 Anna Papadopoulou
Anna Papadopoulou Evangelia Bountouvi
Evangelia Bountouvi- 1Laboratory of Clinical Biochemistry, University General Hospital “Attikon”, Medical School, National & Kapodistrian University of Athens, Athens, Greece
- 2Neonatal Intensive Care Unit, General Hospital “Alexandra”, Athens, Greece
Noonan, Costello and Cardio-facio-cutaneous syndromes belong to a group of disorders named RASopathies due to their common pathogenetic origin that lies on the Ras/MAPK signaling pathway. Genetics has eased, at least in part, the distinction of these entities as they are presented with overlapping clinical features which, sometimes, become more pronounced with age. Distinctive face, cardiac and skeletal defects are among the primary abnormalities seen in these patients. Skeletal dysmorphisms range from mild to severe and may include anterior chest wall anomalies, scoliosis, kyphosis, short stature, hand anomalies, muscle weakness, osteopenia or/and osteoporosis. Patients usually have increased serum concentrations of bone resorption markers, while markers of bone formation are within normal range. The causative molecular defects encompass the members of the Ras/MAPK/ERK pathway and the adjacent cascades, important for the maintenance of normal bone homeostasis. It has been suggested that modulation of the expression of specific molecules involved in the processes of bone remodeling may affect the osteogenic fate decision, potentially, bringing out new pharmaceutical targets. Currently, the laboratory imprint of bone metabolism on the clinical picture of the affected individuals is not clear, maybe due to the rarity of these syndromes, the small number of the recruited patients and the methods used for the description of their clinical and biochemical profiles.
1 Introduction
Since the first description of nine patients with “pulmonary valve stenosis, small stature, hypertelorism, mild intellectual disability, ptosis, undescended testes, and skeletal malformations” (1), the multiple “faces” of Noonan syndrome(NS) came to light stepwise, thanks to the refined clinical observation and elucidation of the causative molecular pathways. Based on new evidence, the term “RASopathies” has been introduced to describe a group of phenotypically similar congenital anomaly disorders associated with pathogenic variants in genes of the Ras/MAPK signaling pathway, a cascade critical for the regulation of cell proliferation and differentiation (2, 3). Although strict genotype–phenotype correlations are hard to be established, some distinctive features have helped in clinical diagnosis and monitoring (4–7). For example, about half of NS patients have mutations in the PTPN11 gene, albeit mutations have also been found in other genes involved in the Ras-cascade (8–17). Costello syndrome(CS) has been associated with mutations in HRAS (18), and cardio-facio-cutaneous syndrome(CFCS) with BRAF, MAP2K1, MAP2K2 and KRAS (11, 19, 20).
Skeletal dysmorphisms, variably and heterogeneously presented, are among the major diagnostic features of RASopathies (6). Prevalent musculoskeletal defects include thoracic wall anomalies, spinal deformities, pes planus, hand anomalies, hip dysplasia, joint laxity or contractures, muscular hypotonia and muscle paucity (21–25). Short stature is also a common clinical sign especially of NS, Costello and CFC syndromes. Notably, growth in NS individuals during childhood and adolescence is often below the 3rd centile (26). Moreover, although still poorly understood, a restricted number of studies have suggested the implication of Ras/MAPK signaling pathway in decreased bone density(BMD) in some RASopathies, -particularly CS (25, 27, 28), NF1 (29–32) and NS (33)- increased bone resorption, as measured by urine degraded crosslinks of mature collagen in NS, Costello and CFC patients (34) and osteoporosis/osteopenia (27, 35–37).
The causative mechanisms behind osseous defects and altered bone metabolism in NS patients remain elusive. The presence of skeletal abnormalities in Ras/MAPK disorders suggest the implication of Ras signal transduction pathway in bone remodeling, while cross-talks with other molecular cascades, such as BMP/Smad/Runx2, may explain the organ-specific pathophysiology (38). The small number of patients included in clinical reports, sometimes without molecular diagnosis of NS, the wide range of patients’ age, the variety of methods and the criteria used for the diagnosis of osteopenia/osteoporosis in children and adults have hampered the deduction of clear inferences so far. Additionally, although skeletal abnormalities are observed in all RASopathies, most focus has been placed on NF1 syndrome. This review summarizes the current knowledge on musculoskeletal phenotypes and biochemical profiles regarding bone metabolism markers seen in NS, CS, CFC syndromes, citing common and distinctive features between these entities, as well as the existing evidence regarding the involvement of the Ras-cascade in bone defects and remodeling.
2 Musculoskeletal features and biochemical bone profile
Noonan, Costello and Cardio-facio-cutaneous syndromes are rare disorders, yet the overall prevalence is nearly 1:2,000. Among them, NS has emerged as the more frequent, while CFC and CS constitute ultra-rare diseases with only some hundreds of affected individuals reported worldwide (39–42). The last years, the musculoskeletal profile of NS/CS/CFC has been outlined in the literature, albeit to a lesser degree compared to other pathological aspects of RASopathies (23–25, 28, 34–36, 43, 44). Despite the overlapping phenotypic imprint and the broad clinical variability -even within the same entity-, some recognizable features may orientate the diagnosis to a certain condition and its distinctive underlying genotype. Stature, bone structure and musculoskeletal deformities -further analyzed below- are the basic aspects of osseous phenotype in RASopathies.
2.1 Short stature
Short stature is a universal component of Rasopathies. It has been reported in roughly half of NS patients and it is a cardinal feature in CFC (>70%) and CS (>90%) (45–51). Typically, growth parameters at birth are within normal range or even above average. However, during the first years of life postnatal growth failure ensues, leading to a low mean height, further compromised by delayed puberty and an attenuated growth spurt (23, 48). Thus, normative growth charts have been developed for NS (52) and CS (53) patients. Along with hormonal factors, musculoskeletal impairment, gastrointestinal abnormalities and increased resting energy expenditure are also implicated in the pathogenesis of short stature (37, 42, 48, 54–56). The above features are more prevalent and severe in CS and CFC and this partially counts for the more profound height restriction observed compared to NS (25, 36, 45, 57, 58). Notably, progressive musculoskeletal deformities exert a constant negative impact on stature even in adulthood (25, 27, 55, 59). Contrary to CFC and CS, some genotype-phenotype correlations have been suggested for NS. Thus, mutations in RAF1, SHOC2 and PTPN11 are associated with more severe growth impairment contrary to SOS1 genotypes (60–62). Of note, PTPN11 pathogenic variations observed in NS with multiple lentigines(NSML) patients have been linked to lower incidence of short stature (51).
The underlying mechanisms of growth failure seem to be multifactorial and not fully elucidated. As discussed below in the text, molecular defects in components or modulators of the Ras/MAPK pathway have been proposed to induce dysregulation at different levels of the cascade leading to height restriction via diverse mechanisms (48). Hence, consequent dysregulation of GH axis can either lead to GH-deficiency and impaired IGF-1 production (37, 45, 63) or GH-resistance/insensitivity at a postreceptor level (64). Besides, a direct effect of Ras/MAPK pathway on growth plate development has been also demonstrated (65, 66). Exogenous GH has been administrated to overcome the subjacent imbalance in GH-signaling, showing good tolerance in NS and CFC patients and some efficiency mainly during the first two years of regimen (45, 48, 67). Notably, growth hormone therapy has been reported to improve final height in the majority of children with NS (68). Typically, dosage is around 50microgram/kg/day or lower in selected cases (68). Since growth hormone replacement therapy in NS prepubertal years has been demonstrated to be important for the final height, therapy introduction is recommended to initiate at the age 4 in most children or earlier in cases of extreme height restriction. Although a clear connection of this therapeutic intervention with hypertrophic cardiomyopathy or malignancy has not been clearly established, patients’ genotyping is mandatory as specific mutations in PTPN1 associated to leukemia or mutations in genes linked to the development of hypertrophic cardiomyopathy (e.g., RIT1) [reviewed in (68, 69)] are among the exclusion criteria. Notably, in children with mutations known to increase leukemia risk (such as p.Asp61Gly and p.Thr73lle in PTPN11), therapy initiation is not recommended before the age of 5 (70). In CS, data are sparse and precautions must be taken due to comorbidities, such as obstructive apnea syndrome and malignancy, apart from the commonly coexisted hypertrophic heart disease (55).
2.2 Osteopenia and bone metabolism
Bone mineral density (BMD) measurement, as a marker of bone health, has been used in NS/CS/CFC studies and low values have been documented in all three syndromes (28, 33–35, 37, 43, 68, 71). Short stature has to be taken into consideration at DEXA evaluation –mostly utilized in current studies-, however lower BMD in NS/CFC/CS patients has been also confirmed after comparison to matched control individuals (33, 35, 43). Current data comes from few cohorts, including a limited number of patients, or case series, thus its frequency is rather difficult to be accurately estimated. Evidence from small NS pediatric cohorts underscores the impairment of bone quality and structure. Noordam et al. reported a marginal decrease in BMD of 16 patients with a slight improvement after 2 years of growth hormone replacement treatment (72). Reduced BMD was also documented in 6 of 12 individuals studied by Choudhry et al. (33). Moreover, approximately 15% of the thirty-five patients enrolled by Baldassare et al. showed compromised bone quality as assessed by phalangeal quantitative ultrasound (QUS) (71) while in a recent retrospective study of 35 individuals, axial and appendicular bone mass were reduced compared to the general population (37). Data in NS adult patients are even scantier. Five over thirty-five individuals have been reported with osteopenia or osteoporosis and two over twenty-six with osteoporosis by Smpokou and Reinker et al, respectively (36, 73). Bone metabolic disease in NS has been supported to be partly primary resulting from an imbalance between osteoclast and osteoblast activity due to RAS/ERK hyperactivation, as discussed below (33, 37, 71). Notably, recent results from experiments in transgenic NS mice revealed an essential role of SHP2 in promoting osteoblastic differentiation and suppressing osteoclastogenesis (74, 75). Moreover, BMD was shown to correlate with decreased muscle mass and low IGF-1 levels (37), while low gonadal steroids also supported to contribute to the final bone phenotype (37, 48, 68, 76).
In CS, osteopenia and osteoporosis have been described as a common feature in a few reports/case series, presenting an increasing incidence with age, which is feasible since bone pathology evolves over time (28, 35, 77). Stevenson et al. described low BMD in seven CS patients (34). Detweiler et al. reported a frequency of 41% (N=34) and 6% (N=33) for osteopenia and osteoporosis, respectively, on a cohort of 43 CS individuals (26 pediatric and 17 adolescent/adult patients) (77). In a series of adult patients, BMD was detected to be impaired in all eight individuals assessed (2 with osteopenia and 6 with osteoporosis). Three of them were symptomatic suggesting clinical significance of osteoporosis in CS (27). Supporting this notion, two of the three infants reported in a small case series suffered by generalized osteoporosis (78). Leoni et al. in 2014 demonstrated reduced BMD in 9 CS individuals compared to age-matched controls (from 5 to 29 years of age) (28). Subsequently, the same group confirmed these results in an extended cohort of 16 individuals over a 4-year follow up study (35). Cakir et al. reported a CS adult patient with a parathyroid adenoma, suffering from osteoporosis since adolescence (79). Many factors have been supported to contribute to final bone phenotype such as decreased mobility, reduced peak bone mass, muscle impairment and severe hypogonadism in some patients (27, 44, 48). The last years, the notion that BMD is also an intrinsic feature of CS has gained ground. Increased RAS/ERK signaling in CS has been proposed to induce osteoclastic hyperactivity and increased bone resorption as supported by studies of bone resorption markers (28, 34).
Data on prevalence of osteopenia or osteoporosis in CFC are sparser (43, 48). Apart from one patient reported with slightly decreased BMD and biochemical evidence of elevated markers of bone resorption (34), Chen et al. recently described one 10 year-old girl suffering from osteoporosis and multiple musculoskeletal deformities in a small series of 3 MAP2K1 CFC patients (80). The first systematic cohort is a recent study of 14 patients who displayed low BMD compared to healthy age-matched controls, despite proper cholecalciferol supplementation. However, only one child met the osteoporosis diagnostic criteria (43).
Overall, the risk of fracture is currently difficult to be determined, yet longitudinal BMD monitoring is recommended. Apart from bone fragility, decreased BMD has been also associated with pain (48, 58), low physical activity (48), low muscle mass, depressed IGF-1 levels (37) and inferior life quality (25, 58). Despite the fact that evidence is lacking pertaining to CFC, upregulated function of RAS/MAPK cascade has also been implied in impaired bone metabolism. Notably, mild increased levels of alkaline phosphatase(ALP) have been reported in CFC patients (25). Besides genetic background, other factors also contribute to bone homeostasis alteration such as lack of autonomous deambulation, sedentary lifestyle and common used medications in CFC (antiepileptics, proton pump inhibitors).
Although pathogenetic mechanisms and association with genetic deficits are not fully understood, emerging data from different groups have underscored the essential role of Ras/MAPK signaling pathway in bone metabolism (23–25, 44, 48). The imbalance between osteoclastic and osteoblastic activity as a result of Ras/MAPK signaling deviation have been proposed to be fundamental in osseous pathophysiology (48). Indeed, preclinical and experimental data have revealed up-regulation of osteoclasts’ development (65, 81), function and survival and, on the other hand, impaired growth and differentiation of osteoblasts along with decreased osteoblasts’ activity (23, 24, 71, 82). The above hypothesis is further supported by biochemical data from NS/CFC/CS individuals; markers of bone resorption such as urine excretion of pyridinium crosslinks (28, 34), and PTH (48) have been elevated while decreased levels of calcitonin –a biomarker of osseous formation and osteoblastic activity- has been determined (48). Constitutional decrease of 25-Vitamin D has been constantly described in all 3 entities, yet no specific correlation with the low BMD has been confirmed (33, 37, 43, 71). Noteworthy, reduced BMD in patients exhibiting a rather normal bone metabolism profile have been also reported (25, 28, 33). Of note, in three different NS pediatric cohorts investigated, all showed normal mean serum calcium, phosphate and ALP concentrations (33, 37, 71). These observations underline the fact that bone homeostasis is a dynamic biological process modulated by complex physiological and environmental influences. Therefore, apart from the intrinsic bone pathology in RASopathies, various environmental and secondary to diseases’ morbidities factors converge into the final bone phenotype. Collectively, deviation of Ras/MAPK pathway, progressive bone deformities, limitations in physical activity, hormonal disturbances such as hypogonadism and perturbed IGF-1 activity, elevated inflammatory cytokines, sun exposure, musculature impairment and medications such as antiepileptics, proton pump inhibitors and corticosteroids are implicated in the altered bone homeostasis observed in RASopathies (23, 44, 48, 83).
2.3 Skeletal deformities
Musculoskeletal deformities have been described as another overlapping component of RASopathy nosologies. As mentioned above, the list of skeletal manifestations, and the incidence of each deformity is rather difficult to be accurately estimated due to the rarity of the disorders, the broad clinical spectrum, the dynamic process of bone development and the heterogeneity among studies focusing on different ages–mainly pediatric-and on diverse diseases’ aspects. Thus, many skeletal findings remain underreported. Although all RASopathies include skeletal defects, CFC and CS have been documented to be characterized by a distinctive osseous phenotype and severe skeletal involvement, especially compared to milder bone manifestations reported in NS patients (23–25). The main skeletal characteristics are presented below and summarized in Table 1.
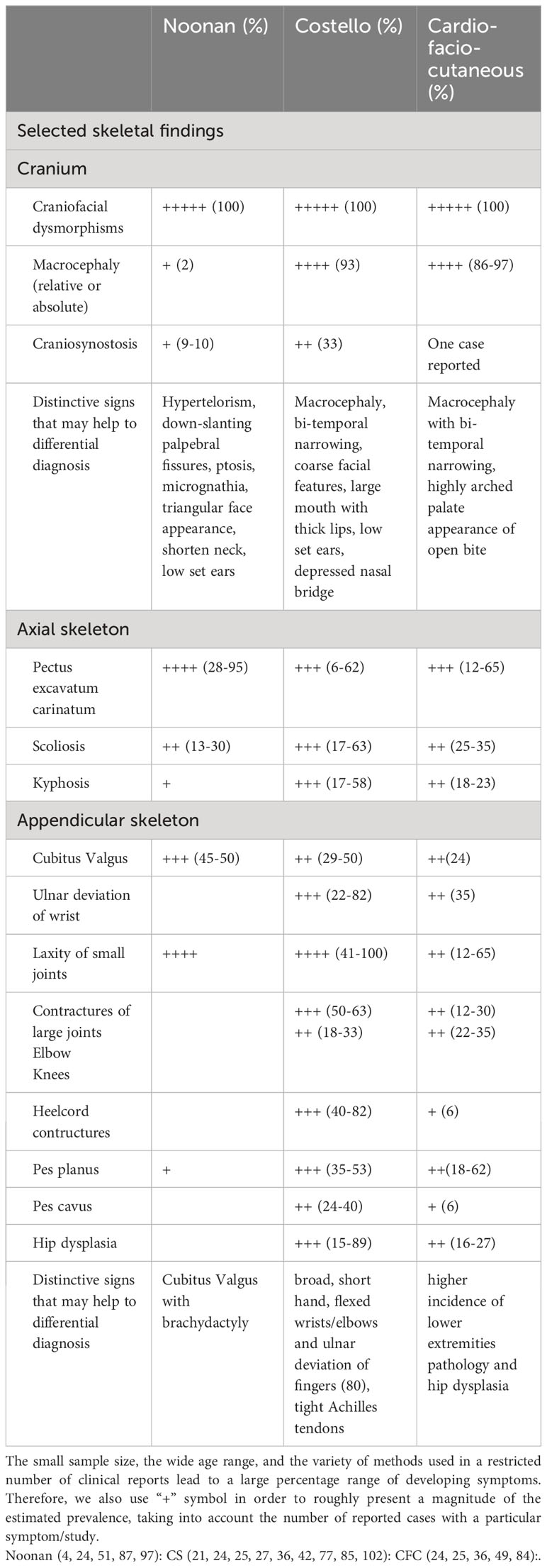
Table 1 Main skeletal clinical characteristics of Noonan, Costello and Cardio-facio-cutaneous syndromes.
Craniofacial features are part of developmental anomalies syndromes and in RASopathies include, among others, relative or absolute macrocephaly with bitemporal constriction, prominent forehead, highly arched palate and dental abnormalities in all three disorders (40, 84, 85). Moreover, pertaining to NS, micrognathia and a triangular face appearance is a distinctive sign, while despite the large appearance of cranium (4, 86, 87), head circumference in adulthood does not differ significantly compared to general population (88). The vast majority of CFC and CS patients do present with macrocephaly (>85% and 93%, respectively) and bi-temporal narrowing. Large mouth with thick lips is a distinctive sign in CS, while appearance of open mouth, partially attributed to the existence of highly arched palate(up to 80%), which is more common in BRAF mutated CFC individuals (41, 84, 89). Recently, RASopathies have been associated to an increased risk of craniosynostosis, expanding the clinical phenotype. In a latest study, it has been suggested that about 33% of CFC and 9% of NS individuals may suffer from craniosynostosis (90). The patients reported owed mutations in BRAF, SHOC2, PTPN11 and KRAS genes, with the latter proposed to be correlated to more severe craniosynostosis (90–94). Craniosynostosis has been also described in a CS patient harboring a duplication in HRAS gene (95). It is worth to mention that in a recent retrospective study late onset synostosis was prominent (92), underscoring both the need for long-term monitoring and the record of this clinical sign.
Deformities of axial skeleton are cardinal features in all three entities (40). Chest wall anomalies, such as pectus excavatum and/or carinatum combined with a broad thorax is distinctive in NS (up to 95%) (23, 24). Similarly, the above pectus abnormalities are common both in CFC and CS and have been reported in up to two-thirds of affected individuals (25, 50, 96). Scoliosis/kyphosis are other universal manifestations reported roughly in 15% of NS (86, 87, 97), 20-35% of CFC and from 17% to more than half of CS individuals (36, 77, 98). Back and chronic pain, especially in adulthood, are more frequently observed in NS patients compared to CS/CFC patients, while kyphosis is not (36, 58).Of note, vertebral reversal with lumbar kyphosis and thoracic lordosis has been described in CS (77). Moreover, in CS individuals, kyphoscoliosis has been recently demonstrated to be slowly progressive in the absence of structural neurological deformities (98).
Appendicular skeleton is also affected (23, 40). Cubitus valgus (ulnar deviation of the elbow) together with brachydactyly is the most common deformity of the upper extremities, presented in nearly half of NS affected individuals (23, 24). Furthermore, CS patients present a more severe abnormal posture including broad, short hand, flexed wrists/elbows and ulnar deviation of fingers. This characteristic posture affects nearly 80% of CS patients, is highly suggestive of the disorder and is partially attributed to laxity of small joints (21, 36). Contrary to hyperextensibility of small joints, large joints are compromised by contractures. Other characteristic features in CS are pes planus, tight Achilles tendons -that even in childhood often require surgical therapy, and hip dysplasia (55). The latter affects 15-89% CS individuals (21, 25, 77) and whereas ligamentous laxity and hypotonia have been proposed as main etiologies of early presented bilateral dysplasia, underlying mechanism of late unilateral onset forms remains elusive (55, 56). All above upper and lower limbs deformities have been also reported in CFC (89). Signs that may differentiate CFC are the higher incidence of lower extremities pathology, such as short and broad feet, knee contractures(~25%), pes planus/cavus (up to two thirds) and hip dysplasia(17-27%) (25, 36, 41, 49). What is more, two case-descriptions of CFC patients have reported on skeletal manifestations, previously unreported; symmetric polyostotic fibrous dysplasia of proximal humerus (99) and symmetrical polyarthritis affecting upper and lower extremities (100). Given the rarity of RASopathies even one case can significantly alter statistics and focus attention on phenotypes that may be under recognized.
Few genotype-phenotype correlations have been proposed. CS-patients harboring the rare mutation Gly13Cys have been demonstrated to exhibit a milder orthopedic phenotype. On contrary, CFC-patients with genetic defects in MAP2K1/2 are presented with more severe osseous manifestations, higher incidence of multiple joint contractures and scoliosis (25).
Since bone homeostasis is a dynamic and multifactorial, process longitudinal monitoring of skeletal manifestations is of high importance as a progressive and worsening pattern is displayed especially in CS and CFC individuals. Hence, compromised independent walking, surgery interventions, limited physical activity are often an issue (25, 55, 89). It is also noteworthy that along with intrinsic bone pathology in RASopathies other factors exert a significant role in final bone phenotype. More precise, some of bones’ features are secondary to other comorbidities such as neurological impairment, intellectual disability, reduced physical activity, hypotonia and lacking musculature (25, 44, 58, 83).
Indeed, generalized muscular hypotonia of varying severity and hypotrophy is a rather consistent feature. Abnormalities on muscle biopsies of CFC/CS individuals, along with data derived from in vitro studies and recent experiments in transgenic mouse models provide further support to the presence of intrinsic myopathy in CS/CFC syndromes (6, 23, 44, 101–103). As a pivotal role of RAS/MAPK signaling cascade in modulation of skeletal myogenesis has been demonstrated, it has been speculated that paucity of muscle fiber and hypotonia could be attributed to impaired myoblast differentiation during the early stages of muscular development (25).
3 Ras/MAPK cascade and bone defects
Research on the molecular etiology of bone defects of NS and other RASopathies focuses mainly on the members of the Ras/MAPK/ERK pathway and, their synergy with other cascades, important for the maintenance of normal bone homeostasis (104) such as Wnt-frizzled/β-catenin, BMP/SMAD/Runx2 and PI3K/AKT (38, 105). Although the effects of the Ras-signaling on osteogenesis are not well defined, emerging evidence suggest that Ras/MAPK pathway is a positive regulator of mesenchymal progenitor cells differentiation into osteoblasts and of osteoblast proliferation during skeletal development (36, 106–108). Recently, it has been demonstrated that inactivation of ERK in osteoprogenitors at an early postnatal stage of skeletal development in mice results in severe bone loss and cleidocranial dysplasia phenotypes (109).
The driver molecules of the Ras transduction signaling are small GTPases of the Ras family(H-ras, K-ras, N-ras), differentially stimulated by diverse upstream effectors, that mediate the translation of extracellular signals in specific genes through downstream cascades such as the well described MAPK axis (Raf-1/mitogen-activated, ERK-activating kinase) (3, 110). Activating mutations in any component of this pathway may result in increased MAPK activity, unrestrained cell proliferation and deviation from the physiological pattern of differentiation.
Among the Ras upstream effectors, SHP2, a protein tyrosine phosphatase encoded by the PTPN11 gene is involved in the regulation of proliferation, differentiation and recruitment of mesenchymal stem cells (MSCs) and of neural crest cells (NCCs) in response to multiple growth factors such as epidermal growth factor (EGF) and platelet derived growth factor (PDGF) via the MAPK pathway (111, 112). It has been shown that MSCs SHP2-deficient mice display multiple abnormal skeletal phenotypes, including postnatal growth retardation, shortened and malformed limbs and pectus excavatum/carinatum (113). Additionally, mice in which SHP2 is deleted in chondrocytes at 4 weeks after birth have scoliosis and kyphosis, a phenotype not seen when the deletion is induced in adult mice (114). In a subsequent study, the lack of SHP2 from hypertrophic chondrocytes showed a significantly decreased number of osteoblasts in the cancellous bone and repressed osteogenic gene expression suggesting a potential role for SHP2 in the regulation of hypertrophic chondrocyte transdifferentiation (115). The significantly diverse phenotypes of the mice lacking SHP2 in proliferating and hypertrophic chondrocytes indicate a developmental stage-specific role for SHP2 in chondrogenesis and bone mineral homeostasis (114), reviewed in (75).
As mentioned earlier, short stature is a common feature of NS. It has been shown that mice bearing PTPN11 gain of function mutations are smaller with increased skull size and hypertelorism (116), while the growth plate length is reduced mainly due to the decrease of the hypertrophic zone (66). NS-causing SHP2 mutants inhibit GH-induced IGF1 release through Ras/MAPK hyperactivation, a mechanism that may lead to growth retardation, suggesting that the short stature seen in PTPN11 NS patients may be alleviated by modulating the activation of this cascade (117).
Activated Ras proteins can also promote or inhibit a number of signaling cascades such as BMP/SMAD/Runx2 and modulate osteogenic gene expression and differentiation. BMP-signaling pathway enhances osteoblast differentiation through SMAD1/5/8, Runx2 and osterix activation (109, 118). In vitro studies on induced pluripotent stem cells from an individual with CFCS that were differentiated into MSC showed enhanced MAPK/ERK activation and SMAD3 hyperactivation leading to an altered osteogenic differentiation with reduced ALP activity and impaired mineralization (82). Similarly, MSC from CS patients showed poor differentiation toward the osteoblastic lineage in vitro, with low ALP and poor mineralization (119). The Wnt/β-catenin (120), PI3K/AKT (121) as well as other pathways involved in bone remodeling are implicated in cross-talk with Ras/MAPK cascade, however the interaction between them is not clear. Some months ago, Yang et al. attributed a new crucial role of KRAS in regulating extracellular matrix accumulation for optimal osteogenesis. It has been shown that stem cells coordinate the expression of small GTPases to modulate autophagy, thereby promoting osteogenic differentiation (122).
The striking complexity of the RAS signaling cascade, the plurality of its molecular components and the interference with various epigenetic factors complicate the understanding of the underlying mechanisms, offering a challenging research field. In this direction, genetic or pharmacologic inhibition of SHP2 has emerged as a critical node for therapeutic applications. Although various SHP2 competitive inhibitors have been reported, only a few allosteric SHP2 inhibitors have been developed and progressed in clinical trials [for detailed review see (75)]. These allosteric SHP2 inhibitors have been extensively studied in cancer (123) and its introduction to other human genetic diseases and musculoskeletal pathologies is currently under investigation (124, 125). Furthermore, a recent study in CS Hras G12V mouse model demonstrated that inhibition of RAS/MAPK signaling pathway using MEK-inhibitor rescued myopathy phenotype both in vitro and in vivo (126). In accordance, Nandi et al. using the same mouse model revealed that mutated Hras contributed to increased osteoclastogenesis, which was reduced by in vitro treatment with MEK and PI3K inhibitors (127). The above results were further supported by another study on an activating Braf L597V CFC mouse model. In particular, Ras/MAPK dysregulation during development inhibited myofiber differentiation which was rescued by MEK inhibition (102). What is more, trametinib, a highly selective reversible allosteric inhibitor of MEK1/2 was shown to restrict myocardial hypertrophy in NS RIT1 mutants (128). Of note, currently the effects of gemline mutations on energy metabolism and mitochondria is being investigated (129). A recent review in multiple Hras CS models highlighted the importance of bioenergetics and mitochondrial proteostasis in heart and skeletal muscles and proposed that CS patients may benefit from regimen with mitochondrial modulators (130). Certainly, high quality clinical studies are warranted to shed light on various aspects and limitations which accompany the novel targeted therapies (23, 75).
4 Conclusions
As evidenced there is significant overlap in addition to discordance of the musculoskeletal phenotypes between Noonan, Costello and CFC syndromes. The rarity of the syndromes and the wide range of patients’ age make it difficult to draw safe conclusions in order to clearly define the clinical and biochemical patients’ profile. The clinical complexity reflects the interactions of an extensive molecular network, with the Ras cascade as a starting point and driver. However, the dissociation of some skeletal manifestations (for example chest deformities) from the Ras/MAPK effect on both osteoclastic and osteoblastic cells suggests a possible impact of this cascade on different cell lines interfering with musculoskeletal development. The elucidation of the biological and genetic background of these diseases may reveal novel target therapies not only for musculoskeletal phenotypes but also for other RASopathies’ clinical manifestations, as well as for different entities, such as cancer and autoimmune disorders.
Author contributions
AP and EB contributed to the conception, design, drafting of the work and revised it critically for important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Noonan JA ED. Associated noncardiac malformations in children with congenital heart disease. Pediatrics (1963) 63:468–70.
2. Tidyman WE, Rauen KA. The rasopathies: developmental syndromes of ras/mapk pathway dysregulation. Curr Opin Genet Dev (2009) 19(3):230–6. doi: 10.1016/j.gde.2009.04.001
3. Zhang W, Liu HT. Mapk signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res (2002) 12(1):9–18. doi: 10.1038/sj.cr.7290105
4. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet (2013) 381(9863):333–42. doi: 10.1016/S0140-6736(12)61023-X
5. The ajmg sequence: Decoding news and trends for the medical genetics community by roxanne nelson. Am J Med Genet A (2019) 179(8):1408–9. doi: 10.1002/ajmg.a.40456. Nelson.
7. Leoni C, Blandino R, Delogu AB, De Rosa G, Onesimo R, Verusio V, et al. Genotype-cardiac phenotype correlations in a large single-center cohort of patients affected by rasopathies: clinical implications and literature review. Am J Med Genet A (2022) 188(2):431–45. doi: 10.1002/ajmg.a.62529
8. Papadopoulos G, Papadopoulou A, Kosma K, Papadimitriou A, Papaevangelou V, Kanaka-Gantenbein C, et al. Molecular and clinical profile of patients referred as noonan or noonan-like syndrome in Greece: A cohort of 86 patients. Eur J Pediatr (2022) 181(10):3691–700. doi: 10.1007/s00431-022-04574-w
9. Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, et al. Gain-of-function sos1 mutations cause a distinctive form of noonan syndrome. Nat Genet (2007) 39(1):75–9. doi: 10.1038/ng1939
10. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in ptpn11, encoding the protein tyrosine phosphatase shp-2, cause noonan syndrome. Nat Genet (2001) 29(4):465–8. doi: 10.1038/ng772
11. Cirstea IC, Gremer L, Dvorsky R, Zhang SC, Piekorz RP, Zenker M, et al. Diverging gain-of-function mechanisms of two novel kras mutations associated with noonan and cardio-facio-cutaneous syndromes. Hum Mol Genet (2013) 22(2):262–70. doi: 10.1093/hmg/dds426
12. Cordeddu V, Di Schiavi E, Pennacchio LA, Ma'ayan A, Sarkozy A, Fodale V, et al. Mutation of shoc2 promotes aberrant protein N-myristoylation and causes noonan-like syndrome with loose anagen hair. Nat Genet (2009) 41(9):1022–6. doi: 10.1038/ng.425
13. Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, et al. Germline kras mutations cause noonan syndrome. Nat Genet (2006) 38(3):331–6. doi: 10.1038/ng1748
14. Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K, et al. Gain-of-function mutations in rit1 cause noonan syndrome, a ras/mapk pathway syndrome. Am J Hum Genet (2013) 93(1):173–80. doi: 10.1016/j.ajhg.2013.05.021
15. Lee ST, Ki CS, Lee HJ. Mutation analysis of the genes involved in the ras-mitogen-activated protein kinase (Mapk) pathway in korean patients with noonan syndrome. Clin Genet (2007) 72(2):150–5. doi: 10.1111/j.1399-0004.2007.00839.x
16. Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, et al. Germline gain-of-function mutations in raf1 cause noonan syndrome. Nat Genet (2007) 39(8):1013–7. doi: 10.1038/ng2078
17. Brand K, Kentsch H, Glashoff C, Rosenberger G. Rasopathy-associated cbl germline mutations cause aberrant ubiquitylation and trafficking of egfr. Hum Mutat (2014) 35(11):1372–81. doi: 10.1002/humu.22682
18. Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, et al. Germline mutations in hras proto-oncogene cause costello syndrome. Nat Genet (2005) 37(10):1038–40. doi: 10.1038/ng1641
19. Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, et al. Germline mutations in genes within the mapk pathway cause cardio-facio-cutaneous syndrome. Science (2006) 311(5765):1287–90. doi: 10.1126/science.1124642
20. Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, et al. Germline kras and braf mutations in cardio-facio-cutaneous syndrome. Nat Genet (2006) 38(3):294–6. doi: 10.1038/ng1749
21. Yassir WK, Grottkau BE, Goldberg MJ. Costello syndrome: orthopaedic manifestations and functional health. J Pediatr Orthop (2003) 23(1):94–8. doi: 10.1097/01241398-200301000-00019
22. Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, Pierpont ME, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics (2010) 126(4):746–59. doi: 10.1542/peds.2009-3207
23. Fowlkes JL, Thrailkill KM, Bunn RC. Rasopathies: the musculoskeletal consequences and their etiology and pathogenesis. Bone (2021) 152:116060. doi: 10.1016/j.bone.2021.116060
24. Stevenson DA, Yang FC. The musculoskeletal phenotype of the rasopathies. Am J Med Genet C Semin Med Genet (2011) 157C(2):90–103. doi: 10.1002/ajmg.c.30296
25. Leoni C, Romeo DM, Pelliccioni M, Di Gia M, Onesimo R, Giorgio V, et al. Musculo-skeletal phenotype of costello syndrome and cardio-facio-cutaneous syndrome: insights on the functional assessment status. Orphanet J Rare Dis (2021) 16(1):43. doi: 10.1186/s13023-021-01674-y
27. White SM, Graham JM Jr., Kerr B, Gripp K, Weksberg R, Cytrynbaum C, et al. The adult phenotype in costello syndrome. Am J Med Genet A (2005) 136(2):128–35. doi: 10.1002/ajmg.a.30747
28. Leoni C, Stevenson DA, Martini L, De Sanctis R, Mascolo G, Pantaleoni F, et al. Decreased bone mineral density in costello syndrome. Mol Genet Metab (2014) 111(1):41–5. doi: 10.1016/j.ymgme.2013.08.007
29. Tucker T, Schnabel C, Hartmann M, Friedrich RE, Frieling I, Kruse HP, et al. Bone health and fracture rate in individuals with neurofibromatosis 1 (Nf1). J Med Genet (2009) 46(4):259–65. doi: 10.1136/jmg.2008.061895
30. Stevenson DA, Moyer-Mileur LJ, Murray M, Slater H, Sheng X, Carey JC, et al. Bone mineral density in children and adolescents with neurofibromatosis type 1. J Pediatr (2007) 150(1):83–8. doi: 10.1016/j.jpeds.2006.10.048
31. Yilmaz K, Ozmen M, Bora Goksan S, Eskiyurt N. Bone mineral density in children with neurofibromatosis 1. Acta Paediatr (2007) 96(8):1220–2. doi: 10.1111/j.1651-2227.2007.00401.x
32. Lammert M, Kappler M, Mautner VF, Lammert K, Storkel S, Friedman JM, et al. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos Int (2005) 16(9):1161–6. doi: 10.1007/s00198-005-1940-2
33. Choudhry KS, Grover M, Tran AA, O'Brian Smith E, Ellis KJ, Lee BH. Decreased bone mineralization in children with noonan syndrome: another consequence of dysregulated ras mapkinase pathway? Mol Genet Metab (2012) 106(2):237–40. doi: 10.1016/j.ymgme.2012.04.003
34. Stevenson DA, Schwarz EL, Carey JC, Viskochil DH, Hanson H, Bauer S, et al. Bone resorption in syndromes of the ras/mapk pathway. Clin Genet (2011) 80(6):566–73. doi: 10.1111/j.1399-0004.2010.01619.x
35. Leoni C, Bisanti C, Viscogliosi G, Onesimo R, Massese M, Giorgio V, et al. Bone tissue homeostasis and risk of fractures in costello syndrome: A 4-year follow-up study. Am J Med Genet A (2022) 188(2):422–30. doi: 10.1002/ajmg.a.62615
36. Reinker KA, Stevenson DA, Tsung A. Orthopaedic conditions in ras/mapk related disorders. J Pediatr Orthop (2011) 31(5):599–605. doi: 10.1097/BPO.0b013e318220396e
37. Delagrange M, Rousseau V, Cessans C, Pienkowski C, Oliver I, Jouret B, et al. Low bone mass in noonan syndrome children correlates with decreased muscle mass and low igf-1 levels. Bone (2021) 153:116170. doi: 10.1016/j.bone.2021.116170
38. Ulici V, Hoenselaar KD, Gillespie JR, Beier F. The pi3k pathway regulates endochondral bone growth through control of hypertrophic chondrocyte differentiation. BMC Dev Biol (2008) 8:40. doi: 10.1186/1471-213X-8-40
39. Gripp KW, Rauen KA. Costello Syndrome. (2006) (Seattle WA: University of Washington, Seattle) 1993–2023.
40. Zenker M. Clinical overview on rasopathies. Am J Med Genet C Semin Med Genet (2022) 190(4):414–24. doi: 10.1002/ajmg.c.32015
42. Choi N, Ko JM, Shin SH, Kim EK, Kim HS, Song MK, et al. Phenotypic and genetic characteristics of five korean patients with costello syndrome. Cytogenet Genome Res (2019) 158(4):184–91. doi: 10.1159/000502045
43. Leoni C, Viscogliosi G, Onesimo R, Bisanti C, Massese M, Giorgio V, et al. Characterization of bone homeostasis in individuals affected by cardio-facio-cutaneous syndrome. Am J Med Genet A (2022) 188(2):414–21. doi: 10.1002/ajmg.a.62588
44. Stevenson DA, Viscogliosi G, Leoni C. Bone health in rasopathies. Am J Med Genet C Semin Med Genet (2022) 190(4):459–70. doi: 10.1002/ajmg.c.32020
45. Lee Y, Choi Y, Seo GH, Kim GH, Choi IH, Keum C, et al. Clinical and molecular spectra of braf-associated rasopathy. J Hum Genet (2021) 66(4):389–99. doi: 10.1038/s10038-020-00852-3
46. Noonan JA, Raaijmakers R, Hall BD. Adult height in noonan syndrome. Am J Med Genet A (2003) 123A(1):68–71. doi: 10.1002/ajmg.a.20502
47. Abe Y, Aoki Y, Kuriyama S, Kawame H, Okamoto N, Kurosawa K, et al. Prevalence and clinical features of costello syndrome and cardio-facio-cutaneous syndrome in Japan: findings from a nationwide epidemiological survey. Am J Med Genet A (2012) 158A(5):1083–94. doi: 10.1002/ajmg.a.35292
48. Siano MA, Pivonello R, Salerno M, Falco M, Mauro C, De Brasi D, et al. Endocrine system involvement in patients with rasopathies: A case series. Front Endocrinol (Lausanne) (2022) 13:1030398. doi: 10.3389/fendo.2022.1030398
49. Armour CM, Allanson JE. Further delineation of cardio-facio-cutaneous syndrome: clinical features of 38 individuals with proven mutations. J Med Genet (2008) 45(4):249–54. doi: 10.1136/jmg.2007.054460
50. Allanson JE, Anneren G, Aoki Y, Armour CM, Bondeson ML, Cave H, et al. Cardio-facio-cutaneous syndrome: does genotype predict phenotype? Am J Med Genet C Semin Med Genet (2011) 157C(2):129–35. doi: 10.1002/ajmg.c.30295
51. Chinton J, Huckstadt V, Moresco A, Gravina LP, Obregon MG. Clinical and molecular characterization of children with noonan syndrome and other rasopathies in Argentina. Arch Argent Pediatr (2019) 117(5):330–7. doi: 10.5546/aap.2019.eng.330
52. Witt DR, Keena BA, Hall JG, Allanson JE. Growth curves for height in noonan syndrome. Clin Genet (1986) 30(3):150–3. doi: 10.1111/j.1399-0004.1986.tb00587.x
53. Sammon MR, Doyle D, Hopkins E, Sol-Church K, Stabley DL, McGready J, et al. Normative growth charts for individuals with costello syndrome. Am J Med Genet A (2012) 158A(11):2692–9. doi: 10.1002/ajmg.a.35534
54. Leoni C, Onesimo R, Giorgio V, Diamanti A, Giorgio D, Martini L, et al. Understanding growth failure in costello syndrome: increased resting energy expenditure. J Pediatr (2016) 170:322–4. doi: 10.1016/j.jpeds.2015.11.076
55. Leoni C, Viscogliosi G, Tartaglia M, Aoki Y, Zampino G. Multidisciplinary management of costello syndrome: current perspectives. J Multidiscip Healthc (2022) 15:1277–96. doi: 10.2147/JMDH.S291757
56. Gripp KW, Morse LA, Axelrad M, Chatfield KC, Chidekel A, Dobyns W, et al. Costello syndrome: clinical phenotype, genotype, and management guidelines. Am J Med Genet A (2019) 179(9):1725–44. doi: 10.1002/ajmg.a.61270
57. Hennekam RC. Costello syndrome: an overview. Am J Med Genet C Semin Med Genet (2003) 117C(1):42–8. doi: 10.1002/ajmg.c.10019
58. Leoni C, Triumbari EKA, Vollono C, Onesimo R, Podagrosi M, Giorgio V, et al. Pain in individuals with rasopathies: prevalence and clinical characterization in a sample of 80 affected patients. Am J Med Genet A (2019) 179(6):940–7. doi: 10.1002/ajmg.a.61111
59. Shikany AR, Baker L, Stabley DL, Robbins K, Doyle D, Gripp KW, et al. Medically actionable comorbidities in adults with costello syndrome. Am J Med Genet A (2020) 182(1):130–6. doi: 10.1002/ajmg.a.61394
60. Tartaglia M, Gelb BD. Noonan syndrome and related disorders: genetics and pathogenesis. Annu Rev Genomics Hum Genet (2005) 6:45–68. doi: 10.1146/annurev.genom.6.080604.162305
61. Lee BH, Kim JM, Jin HY, Kim GH, Choi JH, Yoo HW. Spectrum of mutations in noonan syndrome and their correlation with phenotypes. J Pediatr (2011) 159(6):1029–35. doi: 10.1016/j.jpeds.2011.05.024
62. Kobayashi T, Aoki Y, Niihori T, Cave H, Verloes A, Okamoto N, et al. Molecular and clinical analysis of raf1 in noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum Mutat (2010) 31(3):284–94. doi: 10.1002/humu.21187
63. Mazzanti L, Tamburrino F, Scarano E, Perri A, Vestrucci B, Guidetti M, et al. Gh therapy and first final height data in noonan-like syndrome with loose anagen hair (Mazzanti syndrome). Am J Med Genet A (2013) 161A(11):2756–61. doi: 10.1002/ajmg.a.36255
64. Binder G, Neuer K, Ranke MB, Wittekindt NE. Ptpn11 mutations are associated with mild growth hormone resistance in individuals with noonan syndrome. J Clin Endocrinol Metab (2005) 90(9):5377–81. doi: 10.1210/jc.2005-0995
65. Bauler TJ, Kamiya N, Lapinski PE, Langewisch E, Mishina Y, Wilkinson JE, et al. Development of severe skeletal defects in induced shp-2-deficient adult mice: A model of skeletal malformation in humans with shp-2 mutations. Dis Model Mech (2011) 4(2):228–39. doi: 10.1242/dmm.006130
66. Tajan M, Pernin-Grandjean J, Beton N, Gennero I, Capilla F, Neel BG, et al. Noonan syndrome-causing shp2 mutants impair erk-dependent chondrocyte differentiation during endochondral bone growth. Hum Mol Genet (2018) 27(13):2276–89. doi: 10.1093/hmg/ddy133
67. Sodero G, Cipolla C, Pane LC, Sessa L, Malavolta E, Arzilli F, et al. Efficacy and safety of growth hormone therapy in children with noonan syndrome. Growth Horm IGF Res (2023) 69-70:101532. doi: 10.1016/j.ghir.2023.101532
68. Dahlgren J, Noordam C. Growth, endocrine features, and growth hormone treatment in noonan syndrome. J Clin Med (2022) 11(7):2034. doi: 10.3390/jcm11072034
69. Seo GH, Yoo HW. Growth hormone therapy in patients with noonan syndrome. Ann Pediatr Endocrinol Metab (2018) 23(4):176–81. doi: 10.6065/apem.2018.23.4.176
70. Kratz CP, Niemeyer CM, Castleberry RP, Cetin M, Bergstrasser E, Emanuel PD, et al. The mutational spectrum of ptpn11 in juvenile myelomonocytic leukemia and noonan syndrome/myeloproliferative disease. Blood (2005) 106(6):2183–5. doi: 10.1182/blood-2005-02-0531
71. Baldassarre G, Mussa A, Carli D, Molinatto C, Ferrero GB. Constitutional bone impairment in noonan syndrome. Am J Med Genet A (2017) 173(3):692–8. doi: 10.1002/ajmg.a.38086
72. Noordam C, Span J, van Rijn RR, Gomes-Jardin E, van Kuijk C, Otten BJ. Bone mineral density and body composition in noonan's syndrome: effects of growth hormone treatment. J Pediatr Endocrinol Metab (2002) 15(1):81–7. doi: 10.1515/jpem.2002.15.1.81
73. Smpokou P, Tworog-Dube E, Kucherlapati RS, Roberts AE. Medical complications, clinical findings, and educational outcomes in adults with noonan syndrome. Am J Med Genet A (2012) 158A(12):3106–11. doi: 10.1002/ajmg.a.35639
74. Wang L, Yang H, Huang J, Pei S, Feng JQ, Jing D, et al. Targeted ptpn11 deletion in mice reveals the essential role of shp2 in osteoblast differentiation and skeletal homeostasis. Bone Res (2021) 9(1):6. doi: 10.1038/s41413-020-00129-7
75. Jensen NR, Kelly RR, Kelly KD, Khoo SK, Sidles SJ, LaRue AC. From stem to sternum: the role of shp2 in the skeleton. Calcif Tissue Int (2023) 112(4):403–21. doi: 10.1007/s00223-022-01042-3
76. Takagi M, Miyashita Y, Koga M, Ebara S, Arita N, Kasayama S. Estrogen deficiency is a potential cause for osteopenia in adult male patients with noonan's syndrome. Calcif Tissue Int (2000) 66(3):200–3. doi: 10.1007/s002230010040
77. Detweiler S, Thacker MM, Hopkins E, Conway L, Gripp KW. Orthopedic manifestations and implications for individuals with costello syndrome. Am J Med Genet A (2013) 161A(8):1940–9. doi: 10.1002/ajmg.a.36047
78. Digilio MC, Sarkozy A, Capolino R, Chiarini Testa MB, Esposito G, de Zorzi A, et al. Costello syndrome: clinical diagnosis in the first year of life. Eur J Pediatr (2008) 167(6):621–8. doi: 10.1007/s00431-007-0558-0
79. Cakir M, Arici C, Tacoy S, Karayalcin U. A case of costello with parathyroid adenoma and hyperprolactinemia. Am J Med Genet A (2004) 124A(2):196–9. doi: 10.1002/ajmg.a.20361
80. Chen J, Che L, Xu C, Zhao S, Yang J, Li M, et al. Cardio-facio-cutaneous syndrome-associated pathogenic map2k1 variants activate autophagy. Gene (2020) 733:144369. doi: 10.1016/j.gene.2020.144369
81. Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q, et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature (2013) 499(7459):491–5. doi: 10.1038/nature12396
82. Choi JY, Han KM, Kim D, Lee BH, Yoo HW, Choi JH, et al. Impaired osteogenesis of disease-specific induced pluripotent stem cells derived from a cfc syndrome patient. Int J Mol Sci (2017) 18(12):2591. doi: 10.3390/ijms18122591
83. Johnson B, Goldberg-Strassler D, Gripp K, Thacker M, Leoni C, Stevenson D. Function and disability in children with costello syndrome and cardiofaciocutaneous syndrome. Am J Med Genet A (2015) 167A(1):40–4. doi: 10.1002/ajmg.a.36828
84. Goodwin AF, Oberoi S, Landan M, Charles C, Groth J, Martinez A, et al. Craniofacial and dental development in cardio-facio-cutaneous syndrome: the importance of ras signaling homeostasis. Clin Genet (2013) 83(6):539–44. doi: 10.1111/cge.12005
85. Goodwin AF, Oberoi S, Landan M, Charles C, Massie JC, Fairley C, et al. Craniofacial and dental development in costello syndrome. Am J Med Genet A (2014) 164A(6):1425–30. doi: 10.1002/ajmg.a.36475
86. Zenker M, Edouard T, Blair JC, Cappa M. Noonan syndrome: improving recognition and diagnosis. Arch Dis Child (2022) 107(12):1073–8. doi: 10.1136/archdischild-2021-322858
87. Roberts A. Noonan Syndrome. GeneReviews®. Adam MP, Mirzaa GM, Pagon RA, et al, editors. Seattle (WA: University of Washington, Seattle (2001) p. 1993–2023.
88. Shaw AC, Kalidas K, Crosby AH, Jeffery S, Patton MA. The natural history of noonan syndrome: A long-term follow-up study. Arch Dis Child (2007) 92(2):128–32. doi: 10.1136/adc.2006.104547
89. Pierpont ME, Magoulas PL, Adi S, Kavamura MI, Neri G, Noonan J, et al. Cardio-facio-cutaneous syndrome: clinical features, diagnosis, and management guidelines. Pediatrics (2014) 134(4):e1149–62. doi: 10.1542/peds.2013-3189
90. Ueda K, Yaoita M, Niihori T, Aoki Y, Okamoto N. Craniosynostosis in patients with rasopathies: accumulating clinical evidence for expanding the phenotype. Am J Med Genet A (2017) 173(9):2346–52. doi: 10.1002/ajmg.a.38337
91. Takenouchi T, Sakamoto Y, Miwa T, Torii C, Kosaki R, Kishi K, et al. Severe craniosynostosis with noonan syndrome phenotype associated with shoc2 mutation: clinical evidence of crosslink between fgfr and ras signaling pathways. Am J Med Genet A (2014) 164A(11):2869–72. doi: 10.1002/ajmg.a.36705
92. Davis AA, Zuccoli G, Haredy MM, Losee J, Pollack IF, Madan-Khetarpal S, et al. Rasopathy in patients with isolated sagittal synostosis. Glob Pediatr Health (2019) 6:2333794X19846774. doi: 10.1177/2333794X19846774
93. Addissie YA, Kotecha U, Hart RA, Martinez AF, Kruszka P, Muenke M. Craniosynostosis and noonan syndrome with kras mutations: expanding the phenotype with a case report and review of the literature. Am J Med Genet A (2015) 167A(11):2657–63. doi: 10.1002/ajmg.a.37259
94. Kratz CP, Zampino G, Kriek M, Kant SG, Leoni C, Pantaleoni F, et al. Craniosynostosis in patients with noonan syndrome caused by germline kras mutations. Am J Med Genet A (2009) 149A(5):1036–40. doi: 10.1002/ajmg.a.32786
95. Nagai K, Niihori T, Okamoto N, Kondo A, Suga K, Ohhira T, et al. Duplications in the G3 domain or switch ii region in hras identified in patients with costello syndrome. Hum Mutat (2022) 43(1):3–15. doi: 10.1002/humu.24287
96. Rios-Gonzalez BE, Rodriguez-Ortiz JF, Castro-Martinez AG, Magana-Torres MT, Barros-Nunez P. Clinical and molecular characterization of costello syndrome in unrelated mexican patients. Clin Dysmorphol (2022) 31(2):55–8. doi: 10.1097/MCD.0000000000000405
97. Lee CK, Chang BS, Hong YM, Yang SW, Lee CS, Seo JB. Spinal deformities in noonan syndrome: A clinical review of sixty cases. J Bone Joint Surg Am (2001) 83(10):1495–502. doi: 10.2106/00004623-200110000-00006
98. Machida M, Rocos B, Taira K, Nemoto N, Oikawa N, Ohashi H, et al. Costello syndrome-associated orthopaedic manifestations focussed on kyphoscoliosis: A case series describing the natural course. J Pediatr Orthop B (2022) 32(4):357–62. doi: 10.1097/BPB.0000000000001013
99. Dong X, Png NCY, Fortier MV, Lim JY, Wong KPL, Choo JTL, et al. Fibrous dysplasia in cardio-facio-cutaneous syndrome: A case report and review of literature. Am J Med Genet A (2022) 188(9):2732–7. doi: 10.1002/ajmg.a.62879
100. Bochet P, Ramond F, Touraine R, Thomas T, Marotte H. Arthritis associated to cardio-facio-cutaneous syndrome related to a map2k1 mutation. Joint Bone Spine (2020) 87(2):169. doi: 10.1016/j.jbspin.2019.09.014
101. Tidyman WE, Lee HS, Rauen KA. Skeletal muscle pathology in costello and cardio-facio-cutaneous syndromes: developmental consequences of germline ras/mapk activation on myogenesis. Am J Med Genet C Semin Med Genet (2011) 157C(2):104–14. doi: 10.1002/ajmg.c.30298
102. Maeda Y, Tidyman WE, Ander BP, Pritchard CA, Rauen KA. Ras/mapk dysregulation in development causes a skeletal myopathy in an activating braf(L597v) mouse model for cardio-facio-cutaneous syndrome. Dev Dyn (2021) 250(8):1074–95. doi: 10.1002/dvdy.309
103. de Boode WP, Semmekrot BA, ter Laak HJ, van der Burgt CJ, Draaisma JM, Lommen EJ, et al. Myopathology in patients with a noonan phenotype. Acta Neuropathol (1996) 92(6):597–602. doi: 10.1007/s004010050566
104. Schindeler A, Little DG. Ras-mapk signaling in osteogenic differentiation: friend or foe? J Bone Miner Res (2006) 21(9):1331–8. doi: 10.1359/jbmr.060603
105. McGonnell IM, Grigoriadis AE, Lam EW, Price JS, Sunters A. A specific role for phosphoinositide 3-kinase and akt in osteoblasts? Front Endocrinol (Lausanne) (2012) 3:88. doi: 10.3389/fendo.2012.00088
106. Xiao G, Jiang D, Thomas P, Benson MD, Guan K, Karsenty G, et al. Mapk pathways activate and phosphorylate the osteoblast-specific transcription factor, cbfa1. J Biol Chem (2000) 275(6):4453–9. doi: 10.1074/jbc.275.6.4453
107. Xiao G, Jiang D, Gopalakrishnan R, Franceschi RT. Fibroblast growth factor 2 induction of the osteocalcin gene requires mapk activity and phosphorylation of the osteoblast transcription factor, cbfa1/runx2. J Biol Chem (2002) 277(39):36181–7. doi: 10.1074/jbc.M206057200
108. Papaioannou G, Mirzamohammadi F, Kobayashi T. Ras signaling regulates osteoprogenitor cell proliferation and bone formation. Cell Death Dis (2016) 7(10):e2405. doi: 10.1038/cddis.2016.314
109. Kim JM, Yang YS, Park KH, Oh H, Greenblatt MB, Shim JH. The erk mapk pathway is essential for skeletal development and homeostasis. Int J Mol Sci (2019) 20(8):1803. doi: 10.3390/ijms20081803
110. Seger R, Krebs EG. The mapk signaling cascade. FASEB J (1995) 9(9):726–35. doi: 10.1096/fasebj.9.9.7601337
111. Nakamura T, Gulick J, Colbert MC, Robbins J. Protein tyrosine phosphatase activity in the neural crest is essential for normal heart and skull development. Proc Natl Acad Sci U.S.A. (2009) 106(27):11270–5. doi: 10.1073/pnas.0902230106
112. Agazie YM, Hayman MJ. Molecular mechanism for a role of shp2 in epidermal growth factor receptor signaling. Mol Cell Biol (2003) 23(21):7875–86. doi: 10.1128/MCB.23.21.7875-7886.2003
113. Lapinski PE, Meyer MF, Feng GS, Kamiya N, King PD. Deletion of shp-2 in mesenchymal stem cells causes growth retardation, limb and chest deformity, and calvarial defects in mice. Dis Model Mech (2013) 6(6):1448–58. doi: 10.1242/dmm.012849
114. Wang L, Huang J, Moore DC, Zuo C, Wu Q, Xie L, et al. Shp2 regulates the osteogenic fate of growth plate hypertrophic chondrocytes. Sci Rep (2017) 7(1):12699. doi: 10.1038/s41598-017-12767-9
115. Tajan M, Paccoud R, Branka S, Edouard T, Yart A. The rasopathy family: consequences of germline activation of the ras/mapk pathway. Endocr Rev (2018) 39(5):676–700. doi: 10.1210/er.2017-00232
116. Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, et al. Mouse model of noonan syndrome reveals cell type- and gene dosage-dependent effects of ptpn11 mutation. Nat Med (2004) 10(8):849–57. doi: 10.1038/nm1084
117. De Rocca Serra-Nedelec A, Edouard T, Treguer K, Tajan M, Araki T, Dance M, et al. Noonan syndrome-causing shp2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced erk hyperactivation, which contributes to short stature. Proc Natl Acad Sci U.S.A. (2012) 109(11):4257–62. doi: 10.1073/pnas.1119803109
118. Wu M, Chen G, Li YP. Tgf-beta and bmp signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res (2016) 4:16009. doi: 10.1038/boneres.2016.9
119. Choi JB, Lee J, Kang M, Kim B, Ju Y, Do HS, et al. Dysregulated ecm remodeling proteins lead to aberrant osteogenesis of costello syndrome ipscs. Stem Cell Rep (2021) 16(8):1985–98. doi: 10.1016/j.stemcr.2021.06.007
120. Westendorf JJ, Kahler RA, Schroeder TM. Wnt signaling in osteoblasts and bone diseases. Gene (2004) 341:19–39. doi: 10.1016/j.gene.2004.06.044
121. Edouard T, Combier JP, Nedelec A, Bel-Vialar S, Metrich M, Conte-Auriol F, et al. Functional effects of ptpn11 (Shp2) mutations causing leopard syndrome on epidermal growth factor-induced phosphoinositide 3-kinase/akt/glycogen synthase kinase 3beta signaling. Mol Cell Biol (2010) 30(10):2498–507. doi: 10.1128/MCB.00646-09
122. Yang YY, Soh R, Vera-Colon M, Huang M, Zur Nieden NI, Wang Y. Targeted proteomic profiling revealed roles of small gtpases during osteogenic differentiation. Anal Chem (2023) 95(17):6879–87. doi: 10.1021/acs.analchem.2c05781
123. Kerr DL, Haderk F, Bivona TG. Allosteric shp2 inhibitors in cancer: targeting the intersection of ras, resistance, and the immune microenvironment. Curr Opin Chem Biol (2021) 62:1–12. doi: 10.1016/j.cbpa.2020.11.007
124. Gripp KW, Schill L, Schoyer L, Stronach B, Bennett AM, Blaser S, et al. The sixth international rasopathies symposium: precision medicine-from promise to practice. Am J Med Genet A (2020) 182(3):597–606. doi: 10.1002/ajmg.a.61434
125. Wang J, Huang L, Huang Y, Jiang Y, Zhang L, Feng G, et al. Therapeutic effect of the injectable thermosensitive hydrogel loaded with shp099 on intervertebral disc degeneration. Life Sci (2021) 266:118891. doi: 10.1016/j.lfs.2020.118891
126. Tidyman WE, Goodwin AF, Maeda Y, Klein OD, Rauen KA. Mek-inhibitor-mediated rescue of skeletal myopathy caused by activating hras mutation in a costello syndrome mouse model. Dis Model Mech (2022) 15(2). doi: 10.1242/dmm.049166
127. Nandi S, Chennappan S, Andrasch Y, Fidan M, Engler M, Ahmad M, et al. Increased osteoclastogenesis contributes to bone loss in the costello syndrome hras G12v mouse model. Front Cell Dev Biol (2022) 10:1000575. doi: 10.3389/fcell.2022.1000575
128. Andelfinger G, Marquis C, Raboisson MJ, Theoret Y, Waldmuller S, Wiegand G, et al. Hypertrophic cardiomyopathy in noonan syndrome treated by mek-inhibition. J Am Coll Cardiol (2019) 73(17):2237–9. doi: 10.1016/j.jacc.2019.01.066
129. Bajia D, Bottani E, Derwich K. Effects of noonan syndrome-germline mutations on mitochondria and energy metabolism. Cells (2022) 11(19):3099. doi: 10.3390/cells11193099
Keywords: CFC, bone homeostasis, skeletal dysmorphisms, RAS/MAPK signaling, RASopathies, short stature, Noonan, Costello
Citation: Papadopoulou A and Bountouvi E (2023) Skeletal defects and bone metabolism in Noonan, Costello and cardio-facio-cutaneous syndromes. Front. Endocrinol. 14:1231828. doi: 10.3389/fendo.2023.1231828
Received: 30 May 2023; Accepted: 18 October 2023;
Published: 27 October 2023.
Edited by:
Marco Tartaglia, Bambino Gesù Children’s Hospital (IRCCS), ItalyReviewed by:
Chiara Leoni, Agostino Gemelli University Polyclinic (IRCCS), ItalyDidier Lacombe, Centre Hospitalier Universitaire de Bordeaux, France
Marielle Elizabeth Yohe, National Institutes of Health (NIH), United States
Copyright © 2023 Papadopoulou and Bountouvi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Papadopoulou, YW5wYXBhZG9AbWVkLnVvYS5ncg==
†These authors have contributed equally to this work