the thermogenic pathway. Redox Biol (2017) 12:854–63. doi: 10.1016/j.redox.2017.04.019
173. Banerjee J, Dorfman MD, Fasnacht R, Douglass JD, Wyse-Jackson AC, Barria A, et al. CX3CL1 action on microglia protects from diet-induced obesity by restoring POMC neuronal excitability and melanocortin system activity impaired by high-fat diet feeding. Int J Mol Sci (2022) 23(12):6380. doi: 10.3390/ijms23126380
174. Yi CX, Walter M, Gao Y, Pitra S, Legutko B, Kalin S, et al. TNFalpha drives mitochondrial stress in POMC neurons in obesity. Nat Commun (2017) 8:15143. doi: 10.1038/ncomms15143
175. Yi C-X, Al-Massadi O, Donelan E, Lehti M, Weber J, Ress C, et al. Exercise protects against high-fat diet-induced hypothalamic inflammation. Physiol Behav (2012) 106(4):485–90. doi: 10.1016/j.physbeh.2012.03.021
176. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330(6005):841–5. doi: 10.1126/science.1194637
177. Valdearcos M, Douglass JD, Robblee MM, Dorfman MD, Stifler DR, Bennett ML, et al. Microglial inflammatory signaling orchestrates the hypothalamic immune response to dietary excess and mediates obesity susceptibility. Cell Metab (2017) 26(1):185–97 e3. doi: 10.1016/j.cmet.2017.05.015
178. Jurga AM, Paleczna M, Kuter KZ. Overview of general and discriminating markers of differential microglia phenotypes. Front Cell Neurosci (2020) 14:198. doi: 10.3389/fncel.2020.00198
179. Vargas-Soria M, García-Alloza M, Corraliza-Gómez M. Effects of diabetes on microglial physiology: a systematic review of in vitro, preclinical and clinical studies. J Neuroinflamm (2023) 20(1):57. doi: 10.1186/s12974-023-02740-x
180. Tracy LM, Bergqvist F, Ivanova EV, Jacobsen KT, Iverfeldt K. Exposure to the saturated free fatty acid palmitate alters BV-2 microglia inflammatory response. J Mol Neurosci (2013) 51(3):805–12. doi: 10.1007/s12031-013-0068-7
181. Chowen JA, Argente J, Barrios V, Freire-Regatillo A, Frago LM, Argente-Arizón P, et al. The opposing effects of ghrelin on hypothalamic and systemic inflammatory processes are modulated by its acylation status and food intake in male rats. Endocrinology (2014) 155(8):2868–80. doi: 10.1210/en.2014-1074
182. Maldonado-Ruiz R, Cárdenas-Tueme M, Montalvo-Martínez L, Vidaltamayo R, Garza-Ocañas L, Reséndez-Perez D, et al. Priming of hypothalamic ghrelin signaling and microglia activation exacerbate feeding in rats’ offspring following maternal overnutrition. Nutrients (2019) 11(6):1241. doi: 10.3390/nu11061241
183. Tsunekawa T, Banno R, Mizoguchi A, Sugiyama M, Tominaga T, Onoue T, et al. Deficiency of PTP1B attenuates hypothalamic inflammation via activation of the JAK2-STAT3 pathway in microglia. EBioMedicine (2017) 16:172–83. doi: 10.1016/j.ebiom.2017.01.007
184. Wu Z, Xi P, Zhang Y, Wang H, Xue J, Sun X, et al. LKB1 up-regulation inhibits hypothalamic inflammation and attenuates diet-induced obesity in mice. Metabolism (2021) 116:154694. doi: 10.1016/j.metabol.2020.154694
185. Maldonado-Ruiz R, Montalvo-Martínez L, Fuentes-Mera L, Camacho A. Microglia activation due to obesity programs metabolic failure leading to type two diabetes. Nutr Diabetes (2017) 7(3):e254–4. doi: 10.1038/nutd.2017.10
186. Tang C-H, Lu D-Y, Yang R-S, Tsai H-Y, Kao M-C, Fu W-M, et al. Leptin-induced IL-6 production is mediated by leptin receptor, insulin receptor substrate-1, phosphatidylinositol 3-kinase, akt, nf-κb, and p300 pathway in microglia. J Immunol (2007) 179(2):1292–302. doi: 10.4049/jimmunol.179.2.1292
187. Spencer SJ, Basri B, Sominsky L, Soch A, Ayala MT, Reineck P, et al. High-fat diet worsens the impact of aging on microglial function and morphology in a region-specific manner. Neurobiol Aging (2019) 74:121–34. doi: 10.1016/j.neurobiolaging.2018.10.018
188. Serrats J, Schiltz JC, Garcia-Bueno B, van Rooijen N, Reyes TM, Sawchenko PE. Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron (2010) 65(1):94–106. doi: 10.1016/j.neuron.2009.11.032
189. Polfliet MM, Goede PH, van Kesteren-Hendrikx EM, van Rooijen N, Dijkstra CD, van den Berg TK. A method for the selective depletion of perivascular and meningeal macrophages in the central nervous system. J Neuroimmunol (2001) 116(2):188–95. doi: 10.1016/s0165-5728(01)00282-x
190. Schlich R, Willems M, Greulich S, Ruppe F, Knoefel WT, Ouwens DM, et al. VEGF in the crosstalk between human adipocytes and smooth muscle cells: depot-specific release from visceral and perivascular adipose tissue. Mediators Inflammation (2013) 2013:982458. doi: 10.1155/2013/982458
191. Zhang Q, Fang W, Ma L, Wang ZD, Yang YM, Lu YQ. VEGF levels in plasma in relation to metabolic control, inflammation, and microvascular complications in type-2 diabetes: a cohort study. Med (Baltimore) (2018) 97(15):e0415. doi: 10.1097/MD.0000000000010415
192. Jais A, Solas M, Backes H, Chaurasia B, Kleinridders A, Theurich S, et al. Myeloid-cell-derived vegf maintains brain glucose uptake and limits cognitive impairment in obesity. Cell (2016) 165(4):882–95. doi: 10.1016/j.cell.2016.03.033
193. Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging. aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science (2014) 346(6205):89–93. doi: 10.1126/science.1252945
194. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol (2005) 57(1):67–81. doi: 10.1002/ana.20315
195. Kim S, Hwang Y, Lee D, Webster MJ. Transcriptome sequencing of the choroid plexus in schizophrenia. Transl Psychiatry (2016) 6(11):e964. doi: 10.1038/tp.2016.229
196. Kwon Y-H, Kim J, Kim C-S, Tu TH, Kim M-S, Suk K, et al. Hypothalamic lipid-laden astrocytes induce microglia migration and activation. FEBS Lett (2017) 591(12):1742–51. doi: 10.1002/1873-3468.12691
197. Sugiyama M, Banno R, Yaginuma H, Taki K, Mizoguchi A, Tsunekawa T, et al. Hypothalamic glial cells isolated by MACS reveal that microglia and astrocytes induce hypothalamic inflammation via different processes under high-fat diet conditions. Neurochem Int (2020) 136:104733. doi: 10.1016/j.neuint.2020.104733
198. Zhang Y, Reichel JM, Han C, Zuniga-Hertz JP, Cai D. Astrocytic process plasticity and IKKβ/NF-κB in central control of blood glucose, blood pressure, and body weight. Cell Metab (2017) 25(5):1091–102.e4. doi: 10.1016/j.cmet.2017.04.002
199. Mendes NF, Gaspar JM, Lima-Júnior JC, Donato J, Velloso LA, Araújo EP. TGF-β1 down-regulation in the mediobasal hypothalamus attenuates hypothalamic inflammation and protects against diet-induced obesity. Metabolism (2018) 85:171–82. doi: 10.1016/j.metabol.2018.04.005
200. Buckman LB, Hasty AH, Flaherty DK, Buckman CT, Thompson MM, Matlock BK, et al. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav Immun (2014) 35:33–42. doi: 10.1016/j.bbi.2013.06.007
201. Guillemot-Legris O, Masquelier J, Everard A, Cani PD, Alhouayek M, Muccioli GG. High-fat diet feeding differentially affects the development of inflammation in the central nervous system. J Neuroinflamm (2016) 13(1):206. doi: 10.1186/s12974-016-0666-8
202. Thaler JP, Yi C-X, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest (2012) 122(1):153–62. doi: 10.1172/jci59660
203. Lee A, Dixit VD. Energy sparing orexigenic inflammation of obesity. Cell Metab (2017) 26(1):10–2. doi: 10.1016/j.cmet.2017.06.013
204. Martchenko SE, Martchenko A, Cox BJ, Naismith K, Waller A, Gurges P, et al. Circadian GLP-1 secretion in mice is dependent on the intestinal microbiome for maintenance of diurnal metabolic homeostasis. Diabetes (2020) 69(12):2589–602. doi: 10.2337/db20-0262
205. Vahl TP, Drazen DL, Seeley RJ, D'Alessio DA, Woods SC. Meal-anticipatory glucagon-like peptide-1 secretion in rats. Endocrinology (2010) 151(2):569–75. doi: 10.1210/en.2009-1002
206. Beiroa D, Imbernon M, Gallego R, Senra A, Herranz D, Villarroya F, et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes (2014) 63(10):3346–58. doi: 10.2337/db14-0302
207. Heiss CN, Mannerås-Holm L, Lee YS, Serrano-Lobo J, Håkansson Gladh A, Seeley RJ, et al. The gut microbiota regulates hypothalamic inflammation and leptin sensitivity in Western diet-fed mice via a GLP-1R-dependent mechanism. Cell Rep (2021) 35(8):109163. doi: 10.1016/j.celrep.2021.109163
208. Imbernon M, Saponaro C, Helms HCC, Duquenne M, Fernandois D, Deligia E, et al. Tanycytes control hypothalamic liraglutide uptake and its anti-obesity actions. Cell Metab (2022) 34(7):1054–63.e7. doi: 10.1016/j.cmet.2022.06.002
Keywords: obesity; inflammation, macrophages, adipose tissue, microglia, hypothalamus, small extracellular vesicles
Citation: Mukherjee S, Skrede S, Haugstøyl M, López M and Fernø J (2023) Peripheral and central macrophages in obesity. Front. Endocrinol. 14:1232171. doi: 10.3389/fendo.2023.1232171
Received: 31 May 2023; Accepted: 28 July 2023;
Published: 31 August 2023.
Edited by:
Mar Quiñones, Health Research Institute of Santiago de Compostela (IDIS), SpainReviewed by:
Amaia Rodríguez, University of Navarra, SpainCopyright © 2023 Mukherjee, Skrede, Haugstøyl, López and Fernø. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sayani Mukherjee, c2F5YW5pLm11a2hlcmplZUBoZWxzZS1iZXJnZW4ubm8=; Johan Fernø, am9oYW4uZmVybm9AdWliLm5v
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.