 Jialin Li
Jialin Li Fenglan Zhang
Fenglan Zhang Miao Xu1
Miao Xu1 Li Li
Li Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 13 November 2023
Sec. Pediatric Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1216767
Introduction: 11β-Hydroxylase deficiency (11β-OHD, OMIM#202010) is the second most common form of congenital adrenal hyperplasia (CAH) caused by pathogenic variants in the CYP11B1 gene. Both single nucleotide variations (SNV)/small insertion and deletion and genomic rearrangements of CYP11B1 are important causes of 11β-OHD. Among these variant types, pathogenic CYP11B2/CYP11B1 chimeras only contribute to a minority of cases. Heterozygote cases (chimera combined with SNV) are very rare, and genetic analysis of these cases can be challenging.
Case presentation: We presented a suspected 11β-OHD female patient with incomplete virilization, adrenal hyperplasia, and hypokalemia hypertension. Whole exome sequencing (WES) revealed that the patient carried both a chimeric CYP11B2/CYP11B1 and a novel missense variant, NM_000497.4: c.203T>G, p.Val68Gly (chr8:143961027) in CYP11B1, which were confirmed by CNVplex and Sanger sequencing, respectively. The patient’s manifestations and genetic findings confirmed the diagnosis of 11β-OHD, and oral dexamethasone was administered as a subsequent treatment.
Conclusion: This report showed a rare CYP11B2/CYP11B1 chimera combined with a novel missense variant in a 11β-OHD female patient. The result expands variant spectrum of CYP11B1 and suggests that both chimera and CYP11B1 variant screening should be performed simultaneously in suspected cases of 11β-OHD. To our knowledge, this is the first report about CYP11B2/CYP11B1 chimera detected by WES analysis. WES combined with CNV analysis is an efficient method in the genetic diagnosis of this rare and complex disorder.
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder caused by a deficiency in enzymes required for the synthesis of cortisol from cholesterol (1). The most common form of CAH, accounting for 95% of cases, is 21-hydroxylase deficiency (21-OHD) (2). The second most common form of CAH is 11β-hydroxylase deficiency (11β-OHD), which accounts for approximately 5-8% of cases (3). 11β-hydroxylase converts 11-deoxycortisol and 11-deoxycorticosterone (DOC) to cortisol and corticosterone. Deficiencies in this enzyme lead to increased levels of 11-deoxycortisol and DOC, which are shunted into adrenal androgen synthesis pathways. The accumulation of DOC and testosterone causes hypertension and virilization in females or precocious puberty in males. Inadequate cortisol production stimulates the release of adrenocorticotrophic hormone (ACTH) as a compensatory mechanism, leading to subsequent adrenal hyperplasia (4, 5).
11β-hydroxylase and aldosterone synthase are encoded by the CYP11B1(OMIM#610613) and CYP11B2 (OMIM#124080) genes, respectively, both of which consist of nine exons and share 95% exonic sequence homology and 90% intronic sequence homology. These genes lie tandemly arranged approximately 40 kb apart on chromosome 8q24 (6). To date, more than 200 pathogenic/likely pathogenic alterations of the CYP11B1 gene associated with 11β-OHD have been reported in the ClinVar Database (https://www.ncbi.nlm.nih.gov/clinvar/). Most of the variants are missense, nonsense, frameshift, and splice variants. Moreover, the deletion of CYP11B1 or chimeric CYP11B2/CYP11B1 gene has been found in a few 11β-OHD patients (7).
With the extensive development of next-generation sequencing (NGS), whole exome sequencing (WES) has become the first-line diagnostic test in most monogenic disorders (8). Some algorithms have been designed to detect copy number variations (CNVs) based on the coverage depth of capture sequencing data, enabling the detection of CNVs larger than 200 kb. However, the reliability of these algorithms in detecting smaller CNVs is limited (9). The high degree of sequence similarity between CYP11B1 and its homologous gene CYP11B2 poses unique challenges for detecting small CYP11B1 deletions or chimeric CYP11B2/CYP11B1 through WES.
In this study, we reported a Chinese patient with classical manifestations of 11β-OHD resulting from compound heterozygous variants, including a novel missense variant NM_000497.4: c.203T>G, p.Val68Gly (chr8:143961027) in CYP11B1 and a rare chimeric CYP11B2/CYP11B1. This study expands the variant spectrum of CYP11B1 and demonstrates that a single WES test combined with WES based CNV analysis can be used effectively for the SNV/InDel identification and the chimeric CYP11B2/CYP11B1 analysis.
The patient is a Chinese woman (46, XX) from a non-consanguineous family. She has one healthy younger brother. She was taller than her peers during childhood, but her growth did not accelerate during subsequent adolescence. She did not experience her first menstrual period until the age of 20. She underwent surgical treatment for “abnormal external genitalia” due to sexual dysfunction, but the specific diagnosis and surgical procedure are unknown. She has been unable to conceive since her marriage at the age of 26. In May 2019, she was admitted to the hospital with suspected bilateral adrenal tumors. Abdominal computed tomographic scan revealed bilateral adrenal multiple nodular hyperplasia (Figure 1A). After two surgeries, the left and right adrenal tumors were successfully removed. Partial adrenal was preserved on both sides to minimize the risk of adrenal insufficiency.
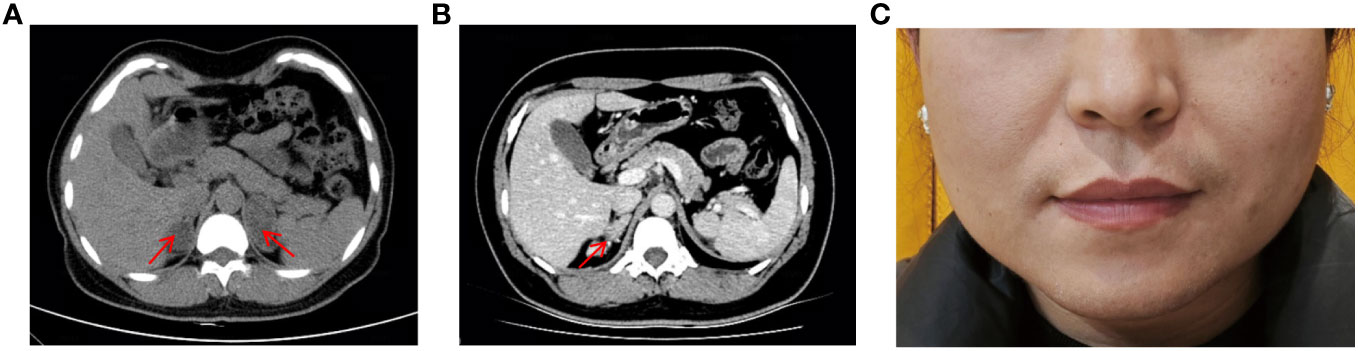
Figure 1 Masculine features and Abdominal CT scan of patient. (A) Abdominal CT scan showed bilateral adrenal hyperplasia, with a nodule on the right approximately 27×22mm and a nodule on the left approximately 42×28mm. (B) Abdominal CT scan revealed recurrence of the right adrenal gland hyperplasia two years after surgery. (C) Physical examination revealed greasy skin pigmentation, facial acne, and slight mustache on the upper lip.
After the second surgery, she experienced irregular menstruation. Two and a half years later, she was admitted to the endocrinology department. Abdominal-enhanced CT scan showed structural disorder in the right adrenal gland area, with spotted and striped shadows (Figure 1B). Physical examination revealed greasy skin pigmentation, facial acne, and slight mustache on the upper lip (Figure 1C). Physical examination showed hypertension (145/100 mmHg), laboratory data showed decreased plasma potassium and aldosterone but elevated levels of adrenocorticotrophic hormone (ACTH), 11-deoxycorticosterone (DOC), 17-hydroxyprogesterone (17-OHP), androstenedione, Dehydroepiandrosterone (DHEA), and testosterone. The results of 1-day medium-dose dexamethasone androgen suppression test showed that 17-OHP, ACTH, androstenedione, DHEA, and testosterone were significantly suppressed (10) (Table 1). The external manifestations and biochemical indicators of patient were all suggestive of an 11β-OHD diagnosis.
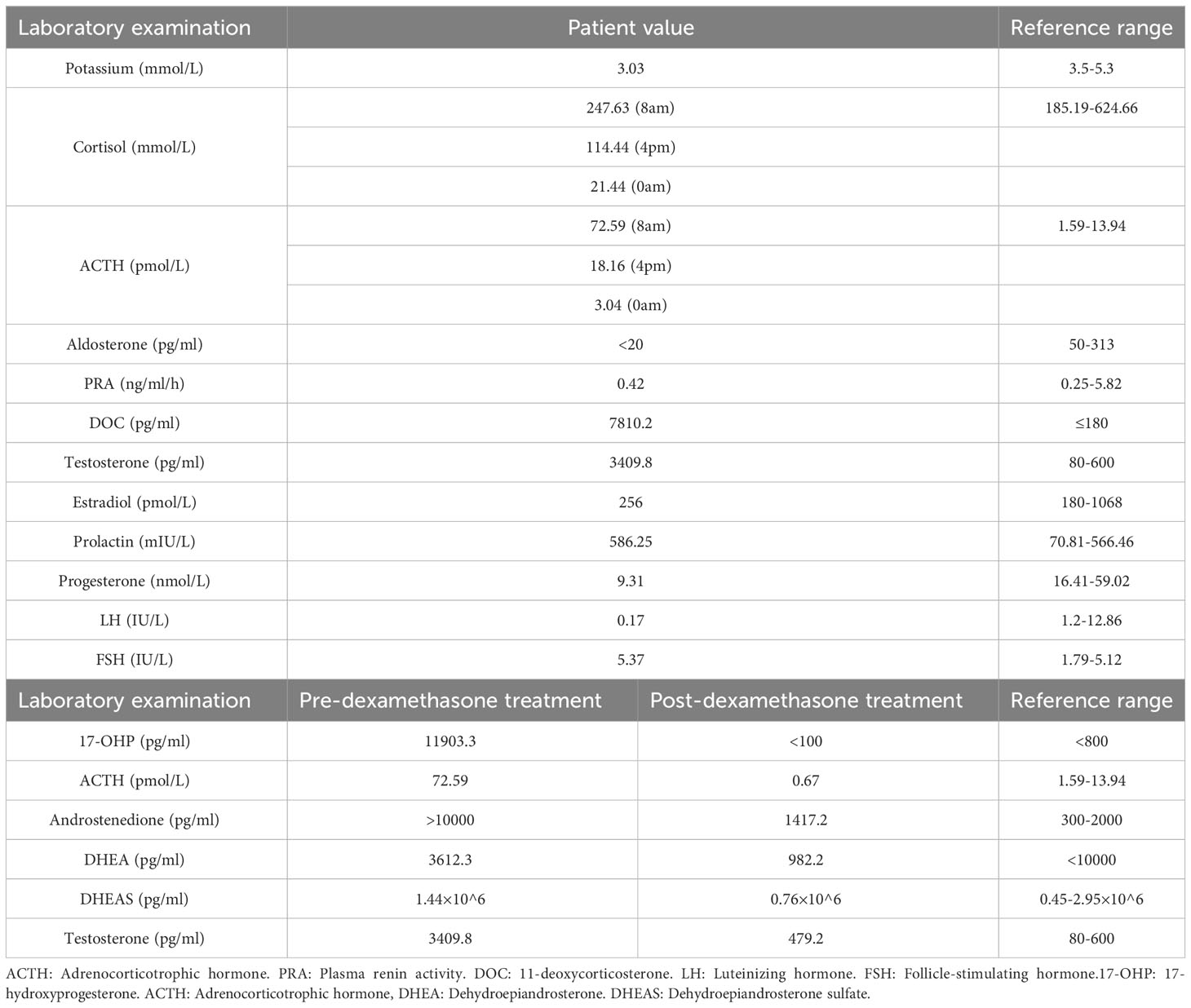
Table 1 The laboratory test of the patient.
To investigate the potential genetic pathogenic mechanism, whole exome sequencing was performed for the patient (Supplementary Materials and Methods). Initially, a novel homozygous missense variant in CYP11B1, NM_000497.4: c.203T>G (p.Val68Gly) was identified (Figure 2A). The newly identified missense variant was in exon 1. Then the variant was further confirmed by sanger sequencing (Figure 2B; Supplementary Materials and Methods). Furthermore, based on our WES-CNV analysis pipeline, a speculative CYP11B1 and CYP11B2 deletion was screened out (chr8:143957127-143994301). The deletion covers exon 1 to exon 6 of CYP11B1 and exon 7 to exon 9 of CYP11B2 (Figure 3A), resulting in the formation of a single hybrid gene consisting of the promoter and exons 1-6 of CYP11B2 and exons 7-9 of CYP11B1 (Figure 3B). Therefore, it’s reasonable to assume that p.Val68Gly of CYP11B1 is a heterozygous variant rather than a homozygous variant.
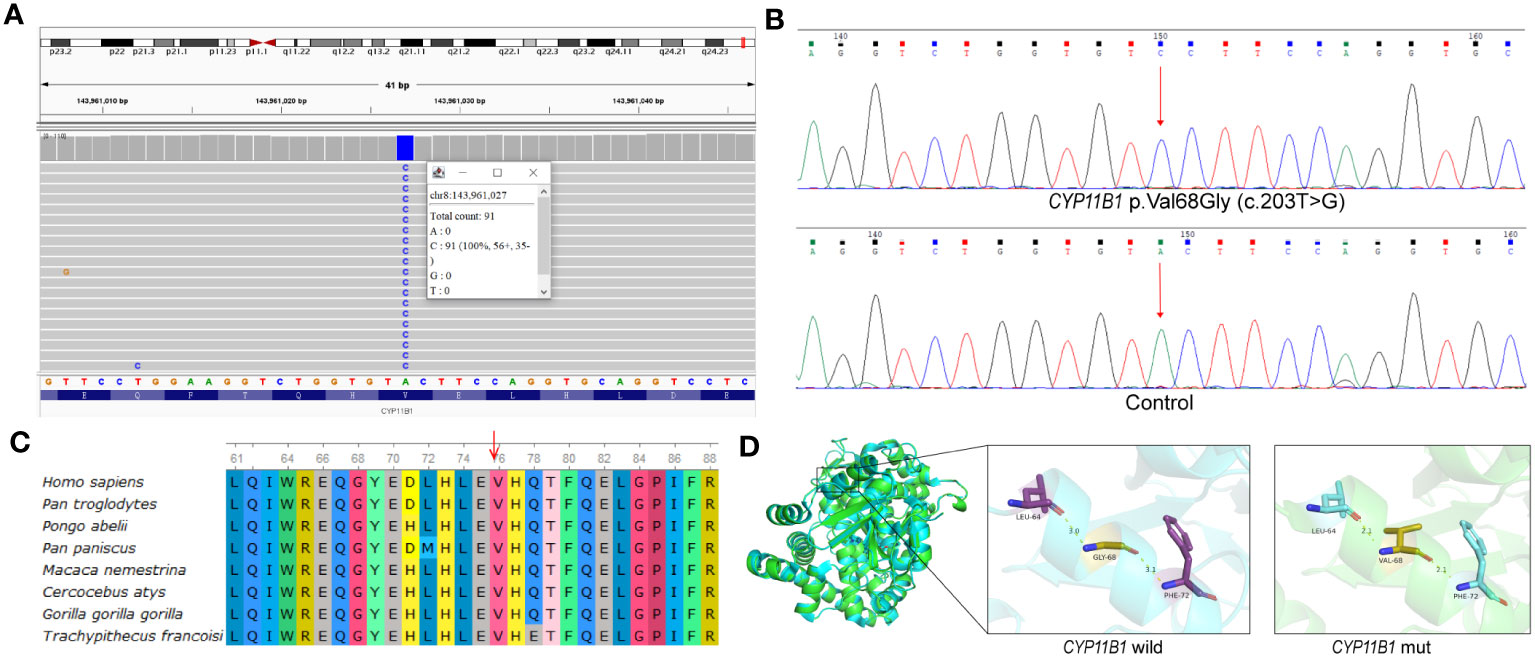
Figure 2 p.Val68Gly of the CYP11B1 in patient. (A) Visualization of variant in CYP11B1 using IGV. Apparently, the patient was a homozygous variant, c.203T>G, located in exon 1. (B) The sequencing chromatogram of the variant in CYP11B1. Arrow indicates mutant nucleotide (c.203T>G). (C) Conservation prediction of this mutant amino acid among different species. (D) Three-dimensional structure of wild-type CYP11B1 and mutant-type CYP11B1. The yellow dotted line represents hydrogen bond.

Figure 3 The chimeric CYP11B2/CYP11B1 gene in patient. (A) The deletion covering exons 1-6 of CYP11B1 and exons 7-9 in CYP11B2 by whole exome sequencing. The violin plot illustrates the distribution of sequencing depth for normal reference samples on each exon, with the ordinate is the Z-score standardization for the sequencing depth. The dotted lines in different colors represent the maximum, average, median and minimum of sequencing depth Z-score in normal references. Solid line represents the sequencing depth Z-score of CYP11B1 and CYP11B2 in the proband sample. Specifically, the sequencing depth Z-score in exons 7-9 of CYP11B2 and exon 1-6 of CYP11B1 is lower than the minimum value in the normal references. (B) Schematic representation of the CYP11B2 gene (exons displayed as grey boxes) and the CYP11B1 gene (exons displayed as white boxes). In the investigated patient, the CYP11B2/CYP11B1 chimera consisting of the promoter and exons 1–6 of CYP11B2 and exons 7–9 of CYP11B1. Arrows indicate the position of the 8 specific probes of CNVplex.(C) Graphic report of CNVplex result in the patient and control. Deletion occurred in the position of seven probes, except for the position of the last probe (located in the intron 2 of CYP11B2).
The p.Val68Gly variant has not been recorded in several databases, including ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), Human Gene Mutation Database (HGMD, https://www.hgmd.cf.ac.uk/), PubMed (https://pubmed.ncbi.nlm.nih.gov/), and MasterMind (https://mastermind.genomenon.com/), indicating that this variant is novel. The Genome Aggregation Database (gnomAD, http://gnomad-sg.org/) does not include the frequency of this variant in normal East Asian populations, which indicates that the allele frequency of this variant is extremely low. To evaluate the pathogenicity of the newly identified variant, several prediction tools were used, in which the REVEL score was 0.339, and the ClinPred score was 0.3473. SIFT and Polyphen2 predicted the variant to be damaging and benign, respectively. Multiple alignments of CYP11B1 suggest that the amino acidic residue, Val68, is conserved at this position across various species (Figure 2C). The prediction of protein three-dimensional structure revealed that Val68 residue locates in an α-helix, and p.Val68Gly causes a change in hydrogen bond length, which may lead to alterations in protein conformation and stability (Figure 2D). According to the ACMG guidelines, the classification for this variant was uncertain significance (PM2+PP3+PP4) (11). Due to the presence of a deletion-type allele, PM3 can be considered. Totally, it is recommended that the variant classification of NM_000497.4: c.203T>G (p.Val68Gly) in CYP11B1 variant can be upgraded to likely pathogenic (PM2+PM3+PP3+PP4).
CNVplex was used to validate the deletion (exon 1 to exon 6 of CYP11B1 and exon 7 to exon 9 in CYP11B2) (Supplementary Materials and Methods). The results showed significant copy number loss in 7 out of 8 groups of probes compared to the control group (Supplementary Table 1), while the other probe showed a normal copy number outside of the potential deletion region (Figure 3C). This result confirmed the reliability of this deletion recognized by WES-CNV analysis.
11β-Hydroxylase deficiency is the second most common cause of congenital adrenal hyperplasia (CAH), accounting for 5–8% after the more prevalent 21-hydroxylase deficiency. The clinical phenotypes of patients with 11β-OHD are complex and nonspecific (12, 13). Patients who do not receive a molecular diagnosis or an appropriate hormonal evaluation may be misdiagnosed as 21-hydroxylase deficiency or other adrenal hyperplasia (14). To determine the CAH classification accurately, a WES analysis and a long-range PCR based CYP21A2 sequencing were ordered simultaneously. However, the results of the long-range PCR for CYP21A2 did not reveal any variants. Whereas a novel p.Val68Gly variant and a chimeric CYP11B2/CYP11B1 were discovered on different alleles of CYP11B1 by WES.
CYP11B1 and CYP11B2 encoded homologues, and have distinct functions in cortisol and aldosterone synthesis, respectively. CYP11B2/CYP11B1 chimeric genes have been shown to arise from unequal crossing over of the CYP11B2 and CYP11B1 during meiosis. The activity deficiency or impaired activity of aldosterone synthase and 11β-hydroxylase resulting from these chimeric genes are important reasons for 11β-OHD (15). After reviewing previous reports on the chimeric CYP11B2/CYP11B1 gene, we collected data on twelve patients with the chimera (Supplementary Table 2). Six of these patients harbored the chimeric CYP11B2/CYP11B1 gene located in intron 6 of CYP11B2. Our patient carried the same pathogenic CYP11B2/CYP11B1 chimera, suggesting that it may be a popular rearrangement event in 11β-OHD patients. Interestingly, the other allele of CYP11B1 contained a new disease-causing variation, p.Val68Gly, in our patient. Although we were unable to obtain blood samples from the patient’s parents and brother for pedigree study, the presence of a deletion-type allele confirmed that the patient harbors a compound heterozygous variation. In the future, further functional analysis for this missense variant will be meaningful.
Previous studies on molecular genetic testing for CYP11B1 variants mainly used CYP11B1-specific PCR with the aid of several key SNPs between CYP11B1 and CYP11B2 (16–18). However, identifying these previous hybrid genes was time-consuming and not feasible for all laboratories, such as southern blot. In 2015, Menabò S used homemade MLPA probes to identify a novel chimeric CYP11B2/CYP11B1 gene in a 11β-OHD patient (19), but this MLPA method has not been widely adopted by genetic laboratories. The first report of a chimeric CYP11B2/CYP11B1 detected by next-generation sequencing, in which 276 genes associated with adrenal diseases were captured, was published in 2022 (20). In this study, whole exome sequencing, a more general method, was used to detect CYP11B1 variants and the chimera simultaneously, which, to our knowledge, is the first report.
In conclusion, a novel missense variant, p.Val68Gly, and a rare chimeric CYP11B2/CYP11B1 gene were simultaneously detected by WES analysis in the suspected 11β-OHD patient, which is consistent with the clinical phenotype. These results indicate that WES is an effective molecular genetic test for detecting SNV/Indel and copy number variations. This study has expanded the variant spectrum of CYP11B1, contributing to early and accurate diagnosis and treatment of 11β-OHD patients, and ultimately promoting better genetic counseling. However, due to the rarity of chimeric variants, there is only one patient in our research, which indicates that further research and validation in larger patient cohorts is still needed in the future.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The studies involving humans were approved by the ethics committee of the First Affiliated Hospital of Ningbo University, China. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
JL and FZ designed the study. JL, MX, CZ, and LL diagnosed the patient, provided follow-up, and acquired clinical data. FZ, HQ, JL, and LQ wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Ningbo Key Clinical Specialty (Endocrinology) (Grant No. 2022-B07).
LQ, FZ, and HQ were employed by Dian Diagnostics Group Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1216767/full#supplementary-material
1. Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, Arlt W, Auchus RJ, Falhammar H, et al. Congenital adrenal hyperplasia-current insights in pathophysiology, diagnostics, and management. Endocr Rev (2022) 43(1):91–159. doi: 10.1210/endrev/bnab016
2. Arriba M, Ezquieta B. Molecular diagnosis of steroid 21-hydroxylase deficiency: A practical approach. Front Endocrinol (Lausanne) (2022) 13:834549. doi: 10.3389/fendo.2022.834549
3. Bulsari K, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Endocr (2017) 55(1):19–36. doi: 10.1007/s12020-016-1189-x
4. Khattab A, Haider S, Kumar A, Dhawan S, Alam D, Romero R, et al. Clinical, genetic, and structural basis of congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Proc Natl Acad Sci U S A (2017) 114(10):E1933–40. doi: 10.1073/pnas.1621082114
5. Nimkarn S, New MI. Steroid 11β- hydroxylase deficiency congenital adrenal hyperplasia. Trends Endocrinol Metab (2008) 19(3):96–9. doi: 10.1016/j.tem.2008.01.002
6. Schiffer L, Anderko S, Hannemann F, Eiden-Plach A, Bernhardt R. The CYP11B subfamily. J Steroid Biochem Mol Biol (2015) 151:38–51. doi: 10.1016/j.jsbmb.2014.10.011
7. Zhao LQ, Han S, Tian HM. Progress in molecular-genetic studies on congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency. World J Pediatr (2008) 4(2):85–90. doi: 10.1007/s12519-008-0016-8
8. Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. The next-generation sequencing revolution and its impact on genomics. Cell (2013) 155(1):27–38. doi: 10.1016/j.cell.2013.09.006
9. Testard Q, Vanhoye X, Yauy K, Naud ME, Vieville G, Rousseau F, et al. Exome sequencing as a first-tier test for copy number variant detection: retrospective evaluation and prospective screening in 2418 cases. J Med Genet (2022) 59(12):1234–40. doi: 10.1136/jmg-2022-108439
10. Hao D, Lin L, Xiaoping X, Linjie W, Lian D, Jun J, et al. Efficacy of medium dose dexamethasone androgen suppression test in the diagnosis of female hyperandrogenism. Natl Med J China (2018) 98(26):2073–7. doi: 10.3760/cma.j.issn.0376-2491.2018.26.003
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
12. Yildiz M, Isik E, Abali ZY, Keskin M, Ozbek MN, Bas F, et al. Clinical and hormonal profiles correlate with molecular characteristics in patients with 11β-hydroxylase deficiency. J Clin Endocrinol Metab (2021) 106(9):e3714–24. doi: 10.1210/clinem/dgab225
13. Parajes S, Loidi L, Reisch N, Dhir V, Rose IT, Hampel R, et al. Functional consequences of seven novel mutations in the CYP11B1 gene: four mutations associated with nonclassic and three mutations causing classic 11{beta}-hydroxylase deficiency. J Clin Endocrinol Metab (2010) 95(2):779–88. doi: 10.1210/jc.2009-0651
14. Wu C, Zhou Q, Wan L, Ni L, Zheng C, Qian Y, et al. Novel homozygous p.R454C mutation in the CYP11B1 gene leads to 11β-hydroxylase deficiency in a Chinese patient. Fertil Steril (2011) 95(3):1122.e3–6. doi: 10.1016/j.fertnstert.2010.09.035
15. Xiong Y, Zeng Z, Liang T, Yang P, Lu Q, Yang J, et al. Unequal crossing over between CYP11B2 and CYP11B1 causes 11 β -hydroxylase deficiency in a consanguineous family. J Steroid Biochem Mol Biol (2023) 233:106375. doi: 10.1016/j.jsbmb.2023.106375
16. Hampf M, Dao NT, Hoan NT, Bernhardt R. Unequal crossing-over between aldosterone synthase and 11beta-hydroxylase genes causes congenital adrenal hyperplasia. J Clin Endocrinol Metab (2001) 86(9):4445–52. doi: 10.1210/jcem.86.9.7820
17. Ezquieta B, Luzuriaga C. Neonatal salt-wasting and 11 β-hydroxylase deficiency in a child carrying a homozygous deletion hybrid CYP11B2 (aldosterone synthase)–CYP11B1 (11 β-hydroxylase). Clin Genet (2004) 66(3):229–35. doi: 10.1111/j.1399-0004.2004.00291.x
18. Portrat S, Mulatero P, Curnow KM, Chaussain JL, Morel Y, Pascoe L. Deletion hybrid genes, due to unequal crossing over between CYP11B1 (11β-hydroxylase) and CYP11B2(Aldosterone synthase) cause steroid 11β-hydroxylase deficiency and congenital adrenal hyperplasia1. J Clin Endocrinol Metab (2001) 86(7):3197–201. doi: 10.1210/jcem.86.7.7671
19. Menabò S, Boccassini S, Gambineri A, Balsamo A, Pasquali R, Prontera O, et al. Improving the diagnosis of 11β-hydroxylase deficiency using home-made MLPA probes: identification of a novel chimeric CYP11B2/CYP11B1 gene in a Sicilian patient. J Endocrinol Invest (2016) 39(3):291–5. doi: 10.1007/s40618-015-0362-z
Keywords: 11β-hydroxylase deficiency, molecular diagnosis, whole exome sequencing, chimeric CYP11B2/CYP11B1, missense variant
Citation: Li J, Zhang F, Xu M, Qiu H, Zhou C, Li L and Qin L (2023) Case Report: A combination of chimeric CYP11B2/CYP11B1 and a novel p.Val68Gly CYP11B1 variant causing 11β-Hydroxylase deficiency in a Chinese patient. Front. Endocrinol. 14:1216767. doi: 10.3389/fendo.2023.1216767
Received: 04 May 2023; Accepted: 25 October 2023;
Published: 13 November 2023.
Edited by:
Antonio Balsamo, Researcher of Alma Mater Studiorum - University Hospital S.Orsola Malpighi, ItalyReviewed by:
Paola Concolino, Agostino Gemelli University Polyclinic (IRCCS), ItalyCopyright © 2023 Li, Zhang, Xu, Qiu, Zhou, Li and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lan Qin, cWlubGFuQGRhemQuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.