 Daniel Benak
Daniel Benak Stepanka Benakova3,4
Stepanka Benakova3,4 Lydie Plecita-Hlavata
Lydie Plecita-Hlavata Marketa Hlavackova
Marketa Hlavackova- 1Laboratory of Developmental Cardiology, Institute of Physiology of the Czech Academy of Sciences, Prague, Czechia
- 2Department of Physiology, Faculty of Science, Charles University, Prague, Czechia
- 3Laboratory of Pancreatic Islet Research, Institute of Physiology of the Czech Academy of Sciences, Prague, Czechia
- 4First Faculty of Medicine, Charles University, Prague, Czechia
The rapidly developing research field of epitranscriptomics has recently emerged into the spotlight of researchers due to its vast regulatory effects on gene expression and thereby cellular physiology and pathophysiology. N6-methyladenosine (m6A) and N6,2’-O-dimethyladenosine (m6Am) are among the most prevalent and well-characterized modified nucleosides in eukaryotic RNA. Both of these modifications are dynamically regulated by a complex set of epitranscriptomic regulators called writers, readers, and erasers. Altered levels of m6A and also several regulatory proteins were already associated with diabetic tissues. This review summarizes the current knowledge and gaps about m6A and m6Am modifications and their respective regulators in the pathophysiology of diabetes mellitus. It focuses mainly on the more prevalent type 2 diabetes mellitus (T2DM) and its treatment by metformin, the first-line antidiabetic agent. A better understanding of epitranscriptomic modifications in this highly prevalent disease deserves further investigation and might reveal clinically relevant discoveries in the future.
1 Introduction
Diabetes mellitus is one of the most common chronic diseases with an increasing prevalence (1). Type 2 diabetes mellitus (T2DM) is more frequent than type 1 diabetes mellitus (T1DM) and accounts for approximately 90% of all cases of diabetes (2). This heterogeneous systemic disorder is mainly characterized by two factors: deficient insulin secretion by pancreatic β-cells and insulin resistance of insulin-sensitive tissues (3). The subsequent chronic hyperglycemia, a hallmark of T2DM, damages glucose-sensitive organs and results in downstream deficits in vital functions (4). Despite a considerable amount of data collected regarding T2DM, the molecular mechanism of its development is still unclear. However, it is known that T2DM is linked with the dysregulation of gene expression profiles in cells (5–7). Epitranscriptomic modifications of RNA are one of the possible mechanisms by which gene expression could be affected during the pathogenesis of T2DM.
To date, over 170 chemical modifications have been described in RNA (8). N6-methyladenosine (m6A) and N6,2’-O-dimethyladenosine (m6Am) are among the most prevalent and well-characterized RNA-modified nucleosides (9–12). The biological effects of these modifications are regulated by proteins called writers (methylation deposition), readers (binding of modified RNA), and erasers (methylation removal). The presence or absence of m6A and m6Am in mRNA affects key stages of its life cycle, including splicing, export, decay, and translation (Figure 1) (13, 14). These dynamic modifications with profound impact on gene expression regulation might thereby play an important role in the pathogenesis of T2DM and become the future targets in the search for the next generation of anti-diabetic drugs.
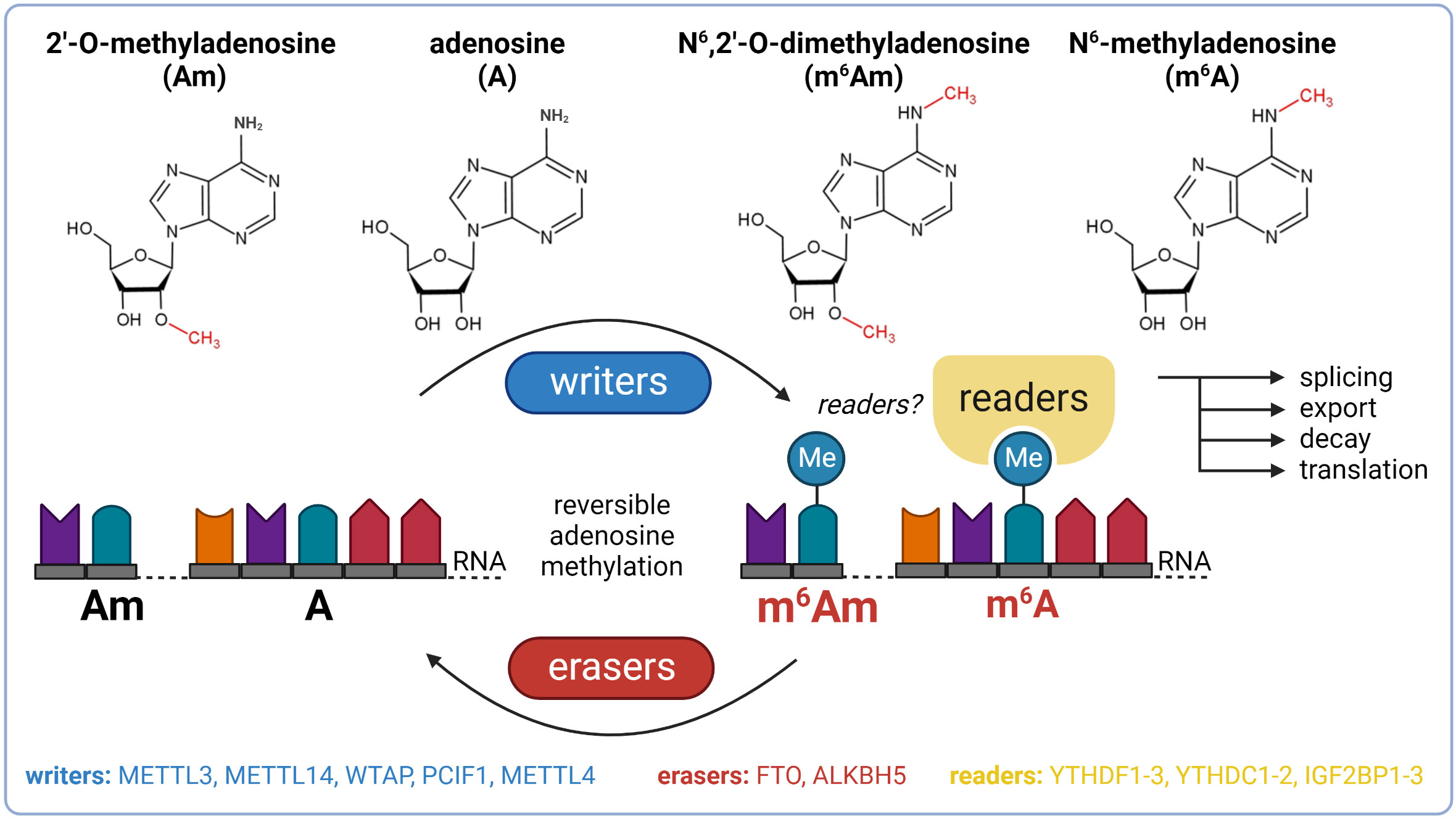
Figure 1 Basic overview of m6A and m6Am epitranscriptomics. ALKBH5, AlkB family member 5; FTO, fat mass and obesity-associated; IGF2BP1-3, insulin-like growth factor 2 mRNA binding proteins 1-3; METTL3, methyltransferase-like 3; METTL14, methyltransferase-like 14; WTAP, Willms’ tumor 1-associating protein; YTHDC1-2, YTH domain-containing protein 1-2; YTHDF1-3, YTH domain-containing family proteins 1-3.
2 N6-methyladenosine
The most prevalent modification in eukaryotic mRNA is m6A (9, 10). Besides mRNA, m6A also occurs in other types of RNA, including ribosomal RNA (rRNA), long non-coding RNA (lncRNA), small nuclear RNA (snRNA), or microRNA (miRNA) (15). The deposition of the methyl group to adenosine (A) is performed by a multicomponent methyltransferase complex (MTC) with a stable core component formed between methyltransferase-like 3 (METTL3) and methyltransferase-like 14 (METTL14). METTL3 functions as a catalytic subunit and METTL14 facilitates RNA binding (16, 17). The third major component of the MTC is the Willms’ tumor 1-associating protein (WTAP) which interacts with the METTL3/METTL14 heterodimer and promotes the localization of the MTC to nuclear speckles (18). The reverse process, demethylation of m6A back to A, is mediated by enzymes called demethylases. In 2011, Fat mass and obesity-associated protein (FTO) was the first described demethylase of m6A (19). This discovery provided evidence of reversible posttranscriptional modifications in mRNAs and renewed the interest of researchers in mRNA modifications (20). After 2 years, alkB homolog 5 (ALKBH5) was reported as another m6A eraser (21). The biological functions of m6A can be mediated by m6A readers which recognize and selectively bind to m6A-decorated RNAs. The most prominent readers are YTH domain-containing family proteins 1-3 (YTHDF1-3) which mediate the degradation of methylated mRNAs, and YTH domain-containing proteins 1-2 (YTHDC1-2) which regulate mRNA splicing and facilitate translation initiation (22–28). In addition to YTH proteins, other readers described include insulin-like growth factor 2 mRNA-binding proteins 1-3 (IGF2BP1-3) which promote the stability of their target mRNAs in an m6A-dependent manner under normal and stress conditions and therefore also affect gene expression output (29).
3 N6,2’-O-dimethyladenosine
m6Am is another prevalent form of modified adenosine, but it is much less studied than m6A. This modification is formed by the methylation of a 2’-O-methyladenosine (Am). It has been described only in mRNA and snRNA. In mRNA, m6Am is found directly downstream to the 7-methylguanosine (m7G), forming the extended cap structure (11, 12). It has been found in at least 30-40% of all transcripts in vertebrate mRNA (11). However, in specific cell lines, m6Am is even more dominant. For instance, HEK293T cells have 92% of 5’ capped mRNAs with m6Am and only 8% with single methylated Am (30). The presence of m6Am in mRNA markedly enhances its stability (31). In snRNA, m6Am is also present at its internal sites and influences pre-mRNA splicing (11, 32). N6-methylation of Am to m6Am is catalyzed by two known writers: phosphorylated CTD interacting factor 1 (PCIF1) and methyltransferase-like 4 (METTL4). PCIF1 has been described as a cap-specific adenosine-N6-methyltransferase (also called CAPAM) which does not methylate adenosine residues in the RNA body (30, 33). However, recently it was reported that PCIF1 also has ancillary methylation activities on internal adenosines (both A and Am), although with lower affinities (34). Importantly, before the recognition of methyltransferase activity of PCIF1, this protein was known to inhibit pancreatic and duodenal homeobox protein 1 (PDX1), a transcription factor crucial for normal pancreas development and function (35, 36). METTL4, the second methyltransferase, is responsible for internal m6Am formation within U2 snRNA (37, 38). The only described m6Am eraser so far is FTO, the well-known m6A demethylase. In 2017, it was reported that FTO preferentially demethylates m6Am rather than m6A (31, 39), but recent studies suggested that the substrate preference of FTO might depend on its cellular localization which varies between cell types. In the nucleus, FTO preferably targets m6A whereas cytosolic FTO demethylates especially m6Am (40, 41). Thus, special attention is needed in FTO research to distinguish the m6A- and m6Am-specific effects of this demethylase (42). No readers of m6Am have been described so far.
4 Pathogenesis of T2DM: the role of m6A and m6Am modifications
4.1 Genetic predisposition to T2DM
The development of T2DM is the result of interaction between environmental factors (e.g. unhealthy diet, sedentary lifestyle, stress) and a strong hereditary component (43). Currently, several hundreds of genetic variants were associated with T2DM, although mostly with only minor effects on disease development (44).
Numerous studies suggested that m6A and m6Am demethylase FTO is among the genes whose variants possess the highest genetic risk of T2DM (44). However, this link is still controversial with significant interethnic differences (45, 46). For instance, the common FTO rs9939609 variant was associated with T2DM in white American, Palestinian, Asian Indian, and obese Iraqi populations, but not in Bengalee Hindu, North Indian, nor Saudi populations (47–57). Also, other genetic polymorphisms in the FTO gene were identified as T2DM risk factors. Carriers of the FTO rs17817449 variant in the Czech-Slavonic and obese Iraqi populations were more susceptible to T2DM and chronic diabetic complications (44, 51, 58). In Iranian obese women, FTO variants rs763967273, rs759031579, rs141115189, rs9926289, rs76804286, and rs9939609 were all related to T2DM (59). On the contrary, African-Americans carrying the rs1421085 C allele were found to be protected against diabetes (54). The polymorphisms in FTO gene seem to regulate the expression level of FTO and its enzymatic function. Detrimental effects of high or low expression of FTO were already confirmed in experimental studies. For instance, it has been shown that FTO depletion activates inflammatory response, one of the main pathogenic features in T2DM patients (60).
Besides FTO, variants of IGF2BP2, an m6A reader, were also associated with a significant risk of T2DM development, namely variant rs4402960 in Asian Indian Sikhs, Czechs, or Italians, and rs11705701 in the Chinese population (44, 55, 61, 62).
Although further studies are needed to unravel the complex polygenic background of T2DM, it seems to be clear that genetic polymorphisms in genes encoding epitranscriptomic regulators are associated both with T2DM and its complications.
4.2 Pancreatic islets
Pancreatic β-cell failure mediated by metabolic stress is the central event in the pathogenesis of T2DM (63). Although the mechanisms underlying β-cell dysfunction are still not fully understood, emerging data suggest an involvement of epigenetic modifications in the adaptation of β-cells to metabolic stress (64).
m6A sequencing in dispersed islets from controls and T2DM patients revealed 6,078 differently methylated sites in 4,155 mRNAs and a higher number of sites with decreased levels of m6A methylation in T2DM compared to controls. Gene ontology analysis of the m6A methylome revealed that the genes affected in T2DM patients are involved in cell-cycle regulation, receptor signaling, insulin secretion, and pancreas development (65). The decreased total m6A levels were observed in Langerhans islets of T2DM patients and also in islets of mice fed with a high-fat diet (a model mimicking T2DM phenotype). Similarly, high glucose conditions (state typical for T2DM) also resulted in lower methylation levels in non-diabetic human pancreatic islets as well as in mouse β-cell line (Min6) (66). Gene expression analysis in whole islets collected from healthy humans and patients with T2DM revealed a down-regulation of several m6A regulators in diabetic individuals – methyltransferase METTL14, demethylases FTO and ALKBH5, and readers YTHDF1 and YTHDF3. In addition to transcripts, protein levels of methyltransferases METTL3 and METTL14 were also decreased (65). The reduction of FTO gene expression and METTL3/14 protein levels in T2DM human islets was observed also in other studies (67–69). RNA-seq datasets (GSE153855; GSE153855) from T2DM and non-T2DM individuals revealed increased gene expression of readers IGF2BP2-3 and decreased gene expression of writer WTAP and readers YTHDF2-3, YTHDC1, and HNRNPC (70–72). m6A reader IGF2BP2 was also up-regulated in β-cells obtained from cadaver pancreases of T2DM patients (73). The current knowledge of diabetic epitranscriptomic changes in human Langerhans islets is summarized in Figure 2. Overall, it seems that the whole epitranscriptomic machinery is attenuated in human diabetic islets. The only up-regulated genes IGF2BP2-3 have also functions unrelated to epitranscriptomics, which might explain their opposite trend.
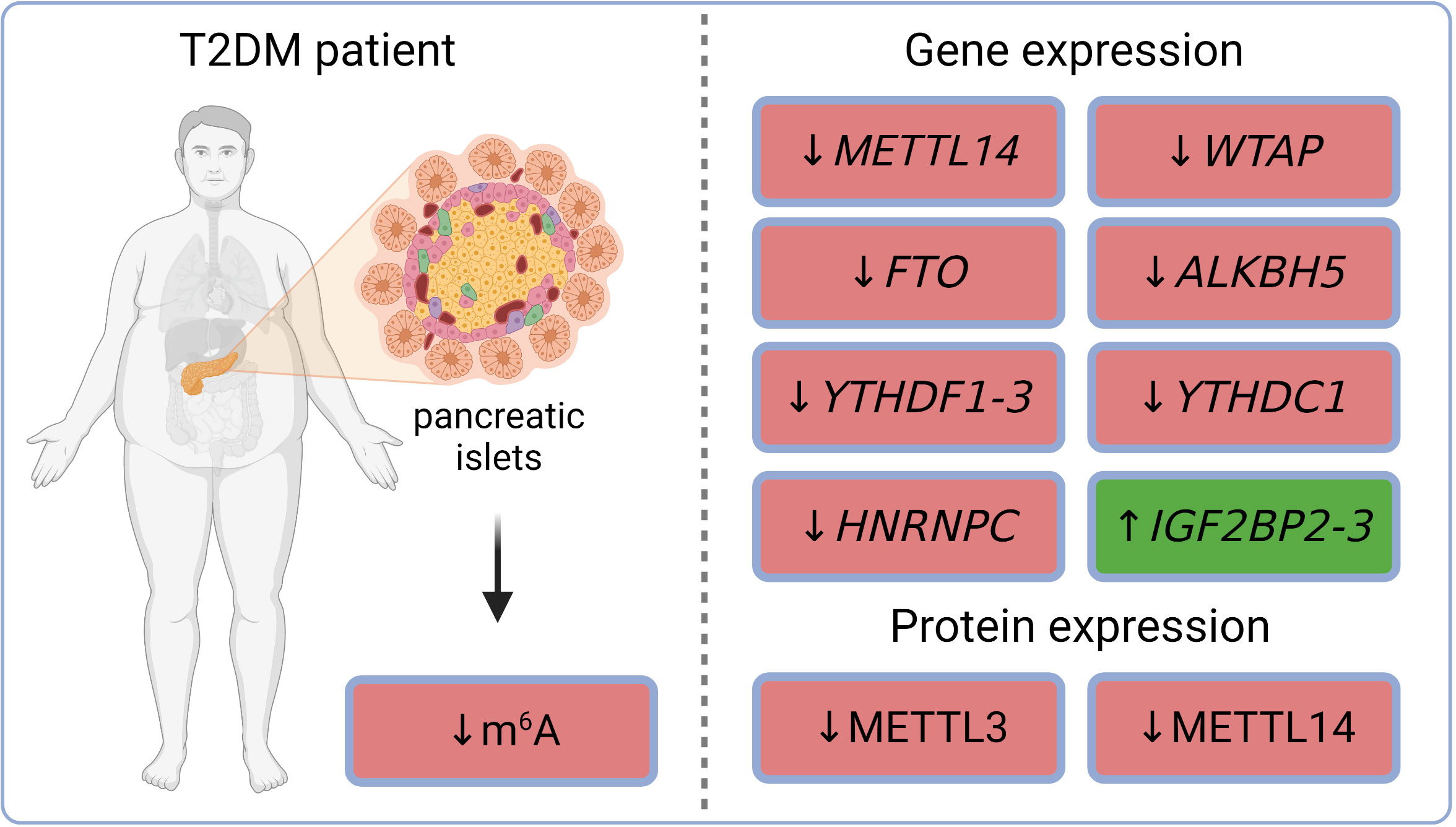
Figure 2 m6A and m6Am regulations in pancreatic islets of T2DM patients. ALKBH5, AlkB family member 5; FTO, fat mass and obesity-associated; HNRNPC, heterogeneous nuclear ribonucleoprotein C; IGF2BP2-3, insulin-like growth factor 2 mRNA binding proteins 2-3; m6A, N6-methyladenosine; METTL3, methyltransferase-like 3; METTL14, methyltransferase-like 14; T2DM, type 2 diabetes mellitus; WTAP, Willms’ tumor 1-associating protein; YTHDC1, YTH domain-containing protein 1; YTHDF1-3, YTH domain-containing family proteins 1-3.
In contrast to these results, Bornaque et al. (66) showed that high glucose concentrations in Min6 cells increased mRNA expression of important m6A regulators – methyltransferase Mettl3 and demethylases Fto and Alkbh5. Glucose treatment also induced a shift in the subcellular protein localization of METTL3 and ALKBH5 (66). Overexpression of FTO in Min6 cells promoted the production of reactive oxygen species (ROS) and led to NF-κB activation, which resulted in the inhibition of insulin secretion (74). These differences between a specific mouse cell line and heterogeneous human islets might be explained by interspecies variation or islet heterogeneity.
MTC specifically regulates the postnatal functional maturation of β-cells. Mice with deletion of Mettl3/14 in Ngn3+ endocrine progenitor cells developed hyperglycemia and hypoinsulinemia 2 weeks after birth. This study also showed that Mettl3/14 deletion silenced the expression of important transcription factors, such as Mafa, Nkx6-1, or Pdx1 (69). Other studies using mouse models with β-cell-specific deletions of MTC subunits (Mettl3, Mettl14, Wtap) also pointed out the importance of MTC in maintaining β-cell function. Deletion of either subunit resulted in decreased m6A levels (65, 70, 75). METTL3 deficiency led to β-cell failure and hyperglycemia (75). METTL14-deficient mice exhibited decreased β-cell mass, reduced insulin secretion, and glucose intolerance (65, 76, 77). Deficiency of WTAP was associated with a reduction of METTL3 levels and resulted in severe hyperglycemia and β-cell failure. Overexpression of Mettl3 in β-cells partially prevented the negative effects of WTAP deficiency (70). Comparing Mettl3-βKO and Wtap-βKO mice revealed down-regulation of β-cell-specific transcription factors (such as Mafa, Nkx6-1, Pdx1, Neurod1, or Foxa2) and insulin secretion-related genes (such as Ins1, Ins2, Brsk2, Cacna1c, Doc2b, Ffar1, G6pc2, Gck, Gipr, Hadh, Ica1, Nnat, Park7, Pclo, Selenot, Serp1, Slc30a8, Stxbp51, Sytl4, Trpm2, Ucn3, and Uqcc2) (70). Besides methyltransferases, also β-cell-specific deletion of reader Ythdc1 resulted in β-cell failure and diabetes (71, 78). This was likely due to the decreased gene expression of β-cell-specific transcription factors (such as Mafa, Nkx6-1, Neurod1, and Hmgn3) and insulin-related genes (such as Ins1, Ins2, Gck, G6pc2, Sytl4, Doc2b, Pclo, Cacna1c, Slc30a8, Ffar1, Gipr, Nnat, and Selenot). Transcription factor MAFA decreased dramatically also on protein level in Ythdc1-βKO islets (71). Yang et al. suggested that YTHDC1 may regulate mRNA splicing and export to modulate glucose metabolism in β-cells by interacting with serine/arginine-rich splicing factor 3 (SRSF3) and cleavage and polyadenylation specific factor 6 (CPSF6) (78).
These data indicate that m6A/m6Am epitranscriptomic machinery vastly affects the biology of pancreatic β-cells and plays a role in the induction of diabetic phenotype. However, the data are still fragmental, and more studies covering more m6A/m6Am regulators are needed to elucidate the exact role of epitranscriptomic regulations in the diabetic pancreas.
4.3 Heart
Cardiovascular disease (CVD) is a common comorbidity and a major cause of mortality among people with T2DM. More than 30% of all T2DM patients are affected by CVD (79). Cardiac dysfunction observed in patients with diabetes that occurs in the absence of other cardiovascular risk factors (such as hypertension, coronary artery disease, or valvular disease) is referred to as diabetic cardiomyopathy (DCM) (80). This condition is characterized by cardiac diastolic dysfunction and later by heart failure (HF) and cardiac death. It is estimated that the risk of HF is 2-3 times higher in individuals with T2DM and that approximately 12% of diabetic patients eventually develop severe HF often leading to death (81). The epitranscriptomic modifications, including m6A, are known to play various roles in the physiology and pathophysiology of the cardiovascular system (20, 82–85). Recent studies have shown that changes in m6A methylation also contribute to HF progression (86–90). However, the role of cardiac m6A and m6Am machinery is not well-characterized in T2DM.
Altered cardiac m6A patterns were detected in db/db mice (model of T2DM and DCM). The differentially methylated transcripts were linked mainly to cardiac fibrosis, myocardial hypertrophy, and myocardial energy metabolism (91). The higher total m6A mass in DCM was associated with the down-regulation of demethylase FTO on both gene and protein levels, while levels of METTL3, METTL14, and ALKBH5 were stable (91). Interestingly, mice with T1DM-induced DCM (C57BL/6 mice injected with streptozotocin) exhibited a different dysregulation of epitranscriptomic machinery (Figure 3). Total m6A levels in the hearts of these mice were decreased. This was linked with an increase of ALKBH5 in the cardiomyocytes of DCM mice and subsequent activation of the Hippo signaling pathway through a YTHDF2-dependent action (92). These results suggest that the two types of diabetes might affect the epitranscriptomic background of DCM differently. It has been reported already that T1DM and T2DM might affect the heart in a different way and result in dissimilar DCM phenotype. This was explained mainly by the different myocardial insulin action (insulin deficiency in T1DM vs insulin resistance and hyperinsulinemia in T2DM) and thus distinct signaling downstream of the insulin receptor (93). Therefore, the contradictory epitranscriptomic results may be explained by the different phenotype between the two types of diabetes. However, further research is needed to resolve this issue.
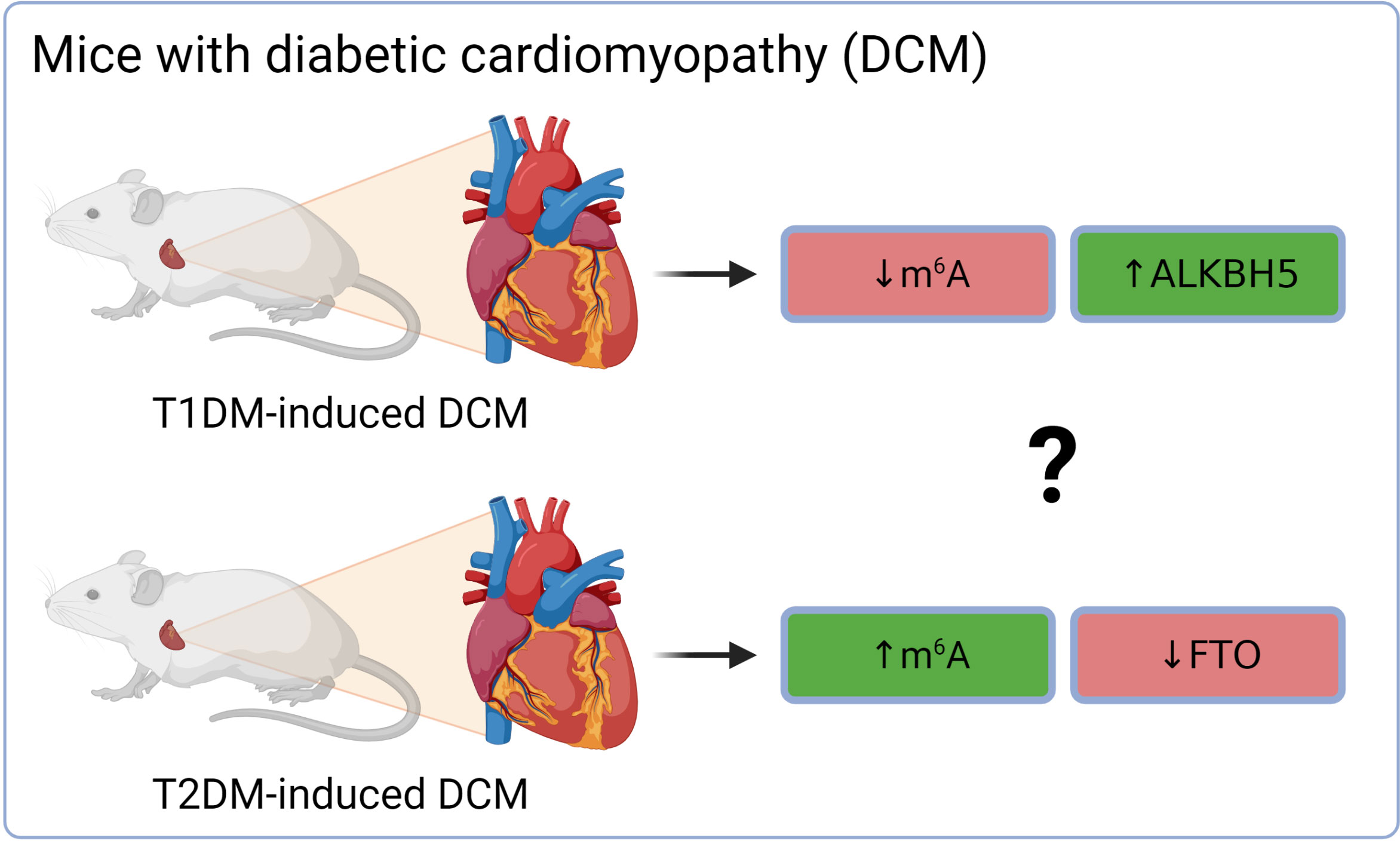
Figure 3 Different epitranscriptomic regulations in DCM on T1DM and T2DM mouse hearts. ALKBH5, AlkB family member 5; FTO, fat mass and obesity-associated; m6A, N6-methyladenosine; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus.
Most of the studies dealing with m6A/m6Am regulations in DCM have been executed on T1DM animal models. Pyroptosis, a type of proinflammatory cell death, is tightly involved in DCM progression. Methyltransferase METTL14 was down-regulated in the hearts of rats with DCM (T1DM-induced) and enhancement of its expression inhibited pyroptosis in myocardial tissues and improved systolic function (increased fractional shortening and ejection fraction) via down-regulation of lncRNA Tincr. The expression of Tincr was regulated in a YTHDF2-dependent manner (94). Peng et al. (95) reported that lncRNA Airn ameliorated diabetes-induced (T1DM) cardiac dysfunction caused by cardiac fibrosis. Their data showed that Airn binds to m6A reader IGF2BP2 and protects it from ubiquitin-proteasome-dependent degradation, leading to an m6A-dependent stabilization of p53 mRNA by IGF2BP2 and subsequent reduction in cardiac fibrosis (95).
Despite the limited amount of data available, it is becoming evident that epitranscriptomic dysregulations in diabetic cardiac tissue might have a significant effect on the function of the heart. However, the exact role of m6A and m6Am in DCM induced by each type of T2DM is yet to be deciphered.
4.4 Kidneys
Diabetic nephropathy (DN), also known as diabetic kidney disease, is a prevalent microvascular complication of T2DM often leading to end-stage renal disease, a life-threatening condition (96). According to the International Diabetes Federation reports, up to 40% of diabetic patients might develop DN (97).
Xu et al. reported, that human kidney 2 (HK-2) cells stimulated with high glucose decreased total m6A methylation level and also methyltransferases METTL3 and METTL14 (98). Interestingly, Jiang et al. observed increased m6A modification in diabetic mice which was caused by elevated levels of METTL3. They also found increased METTL3 levels in renal biopsies from DN patients. Further experiments showed that METTL3 exerted pro-inflammatory and pro-apoptotic effects in an IGF2BP2-dependent manner and that targeting METTL3 alleviated the DN injury (99). A negative effect of METTL3 in DN was reported also by Tang et al. (100). METTL14 was also highly expressed in the kidneys of DN patients and HRGEC (high glucose-induced human renal glomerular endothelial cells). METTL14 worsened renal injury and inflammation was reported in db/db mice (101). Lu et al. also reported high levels of METTL14 in renal biopsy samples from patients with glomerulosclerosis and DN. Mice with podocyte-specific METTL14 deletion were then associated with improved glomerular function and alleviated podocyte injury compared to wild-type nephropathic mice (102). Also the third component of the MTC – WTAP – was reported to induce pyroptosis and inflammation in high glucose-treated HK-2 cells (103). Besides the methyltransferases, FTO was described to promote the progression of DN (104). However, several SNPs in the FTO gene were associated with a significantly lower risk of nephropathy in T2DM patients (62). Urine levels of m6A were decreased in patients with T2DM and even more with DN (105).
The existing data indicate that m6A machinery is affected in DN and that its dysregulation has a negative outcome on the progression of the pathology.
4.5 Liver
Liver disease ranks among notable causes of death in T2DM patients (106). Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease and is strongly associated with T2DM (107–109). The prevalence of this comorbidity among T2DM patients reaches up to 70% (110). It has been described that NAFLD is promoted by m6A modification dysregulation (111–116). Moreover, liver tissues from T2DM patients and mice on HFD showed elevated levels of m6A and also METTL3. Hepatocyte-specific knockout of Mettl3 in mice then led to improved insulin sensitivity and decreased fatty acid synthesis (117). Jiang et al. also reported that baicalin – a flavonoid glycoside used in traditional Chinese medicine – suppressed T2DM-induced liver tumor progression in a METT3/m6A-dependent manner (118).
4.6 Eyes
Chronic exposure to hyperglycemia affects the microvasculature, eventually leading to diabetic retinopathy (DR), the main cause of blindness in the developed world. It has been described that m6A modification is regulated by various risk factors associated with DR, such as inflammation, oxidative stress, angiogenesis, or glucose and lipid metabolism (119). FTO polymorphism (rs8050136) was associated with a higher risk of DR (120). In retinal pigment epithelium (RPE) cells, high-glucose conditions down-regulated the expression of METTL3 on both transcript and protein levels. Further experiments showed that METTL3 overexpression alleviated the cytotoxic effects of high-glucose on RPE cells, while METTL3 depletion had the opposite effect (121). Conversely, diabetic stress-induced up-regulation of METTL3 and subsequent increase of m6A levels in human retinal pericytes and also mouse retinas. Specific depletion of METTL3 in pericytes suppressed diabetes-induced pericyte dysfunction and vascular complication in vivo (122). A recent study showed down-regulation of METTL3 in vitreous humor samples from patients with DR, a mouse model of DR, and also high glucose-induced human retinal microvascular endothelial cells (123).
Despite these conflicting data on METTL3 expression, it seems to be clear that epitranscriptomic regulations are affected in DR, but the exact role of m6A in the pathogenesis remains to be elucidated in the future.
4.7 Skin
Dysregulation of autophagy is a contributing factor for delayed wound healing in diabetic skin. YTHDC1, an m6A reader, has been described as a modulator of autophagy in diabetic keratinocytes which regulates the mRNA stability of an autophagy receptor (124). Interestingly, YTHDC1 interacted and cooperated with ELAVL1 (ELAV-like RNA binding protein 1), a well-established RNA stabilizer also linked to m6A methylation. It has been described previously that loss of m6A methylation enhances ELAVL1 RNA binding to increase RNA stability (125).
4.8 Blood
Decreased m6A methylation levels were detected in RNA isolated from the peripheral blood of T2DM patients and also diabetic rats (126, 127). In accordance with these results, significantly higher gene expression of FTO (and not ALKBH5) in peripheral blood from T2DM patients was detected (126). However, Onalan et al. (127) observed an up-regulated expression of both demethylases in venous blood samples from T2DM patients. The increased expression of FTO on both gene and protein levels was later confirmed by another study which pointed out the correlation between high FTO levels and T2DM severity (128). The gene expression of FTO was also up-regulated in white blood cells from T2DM patients compared to healthy individuals and the expression level of FTO was positively correlated with fasting glucose concentration (129). Besides erasers, METTL3 mRNA was down-regulated in serum samples from T2DM patients (121). Progressively higher T2DM risk was associated with low serum IGF2BP3 levels (72).
Taken together, the content of m6A or its regulators in the peripheral blood may serve as novel potential biomarkers of T2DM in the future (126).
4.9 Treatment of T2DM: the role of m6A and m6Am modifications
Metformin is the first-line therapy for the treatment of T2DM, yet its molecular mechanisms of action are not fully understood (130, 131). The main effect of metformin treatment is inhibition of hepatic gluconeogenesis. At the molecular level, several mechanisms have been proposed to explain this phenomenon, such as inhibition of mitochondrial complex I activity, activation of AMPK, or increase in hepatocellular redox state due to inhibition of GPD2 (glycerol-3-phosphate dehydrogenase 2). The secondary effects of metformin treatment include an increase in muscle glucose uptake, a decrease in intestinal glucose absorption, and a change in the composition of the gut microbiome (130).
According to recent studies, metformin also affects epitranscriptomic regulations, including m6A machinery. Metformin was shown to reduce m6A methylation via the down-regulation of methyltransferase METTL3 in breast cancer cells (132). In hepatocellular carcinoma, metformin treatment was associated with METTL3 inhibition (133). Metformin also attenuated multiple myeloma cell proliferation and encouraged apoptosis by suppressing METTL3-mediated m6A methylation of its targets (134). Surprisingly, METTL3 expression was up-regulated after metformin treatment in adenocarcinoma cells (135). YTHDC2, a key m6A reader, is an important target of metformin in preventing the progression of vascular smooth muscle cell (VSMC) dysfunction under high glucose, a simulation of VSMC dysfunction caused by T2DM (136). Recently, Liao et al. (137) showed that metformin combats obesity by targeting FTO in an m6A-YTHDF2-dependent manner. This study suggests that metformin inhibited the protein expression of FTO, resulting in higher m6A methylation in mRNAs of crucial cell cycle regulators. The binding of YTHDF2 to modified transcripts then triggered mRNA decay and subsequent decrease of protein expression. In consequence, the mitotic clonal expansion process was blocked and adipogenesis was inhibited.
This fragmentary information suggests that metformin may both decrease and increase m6A methylation and that the target tissue or cell type may be the determining factor. However, in vivo studies focusing on the epitranscriptomic effect of metformin are needed to decipher this phenomenon, as the in vivo and in vitro response may also differ, especially if the primary target of metformin treatment is the liver. Despite these ambiguities, the association between epitranscriptomics and metformin is revealing itself, however, the role of m6A modification in the treatment of diabetes remains unclear.
5 Conclusion and perspectives
The significant role of epitranscriptomics in cellular physiology and pathophysiology has been widely accepted by the scientific community in the past few years. However, despite the increased interest of researchers in RNA modifications, the complex epitranscriptomic regulations are still not fully understood. Our review focused on two of the most prevalent modifications – m6A and m6Am – in the pathogenesis of T2DM. The fragmental current knowledge indicates that diabetic tissues are associated with the dysregulation of epitranscriptomic machinery (summarized in Figure 4). However, it is essential to correctly distinguish whether these dysregulations contribute to the development of the disease or are merely a consequence of it. Several studies already showed that a deficiency of epitranscriptomic regulators can promote the pathological conditions typical for T2DM. Thus, targeting the epitranscriptomic regulations might have future applications in the clinic and consequently reduce the morbidity and mortality of T2DM patients.
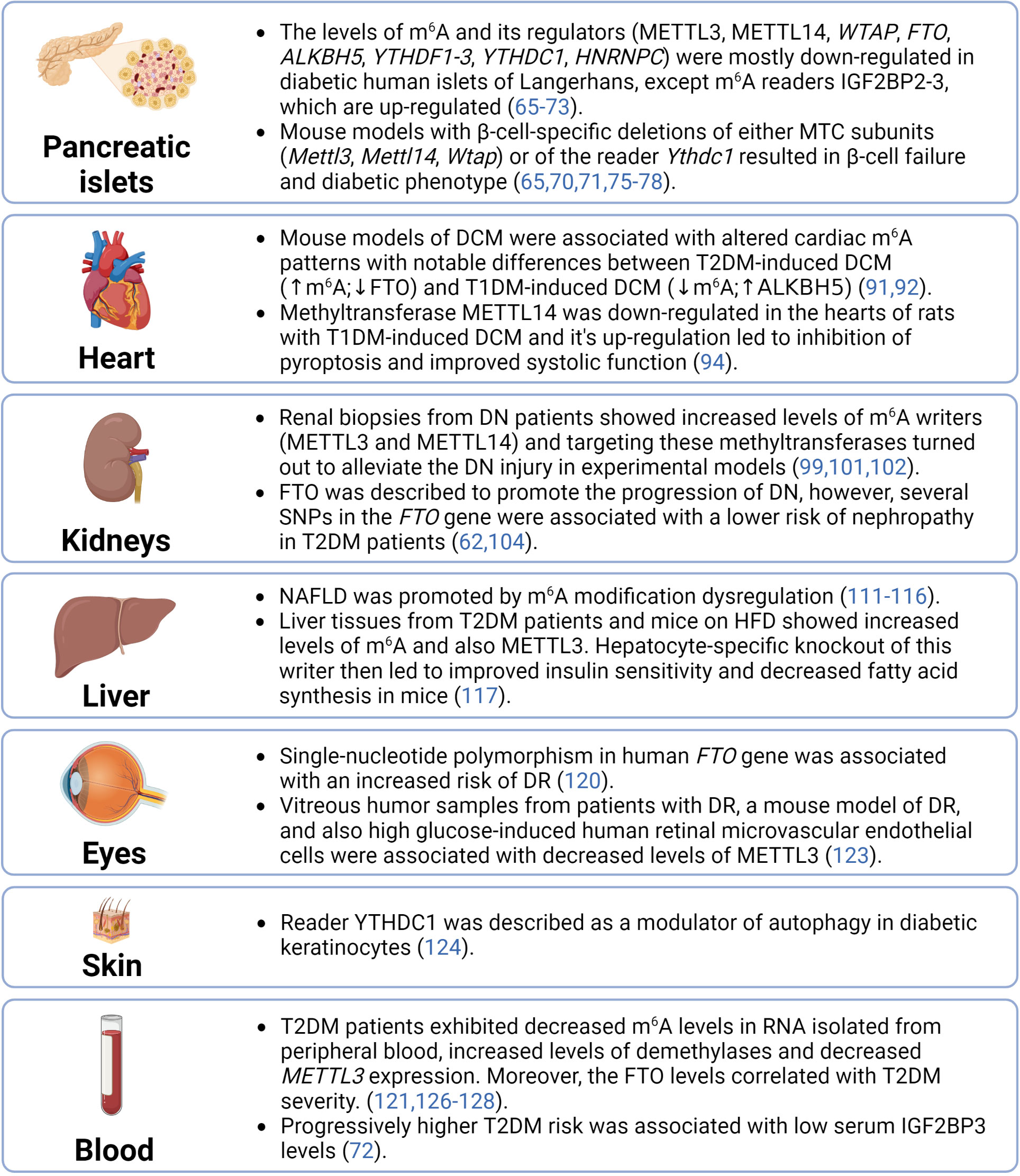
Figure 4 Summary of epitranscriptomic regulations in diabetic tissues. ALKBH5, AlkB family member 5; DCM, diabetic cardiomyopathy; DN, diabetic nephrophathy; DR, diabetic retinopathy; FTO, fat mass and obesity-associated; HFD, high-fat diet; HNRNPC, heterogeneous nuclear ribonucleoprotein C; IGF2BP2-3, insulin-like growth factor 2 mRNA binding proteins 2-3; m6A, N6-methyladenosine; METTL14, methyltransferase-like 14; METTL3, methyltransferase-like 3; MTC, multicomponent methyltransferase complex; NAFLD, non-alcoholic fatty liver disease; SNPs, single-nucleotide polymorphisms; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; WTAP, Willms’ tumor 1-associating protein; YTHDC1, YTH domain-containing protein 1; YTHDF1-3, YTH domain-containing family proteins 1-3.
Author contributions
DB and SB drafted the article, LP-H and MH provided substantive revisions. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Charles University Grant Agency (grant number GA UK 243423) to SB; the Czech Science Foundation (grant number 19-04790Y) to MH; the Czech Science Foundation (grant number 22-11439S) to LP-H; and the project National Institute for Research of Metabolic and Cardiovascular Diseases (Programme EXCELES, ID project No. LX22NPO5104) – Funded by the European Union – Next Generation EU.
Acknowledgments
Figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. WHO. Diabetes (2022). Available at: https://www.who.int/news-room/fact-sheets/detail/diabetes.
2. Goyal R, Jialal I. Diabetes mellitus type 2. Treasure Island, Florida StatPearls Publishing (2023).
3. Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, et al. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci (2020) 21(17):6275. doi: 10.3390/ijms21176275
4. Malone JI, Hansen BC. Does obesity cause type 2 diabetes mellitus (T2DM)? or is it the opposite? Pediatr Diabetes (2019) 20(1):5–9. doi: 10.1111/pedi.12787
5. Ottosson-Laakso E, Krus U, Storm P, Prasad RB, Oskolkov N, Ahlqvist E, et al. Glucose-induced changes in gene expression in human pancreatic islets: causes or consequences of chronic hyperglycemia. Diabetes (2017) 66(12):3013–28. doi: 10.2337/db17-0311
6. Lother A, Bondareva O, Saadatmand AR, Pollmeier L, Härdtner C, Hilgendorf I, et al. Diabetes changes gene expression but not DNA methylation in cardiac cells. J Mol Cell Cardiol (2021) 151:74–87. doi: 10.1016/j.yjmcc.2020.11.004
7. Patti ME. Gene expression in the pathophysiology of type 2 diabetes mellitus. Curr Diabetes Rep (2004) 4(3):176–81. doi: 10.1007/s11892-004-0020-x
8. Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res (2018) 46(D1):D303–d7. doi: 10.1093/nar/gkx1030
9. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from novikoff hepatoma cells. Proc Natl Acad Sci USA (1974) 71(10):3971–5. doi: 10.1073/pnas.71.10.3971
10. Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N, Rechavi G. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat Protoc (2013) 8(1):176–89. doi: 10.1038/nprot.2012.148
11. Wei C, Gershowitz A, Moss B. N6, O2'-dimethyladenosine a novel methylated ribonucleoside next to the 5' terminal of animal cell and virus mRNAs. Nature (1975) 257(5523):251–3. doi: 10.1038/257251a0
12. Bokar JA. The biosynthesis and functional roles of methylated nucleosides in eukaryotic mRNA. In: Grosjean H, editor. Fine-tuning of RNA functions by modification and editing. Berlin, Heidelberg: Springer Berlin Heidelberg (2005). p. 141–77.
13. Lee Y, Choe J, Park OH, Kim YK. Molecular mechanisms driving mRNA degradation by m(6)A modification. Trends Genet (2020) 36(3):177–88. doi: 10.1016/j.tig.2019.12.007
14. Boo SH, Kim YK. The emerging role of RNA modifications in the regulation of mRNA stability. Exp Mol Med (2020) 52(3):400–8. doi: 10.1038/s12276-020-0407-z
15. Oerum S, Meynier V, Catala M, Tisné C. A comprehensive review of m6A/m6Am RNA methyltransferase structures. Nucleic Acids Res (2021) 49(13):7239–55. doi: 10.1093/nar/gkab378
16. Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell (2016) 63(2):306–17. doi: 10.1016/j.molcel.2016.05.041
17. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature (2016) 534(7608):575–8. doi: 10.1038/nature18298
18. Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res (2014) 24(2):177–89. doi: 10.1038/cr.2014.3
19. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol (2011) 7(12):885–7. doi: 10.1038/nchembio.687
20. Dieterich C, Völkers M. Chapter 6 - RNA modifications in cardiovascular disease–an experimental and computational perspective. In: Devaux Y, Robinson EL, editors. Epigenetics in cardiovascular disease. London, United Kingdom: Academic Press (2021). p. 113–25.
21. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell (2013) 49(1):18–29. doi: 10.1016/j.molcel.2012.10.015
22. Zaccara S, Jaffrey SR. A unified model for the function of YTHDF proteins in regulating m(6)A-modified mRNA. Cell (2020) 181(7):1582–95.e18. doi: 10.1016/j.cell.2020.05.012
23. Lasman L, Krupalnik V, Viukov S, Mor N, Aguilera-Castrejon A, Schneir D, et al. Context-dependent functional compensation between ythdf m6A reader proteins. Genes Dev (2020) 34(19-20):1373–91. doi: 10.1101/gad.340695.120
24. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature (2014) 505(7481):117–20. doi: 10.1038/nature12730
25. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell (2015) 161(6):1388–99. doi: 10.1016/j.cell.2015.05.014
26. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell (2016) 61(4):507–19. doi: 10.1016/j.molcel.2016.01.012
27. Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res (2017) 27(9):1115–27. doi: 10.1038/cr.2017.99
28. Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res (2017) 27(3):315–28. doi: 10.1038/cr.2017.15
29. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol (2018) 20(3):285–95. doi: 10.1038/s41556-018-0045-z
30. Akichika S, Hirano S, Shichino Y, Suzuki T, Nishimasu H, Ishitani R, et al. Cap-specific terminal n (6)-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science (2019) 363(6423):eaav0080. doi: 10.1126/science.aav0080
31. Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible methylation of m(6)A(m) in the 5' cap controls mRNA stability. Nature (2017) 541(7637):371–5. doi: 10.1038/nature21022
32. Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur JJ, et al. FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat Chem Biol (2019) 15(4):340–7. doi: 10.1038/s41589-019-0231-8
33. Sun H, Zhang M, Li K, Bai D, Yi C. Cap-specific, terminal N(6)-methylation by a mammalian m(6)Am methyltransferase. Cell Res (2019) 29(1):80–2. doi: 10.1038/s41422-018-0117-4
34. Yu D, Dai N, Wolf EJ, Corrêa IR Jr., Zhou J, Wu T, et al. Enzymatic characterization of mRNA cap adenosine-N6 methyltransferase PCIF1 activity on uncapped RNAs. J Biol Chem (2022) 298(4):101751. doi: 10.1016/j.jbc.2022.101751
35. Liu A, Desai BM, Stoffers DA. Identification of PCIF1, a POZ domain protein that inhibits PDX-1 (MODY4) transcriptional activity. Mol Cell Biol (2004) 24(10):4372–83. doi: 10.1128/MCB.24.10.4372-4383.2004
36. Claiborn KC, Sachdeva MM, Cannon CE, Groff DN, Singer JD, Stoffers DA. Pcif1 modulates Pdx1 protein stability and pancreatic β cell function and survival in mice. J Clin Invest (2010) 120(10):3713–21. doi: 10.1172/JCI40440
37. Chen H, Gu L, Orellana EA, Wang Y, Guo J, Liu Q, et al. METTL4 is an snRNA m(6)Am methyltransferase that regulates RNA splicing. Cell Res (2020) 30(6):544–7. doi: 10.1038/s41422-019-0270-4
38. Goh YT, Koh CWQ, Sim DY, Roca X, Goh WSS. METTL4 catalyzes m6Am methylation in U2 snRNA to regulate pre-mRNA splicing. Nucleic Acids Res (2020) 48(16):9250–61. doi: 10.1093/nar/gkaa684
39. Mauer J, Jaffrey SR. FTO, m6Am, and the hypothesis of reversible epitranscriptomic mRNA modifications. FEBS Lett (2018) 592(12):2012–22. doi: 10.1002/1873-3468.13092
40. Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. Differential m(6)A, m(6)A(m), and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell (2018) 71(6):973–85.e5. doi: 10.1016/j.molcel.2018.08.011
41. Relier S, Ripoll J, Guillorit H, Amalric A, Achour C, Boissière F, et al. FTO-mediated cytoplasmic m(6)A(m) demethylation adjusts stem-like properties in colorectal cancer cell. Nat Commun (2021) 12(1):1716. doi: 10.1038/s41467-021-21758-4
42. Benak D, Kolar F, Zhang L, Devaux Y, Hlavackova M. RNA Modification m(6)Am: the role in cardiac biology. Epigenetics (2023) 18(1):2218771. doi: 10.1080/15592294.2023.2218771
43. Ali O. Genetics of type 2 diabetes. World J Diabetes (2013) 4(4):114–23. doi: 10.4239/wjd.v4.i4.114
44. Hubacek JA, Dlouha L, Adamkova V, Dlouha D, Pacal L, Kankova K, et al. Genetic risk score is associated with T2DM and diabetes complications risks. Gene (2023) 849:146921. doi: 10.1016/j.gene.2022.146921
45. Yang Y, Liu B, Xia W, Yan J, Liu HY, Hu L, et al. FTO genotype and type 2 diabetes mellitus: spatial analysis and meta-analysis of 62 case-control studies from different regions. Genes (Basel) (2017) 8(2):70. doi: 10.3390/genes8020070
46. Hubáček JA, Šedová L, Olišarová V, Adámková V, Tóthová V. Different prevalence of T2DM risk alleles in Roma population in comparison with the majority Czech population. Mol Genet Genomic Med (2020) 8(9):e1361. doi: 10.1002/mgg3.1361
47. Sabarneh A, Ereqat S, Cauchi S, AbuShamma O, Abdelhafez M, Ibrahim M, et al. Common FTO rs9939609 variant and risk of type 2 diabetes in Palestine. BMC Med Genet (2018) 19(1):156. doi: 10.1186/s12881-018-0668-8
48. Sarkar P, Chatterjee D, Bandyopadhyay AR. Effect of MTHFR (rs1801133) and FTO (rs9939609) genetic polymorphisms and obesity in T2DM: a study among bengalee Hindu caste population of West Bengal, India. Ann Hum Biol (2021) 48(1):62–5. doi: 10.1080/03014460.2021.1876920
49. Bakhashab S, Filimban N, Altall RM, Nassir R, Qusti SY, Alqahtani MH, et al. The effect sizes of PPARγ rs1801282, FTO rs9939609, and MC4R rs2229616 variants on type 2 diabetes mellitus risk among the Western Saudi population: a cross-sectional prospective study. Genes (Basel) (2020) 11(1):98. doi: 10.3390/genes11010098
50. Bazzi MD, Nasr FA, Alanazi MS, Alamri A, Turjoman AA, Moustafa AS, et al. MC4R, SLC30A8, and KCNQ1 gene variants and type 2 diabetes in Saudi population. Genet Mol Res (2014) 13(4):10194–203. doi: 10.4238/2014.December.4.14
51. Younus LA, Algenabi AHA, Abdul-Zhara MS, Hussein MK. FTO gene polymorphisms (rs9939609 and rs17817449) as predictors of type 2 diabetes mellitus in obese Iraqi population. Gene (2017) 627:79–84. doi: 10.1016/j.gene.2017.06.005
52. Nasser FA, Algenabi AA, Hadi NR, Hussein MK, Fatima G, Al-Aubaidy HA. The association of the common fat mass and obesity associated gene polymorphisms with type 2 diabetes in obese Iraqi population. Diabetes Metab Syndr (2019) 13(4):2451–5. doi: 10.1016/j.dsx.2019.06.024
53. Chauhan G, Tabassum R, Mahajan A, Dwivedi OP, Mahendran Y, Kaur I, et al. Common variants of FTO and the risk of obesity and type 2 diabetes in indians. J Hum Genet (2011) 56(10):720–6. doi: 10.1038/jhg.2011.87
54. Bressler J, Kao WH, Pankow JS, Boerwinkle E. Risk of type 2 diabetes and obesity is differentially associated with variation in FTO in whites and African-americans in the ARIC study. PloS One (2010) 5(5):e10521. doi: 10.1371/journal.pone.0010521
55. Sanghera DK, Ortega L, Han S, Singh J, Ralhan SK, Wander GS, et al. Impact of nine common type 2 diabetes risk polymorphisms in Asian Indian sikhs: PPARG2 (Pro12Ala), IGF2BP2, TCF7L2 and FTO variants confer a significant risk. BMC Med Genet (2008) 9:59. doi: 10.1186/1471-2350-9-59
56. Vasan SK, Karpe F, Gu HF, Brismar K, Fall CH, Ingelsson E, et al. FTO genetic variants and risk of obesity and type 2 diabetes: a meta-analysis of 28,394 indians. Obes (Silver Spring) (2014) 22(3):964–70. doi: 10.1002/oby.20606
57. Yajnik CS, Janipalli CS, Bhaskar S, Kulkarni SR, Freathy RM, Prakash S, et al. FTO gene variants are strongly associated with type 2 diabetes in south Asian indians. Diabetologia (2009) 52(2):247–52. doi: 10.1007/s00125-008-1186-6
58. Hubacek JA, Dlouha D, Klementova M, Lanska V, Neskudla T, Pelikanova T. The FTO variant is associated with chronic complications of diabetes mellitus in Czech population. Gene (2018) 642:220–4. doi: 10.1016/j.gene.2017.11.040
59. Ghafarian-Alipour F, Ziaee S, Ashoori MR, Zakeri MS, Boroumand MA, Aghamohammadzadeh N, et al. Association between FTO gene polymorphisms and type 2 diabetes mellitus, serum levels of apelin and androgen hormones among Iranian obese women. Gene (2018) 641:361–6. doi: 10.1016/j.gene.2017.10.082
60. McFadden MJ, Sacco MT, Murphy KA, Park M, Gokhale NS, Somfleth KY, et al. FTO suppresses STAT3 activation and modulates proinflammatory interferon-stimulated gene expression. J Mol Biol (2022) 434(6):167247. doi: 10.1016/j.jmb.2021.167247
61. Han L, Li Y, Tang L, Chen Z, Zhang T, Chen S, et al. IGF2BP2 rs11705701 polymorphisms are associated with prediabetes in a Chinese population: a population-based case-control study. Exp Ther Med (2016) 12(3):1849–56. doi: 10.3892/etm.2016.3554
62. Montesanto A, Bonfigli AR, Crocco P, Garagnani P, De Luca M, Boemi M, et al. Genes associated with type 2 diabetes and vascular complications. Aging (Albany NY) (2018) 10(2):178–96. doi: 10.18632/aging.101375
63. Eizirik DL, Pasquali L, Cnop M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol (2020) 16(7):349–62. doi: 10.1038/s41574-020-0355-7
64. De Jesus DF, Kulkarni RN. Epigenetic modifiers of islet function and mass. Trends Endocrinol Metab (2014) 25(12):628–36. doi: 10.1016/j.tem.2014.08.006
65. De Jesus DF, Zhang Z, Kahraman S, Brown NK, Chen M, Hu J, et al. m(6)A mRNA methylation regulates human β-cell biology in physiological states and in type 2 diabetes. Nat Metab (2019) 1(8):765–74. doi: 10.1038/s42255-019-0089-9
66. Bornaque F, Delannoy CP, Courty E, Rabhi N, Carney C, Rolland L, et al. Glucose regulates m(6)A methylation of RNA in pancreatic islets. Cells (2022) 11(2):291. doi: 10.3390/cells11020291
67. Taneera J, Prasad RB, Dhaiban S, Mohammed AK, Haataja L, Arvan P, et al. Silencing of the FTO gene inhibits insulin secretion: an in vitro study using GRINCH cells. Mol Cell Endocrinol (2018) 472:10–7. doi: 10.1016/j.mce.2018.06.003
68. Kirkpatrick CL, Marchetti P, Purrello F, Piro S, Bugliani M, Bosco D, et al. Type 2 diabetes susceptibility gene expression in normal or diabetic sorted human alpha and beta cells: correlations with age or BMI of islet donors. PloS One (2010) 5(6):e11053. doi: 10.1371/journal.pone.0011053
69. Wang Y, Sun J, Lin Z, Zhang W, Wang S, Wang W, et al. m(6)A mRNA methylation controls functional maturation in neonatal murine β-cells. Diabetes (2020) 69(8):1708–22. doi: 10.2337/db19-0906
70. Li X, Yang Y, Li Z, Wang Y, Qiao J, Chen Z. Deficiency of WTAP in islet beta cells results in beta cell failure and diabetes in mice. Diabetologia (2023) 66(6):1084–96. doi: 10.1007/s00125-023-05900-z
71. Li X, Yang Y, Chen Z. Downregulation of the m(6)A reader protein YTHDC1 leads to islet β-cell failure and diabetes. Metabolism (2023) 138:155339. doi: 10.1016/j.metabol.2022.155339
72. Wu X, Wang W, Fan S, You L, Li F, Zhang X, et al. U-Shaped association between serum IGF2BP3 and T2DM: a cross-sectional study in Chinese population. J Diabetes (2023) 15(4):349–61. doi: 10.1111/1753-0407.13378
73. Marselli L, Thorne J, Dahiya S, Sgroi DC, Sharma A, Bonner-Weir S, et al. Gene expression profiles of beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PloS One (2010) 5(7):e11499. doi: 10.1371/journal.pone.0011499
74. Fan HQ, He W, Xu KF, Wang ZX, Xu XY, Chen H. FTO inhibits insulin secretion and promotes NF-κB activation through positively regulating ROS production in pancreatic β cells. PloS One (2015) 10(5):e0127705. doi: 10.1371/journal.pone.0127705
75. Li X, Jiang Y, Sun X, Wu Y, Chen Z. METTL3 is required for maintaining β-cell function. Metabolism (2021) 116:154702. doi: 10.1016/j.metabol.2021.154702
76. Liu J, Luo G, Sun J, Men L, Ye H, He C, et al. METTL14 is essential for β-cell survival and insulin secretion. Biochim Biophys Acta Mol Basis Dis (2019) 1865(9):2138–48. doi: 10.1016/j.bbadis.2019.04.011
77. Men L, Sun J, Luo G, Ren D. Acute deletion of METTL14 in β-cells of adult mice results in glucose intolerance. Endocrinology (2019) 160(10):2388–94. doi: 10.1210/en.2019-00350
78. Yang K, Sun J, Zhang Z, Xiao M, Ren D, Liu SM. Reduction of mRNA m(6)A associates with glucose metabolism via YTHDC1 in human and mice. Diabetes Res Clin Pract (2023) 198:110607. doi: 10.1016/j.diabres.2023.110607
79. Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007-2017. Cardiovasc Diabetol (2018) 17(1):83. doi: 10.1186/s12933-018-0728-6
80. Paolillo S, Marsico F, Prastaro M, Renga F, Esposito L, De Martino F, et al. Diabetic cardiomyopathy: definition, diagnosis, and therapeutic implications. Heart Fail Clin (2019) 15(3):341–7. doi: 10.1016/j.hfc.2019.02.003
81. Geng X, Li Z, Yang Y. Emerging role of epitranscriptomics in diabetes mellitus and its complications. Front Endocrinol (Lausanne) (2022) 13:907060. doi: 10.3389/fendo.2022.907060
82. Longenecker JZ, Gilbert CJ, Golubeva VA, Martens CR, Accornero F. Epitranscriptomics in the heart: a focus on m(6)A. Curr Heart Fail Rep (2020) 17(5):205–12. doi: 10.1007/s11897-020-00473-z
83. Wu S, Zhang S, Wu X, Zhou X. m(6)A RNA methylation in cardiovascular diseases. Mol Ther (2020) 28(10):2111–9. doi: 10.1016/j.ymthe.2020.08.010
84. Kumari R, Ranjan P, Suleiman ZG, Goswami SK, Li J, Prasad R, et al. mRNA modifications in cardiovascular biology and disease: with a focus on m6A modification. Cardiovasc Res (2022) 118(7):1680–92. doi: 10.1093/cvr/cvab160
85. Semenovykh D, Benak D, Holzerova K, Cerna B, Telensky P, Vavrikova T, et al. Myocardial m6A regulators in postnatal development: effect of sex. Physiol Res (2022) 71(6):877–82. doi: 10.33549/physiolres.934970
86. Kmietczyk V, Riechert E, Kalinski L, Boileau E, Malovrh E, Malone B, et al. m(6)A-mRNA methylation regulates cardiac gene expression and cellular growth. Life Sci Alliance (2019) 2(2):e201800233. doi: 10.26508/lsa.201800233
87. Mathiyalagan P, Adamiak M, Mayourian J, Sassi Y, Liang Y, Agarwal N, et al. FTO-dependent N(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation (2019) 139(4):518–32. doi: 10.1161/CIRCULATIONAHA.118.033794
88. Berulava T, Buchholz E, Elerdashvili V, Pena T, Islam MR, Lbik D, et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur J Heart Fail (2020) 22(1):54–66. doi: 10.1002/ejhf.1672
89. Zhang B, Xu Y, Cui X, Jiang H, Luo W, Weng X, et al. Alteration of m6A RNA methylation in heart failure with preserved ejection fraction. Front Cardiovasc Med (2021) 8:647806. doi: 10.3389/fcvm.2021.647806
90. Zhang B, Jiang H, Wu J, Cai Y, Dong Z, Zhao Y, et al. m6A demethylase FTO attenuates cardiac dysfunction by regulating glucose uptake and glycolysis in mice with pressure overload-induced heart failure. Signal Transduct Target Ther (2021) 6(1):377. doi: 10.1038/s41392-021-00699-w
91. Ju W, Liu K, Ouyang S, Liu Z, He F, Wu J. Changes in N6-methyladenosine modification modulate diabetic cardiomyopathy by reducing myocardial fibrosis and myocyte hypertrophy. Front Cell Dev Biol (2021) 9:702579. doi: 10.3389/fcell.2021.702579
92. Shao Y, Li M, Yu Q, Gong M, Wang Y, Yang X, et al. CircRNA CDR1as promotes cardiomyocyte apoptosis through activating hippo signaling pathway in diabetic cardiomyopathy. Eur J Pharmacol (2022) 922:174915. doi: 10.1016/j.ejphar.2022.174915
93. Hölscher ME, Bode C, Bugger H. Diabetic cardiomyopathy: does the type of diabetes matter? Int J Mol Sci (2016) 17(12):2136. doi: 10.3390/ijms17122136
94. Meng L, Lin H, Huang X, Weng J, Peng F, Wu S. METTL14 suppresses pyroptosis and diabetic cardiomyopathy by downregulating TINCR lncRNA. Cell Death Dis (2022) 13(1):38. doi: 10.1038/s41419-021-04484-z
95. Peng T, Liu M, Hu L, Guo D, Wang D, Qi B, et al. LncRNA airn alleviates diabetic cardiac fibrosis by inhibiting activation of cardiac fibroblasts via a m6A-IMP2-p53 axis. Biol Direct (2022) 17(1):32. doi: 10.1186/s13062-022-00346-6
96. Faselis C, Katsimardou A, Imprialos K, Deligkaris P, Kallistratos M, Dimitriadis K. Microvascular complications of type 2 diabetes mellitus. Curr Vasc Pharmacol (2020) 18(2):117–24. doi: 10.2174/1570161117666190502103733
97. Natesan V, Kim SJ. Diabetic nephropathy - a review of risk factors, progression, mechanism, and dietary management. Biomol Ther (Seoul) (2021) 29(4):365–72. doi: 10.4062/biomolther.2020.204
98. Xu Z, Jia K, Wang H, Gao F, Zhao S, Li F, et al. METTL14-regulated PI3K/Akt signaling pathway via PTEN affects HDAC5-mediated epithelial-mesenchymal transition of renal tubular cells in diabetic kidney disease. Cell Death Dis (2021) 12(1):32. doi: 10.1038/s41419-020-03312-0
99. Jiang L, Liu X, Hu X, Gao L, Zeng H, Wang X, et al. METTL3-mediated m(6)A modification of TIMP2 mRNA promotes podocyte injury in diabetic nephropathy. Mol Ther (2022) 30(4):1721–40. doi: 10.1016/j.ymthe.2022.01.002
100. Tang W, Zhao Y, Zhang H, Peng Y, Rui Z. METTL3 enhances NSD2 mRNA stability to reduce renal impairment and interstitial fibrosis in mice with diabetic nephropathy. BMC Nephrol (2022) 23(1):124. doi: 10.1186/s12882-022-02753-3
101. Li M, Deng L, Xu G. METTL14 promotes glomerular endothelial cell injury and diabetic nephropathy via m6A modification of α-klotho. Mol Med (2021) 27(1):106. doi: 10.1186/s10020-021-00365-5
102. Lu Z, Liu H, Song N, Liang Y, Zhu J, Chen J, et al. METTL14 aggravates podocyte injury and glomerulopathy progression through N(6)-methyladenosine-dependent downregulating of Sirt1. Cell Death Dis (2021) 12(10):881. doi: 10.1038/s41419-021-04156-y
103. Lan J, Xu B, Shi X, Pan Q, Tao Q. WTAP-mediated N(6)-methyladenosine modification of NLRP3 mRNA in kidney injury of diabetic nephropathy. Cell Mol Biol Lett (2022) 27(1):51. doi: 10.1186/s11658-022-00350-8
104. Sun Q, Geng H, Zhao M, Li Y, Chen X, Sha Q, et al. FTO-mediated m(6) a modification of SOCS1 mRNA promotes the progression of diabetic kidney disease. Clin Transl Med (2022) 12(6):e942. doi: 10.1002/ctm2.942
105. Wan SJ, Hua Q, Xing YJ, Cheng Y, Zhou SM, Sun Y, et al. Decreased urine N6-methyladenosine level is closely associated with the presence of diabetic nephropathy in type 2 diabetes mellitus. Front Endocrinol (Lausanne) (2022) 13:986419. doi: 10.3389/fendo.2022.986419
106. Tolman KG, Fonseca V, Dalpiaz A, Tan MH. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care (2007) 30(3):734–43. doi: 10.2337/dc06-1539
107. Loria P, Lonardo A, Anania F. Liver and diabetes. A vicious circle. Hepatol Res (2013) 43(1):51–64. doi: 10.1111/j.1872-034X.2012.01031.x
108. Powell EE, Wong VW, Rinella M. Non-alcoholic fatty liver disease. Lancet (2021) 397(10290):2212–24. doi: 10.1016/S0140-6736(20)32511-3
109. Akshintala D, Chugh R, Amer F, Cusi K. Nonalcoholic fatty liver disease: the overlooked complication of type 2 diabetes. Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, editors. South Dartmouth (MA: MDText.com, Inc) (2000).
110. Anstee QM, McPherson S, Day CP. How big a problem is non-alcoholic fatty liver disease? Bmj (2011) 343:d3897. doi: 10.1136/bmj.d3897
111. Yang Y, Cai J, Yang X, Wang K, Sun K, Yang Z, et al. Dysregulated m6A modification promotes lipogenesis and development of non-alcoholic fatty liver disease and hepatocellular carcinoma. Mol Ther (2022) 30(6):2342–53. doi: 10.1016/j.ymthe.2022.02.021
112. Tang J, Zhao X, Wei W, Liu W, Fan H, Liu XP, et al. METTL16-mediated translation of CIDEA promotes non-alcoholic fatty liver disease progression via m6A-dependent manner. PeerJ (2022) 10:e14379. doi: 10.7717/peerj.14379
113. Peng Z, Gong Y, Wang X, He W, Wu L, Zhang L, et al. METTL3-m(6)A-Rubicon axis inhibits autophagy in nonalcoholic fatty liver disease. Mol Ther (2022) 30(2):932–46. doi: 10.1016/j.ymthe.2021.09.016
114. Chen X, Gao Y, Yang X, Zhang H, Mo Z, Tan A. Relationship of FTO gene variations with NAFLD risk in Chinese men. Open Life Sci (2020) 15(1):860–7. doi: 10.1515/biol-2020-0081
115. Cheng W, Li M, Zhang L, Zhou C, Yu S, Peng X, et al. New roles of N6-methyladenosine methylation system regulating the occurrence of non-alcoholic fatty liver disease with N6-methyladenosine-modified MYC. Front Pharmacol (2022) 13:973116. doi: 10.3389/fphar.2022.973116
116. Wei X, Zhang J, Tang M, Wang X, Fan N, Peng Y. Fat mass and obesity-associated protein promotes liver steatosis by targeting PPARα. Lipids Health Dis (2022) 21(1):29. doi: 10.1186/s12944-022-01640-y
117. Xie W, Ma LL, Xu YQ, Wang BH, Li SM. METTL3 inhibits hepatic insulin sensitivity via N6-methyladenosine modification of fasn mRNA and promoting fatty acid metabolism. Biochem Biophys Res Commun (2019) 518(1):120–6. doi: 10.1016/j.bbrc.2019.08.018
118. Jiang H, Yao Q, An Y, Fan L, Wang J, Li H. Baicalin suppresses the progression of type 2 diabetes-induced liver tumor through regulating METTL3/m(6)A/HKDC1 axis and downstream p-JAK2/STAT1/clevaged Capase3 pathway. Phytomedicine (2022) 94:153823. doi: 10.1016/j.phymed.2021.153823
119. Kumari N, Karmakar A, Ahamad Khan MM, Ganesan SK. The potential role of m6A RNA methylation in diabetic retinopathy. Exp Eye Res (2021) 208:108616. doi: 10.1016/j.exer.2021.108616
120. Hsiao YT, Shen FC, Weng SW, Wang PW, Chen YJ, Lee JJ. Multiple single nucleotide polymorphism testing improves the prediction of diabetic retinopathy risk with type 2 diabetes mellitus. J Pers Med (2021) 11(8):689. doi: 10.3390/jpm11080689
121. Zha X, Xi X, Fan X, Ma M, Zhang Y, Yang Y. Overexpression of METTL3 attenuates high-glucose induced RPE cell pyroptosis by regulating miR-25-3p/PTEN/Akt signaling cascade through DGCR8. Aging (Albany NY) (2020) 12(9):8137–50. doi: 10.18632/aging.103130
122. Suo L, Liu C, Zhang QY, Yao MD, Ma Y, Yao J, et al. METTL3-mediated N(6)-methyladenosine modification governs pericyte dysfunction during diabetes-induced retinal vascular complication. Theranostics (2022) 12(1):277–89. doi: 10.7150/thno.63441
123. Cao X, Song Y, Huang LL, Tian YJ, Wang XL, Hua LY. m(6)A transferase METTL3 regulates endothelial-mesenchymal transition in diabetic retinopathy via lncRNA SNHG7/KHSRP/MKL1 axis. Genomics (2022) 114(6):110498. doi: 10.1016/j.ygeno.2022.110498
124. Liang D, Lin WJ, Ren M, Qiu J, Yang C, Wang X, et al. m(6)A reader YTHDC1 modulates autophagy by targeting SQSTM1 in diabetic skin. Autophagy (2022) 18(6):1318–37. doi: 10.1080/15548627.2021.1974175
125. Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol (2014) 16(2):191–8. doi: 10.1038/ncb2902
126. Shen F, Huang W, Huang JT, Xiong J, Yang Y, Wu K, et al. Decreased N(6)-methyladenosine in peripheral blood RNA from diabetic patients is associated with FTO expression rather than ALKBH5. J Clin Endocrinol Metab (2015) 100(1):E148–54. doi: 10.1210/jc.2014-1893
127. Onalan E, Yakar B, Onalan EE, Karakulak K, Kaymaz T, Donder E. m(6)A RNA, FTO, ALKBH5 expression in type 2 diabetic and obesity patients. J Coll Physicians Surg Pak (2022) 32(9):1143–8. doi: 10.29271/jcpsp.2022.09.1143
128. Masoud Abd El Gayed E, Kamal El Din Zewain S, Ragheb A, ElNaidany SS. Fat mass and obesity-associated gene expression and disease severity in type 2 diabetes mellitus. Steroids (2021) 174:108897. doi: 10.1016/j.steroids.2021.108897
129. Yang Y, Shen F, Huang W, Qin S, Huang JT, Sergi C, et al. Glucose is involved in the dynamic regulation of m6A in patients with type 2 diabetes. J Clin Endocrinol Metab (2019) 104(3):665–73. doi: 10.1210/jc.2018-00619
130. LaMoia TE, Shulman GI. Cellular and molecular mechanisms of metformin action. Endocr Rev (2021) 42(1):77–96. doi: 10.1210/endrev/bnaa023
131. Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab (2014) 20(6):953–66. doi: 10.1016/j.cmet.2014.09.018
132. Cheng L, Zhang X, Huang YZ, Zhu YL, Xu LY, Li Z, et al. Metformin exhibits antiproliferation activity in breast cancer via miR-483-3p/METTL3/m(6)A/p21 pathway. Oncogenesis (2021) 10(1):7. doi: 10.1038/s41389-020-00290-y
133. Zhang Q, Xiong L, Wei T, Liu Q, Yan L, Chen J, et al. Hypoxia-responsive PPARGC1A/BAMBI/ACSL5 axis promotes progression and resistance to lenvatinib in hepatocellular carcinoma. Oncogene (2023) 42(19):1509–23. doi: 10.1038/s41388-023-02665-y
134. Chen CJ, Huang JY, Huang JQ, Deng JY, Shangguan XH, Chen AZ, et al. Metformin attenuates multiple myeloma cell proliferation and encourages apoptosis by suppressing METTL3-mediated m6A methylation of THRAP3, RBM25, and USP4. Cell Cycle (2023) 22(8):986–1004. doi: 10.1080/15384101.2023.2170521
135. Li K, Gao S, Ma L, Sun Y, Peng ZY, Wu J, et al. Stimulation of let-7 maturation by metformin improved the response to tyrosine kinase inhibitor therapy in an m6A dependent manner. Front Oncol (2021) 11:731561. doi: 10.3389/fonc.2021.731561
136. Yuan J, Liu Y, Zhou L, Xue Y, Lu Z, Gan J. YTHDC2-mediated circYTHDC2 N6-methyladenosine modification promotes vascular smooth muscle cells dysfunction through inhibiting ten-eleven translocation 2. Front Cardiovasc Med (2021) 8:686293. doi: 10.3389/fcvm.2021.686293
Keywords: type 2 diabetes mellitus, T2DM, diabetes, RNA, epigenetics, epitranscriptomics, m6A, m6Am
Citation: Benak D, Benakova S, Plecita-Hlavata L and Hlavackova M (2023) The role of m6A and m6Am RNA modifications in the pathogenesis of diabetes mellitus. Front. Endocrinol. 14:1223583. doi: 10.3389/fendo.2023.1223583
Received: 16 May 2023; Accepted: 26 June 2023;
Published: 07 July 2023.
Edited by:
Cecil Jack Weale, Cape Peninsula University of Technology, South AfricaReviewed by:
Laura Marroqui, Miguel Hernández University of Elche, SpainMridusmita Saikia, Cornell University, United States
Copyright © 2023 Benak, Benakova, Plecita-Hlavata and Hlavackova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marketa Hlavackova, bWFya2V0YS5obGF2YWNrb3ZhQGZndS5jYXMuY3o=