 Julie M. Hughes
Julie M. Hughes Katelyn I. Guerriere
Katelyn I. Guerriere Kristin L. Popp1,2
Kristin L. Popp1,2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 18 September 2023
Sec. Translational and Clinical Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1219454
This article is part of the Research TopicIntegrative Exercise EndocrinologyView all 5 articles
Hormones and mechanical loading co-regulate bone throughout the lifespan. In this review, we posit that times of increased hormonal influence on bone provide opportunities for exercise to optimize bone strength and prevent fragility. Examples include endogenous secretion of growth hormones and sex steroids that modulate adolescent growth and exogenous administration of osteoanabolic drugs like teriparatide, which increase bone stiffness, or its resistance to external forces. We review evidence that after bone stiffness is increased due to hormonal stimuli, mechanoadaptive processes follow. Specifically, exercise provides the mechanical stimulus necessary to offset adaptive bone resorption or promote adaptive bone formation. The collective effects of both decreased bone resorption and increased bone formation optimize bone strength during youth and preserve it later in life. These theoretical constructs provide physiologic foundations for promoting exercise throughout life.
In an editorial published in 2002, Dr. Ego Seeman wrote, “Each region of the axial and appendicular skeleton, each point on the external and internal surfaces along a bone’s length and circumference, is fashioned by genetic factors, local mechanical and hormonal factors, into a structure adapted to loading (1).” Of these factors, mechanical and hormonal factors are always in flux, and their interconnectedness in bone functional adaptation throughout life can reveal practical solutions for promoting bone strength and preventing bone fragility. In this paper, we review a series of well-established observations of hormonal effects on bone throughout life and interpret these within the context of synergistic and beneficial interactions with mechanical regulation of bone by exercise.
Classic examples of hormonal effects on bone that we discuss include bone growth from anabolic sex hormone secretion during puberty and subsequent functional adaptation of bone that helps explain sex-based differences in bone properties during adulthood. Contemporary examples include bone resorption that follows exogenous osteoanabolic hormone treatment for osteoporosis. In each of these scenarios, we use our working model of bone functional adaptation (2) to help decipher the synergistic physiological pathways by which hormones and exercise co-regulate bone and the modulating effects that exercise has on these processes.
We focus the review on physiologic responses of bone to hormone-induced increases in bone stiffness, which is a mechanical property that reflects the ability of bone to resist external forces without permanent changes to its structure. We begin by reviewing hormone-induced increases in bone stiffness and then discuss the role exercise serves in offsetting bone resorption or promoting bone formation. We conclude by discussing practical implications of exercise for maintaining and promoting bone health, concurrent with hormone-induced increases in bone stiffness throughout life.
Beneficial effects of exercise on bone mechanical properties have long been recognized (3), as is the co-regulation of bone by hormones and mechanical loading (4, 5). Recently, we posited that bone stiffness, rather than the traditionally referenced mass or strength, is the mechanical property that governs bone adaptation to mechanical loading (2). Classic studies revealed a narrow homeostatic range of 2,000-3,000 microstrain (6, 7) in loaded bones is maintained across many different species during peak functional activities. Therefore, it can be argued that stiffness, which is the ability of bone to resist applied loads and return to its original form, is regulated by bone functional adaptation, and not mass (quantity of bone) or strength (ultimate force at which the bone breaks). The corollary is that when bone stiffness is altered for non-mechanical reasons such as hormonal stimulation, the new bone stiffness must be interpreted within the context of the prevailing mechanical environment, and as a result, bone functional adaptation may be stimulated.
To decipher the complex interactions between mechanical and hormonal interactions in the regulation of bone stiffness, and the critical role that exercise serves in these processes, we first begin with a physiologic guide for interpreting bone mechanoadaptation after increased or decreased mechanical loading. We then layer on the interaction of hormone-driven changes to the mechanical environment. This tiered approach can help provide a physiological foundation from which the significance of exercise for promoting bone health during times of hormonal and mechanical interactions can be appreciated.
We recently proposed a theoretical model for mechanical regulation of bone stiffness (2), based on classic models (3, 8–10), that provides a guide for deciphering tissue-level responses of bone to exercise and also disuse. This model is akin to the classic model of bone functional adaptation proposed by Dr. Harold Frost, known as the Mechanostat. Our model differs in its focus on regulating bone stiffness instead of mass or strength and in recognition of four distinct mechanoadaptive pathways (Figures 1A–D) consisting of bone modeling (the independent action of osteoclasts or osteoblasts) or remodeling (the couple actions of osteoclasts and osteoblasts in a remodeling unit) acting in a negative feedback system. Briefly, when mechanical loading (Figure 2.1) on a bone of a given stiffness (Figure 2.2) is greater than customary (up until the point of structural failure), the bone matrix experiences strain (Figure 2.3) that initiates both formation modeling (Figures 1A, 2.4) and targeted remodeling (Figures 1B, 2.5). However, when mechanical loading is less than the bone is accustomed to, lower strain stimuli can initiate disuse-mediated bone remodeling (Figures 1C, 2.6) and resorption modeling (Figures 1D, 2.7).

Figure 1 Four pathways of bone functional adaptation: Schematic of the four mechanoadaptive responses at the cellular level. Formation modeling (A): osteocyte perturbation by mechanical loading induces osteoblastic bone formation on a surface. Targeted remodeling (B): microcracks generated during loading stimulate osteocyte apoptosis and targeted removal of bone by osteoclasts and subsequent formation of bone by osteoblasts. Disuse-mediated remodeling (C): osteocyte apoptosis with disuse stimulates bone resorption and coupled formation. Also depicted is the negative bone balance within each remodeling unit that can accompany disuse-mediated remodeling. Resorption modeling (D): disuse-mediated osteocyte apoptosis stimulates osteoclastic bone resorption on a surface. Reprinted with permission from Hughes et al., Exercise and Sport Sciences Reviews, 2020 (2).
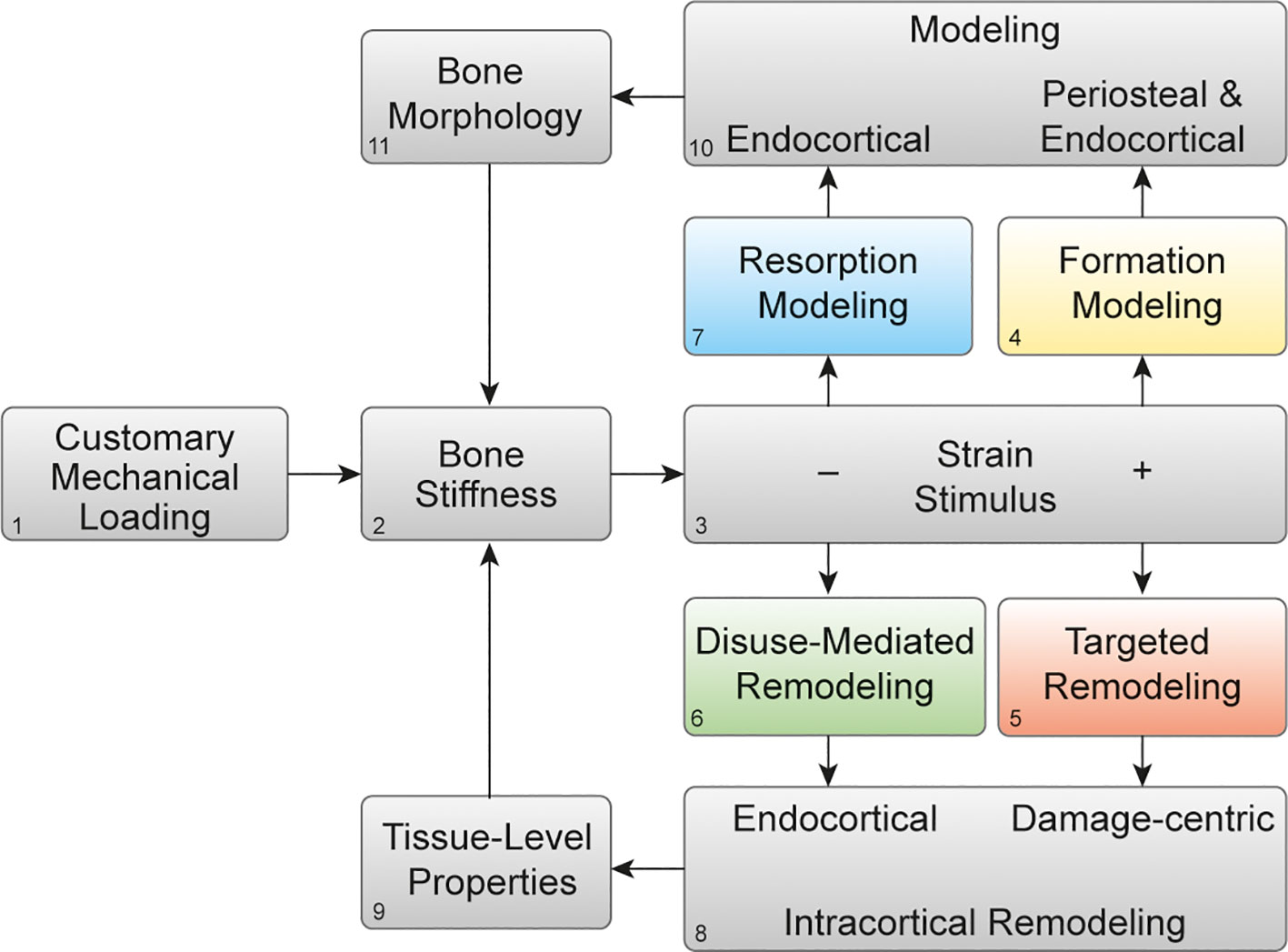
Figure 2 Mechanoadaptation of bone: Theoretical framework of bone functional adaptation at the diaphysis. Mechanical loading (1) on a bone of a given stiffness (2) will produce a strain stimulus (3), which, if greater than customary, may elicit bone formation modeling (4) or targeted remodeling (5). In the case of disuse and a lower than customary strain stimulus, disuse-mediated remodeling (6) and resorption modeling (7) may occur. Remodeling near damage or the endocortical surface (8) primarily alters tissue-level mechanical properties (9), whereas modeling on the periosteal or endocortical surfaces (10) alters the bone morphology (11). Bone morphology and tissue-level properties collectively determine whole-bone stiffness (2), which then influences the strain response to subsequent bouts of mechanical loading. Reprinted with permission from Hughes et al., Exercise and Sport Sciences Reviews, 2020 (2).
Targeted remodeling is damage-centric and can occur following osteocyte expression of genes associated with osteoclastogenesis within 100 and 300 microns of linear microdamage (11) in cortical and trabecular bone. Disuse-mediated remodeling occurs near the endocortical surface (Figure 2.8), and both types of remodeling will influence the tissue-level properties of the bone (Figure 2.9). Bone modeling which occurs on the periosteal and endocortical surfaces of the long bone diaphysis in formation modeling and on the endocortical surface in resorption modeling (Figure 2.10), and by altering the size, shape, and cortical thickness of the bone, will ultimately influence the morphology (Figure 2.11). In turn, changes in the bone morphology and tissue-level properties will together alter bone stiffness in a negative feedback fashion (Figure 2.2). We propose that this simple negative feedback model can help decipher the intricate interactions between mechanical and hormonal regulation of bone stiffness, as we discuss below.
There are three tissue-level modes of increasing bone stiffness in response to hormonal stimulation. The first mode is through formation modeling on trabecular, endocortical, or periosteal surfaces which can increase trabecular and cortical thickness (12). A second mode for increasing bone stiffness is by enhancing osteoblastic activity within existing remodeling units (13), and a third mode is by preventing bone resorption altogether (14), whether by suppressing the activation of new remodeling cycles or preventing resorption modeling. These mechanisms, whether through bone anabolism or prevention of resorption, by increasing bone stiffness, will alter the customary strain stimulus and initiate bone functional adaptation. In each of the examples below, we begin with well-established observations of hormone-mediated increases in bone stiffness. We then review adaptive phenomena that occur after hormone-mediated increases in stiffness, whose foundations in mechanical regulation of bone are not always recognized. We conclude each example by highlighting the potential of exercise to offset bone resorption or promote bone formation that can accompany bone functional adaptation after hormone-induced increases in bone stiffness.
During growth, long bones increase in length by endochondral ossification, a process that is largely driven by pulsatile secretion of growth hormone (GH) and other growth factors (4, 15). Endochondral ossification concludes by estrogen-induced closure of the growth plates in girls two years earlier than in boys, who experience a rise in estrogen via the aromatase pathways (16). Recently, animal studies have demonstrated that sex-based differences in GH secretion, favoring males, may have epigenetic foundations (17–20). The longitudinal pubertal growth in bone may provide some of the mechanical stimulus that drives radial growth through deposition on the subperiosteal surface, and radial growth also has roots in hormonal stimulation (4, 21). During puberty, periosteal apposition is particularly accelerated in boys relative to girls (22), and the resultant structural advantage of greater bone size is attributed to exposure to greater testosterone, growth hormone, and insulin-like growth factor-1 (IGF-1) concentrations during puberty in boys than in girls (23, 24). However, testosterone could also indirectly increase bone size through stimulating increases in muscle mass leading to greater mechanical loading (25). Bone stiffness increases disproportionately relative to the amount of new bone mass that is formed when formation modeling occurs on the periosteal surface of the diaphysis of long bones (26). This is because periosteal apposition occurs on the surface furthest from the neutral axis in bending and therefore increases the cross-sectional moment of inertia (26, 27). Therefore, formation modeling that leads to periosteal apposition in an accelerated manner in boys during puberty should disproportionately increase bone stiffness in boys relative to girls. There are mechanical advantages that accompany the increase in stiffness, with boys attaining 28% to 63% greater strength of the appendicular long bones across longitudinal growth, compared to biologically age-matched girls (28).
Although boys have a mechanical advantage in terms of overall bone strength, they also have 28% to 80% more porous cortices than girls (28). The physiologic foundations for the greater increases in cortical porosity throughout growth in boys, relative to girls, is unknown but has been attributed to the need for bones to not only remain stiff and strong but also minimally massive (29). Indeed, bone tissue is approximately twice as dense as other body tissues and therefore requires more energy to move during locomotion (3). We propose a novel way to at least partially explain the observations that men not only have wider bones but also more porous bones on average, compared to women. Specifically, we offer that increased porosity could be a compensatory mechanism that follows periosteal apposition (Figure 3). When forces from habitual mechanical loading are applied to a bone that has increased stiffness due to hormone-driven periosteal apposition (Figure 3.1), the stiffer structure will deform less, and strain stimuli will fall below customary ranges (Figure 3.2). This will elicit disuse-mediated remodeling (Figure 3.3), predominantly near the endocortical surface (Figure 3.4). Intracortical remodeling with a negative bone balance will increase tissue porosity (Figure 3.5), leading to decreased bone stiffness (Figure 3.1). Decreased strain stimuli will simultaneously elicit resorption modeling (Figure 3.6), also predominantly at the endocortical surface (Figure 3.7). This resorption, independent of formation, will expand the marrow cavity (Figure 3.8). Together, increased tissue porosity and endocortical expansion will decrease bone stiffness (Figure 3.2) and subsequent strain stimuli will return to equilibrium, as is the hallmark of negative feedback loops. Thus, bone loss after periosteal apposition during growth may occur at least partially because of a perceived disuse that accompanies increases in bone stiffness without parallel increases in mechanical loading.
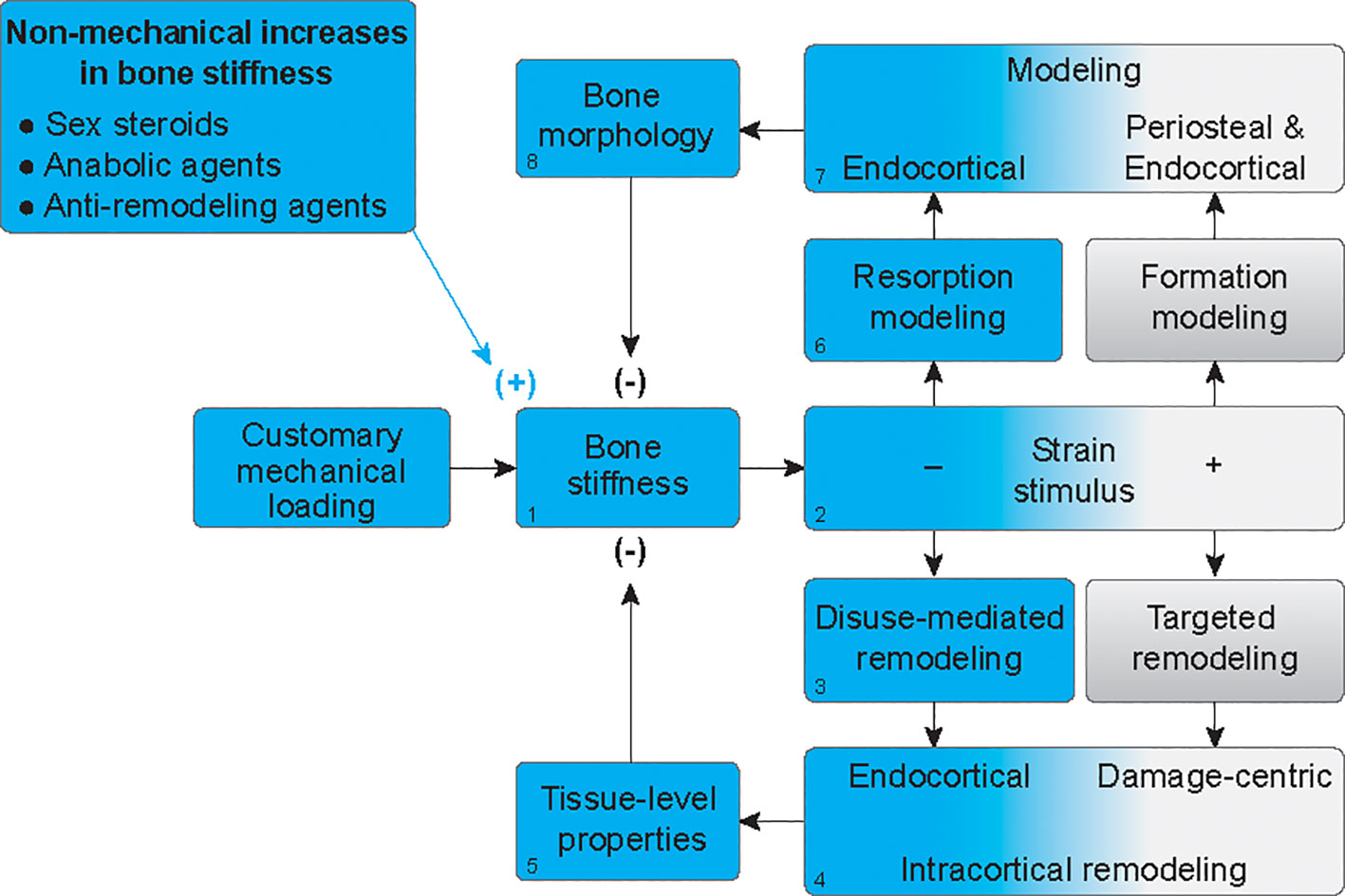
Figure 3 Mechanoadaptation of bone after hormone-mediated increases in bone stiffness. In the absence of proportionally greater increases in customary mechanical loading, when bone stiffness is increased by hormonal stimuli (1), strain stimuli will fall below customary ranges (blue gradient) (2), thus eliciting disuse-mediated remodeling (3), predominantly near the endocortical surface (4). Intracortical remodeling with a negative bone balance, will increase tissue porosity (5), leading to decreased bone stiffness (1). Decreased strain stimuli will simultaneously elicit resorption modeling (6), also predominantly at the endocortical surface (7). This resorption, independent of formation, will expand the marrow cavity (8). Together, increased tissue porosity and endocortical expansion will decrease bone stiffness to homeostatic levels (1).
While boys experience endocortical expansion during adolescence, they do so to a greater degree than girls (22), leading to narrower medullary cavities in adult women, on average, than in men. Not only are girls not exposed to the concentrations of androgens and growth hormones that lead to accelerated periosteal expansion in boys, but girls also experience greater increases in concentrations of estrogen (30). The anti-resorptive effects of estrogen are well-established (30), and the physiologic model in Figure 3 would predict that inhibition of bone resorption by estrogen, combined with less periosteal expansion than that observed in boys, would preserve bone stiffness and maintain customary ranges of strain stimuli during mechanical loading. Ultimately, this would lead to a lack of stimulus for endocortical expansion in girls compared to boys. Collectively, hormonal and mechanical co-regulation of bone stiffness can provide physiological context for sex-based differences in the adult skeleton, with men having wider, more porous bones and expanded medullary cavities than women (22, 31).
The perceived disuse that may accompany increases in bone stiffness from periosteal apposition in boys is counterintuitive because it does not include classic disuse conditions such as bed rest, limb immobilization, or microgravity and occurs in young healthy adolescents, absent of disease. Nonetheless, both traditional and perceived disuse occur when strain stimuli fall below customary levels, stimulating bone resorption. The practical implication of recognizing adaptive resorption is that increasing the habitual mechanical forces on bone through exercise could prevent it (Figure 4). If bone stiffness is increased from non-mechanical stimuli such as hormonally driven periosteal apposition, but there is a simultaneous increase in the mechanical loads placed on bone with exercise, this may maintain strain stimuli within a customary range and prevent disuse-mediated resorption.
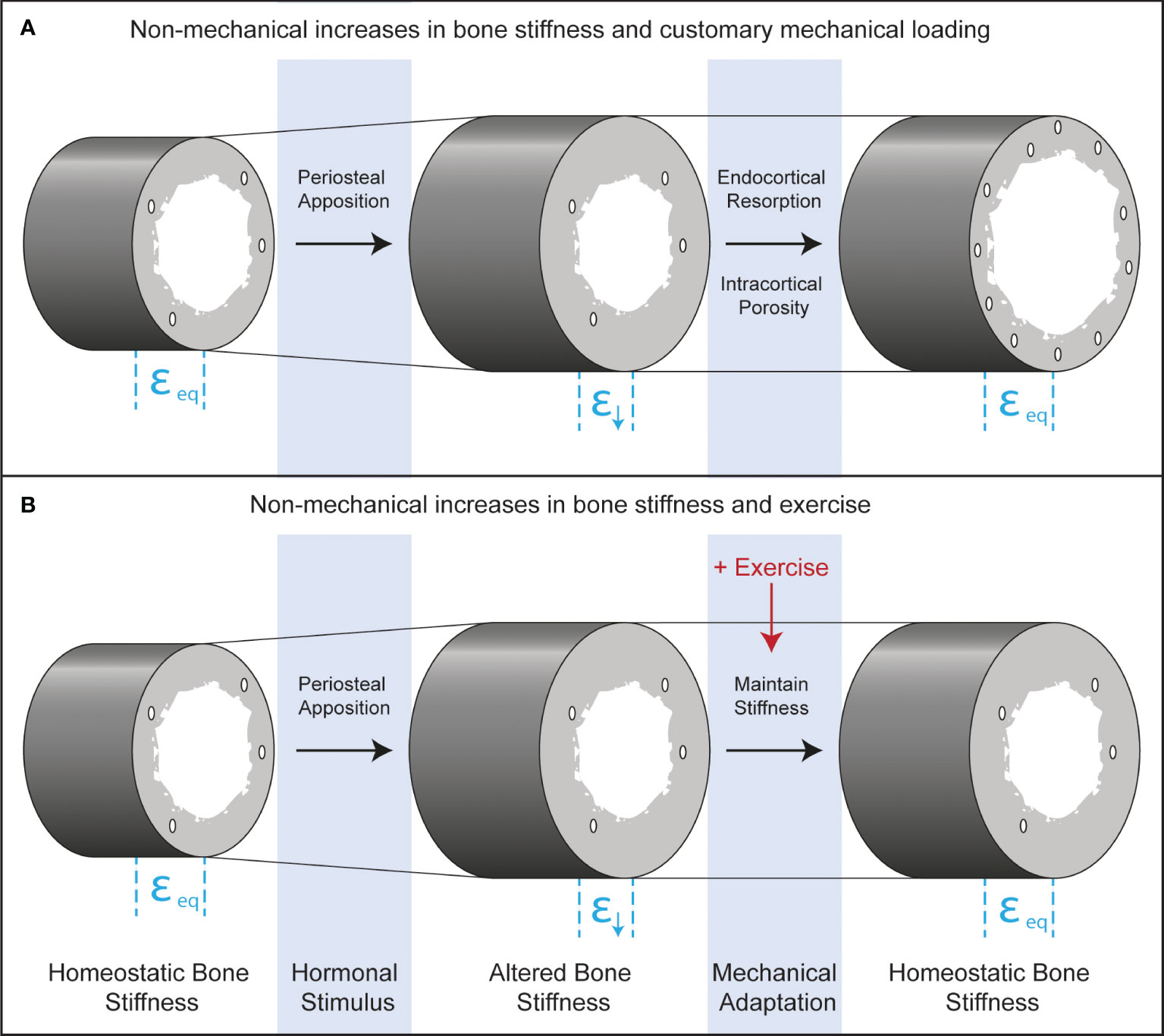
Figure 4 Longitudinal bone cross-sections depicting endocrine and mechanical co-regulation of bone stiffness after androgen- and TPTD-driven periosteal apposition. Before hormonal stimulation, strains during customary mechanical loading will be at equilibrium (εeq). Periosteal apposition increases the diameter of the bone and therefore the stiffness. Increased stiffness leads to declines in strain during customary mechanical loading (ε↓), which could lead to functional adaptation of bone through increased resorption at the endocortical surface or through intracortical remodeling that increases porosity. These adaptive responses will bring strains to homeostatic levels. Exercise may inhibit adaptive bone resorption, leading to maximal gains in bone formation from the initiating hormonal stimulus.
For girls, exercise could capitalize on the increased mechanosensitivity of bone that accompanies estrogen exposure (32–34) and ultimately help attain a stronger adult skeleton. Promoting exercise during adolescent growth may hold other advantages for both sexes beyond preventing perceived disuse in boys and capitalizing on increased mechanosensitivity in girls. During puberty, growth in width lags behind growth in length, potentially creating a vulnerable window for increased risk of fracture in the long bones of the extremities (35). It is conceivable, though not yet experimentally demonstrated that exercise during this vulnerable period could stimulate more rapid periosteal expansion and help offset increased fracture risk. Finally, adolescence may be an opportune time to participate in exercise to stimulate adaptive bone formation during growth, because mechanosensitive osteoblasts are active along much of the bone surface due to longitudinal and appositional growth than during other times in life. There is a large body of literature supporting these concepts that exercise when young holds promise for building strong bones and offsetting the risk of osteoporosis and related fractures in adulthood for men and women (36–39). Even though men have greater bone strength and lower risk of osteoporotic fractures than women, osteoporotic fractures are still common in men who account for 30% of hip fractures (40). Therefore, prevention of disuse-mediated bone resorption during growth in boys and capitalizing on increased mechanosensitivity in girls with exercise may be an important strategy to decrease fracture risk later in life in both men and women.
Daily injections of PTH (1-84) or its analog, TPTD [PTH (1-34)], are osteoanabolic. The mechanisms whereby PTH or its analogs influence bone cells are numerous (13, 41). TPTD was the first anabolic drug available for treatment of osteoporosis, and histomorphometric evaluation of transiliac crest biopsies revealed that TPTD stimulates bone formation on all four primary bone surfaces (12). A classic study provided evidence that TPTD may initiate bone formation from bone lining cells, which are flattened osteoblastic cells aligned along quiescent surfaces (42). This theory was confirmed in a lineage tracing study reporting that three days of PTH administration resulted in in vivo conversion of normally flattened bone lining cells into cuboidal, mature osteoblasts (43). Thus, early anabolic action of intermittent PTH and TPTD is likely modeling-based. In cortical bone, formation modeling with TPTD can occur on both the endocortical and periosteal surfaces (12, 44). Biopsy studies also report increases in bone formation within existing remodeling units (12, 45, 46) and even reveal a novel remodeling phenomenon by which TPTD stimulates bone formation within the remodeling unit in such an accelerated manner that “overflow remodeling” can occur. In overflow remodeling, bone formation not only fills the resorption lacunae previously hollowed out by osteoclasts but also pours out over the lacunar boundary and onto previously quiescent surfaces (12). Collectively, these three modes of bone formation increase bone stiffness (47) with TPTD administration.
While bone formation is the hallmark of osteoanabolic drugs, much less is known about the mechanistic foundations for a delayed resorption after initiation of treatment with an intermittent PTH analog. Biopsy studies and serum biochemical markers of bone formation reveal that bone formation activity increases rapidly once TPTD is initiated, peaks within six to 12 months, and remains elevated above baseline values for at least 3 years of treatment (12, 45, 46, 48). After the first month of TPTD treatment, biochemical markers of bone resorption also begin to increase, and like markers of bone formation, also peak at 6-12 months, followed by a steady decline, but remaining elevated over baseline (49, 50).
The physiological foundations for increasing bone resorption after TPTD administration are not fully delineated. Coupling mechanisms between osteoblasts and osteoclasts such as secretion of RANKL, which promotes differentiation of osteoclasts (51), may partially explain increases in bone resorption. Co-regulation of bone stiffness by hormonal and mechanical stimuli can also account for these observations. When TPTD increases bone formation, bone stiffness is also increased (Figure 3.1). Without a simultaneous increase in habitual mechanical loading, strain stimuli will fall below customary levels (Figure 3.2), thus eliciting disuse-mediated bone remodeling (Figure 3.3), particularly at the endocortical surface (Figure 3.4). If TPTD continues to increase bone stiffness, independent of increases in mechanical loading or changes to sex steroids, bone remodeling should continue. When complete, disuse-mediated remodeling results in a negative bone balance and therefore increased porosity (Figure 3.5), leading to decreased bone stiffness and equilibrium (Figure 3.2). Resorption modeling, the other physiologic response to lower than customary strain stimuli, is not observed with TPTD, likely because endocortical lining cells that have converted to active osteoblasts may be actively forming bone along the surface, and during disuse, osteoclasts appear to avoid regions of ongoing osteoblast activity (52). Thus, we propose that increased but delayed bone resorption after TPTD treatment is partially due to a disuse-mediated increase in bone remodeling.
Besides anabolic drugs, other pharmacologic agents can increase bone stiffness. These include anti-resorptive drugs such as bisphosphonates, which are incorporated into the mineralized matrix and promote apoptosis of mature osteoclasts when released by the acid environment underneath active osteoclasts (53). After long-term treatment with bisphosphonates, brief periods of cessation can be introduced to prevent rare negative consequences of long-term remodeling suppression (54). Another anti-resorptive drug is denosumab which is not accumulated into the bone matrix and inhibits osteoclast differentiation, activity, and survival of osteoclasts (53). Inhibiting bone resorption by these drugs results in increased bone density, stiffness, strength, and ultimately, prevention of osteoporotic fractures (55). Discontinuation of drugs that increase bone stiffness can lead to increased bone resorption, which can reverse the drug’s effect on bone and fracture risk (54, 56–58). We propose this increased activation of bone resorption is in part due to adaptive bone resorption. If anabolic or anti-resorptive drugs increase bone stiffness (Figure 3.1), absent of proportional increases in customary mechanical loading, strain stimuli will be decreased (Figure 3.2), and bone functional adaption in the mode of disuse-mediated bone remodeling (Figure 3.3) or resorption modeling (Figure 3.6) will ensue. These processes would lead to increased porosity (Figure 3.5) and endocortical resorption (Figure 3.7), respectively, that would decrease bone stiffness (Figure 3.1) until homeostasis is achieved (Figure 3.2).
One strategy that has been effective for counteracting bone resorption after drug discontinuation is sequencing anabolic followed by anti-resorptive drugs (59–61). By providing drugs that inhibit resorption, loss from perceived disuse following non-mechanical increases in stiffness can be prevented. Although studies have demonstrated a benefit of sequencing anabolic therapy first, followed by anti-resorptive therapy, an unfortunate extension of our model would be that if the anti-resorptive therapy is eventually ceased, functional adaptation of bone would result in disuse-mediated bone loss. Relatively new therapies that both induce bone anabolism and inhibit bone remodeling, like the anti-sclerostin antibody, romosozumab, may help prevent bone resorption that accompanies cessation of drug therapy (62).
Increasing customary mechanical loading with exercise is a non-pharmacologic means for blunting disuse-mediated bone resorption after increases in bone stiffness. Increases in bone-loading exercise also holds promise as a companion to osteoporosis drugs for offsetting disuse-mediated bone loss. Prevention of disuse-mediate bone resorption by exercise has been demonstrated in classic disuse scenarios such as spinal cord injury, space flight, and bedrest (63–65). Our model would predict that a combination of exercise and anti-resorptive drugs would synergistically attenuate bone loss during disuse, as has been demonstrated in animal models with hindlimb unloading (66) and in astronauts due to microgravity (67). Combining exercise with osteoanabolic therapy may be successful in offsetting resorption that accompanies drugs that increase bone stiffness. In a rodent model of type 2 diabetes, TPTD and exercise separately increased lumbar spine BMD, but only the two combined improved trabecular and cortical bone microarchitecture and breaking strength (68). Similarly, combined TPTD and exercise improved bone structure and strength of cortical bone at the femoral diaphysis in ovariectomized and tail-suspended rats (69). In postmenopausal women with osteoporosis, TPTD and whole-body vibration exercise had synergistic effects in increasing lumbar spine BMD, compared to TPTD alone (70).
Exercise may also attenuate the rapid increase in bone resorption with cessation of osteoporosis therapy. TPTD administered for 12 weeks partially prevented bone loss in ovariectomized rats. After cessation of TPTD, eight weeks of treadmill exercise maintained the TPTD-stimulated bone anabolism, compared to non-exercise controls whose bone mass returned to pre-TPTD levels in eight weeks (71). Whether exercise can combat bone loss after cessation of osteoporosis therapies in humans remains to be determined. Studies of exercise in women and men with osteoporosis who cease osteoporotic drug therapies are needed to determine if exercise can indeed prevent bone resorption.
In this review, we discussed the concept that when bone stiffness is increased due to hormonal stimuli, without simultaneous increases in mechanical loading, disuse-mediated resorption can occur and offset some of the mechanical advantages of hormone-induced gains in bone tissue. Recognition of these integrated physiological processes provides a framework for interpreting successive adaptions. Recognizing these interactions also highlights the benefits of exercise for preventing disuse-mediated bone resorption that can follow increases in bone stiffness during adolescence, osteoanabolic drug therapy, and following cessation of osteoporosis treatments, thus providing further support for the important role for exercise in offsetting skeletal fragility throughout life.
JH conceived of the manuscript. CC designed the figures. SP revised the manuscript. All authors contributed to the article and approved the submitted version.
Research supported in part by an appointment to the Postgraduate Research Participation Program funded by USARIEM & administered by Oak Ridge Institute for Science and Engineering (KLP and CC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Seeman E. An exercise in geometry. J Bone Miner Res (2002) 17:373–80. doi: 10.1359/jbmr.2002.17.3.373
2. Hughes JM, Castellani CM, Popp KL, Guerriere KI, Matheny RW Jr., Nindl BC, et al. The central role of osteocytes in the four adaptive pathways of bone's mechanostat. Exerc Sport Sci Rev (2020) 48:140–8. doi: 10.1249/JES.0000000000000225
3. Martin RB. The importance of mechanical loading in bone biology and medicine. J Musculoskelet Neuronal Interact (2007) 7:48–53.
4. Borer KT. Physical activity in the prevention and amelioration of osteoporosis in women: Interaction of mechanical, hormonal and dietary factors. Sports Med (2005) 35:779–830. doi: 10.2165/00007256-200535090-00004
5. Price JS, Sugiyama T, Galea GL, Meakin LB, Sunters A, Lanyon LE. Role of endocrine and paracrine factors in the adaptation of bone to mechanical loading. Curr Osteoporosis Rep (2011) 9:76–82. doi: 10.1007/s11914-011-0050-7
6. Fritton SP, McLeod KJ, Rubin CT. Quantifying the strain history of bone: Spatial uniformity and self-similarity of low-magnitude strains. J Biomech (2000) 33:317–25. doi: 10.1016/S0021-9290(99)00210-9
7. Rubin CT. Skeletal strain and the functional significance of bone architecture. Calcif Tissue Int (1984) 36 Suppl 1:S11–8. doi: 10.1007/BF02406128
8. Frost HM. Bone "mass" and the "mechanostat": A proposal. Anat Rec (1987) 219:1–9. doi: 10.1002/ar.1092190104
9. Frost HM. Bone's mechanostat: A 2003 update. Anat Rec A Discovery Mol Cell Evol Biol (2003) 275:1081–101. doi: 10.1002/ar.a.10119
11. Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone (2012) 50:1115–22. doi: 10.1016/j.bone.2012.01.025
12. Dempster DW, Zhou H, Recker RR, Brown JP, Bolognese MA, Recknor CP, et al. A longitudinal study of skeletal histomorphometry at 6 and 24 months across four bone envelopes in postmenopausal women with osteoporosis receiving teriparatide or zoledronic acid in the SHOTZ trial. J Bone Miner Res (2016) 31:1429–39. doi: 10.1002/jbmr.2804
13. Tabacco G, Bilezikian JP. Osteoanabolic and dual action drugs. Br J Clin Pharmacol (2019) 85:1084–94. doi: 10.1111/bcp.13766
14. Kohrt WM, Wherry SJ, Wolfe P, Sherk VD, Wellington T, Swanson CM, et al. Maintenance of serum ionized calcium during exercise attenuates parathyroid hormone and bone resorption responses. J Bone Miner Res (2018) 33:1326–34. doi: 10.1002/jbmr.3428
15. Mauras N, Blizzard RM, Link K, Johnson ML, Rogol AD, Veldhuis JD. Augmentation of growth hormone secretion during puberty: Evidence for a pulse amplitude-modulated phenomenon. J Clin Endocrinol Metab (1987) 64:596–601. doi: 10.1210/jcem-64-3-596
16. Nilsson O, Baron J. Fundamental limits on longitudinal bone growth: growth plate senescence and epiphyseal fusion. Trends Endocrinol Metabolism: TEM (2004) 15:370–4. doi: 10.1016/j.tem.2004.08.004
17. Wauthier V, Sugathan A, Meyer RD, Dombkowski AA, Waxman DJ. Intrinsic sex differences in the early growth hormone responsiveness of sex-specific genes in mouse liver. Mol Endocrinol (2010) 24:667–78. doi: 10.1210/me.2009-0454
18. Brie B, Ornstein A, Ramirez MC, Lacau-Mengido I, Becu-Villalobos D. Epigenetic modifications in the GH-dependent Prlr, Hnf6, Cyp7b1, Adh1 and Cyp2a4 genes. J Mol Endocrinol (2020) 64:165–79. doi: 10.1530/JME-19-0205
19. Reynolds CM, Perry JK, Vickers MH. Manipulation of the growth hormone-insulin-like growth factor (GH-IGF) axis: A treatment strategy to reverse the effects of early life developmental programming. Int J Mol Sci (2017) 18:1729. doi: 10.3390/ijms18081729
20. Zhong H, Xiao J, Chen W, Zhou Y, Tang Z, Guo Z, et al. DNA methylation of pituitary growth hormone is involved in male growth superiority of Nile tilapia (Oreochromis niloticus). Comp Biochem Physiol B Biochem Mol Biol (2014) 171:42–8. doi: 10.1016/j.cbpb.2014.03.006
21. Seeman E. Structural basis of growth-related gain and age-related loss of bone strength. Rheumatol (Oxford) (2008) 47 Suppl 4:iv2–8. doi: 10.1093/rheumatology/ken177
22. Gabel L, Nettlefold L, Brasher PM, Moore SA, Ahamed Y, Macdonald HM, et al. Reexamining the surfaces of bone in boys and girls during adolescent growth: A 12-year mixed longitudinal pQCT study. J Bone Miner Res (2015) 30:2158–67. doi: 10.1002/jbmr.2570
23. Giustina A, Mazziotti G, Canalis E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr Rev (2008) 29:535–59. doi: 10.1210/er.2007-0036
24. Monson JP, Drake WM, Carroll PV, Weaver JU, Rodriguez-Arnao J, Savage MO. Influence of growth hormone on accretion of bone mass. Horm Res (2002) 58 Suppl 1:52–6. doi: 10.1159/000064765
25. Schoenau E. Bone mass increase in puberty: what makes it happen? Horm Res (2006) 65 Suppl 2:2–10. doi: 10.1159/000091748
26. Seeman E. Periosteal bone formation–a neglected determinant of bone strength. N Engl J Med (2003) 349:320–3. doi: 10.1056/NEJMp038101
27. Ruff CB, Hayes WC. Sex differences in age-related remodeling of the femur and tibia. J Orthop Res (1988) 6:886–96. doi: 10.1002/jor.1100060613
28. Gabel L, Macdonald HM, McKay HA. Sex differences and growth-related adaptations in bone microarchitecture, geometry, density, and strength from childhood to early adulthood: A mixed longitudinal HR-pQCT study. J Bone Miner Res (2016) 32(2):250–63. doi: 10.1002/jbmr.2982
29. Seeman E. The structural and biomechanical basis of the gain and loss of bone strength in women and men. Endocrinol Metab Clin North Am (2003) 32:25–38. doi: 10.1016/S0889-8529(02)00078-6
30. Khosla S, Oursler MJ, Monroe DG. Estrogen and the skeleton. Trends Endocrinol Metab (2012) 23:576–81. doi: 10.1016/j.tem.2012.03.008
31. Jepsen KJ, Centi A, Duarte GF, Galloway K, Goldman H, Hampson N, et al. Biological constraints that limit compensation of a common skeletal trait variant lead to inequivalence of tibial function among healthy young adults. J Bone Miner Res (2011) 26:2872–85. doi: 10.1002/jbmr.497
32. Lewis KJ, Cabahug-Zuckerman P, Boorman-Padgett JF, Basta-Pljakic J, Louie J, Stephen S, et al. Estrogen depletion on In vivo osteocyte calcium signaling responses to mechanical loading. Bone (2021) 152:116072. doi: 10.1016/j.bone.2021.116072
33. Cheng MZ, Zaman G, Rawlinson SC, Pitsillides AA, Suswillo RF, Lanyon LE. Enhancement by sex hormones of the osteoregulatory effects of mechanical loading and prostaglandins in explants of rat ulnae. J Bone Miner Res (1997) 12:1424–30. doi: 10.1359/jbmr.1997.12.9.1424
34. Joldersma M, Klein-Nulend J, Oleksik AM, Heyligers IC, Burger EH. Estrogen enhances mechanical stress-induced prostaglandin production by bone cells from elderly women. Am J Physiol Endocrinol Metab (2001) 280:E436–42. doi: 10.1152/ajpendo.2001.280.3.E436
35. Rauch F. Bone growth in length and width: the Yin and Yang of bone stability. J Musculoskelet Neuronal Interact (2005) 5:194–201.
36. Warden SJ, Fuchs RK, Castillo AB, Nelson IR, Turner CH. Exercise when young provides lifelong benefits to bone structure and strength. J Bone Miner Res (2007) 22:251–9. doi: 10.1359/jbmr.061107
37. Nikander R, Sievanen H, Heinonen A, Daly RM, Uusi-Rasi K, Kannus P. Targeted exercise against osteoporosis: A systematic review and meta-analysis for optimising bone strength throughout life. BMC Med (2010) 8:47. doi: 10.1186/1741-7015-8-47
38. Kontulainen S, Sievanen H, Kannus P, Pasanen M, Vuori I. Effect of long-term impact-loading on mass, size, and estimated strength of humerus and radius of female racquet-sports players: a peripheral quantitative computed tomography study between young and old starters and controls. J Bone Miner Res (2002) 17:2281–9. doi: 10.1359/jbmr.2002.17.12.2281
39. Daily RM, Petit MA. Optimizing bone mass and strength: The role of physical activity and nutrition during growth (Medicine & Sport science, vol 51). J Sports Sci Med (2007) 51:381. doi: 10.1159/isbn.978-3-318-01459-4
40. Cauley JA, Cawthon PM, Peters KE, Cummings SR, Ensrud KE, Bauer DC, et al. Risk factors for hip fracture in older men: the osteoporotic fractures in men study (MrOS). J Bone Miner Res (2016) 31:1810–9. doi: 10.1002/jbmr.2836
41. Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone (2007) 40:1434–46. doi: 10.1016/j.bone.2007.03.017
42. Dobnig H, Turner RT. Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology (1995) 136:3632–8. doi: 10.1210/endo.136.8.7628403
43. Kim SW, Pajevic PD, Selig M, Barry KJ, Yang JY, Shin CS, et al. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J Bone Miner Res (2012) 27:2075–84. doi: 10.1002/jbmr.1665
44. Rooney AM, Dempster DW, Nieves JW, Zhou H, Bostrom MPG, Cosman F. Effects of teriparatide and loading modality on modeling-based and remodeling-based bone formation in the human femoral neck. Bone (2022) 157:116342. doi: 10.1016/j.bone.2022.116342
45. Cosman F, Dempster DW, Nieves JW, Zhou H, Zion M, Roimisher C, et al. Effect of teriparatide on bone formation in the human femoral neck. J Clin Endocrinol Metab (2016) 101:1498–505. doi: 10.1210/jc.2015-3698
46. Dempster DW, Zhou H, Recker RR, Brown JP, Recknor CP, Lewiecki EM, et al. Remodeling- and modeling-based bone formation with teriparatide versus denosumab: A longitudinal analysis from baseline to 3 months in the AVA study. J Bone Miner Res (2018) 33:298–306. doi: 10.1002/jbmr.3309
47. Agarwal S, Shiau S, Kamanda-Kosseh M, Bucovsky M, Kil N, Lappe JM, et al. Teriparatide followed by denosumab in premenopausal idiopathic osteoporosis: Bone microstructure and strength by HR-pQCT. J Bone Miner Res (2022) 38(1):35–47. doi: 10.1002/jbmr.4739
48. Miyauchi A, Matsumoto T, Sugimoto T, Tsujimoto M, Warner MR, Nakamura T. Effects of teriparatide on bone mineral density and bone turnover markers in Japanese subjects with osteoporosis at high risk of fracture in a 24-month clinical study: 12-month, randomized, placebo-controlled, double-blind and 12-month open-label phases. Bone (2010) 47:493–502. doi: 10.1016/j.bone.2010.05.022
49. Stepan JJ, Burr DB, Li J, Ma YL, Petto H, Sipos A, et al. Histomorphometric changes by teriparatide in alendronate-pretreated women with osteoporosis. Osteoporos Int (2010) 21:2027–36. doi: 10.1007/s00198-009-1168-7
50. Lindsay R, Krege JH, Marin F, Jin L, Stepan JJ. Teriparatide for osteoporosis: importance of the full course. Osteoporos Int (2016) 27:2395–410. doi: 10.1007/s00198-016-3534-6
51. Sims NA, Martin TJ. Coupling Signals between the Osteoclast and Osteoblast: How are Messages Transmitted between These Temporary Visitors to the Bone Surface? Front Endocrinol (Lausanne) (2015) 6:41. doi: 10.3389/fendo.2015.00041
52. Ausk BJ, Huber P, Srinivasan S, Bain SD, Kwon RY, McNamara EA, et al. Metaphyseal and diaphyseal bone loss in the tibia following transient muscle paralysis are spatiotemporally distinct resorption events. Bone (2013) 57:413–22. doi: 10.1016/j.bone.2013.09.009
53. Fukumoto S, Matsumoto T. Recent advances in the management of osteoporosis. F1000Res (2017) 6:625. doi: 10.12688/f1000research.10682.1
54. Wang M, Wu YF, Girgis CM. Bisphosphonate drug holidays: Evidence from clinical trials and real-world studies. JBMR Plus (2022) 6:e10629. doi: 10.1002/jbm4.10629
55. Black DM, Rosen CJ. Clinical practice. Postmenopausal Osteoporosis. N Engl J Med (2016) 374:254–62. doi: 10.1056/NEJMcp1513724
56. Leder BZ, Neer RM, Wyland JJ, Lee HW, Burnett-Bowie SM, Finkelstein JS. Effects of teriparatide treatment and discontinuation in postmenopausal women and eugonadal men with osteoporosis. J Clin Endocrinol Metab (2009) 94:2915–21. doi: 10.1210/jc.2008-2630
57. McClung MR, Wagman RB, Miller PD, Wang A, Lewiecki EM. Observations following discontinuation of long-term denosumab therapy. Osteoporos Int (2017) 28:1723–32. doi: 10.1007/s00198-017-3919-1
58. Cummings SR, Ferrari S, Eastell R, Gilchrist N, Jensen JB, McClung M, et al. Vertebral fractures after discontinuation of denosumab: A post hoc analysis of the randomized placebo-controlled FREEDOM trial and its extension. J Bone Miner Res (2018) 33:190–8. doi: 10.1002/jbmr.3337
59. Tsai JN, Uihlein AV, Lee H, Kumbhani R, Siwila-Sackman E, McKay EA, et al. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: The DATA study randomised trial. Lancet (2013) 382:50–6. doi: 10.1016/S0140-6736(13)60856-9
60. Tsai JN, Uihlein AV, Burnett-Bowie SM, Neer RM, Derrico NP, Lee H, et al. Effects of two years of teriparatide, denosumab, or both on bone microarchitecture and strength (DATA-HRpQCT study). J Clin Endocrinol Metab (2016) 101:2023–30. doi: 10.1210/jc.2016-1160
61. Tsai JN, Uihlein AV, Burnett-Bowie SA, Neer RM, Zhu Y, Derrico N, et al. Comparative effects of teriparatide, denosumab, and combination therapy on peripheral compartmental bone density, microarchitecture, and estimated strength: the DATA-HRpQCT Study. J Bone Miner Res (2015) 30:39–45. doi: 10.1002/jbmr.2315
62. Ramchand SK, Seeman E. Reduced bone modeling and unbalanced bone remodeling: Targets for antiresorptive and anabolic therapy. Handb Exp Pharmacol (2020) 262:423–50. doi: 10.1007/164_2020_354
63. Fang Y, Morse LR, Nguyen N, Battaglino RA, Goldstein RF, Troy KL. Functional electrical stimulation (FES)-assisted rowing combined with zoledronic acid, but not alone, preserves distal femur strength and stiffness in people with chronic spinal cord injury. Osteoporos Int (2021) 32:549–58. doi: 10.1007/s00198-020-05610-x
64. Sibonga J, Matsumoto T, Jones J, Shapiro J, Lang T, Shackelford L, et al. Resistive exercise in astronauts on prolonged spaceflights provides partial protection against spaceflight-induced bone loss. Bone (2019) 128:112037. doi: 10.1016/j.bone.2019.07.013
65. Konda NN, Karri RS, Winnard A, Nasser M, Evetts S, Boudreau E, et al. A comparison of exercise interventions from bed rest studies for the prevention of musculoskeletal loss. NPJ Microgravity (2019) 5:12. doi: 10.1038/s41526-019-0073-4
66. Swift JM, Swift SN, Nilsson MI, Hogan HA, Bouse SD, Bloomfield SA. Cancellous bone formation response to simulated resistance training during disuse is blunted by concurrent alendronate treatment. J Bone Miner Res (2011) 26:2140–50. doi: 10.1002/jbmr.407
67. Leblanc A, Matsumoto T, Jones J, Shapiro J, Lang T, Shackelford L, et al. Bisphosphonates as a supplement to exercise to protect bone during long-duration spaceflight. Osteoporos Int (2013) 24:2105–14. doi: 10.1007/s00198-012-2243-z
68. Abe K, Miyakoshi N, Kasukawa Y, Nozaka K, Tsuchie H, Sato C, et al. Effects of teriparatide and low-intensity aerobic exercise on osteopenia in type 2 diabetes mellitus rats. J Bone Miner Metab (2022) 40:229–39. doi: 10.1007/s00774-021-01289-0
69. Sato C, Miyakoshi N, Kasukawa Y, Nozaka K, Tsuchie H, Nagahata I, et al. Teriparatide and exercise improve bone, skeletal muscle, and fat parameters in ovariectomized and tail-suspended rats. J Bone Miner Metab (2021) 39:385–95. doi: 10.1007/s00774-020-01184-0
70. Jepsen DB, Ryg J, Hansen S, Jørgensen NR, Gram J, Masud T. The combined effect of Parathyroid hormone (1-34) and whole-body Vibration exercise in the treatment of postmenopausal OSteoporosis (PaVOS study): a randomized controlled trial. Osteoporos Int (2019) 30:1827–36. doi: 10.1007/s00198-019-05029-z
Keywords: endocrine, mechanobiology, mechanostat, hormones, growth, osteoanabolic therapy
Citation: Hughes JM, Guerriere KI, Popp KL, Castellani CM and Pasiakos SM (2023) Exercise for optimizing bone health after hormone-induced increases in bone stiffness. Front. Endocrinol. 14:1219454. doi: 10.3389/fendo.2023.1219454
Received: 09 May 2023; Accepted: 17 August 2023;
Published: 18 September 2023.
Edited by:
Katarina Tomljenovic Borer, The University of Michigan, United StatesReviewed by:
Gregory Clines, University of Michigan, United StatesCopyright © 2023 Hughes, Guerriere, Popp, Castellani and Pasiakos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie M. Hughes, SnVsaWUubS5odWdoZXMxNy5jaXZAaGVhbHRoLm1pbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.