 Sandra Pihlström1,2*†
Sandra Pihlström1,2*† Sampo Richardt1,2†
Sampo Richardt1,2† Kirsi Määttä1,2
Kirsi Määttä1,2 Minna Pekkinen1,2,3
Minna Pekkinen1,2,3 Vesa M. Olkkonen4,5
Vesa M. Olkkonen4,5 Outi Mäkitie1,2,3,6
Outi Mäkitie1,2,3,6 Riikka E. Mäkitie1,2,7
Riikka E. Mäkitie1,2,7- 1Folkhälsan Institute of Genetics, Helsinki, Finland
- 2Research Program for Clinical and Molecular Metabolism, Faculty of Medicine, University of Helsinki, Helsinki, Finland
- 3Children´s Hospital, University of Helsinki and Helsinki University Hospital, Helsinki, Finland
- 4Minerva Foundation Institute for Medical Research, Helsinki, Finland
- 5Department of Anatomy, Faculty of Medicine, University of Helsinki, Helsinki, Finland
- 6Department of Molecular Medicine and Surgery and Center for Molecular Medicine, Karolinska Institutet, Stockholm, Sweden
- 7Department of Otorhinolaryngology – Head and Neck Surgery, Helsinki University Hospital and University of Helsinki, Helsinki, Finland
Pathogenic heterozygous variants in SGMS2 cause a rare monogenic form of osteoporosis known as calvarial doughnut lesions with bone fragility (CDL). The clinical presentations of SGMS2-related bone pathology range from childhood-onset osteoporosis with low bone mineral density and sclerotic doughnut-shaped lesions in the skull to a severe spondylometaphyseal dysplasia with neonatal fractures, long-bone deformities, and short stature. In addition, neurological manifestations occur in some patients. SGMS2 encodes sphingomyelin synthase 2 (SMS2), an enzyme involved in the production of sphingomyelin (SM). This review describes the biochemical structure of SM, SM metabolism, and their molecular actions in skeletal and neural tissue. We postulate how disrupted SM gradient can influence bone formation and how animal models may facilitate a better understanding of SGMS2-related osteoporosis.
Introduction
Osteoporosis is a chronic bone disease with a significant global impact on morbidity and mortality. The defining characteristics are low bone mineral density (BMD) and disturbed bone microarchitecture, which enhance the risk of fragility fractures (1). Most often, polygenetic factors rather than single gene abnormalities are thought to influence a person’s bone health and risk of osteoporosis (2). Nevertheless, several uncommon monogenic types of osteoporosis have been found (1–3).
One of the most recently identified genes to cause a rare monogenic form of osteoporosis is SGMS2, which codes for the enzyme sphingomyelin synthase 2 (SMS2) (4). SMS2 catalyzes the production of sphingomyelin (SM), a type of sphingolipid that serves as a major component of the cell and Golgi membranes. Heterozygous mutations in the SGMS2 gene (p.Arg50*, p.Ile62Ser, p.Met64Arg) cause a rare skeletal disorder termed calvarial doughnut lesions with bone fragility (CDL) with or without spondylometaphyseal dysplasia, with low BMD, neonatal fractures, long-bone deformities, and short stature (OMIM #126550). In addition to the skeletal manifestations, several patients experience neurological symptoms, the most frequent being transitory, spontaneously resolving, and recurrent cranial nerve palsies (4–8). The clinical presentation and disease severity is highly variable and dependent on the underlying SGMS2 variant. Therefore, it is likely that the SGMS2 variants could be causal in further primary osteoporosis patients with yet an unidentified genetic cause and the range of phenotypic manifestations significantly greater than has been previously described. Hence, it is of great importance to further understand how SM metabolism and lipid distribution affect bone development and metabolism. In this review, we aim to provide a comprehensive overview of the research topic and bring the latest knowledge of SMS2 and SM metabolism in skeletal and neural tissue to clinicians and researchers working in skeletal and neurological research fields. In addition, we underline the importance of developing sgms2 modified animal models for studying molecular and cellular mechanisms underlying SGMS2-related bone fragility with neurological features.
Sphingomyelin, a type of sphingolipid
Sphingolipids are fundamental structural components in cell membranes, including the plasma membrane, Golgi apparatus and endosome membrane. Sphingolipids contribute to the characteristic key properties of these membranes including the protective barrier function of the plasma membrane (9). Sphingolipids are essential in cell signaling, by both forming lipid rafts that play a crucial role in protein sorting and receptor‐mediated signal transduction and by serving as stores for signaling molecules. Sphingolipid metabolites are for instance important mediators in the signaling cascades involved in differentiation, apoptosis, proliferation, inflammation, and senescence (10).
Sphingolipids have a structural feature of a sphingosine backbone that is comprised of an alkyl chain of 18 carbon atoms with one to three hydroxyl groups and one amino group (Figure 1A). To the amino group, different functional groups can bind to yield e.g., sphingosine-1-phosphate (S1P), SM, and ceramide (11). The most abundant sphingolipid in majority of mammalian cells, representing 85% of all sphingolipids, is SM (12). In SM, the sphingosine backbone is bound to a fatty acid tail via the amino group and to a phosphocholine group via the terminal hydroxyl group (Figure 1A). SM is produced in the luminal leaflet of trans-Golgi lumen membranes from ceramide, which is provided by the endoplasmic reticulum (ER). SM is transported to the plasma membrane by vesicular traffic, where it accumulates in the exoplasmic leaflet (13). Except for maintaining plasma membrane structure, SM is enriched in the endocytic recycling compartment and the trans-Golgi network, and can control the actions of growth factor receptors and matrix proteins as well as serve as a binding site for different micro-organisms (14). SM is also a binding partner for cholesterol, influencing cholesterol homeostasis and forming a SM/sterol concentration gradient along the secretory pathways (15). In addition, SM may be a critical source of phosphocholine needed for mineralization (4, 10). Several investigations have demonstrated that abnormal SM metabolism results in abnormalities in the mineralization of the bone matrix (4, 16, 17).
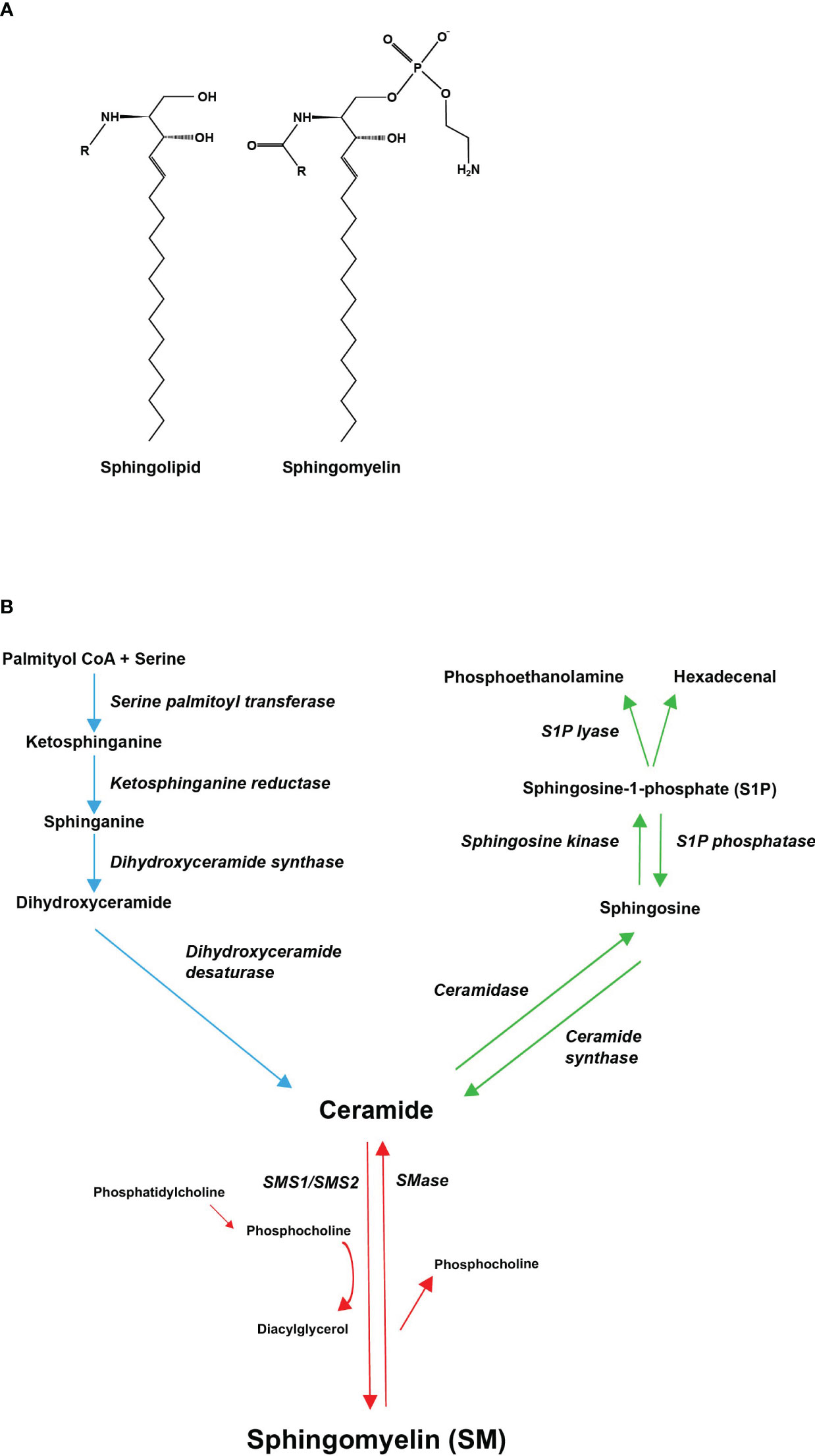
Figure 1 Molecular structures of sphingolipids and the metabolic pathway of sphingomyelin. (A) Every sphingolipid has a single sphingosine backbone, which is an 18-carbon alkyl chain with one to three hydroxyl groups and one amino group. Different functional groups (R) can bind to the amino group. Sphingomyelin is composed of the sphingosine backbone bound to a fatty acid tail and a phosphocholine group via its amino group and terminal hydroxyl group, respectively. (B) Sphingomyelin can be synthesized de novo, through ceramide, from saturated fatty acids (palmitic acid) (demonstrated with blue arrows). Ceramide serves as a substrate for sphingomyelin synthase (SMS), which catalyzes the transfer of phosphocholine, cleaved from phosphatidylcholine (PC), onto ceramide generating SM and diacylglycerol (DAG) (demonstrated with red arrows). Ceramide can also be converted to sphingosine-1-phosphate (S1P) through hydrolysis of its fatty acid residue and subsequent phosphorylation of its terminal hydroxyl group (demonstrated with green arrows). All pathways converge in ceramides.
Sphingomyelin metabolism
The metabolism of SM is highly regulated and involves multiple bioactive sphingolipids. SM can be synthesized de novo, through ceramide, from precursors such as palmitoyl CoA and serine (Figure 1B, blue pathway) (12). Production of ceramide takes place on the cytosolic surface of the ER (13). Ceramide is then transported to the Golgi complex, where it serves as a substrate for sphingomyelin synthase (SMS) and other sphingolipid-generating enzymes (10). Both vesicular and nonvesicular transport mechanisms can mediate ceramide transport (13).
SMS is a membrane-bound enzyme that has two isoforms, SMS1 and SMS2. SMS2 is found in the plasma membrane and in the Golgi apparatus, whereas SMS1 is localized only in the Golgi (18). SMS catalyzes the transfer of phosphocholine, cleaved from phosphatidylcholine (PC), onto ceramide, generating SM and diacylglycerol (DAG) (Figure 1B, red pathway) (12). SM is then delivered by vesicular transport to the plasma membrane (19). SMS2 is also able to catalyze the reverse reaction. However, SMS2 can only regenerate ceramide but is unable to release phosphocholine, instead a histidine-phosphocholine intermediate is formed. Regeneration of phosphocholine from sphingomyelin is done by sphingomyelinases (SMases). SMases fall into three categories depending on their pH optima: acidic, alkaline, and neutral (10, 12). Sphingomyelin phosphodiesterase 3 (SMPD3), one of the four neutral SMases, is largely confined to the bone, cartilage, and brain tissue (17). Ceramide can also be converted to sphingosine-1-phosphate (S1P) through hydrolysis of its fatty acid residue and subsequent phosphorylation of its terminal hydroxyl group (Figure 1B, green pathway) (10). For lipids exiting the sphingolipid pool, only a single irreversible catalytic pathway exists: Sphingosine-1-phosphate lyase (S1P lyase) breaks the sphingosine backbone of S1P generating non-sphingolipids (20).
The generation and degradation of SM and the related bioactive lipids are intertwined. Hence, the regulation of these lipids could be affected by the enzymes involved in the metabolism of SM. Due to the interconnectedness of these lipids, changes in one causes a “ripple” effect in the others as a new equilibrium between the substrates sets. Furthermore, there are great variations in these lipids’ concentrations. Since SM has a tenfold higher concentration than ceramide, for instance, even minimal changes in SM can have a large impact on ceramide levels (21).
SM is the preferred binding partner of cholesterol. SM produced in the lumen of the trans-Golgi and at the outer leaflet of the plasma membrane provides a thermodynamic trap for cholesterol synthesized in the ER, contributing to the formation of a SM/sterol gradient along the secretory pathway (15). ER and cis-Golgi membranes are characterized by low sphingolipid and sterol content while the plasma membrane and trans-Golgi have a high sphingolipid and sterol content (22). This nonrandom SM gradient is important for maintaining ER- and plasma membrane specific lipid composition and fundamental for physical membrane properties that help specify organelle identity and function.
Osteoporosis
Recent research has identified a wide range of illnesses affecting skeletal homeostasis, and often with a genetic basis. Monogenic disorders are caused by a single-gene mutation, which is usually germline but occasionally somatic, while oligogenic or polygenic conditions involve multiple genetic variants (23). Osteoporosis is most commonly polygenic and related to aging, or secondary to other illnesses. However, primary osteoporosis may present already in childhood and is then usually a monogenic disease (24). SGMS2-related osteoporosis belongs to this group of monogenic metabolic bone disorders (25).
SGMS2-related osteoporosis
The rare autosomal dominant inherited bone disease named calvarial doughnut lesions with bone fragility (CDL) with or without spondylometaphyseal dysplasia (OMIM #126550) was described more than 50 years ago (26, 27). However, its genetic cause – mutation in SGMS2 – was identified only in 2019 (4). In humans, SGMS2 is located on chromosome 4 and codes for a 365 amino acid protein – sphingomyelin synthase 2 (SMS2). So far, three heterozygous variants have been detected by next-generation sequencing and confirmed by Sanger sequencing in 32 affected subjects from 12 unrelated families (4–8). The reported variants include a c.148C>T variant, which introduces a premature stop codon in exon 2 (p.Arg50*) and yields a truncated enzyme, and two missense variants, c.185T>G (p.Ile62Ser) and c.191T>G (p.Met64Arg) (4, 5). The study by Pekkinen et al. (4) was the first to link aberrant SM metabolism to a bone disease, highlighting the importance of sphingolipids for bone growth and development.
The clinical presentations of SGMS2-related osteoporosis range from childhood-onset osteoporosis with low BMD and skeletal fragility with or without sclerotic doughnut-shaped lesions in the skull to a severe spondylometaphyseal dysplasia with neonatal fractures, long-bone deformities, and short stature (4–8, 26). Additionally, glaucoma was diagnosed in two individuals from one affected family harboring a p.Arg50* mutation, as described by Pekkinen et al. (4). Interestingly, the association of glaucoma with SGMS2 was further strengthened when Collantes and coworkers described a Filipino family harboring the SGMS2 p.Arg50* mutation, with characteristic skull lesions and juvenile onset open angle glaucoma (7).
Thus far, several single patients as well as larger multigenerational families with heterozygous SGMS2 variants have been reported. Mutations p.Ile62Ser and p.Met64Arg, that give rise to a more severe phenotype with neonatal fractures, severe short stature, and spondylometaphyseal dysplasia, have been reported in 3 affected subjects in 2 families (4). The p.Arg50* variant, associated with a milder phenotype, is more common and has been described in 29 subjects in 10 families. A more detailed description of the clinical data of the patients with a SGMS2 p.Arg50* mutation is summarized in Tables 1A, 1B, and presented separately for each family. In addition to the skeletal phenotype, patients portray various neurological manifestations, which will be covered in more detail later in the text. It remains partly unclear how the SGMS2 variants lead to skeletal fragility and what explains the significant phenotypic differences between patients with the more common p.Arg50* variant and those with the missense variants.
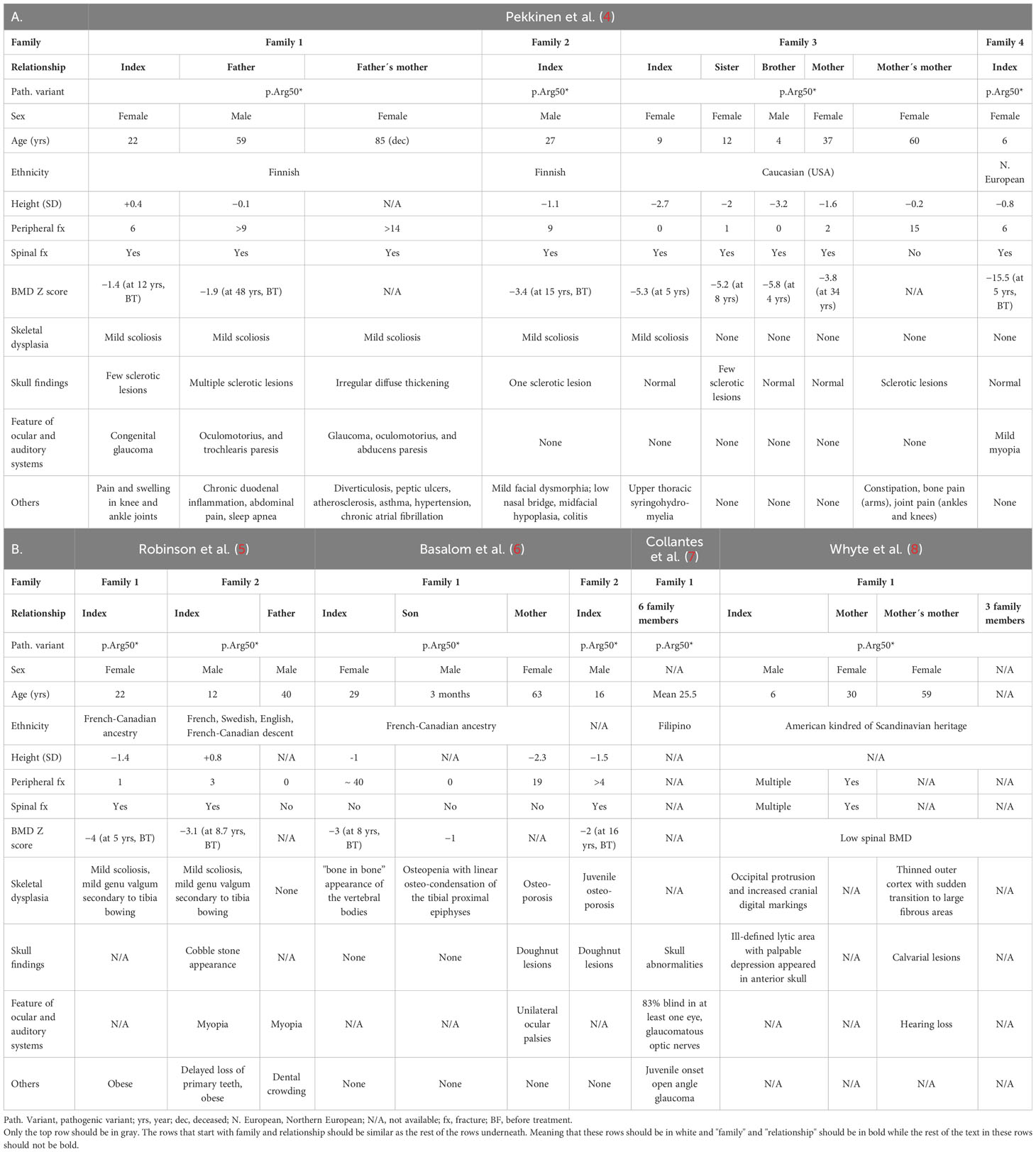
Table 1 Clinical findings in 29 subjects with a SGMS2 p. Arg50* variant.
Bone tissue characteristics and expression of SGMS2 in tissues and cells
Transiliac and femoral bone biopsy samples from patients with SGMS2-related osteoporosis reveal reduced mineral content and decreased bone volume with unorganized collagenous network (4, 5, 8). Mäkitie et al. have demonstrated that patients harboring a p.Arg50* mutation have a discorded collagenous apposition, their osteocyte lacunae appear too large and the lacuna-canalicular network is extremely distorted and short spanned (28). In human tissues, SGMS2 transcripts have been detected in brain, heart, kidney, liver, muscle, and stomach (29). SGMS2 expression has also been detected in primary chondrocytes isolated from patients with osteoarthritis (30). In mice, sgms2 is highly expressed in cortical bone, vertebrae, kidney, and liver (4). In vitro studies performed by Pekkinen et al. showed that cultured murine osteoblasts, bone marrow macrophages and osteoclasts expressed sgms2 at similar levels (4). Results on patients’ bone biopsies in the Pekkinen et al. study also suggested that osteoclast numbers may be increased based on bone resorption parameters. However, osteoclast formation and function in vitro were normal, as analyzed from peripheral blood monocytes from 2 patients with a p.Arg50*mutation (4).
Enzymatic activity and cellular location of the SMS2 variants
SMS2 is a multi-membrane spanning protein that primarily contributes to sphingomyelin synthesis and homeostasis at the plasma membrane. The three pathogenic variants of SGMS2 (p.Arg50*, p.Ile62Ser and p.Met64Arg) are all located in the N-terminal part of the protein in the region immediately upstream of transmembrane domain 1 (TMD1) (Figure 2) (4). Variants p.Ile62Ser and p.Met64Arg do not have an effect on SMS2 enzymatic activity. Instead, due to the missense variants, SMS2 is unable to exit the ER because their N-terminal cytosolic tails lack a functioning independent ER export signal (31). Sokoya et al. demonstrated that isoleucine at position 62 and methionine at position 64 in SMS2 are part of a conserved sequence motif, IXMP, which is located 13–14 residues upstream of the first membrane span and is part of this ER export signal (Figure 2). By transfecting SMS2I62S and SMS2M64R constructs into Hela cells, they detected the subcellular location of the SMS2 variants with immunofluorescence microscopy, and revealed that SMS2I62S and SMS2M64R were both retained in the ER, while wild type SMS2 localized to the Golgi and the plasma membrane (31). The SMS2 p.Arg50* variant is predicted to result in a truncated enzyme lacking the entire transmembrane helices including the active sites of the enzyme (Figure 2) (4). However, Sokoya and co-workers have hypothesized that the nonsense p.Arg50* variant produces a shortened yet functional enzyme with methionine at position 64 serving as an alternative translation initiation site (31). In addition, they hypothesized that the p.Arg50* variant is exported out of ER but fails to reach the plasma membrane and mislocalizes to the cis/medial Golgi (31).
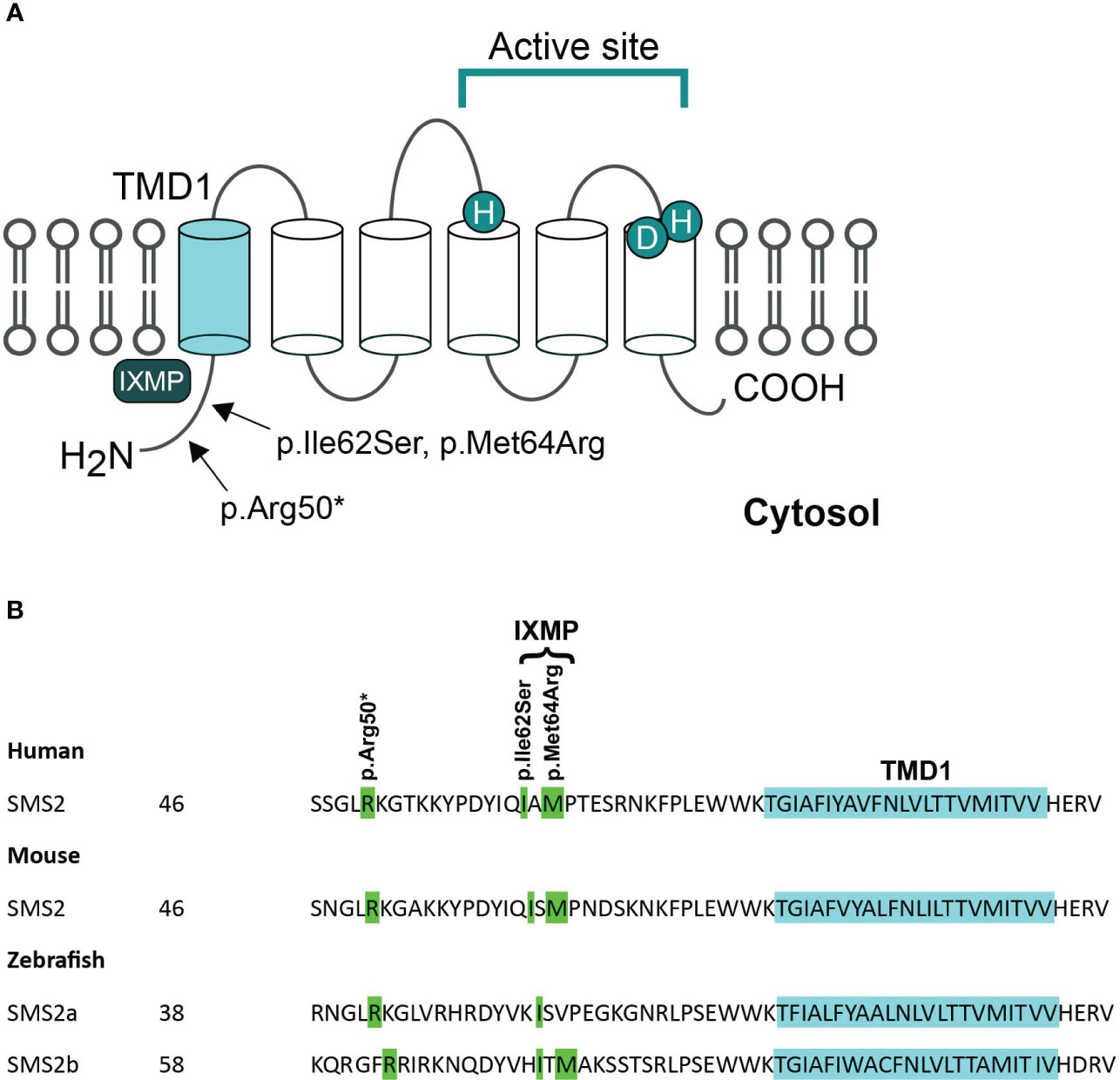
Figure 2 Pathogenic variants of SMS2. (A) Predicted membrane topology of SMS2 indicating active site residues and positions of 3 residues substituted in pathogenic SMS2 variants (p.Arg50*, Ile62Ser, Met64Arg). (B) SMS2 sequence alignment of the region immediately upstream of transmembrane domain 1 (TMD1) in human, mouse, and zebrafish. Pathogenic SMS2 variants and the ER export defected by Ile62Ser- and Met64Arg-variations are indicated. Database accession numbers for the sequences are human SMS2, Q8NHU3; mouse SMS2, Q9D4B1; zebrafish SMS2a, B8A5Q0; zebrafish SMS2b, Q6DEI3. Adapted and reprinted by permission from JCI Insight (Creative Commons Attribution 4.0 International License (CC BY 4.0)) (Pekkinen et al., (4), copyright 2019).
The SMS2 missense variants also enhance de novo SM biosynthesis, based on elevated triacylglycerol levels in SGMS2-mutated patient-derived fibroblasts (4). The elevated triacylglycerol levels are likely to result from rapid conversion of the SM synthesis byproduct, DAG, to triacylglycerol. Therefore, it is anticipated that the onset of the disease is a result of improperly targeted bulk SM production rather than a decreased ability to synthesize SM (4, 31). This finding implies that pathogenic SMS2 variants accumulate SM in the ER and display a disrupted SM asymmetry at the plasma membrane due to altered subcellular organization of SM and cholesterol. In addition, Sokoya and co-workers discovered that pathogenic SMS2 variants significantly alter the ER glycerophospholipid profile (31). These changes include an increased degree of phospholipid desaturation and an increase in cone-shaped ethanolamine-containing phospholipids, which may be a cellular adaptation to the SM-mediated rigidification of the ER bilayer (31). Pathogenic SMS2 mutations may therefore severely impair the ability of cells to maintain nonrandom lipid distributions in the secretory pathway, which may be essential for osteogenic cells’ ability to form bone (31). However, it remains unknown exactly how the pathogenic SMS2 variants affect the subcellular organization of SM and how cholesterol contributes to the development of osteoporosis and CDL in affected patients.
Potential impact of disrupted SM gradients on bone formation
Based on the Pekkinen et al. study, SGMS2 transcript levels are highest in cortical bone and vertebrae in murine model (4), indicating that the effect of pathogenic variants on the lipid composition of secretory organelles could be severe in bone cells. Bone is formed when collagen fibrils are deposits into a matrix that will mineralize, in the presence of Ca2+ and inorganic phosphate (Pi), when hydroxyapatite crystals grow within and between the newly synthesized collagen fibrils (32). Collagen synthesis begins in the ER as pre-collagen, which leaves the ER as pro-collagen through coat protein complex type II (COPII) vesicles. Pro-collagen’s ability to leave the ER is dependent on the COPII coat, which is made up of the essential elements Sar1, Sec23/24, and Sec13/31. In addition, TANGO1, an ER-resident transmembrane protein, is required for packaging pro-collagen fibers into COPII vesicles (Figure 3A) (34). Mutations in COPII components and TANGO1 have been reported to selectively disrupt procollagen export from the ER and cause insufficient bone mineralization (35–37). One scenario is that SGMS2 variants could impair the formation of secretory vesicles containing pro-collagen due to the rigidifying effect of SM on both leaflets of the ER bilayer (Figure 3A). This would prevent proper export of collagen from the ER and impact bone formation.
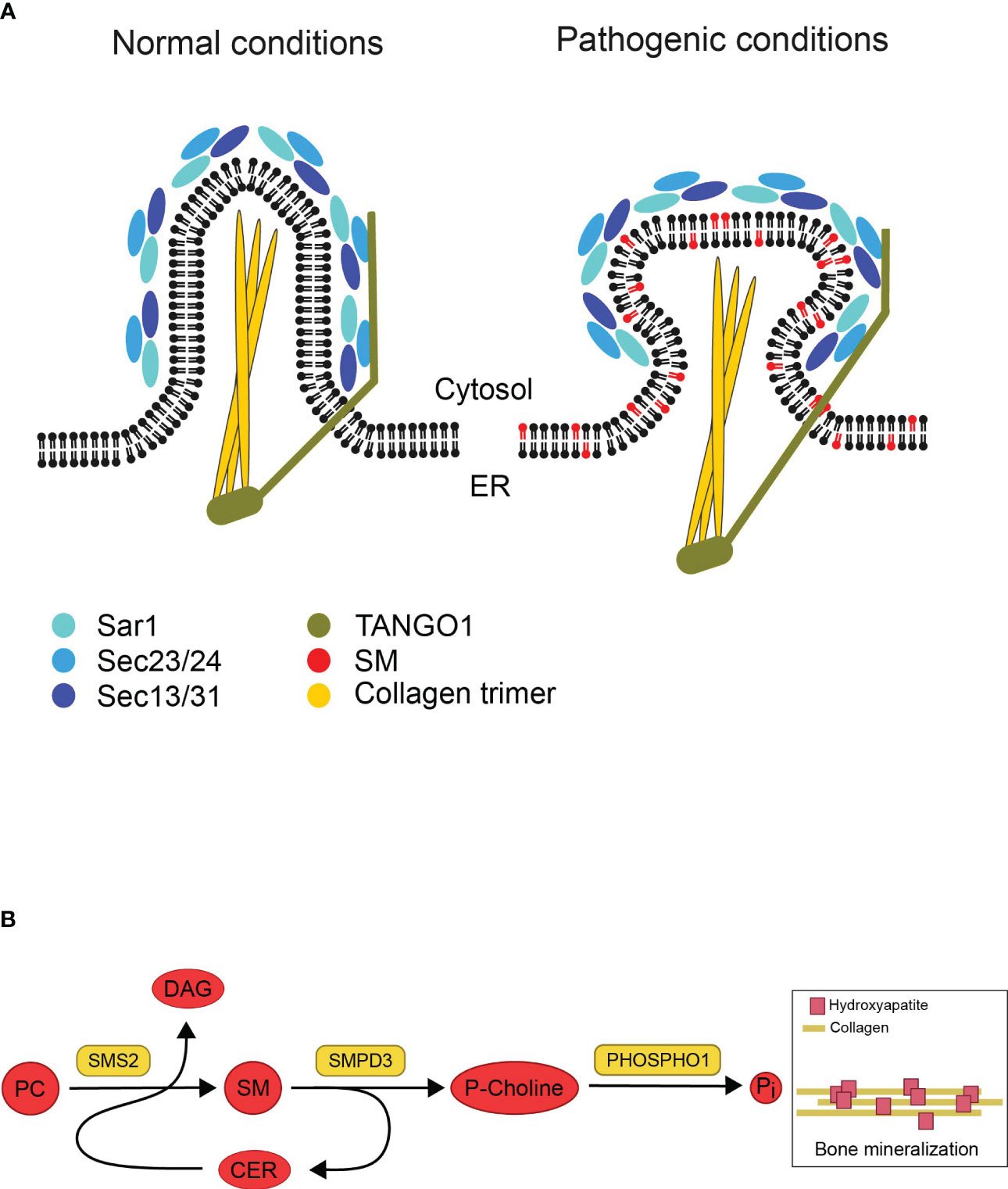
Figure 3 Illustrations based on potential impact of disrupted SM gradients on bone formation. (A) Model showing potential effects of pathogenic SMS2 variants on collagen secretion during bone formation. Collagen synthesis starts in the ER. Under normal conditions, the COPII coat proteins (Sar1, Sec23/24, Sec13/31) and the accessory protein TANGO1 are assembled as pro-collagen trimers leave the ER in secretory vesicles. The rigidifying impact of SM on both ER bilayer leaflets would hinder the development of these big cargos (pathogenic conditions). This, in turn, would stop collagen trimers from being properly exported from the ER, which would negatively impact bone formation. Adapted and reprinted from Gillon et al. (33) (Biochim Biophys Acta; 1821(8): 1040–1049). (B) Model describing how sphingomyelin metabolic enzymes contribute to the mineralization of bone. In the exoplasmic leaflet of the plasma membrane, sphingomyelin (SM) is broken down by the enzyme sphingomyelin phosphodiesterase 3 (SMPD3) to produce ceramide (Cer) and phosphocholine (P-Choline). Phosphatase (PHOSPHO1) uses P-Choline as a substrate to release phosphate and promote bone mineralization. The plasma membrane-resident sphingomyelin synthase SMS2 produces diacylglycerol (DAG) as a byproduct while regenerating SM from Cer released by SMPD3 utilizing phosphatidylcholine (PC) as a head group donor. The osteoblast surface SMPD3 and SMS2 enzymatic activities work together to provide the constant flow of phosphocholine needed for normal bone mineralization. Adapted and reprinted by permission from JCI Insight (Creative Commons Attribution 4.0 International (CC BY 4.0)) (Pekkinen et al., (4), copyright 2019).
Another possible explanation is that bone mineralization is adversely affected by pathogenic SMS2 variant due to disturbed SM asymmetry at the plasma membrane in osteogenic cells. When bone mineralizes, matrix vesicles bud off from osteoblasts’ apical membrane and deposit their phosphate- and Ca2+-rich contents at the mineralization site (32). Both SMS2 and SMases can break down SM into ceramide in the cytosolic leaflet of the plasma membrane, but unlike SMases, SMS2 is unable to release phosphocholine (12). Phosphocholine can be utilized to produce Pi for the mineralization process, where Pi precipitates into hydroxyapatite when combined with Ca2+ (Figure 3B) (10). During cartilage mineralization, matrix vesicles, the initial sites of mineral production, have been seen to degrade SM rapidly. SMases like SMPD3, which are present in matrix vesicles, are likely responsible for the reduction of SM. Due to this degradation, phosphocholine (P-Choline) is produced, which PHOSPHO1 can employ to liberate phosphate (10, 38, 39). Even though SMS2 itself cannot release phosphocholine, it may regenerate SM from ceramide released by SMPD3, and refill the SM pool used by SMPD3 to set free phosphocholine during bone mineralization. The regeneration of SM by SMS activity would guarantee a steady supply of phosphate (4). This concept could explain how pathogenic SMS2 variants may cause a premature exhaustion of the lipid-based phosphate store thus interfering with normal bone mineralization.
Sphingomyelin synthase and SM metabolism in the skeletal system
In addition to the study by Pekkinen et al. (4), several other studies have demonstrated sphingomyelin synthases’ various key roles in bone homeostasis. Matsumoto and collaborators, who examined the role of sphingomyelin synthases in the skeletal development of mice, discovered that whereas SMS2 deficit did not affect bone formation, SMS1 deficiency did (16). Compared to control mice, SMS1-deficient mice had decreased cortical and trabecular bone mass, lower BMD, and delayed mineral deposition. The osteoid volume and the osteoid development in these animals increased dramatically, a further evidence for impaired mineralization. Also, when stimulated with bone morphogenic protein 2 (BMP2), tamoxifen-inducible SMS1-deficient calvarial osteoblasts demonstrated a significant decrease in the expression of several bone structural components and reduced mineralization (16).
Even though the nature of SMS2’s effect on bone is uncertain, it has been suggested that SMS2 indirectly affects osteoclast differentiation through osteoblasts (40). Osteoclasts are of hematopoietic lineage origin and their synthesis is being controlled by macrophage colony-stimulating factor (M-CSF) and nuclear factor b ligand (RANKL). RANKL, secreted by osteoblasts, regulates factors that control bone resorption, including parathyroid hormone (PTH), 1,25(OH)2 vitamin D, and interleukins 6 and 11 (IL-6, IL-11) (41). Yoshikawa and collaborators showed that deletion of SMS2 with siRNA affected osteoclastogenesis through the 1,25(OH)2D pathway. 1,25(OH)2D binds to vitamin D receptor (VDR) which then dimerizes with retinoid-X-receptor-α (RXRα) to regulate gene expression, including RANKL. RXRα is a receptor that exerts its action by binding, as homo- or heterodimers, to specific sequences in the promoters of target genes and regulating their transcription (42). A siRNA-mediated SMS2 knockdown in mouse primary osteoblasts reduced the expression of RXRα mRNA and, as expected, the expression of RANKL after 1,25(OH)2D stimulation. In consequence, the number of differentiated osteoclasts was significantly reduced (40). To date, the mechanism behind RXR downregulation in these cells is still unknown. Interestingly, the suppression of osteoclastogenesis would increase bone mass (43), not decrease it as SMS-deficiency studies show (4, 16). As earlier described, bone biopsies from the patients harboring a SGMS2-variant in the Pekkinen et al. study also suggest that osteoclast numbers might be increased based on bone resorption parameters (4). However, the in vitro study on peripheral blood monocytes showed normal osteoclast formation and function (4). These findings imply that alterations in the mineralization process itself rather than an abnormally high bone turnover might cause skeletal abnormalities in SMS-deficient conditions.
Abnormal activity of other sphingomyelin metabolizing enzymes has been linked to bone abnormalities in mice. Sphingomyelin phosphodiesterase 3 (SMPD3) has been recognized as an essential regulator of development in skeletal and cartilaginous tissues (10). Deletion of Smpd3 in mice leads to severe skeletal abnormalities, poor mineralization of bone and cartilage, and features consistent with severe osteogenesis imperfecta (44, 45). S1P has bone-specific roles, specifically in osteoblasts and osteoclasts where S1P acts as an osteoclast-osteoblast coupling factor to coordinate bone resorption and bone formation (46). According to previous studies, the S1P balance in bone is essential for maintaining skeletal homeostasis since disturbance of this balance in mice results in osteopetrosis and osteoporosis (47). Moreover, DAG, the byproduct of sphingomyelin synthase, activates protein kinase C (PKC) in cells (48). It has been shown that PKCδ, a novel isoform of PKC, is essential for the signaling of Wnt3a-induced osteoblastogenesis since PKC mutant mice exhibit inadequate embryonic skeletal development (49). The PKC pathway is also used by Wnt7b to stimulate osteoblast differentiation (49). However, the DAG itself has not been implicated in bone formation.
Neurological findings in SGMS2-related osteoporosis
In addition to the bone phenotype, several patients with a pathogenic SGMS2 variant exhibit neurological symptoms (Table 2) (4–6, 8). The most prevalent findings were isolated cranial nerve palsies that are transient, recurrent, and spontaneously remitting. Most commonly, these have been peripheral facial nerve palsies but oculomotor, trochlear and abducens nerves have also been affected. Diagnostics of clinical evaluations were repeatedly normal, showing no indication as to what these palsies could be attributed to. Other reported neurological findings included migraines, depression, dystonia, trigeminal neuralgia, sensory neuropathy, ataxia and absent or decreased reflexes (4–6, 8).
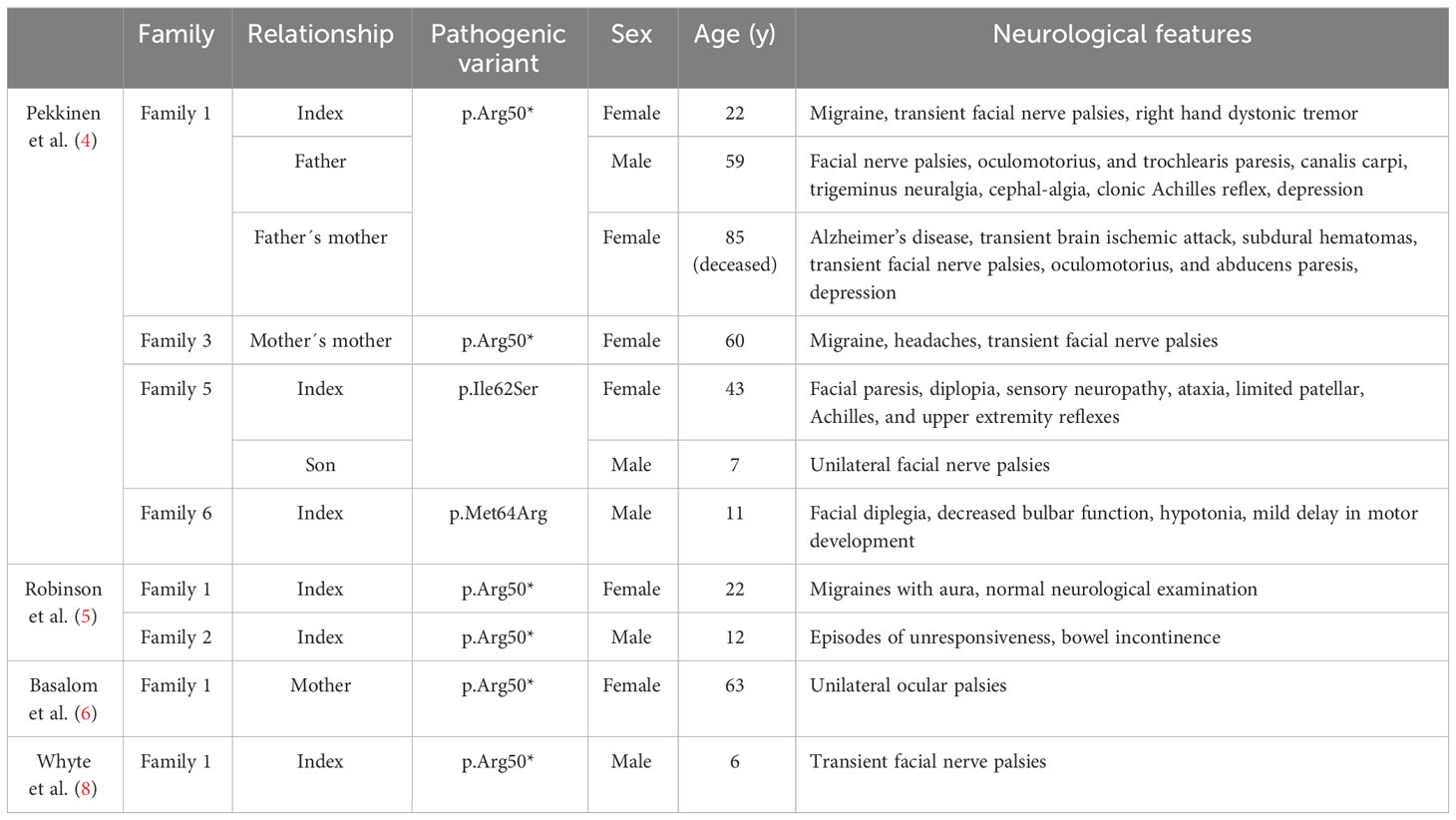
Table 2 Nerological features in 11 subjects with a pathogenic SGMS2 variant.
Sphingomyelin metabolism in neurological diseases
In the mammalian body, the nervous system is one of the tissues with the highest lipid complexity and content. The formation and preservation of the functional integrity of the central nervous system (CNS) depends on sphingolipids, which are particularly abundant in the brain (50). Alterations in neural membrane glycerophospho- and sphingolipid composition can influence crucial intra- and intercellular signaling and alter the membrane’s properties (51). In the CNS, SM is enriched in oligodendrocytes and myelin, and plenty of evidence indicates that SM metabolism plays an important role in neurodegenerative and psychiatric diseases (50).
Cerebral ischemia, a condition where the brain is deprived of its blood supply and causes neurodegeneration, has been linked to SM metabolism (12). After brain ischemia, SM hydrolysis and consequent pro-apoptotic ceramide production take place (52, 53). Functional studies also demonstrate that inhibition of neutral SMases suppresses the ischemia-related apoptotic process both in vitro and in vivo (54).
The role of lipids in Alzheimer’s disease, characterized by deposition of amyloid-beta (Aβ) plaques, has been studied extensively and data suggests that lipid composition of the brain may be involved in neurodegenerative processes (55). Deregulation of SM and ceramide, in particular, have been associated with the disease (12) and may promote abnormal amyloid processing (56). Early pathogenesis of Alzheimer´s disease has been suggested to associate with SM located in the cerebrospinal fluid (CSF), since CSF SM is positively correlated with Aβ and tau levels in high-risk healthy patients and in patients in the prodromal stage of Alzheimer’s disease (57, 58). Decreased SM levels and increased ceramide levels, together with an elevated expression of acid SMase, have also been consistently observed in Alzheimer’s disease brains (59, 60).
Clinical hallmarks of Parkinson´s disease are Lewy bodies or α-synuclein (61). Lysosomal pathways have been found to degrade α-synuclein, leading to the hypothesis that their impairment may play a role in the development of Parkinson disease (62). Two variants of the lysosomal acid SMase have been reported to increase the risk of Parkinson disease significantly, suggesting that a disturbed SM metabolism may play a role (63, 64).
Model organisms with SGMS2 variations
Since the exact mechanism by which SGMS2 variants alter SM metabolism in bone remains unclear, genetically modified animal models are a good method to mimic the disease and characterize skeletal pathology. Sms2 knockout (KO) mice are available (65, 66). Hailemariam and coworkers developed Sms2 KO mice already in 2008 and 3 years later, in 2011, Mitsutake and coworkers followed their lead (65, 66). These Sms2 KO mice have been utilized in multiple studies during the last decade (Table 3). Mitsutake and coworkers showed that by removing Sms2 in mice, diet-induced obesity and insulin resistance are reduced (65). Hailemariam and coworkers revealed that sgms2 KO mice had a diminished NFκB response to inflammatory/immunologic stimuli (66). However, neither study reported any obvious bone abnormalities (65, 66). Despite this, a bone phenotype may have gone unnoticed. On the other hand, it is possible that removal of SMS2 is not sufficient to cause a bone phenotype, since SMS1 also produces SM. Instead, it may be necessary to induce pathogenic variants of sgms2, to provoke mislocalization of Sms2 in the cells, to be able to engender bone abnormalities. Such a study has not yet been reported. No neurological manifestations in sgms2 KO mice have been described (65, 66).
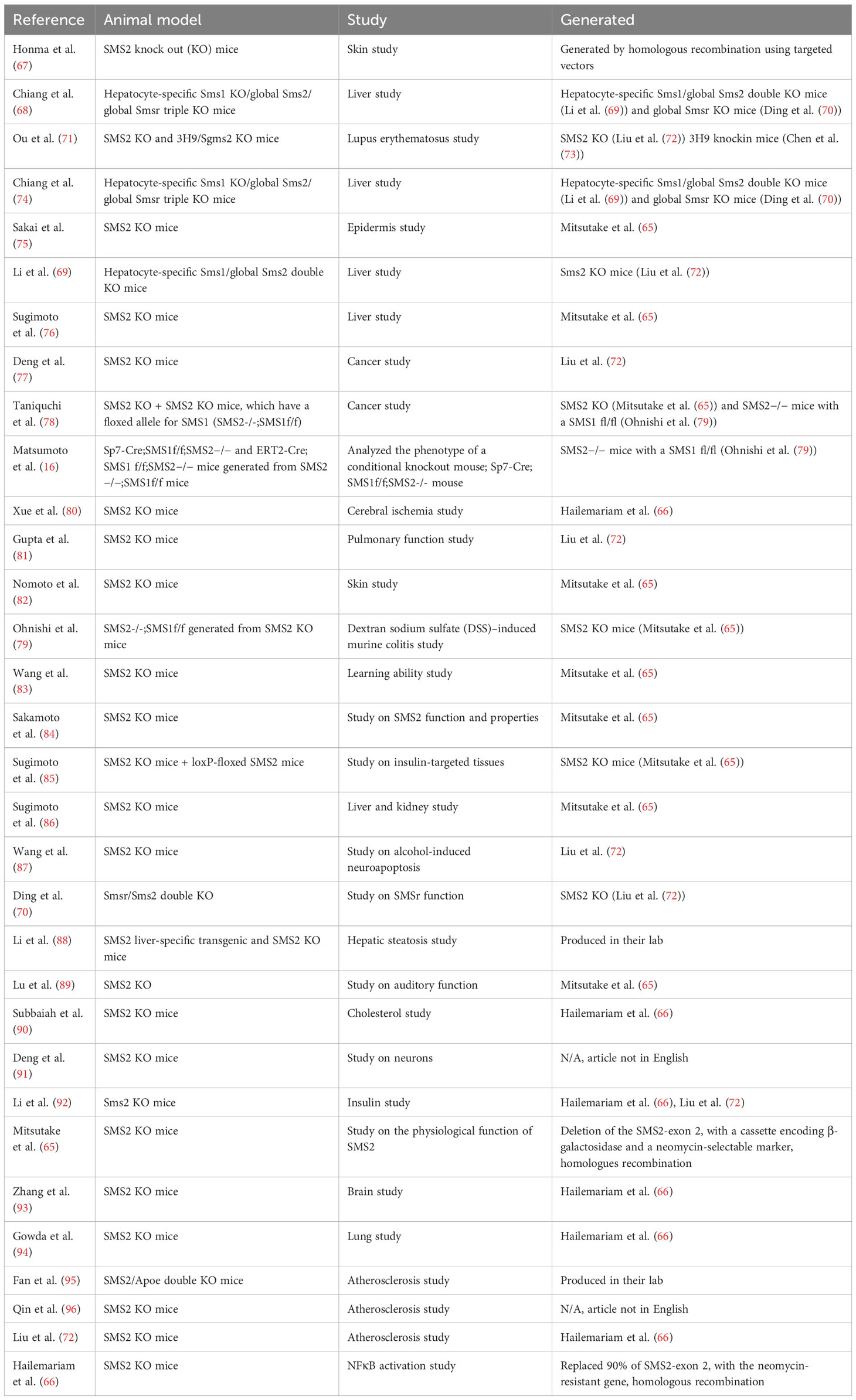
Table 3 SMS2 modified animal models in various mouse studies.
Zebrafish is a good vertebrate model to study human skeletal diseases, since zebrafish share similar skeletal elements and ossification types with mammals (97). However, there are several differences between zebrafish and mammals related to bone morphology and function, which need to be taken into account when using zebrafish as a model for skeletal diseases (98). The zebrafish has two orthologues of human SGMS2, sgms2a and sgms2b (99, 100). To date, no sgms2 KO zebrafish models have been reported. However, our recent work demonstrates that knockdown of sgms2a, sgms2b and sgms2a+b by CRISPR-Cas13d result in defective cartilage and early skeletal element development in comparison to control fish (101). To further elucidate the skeletal abnormalities caused by sgms2 knockdowns, sgms2 knockouts and perhaps pathogenic sgms2 knockins mimicking human pathogenic mutations need to be established. Our study also revealed that sgms2a, sgms2b, sgms2 a+b knockdown zebrafish (6 days post fertilization) showed altered locomotor activity and behavioral response to light/dark transition test compared to controls, indicating a possible role of sgms2 in brain and nervous system function in zebrafish (101).
Conclusions
Recent human studies indicate that heterozygous variants in SGMS2 lead to a spectrum of skeletal disorders in which skeletal fragility is the leading manifestation. On the bone tissue level, at least the p.Arg50* variant leads to greatly altered bone architecture and defective mineralization. The molecular and cellular mechanisms behind SGMS2-linked osteoporosis are not fully understood, but it is believed that the onset of the disease is a result of improperly targeted bulk SM production rather than a diminished capacity to synthesize SM. The SGMS2 variants are anticipated to disturb the export of SMS2 from the ER. The missense variants cause the SMS2 protein to be retained in the ER while the p.Arg50* variant is hypothesized to mislocalize the protein to the cis/medial Golgi. This mistargeted SM production results in significant deviations in organellar lipid compositions and membrane properties along the secretory pathway. Different targeted SM production between the missense and the p.Arg50* variants could, therefore, explain the phenotypic differences seen between patients with different SGMS2 variants.
The relationship between the abnormal subcellular organization of SM and the development of osteoporosis in affected patients remains unknown. One possibility is that pathogenic SMS2 variants affect lipid composition of secretory organelles and prevent proper export of collagen from the ER, affecting bone formation (32). On the other hand, pathogenic SMS2 variants may disturb the SM asymmetry of the plasma membrane in osteogenic cells and negatively affect bone mineralization (31). In cells expressing pathogenic SMS2 variants, it is possible that cytosolically exposed SM is constitutively converted to ceramide prematurely, unintentionally diminishing the fuel that powers matrix vesicle formation. This may hamper a continuous supply of phosphocholine required for normal bone mineralization (4).
The role of SM metabolism in the CNS remains less well characterized. Several CNS disorders, including cerebral ischemia, neurodegenerative diseases, and psychiatric illnesses, have been linked to altered SM metabolism. Extensive clinical evaluation of patients has revealed no apparent cause of patients’ neurological symptoms and they are therefore presumed to be secondary to the altered SMS2 function. However, it is unknown whether these arise directly from changed SM metabolism or if they are merely the outcome of cellular circumstances that are otherwise pathologically changed.
Further research is needed to shed light on the molecular mechanisms leading from genetic variants to bone fragility and whether SM metabolism may provide novel targets for therapeutic intervention. Also, new sgms2 stable mutant zebrafish lines could be utilized in drug discovery and screening platform. Targeted treatments may also be relevant in other forms of osteoporosis in the general population.
Author contributions
All authors contributed to the article and approved the submitted version.
Funding
Academy of Finland (318137, 322647), Sigrid Juselius Foundation, Folkhälsan Research Foundation, Foundation for Pediatric Research (190155, 200196), Nylands Nation at University of Helsinki, Sigrid Jusélius stiftelse, Novo Nordisk Foundation (NNF180C0034982), HUS EVO at Helsinki University Hospital (TYH2021221), Finnish ORL–HNS Foundation, Finnish Medical Foundation.
Conflict of interest
OM declares consultancy to Kyowa Kirin, Alexion, Merck and Sandoz.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mäkitie RE, Costantini A, Kämpe A, Alm JJ, Mäkitie O. New insights into monogenic causes of osteoporosis. Front Endocrinol (Lausanne) (2019) 10:70. doi: 10.3389/fendo.2019.00070
2. Mäkitie RE, Kämpe AJ, Taylan F, Mäkitie O. Recent discoveries in monogenic disorders of childhood bone fragility. Curr Osteoporos Rep (2017) 15(4):303–10. doi: 10.1007/s11914-017-0388-6
3. Peacock M, Turner CH, Econs MJ, Foroud T. Genetics of osteoporosis. Endocrine Rev (2002) 23(3):303–26. doi: 10.1210/edrv.23.3.0464
4. Pekkinen M, Terhal PA, Botto LD, Henning P, Mäkitie RE, Roschger P, et al. Osteoporosis and skeletal dysplasia caused by pathogenic variants in SGMS2. JCI Insight (2019) 4(7):e126180. doi: 10.1172/jci.insight.126180
5. Robinson M-E, Bardai G, Veilleux L-N, Glorieux FH, Rauch F. Musculoskeletal phenotype in two unrelated individuals with a recurrent nonsense variant in SGMS2. Bone (2020) 134:115261. doi: 10.1016/j.bone.2020.115261
6. Basalom S, Fiscaletti M, Miranda V, Huber C, Couture G, Drouin R, et al. Calvarial doughnut lesions with bone fragility in a French-Canadian family; case report and review of the literature. Bone Rep (2021) 15:101121. doi: 10.1016/j.bonr.2021.101121
7. Collantes ER, Flores-Anotado JF, Guevarra MC, Dumdum A, Futolan J, de Guzman MHP, et al. SGMS2 mutation in a large Filipino family with calvarial doughnut lesions with bone fragility and juvenile-onset open angle glaucoma. Invest Ophthalmol Visual Sci (2022) 63(7):4019–A0361. Presented at the 2022 ARVO Annual Meeting, held in Denver, CO, USA, May 1-4.
8. Whyte MPL, Michael McAlister E, Gottesman WH, Nenninger GS, Wenkert A, Mumm D, et al. Sphingomyelin synthase 2 mutation arg50* Causes autosomal dominant osteoporosis with “Doughnut” Lesions of the skull (OMIM %126550), in: A Multigenerational American Kindred. ASBMR 2019 Annual meeting, Orlando, FL, USA, September 20-23, 2019.
9. Aguilera-Romero A, Gehin C, Riezman H. Sphingolipid homeostasis in the web of metabolic routes. Biochim Biophys Acta (BBA) Mol Cell Biol Lipids (2014) 1841(5):647–56. doi: 10.1016/j.bbalip.2013.10.014
10. Khavandgar Z, Murshed M. Sphingolipid metabolism and its role in the skeletal tissues. Cell Mol Life Sci (2015) 72(5):959–69. doi: 10.1007/s00018-014-1778-x
11. Merrill AH Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev (2011) 111(10):6387–422. doi: 10.1021/cr2002917
12. Bienias K, Fiedorowicz A, Sadowska A, Prokopiuk S, Car H. Regulation of sphingomyelin metabolism. Pharmacol Rep (2016) 68(3):570–81. doi: 10.1016/j.pharep.2015.12.008
13. Pralhada Rao R, Vaidyanathan N, Rengasamy M, Mammen Oommen A, Somaiya N, Jagannath MR. Sphingolipid metabolic pathway: an overview of major roles played in human diseases. J Lipids (2013) 2013:178910. doi: 10.1155/2013/178910
14. Slotte JP. Biological functions of sphingomyelins. Prog Lipid Res (2013) 52(4):424–37. doi: 10.1016/j.plipres.2013.05.001
15. Slotte JP. Molecular properties of various structurally defined sphingomyelins – Correlation of structure with function. Prog Lipid Res (2013) 52(2):206–19. doi: 10.1016/j.plipres.2012.12.001
16. Matsumoto G, Hashizume C, Watanabe K, Taniguchi M, Okazaki T. Deficiency of sphingomyelin synthase 1 but not sphingomyelin synthase 2 reduces bone formation due to impaired osteoblast differentiation. Mol Med (2019) 25(1):56. doi: 10.1186/s10020-019-0123-0
17. Li J, Manickam G, Ray S, Oh CD, Yasuda H, Moffatt P, et al. Smpd3 expression in both chondrocytes and osteoblasts is required for normal endochondral bone development. Mol Cell Biol (2016) 36(17):2282–99. doi: 10.1128/MCB.01077-15
18. Milhas D, Clarke CJ, Hannun YA. Sphingomyelin metabolism at the plasma membrane: implications for bioactive sphingolipids. FEBS Lett (2010) 584(9):1887–94. doi: 10.1016/j.febslet.2009.10.058
19. van Meer G, Holthuis JCM. Sphingolipid transport in eukaryotic cells. Biochim Biophys Acta (BBA) Mol Cell Biol Lipids (2000) 1486(1):145–70. doi: 10.1016/s1388-1981(00)00054-8
20. Taha TA, Mullen TD, Obeid LM. A house divided: ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim Biophys Acta (2006) 1758(12):2027–36. doi: 10.1016/j.bbamem.2006.10.018
21. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol (2008) 9(2):139–50. doi: 10.1038/nrm2329
22. Holthuis JCM, Menon AK. Lipid landscapes and pipelines in membrane homeostasis. Nature (2014) 510(7503):48–57. doi: 10.1038/nature13474
23. Unger S, Ferreira CR, Mortier GR, Ali H, Bertola DR, Calder A, et al. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet Part A (2023) 191(5):1164–209. doi: 10.1002/ajmg.a.63132
24. Costantini A, Mäkitie RE, Hartmann MA, Fratzl-Zelman N, Zillikens MC, Kornak U, et al. Early-onset osteoporosis: rare monogenic forms elucidate the complexity of disease pathogenesis beyond type I collagen. J Bone Miner Res (2022) 37(9):1623–41. doi: 10.1002/jbmr.4668
25. Hannan FM, Newey PJ, Whyte MP, Thakker RV. Genetics of skeletal disorders. Handb Exp Pharmacol (2020) 262:325–51. doi: 10.1007/164_2020_350
26. Jaakkola E, Laine CM, Mäyränpää MK, Falck A, Ignatius J, Mäkitie O. Calvarial doughnut lesions and osteoporosis: a new three-generation family and review. Am J Med Genet A (2009) 149a(11):2371–7. doi: 10.1002/ajmg.a.33040
27. Keats TE, Holt JF. The calvarial "doughnut lesion": a previously undescribed entity. Am J Roentgenol Radium Ther Nucl Med (1969) 105(2):314–8. doi: 10.2214/ajr.105.2.314
28. Mäkitie RE, Blouin S, Välimäki VV, Pihlström S, Määttä K, Pekkinen M, et al. Abnormal bone tissue organization and osteocyte lacunocanalicular network in early-onset osteoporosis due to SGMS2 mutations. JBMR Plus (2021) 5(11):e10537. doi: 10.1002/jbm4.10537
29. Huitema K, van den Dikkenberg J, Brouwers JF, Holthuis JC. Identification of a family of animal sphingomyelin synthases. EMBO J (2004) 23(1):33–44. doi: 10.1038/sj.emboj.7600034
30. Steinberg J, Ritchie GRS, Roumeliotis TI, Jayasuriya RL, Clark MJ, Brooks RA, et al. Integrative epigenomics, transcriptomics and proteomics of patient chondrocytes reveal genes and pathways involved in osteoarthritis. Sci Rep (2017) 7(1):8935. doi: 10.1038/s41598-017-09335-6
31. Sokoya T, Parolek J, Foged MM, Danylchuk DI, Bozan M, Sarkar B, et al. Pathogenic variants of sphingomyelin synthase SMS2 disrupt lipid landscapes in the secretory pathway. Elife (2022) 11:e79278. doi: 10.7554/eLife.79278
32. Murshed M. Mechanism of bone mineralization. Cold Spring Harb Perspect Med (2018) 8(12):a031229. doi: 10.1101/cshperspect.a031229
33. Gillon AD, Latham CF, Miller EA. Vesicle-mediated ER export of proteins and lipids. Biochim Biophys Acta - Mol Cell Biol (2012). 1821(8):1040–9. doi: 10.1016/j.bbalip.2012.01.005
34. McCaughey J, Stevenson NL, Cross S, Stephens DJ. ER-to-Golgi trafficking of procollagen in the absence of large carriers. J Cell Biol (2018) 218(3):929–48. doi: 10.1083/jcb.201806035
35. Townley AK, Feng Y, Schmidt K, Carter DA, Porter R, Verkade P, et al. Efficient coupling of Sec23-Sec24 to Sec13-Sec31 drives COPII-dependent collagen secretion and is essential for normal craniofacial development. J Cell Sci (2008) 121(18):3025–34. doi: 10.1242/jcs.031070
36. Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, Hur DJ, et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet (2006) 38(10):1192–7. doi: 10.1038/ng1876
37. Guillemyn B, Nampoothiri S, Syx D, Malfait F, Symoens S. Loss of TANGO1 leads to absence of bone mineralization. JBMR Plus (2021) 5(3):e10451. doi: 10.1002/jbm4.10451
38. Roberts S, Narisawa S, Harmey D, Millán JL, Farquharson C. Functional involvement of PHOSPHO1 in matrix vesicle–mediated skeletal mineralization. J Bone Mineral Res (2007) 22(4):617–27. doi: 10.1359/jbmr.070108
39. Golub EE. Role of matrix vesicles in biomineralization. Biochim Biophys Acta (2009) 1790(12):1592–8. doi: 10.1016/j.bbagen.2009.09.006
40. Yoshikawa Y, Yoshizawa T, Domae E, Hirai Y, Kamada A, Okazaki T, et al. Knockdown of sphingomyelin synthase 2 inhibits osteoclastogenesis by decreasing RANKL expression in mouse primary osteoblasts. BioMed Res (2019) 40(5):189–96. doi: 10.2220/biomedres.40.189
41. Yavropoulou MP, Yovos JG. Osteoclastogenesis–current knowledge and future perspectives. J Musculoskelet Neuronal Interact (2008) 8(3):204–16.
42. Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene (2004) 328:1–16. doi: 10.1016/j.gene.2003.12.005
43. Oshima K, Nampei A, Matsuda M, Iwaki M, Fukuhara A, Hashimoto J, et al. Adiponectin increases bone mass by suppressing osteoclast and activating osteoblast. Biochem Biophys Res Commun (2005) 331(2):520–6. doi: 10.1016/j.bbrc.2005.03.210
44. Sillence DO, Ritchie HE, Dibbayawan T, Eteson D, Brown K. Fragilitas ossium (fro/fro) in the mouse: a model for a recessively inherited type of osteogenesis imperfecta. Am J Med Genet (1993) 45(2):276–83. doi: 10.1002/ajmg.1320450227
45. Aubin I, Adams CP, Opsahl S, Septier D, Bishop CE, Auge N, et al. A deletion in the gene encoding sphingomyelin phosphodiesterase 3 (Smpd3) results in osteogenesis and dentinogenesis imperfecta in the mouse. Nat Genet (2005) 37(8):803–5. doi: 10.1038/ng1603
46. Grewe JM, Knapstein PR, Donat A, Jiang S, Smit DJ, Xie W, et al. The role of sphingosine-1-phosphate in bone remodeling and osteoporosis. Bone Res (2022) 10(1):34. doi: 10.1038/s41413-022-00205-0
47. Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol (2012) 22(1):50–60. doi: 10.1016/j.tcb.2011.09.003
48. Gallegos LL, Newton AC. Spatiotemporal dynamics of lipid signaling: protein kinase C as a paradigm. IUBMB Life (2008) 60(12):782–9. doi: 10.1002/iub.122
49. Tu X, Joeng KS, Nakayama KI, Nakayama K, Rajagopal J, Carroll TJ, et al. Noncanonical Wnt signaling through G protein-linked PKCdelta activation promotes bone formation. Dev Cell (2007) 12(1):113–27. doi: 10.1016/j.devcel.2006.11.003
50. Olsen ASB, Færgeman NJ. Sphingolipids: membrane microdomains in brain development, function and neurological diseases. Open Biol (2017) 7(5):170069. doi: 10.1098/rsob.170069
51. Demirkan A, Isaacs A, Ugocsai P, Liebisch G, Struchalin M, Rudan I, et al. Plasma phosphatidylcholine and sphingomyelin concentrations are associated with depression and anxiety symptoms in a Dutch family-based lipidomics study. J Psychiatr Res (2013) 47(3):357–62. doi: 10.1016/j.jpsychires.2012.11.001
52. Kubota M, Narita K, Nakagomi T, Tamura A, Shimasaki H, Ueta N, et al. Sphingomyelin changes in rat cerebral cortex during focal ischemia. Neurol Res (1996) 18(4):337–41. doi: 10.1080/01616412.1996.11740432
53. Sun X, Liu C, Qian M, Zhao Z, Guo J. Ceramide from sphingomyelin hydrolysis differentially mediates mitogen-activated protein kinases (MAPKs) activation following cerebral ischemia in rat hippocampal CA1 subregion. J BioMed Res (2010) 24(2):132–7. doi: 10.1016/S1674-8301(10)60021-8
54. Soeda S, Tsuji Y, Ochiai T, Mishima K, Iwasaki K, Fujiwara M, et al. Inhibition of sphingomyelinase activity helps to prevent neuron death caused by ischemic stress. Neurochem Int (2004) 45(5):619–26. doi: 10.1016/j.neuint.2004.04.001
55. Chew H, Solomon VA, Fonteh AN. Involvement of lipids in alzheimer's disease pathology and potential therapies. Front Physiol (2020) 11:598. doi: 10.3389/fphys.2020.00598
56. Haughey NJ, Bandaru VV, Bae M, Mattson MP. Roles for dysfunctional sphingolipid metabolism in Alzheimer's disease neuropathogenesis. Biochim Biophys Acta (2010) 1801(8):878–86. doi: 10.1016/j.bbalip.2010.05.003
57. Mielke MM, Haughey NJ, Bandaru VVR, Zetterberg H, Blennow K, Andreasson U, et al. Cerebrospinal fluid sphingolipids, β-amyloid, and tau in adults at risk for Alzheimer's disease. Neurobiol Aging (2014) 35(11):2486–94. doi: 10.1016/j.neurobiolaging.2014.05.019
58. Kosicek M, Zetterberg H, Andreasen N, Peter-Katalinic J, Hecimovic S. Elevated cerebrospinal fluid sphingomyelin levels in prodromal Alzheimer's disease. Neurosci Lett (2012) 516(2):302–5. doi: 10.1016/j.neulet.2012.04.019
59. He X, Huang Y, Li B, Gong CX, Schuchman EH. Deregulation of sphingolipid metabolism in Alzheimer's disease. Neurobiol Aging (2010) 31(3):398–408. doi: 10.1016/j.neurobiolaging.2008.05.010
60. Satoi H, Tomimoto H, Ohtani R, Kitano T, Kondo T, Watanabe M, et al. Astroglial expression of ceramide in Alzheimer's disease brains: a role during neuronal apoptosis. Neuroscience (2005) 130(3):657–66. doi: 10.1016/j.neuroscience.2004.08.056
61. Stefanis L. α-synuclein in parkinson's disease. Cold Spring Harb Perspect Med (2012) 2(2):a009399. doi: 10.1101/cshperspect.a009399
62. Xilouri M, Vogiatzi T, Vekrellis K, Stefanis L. alpha-synuclein degradation by autophagic pathways: a potential key to Parkinson's disease pathogenesis. Autophagy (2008) 4(7):917–9. doi: 10.4161/auto.6685
63. Foo JN, Liany H, Bei JX, Yu XQ, Liu J, Au WL, et al. Rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson's disease. Neurobiol Aging (2013) 34(12):2890. doi: 10.1016/j.neurobiolaging.2013.06.010
64. Gan-Or Z, Ozelius LJ, Bar-Shira A, Saunders-Pullman R, Mirelman A, Kornreich R, et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology (2013) 80(17):1606–10. doi: 10.1212/WNL.0b013e31828f180e
65. Mitsutake S, Zama K, Yokota H, Yoshida T, Tanaka M, Mitsui M, et al. Dynamic modification of sphingomyelin in lipid microdomains controls development of obesity, fatty liver, and type 2 diabetes. J Biol Chem (2011) 286(32):28544–55. doi: 10.1074/jbc.M111.255646
66. Hailemariam TK, Huan C, Liu J, Li Z, Roman C, Kalbfeisch M, et al. Sphingomyelin synthase 2 deficiency attenuates NFκB activation. Arteriosclerosis Thrombosis Vasc Biol (2008) 28(8):1519–26. doi: 10.1161/ATVBAHA.108.168682
67. Honma N, Hatta I, Okazaki T, Tokudome Y. Modulation of function and structure of stratum corneum in sphingomyelin synthase 2-deficient mice. Chem Phys Lipids (2022) 249:105255. doi: 10.21203/rs.3.rs-1627640/v1
68. Chiang YP, Li Z, Chen Y, Cao Y, Jiang XC. Sphingomyelin synthases 1 and 2 exhibit phosphatidylcholine phospholipase C activity. J Biol Chem (2021) 297(6):101398. doi: 10.1016/j.jbc.2021.101398
69. Li Z, Chiang YP, He M, Zhang K, Zheng J, Wu W, et al. Effect of liver total sphingomyelin synthase deficiency on plasma lipid metabolism. Biochim Biophys Acta Mol Cell Biol Lipids (2021) 1866(5):158898. doi: 10.1016/j.bbalip.2021.158898
70. Ding T, Kabir I, Li Y, Lou C, Yazdanyar A, Xu J, et al. All members in the sphingomyelin synthase gene family have ceramide phosphoethanolamine synthase activity. J Lipid Res (2015) 56(3):537–45. doi: 10.1194/jlr.M054627
71. Ou P, Stanek A, Huan Z, Roman CAJ, Huan C. SMS2 deficiency impairs PKCδ-regulated B cell tolerance in the germinal center. Cell Rep (2021) 36(9):109624. doi: 10.1016/j.celrep.2021.109624
72. Liu J, Huan C, Chakraborty M, Zhang H, Lu D, Kuo MS, et al. Macrophage sphingomyelin synthase 2 deficiency decreases atherosclerosis in mice. Circ Res (2009) 105(3):295–303. doi: 10.1161/CIRCRESAHA.109.194613
73. Chen C, Nagy Z, Luning Prak E, Weigert M. Immunoglobulin heavy chain gene replacement: A mechanism of receptor editing. Immunity (1995) 3(6):747–55. doi: 10.1016/1074-7613(95)90064-0
74. Chiang YP, Li Z, Chen Y, Cao Y, Jiang XC. Sphingomyelin synthase related protein is a mammalian phosphatidylethanolamine phospholipase C. Biochim Biophys Acta Mol Cell Biol Lipids (2021) 1866(11):159017. doi: 10.1016/j.bbalip.2021.159017
75. Sakai S, Makino A, Nishi A, Ichikawa T, Yamashita T, Taniguchi M, et al. [Pathogenic and compensatory mechanisms in epidermis of sphingomyelin synthase 2-deficient mice]. Skin Pharmacol Physiol (2021) 34(5):246–52. doi: 10.1159/000515608
76. Sugimoto M, Hamada T, Wakabayasi M, Yoshioka T, Kato H, Konishi H, et al. Sphingomyelin synthase 2 loss suppresses steatosis but exacerbates fibrosis in the liver of mice fed with choline-deficient, L-amino acid-defined, high-fat diet. Biochem Biophys Res Commun (2020) 533(4):1269–75. doi: 10.1016/j.bbrc.2020.09.142
77. Deng Y, Hu JC, He SH, Lou B, Ding TB, Yang JT, et al. Sphingomyelin synthase 2 facilitates M2-like macrophage polarization and tumor progression in a mouse model of triple-negative breast cancer. Acta Pharmacol Sin (2021) 42(1):149–59. doi: 10.1038/s41401-020-0419-1
78. Taniguchi M, Ueda Y, Matsushita M, Nagaya S, Hashizume C, Arai K, et al. Deficiency of sphingomyelin synthase 2 prolongs survival by the inhibition of lymphoma infiltration through ICAM-1 reduction. FASEB J (2020) 34(3):3838–54. doi: 10.1096/fj.201901783RR
79. Ohnishi T, Hashizume C, Taniguchi M, Furumoto H, Han J, Gao R, et al. Sphingomyelin synthase 2 deficiency inhibits the induction of murine colitis-associated colon cancer. FASEB J (2017) 31(9):3816–30. doi: 10.1096/fj.201601225RR
80. Xue J, Yu Y, Zhang X, Zhang C, Zhao Y, Liu B, et al. Sphingomyelin synthase 2 inhibition ameliorates cerebral ischemic reperfusion injury through reducing the recruitment of toll-like receptor 4 to lipid rafts. J Am Heart Assoc (2019) 8(22):e012885. doi: 10.1161/JAHA.119.012885
81. Gupta G, Baumlin N, Poon J, Ahmed B, Chiang YP, Railwah C, et al. Airway resistance caused by sphingomyelin synthase 2 insufficiency in response to cigarette smoke. Am J Respir Cell Mol Biol (2020) 62(3):342–53. doi: 10.1165/rcmb.2019-0133OC
82. Nomoto K, Itaya Y, Watanabe K, Yamashita T, Okazaki T, Tokudome Y. Epidermal permeability barrier function and sphingolipid content in the skin of sphingomyelin synthase 2 deficient mice. Exp Dermatol (2018) 27(8):827–32. doi: 10.1111/exd.13497
83. Wang M, Uchiumi O, Ogiso H, Shui Y, Zou J, Hashizume C, et al. Stressful learning paradigm precludes manifestation of cognitive ability in sphingomyelin synthase-2 knockout mice. Behav Brain Res (2017) 319:25–30. doi: 10.1016/j.bbr.2016.11.010
84. Sakamoto H, Yoshida T, Sanaki T, Shigaki S, Morita H, Oyama M, et al. Possible roles of long-chain sphingomyelines and sphingomyelin synthase 2 in mouse macrophage inflammatory response. Biochem Biophys Res Commun (2017) 482(2):202–7. doi: 10.1016/j.bbrc.2016.11.041
85. Sugimoto M, Shimizu Y, Zhao S, Ukon N, Nishijima K-i, Wakabayashi M, et al. Characterization of the role of sphingomyelin synthase 2 in glucose metabolism in whole-body and peripheral tissues in mice. Biochim Biophys Acta (BBA) Mol Cell Biol Lipids (2016) 1861(8, Part A):688–702. doi: 10.1016/j.bbalip.2016.04.019
86. Sugimoto M, Wakabayashi M, Shimizu Y, Yoshioka T, Higashino K, Numata Y, et al. Imaging mass spectrometry reveals acyl-chain- and region-specific sphingolipid metabolism in the kidneys of sphingomyelin synthase 2-deficient mice. PloS One (2016) 11(3):e0152191. doi: 10.1371/journal.pone.0152191
87. Wang L, Wu L, Wang X, Deng J, Ma Z, Fan W, et al. Prenatal alcohol exposure inducing the apoptosis of mossy cells in hippocampus of SMS2–/– mice. Environ Toxicol Pharmacol (2015) 40(3):975–82. doi: 10.1016/j.etap.2015.10.004
88. Li Y, Dong J, Ding T, Kuo MS, Cao G, Jiang XC, et al. Sphingomyelin synthase 2 activity and liver steatosis: an effect of ceramide-mediated peroxisome proliferator-activated receptor γ2 suppression. Arterioscler Thromb Vasc Biol (2013) 33(7):1513–20. doi: 10.1161/ATVBAHA.113.301498
89. Lu MH, Takemoto M, Watanabe K, Luo H, Nishimura M, Yano M, et al. Deficiency of sphingomyelin synthase-1 but not sphingomyelin synthase-2 causes hearing impairments in mice. J Physiol (2012) 590(16):4029–44. doi: 10.1113/jphysiol.2012.235846
90. Subbaiah PV, Jiang XC, Belikova NA, Aizezi B, Huang ZH, Reardon CA. Regulation of plasma cholesterol esterification by sphingomyelin: effect of physiological variations of plasma sphingomyelin on lecithin-cholesterol acyltransferase activity. Biochim Biophys Acta (2012) 1821(6):908–13. doi: 10.1016/j.bbalip.2012.02.007
91. Deng TX, Wang ZX, Gao XQ, Shi YY, Ma ZY, Jin HX, et al. [Alcohol-induced proliferation of neurons in mouse hippocampal dentate gyrus: a possible role of ceramide]. Sheng Li Xue Bao (2011) 63(6):479–90.
92. Li Z, Zhang H, Liu J, Liang CP, Li Y, Li Y, et al. Reducing plasma membrane sphingomyelin increases insulin sensitivity. Mol Cell Biol (2011) 31(20):4205–18. doi: 10.1128/MCB.05893-11
93. Zhang Y, Dong J, Zhu X, Wang W, Yang Q. The effect of sphingomyelin synthase 2 (SMS2) deficiency on the expression of drug transporters in mouse brain. Biochem Pharmacol (2011) 82(3):287–94. doi: 10.1016/j.bcp.2011.04.009
94. Gowda S, Yeang C, Wadgaonkar S, Anjum F, Grinkina N, Cutaia M, et al. Sphingomyelin synthase 2 (SMS2) deficiency attenuates LPS-induced lung injury. Am J Physiol Lung Cell Mol Physiol (2011) 300(3):L430–40. doi: 10.1152/ajplung.00208.2010
95. Fan Y, Shi F, Liu J, Dong J, Bui HH, Peake DA, et al. Selective reduction in the sphingomyelin content of atherogenic lipoproteins inhibits their retention in murine aortas and the subsequent development of atherosclerosis. Arterioscler Thromb Vasc Biol (2010) 30(11):2114–20. doi: 10.1161/ATVBAHA.110.213363
96. Qin R, Chen ML, Zhu K, Deng JB, Shi YY. [Sphingomyelin synthase 2 deficiency decreases atherosclerosis and inhibits inflammation in mice]. Sheng Li Xue Bao (2010) 62(4):333–8.
97. Tonelli F, Bek JW, Besio R, De Clercq A, Leoni L, Salmon P, et al. Zebrafish: A resourceful vertebrate model to investigate skeletal disorders. Front Endocrinol (Lausanne) (2020) 11:489. doi: 10.3389/fendo.2020.00489
98. Dietrich K, Fiedler IAK, Kurzyukova A, López-Delgado AC, McGowan LM, Geurtzen K, et al. Skeletal biology and disease modeling in zebrafish. J Bone Mineral Res (2021) 36(3):436–58. doi: 10.1002/jbmr.4256
99. Postlethwait JH, Woods IG, Ngo-Hazelett P, Yan YL, Kelly PD, Chu F, et al. Zebrafish comparative genomics and the origins of vertebrate chromosomes. Genome Res (2000) 10(12):1890–902. doi: 10.1101/gr.164800
100. Bradford YM, Van Slyke CE, Ruzicka L, Singer A, Eagle A, Fashena D, et al. Zebrafish information network, the knowledgebase for Danio rerio research. Genetics (2022) 220(4):iyac016. doi: 10.1093/genetics/iyac016
Keywords: SMS2, SGMS2-related osteoporosis, sphingomyelin metabolism, bone and neural tissue, sphingolipids
Citation: Pihlström S, Richardt S, Määttä K, Pekkinen M, Olkkonen VM, Mäkitie O and Mäkitie RE (2023) SGMS2 in primary osteoporosis with facial nerve palsy. Front. Endocrinol. 14:1224318. doi: 10.3389/fendo.2023.1224318
Received: 25 May 2023; Accepted: 18 September 2023;
Published: 11 October 2023.
Edited by:
Gudrun Stenbeck, Brunel University London, United KingdomReviewed by:
Patricia Canto, National Autonomous University of Mexico, MexicoRamón Coral, National Polytechnic Institute (IPN), Mexico
Copyright © 2023 Pihlström, Richardt, Määttä, Pekkinen, Olkkonen, Mäkitie and Mäkitie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Pihlström, c2FuZHJhLnBpaGxzdHJvbUBoZWxzaW5raS5maQ==
†These authors have contributed equally to this work