 Xuan Zhao
Xuan Zhao Lu-Yao Bie2
Lu-Yao Bie2 Dao-Ran Pang
Dao-Ran Pang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 21 September 2023
Sec. Diabetes: Molecular Mechanisms
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1228045
This article is part of the Research Topic Metaflammation in Obesity and Diabetes View all 8 articles
Type II diabetes mellitus (T2DM) is a chronic metabolic disease characterized by prolonged hyperglycemia and insulin resistance (IR). Its incidence is increasing annually, posing a significant threat to human life and health. Consequently, there is an urgent requirement to discover effective drugs and investigate the pathogenesis of T2DM. Autophagy plays a crucial role in maintaining normal islet structure. However, in a state of high glucose, autophagy is inhibited, resulting in impaired islet function, insulin resistance, and complications. Studies have shown that modulating autophagy through activation or inhibition can have a positive impact on the treatment of T2DM and its complications. However, it is important to note that the specific regulatory mechanisms vary depending on the target organ. This review explores the role of autophagy in the pathogenesis of T2DM, taking into account both genetic and external factors. It also provides a summary of reported chemical drugs and traditional Chinese medicine that target the autophagic pathway for the treatment of T2DM and its complications.
Autophagy, also known as self-cleaning and self-eating, is a conserved biological process in eukaryotes (1). Autophagy can provide nutrients to maintain cellular function by breaking down macromolecules, organelles, proteins, and end products, under starvation conditions. Moreover, it also helps in maintaining cellular homeostasis by eliminating damaged organelles, misfolded proteins, and lipid droplets (2). There are two types of autophagy: selective and non-selective, which are triggered by different signals. While selective and non-selective autophagy respond to different signals, both pathways converge to initiate autophagy (3). In mammals, autophagy initiation is linked to the omegasome, a functional domain of the endoplasmic reticulum (ER) that is rich in the lipid phosphatidylinositol 3-phosphate (PI3P). Starting from the omegasome, the isolation membrane elongates into a cup-shaped structure and initiates the process of phagocytosing intracellular material (4). Overall, autophagy is a process that can be divided into four steps (Figure 1). The first step is initiation, where the ULK1-dominant protein complex triggers the formation of another important protein complex called PI3KC3-C1. These two complexes are then recruited to the phagophore assembly site, where they assist in the formation of autophagosomes. The second step is extension. ATG12 is activated by ATG7, leading to the formation of an ATG12-ATG5 complex. This complex then interacts with either ATG16 or ATG16L1 to form the ATG12-ATG5-ATG16L complex. Simultaneously, ATG7 facilitates the binding of phosphatidylethanolamine (PE) to LC3-I, resulting in the formation of LC3-II. The sequential assembly of these protein-protein and protein-lipid complexes enables the extension of the autophagosome bilayer. The third step involves the maturation of autophagosomes, where closed autophagosomes are formed. In the fourth step, autophagosomes fuse sequentially with lysosomes to form autophagic lysosomes. Subsequently, the contents are digested by lysosomal hydrolases, and the resulting metabolites become available for recycling. The identification of crucial genes for autophagy in yeast has revolutionized our comprehension of mammalian physiology and human pathophysiology. To date, over 35 autophagy-related genes have been identified in yeast (5). Among these genes, the 15 core genes necessary for starvation-induced autophagy and selective autophagy are highly conserved in mammals (6). Autophagy-associated gene mutations have been linked to numerous human diseases (7). During the progression of chronic diseases, the accumulation of damaged organelles, protein aggregates, lipid droplets, and aging cells has been observed, which is speculated to be related to the disruption of autophagy (8, 9).
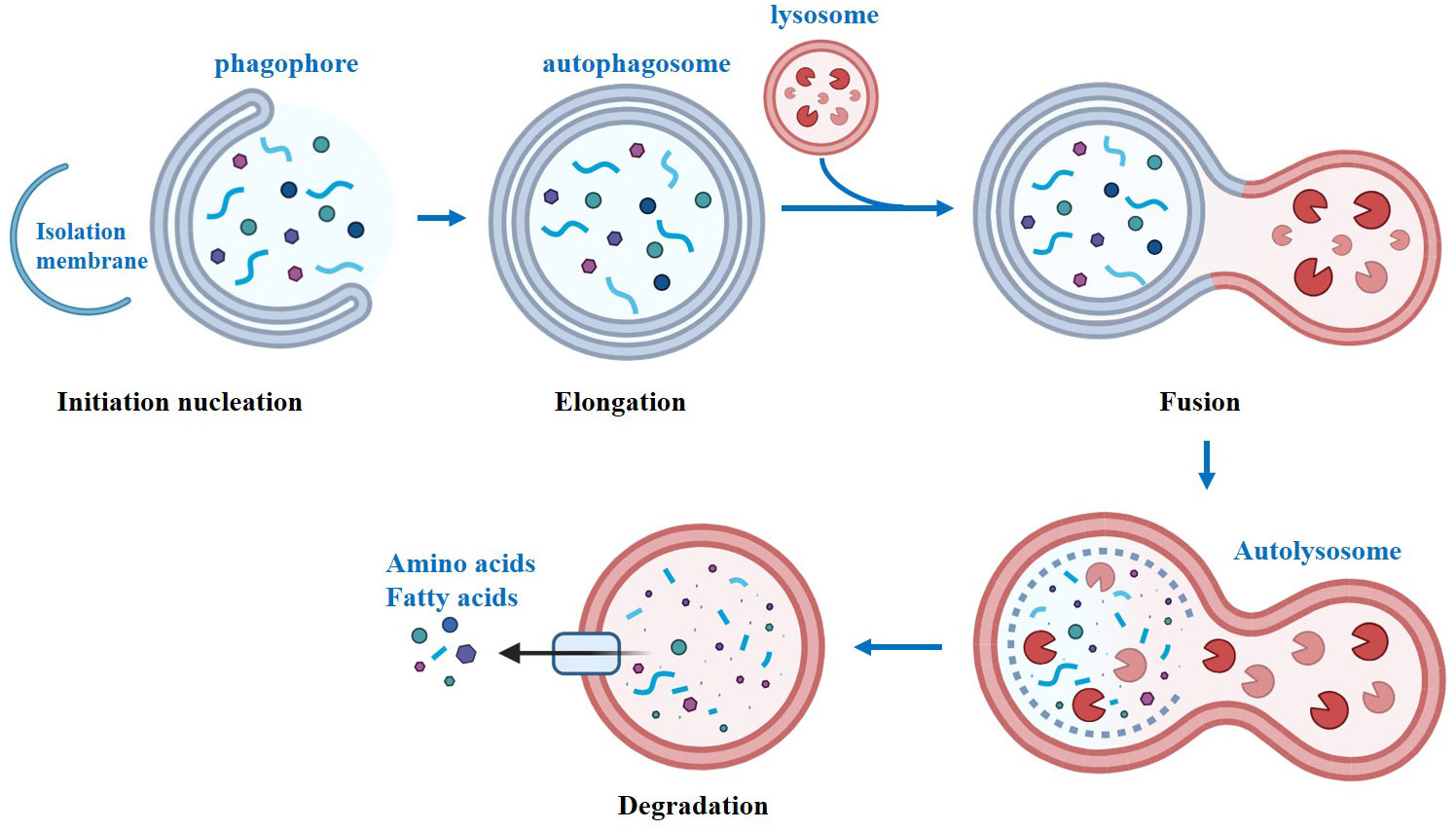
Figure 1 The classical process of autophagy.
Diabetes mellitus (DM) is a metabolic disease characterized by chronic hyperglycemia. Type II diabetes mellitus (T2DM) is the most common type of DM, accounting for over 90% of diabetic patients (10). Typically, the elderly population is more susceptible to T2DM. However, due to improved living standards and changing lifestyles, the incidence of T2DM is increasing among children, teenagers, and young adults. According to data from the International Diabetes Federation (IDF), there are currently 537 million T2DM patients worldwide as of November 2021 (11) (Figure 2). The IDF predicts that this number will rise to 643 million by 2030 and 784 million by 2045 (11). The prevalence of diabetes in China is approximately 10%, with a total of 114 million diabetic patients, representing one-third of the world’s diabetic population (12). Insulin resistance (IR) is a characteristic of Type 2 Diabetes Mellitus (T2DM) where the efficiency of insulin to absorb and utilize glucose is reduced. This results in the compensatory secretion of more insulin, leading to hyperinsulinemia (13). The overuse of islet cells eventually exhausts the pancreas, reducing its ability to produce insulin and ultimately leading to diabetes mellitus (13). According to the United Kingdom Prospective Diabetes Study (UKPDS), around 50% of individuals with T2DM have impaired islet cells, with more than 90% of these individuals also experiencing Insulin Resistance (IR) (14). In addition to high blood sugar levels, diabetic patients are at an increased risk of developing various health complications. Prolonged high blood sugar can result in a decline in organ function and severe complications affecting the heart, blood, eyes, kidneys, nerves, and teeth. It can even cause serious cardiovascular disease, blindness, renal failure, and amputation (15). Diabetes mellitus has become a silent threat in modern life, significantly impacting people’s daily lives and reducing their quality of life. Diabetes mellitus imposes a significant psychological burden on patients and a substantial economic burden on both society and families (16).
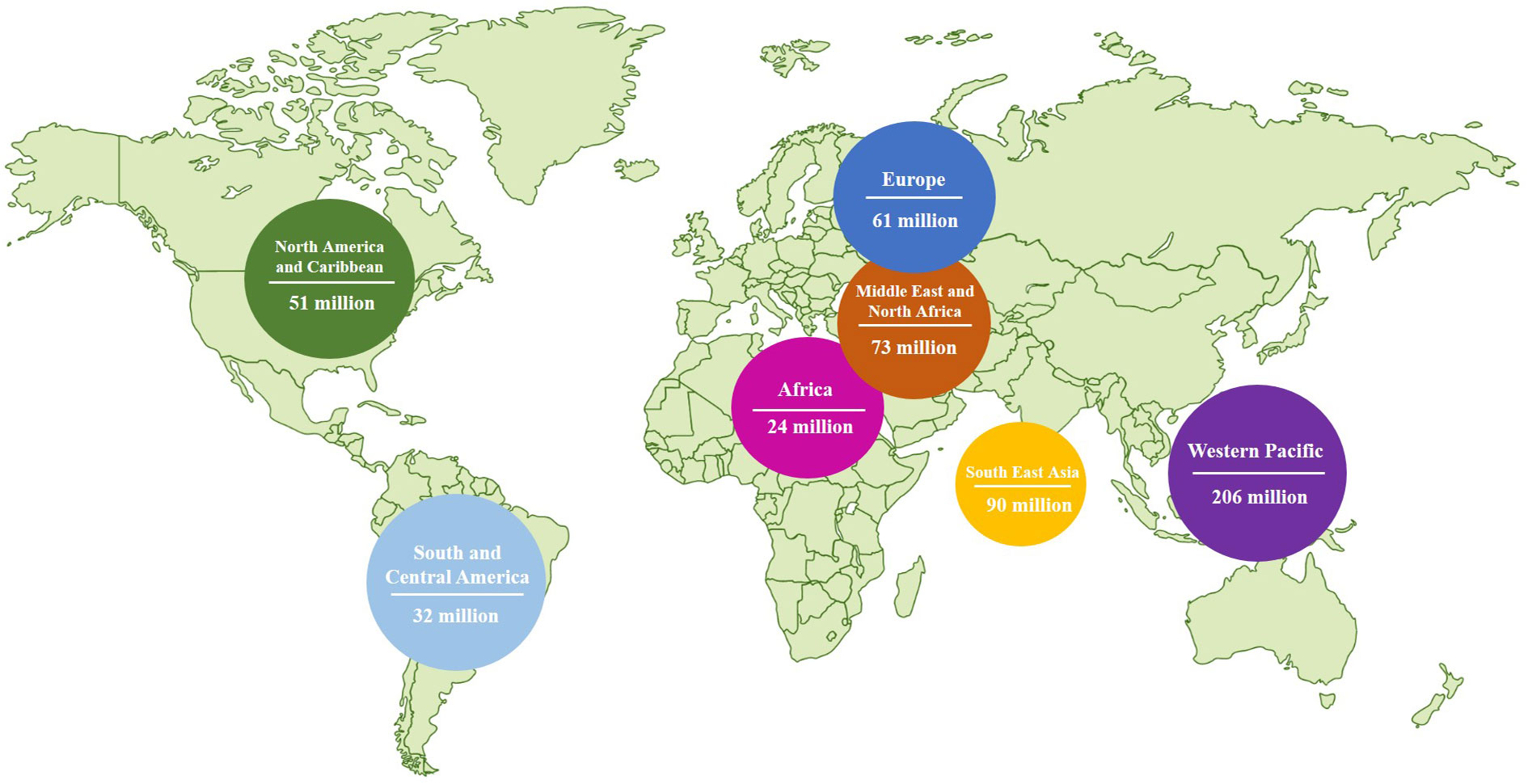
Figure 2 Diabetes around the world in 2021 (data was from IDF).
Some studies indicate that autophagy plays a significant role in the study of human diseases (2), particularly in glucose metabolism (17, 18) (Figure 3). Understanding the mechanism of autophagy is crucial in researching diabetes mellitus and its complications. This review examines the regulation of autophagy in diabetes and its complications, specifically focusing on the changes in autophagy caused by high-glucose and high-fat conditions. It also explores the effects of nutrient deficiencies and excesses on autophagy. Additionally, the review investigates how autophagy regulates endoplasmic reticulum stress and oxidative stress in diabetic states. Furthermore, it discusses the role of autophagy in the regulation of diabetic complications and explores how both chemical and traditional Chinese medicines target autophagy in the treatment of diabetes and its complications. This review’s aim is to provide some inspiration for the treatment of T2DM.
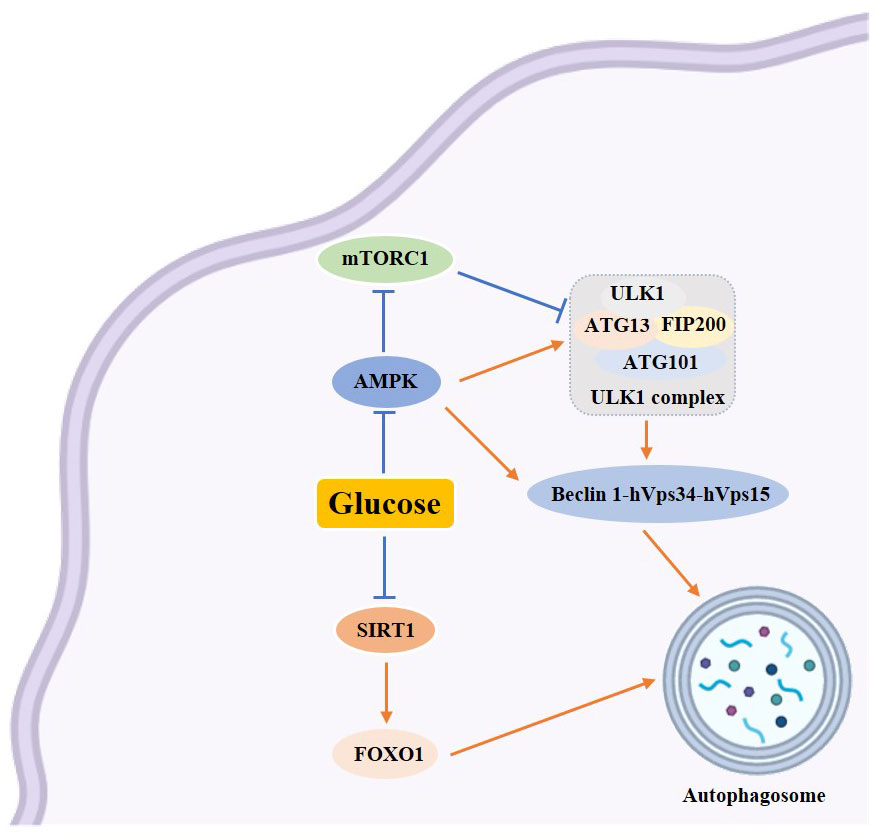
Figure 3 Schematic diagram of the relationship between autophagy and glucose metabolism. →: active; ⟞ :inhibit.
Autophagy is intricately linked to glycolipid levels within the body, and it exerts a protective effect on pancreatic β-cells (19). Studies have shown that a prolonged high-glucose-high-fat diet can hinder autophagy while fasting and restricting calorie intake can activate it (20). Sustained hyperglycemia and excess free fatty acids can lead to an increase in mitochondrial oxygen consumption, resulting in an elevation of reactive oxygen species (ROS) and subsequent abnormal oxidative stress (21). Additionally, ROS can induce autophagy through various mechanisms, including the modulation of autophagy via the mTOR and MAPK signaling pathways, the regulation of autophagy by ROS through the PI3K/Akt, AMPK, and ERK, JNK signaling pathways, and the oxidation of ATG4 by ROS to regulate autophagy (22–24). To summarize, oxidative stress activates autophagy, which helps eliminate cellular damage caused by oxidative stress and maintain cellular homeostasis. Excessive levels of ROS can lead to endoplasmic reticulum stress, endoplasmic reticulum stress can also be induced by insulin resistance. Evidence suggests that insulin inhibits autophagy, while glucagon activates it (25). Insulin, the sole hormone responsible for lowering glucose levels in the body, is synthesized within the endoplasmic reticulum. The endoplasmic reticulum holds significant importance within the cell as it is involved in various processes such as protein synthesis, transport, folding, and degradation. Maintaining homeostasis within the endoplasmic reticulum is crucial for the proper functioning of β-cells. When pancreatic β-cells are stimulated by prolonged high glucose levels, it disrupts homeostasis and leads to an accumulation of unfolded and misfolded proteins in the endoplasmic reticulum of the cells. In order to ensure cell survival, these misfolded proteins need to be eliminated, triggering endoplasmic reticulum stress (26). When endoplasmic reticulum stress is induced by strong intracellular and extracellular stimuli, the endoplasmic reticulum has two mechanisms to deal with the accumulation of misfolded proteins. One is the unfolded protein response (UPS), and the other is endoplasmic reticulum associated degradation (ERAD). However, in cases where both UPS and ERAD, activated by endoplasmic reticulum stress, are insufficient to restore the endoplasmic reticulum to its normal state, autophagy becomes the last resort to restore endoplasmic reticulum homeostasis. Autophagic vesicles engulf the damaged endoplasmic reticulum, which is then transported to lysosomes for degradation. Studies have shown that ERS and dysregulated autophagy are interconnected features of diabetes in human pancreatic islet β-cells (26). Endoplasmic reticulum stress can restore the normal function of the endoplasmic reticulum through cellular autophagy. This process involves inducing cellular autophagy through unfolded proteins response (UPR) (27), promoting Ca2+ influx into the cytoplasm to induce cellular autophagy (28), and inhibiting Bcl-2 to induce endoplasmic reticulum autophagy (29) (Figure 4). The occurrence of autophagy has both positive and negative effects on the endoplasmic reticulum. Moderate autophagy can reduce endoplasmic reticulum swelling and alleviate the pressure caused by the accumulation of faulty proteins. However, excessive autophagy can result in cell death (30).
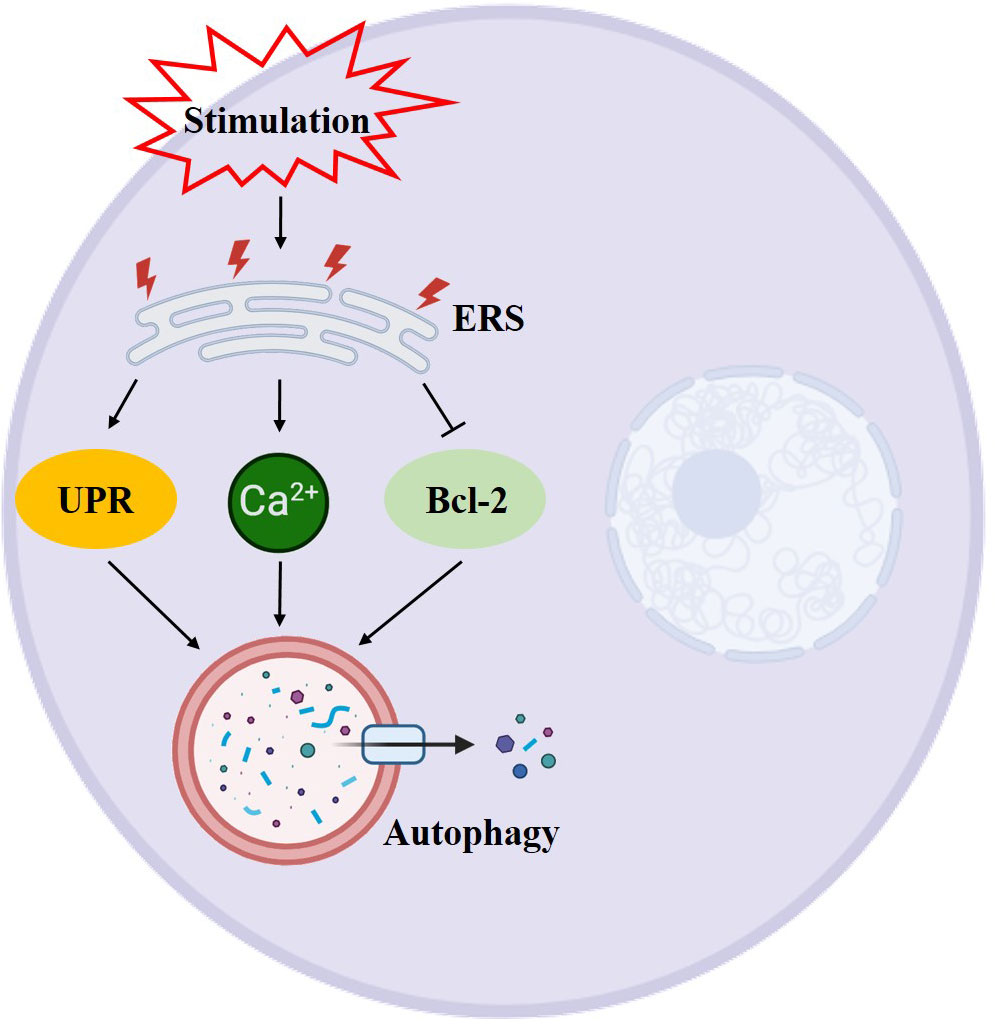
Figure 4 Schematic diagram of the relationship between ERS and autophagy. →: active; ⟞: inhibit.
In recent years, there has been a growing body of research highlighting the significance of autophagy in the development of diabetes and its associated complications (Figure 5). This section aims to shed light on the role of autophagy in the progression of diabetes and its complications and treatment.
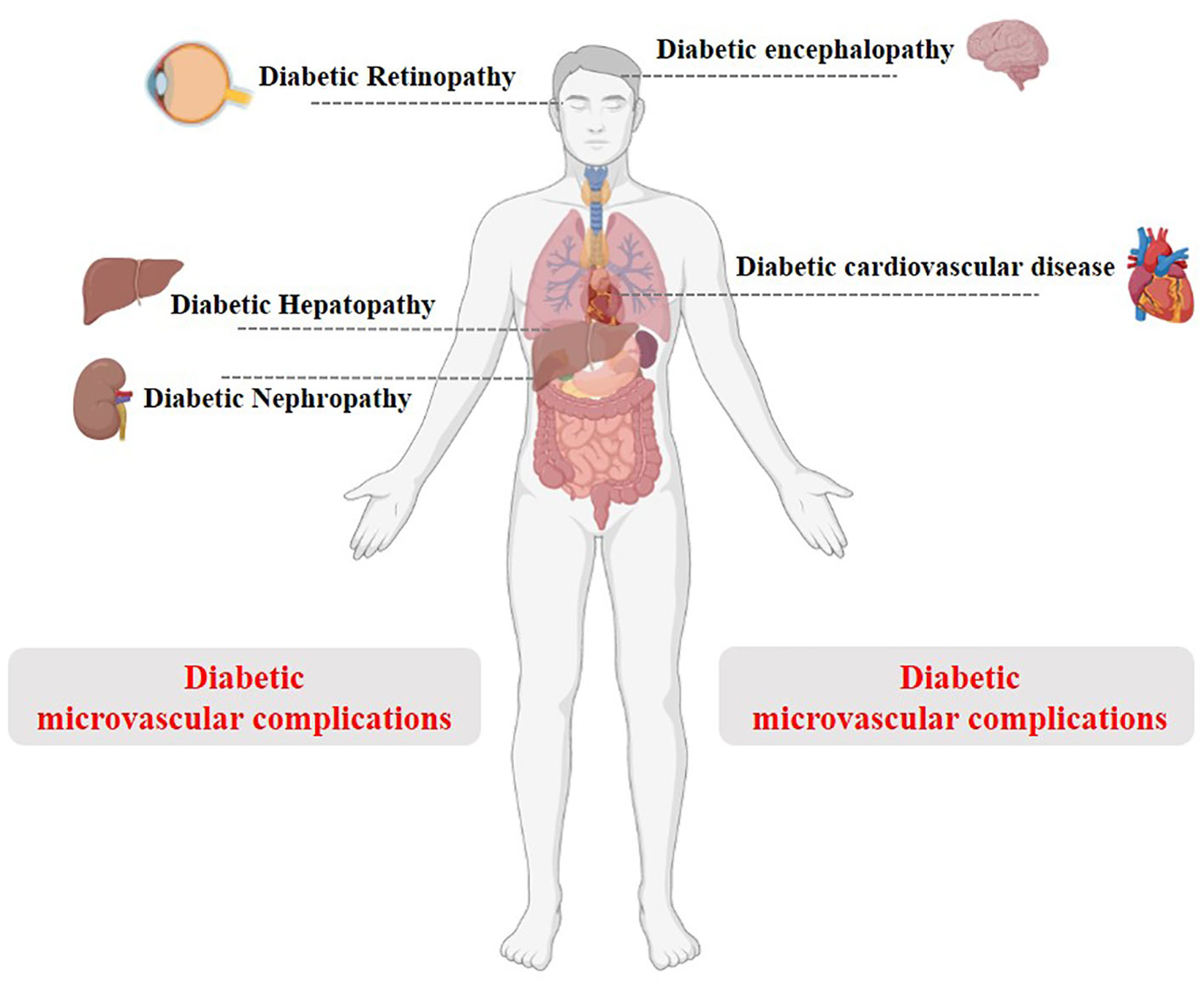
Figure 5 Schematic diagram of T2DM and its complications.
T2DM is a progressive disease that initially presents as insulin resistance. In order to achieve the desired blood glucose level, increased insulin secretion by pancreatic β-cells is required to compensate (13). However, over time, the pancreatic β-cells become overwhelmed by the increased workload and begin to fail, resulting in reduced insulin secretion to compensate for insulin resistance (13, 31). The mechanisms that regulate pancreatic β-cell fate in T2DM are not yet fully understood. However, studies have shown that autophagy plays a crucial role in maintaining normal islet architecture (32). Human pancreatic cells with impaired autophagy regulation have been observed to have an increased association with β-cell dysfunction and failure (33–37). On the other hand, increased autophagy has been found to improve insulin resistance (38–41), and it may serve as a survival mechanism for β-cells (42, 43).
This section delves into the significance of autophagy in pancreatic β-cells, particularly in the context of T2DM. The mTORC1 is a natural inhibitor of autophagy and is crucial for promoting the growth and survival of islet β-cells during the early stages of insulin resistance (44). However, excessive mTORC1 activity due to a high-calorie diet can disrupt autophagy (45). Conversely, inhibiting mTORC1 can enhance autophagy and aid in the removal of misfolded proteins and damaged organelles (46). Bartolomé A et al. (47) demonstrated that chronic overactivation of mTORC1 in a mouse model (β- tsc2 -/-) led to increased pancreatic β-cell death and impaired autophagy. In a separate study, Matthew R. Brown et al. (48) found that negative regulation of REV-ERBα may improve β-cell function under glucotoxicity by enhancing autophagy. This was achieved by investigating the link between the core circadian clock nuclear receptor REV-ERBα, autophagy, and β-cell failure. Rui Liang et al.’s research (49) discovered that in both healthy and diabetic pancreases, the autophagy regulators LC3 and p62/SQSTM1 were found to be expressed more significantly in β cells than in non-β endocrine cells. Additionally, the expression of LC3 and p62/SQSTM1 was significantly reduced in T2DM patients and was inversely correlated with HbA1c levels, suggesting that the autophagic ability of β-cells is impaired as the disease progresses. Tanima Chatterjee et al.’s research (50) discovered that inhibiting autophagy regulation in pancreatic β-cells resulted in apoptosis, ultimately leading to cell death. In a separate study, pancreatic β-cell lines and human islets were exposed to high levels of glucose, leading to an accumulation of autophagosomes, impaired mitochondria, and increased mTOR expression (51). These findings suggest that high glucose levels block autophagic flux and lead to cell death. However, when treated with rapamycin, the changes in autophagic flux as well as glucose-induced cell death were reversed.
Several chemical drugs have also demonstrated good regulation of autophagy to treat diabetes. Liraglutide, a GLP-1 analog (52), has demonstrated effective regulation of autophagy for the treatment of diabetes. Liraglutide has been found to upregulate autophagy mediated by FoxO1, which helps to improve pancreatic β-cell injury (53). Additionally, Liraglutide has been reported to protect INS-1 cells from apoptosis induced by high glucose levels and significantly increase cellular autophagy (54). These findings suggest the potential of Liraglutide in targeting autophagy to prevent β-cell apoptosis. Another drug, Exendin-4, a GLP-1 receptor agonist, has also been found to prevent Tac-induced islet injury by activating autophagosome clearance to reduce autophagosome burden (55). Metformin, on the other hand, inhibits MIN6 β cell proliferation and promotes apoptosis through AMPK-dependent and autophagy-mediated mechanisms (56). Additionally, studies have shown that vitamin B6 can protect RIN-m5f cells from apoptosis caused by high glucose levels through the mTOR pathway-mediated autophagy (57).
In addition to chemical drugs, traditional Chinese compounds and extracts have been found to improve the course of T2DM by regulating autophagy. Yunpi Heluo decoction (YPHLD) has been reported to regulate the SIRT1-FoxO1 signaling pathway in skeletal muscle, improve lipid metabolism, increase autophagy levels, and attenuate insulin resistance, potentially making it an effective treatment for diabetes (58). Xiaokeping (XKP) has also shown potential in protecting islet β-cells from high glucose toxicity by inducing mTOR-mediated autophagy and reducing pancreatic β-cell apoptosis (59). Additionally, Morus alba leaves ethanol extract has been found to protect islet cells from dysfunction and death by inducing AMPK/mTOR-mediated autophagy (60). Finally, Dioscin has been shown to significantly attenuate insulin resistance in adipose tissue through the IRS-1/PI3K/Akt pathway (61). Kaempferol has been shown to activate autophagy through the AMPK/mTOR pathway, resulting in pancreatic β-cell protection in the treatment of T2DM (62). Similarly, Silymarin, a flavonoid found in the fruit of Silybum marianum, has been found to increase autophagy and protect INS-1 cells from TNFα or IL-1β-induced death. This is achieved through the activation of autophagy-dependent ERs, which help maintain cellular energy homeostasis (63).
In addition to drug therapy, certain experimental chemicals have shown promise in modulating autophagy to alleviate T2DM. Specifically, 4-Phenylbutyric acid and rapamycin have been found to induce autophagy and increase the autophagic flux, ultimately preventing β-cell apoptosis (64) (Figure 6).
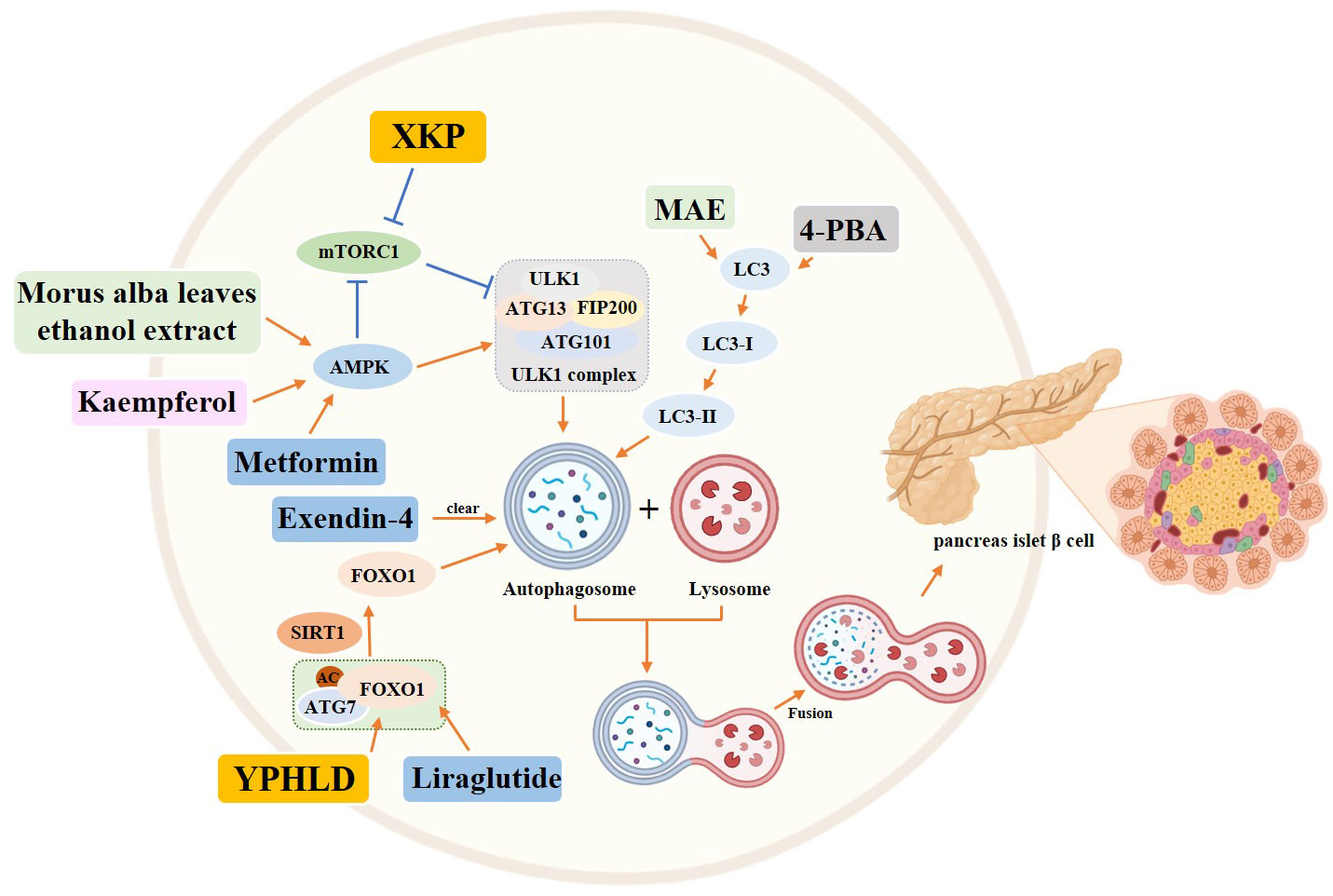
Figure 6 Role of autophagy in pancreatic islet β cells in the diabetic state. Yellow rectangle: Traditional Chinese compounds; Blue rectangle: Chemical drugs; Pink rectangle: Monomers from Chinese Herbal; Gray rectangle: Experimental Chemicals. →: activate: ⟞: inhibit.
Exercise has been found to promote autophagy by activating the AMPK/PGC1α pathway, which can help reduce insulin resistance in mice (65).
This discovery suggests that increasing autophagy could potentially delay disease progression and maintain β-cell function in T2DM. Therefore, inducing autophagy in islet cells through exogenous drugs or other techniques could be a promising approach for preventing or treating T2DM.
The kidney is highly susceptible to diabetic microvascular injury due to prolonged high blood glucose levels (66–68). This can lead to cellular glucotoxicity and increased oxidative stress, resulting in irreversible kidney damage and end-stage renal disease, known as diabetic nephropathy (DN) (69). Diabetic nephropathy is a frequent complication of diabetes, affecting around 50% of diabetic patients and being the primary cause of death in those with type I and type II diabetes (66, 70–72).
Research indicates that traditional therapies are ineffective in slowing down the advancement of diabetic nephropathy (73–75). Additionally, there is mounting evidence that autophagy is a crucial factor in maintaining kidney health, preventing disease, and slowing the aging process. Autophagy has been shown to contribute to the development of DN (68, 76–79), and targeting it may have a positive impact on kidney function (80). This section will delve into the role of autophagy in the progression of diabetic nephropathy.
Renal podocytes are highly specialized, terminally differentiated and nonproliferative cells that exhibit high autophagic activity under non-stress conditions (81). Alterations in autophagy have been observed in diabetic podocytes (82, 83), indicating that regulating autophagy to maintain homeostasis could be a potential target for treating diabetic nephropathy. In a study by Olivia Lenoir et al. (84), renal podocytes of mice were exposed to high sugar levels for 48 hours, which induced autophagy. After being exposed to high glucose levels for 15 days, autophagy was inhibited. In vivo experiments on mice induced with STZ to develop T1DM showed that autophagy in renal podocytes was induced at 4 weeks, but became inhibited and resulted in renal podocyte damage at 8 weeks. Knocking down the Atg5 gene in renal podocytes accelerated diabetes-induced deterioration, leading to glomerular filtration barrier leakage and glomerulosclerosis. In diabetic conditions, the expression of Beclin-1, Atg12-5 and LC-3 in renal podocytes was found to be inhibited both in vitro and in vivo, leading to damage in renal podocytes due to the inhibition of autophagy (85). In a high-fat diet (HFD)-induced diabetes model, podocyte-specific autophagy-deficient mice showed a loss of renal podocytes and massive proteinuria (86). Markus Gödel et al. (87) discovered that mTORC1 was highly activated and autophagy was inhibited in renal podocytes of T2DM mice and patients. However, when mTORC1 gene copies were reduced during the experiment, the disease process in diabetic nephropathy was significantly improved. Wei et al. (88) observed that in a high glucose state, autophagy levels were reduced in human renal podocytes. However, when treated with the autophagy activator rapamycin, autophagy was activated and insulin response was enhanced. This suggests that autophagy regulates insulin responsiveness and that activation of autophagy can improve cellular damage in human podocytes. The use of the mTORC1 inhibitor rapamycin has shown significant improvement in the progression of diabetic nephropathy in T2D rats (89). Further studies by Xiao et al. (90) on STZ-induced T1DM mice have also shown that rapamycin treatment increases autophagosomes and attenuates renal podocyte fusion, indicating that rapamycin improves renal injury in diabetic mice by increasing autophagic activity and inhibiting podocyte apoptosis. However, it is important to note that AGEs inhibit autophagosome formation and renewal in renal podocytes by activating mTOR and inhibiting nuclear translocation of the pro-autophagic TFEB, ultimately causing renal podocyte injury (91). These experimental results suggest that long-term high glucose levels reduce the autophagy level of renal podocytes, leading to renal podocyte damage and the eventual development of diabetic nephropathy.
In addition to renal podocytes, proximal tubular epithelial cells also have a significant role in maintaining renal function. During the course of diabetes, autophagy in proximal tubular epithelial cells undergoes significant changes (82, 92–94). In 1992, Barbosa et al. discovered that in STZ-induced rats, the volume and density of autophagic vesicles in proximal tubular cells were significantly reduced, which led to the accumulation of autophagic substrates (95). When the autophagy-associated gene 7 (Atg7) in the proximal tubular cells was removed, diabetic mice experienced defective autophagy, resulting in more severe renal hypertrophy, tubular damage, inflammation, fibrosis, and proteinuria (96). Kosuke Yamahara et al. (97) discovered that kidney biopsy specimens from type II diabetic patients exhibited deficient autophagy. Additionally, high glucose and obesity were found to significantly inhibit the protective role of autophagy in proximal renal tubular cells. The overactivation of mTORC1 resulted in defective autophagy, which induced proximal tubular cell injury. However, when the diet of diabetic animals was restricted or altered, autophagy was significantly restored, leading to an improvement in diabetic nephropathy (98–100).
Some drug, such as metformin, has been found to reduce renal injury in diabetic rats by regulating autophagy through the Sirt1/FoxO1 signaling pathway (101, 102). Prostaglandin E1 (PGE1) has also been shown to restore autophagy and insulin resistance in the kidney of type II diabetic (T2DM) rats and promote autophagy-related fibroblast growth factor 21 (FGF21) protein expression, thereby reducing insulin resistance (103). In addition, experimental data from liraglutide treated Male 8-week-old spontaneously diabetic Torii (SDT) fatty rats showed increased expression of phosphorylated (p)-eNOS and p-AMPK in glomeruli, downregulated expression of p-mTOR, and increased expression of LC3B-II, indicating that liraglutide plays a protective role in the kidney by activating autophagy (104). The sodium-dependent glucose transporters 2 (SGLT2) located in the proximal tubule are responsible for reabsorbing 90% of the glucose that is filtered through the glomerulus (105). Studies have shown that SGLT2 inhibitors induce AMPK and SIRT1 to stimulate autophagy, which helps to alleviate cellular stress, as well as glomerular and tubular injury (106). In a study involving 8-week-old male db/db mice, administration of SGLT2 inhibitor Empagliflozin and DPP4 inhibitor Linagliptin resulted in enhanced autophagy in renal podocytes, indicating that both drugs can attenuate the progression of diabetic nephropathy through the autophagic pathway. This resulted in the attenuation of thylakoid expansion, podocyte foot process loss, and urinary albumin excretion (107). Recent studies have demonstrated that Empagliflozin can enhance autophagic activity in renal tubular cells exposed to high glucose levels, thereby providing a protective effect on the kidneys (108). Additionally, GLP-1, a crucial pro-intestinal hormone, has been found to stimulate autophagy through the AMPK/mTOR signaling pathway and related proteins, ultimately alleviating diabetic nephropathy (109).
Botanicals have been found to have a protective effect on the kidneys in diabetic individuals by inducing autophagy (110, 111). A study conducted on STZ-induced diabetic rats showed that Korean red ginseng treatment resulted in an upregulation of LC3 and a downregulation of p62 in rat kidney cells. Additionally, there was an increase in the levels of ATG7 and inhibition of mTOR, which suggests the activation of autophagy and subsequent recovery from kidney injury (110). The effectiveness of resveratrol in reducing high glucose-induced apoptosis was investigated in db/db mice and renal podocytes (112). The activation of autophagy involving miR-383-5p was found to be the mechanism behind this attenuation. Additionally, triptolide was found to restore autophagy through the miR-141-3p/PTEN/Akt/mTOR pathway, which resulted in the attenuation of fibrosis (113). According to research, Astragaloside IV (AS-IV) has the ability to induce autophagy through the AMPKα pathway (114) or SIRT1 deacetylation of NF-κB p65 subunit (115), which can help alleviate the symptoms of diabetic nephropathy. Another study found that Ginsenoside Rg1 can attenuate podocyte EMT by enhancing AKT/GSK3β/β-linked protein pathway-mediated autophagy, indicating its potential therapeutic benefits for DN (116) (Figure 7).
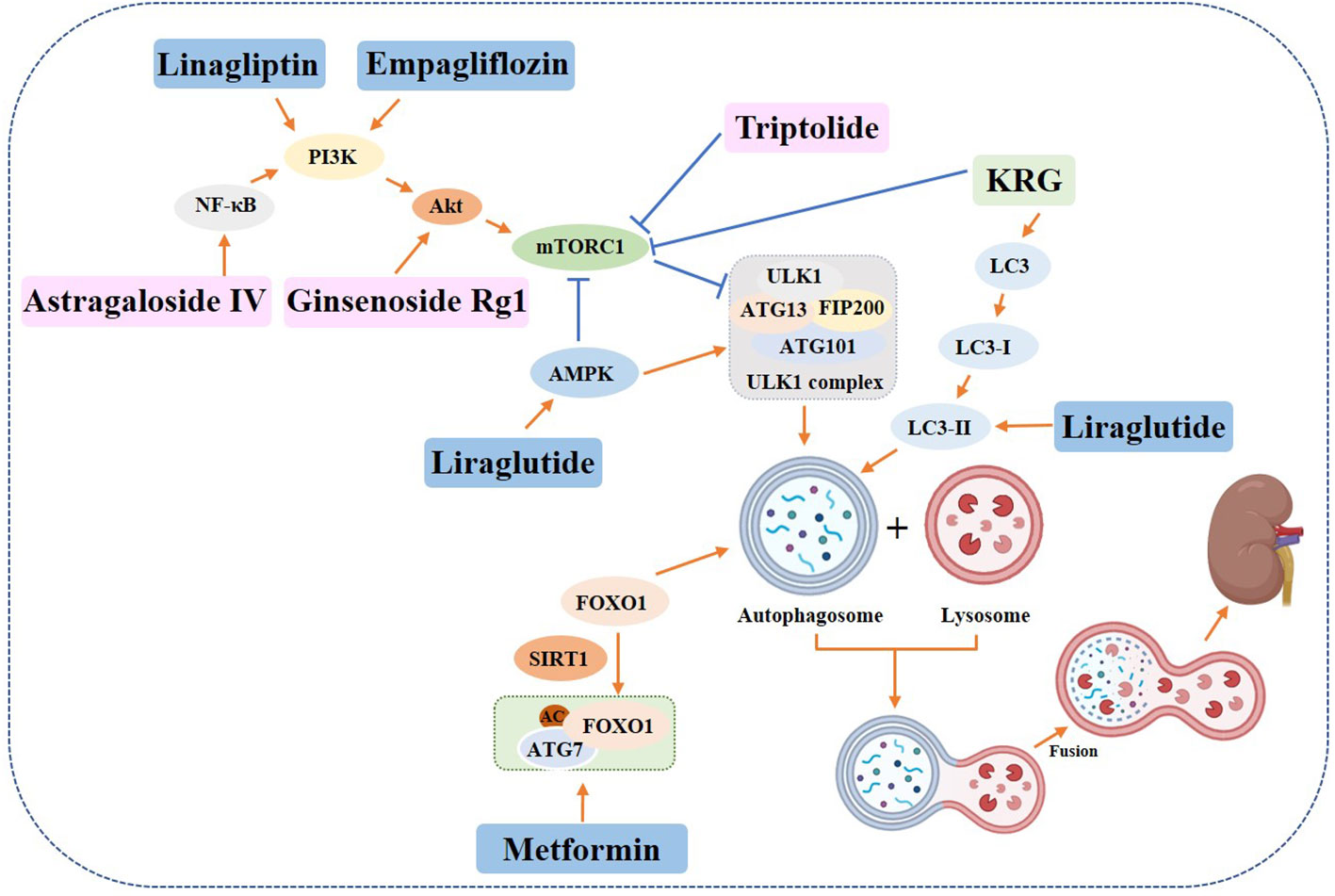
Figure 7 Role of autophagy in diabetic nephropathy. Yellow rectangle: Traditional Chinese compounds; Blue rectangle: Chemical drugs; Pink rectangle: Monomers from Chinese Herbal; Gray rectangle: Experimental Chemicals. →: activate: ⟞: inhibit.
The results indicate that enhancing autophagic activity could be a promising approach to treating diabetic nephropathy. Nevertheless, some studies suggest that reduced autophagy may also have a protective effect on the kidneys against T2DM damage (117). Further experiments are needed to confirm the exact role of autophagy in this context.
The liver is an important organ involved in glucose regulation, lipid metabolism, and insulin action. NAFLD is a widespread liver disease (118), and its disease progression is closely related to T2DM as well as IR (119). As liver disease progresses, IR is exacerbated, and IR is considered a critical event affecting T2DM and NAFLD (120). Global epidemiological statistics for 2019 (121) showed that 55.5% of patients with T2DM worldwide develop NAFLD. As early as 2016, the European Association for the Study of the Liver, the European Association for the Study of Diabetes, and the European Association for the Study of Obesity strongly recommended: NAFLD screening was performed in patients with established T2DM and T2DM screening was performed in patients with NAFLD (122). These two pathological states of T2DM and NAFLD seem to be difficult to say who is the cause and who is the effect, but they share similar complex pathophysiological mechanisms, such as insulin resistance, chronic hyperglycemia, lipotoxicity, low-grade inflammation and increased oxidative stress, and both act synergistically to increase adverse clinical outcomes (123, 124). Diabetics are two-fold more likely to develop NAFLD and vice versa (125). Moreover, T2DM accelerates the progression of NAFLD, allowing NAFLD to rapidly progress to nonalcoholic steatohepatitis (NASH), cirrhosis, and hepatocellular carcinoma (126). In 2020, international experts from 31 organizations proposed the definition of MAFLD (Metabolic dysfunction-associated fatty liver disease) to replace NAFLD, where one of the diagnostic criteria is: confirmed T2DM (127). Although the change in terminology from NAFLD to MAFLD is still under intense debate, it largely demonstrates the close relationship between liver disease and T2DM and the underlying metabolic dysfunction. Studies have shown that autophagy is closely related to NAFLD during its development (128). Impaired autophagic flux was observed in the liver of NAFLD patients, NAFLD mouse models and lipid-overloaded human hepatocytes (129). Autophagy is closely related to hepatic lipid metabolism, and inhibition of autophagy increases triglyceride and lipid droplets in vitro and in vivo, promoting lipid accumulation, which further inhibits autophagy and increases lipid retention (130). Chloroquine (an autophagy inhibitor) exacerbates hepatic steatosis and liver injury, while carbamazepine and rapamycin (2 autophagy activators) enhance ethanol-induced macroautophagy in hepatocytes in vitro and in vivo (131). Inhibition of the autophagy-associated gene Atg7 in vitro and in vivo resulted in defective insulin signaling and elevated endoplasmic reticulum stress. In contrast, restoration of Atg7 expression in the liver resulted in reduced endoplasmic reticulum stress, enhanced hepatic insulin action, and increased systemic glucose tolerance in obese mice (132). Branched-chain amino acids (BCAAs, including leucine, isoleucine, and valine), which are abundant in high-protein foods, are thought to be closely associated with the NALFD-related metabolic disease T2DM. Excess circulating BCAAs activate mTOR and inhibit autophagy, and the blockage of autophagy hinders the self-repair mechanism of hepatic lipotoxicity and increases apoptosis (133). Most of the drugs currently used to treat diabetic liver injury are single hypoglycemic and hepatoprotective drugs, which are ineffective and have significant side effects.
Some chemical drugs can alleviate the symptoms of diabetic liver disease by regulating autophagy. Studies have shown that Empagliflozin, an SGLT-2 inhibitor, in addition to its significant hypoglycemic effect, can also enhance autophagy in liver macrophages via the AMPK/mTOR signaling pathway and further inhibit the IL-17/IL-23 axis-mediated inflammatory response, thereby significantly ameliorating liver injury in mice with T2DM combined with NAFLD (134). It has also been shown that Empagliflozin can improve hepatic steatosis through the AMPK-TET2 autophagic pathway (135). Liraglutide promotes autophagy through the AMPK/mTOR pathway to reduce lipid accumulation and exert anti-lipotoxic effects (136, 137). Vitamin D3 may improve hepatic lipid abnormalities by activating the autophagy-regulated AMPK/Akt-mTOR signaling pathway in diabetics (138). A report on the active form of vitamin D, 1,25(OH)2D3, suggests that it can prevent hepatic lipid degradation by stimulating ATG16L1-mediated autophagy (139).
Plant-derived monomeric compounds also protect against liver injury caused by T2DM. Punicalagin (PU) administration to C57BL/6J mice (at a dose of 20 mg/kg/day) and HepG2 cells (at doses of 5, 10, and 20 μM) significantly improved liver histology, reversed serum biochemical abnormalities and increased the number of autophagosomes in the liver of T2DM mice. The experimental results showed that PU restored autophagy through the Akt/FoxO3a signaling pathway, thereby protecting against T2DM-induced liver injury (140). Astragaloside alleviated liver injury in type 2 diabetes by promoting AMPK/mTOR-mediated autophagy (141) (Figure 8).
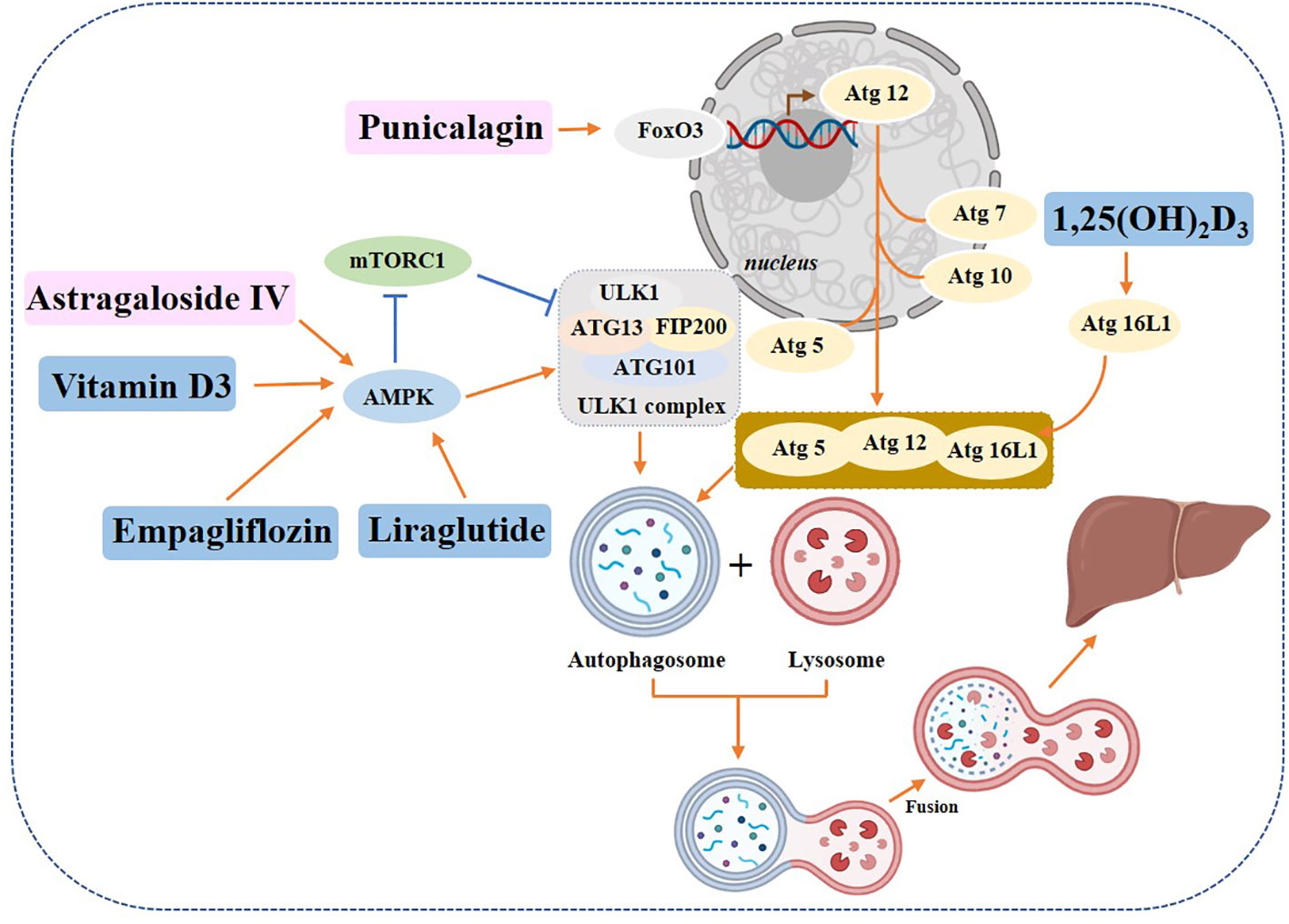
Figure 8 Role of autophagy in diabetic hepatopathy. Yellow rectangle: Traditional Chinese compounds; Blue rectangle: Chemical drugs; Pink rectangle: Monomers from Chinese Herbal; Gray rectangle: Experimental Chemicals. →: activate; ⟞: inhibit.
Taken together, therapies that restore autophagic flux may attenuate or prevent the progression of T2DM liver disease.
Diabetic Cardiomyopathy is divided into two main categories: one affecting the large vessels (large arteries, including the aorta, femoral arteries and coronary arteries) and the other affecting the microvasculature (small vessels, including capillaries of the eyes, kidneys and nerves) (142). People with diabetes have a 3 to 10-fold increased risk of cardiovascular complications compared with those with normal blood glucose (143). And cardiovascular complications of diabetes increase the risk of myocardial infarction, stroke, and limb loss. The lethality of cardiovascular complications of diabetes is extremely high and is the leading cause of death (144). The myocardium is particularly vulnerable to the negative effects of the hyperglycemic state because it is both an insulin-sensitive and glycolysis-dependent tissue (145). Persistent hyperglycemia inhibits cardiomyocyte autophagy (146, 147). Studies have shown that abnormal cellular metabolism and accumulation of damaged/defective organelles have been observed in cardiomyocytes with diabetic cardiomyopathy (148) and abnormal regulation of autophagy has been observed in preclinical trials in the diabetic heart (149). There is growing evidence that autophagy plays an important role in cardiovascular disease (150–153), restoration of autophagy is cardioprotective and impaired autophagy leads to cardiac damage (154, 155). In research on diabetic cardiomyopathy, modulation of autophagy (whether it is enhanced or diminished needs to be confirmed) improved cardiac function in T2DM rats (156). SGLT2 inhibitors reduced the incidence of cardiovascular death and hospitalization for heart failure by activating autophagy (157). However, it has also been shown that the inhibition of autophagy in cardiomyocytes by sustained hyperglycemia is an adaptively protective mechanism that limits the cytotoxicity of high glucose. When cardiomyocytes in a high-glucose state were treated with an autophagy inhibitor (3-MA) or silencing of autophagy-associated genes (Atg7), autophagy was inhibited and high glucose-induced cardiomyocyte death was attenuated. Contrary to expectations, increasing autophagy through the use of autophagy activators such as rapamycin or by overexpressing Beclin-1 or Atg7 actually made cardiomyocytes more susceptible to the toxic effects of hyperglycemia (146). There are also studies with the same findings that suggest that insulin resistance overactivated autophagy in cardiomyocytes, thus preventing the survival of cardiac cells in diabetics. When autophagy in diabetic cardiomyocytes is inhibited, the survival rate of cardiomyocytes is raised (145, 158, 159). Feng et al. (160) showed that high glucose levels increased diabetic cardiomyopathy-related factor (DCRF) expression and induced cardiomyocyte autophagy and lowering DCRF expression reduced cardiomyocyte autophagy, reduced myocardial fibrosis, and improved cardiac function in diabetic rats.
Several chemical agents, as well as plant-derived monomers, have been reported to exert therapeutic effects by modulating autophagy. Zinc, a protective factor in diabetic cardiomyopathy, may protect the heart by inhibiting autophagy (161). Metformin has also been verified to improve the course of experimental cardiomyopathy by activating autophagy directly or indirectly through the activation of AMPK and SIRT1 (162, 163). Some botanicals can also protect the heart of diabetics by regulating autophagy. Carnosic acid reduces diabetic myocardial ischemia-reperfusion injury by enhancing autophagy (164). Polyphenols from green tea extract can protect rat hearts by improving autophagy (165). Quercitrin has been demonstrated to activate the ERK signaling pathway and enhance cellular autophagy in endothelial progenitor cells in a dose-dependent manner in vitro experiments and thus may be used as a therapy for a variety of diseases caused by impaired endothelial function (166). Resveratrol enhances autophagy via SIRT1/FOXO1/Rab7 axis in vivo and in vitro and ameliorates myocardial oxidative stress injury in diabetic mice (167). β-carotene can inhibit autophagy through activation of PI3K/Akt/mTOR, which in turn exerts cardioprotective effects, and can protect H2c9 cells damaged by advanced glycosylation products (168). Curcumin ameliorates diabetic cardiomyopathy by activating AMPK and JNK1, phosphorylated Bcl-2 and Bim and subsequently disrupted their interactions with Beclin1, promoting autophagy and attenuating apoptosis in vivo and in vitro diabetic models (169).
In addition to diabetic cardiomyopathy, drugs have also been reported to exert therapeutic effects by modulating autophagy in studies related to diabetes combined with atherosclerosis. α-lipoic Acid sulfate, a fatty acid present in the human body, protects vascular smooth muscle cells from atherosclerosis in T2DM patients by downregulating autophagy through the AMPK/mTOR pathway (170), but Sirt6-mediated Caveolin acetylation 1 is the key factor in atherosclerosis in T2DM through activation of autophagy (171).
Based on the above findings, in terms of the relationship between autophagy and diabetic macrovascular complications (mainly diabetic cardiomyopathy), some scholars believe that the role of autophagy in diabetic cardiomyopathy is more like the “Goldilocks” phenomenon, i.e. there is a narrow “optimal” autophagic region, and when we adjust autophagy within the “optimal range”, the best therapeutic effect will be achieved (172, 173).
Diabetes-associated cognitive decline (DACD) is one of the common complications in diabetics (174). DACD refers to the decline in cognitive function in diabetics, manifested by deficits in learning, memory, reasoning and verbal expression. The long-term hyperglycemia of T2DM significantly increases brain glucose flow (175), producing damage to the central nervous system (176, 177), affecting several cognitive domains and increasing the risk of cognitive dysfunction (178–180). The longer the duration of T2DM, the more likely it is to develop cognitive dysfunction (181). In general, DACD is difficult to reverse. Without effective intervention, diabetes will greatly accelerate the progression from mild cognitive impairment to dementia (182). Here we have to mention that Alzheimer’s disease, a form of cognitive impairment, has overlapping molecular and biochemical features with T2DM (183), hence Alzheimer’s disease is also called type III diabetes (184). In recent years, an increasing number of scientific studies have confirmed that autophagy plays an important role in the molecular basis of T2DM and cognitive impairment (185, 186). However, findings vary on how autophagy regulates the relationship between diabetes and cognitive impairment. Some findings suggest that T2DM causes enhanced autophagy, which in turn causes deposition of Aβ in the brain and exacerbates cognitive impairment. For example, Son et al. (187) found that insulin resistance promotes Aβ production in the brain by altering insulin signaling and increasing autophagic flux. Ma et al. (188) found that diabetes activates autophagy, but the autophagy-lysosome function is impaired, and the resulting of autophagy-lysosome dysfunction caused Aβ deposition in diabetic cognitive impairment. However, more voices contradict this, and most findings suggest that the development of diabetic cognitive impairment is associated with the reduced autophagic activity. For example, Wu et al. (189) found that reduced autophagic activity of db/db diabetic mice is associated with the accumulation of Aβ, the classic marker of Alzheimer’s disease. Decreased spatial learning, as well as memory capacity in aged diabetic mice, is associated with the inhibition of autophagy marked by the upregulation of p62 and the downregulation of Beclin protein in the hippocampus (190, 191). Excessive activation of the PI3k/Akt/mTOR signaling pathway and inhibition of autophagy can be observed in macrophages of the brains of diabetic animals (192, 193). Up to now, some Chinese traditional herbs’ monomers and compounds, and chemical drugs in the market, can improve diabetic cognitive impairment by modulating autophagy. We’ll cover each in the following.
Gliflozins, an oral antidiabetic drug, can restore mTOR to its activated physiological state and prevent the development of neurodegenerative diseases (194). Metformin improves cognitive impairment in T2DM by regulating autophagy through an AMPK-dependent pathway (195). A high dose of liraglutide (200 μg/kg) alleviated learning and cognitive impairment in T2DM rats enhanced autophagic signaling and improved cognitive function by activating PI3K/Akt and AMPK pathways to inhibit p-mTOR expression (196). Exendin-4 activated PKA and PI3K/Akt signaling pathways in rat cerebral cortex to promote autophagy and inhibit the onset of apoptosis (197). Melatonin activates autophagy through the TLR4/Akt/mTOR pathway and improves learning and memory in type II diabetic mice (198).
In addition to chemical drugs, there are also reports of Chinese herbal monomers and compounds targeting autophagy to improve cognitive function in diabetics. Huanglian Jiedu Decoction can inhibit the activation of inflammasome NLRP and upregulate autophagy (199). ZDF rats administered with ZiBuPiYin Recipe were found to obtain improvement in learning memory impairment, presumably achieved by inhibiting mTOR upregulation, enhancing autophagy, and promoting Aβ clearance (200). Besides traditional Chinese compounds, the traditional Chinese monomer was reported to have the same biological activities. Berberine, a quaternary alkaloid isolated from Coptis, a traditional Chinese medicine, enhances cellular autophagy and attenuates neuronal apoptosis by activating the AMPK/mTOR pathway (201) (Figure 9).
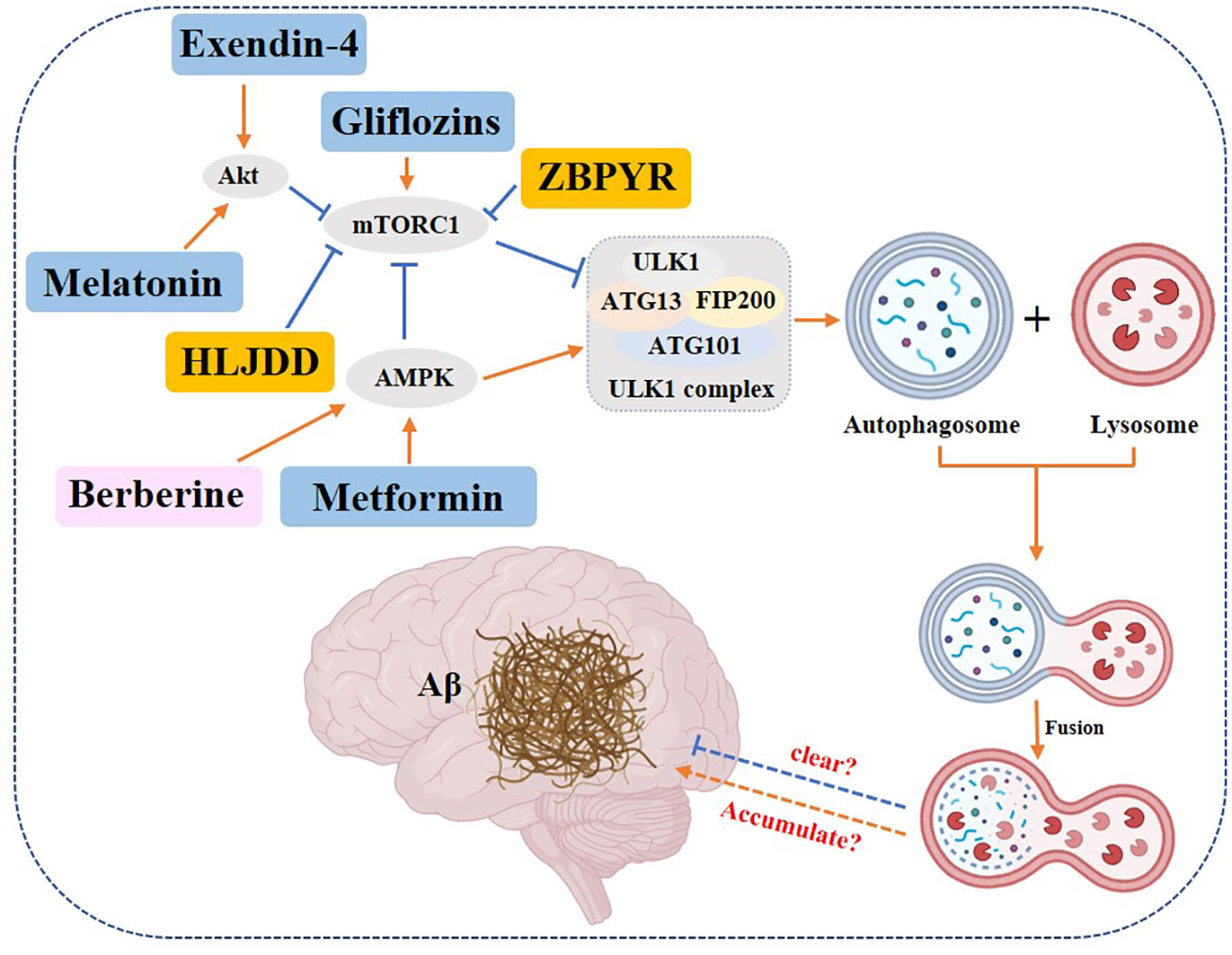
Figure 9 Role of autophagy in DACD. Yellow rectangle: Traditional Chinese compounds; Blue rectangle: Chemical drugs; Pink rectangle: Monomers from Chinese Herbal; Gray rectangle: Experimental Chemicals. → activate; ⟞: inhibit.
In summary, most of the studies concluded that enhanced autophagy can promote the clearance of Aβ and thus improve the condition of DACD.
Diabetic retinopathy (DR) is a chronic and progressive complication of type 2 diabetes mellitus (T2DM), primarily resulting from prolonged hyperglycemia (202). It is also the leading cause of blindness in diabetic patients. The incidence of DR has significantly increased due to the elongation of human lifespan and the rising prevalence of T2DM. According to WHO statistics (203), it is projected that approximately one-third of diabetic patients worldwide will develop diabetic retinopathy by 2025. Recent scientific research has revealed that diabetic retinopathy is not only a vascular condition but also a neurodegenerative disease (204). Autophagy has been found to have a significant impact on the development and treatment of diabetic retinopathy (205–207). For instance, The dysregulation of mTOR-dependent autophagy is a contributing factor to the loss of retinal ganglion cells in streptozotocin-induced diabetic retinopathy. This loss further worsens the condition of diabetic retinopathy (208). Autophagy-related proteins are highly expressed in the retinas of both normal individuals and diabetic patients (209, 210). Notably, the Atg16L1 gene shows a significant up-regulation, indicating its involvement in the cell death process associated with diabetic retinopathy (211). An experiment was conducted to observe the effects of high glucose conditions on human retinal pigment epithelial cells (ARPE-19). The cells were exposed to 30 mmol/L D-glucose and subsequently observed. Subsequently, the observed results indicated a reduction in ARPE-19 cell viability, an increase in apoptosis, and elevated protein expression of Bax, Caspase-3, and LC3-II/LC3-I. Conversely, the expression of Bcl-2, p62, and p-mTOR was decreased. Notably, the activation of autophagy was observed. Moreover, the effects induced by high glucose were reversed upon treatment with the autophagy inhibitor 3-MA (212). The same results were obtained in another experiment. The expression of LC3-I in ARPE-19 cells was significantly reduced under high glucose stress. Additionally, intracellular ROS levels were significantly elevated, and the number of autophagosomes was significantly increased. These findings indicate that high glucose activates autophagy (213). Contrary experimental results have also been reported regarding the regulation of autophagy in response to high glucose. When mouse retinal explants were treated with high glucose (HG) for 10 days, it was observed that there was a significant increase in explant apoptosis, while autophagic flux showed a significant decrease. The findings indicate a potential relationship between autophagy dysfunction and the mechanism of DR. There have been reports of drugs targeting autophagy for the treatment of DR, which are described below.
Treatment of retinal explants treated with the growth hormone analog octreotide (OCT) demonstrated that the explants were shielded from apoptosis and exhibited enhanced autophagic activity. Furthermore, the addition of the autophagy inhibitor chloroquine completely negated the anti-apoptotic effect exerted by OCT (214). The study on Glucagon-like peptide-1 (GLP-1) treatment in rats with type II diabetes demonstrates that GLP-1 reduces oxidative stress-induced autophagy by activating the GLP-1R-ERK1/2-HDAC6 signaling pathway. This mechanism ultimately leads to an improvement in diabetic retinopathy (215). PG545, a heparanase inhibitor, was administered to diabetic mice via intraperitoneal injection at a dose of 20 mg/kg/d. This treatment effectively slowed down the diabetes-induced changes in body weight and reduced fasting blood glucose levels in mice. Additionally, PG545 was observed to mitigate the reduction in retinal thickness and the formation of microaneurysms both in vivo and in vitro. These beneficial effects were attributed to the promotion of autophagy and the inhibition of inflammatory responses by PG545 (216). β-hydroxybutyrate (BHB) has been previously employed as a trophic agent for brain-derived neurotrophic factor (BDNF). In experimental studies, the administration of BHB (25-50-100 mg/kg) to treat STZ-induced C57BL/6J diabetic mice resulted in a noteworthy reduction in autophagy activation in M1 microglia, while simultaneously increasing retinal BDNF levels (217). Arjunolic acid, an active ingredient derived from Cyclocarya paliurus, was investigated in a rat model of STZ-induced diabetes mellitus. The experimental animals were orally administered doses of 10 mg/kg and 30 mg/kg of Arjunolic acid for a duration of 10 weeks. The study revealed that Arjunolic acid effectively safeguards retinal cells against oxidative stress and inflammation induced by STZ. This protection is mediated through the autophagy pathway, which is regulated by AMPK/mTOR/HO-1 (218). Artesunate (ART), an active ingredient derived from the traditional Chinese medicine Artemisia annua L., was intravenously injected into STZ-induced diabetic rats. The study demonstrated that ART improved the physiological state of the retina in a positive manner. The findings indicated that the expression of Beclin-1 and the ratio of LC3II/I were up-regulated, while p62 was down-regulated. Additionally, the AMPK/SIRT1 pathway was activated, suggesting that ART can activate autophagy and potentially play a therapeutic role (219). Similarly, quercetin treatment of human retinal microvascular endothelial cells (HRMECs) cultured in vitro at high glucose levels showed a dose-dependent inhibition of autophagy (220). On the other hand, Gypenoside XVII (Gyp-17), a natural product isolated from the traditional Chinese medicine Panax ginseng, was used for the treatment of early diabetic retinopathy (DR) in db/db mice and Müller cells. The results demonstrated that Gyp-17 significantly increased the expression of pro-autophagy-related proteins and could prevent early DR by enhancing autophagy (221). Norkurarinone and isoxanthohumol have been shown to enhance cellular oxidative stress, activate the PI3K/AKT/mTOR signaling pathway, and regulate autophagy dysregulation. These effects have been found to protect human retinal microvascular endothelial cells under conditions of high glucose and hypoxia (222). Procyanidin, a polyphenolic compound, also exhibits anti-diabetic properties. In an experimental study, retinal pigment epithelial cells were initially exposed to high glucose, resulting in reduced cell viability, increased apoptosis rate, and enhanced autophagic flux. However, subsequent administration of procyanidin reversed these changes induced by high sugar levels. Furthermore, when the autophagy agonist rapamycin was reintroduced, retinal pigment epithelial cells exhibited decreased activity and increased apoptosis. This suggests that PC protects retinal pigment epithelial cells by inhibiting autophagy (223). In type II diabetic rat model, it was discovered that Mingmu Xiaomeng Tablets (MMXM) administration protected the retina in the diabetic state. This protection was achieved by decreasing the protein expression of LC3-II and p62, reducing the phosphorylation of PI3K, Akt, and mTOR, inhibiting PI3K/Akt/mTOR signaling, and promoting autophagy (224).
This section highlights the dual role of autophagy in DR and its potential as a target for drug therapy. The study reveals that certain drugs can activate autophagy to positively treat DR, while others can inhibit autophagy. However, the exact mechanism of action is still not fully understood and requires further investigation.
Diabetes mellitus is an endocrine metabolic disease characterized by disorders in glucose and lipid metabolism, leading to increased plasma glucose levels. Its pathophysiology is characterized by a deficiency in insulin secretion, either absolute or relative, with or without increased glucagon activity (15). With accelerated economic development and industrialization, the prevalence of diabetes and the number of diabetics have risen sharply, and diabetes itself and its complications have placed a heavy burden on human health and social development (225, 226). The pathogenesis of diabetes is complex, including both genetic and external environmental factors, and there is an intricate link between diabetes and diabetes complications, and between complications and complications. Autophagy, as a strictly regulated intracellular catabolic process, is closely related to glucose metabolism. One type of autophagy is non-selective autophagy, which refers to autophagy initiated in the starved state. The other type of autophagy is selective autophagy, which removes and recycles harmful or unwanted substances from the cell, such as protein aggregates, damaged mitochondria, accumulated peroxisomes, excess ribosomes, endoplasmic reticulum, and other cellular phases, which, if left to accumulate without timely removal, can lead to human diseases (3). Studies have shown that autophagy is disrupted under a high-calorie diet (51, 88, 97). Impaired autophagy function in pancreatic islet beta cells is associated with beta cell dysfunction and failure. Activation of autophagy improves insulin resistance in the diabetic process, and inhibition of autophagy accelerates the death of islet beta cells. In studies on the relationship between autophagy and diabetic complication, it was found that autophagy is involved in the development of diabetic nephropathy, diabetic hepatopathy, diabetic cardiomyopathy, diabetic cognitive dysfunction, and so on, with different mechanisms of upregulation or downregulation of autophagy, which are briefly summarized here.
Both in vivo and in vitro experiments showed that short-term (48 hours in vitro, 4 weeks in vivo) high-sugar exposure activated autophagy in renal podocytes, while long-term (15 days in vitro, 8 weeks in vivo) high-sugar exposure inhibited autophagy (84). And the physiological characteristics of T2DM are precisely the chronic hyperglycemia levels. Experiments have shown that both chronic high blood glucose levels (88), and the accumulation of AGEs (91), as well as the knockout deletion of autophagy-related genes (Atg5-12/Beclin-1/LC-3) (84–86, 96), eventually lead to damage of renal podocytes and proximal tubular epithelial cells by inhibition of autophagy, which in turn causes renal inflammation, fibrosis, proteinuria, and other symptoms of diabetic nephropathy. When the autophagy inhibitor (rapamycin) was used to treat the diabetic nephropathy cells, the autophagic flux was increased and the course of diabetic nephropathy was improved (89, 90). In conclusion, the development of diabetic nephropathy is associated with the inhibition of autophagy, and activation of autophagy can restore the viability of renal podocytes and proximal renal tubular cells, and thus treat diabetic nephropathy.
As an important organ for regulating glucose and lipid metabolism, the progression of liver disease and T2DM are both closely related to metabolic dysfunction. Impaired autophagy has been demonstrated to be present in diabetic liver disease (129). A high protein diet (133), use of autophagy inhibitors (chloroquine) (131), and autophagy gene deletion (Atg7) (132) all lead to disruption of autophagy, which in turn leads to obstruction of hepatic lipotoxic self-repair mechanisms and increases apoptosis, resulting in the development and progression of diabetic liver disease. When autophagy activators are used and expression of the autophagy gene (Atg7) is restored, a positive effect on the treatment of the disease is demonstrated (132). In conclusion, autophagy inhibition plays a negative role in the development and progression of diabetic liver disease, and activation of autophagy may be a new target for the treatment of diabetic liver disease.
Cardiac muscle cells are insulin-sensitive tissues and glycolysis-dependent tissues, making them vulnerable to hyperglycemia. Studies have shown that persistent hyperglycemia inhibits autophagy in cardiac muscle cells (146, 147). However, this autophagy inhibition does not always have a negative effect on cardiac muscle cells. When cardiac muscle cells in the high-glucose state were treated with an autophagy inhibitor (3-MA) or silencing of autophagy-related genes (Atg7), autophagy was inhibited and high-glucose-induced cardiac muscle cell death was reduced. Meanwhile, when autophagy is activated with an autophagy activator (rapamycin), the cells are prone to suffer from high glucose toxicity. It has also been shown that insulin resistance over-activates autophagy in cardiomyocytes and prevents cardiomyocyte survival. Therefore, inhibition of autophagy in cardiac muscle cells may enhance their survival rate. The role of autophagy in diabetic cardiomyopathy is considered by some scholars a “Goldilocks” phenomenon, which means that excessive activation or inhibition of autophagy can lead to the development and progression of diabetes and its complications. In order for autophagy to play a positive role, it may be necessary to find an “optimal range” of autophagy, within which autophagy is triggered to obtain the best positive feedback (172, 173).
Prolonged high sugar levels can produce damage to the central nervous system, affecting cognitive domains and increasing the risk of cognitive dysfunction. The longer the duration of T2DM, the more likely it is that cognitive dysfunction will occur. It has been confirmed that autophagy plays a role in DACD. However, the specific trends of the roles are not the same. Most studies suggest that the occurrence of DACD is related to the inhibition of autophagy, which is suppressed and Aβ accumulates (187). However, some studies have suggested that T2DM activates autophagy, which in turn causes Aβ deposition in the brain. Therefore, we wonder whether the role of autophagy in DACD is similar to that of the “Goldilocks” phenomenon, in which excessive activation or inhibition can lead to accelerated disease progression, and the most appropriate treatment point is within a narrow range.
As a vasculopathy and neuropathy, DR is the leading cause of blindness in diabetic patients. Research studies have identified autophagy as a potential target for the treatment of DR. In human retinal pigment epithelial cells, prolonged high glucose levels activate autophagy, while in mice retinal explants, they inhibit autophagy. Therefore, dysregulation of autophagy worsens DR. When considering autophagy as a therapeutic target for DR, certain drugs have shown the ability to prevent or treat DR by activating autophagy and reducing oxidative stress. However, other drugs have beneficial effects by inhibiting autophagy. It is important to note that the role of autophagy in DR is dose-dependent, acting protectively at low doses but promoting cell death at high doses.
Oral hypoglycemic drugs currently used in clinical practice are divided into seven major classes: sulfonylureas, biguanides, alpha-glucosidase inhibitors, thiazolidinediones, glinides, glucagon-like peptide-1 (GLP-1) and its analogs, and DPP-4 inhibitors. Some chemical drugs are mentioned in this review that can treat T2DM and its complications by regulating autophagy. For example, metformin (a biguanide hypoglycemic agent) can exert therapeutic effects on pancreatic β-cell protection (56), diabetic nephropathy (102, 103), cardiomyopathy (163, 164), and DACD (196) by activating autophagy. Liraglutide, a GLP-1 analog, has been shown to enhance autophagy, providing protection to pancreatic β cells (53, 54) and potential treatment for conditions such as diabetic nephropathy and diabetic hepatopathy. However, it is important to note that GLP-1 inhibits autophagy and improves diabetic retinopathy (215). Additionally, Exendin-4 and Linagliptin have been found to activate autophagy, leading to potential benefits in protecting pancreatic β-cells and improving conditions like diabetic nephropathy and DACD (55, 197). Empagliflozin (SGLT2 inhibitor) can be used to treat diabetic nephropathy and diabetic hepatopathy by enhancing autophagy (107, 108, 134, 135), but Gliflozins (SGLT2 inhibitor) is used to alleviate DACD by inhibiting autophagy (194). In addition to the commonly used glucose-lowering drugs in clinical practice, there are alprostadil (prostaglandins), vitamin D3 and 1,25(OH)2D3 (Vitamin), α-lipoic acid (a fatty acid in the human body), melatonin (Indole heterocycles), zinc, octreotide (growth hormone), PG545 (heparanase inhibitor), β-hydroxybutyrate have also been shown in experiments to treat diabetes-related complications by modulating autophagy (138, 139, 161, 170, 214, 216, 217). Among them, zinc, α-lipoic acid, GLP-1 and β-hydroxybutyrat alleviate the symptoms of diabetic cardiovascular complications by inhibiting autophagy, while the rest of the drugs play a therapeutic role in treating the disease by upregulating autophagy.
Most chemical drugs are single drug components, and some of them can produce unexpected adverse effects. For example, mTOR inhibitors can activate autophagy to provide kidney protection, but at the same time, they can inhibit cell growth and hinder kidney repair. Considering that diabetes and diabetic complications, as well as diabetic complications and complications, do not exist independently but are complexly connected and closely linked, it is promising to consider the application of herbal medicine for the treatment of metabolic diseases. In traditional Chinese medicine formula, Yunpi Heluo Decoction can activate autophagy by regulating the SIRT1-FoxO1 signaling pathway in skeletal muscle, while improving lipid metabolism, attenuating insulin resistance and exerting a protective effect on pancreatic β cells (58). Huanglian Jiedu Decoction can improve DACD by inhibiting the activation of NLRP3 inflammatory vesicles upregulating autophagy (199). Zibu Piyin Recipe improves DACD by inhibiting mTOR upregulation, enhancing autophagy, and promoting Aβ clearance (200). Xiaokeping induces mTOR-mediated autophagy and reduces apoptosis in pancreatic β-cells, with a potential protective effect on pancreatic β-cells in high glucose toxicity (59). Mingmu Xiaomeng Tablets exerts a protective effect on the retina during diabetic states by inhibiting the PI3K/Akt/mTOR signaling pathway and promoting autophagy (224). Besides the TCM formula, herbal extracts have also shown good efficacy in animal and cellular models. For example, Morus alba leaves ethanol extract could protect islet cells by inducing AMPK/mTOR-mediated autophagy and by upregulating LC3-activated autophagy, respectively (60). Ginseng extract could treat diabetic nephropathy by upregulating LC3 and downregulating p62 (110). Green tea extract polyphenols could also treat diabetic cardiomyopathy in rats by improving autophagy (165). Monomers from TCM, such as kaempferol and silymarin can maintain cellular energy homeostasis, increase autophagy, and exert insulin β-cell protection through AMPK/mTOR pathway (62), activating autophagy-dependent ERs (63) respectively. Resveratrol, triptolide, astragaloside IV, ginsenoside Rg1, and ferulic acid can exert their effects in the treatment of diabetic nephropathy through activation of miR-383-5p (112), ERK signaling, miR-141-3p/PTEN/Akt/mTOR pathway (113), and AKT/GSK3β/β-linked protein pathway-mediated autophagy (116), respectively. Among them, astragaloside IV can improve diabetic hepatopathy by promoting AMPK/mTOR-mediated autophagy (114, 115), and PU can restore autophagy through Akt/FoxO3a signaling pathway to treat diabetic hepatopathy (140). Resveratrol, carnosic acid, beta carotene, and curcumin can activate autophagy through the SIRT1/FOXO1/Rab7 axis (167), PI3K/Akt/mTOR (168), activate AMPK and JNK1, phosphorylated Bcl-2 and Bim and subsequently disrupted their interactions with Beclin1 to treat diabetic cardiomyopathy (169). Quercetin can activate the ERK signaling pathway in a dose-dependent manner and enhance cellular autophagy in endothelial progenitor cells (166). Berberine enhances cellular autophagy, attenuates apoptosis and improves DACD by activating the AMPK/mTOR pathway (201). Arjunolic acid, artesunate, quercetin, gypenoside XVII, norkurarinone and isoxanthohumol, procyanidin treat diabetic retinopathy through activating or inhibiting autophagy (218–223). All the above-mentioned drugs have been summarized in Supplementary Table 1.
In the mechanistic exploration of complex metabolic diseases T2DM as well as drug development, as a mechanism to regulate energy metabolic homeostasis autophagy has now been shown to play a role in the treatment of diabetes. Targeting the regulation of autophagy is likely to have great advantages for the treatment of T2DM and its complications. It is however important to note that autophagy is inherently two-sided, and its two-sidedness is even more evident in its treatment of diabetes and its complications. Therefore, elucidating the range within which autophagy can be regulated for optimal therapeutic effect is the focus of future research.
XZ drafted this manuscript. L-YB made additions to the text and made changes for contextualization. D-RP drew the structural formula for the compounds. XL and L-FY consulted references. D-DC and YRW organized the references. YG reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of Shandong Province (No. ZR2022QH299), The Key Research and Development Program of Shandong Province (No. 2021CXGC010511), National Natural Science Foundation of China (No. 822046560), Traditional Chinese Medicine Science and Technology Project of Shandong Province (No. Q-2022055), Youth Innovation Team Program in University of Shandong Province (No. 2022KJ341), Higher Education Development Planning Research Project of Shandong University of Traditional Chinese Medicine (No. GJYJZ202210). The authors appreciate their help in earnest.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1228045/full#supplementary-material
DM, Diabetes mellitus; T2DM, Type II diabetes mellitus; DN, Diabetic Nephropathy; DACD, Diabetes-associated cognitive decline; DH, Diabetic Hepatopathy; DC, Diabetic Cardiomyopathy; DR, Diabetic Retinopathy; IR, Insulin Resistance; IDF, International Diabetes Federation; UKPDS, The United Kingdom Prospective Diabetes Study; ERS, Endoplasmic reticulum stress; YPHLD, Yunpi Heluo decoction; XKP, Xiaokeping.
1. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) 1. Autophagy (2021) 17(1):1–382. doi: 10.1080/15548627.2020.1797280
2. Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med (2020) 383(16):1564–76. doi: 10.1056/NEJMra2022774
3. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol (2023) 24(3):167–85. doi: 10.1038/s41580-022-00542-2
4. Hurley JH, Young LN. Mechanisms of autophagy initiation. Annu Rev Biochem (2017) 86:225–44. doi: 10.1146/annurev-biochem-061516-044820
5. Levine B, Kroemer G. Biological functions of autophagy genes: A disease perspective. Cell (2019) 176(1-2):11–42. doi: 10.1016/j.cell.2018.09.048
6. Ohsumi Y. Historical landmarks of autophagy research. Cell Res (2014) 24(1):9–23. doi: 10.1038/cr.2013.169
7. Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science (2004) 306(5698):990–5. doi: 10.1126/science.1099993
8. Kitada M, Koya D. Autophagy in metabolic disease and ageing. Nat Rev Endocrinol (2021) 17(11):647–61. doi: 10.1038/s41574-021-00551-9
9. Yamamoto H, Zhang S, Mizushima N. Autophagy genes in biology and disease. Nat Rev Genet (2023) 24(6):382–400. doi: 10.1038/s41576-022-00562-w
10. Cheng AYY, Gomes MB, Kalra S, Kengne AP, Mathieu C, Shaw JE, et al. Applying the WHO global targets for diabetes mellitus. Nat Rev Endocrinol (2023) 19(4):194–200. doi: 10.1038/s41574-022-00793-1
11. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract (2021) 109119. doi: 10.1016/j.diabres.2021.109119
12. Aschner P, Karuranga S, James S, Simmons D, Basit A, Shaw JE, et al. The International Diabetes Federation's guide for diabetes epidemiological studies. Diabetes Res Clin Pract (2021) 172:108630. doi: 10.1016/j.diabres.2020.108630
13. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med (2017) 23(7):804–14. doi: 10.1038/nm.4350
14. Bailey CJ, Grant PJ. The UK prospective diabetes study. Lancet (1998) 352(9144):1932. doi: 10.1016/S0140-6736(98)00090-7
15. Ahmad E, Lim S, Lamptey R, Webb DR, Davies MJ. Type 2 diabetes. Lancet (2022) 400(10365):1803–20. doi: 10.1016/S0140-6736(22)01655-5
16. Chan JCN, Lim LL, Wareham NJ, Shaw JE, Orchard TJ, Zhang P, et al. The Lancet Commission on diabetes: using data to transform diabetes care and patient lives. Lancet (2021) 396(10267):2019–82. doi: 10.1016/S0140-6736(20)32374-6
17. Marasco MR, Linnemann AK. β-cell autophagy in diabetes pathogenesis. Endocrinology (2018) 159(5):2127–41. doi: 10.1210/en.2017-03273
18. Ha J, Guan KL, Kim J. AMPK and autophagy in glucose/glycogen metabolism. Mol Aspects Med (2015) 46:46–62. doi: 10.1016/j.mam.2015.08.002
19. Riahi Y, Wikstrom JD, Bachar-Wikstrom E, Polin N, Zucker H, Lee MS, et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia (2016) 59(7):1480–91. doi: 10.1007/s00125-016-3868-9
20. Bagherniya M, Butler AE, Barreto GE, Sahebkar A. The effect of fasting or calorie restriction on autophagy induction: A review of the literature. Ageing Res Rev (2018) 47:183–97. doi: 10.1016/j.arr.2018.08.004
21. Zhang P, Li T, Wu X, Nice EC, Huang C, Zhang Y. Oxidative stress and diabetes: antioxidative strategies. Front Med (2020) 14(5):583–600. doi: 10.1007/s11684-019-0729-1
22. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ (2015) 22(3):377–88. doi: 10.1038/cdd.2014.150
23. Gao Q. Oxidative stress and autophagy. Adv Exp Med Biol (2019) 1206:179–98. doi: 10.1007/978-981-15-0602-4_9
24. Yun HR, Jo YH, Kim J, Shin Y, Kim SS, Choi TG. Roles of autophagy in oxidative stress. Int J Mol Sci (2020) 21(9):3289. doi: 10.3390/ijms21093289. null.
25. Deter RL, De Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol (1967) 33(2):437–49. doi: 10.1083/jcb.33.2.437
26. Yong J, Johnson JD, Arvan P, Han J, Kaufman RJ. Therapeutic opportunities for pancreatic β-cell ER stress in diabetes mellitus. Nat Rev Endocrinol (2021) 17(8):455–67. doi: 10.1038/s41574-021-00510-4
27. Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: Autophagy induction, inhibition and selection. Autophagy (2015) 11(11):1956–77. doi: 10.1080/15548627.2015.1091141
28. Zhang IX, Hermann A, Leon J, Jeyarajan S, Arunagiri A, Arvan P. ER stress increases expression of intracellular calcium channel RyR1 to modify Ca2+ homeostasis in pancreatic beta cells. J Biol Chem (2023) 299(8):105065. doi: 10.1016/j.jbc.2023.105065
29. Chiu WT, Chang HA, Lin YH, Lin YS, Chang HT, Lin HH. Bcl-2 regulates store-operated Ca2+ entry to modulate ER stress-induced apoptosis. Cell Death Discovery (2018) 4:37. doi: 10.1038/s41420-018-0039-4
30. Popat A, Patel AA, Warnes G. A flow cytometric study of ER stress and autophagy. Cytometry A (2019) 95(6):672–82. doi: 10.1002/cyto.a.23665
31. Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia (2003) 46(1):3–19. doi: 10.1007/s00125-002-1009-0
32. Lee MS. Role of islet β cell autophagy in the pathogenesis of diabetes. Trends Endocrinol Metab (2014) 25(12):620–7. doi: 10.1016/j.tem.2014.08.005
33. Yuan T, Rafizadeh S, Gorrepati KD, Lupse B, Oberholzer J, Maedler K, et al. Reciprocal regulation of mTOR complexes in pancreatic islets from humans with type 2 diabetes. Diabetologia (2017) 60(4):668–78. doi: 10.1007/s00125-016-4188-9
34. Marchetti P, Masini M. Autophagy and the pancreatic beta-cell in human type 2 diabetes. Autophagy (2009) 5(7):1055–6. doi: 10.4161/auto.5.7.9511
35. Rivera JF, Costes S, Gurlo T, Gurlo T, Glabe CG, Butler PC. Autophagy defends pancreatic β cells from human islet amyloid polypeptide-induced toxicity. J Clin Invest (2014) 124(8):3489–500. doi: 10.1172/JCI71981
36. Sheng Q, Xiao X, Prasadan K, Chen C, Ming Y, Fusco J, et al. Autophagy protects pancreatic beta cell mass and function in the setting of a high-fat and high-glucose diet. Sci Rep (2017) 7(1):16348. doi: 10.1038/s41598-017-16485-0
37. Lambelet M, Terra LF, Fukaya M, Meyerovich K, Labriola L, Cardozo AK, et al. Dysfunctional autophagy following exposure to pro-inflammatory cytokines contributes to pancreatic β-cell apoptosis. Cell Death Dis (2018) 9(2):96. doi: 10.1038/s41419-017-0121-5
38. Yang L, Lin H, Lin W, Xu X. Exercise ameliorates insulin resistance of type 2 diabetes through motivating short-chain fatty acid-mediated skeletal muscle cell autophagy. Biol (Basel) (2020) 9(8):203. doi: 10.3390/biology9080203
39. Fujitani Y, Kawamori R, Watada H. The role of autophagy in pancreatic beta-cell and diabetes. Autophagy (2009) 5(2):280–2. doi: 10.4161/auto.5.2.7656
40. Frendo-Cumbo S, Jaldin-Fincati JR, Coyaud E, Laurent EMN, Townsend LK, Tan JMJ, et al. Deficiency of the autophagy gene ATG16L1 induces insulin resistance through KLHL9/KLHL13/CUL3-mediated IRS1 degradation. J Biol Chem (2019) 294(44):16172–85. doi: 10.1074/jbc.RA119.009110
41. Cai J, Pires KM, Ferhat M, Chaurasia B, Buffolo MA, Smalling R, et al. Autophagy ablation in adipocytes induces insulin resistance and reveals roles for lipid peroxide and Nrf2 signaling in adipose-liver crosstalk. Cell Rep (2018) 25(7):1708–17.e5. doi: 10.1016/j.celrep.2018.10.040
42. Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab (2008) 8(4):325–32. doi: 10.1016/j.cmet.2008.08.009
43. Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab (2008) 8(4):318–24. doi: 10.1016/j.cmet.2008.08.013
44. Guillén C, Benito M. mTORC1 overactivation as a key aging factor in the progression to type 2 diabetes mellitus. Front Endocrinol (Lausanne) (2018) 9:621. doi: 10.3389/fendo.2018.00621
45. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
46. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi: 10.1016/j.cell.2012.03.017
47. Bartolomé A, Kimura-Koyanagi M, Asahara S, Guillén C, Inoue H, Teruyama K, et al. Pancreatic β-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes (2014) 63(9):2996–3008. doi: 10.2337/db13-0970
48. Brown MR, Laouteouet D, Delobel M, Villard O, Broca C, Bertrand G, et al. The nuclear receptor REV-ERBα is implicated in the alteration of β-cell autophagy and survival under diabetogenic conditions. Cell Death Dis (2022) 13(4):353. doi: 10.1038/s41419-022-04767-z
49. Liang R, Liu N, Cao J, Liu T, Sun P, Cai X, et al. HIF-1α/FOXO1 axis regulated autophagy is protective for β cell survival under hypoxia in human islets. Biochim Biophys Acta Mol Basis Dis (2022) 1868(5):166356. doi: 10.1016/j.bbadis.2022.166356
50. Chatterjee T, Pattanayak R, Ukil A, Chowdhury S, Bhattacharyya M. Autophagy protects peripheral blood mononuclear cells against inflammation, oxidative and nitrosative stress in diabetic dyslipidemia. Free Radic Biol Med (2019) 143:309–23. doi: 10.1016/j.freeradbiomed.2019.07.034
51. Mir SU, George NM, Zahoor L, Harms R, Guinn Z, Sarvetnick NE. Inhibition of autophagic turnover in β-cells by fatty acids and glucose leads to apoptotic cell death. J Biol Chem (2015) 290(10):6071–85. doi: 10.1074/jbc.M114.605345
52. Rossi MC, Nicolucci A. Liraglutide in type 2 diabetes: from pharmacological development to clinical practice. Acta BioMed (2009) 80(2):93–101.
53. Li XD, He SS, Wan TT, Li YB. Liraglutide protects palmitate-induced INS-1 cell injury by enhancing autophagy mediated via FoxO1. Mol Med Rep (2021) 23(2):1. doi: 10.3892/mmr.2020.11786
54. Chen ZF, Li YB, Han JY, Yin JJ, Wang Y, Zhu LB, et al. Liraglutide prevents high glucose level induced insulinoma cells apoptosis by targeting autophagy. Chin Med J (Engl) (2013) 126(5):937–41. doi: 10.3760/cma.j.issn.0366-6999.20122331
55. Lim SW, Jin L, Jin J, Yang CW. Effect of exendin-4 on autophagy clearance in beta cell of rats with tacrolimus-induced diabetes mellitus. Sci Rep (2016) 6:29921. doi: 10.1038/srep29921
56. Jiang Y, Huang W, Wang J, Xu Z, He J, Lin X, et al. Metformin plays a dual role in MIN6 pancreatic β cell function through AMPK-dependent autophagy. Int J Biol Sci (2014) 10(3):268–77. doi: 10.7150/ijbs.7929
57. Zhang Y, Zhou XA, Liu C, Shen QW, Wu YY. Vitamin B6 inhibits high glucose-induced islet β Cell apoptosis by upregulating autophagy. Metabolites (2022) 12(11):1048. doi: 10.3390/metabo12111048
58. Mao ZJ, Xia WS, Chai F. Yunpi Heluo decoction attenuates insulin resistance by regulating SIRT1-FoxO1 autophagy pathway in skeletal muscle of Zucker diabetic fatty rats. J Ethnopharmacol (2021) 270:113828. doi: 10.1016/j.jep.2021.113828
59. Wu Y, Hu Y, Zhou H, Wei Y, Kang X, Liu D. Xiaokeping-induced autophagy protects pancreatic β-cells against apoptosis under high glucose stress. BioMed Pharmacother (2018) 105:407–12. doi: 10.1016/j.biopha.2018.05.147
60. Ji S, Zhu C, Gao S, Shao X, Chen X, Zhang H, et al. Morus alba leaves ethanol extract protects pancreatic islet cells against dysfunction and death by inducing autophagy in type 2 diabetes. Phytomedicine (2021) 83:153478. doi: 10.1016/j.phymed.2021.153478
61. Li H, Yu L, Zhao C. Dioscin attenuates high−fat diet−induced insulin resistance of adipose tissue through the IRS−1/PI3K/Akt signaling pathway. Mol Med Rep (2019) 19(2):1230–7. doi: 10.3892/mmr.2018.9700
62. Varshney R, Gupta S, Roy P. Cytoprotective effect of kaempferol against palmitic acid-induced pancreatic β-cell death through modulation of autophagy via AMPK/mTOR signaling pathway. Mol Cell Endocrinol (2017) 448:1–20. doi: 10.1016/j.mce.2017.02.033
63. Yang J, Sun Y, Xu F, Liu W, Hayashi T, Onodera S, et al. Involvement of estrogen receptors in silibinin protection of pancreatic β-cells from TNFα- or IL-1β-induced cytotoxicity. BioMed Pharmacother (2018) 102:344–53. doi: 10.1016/j.biopha.2018.01.128
64. Gadallah SH, Ghanem HM, Abdel-Ghaffar A, Metwaly FG, Hanafy LK, Ahmed EK. 4-Phenylbutyric acid and rapamycin improved diabetic status in high fat diet/streptozotocin-induced type 2 diabetes through activation of autophagy. Arch Physiol Biochem (2021) 127(3):235–44. doi: 10.1080/13813455.2019.1628069
65. Cheng F, Dun Y, Cheng J, Ripley-Gonzalez JW, Jiang W, You B, et al. Exercise activates autophagy and regulates endoplasmic reticulum stress in muscle of high-fat diet mice to alleviate insulin resistance. Biochem Biophys Res Commun (2022) 601:45–51. doi: 10.1016/j.bbrc.2022.02.058
66. Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KA, Zoungas S, et al. Diabetic kidney disease. Nat Rev Dis Primers (2015) 1:15018. doi: 10.1038/nrdp.2015.18
67. Rico-Fontalvo J, Aroca G, Cabrales J, Daza-Arnedo R, Yánez-Rodríguez T, Martínez-Ávila MC, et al. Molecular mechanisms of diabetic kidney disease. Int J Mol Sci (2022) 23(15):8668. doi: 10.3390/ijms23158668
68. Yang D, Livingston MJ, Liu Z, Dong G, Zhang M, Chen JK, et al. Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential. Cell Mol Life Sci (2018) 75(4):669–88. doi: 10.1007/s00018-017-2639-1
69. Atkins RC, Zimmet PZ. Diabetic kidney disease: act now or pay later. Med J Aust (2010) 192(5):272–4. doi: 10.5694/j.1326-5377.2010.tb03506.x
70. Saad R, Tadmor H, Ertracht O, Nakhoul N, Nakhoul F, Evgeny F, et al. The molecular effects of SGLT2i empagliflozin on the autophagy pathway in diabetes mellitus type 2 and its complications. J Diabetes Res (2022) 2022:8337823. doi: 10.1155/2022/8337823
71. Fineberg D, Jandeleit-Dahm KA, Cooper ME. Diabetic nephropathy: diagnosis and treatment. Nat Rev Endocrinol (2013) 9(12):713–23. doi: 10.1038/nrendo.2013.184
72. Hu Q, Jiang L, Yan Q, Zeng J, Ma X, Zhao Y. A natural products solution to diabetic nephropathy therapy. Pharmacol Ther (2023) 241:108314. doi: 10.1016/j.pharmthera.2022.108314
73. Nathan DM. Finding new treatments for diabetes–how many, how fast. how good? N Engl J Med (2007) 356(5):437–40. doi: 10.1056/NEJMp068294
74. Lin YC, Chang YH, Yang SY, Wu KD, Chu TS. Update of pathophysiology and management of diabetic kidney disease. J Formos Med Assoc (2018) 117(8):662–75. doi: 10.1016/j.jfma.2018.02.007
75. McGrath K, Edi R. Diabetic kidney disease: diagnosis, treatment, and prevention. Am Fam Physician (2019) 99(12):751–9.
76. Choi ME. Autophagy in kidney disease. Annu Rev Physiol (2020) 82:297–322. doi: 10.1146/annurev-physiol-021119-034658
77. Lin TA, Wu VC, Wang CY. Autophagy in chronic kidney diseases. Cells (2019) 8(1):61. doi: 10.3390/cells8010061
78. Kaushal GP, Chandrashekar K, Juncos LA, Shah SV. Autophagy function and regulation in kidney disease. Biomolecules (2020) 10(1):100. doi: 10.3390/biom10010100
79. Koch EAT, Nakhoul R, Nakhoul F, Nakhoul N. Autophagy in diabetic nephropathy: a review. Int Urol Nephrol (2020) 52(9):1705–12. doi: 10.1007/s11255-020-02545-4
80. Liu WJ, Gan Y, Huang WF, Wu HL, Zhang XQ, Zheng HJ, et al. Lysosome restoration to activate podocyte autophagy: a new therapeutic strategy for diabetic kidney disease. Cell Death Dis (2019) 10(11):806. doi: 10.1038/s41419-019-2002-6
81. Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest (2010) 120(4):1084–96. doi: 10.1172/JCI39492
82. Gonzalez CD, Carro Negueruela MP, Nicora Santamarina C, Resnik R, Vaccaro MI. Autophagy dysregulation in diabetic kidney disease: from pathophysiology to pharmacological interventions. Cells (2021) 10(9):2497. doi: 10.3390/cells10092497
83. Liu N, Xu L, Shi Y, Zhuang S. Podocyte autophagy: A potential therapeutic target to prevent the progression of diabetic nephropathy. J Diabetes Res (2017) 2017:3560238. doi: 10.1155/2017/3560238
84. Lenoir O, Jasiek M, Hénique C, Guyonnet L, Hartleben B, Bork T, et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy (2015) 11(7):1130–45. doi: 10.1080/15548627.2015.1049799
85. Fang L, Zhou Y, Cao H, Wen P, Jiang L, He W, et al. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PloS One (2013) 8(4):e60546. doi: 10.1371/journal.pone.0060546
86. Tagawa A, Yasuda M, Kume S, Yamahara K, Nakazawa J, Chin-Kanasaki M, et al. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes (2016) 65(3):755–67. doi: 10.2337/db15-0473
87. Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest (2011) 121(6):2197–209. doi: 10.1172/JCI44774
88. Xin W, Li Z, Xu Y, Yu Y, Zhou Q, Chen L, et al. Autophagy protects human podocytes from high glucose-induced injury by preventing insulin resistance. Metabolism (2016) 65(9):1307–15. doi: 10.1016/j.metabol.2016.05.015
89. Yang Y, Wang J, Qin L, Shou Z, Zhao J, Wang H, et al. Rapamycin prevents early steps of the development of diabetic nephropathy in rats. Am J Nephrol (2007) 27(5):495–502. doi: 10.1159/000106782
90. Xiao T, Guan X, Nie L, Wang S, Sun L, He T, et al. Rapamycin promotes podocyte autophagy and ameliorates renal injury in diabetic mice. Mol Cell Biochem (2014) 394(1-2):145–54. doi: 10.1007/s11010-014-2090-7
91. Zhao X, Chen Y, Tan X, Zhang L, Zhang H, Li Z, et al. Advanced glycation end-products suppress autophagic flux in podocytes by activating mamMalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. J Pathol (2018) 245(2):235–48. doi: 10.1002/path.5077
92. Sakai S, Yamamoto T, Takabatake Y, Takahashi A, Namba-Hamano T, Minami S, et al. Proximal tubule autophagy differs in type 1 and 2 diabetes. J Am Soc Nephrol (2019) 30(6):929–45. doi: 10.1681/ASN.2018100983
93. Liu WJ, Shen TT, Chen RH, Wu HL, Wang YJ, Deng JK, et al. Autophagy-lysosome pathway in renal tubular epithelial cells is disrupted by advanced glycation end products in diabetic nephropathy. J Biol Chem (2015) 290(33):20499–510. doi: 10.1074/jbc.M115.666354
94. Xu Y, Liu L, Xin W, Zhao X, Chen L, Zhen J, et al. The renoprotective role of autophagy activation in proximal tubular epithelial cells in diabetic nephropathy. J Diabetes Complications (2015) 29(8):976–83. doi: 10.1016/j.jdiacomp.2015.07.021
95. Barbosa Júnior Ade A, Zhou H, Hültenschmidt D, Totovic V, Jurilj N, Pfeifer U. Inhibition of cellular autophagy in proximal tubular cells of the kidney in streptozotocin-diabetic and uninephrectomized rats. Virchows Arch B Cell Pathol Incl Mol Pathol (1992) 61(6):359–66. doi: 10.1007/BF02890439
96. Ma Z, Li L, Livingston MJ, Zhang D, Mi Q, Zhang M, et al. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J Clin Invest (2020) 130(9):5011–26. doi: 10.1172/JCI135536
97. Yamahara K, Kume S, Koya D, Tanaka Y, Morita Y, Chin-Kanasaki M, et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J Am Soc Nephrol (2013) 24(11):1769–81. doi: 10.1681/ASN.2012111080
98. Kitada M, Takeda A, Nagai T, Ito H, Kanasaki K, Koya D. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp Diabetes Res (2011) 2011:908185. doi: 10.1155/2011/908185
99. Kitada M, Ogura Y, Suzuki T, Sen S, Lee SM, Kanasaki K, et al. A very-low-protein diet ameliorates advanced diabetic nephropathy through autophagy induction by suppression of the mTORC1 pathway in Wistar fatty rats, an animal model of type 2 diabetes and obesity. Diabetologia (2016) 59(6):1307–17. doi: 10.1007/s00125-016-3925-4
100. Calvo-Rubio M, Burón MI, López-Lluch G, Navas P, Cabo R, Ramsey JJ, et al. Dietary fat composition influences glomerular and proximal convoluted tubule cell structure and autophagic processes in kidneys from calorie-restricted mice. Aging Cell (2016) 15(3):477–87. doi: 10.1111/acel.12451
101. Ren H, Shao Y, Wu C, Ma X, Lv C, Wang Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol Cell Endocrinol (2020) 500:110628. doi: 10.1016/j.mce.2019.110628
102. Xu J, Liu LQ, Xu LL, Xing Y, Ye S. Metformin alleviates renal injury in diabetic rats by inducing Sirt1/FoxO1 autophagic signal axis. Clin Exp Pharmacol Physiol (2020) 47(4):599–608. doi: 10.1111/1440-1681.13226
103. Wei W, An XR, Jin SJ, Li XX, Xu M. Inhibition of insulin resistance by PGE1 via autophagy-dependent FGF21 pathway in diabetic nephropathy. Sci Rep (2018) 8(1):9. doi: 10.1038/s41598-017-18427-2
104. Yamada S, Tanabe J, Ogura Y, Nagai Y, Sugaya T, Ohata K, et al. Renoprotective effect of GLP-1 receptor agonist, liraglutide, in early-phase diabetic kidney disease in spontaneously diabetic Torii fatty rats. Clin Exp Nephrol (2021) 25(4):365–75. doi: 10.1007/s10157-020-02007-2
105. Rieg T, Vallon V. Development of SGLT1 and SGLT2 inhibitors. Diabetologia (2018) 61(10):2079–86. doi: 10.1007/s00125-018-4654-7
106. Packer M. Interplay of adenosine monophosphate-activated protein kinase/sirtuin-1 activation and sodium influx inhibition mediates the renal benefits of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes: A novel conceptual framework. Diabetes Obes Metab (2020) 22(5):734–42. doi: 10.1111/dom.13961
107. Korbut AI, Taskaeva IS, Bgatova NP, Muraleva NA, Orlov NB, Dashkin MV, et al. SGLT2 inhibitor empagliflozin and DPP4 inhibitor linagliptin reactivate glomerular autophagy in db/db mice, a model of type 2 diabetes. Int J Mol Sci (2020) 21(8):2987. doi: 10.3390/ijms21082987
108. Lee YH, Kim SH, Kang JM, Heo JH, Kim DJ, Park SH, et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am J Physiol Renal Physiol (2019) 317(4):F767–80. doi: 10.1152/ajprenal.00565.2018
109. Yang S, Lin C, Zhuo X, Wang J, Rao S, Xu W, et al. Glucagon-like peptide-1 alleviates diabetic kidney disease through activation of autophagy by regulating AMP-activated protein kinase-mamMalian target of rapamycin pathway. Am J Physiol Endocrinol Metab (2020) 319(6):E1019–30. doi: 10.1152/ajpendo.00195.2019
110. Karunasagara S, Hong GL, Park SR, Lee NH, Jung DY, Kim TW, et al. Korean red ginseng attenuates hyperglycemia-induced renal inflammation and fibrosis via accelerated autophagy and protects against diabetic kidney disease. J Ethnopharmacol (2020) 254:112693. doi: 10.1016/j.jep.2020.112693
111. Chowdhury S, Ghosh S, Das AK, Sil PC. Ferulic acid protects hyperglycemia-induced kidney damage by regulating oxidative insult, inflammation and autophagy. Front Pharmacol (2019) 10:27. doi: 10.3389/fphar.2019.00027
112. Huang SS, Ding DF, Chen S, Dong CL, Ye XL, Yuan YG, et al. Resveratrol protects podocytes against apoptosis via stimulation of autophagy in a mouse model of diabetic nephropathy. Sci Rep (2017) 7:45692. doi: 10.1038/srep45692
113. Li XY, Wang SS, Han Z, Han F, Chang YP, Yang Y, et al. Triptolide Restores Autophagy to Alleviate Diabetic Renal Fibrosis through the miR-141-3p/PTEN/Akt/mTOR Pathway. Mol Ther Nucleic Acids (2017) 9:48–56. doi: 10.1016/j.omtn.2017.08.011
114. Guo H, Wang Y, Zhang X, Zang Y, Zhang Y, Wang L, et al. Astragaloside IV protects against podocyte injury via SERCA2-dependent ER stress reduction and AMPKα-regulated autophagy induction in streptozotocin-induced diabetic nephropathy. Sci Rep (2017) 7(1):6852. doi: 10.1038/s41598-017-07061-7
115. Wang X, Gao Y, Tian N, Zhu Z, Wang T, Xu J, et al. Astragaloside IV represses high glucose-induced mesangial cells activation by enhancing autophagy via SIRT1 deacetylation of NF-κB p65 subunit. Drug Des Devel Ther (2018) 12:2971–80. doi: 10.2147/DDDT.S174058
116. Shi Y, Gao Y, Wang T, Wang X, He J, Xu J, et al. Ginsenoside rg1 alleviates podocyte EMT passage by regulating AKT/GSK3 β / β -catenin pathway by restoring autophagic activity. Evid Based Complement Alternat Med (2020) 2020:1903627. doi: 10.1155/2020/1903627
117. Lin CF, Kuo YT, Chen TY, Chien CT. Quercetin-rich guava (Psidium guajava) juice in combination with trehalose reduces autophagy, apoptosis and pyroptosis formation in the kidney and pancreas of type II diabetic rats. Molecules (2016) 21(3):334. doi: 10.3390/molecules21030334
118. Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M, Richardson PA, et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology (2018) 155(6):1828–1837.e2. doi: 10.1053/j.gastro.2018.08.024
119. Stefan N, Cusi K. A global view of the interplay between non-alcoholic fatty liver disease and diabetes. Lancet Diabetes Endocrinol (2022) 10(4):284–96. doi: 10.1016/S2213-8587(22)00003-1
120. Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest (2020) 130(3):1453–60. doi: 10.1172/JCI134165
121. Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J Hepatol (2019) 71(4):793–801. doi: 10.1016/j.jhep.2019.06.021
122. EASL, EASD, EASO. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol (2016) 64(6):1388–402. doi: 10.1016/j.jhep.2015.11.004
123. Targher G, Corey KE, Byrne CD, Roden M. The complex link between NAFLD and type 2 diabetes mellitus - mechanisms and treatments. Nat Rev Gastroenterol Hepatol (2021) 18(9):599–612. doi: 10.1038/s41575-021-00448-y
124. Powell EE, Wong VW, Rinella M. Non-alcoholic fatty liver disease. Lancet (2021) 397(10290):2212–24. doi: 10.1016/S0140-6736(20)32511-3
125. Hamed AE, Elwan N, Naguib M, Elwakil R, Esmat G, Kassas ME, et al. Diabetes association with liver diseases: an overview for clinicians. Endocr Metab Immune Disord Drug Targets (2019) 19(3):274–80. doi: 10.2174/1871530318666181116111945
126. Mantovani A, Petracca G, Beatrice G, Tilg H, Byrne CD, Targher G. Non-alcoholic fatty liver disease and risk of incident diabetes mellitus: an updated meta-analysis of 501 022 adult individuals. Gut (2021) 70(5):962–9. doi: 10.1136/gutjnl-2020-322572
127. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol (2020) 73(1):202–9. doi: 10.1016/j.jhep.2020.03.039
128. Fukuo Y, Yamashina S, Sonoue H, Arakawa A, Nakadera E, Aoyama T, et al. AbnorMality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease. Hepatol Res (2014) 44(9):1026–36. doi: 10.1111/hepr.12282
129. González-Rodríguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis (2014) 5:e1179. doi: 10.1038/cddis.2014.162
130. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature (2009) 458(7242):1131–5. doi: 10.1038/nature07976
131. Lin CW, Zhang H, Li M, Xiong X, Chen X, Dong XC, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol (2013) 58(5):993–9. doi: 10.1016/j.jhep.2013.01.011
132. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab (2010) 11(6):467–78. doi: 10.1016/j.cmet.2010.04.005
133. Zhang F, Zhao S, Yan W, Xia Y, Chen X, Wang W, et al. Branched chain amino acids cause liver injury in obese/diabetic mice by promoting adipocyte lipolysis and inhibiting hepatic autophagy. EBioMedicine (2016) 13:157–67. doi: 10.1016/j.ebiom.2016.10.013
134. Meng Z, Liu X, Li T, Fang T, Cheng Y, Han L, et al. The SGLT2 inhibitor empagliflozin negatively regulates IL-17/IL-23 axis-mediated inflammatory responses in T2DM with NAFLD via the AMPK/mTOR/autophagy pathway. Int Immunopharmacol (2021) 94:107492. doi: 10.1016/j.intimp.2021.107492
135. Li T, Fang T, Xu L, Liu X, Li X, Xue M, et al. Empagliflozin Alleviates Hepatic Steatosis by Activating the AMPK-TET2-Autophagy Pathway in vivo and in vitro. Front Pharmacol (2020) 11:622153. doi: 10.3389/fphar.2020.622153
136. He Y, Ao N, Yang J, Wang X, Jin S, Du J. The preventive effect of liraglutide on the lipotoxic liver injury via increasing autophagy. Ann Hepatol (2020) 19(1):44–52. doi: 10.1016/j.aohep.2019.06.023
137. He Q, Sha S, Sun L, Zhang J, Dong M. GLP-1 analogue improves hepatic lipid accumulation by inducing autophagy via AMPK/mTOR pathway. Biochem Biophys Res Commun (2016) 476(4):196–203. doi: 10.1016/j.bbrc.2016.05.086
138. Lim H, Lee H, Lim Y. Effect of vitamin D3 supplementation on hepatic lipid dysregulation associated with autophagy regulatory AMPK/Akt-mTOR signaling in type 2 diabetic mice. Exp Biol Med (Maywood) (2021) 246(10):1139–47. doi: 10.1177/1535370220987524
139. Li R, Guo E, Yang J, Li A, Yang Y, Liu S, et al. 1,25(OH)2D3 attenuates hepatic steatosis by inducing autophagy in mice. Obes (Silver Spring) (2017) 25(3):561–71. doi: 10.1002/oby.21757
140. Zhang Y, Cao Y, Chen J, Qin H, Yang L. A New Possible Mechanism by Which Punicalagin Protects against Liver Injury Induced by Type 2 Diabetes Mellitus: Upregulation of Autophagy via the Akt/FoxO3a Signaling Pathway. J Agric Food Chem (2019) 67(50):13948–59. doi: 10.1021/acs.jafc.9b05910
141. Zhu Y, Su Y, Zhang J, Zhang Y, Li Y, Han Y, et al. Astragaloside IV alleviates liver injury in type 2 diabetes due to promotion of AMPK/mTOR−mediated autophagy. Mol Med Rep (2021) 23(6):437. doi: 10.3892/mmr.2021.12076
142. Beckman JA, Paneni F, Cosentino F, Creager MA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part II. Eur Heart J (2013) 34(31):2444–52. doi: 10.1093/eurheartj/eht142
143. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care (2004) 27(5):1047–53. doi: 10.2337/diacare.27.5.1047
144. Vedantham S, Ananthakrishnan R, Schmidt AM, Ramasamy R. Aldose reductase, oxidative stress and diabetic cardiovascular complications. Cardiovasc Hematol Agents Med Chem (2012) 10(3):234–40. doi: 10.2174/187152512802651097
145. Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LMD. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol (2011) 50(6):1035–43. doi: 10.1016/j.yjmcc.2011.03.002
146. Kobayashi S, Xu X, Chen K, Liang Q. Suppression of autophagy is protective in high glucose-induced cardiomyocyte injury. Autophagy (2012) 8(4):577–92. doi: 10.4161/auto.18980
147. Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy (2015) 11(7):1146–60. doi: 10.1080/15548627.2015.1051295
148. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res (2018) 122(4):624–38. doi: 10.1161/CIRCRESAHA.117.311586
149. Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta (2015) 1852(2):252–61. doi: 10.1016/j.bbadis.2014.05.020
150. Martinet W, De Meyer GR. Autophagy in atherosclerosis. Curr Atheroscler Rep (2008) 10(3):216–23. doi: 10.1007/s11883-008-0034-y
151. Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab (2012) 15(4):534–44. doi: 10.1016/j.cmet.2012.02.011
152. Packer M. Autophagy-dependent and -independent modulation of oxidative and organellar stress in the diabetic heart by glucose-lowering drugs. Cardiovasc Diabetol (2020) 19(1):62. doi: 10.1186/s12933-020-01041-4
153. Dewanjee S, Vallamkondu J, Kalra RS, John A, Reddy PH, Kandimalla R. Autophagy in the diabetic heart: A potential pharmacotherapeutic target in diabetic cardiomyopathy. Ageing Res Rev (2021) 68:101338. doi: 10.1016/j.arr.2021.101338
154. Orogo AM, Gustafsson ÅB. Therapeutic targeting of autophagy: potential and concerns in treating cardiovascular disease. Circ Res (2015) 116(3):489–503. doi: 10.1161/CIRCRESAHA.116.303791
155. Sciarretta S, Boppana VS, Umapathi M, Frati G, Sadoshima J. Boosting autophagy in the diabetic heart: a translational perspective. Cardiovasc Diagn Ther (2015) 5(5):394–402. doi: 10.3978/j.issn.2223-3652.2015.07.02
156. Lee TI, Bai KJ, Chen YC, Lee TW, Chung CC, Tsai WC, et al. Histone deacetylase inhibition of cardiac autophagy in rats on a high−fat diet with low−dose streptozotocin-induced type 2 diabetes mellitus. Mol Med Rep (2018) 17(1):594–601. doi: 10.3892/mmr.2017.7905
157. Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose co-transporter 2 (SGLT2) inhibitors: A state-of-the-art review. JACC Basic Transl Sci (2020) 5(6):632–44. doi: 10.1016/j.jacbts.2020.02.004
158. Chen C, Yang S, Li H, Yin Z, Fan J, Zhao Y, et al. Mir30c Is Involved in Diabetic Cardiomyopathy through Regulation of Cardiac Autophagy via BECN1. Mol Ther Nucleic Acids (2017) 7:127–39. doi: 10.1016/j.omtn.2017.03.005
159. Munasinghe PE, Riu F, Dixit P, Edamatsu M, Saxena P, Hamer NS, et al. Type-2 diabetes increases autophagy in the human heart through promotion of Beclin-1 mediated pathway. Int J Cardiol (2016) 202:13–20. doi: 10.1016/j.ijcard.2015.08.111
160. Feng Y, Xu W, Zhang W, Wang W, Liu T, Zhou X. LncRNA DCRF regulates cardiomyocyte autophagy by targeting miR-551b-5p in diabetic cardiomyopathy. Theranostics (2019) 9(15):4558–66. doi: 10.7150/thno.31052
161. Lu Y, Liu Y, Li H, Wang X, Wu W, Gao L. Effect and mechanisms of zinc supplementation in protecting against diabetic cardiomyopathy in a rat model of type 2 diabetes. Bosn J Basic Med Sci (2015) 15(1):14–20. doi: 10.17305/bjbms.2015.63
162. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes (2011) 60(6):1770–8. doi: 10.2337/db10-0351
163. Xie Z, He C, Zou MH. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy (2011) 7(10):1254–5. doi: 10.4161/auto.7.10.16740
164. Hu M, Li T, Bo Z, Xiang F. The protective role of carnosic acid in ischemic/reperfusion injury through regulation of autophagy under T2DM. Exp Biol Med (Maywood) (2019) 244(7):602–11. doi: 10.1177/1535370219840987
165. Liu J, Tang Y, Feng Z, Liu J, Liu J, Long J. (-)-Epigallocatechin-3-gallate attenuated myocardial mitochondrial dysfunction and autophagy in diabetic Goto-Kakizaki rats. Free Radic Res (2014) 48(8):898–906. doi: 10.3109/10715762.2014.920955
166. Zhi K, Li M, Bai J, Wu Y, Zhou S, Zhang X, et al. Quercitrin treatment protects endothelial progenitor cells from oxidative damage via inducing autophagy through extracellular signal-regulated kinase. Angiogenesis (2016) 19(3):311–24. doi: 10.1007/s10456-016-9504-y
167. Wang B, Yang Q, Sun YY, Xing YF, Wang YB, Lu XT, et al. Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J Cell Mol Med (2014) 18(8):1599–611. doi: 10.1111/jcmm.12312
168. Zhao G, Zhang X, Wang H, Chen Z. Beta carotene protects H9c2 cardiomyocytes from advanced glycation end product-induced endoplasmic reticulum stress, apoptosis, and autophagy via the PI3K/Akt/mTOR signaling pathway. Ann Transl Med (2020) 8(10):647. doi: 10.21037/atm-20-3768
169. Yao Q, Ke ZQ, Guo S, Yang XS, Zhang FX, Liu XF, et al. Curcumin protects against diabetic cardiomyopathy by promoting autophagy and alleviating apoptosis. J Mol Cell Cardiol (2018) 124:26–34. doi: 10.1016/j.yjmcc.2018.10.004
170. Qiu X, Liu K, Xiao L, Jin S, Dong J, Teng X, et al. Alpha-lipoic acid regulates the autophagy of vascular smooth muscle cells in diabetes by elevating hydrogen sulfide level. Biochim Biophys Acta Mol Basis Dis (2018) 1864(11):3723–38. doi: 10.1016/j.bbadis.2018.09.005
171. Zhao Y, Jia X, Yang X, Bai X, Lu Y, Zhu L, et al. Deacetylation of Caveolin-1 by Sirt6 induces autophagy and retards high glucose-stimulated LDL transcytosis and atherosclerosis formation. Metabolism (2022) 131:155162. doi: 10.1016/j.metabol.2022.155162
172. Xie M, Morales CR, Lavandero S, Hill JA. Tuning flux: autophagy as a target of heart disease therapy. Curr Opin Cardiol (2011) 26(3):216–22. doi: 10.1097/HCO.0b013e328345980a
173. Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, et al. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest (2012) 122(11):3919–30. doi: 10.1172/JCI63888
174. Karanth SD, Abner EL, Braithwaite D, Fardo DW, Nelson PT, Katsumata Y. Type 2 Diabetes and association with neuropathological changes and domain specific cognitive decline. Alzheimers Dement (2021) 17 Suppl 2:e058476. doi: 10.1002/alz.058476
175. Fournet M, Bonté F, Desmoulière A. Glycation damage: A possible hub for major pathophysiological disorders and aging. Aging Dis (2018) 9(5):880–900. doi: 10.14336/AD.2017.1121
176. Etchegoyen M, Nobile MH, Baez F, Posesorski B, González J, Lago N, et al. Metabolic syndrome and neuroprotection. Front Neurosci (2018) 12:196. doi: 10.3389/fnins.2018.00196
177. Banks WA, Rhea EM. The blood-brain barrier, oxidative stress, and insulin resistance. Antioxidants (Basel) (2021) 10(11):1695. doi: 10.3390/antiox10111695
178. Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology (1999) 53(9):1937–42. doi: 10.1212/wnl.53.9.1937
179. Umegaki H, Hayashi T, Nomura H, Yanagawa M, Nonogaki Z, Nakshima H, et al. Cognitive dysfunction: an emerging concept of a new diabetic complication in the elderly. Geriatr Gerontol Int (2013) 13(1):28–34. doi: 10.1111/j.1447-0594.2012.00922.x
180. Casagrande SS, Lee C, Stoeckel LE, Stoeckel LE, Menke A, Cowie CC. Cognitive function among older adults with diabetes and prediabetes, NHANES 2011-2014. Diabetes Res Clin Pract (2021) 178:108939. doi: 10.1016/j.diabres.2021.108939
181. Rawlings AM, Sharrett AR, Albert MS, Coresh J, Windham BG, Power MC, et al. The association of late-life diabetes status and hyperglycemia with incident mild cognitive impairment and dementia: the ARIC study. Diabetes Care (2019) 42(7):1248–54. doi: 10.2337/dc19-0120
182. Xu W, Caracciolo B, Wang HX, Winblad B, Bäckman L, Qiu C, et al. Accelerated progression from mild cognitive impairment to dementia in people with diabetes. Diabetes (2010) 59(11):2928–35. doi: 10.2337/db10-0539
183. Nicolls MR. The clinical and biological relationship between Type II diabetes mellitus and Alzheimer's disease. Curr Alzheimer Res (2004) 1(1):47–54. doi: 10.2174/1567205043480555
184. González A, Camila C, Maccioni RB. Alzheimer's disease: A potential diabetes type 3. Alzheimers Dement (2021) 17 Suppl 2:e058533. doi: 10.1002/alz.058533
185. Caberlotto L, Nguyen TP, Lauria M, Priami C, Rimondini R, Maioli S, et al. Cross-disease analysis of Alzheimer's disease and type-2 Diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases. Sci Rep (2019) 9(1):3965. doi: 10.1038/s41598-019-39828-5
186. Miao Y, Guo D, Li W, Zhong Y. Diabetes promotes development of Alzheimer's disease through suppression of autophagy. J Alzheimers Dis (2019) 69(1):289–96. doi: 10.3233/JAD-190156
187. Son SM, Song H, Byun J, Park KS, Jang HC, Park YJ, et al. Accumulation of autophagosomes contributes to enhanced amyloidogenic APP processing under insulin-resistant conditions. Autophagy (2012) 8(12):1842–4. doi: 10.4161/auto.21861
188. Ma LY, Lv YL, Huo K, Liu J, Shang SH, Fei YL, et al. Autophagy-lysosome dysfunction is involved in Aβ deposition in STZ-induced diabetic rats. Behav Brain Res (2017) 320:484–93. doi: 10.1016/j.bbr.2016.10.031
189. Wu Y, Ye L, Yuan Y, Jiang T, Guo X, Wang Z, et al. Autophagy activation is associated with neuroprotection in diabetes-associated cognitive decline. Aging Dis (2019) 10(6):1233–45. doi: 10.14336/AD.2018.1024
190. Guan ZF, Zhou XL, Zhang XM, Zhang Y, Wang YM, Guo QL, et al. Beclin-1- mediated autophagy may be involved in the elderly cognitive and affective disorders in streptozotocin-induced diabetic mice. Transl Neurodegener (2016) 5:22. doi: 10.1186/s40035-016-0070-4
191. Guan ZF, Tao YH, Zhang XM, Guo QQL, Liu YC, Zhang Y, et al. G-CSF and cognitive dysfunction in elderly diabetic mice with cerebral small vessel disease: Preventive intervention effects and underlying mechanisms. CNS Neurosci Ther (2017) 23(6):462–74. doi: 10.1111/cns.12691
192. Wang B, Zhong Y, Li Q, Cui L, Hunag G. Autophagy of macrophages is regulated by PI3k/Akt/mTOR signalling in the development of diabetic encephalopathy. Aging (Albany NY) (2018) 10(10):2772–82. doi: 10.18632/aging.101586
193. Bathina S, Das UN. Dysregulation of PI3K-Akt-mTOR pathway in brain of streptozotocin-induced type 2 diabetes mellitus in Wistar rats. Lipids Health Dis (2018) 17(1):168. doi: 10.1186/s12944-018-0809-2
194. Rizzo MR, Di Meo I, Polito R, Auriemma MC, Gambardella A, di Mauro G, et al. Cognitive impairment and type 2 diabetes mellitus: Focus of SGLT2 inhibitors treatment. Pharmacol Res (2022) 176:106062. doi: 10.1016/j.phrs.2022.106062
195. Chen JL, Luo C, Pu D, Zhang GQ, Zhao YX, Sun Y, et al. Metformin attenuates diabetes-induced tau hyperphosphorylation in vitro and in vivo by enhancing autophagic clearance. Exp Neurol (2019) 311:44–56. doi: 10.1016/j.expneurol.2018.09.008
196. Yang Y, Fang H, Xu G, Zhen Y, Zhang Y, Tian J, et al. Liraglutide improves cognitive impairment via the AMPK and PI3K/Akt signaling pathways in type 2 diabetic rats. Mol Med Rep (2018) 18(2):2449–57. doi: 10.3892/mmr.2018.9180
197. Candeias E, Sebastião I, Cardoso S, Carvalho C, Santos MS, Oliveira CR, et al. Brain GLP-1/IGF-1 signaling and autophagy mediate exendin-4 protection against apoptosis in type 2 diabetic rats. Mol Neurobiol (2018) 55(5):4030–50. doi: 10.1007/s12035-017-0622-3
198. Cui Y, Yang M, Wang Y, Ren J, Lin P, Cui C, et al. Melatonin prevents diabetes-associated cognitive dysfunction from microglia-mediated neuroinflammation by activating autophagy via TLR4/Akt/mTOR pathway. FASEB J (2021) 35(4):e21485. doi: 10.1096/fj.202002247RR
199. Tian R, Liu X, Jing L, Yang L, Xie N, Hou Y, et al. Huang-Lian-Jie-Du decoction attenuates cognitive dysfunction of rats with type 2 diabetes by regulating autophagy and NLRP3 inflammasome activation. J Ethnopharmacol (2022) 292:115196. doi: 10.1016/j.jep.2022.115196
200. Bi T, Zhan L, Zhou W, Sui H. Effect of the ziBuPiYin recipe on diabetes-associated cognitive decline in zucker diabetic fatty rats after chronic psychological stress. Front Psychiatry (2020) 11:272. doi: 10.3389/fpsyt.2020.00272
201. Xue H, Ji Y, Wei S, Yu Y, Yan X, Liu S, et al. HGSD attenuates neuronal apoptosis through enhancing neuronal autophagy in the brain of diabetic mice: The role of AMP-activated protein kinase. Life Sci (2016) 153:23–34. doi: 10.1016/j.lfs.2016.04.004
202. Simó-Servat O, Hernández C, Simó R. Diabetic retinopathy in the context of patients with diabetes. Ophthalmic Res (2019) 62(4):211–7. doi: 10.1159/000499541
203. Yau JW, Rogers SL, Kawasaki R, Lamoureux EL, Kowalski JW, Bek T, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care (2012) 35(3):556–64. doi: 10.2337/dc11-1909
204. Forrester JV, Kuffova L, Delibegovic M. The role of inflammation in diabetic retinopathy. Front Immunol (2020) 11:583687. doi: 10.3389/fimmu.2020.583687
205. Boya P, Esteban-Martínez L, Serrano-Puebla A, Gómez-Sintes R, Villarejo-Zori B. Autophagy in the eye: Development, degeneration, and aging. Prog Retin Eye Res (2016) 55:206–45. doi: 10.1016/j.preteyeres.2016.08.001
206. Yang X, Huang Z, Xu M, Chen Y, Cao M, Yi G, et al. Autophagy in the retinal neurovascular unit: New perspectives into diabetic retinopathy. J Diabetes (2023) 15(5):382–96. doi: 10.1111/1753-0407.13373
207. Piano I, Novelli E, Della Santina L, Strettoi E, Cervetto L, Gargini C. Involvement of autophagic pathway in the progression of retinal degeneration in a mouse model of diabetes. Front Cell Neurosci (2016) 10:42. doi: 10.3389/fncel.2016.00042
208. Madrakhimov SB, Yang JY, Kim JH, Han JW, Park TK. mTOR-dependent dysregulation of autophagy contributes to the retinal ganglion cell loss in streptozotocin-induced diabetic retinopathy. Cell Commun Signal (2021) 19(1):29. doi: 10.1186/s12964-020-00698-4
209. Peng H, Han W, Ma B, Dai S, Long J, Zhou S, et al. Autophagy and senescence of rat retinal precursor cells under high glucose. Front Endocrinol (Lausanne) (2022) 13:1047642. doi: 10.3389/fendo.2022.1047642
210. Wang N, Wei L, Liu D, Zhang Q, Xia X, Ding L, et al. Identification and validation of autophagy-related genes in diabetic retinopathy. Front Endocrinol (Lausanne) (2022) 13:867600. doi: 10.3389/fendo.2022.867600
211. Gao X, Du Y, Lau WB, Li Y, Zhu S, Ma XL. Atg16L1 as a novel biomarker and autophagy gene for diabetic retinopathy. J Diabetes Res (2021) 2021:5398645. doi: 10.1155/2021/5398645
212. Zhang Q, Li HS, Li R, Du JH, Jiao C. Autophagy dysregulation mediates the damage of high glucose to retinal pigment epithelium cells. Int J Ophthalmol (2021) 14(6):805–11. doi: 10.18240/ijo.2021.06.04
213. Huang C, Lu H, Xu J, Yu H, Wang X, Zhang X. Protective roles of autophagy in retinal pigment epithelium under high glucose condition via regulating PINK1/Parkin pathway and BNIP3L. Biol Res (2018) 51(1):22. doi: 10.1186/s40659-018-0169-4
214. Amato R, Catalani E, Dal Monte M, Cammalleri M, Di Renzo I, Perrotta C, et al. Autophagy-mediated neuroprotection induced by octreotide in an ex vivo model of early diabetic retinopathy. Pharmacol Res (2018) 128:167–78. doi: 10.1016/j.phrs.2017.09.022
215. Cai X, Li J, Wang M, She M, Tang Y, Li J, et al. GLP-1 treatment improves diabetic retinopathy by alleviating autophagy through GLP-1R-ERK1/2-HDAC6 signaling pathway. Int J Med Sci (2017) 14(12):1203–12. doi: 10.7150/ijms.20962
216. Wang Y, Liu X, Zhu L, Li W, Li Z, Lu X, et al. PG545 alleviates diabetic retinopathy by promoting retinal Müller cell autophagy to inhibit the inflammatory response. Biochem Biophys Res Commun (2020) 531(4):452–8. doi: 10.1016/j.bbrc.2020.07.134
217. Trotta MC, Gesualdo C, Herman H, Gharbia S, Balta C, Lepre CC, et al. Systemic beta-hydroxybutyrate affects BDNF and autophagy into the retina of diabetic mice. Int J Mol Sci (2022) 23(17):10184. doi: 10.3390/ijms231710184
218. Zhang XX, Ji YL, Zhu LP, Wang ZH, Fang CQ, Jiang CH, et al. Arjunolic acid from Cyclocarya paliurus ameliorates diabetic retinopathy through AMPK/mTOR/HO-1 regulated autophagy pathway. J Ethnopharmacol (2022) 284:114772. doi: 10.1016/j.jep.2021.114772
219. Li L, Chen J, Zhou Y, Zhang J, Chen L. Artesunate alleviates diabetic retinopathy by activating autophagy via the regulation of AMPK/SIRT1 pathway. Arch Physiol Biochem (2023) 129(4):943–50. doi: 10.1080/13813455.2021.1887266
220. Li R, Chen L, Yao GM, Yan HL, Wang L. Effects of quercetin on diabetic retinopathy and its association with NLRP3 inflammasome and autophagy. Int J Ophthalmol (2021) 14(1):42–9. doi: 10.18240/ijo.2021.01.06
221. Luo Y, Dong X, Lu S, Gao Y, Sun G, Sun X. Gypenoside XVII alleviates early diabetic retinopathy by regulating Müller cell apoptosis and autophagy in db/db mice. Eur J Pharmacol (2021) 895:173893. doi: 10.1016/j.ejphar.2021.173893
222. Zhao K, Li Y, Qiu Y, Huang R, Lin M, Chen L, et al. Norkurarinone and isoxanthohumol inhibit high glucose and hypoxia-induced angiogenesis via improving oxidative stress and regulating autophagy in human retinal microvascular endothelial cells. Biochem Biophys Res Commun (2022) 634:20–9. doi: 10.1016/j.bbrc.2022.09.095
223. Li R, Li H, Zhang Q. Procyanidin protects human retinal pigment epithelial cells from high glucose by inhibiting autophagy. Environ Toxicol (2022) 37(2):201–11. doi: 10.1002/tox.23389
224. Fang Y, Shi K, Lu H, Lu L, Qiu B. Mingmu xiaomeng tablets restore autophagy and alleviate diabetic retinopathy by inhibiting PI3K/Akt/mTOR signaling. Front Pharmacol (2021) 12:632040. doi: 10.3389/fphar.2021.632040
225. Bjornstad P, Chao LC, Cree-Green M, Dart AB, King M, Looker HC, et al. Youth-onset type 2 diabetes mellitus: an urgent challenge. Nat Rev Nephrol (2023) 19(3):168–84. doi: 10.1038/s41581-022-00645-1
Keywords: Type II diabetes mellitus (T2DM), autophagy, complications, hypoglycemic agents, traditional Chinese medicine
Citation: Zhao X, Bie L-Y, Pang D-R, Li X, Yang L-F, Chen D-D, Wang Y-R and Gao Y (2023) The role of autophagy in the treatment of type II diabetes and its complications: a review. Front. Endocrinol. 14:1228045. doi: 10.3389/fendo.2023.1228045
Received: 27 May 2023; Accepted: 24 August 2023;
Published: 21 September 2023.
Edited by:
Oleg Gorbatyuk, University of Alabama at Birmingham, United StatesReviewed by:
Vivek Kumar Pandey, University of Kentucky, United StatesCopyright © 2023 Zhao, Bie, Pang, Li, Yang, Chen, Wang and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Gao, NjAyMzAwMDNAc2R1dGNtLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.