 Sanjay Basak
Sanjay Basak Saikanth Varma
Saikanth Varma Asim K. Duttaroy
Asim K. Duttaroy- 1Molecular Biology Division, ICMR-National Institute of Nutrition, Indian Council of Medical Research, Hyderabad, India
- 2Department of Nutrition, Institute of Basic Medical Sciences, Faculty of Medicine, University of Oslo, Oslo, Norway
Maternal endocrine homeostasis is vital to a successful pregnancy, regulated by several hormones such as human chorionic gonadotropin, estrogen, leptin, glucocorticoid, insulin, prostaglandin, and others. Endocrine stress during pregnancy can modulate nutrient availability from mother to fetus, alter fetoplacental growth and reproductive functions. Endocrine disrupters such as bisphenols (BPs) and phthalates are exposed in our daily life's highest volume. Therefore, they are extensively scrutinized for their effects on metabolism, steroidogenesis, insulin signaling, and inflammation involving obesity, diabetes, and the reproductive system. BPs have their structural similarity to 17-β estradiol and their ability to bind as an agonist or antagonist to estrogen receptors to elicit an adverse response to the function of the endocrine and reproductive system. While adults can negate the adverse effects of these endocrine-disrupting chemicals (EDCs), fetuses do not equip themselves with enzymatic machinery to catabolize their conjugates. Therefore, EDC exposure makes the fetoplacental developmental window vulnerable to programming in utero. On the one hand prenatal BPs and phthalates exposure can impair the structure and function of the ovary and uterus, resulting in placental vascular defects, inappropriate placental expression of angiogenic growth factors due to altered hypothalamic response, expression of nutrient transporters, and epigenetic changes associated with maternal endocrine stress. On the other, their exposure during pregnancy can affect the offspring's metabolic, endocrine and reproductive functions by altering fetoplacental programming. This review highlights the latest development in maternal metabolic and endocrine modulations from exposure to estrogenic mimic chemicals on subcellular and transgenerational changes in placental development and its effects on fetal growth, size, and metabolic & reproductive functions.
1 Introduction
Pregnancy hormones such as estrogen, human chorionic gonadotropin (hCG), glucocorticoid, leptin, insulin, prostaglandin, and others are vital to a successful pregnancy. There are several risks associated with pregnancy-related complications, but one among them is the exposure of the mother and fetus to the modulators of pregnancy hormones by endocrine disruptors (1). Endocrine-disrupting chemicals (EDCs) can mimic the body’s hormonal response over life-long exposure. Among these thousand chemicals, endocrine disruptors such as bisphenols (BPs) and phthalates are abundant and present in substances contaminated daily, including food surfaces, cosmetics, and many more. The mechanisms underlying the endocrine stressors during critical development phases on predisposing adult disease risk are evolving (2). Epigenetic modification ensures developmental plasticity that allows adaptive regulation of the developing fetus at the expense of altered placental functions in response to EDC exposure. Normal placentation is the key to a healthy fetus. At the same time, exposure to EDCs can modulate placental functions by altering placental invasion and angiogenesis required for successful placentation (3). Placenta is the active barrier between the mother and fetus and involves waste disposal, gaseous exchange, immunogenic barrier, and hormone production. EDCs can affect placental barrier functions, structure, and stability of gene expression due to maternal exposure during pregnancy that modulate maternal endocrine balance, implantation processes, and fetus organ development (4).
BPA is an estrogenic chemical that disrupts the development programming of reproductive function through changes in gonadal hormones (5). Prenatal phthalate exposure disrupts the hormonal balance in pregnant women and transmits its risks to their offspring (6). In addition, exposure to EDCs predisposes the risk of preterm birth and altered intrauterine growth, gestational length, birth weight, ponderal index, femur length, abdominal circumference, head circumference, and anogenital index in newborns. Scientific investigations have consistently shown the interplay between epigenetics, lifestyle exposure, and the gestational environment. The epigenetic changes due to exposure to EDCs during critical windows of gestation and its effects on gastrulation, fetal organogenesis, and overall growth and development are emerging. The critical window of EDC exposure during pregnancy can disrupt the programming of organ development that could have long-lasting effects. Fetal exposure to these EDCs, such as BPs (bisphenol A, bisphenol S), Phthalates (diethylhexyl phthalate, DEHP and their metabolites) are suspected of altering the programming of normal biological functions, including endocrine imbalance and fertility. This review consolidates pregnancy-linked hormonal balance in response to EDC exposure, the endocrine effects on placentation, epigenetic changes, and maternal endocrine regulation on fetal growth and reproductive development.
2 Exposure to endocrine disruptors affects maternal endocrine balance and fetal growth, development, and pregnancy outcomes
Pregnancy is a complex physiological process regulated by various hormones and signalling molecules within the endocrine system. The maternal endocrine system changes during pregnancy to support fetal growth and development that involves changes in several axes, including hypothalamic-pituitary-adrenal (HPA), thyroid, and gonads. The HPA axis is crucial in regulating the maternal endocrine system during pregnancy. The hypothalamus secretes the corticotrophin-releasing hormone, which stimulates the pituitary to release adrenocorticotropic hormone (ACTH). ACTH, in turn, stimulates the adrenal glands to produce cortisol, an essential hormone for fetal development. However, excessive cortisol production can lead to adverse pregnancy outcomes, such as premature birth and low birth weight (7). The thyroid regulates metabolism, growth, and development during pregnancy. The thyroid gland produces two hormones, triiodothyronine (T3) and thyroxine (T4), essential for fetal brain development. The demand for thyroid hormones increases during pregnancy, and the maternal thyroid gland enlarges to meet this demand. In addition, the placenta produces hCG, which stimulates the maternal thyroid gland to produce more thyroid hormones. However, excessive thyroid production can lead to hyperthyroidism, negatively affecting the mother and fetus. Therefore, tight regulation of the thyroid axis is essential in protecting the mother during pregnancy (8).
The reproductive organs undergo significant changes during pregnancy to support fetal growth. The ovaries stop producing eggs, and the placenta takes over the production of estrogen and progesterone, essential for maintaining pregnancy. Placenta does not have all the necessary enzymes to convert cholesterol to estrogen or make progesterone (9). Estrogen and progesterone promote the growth of the uterus and help maintain the pregnancy by relaxing the uterus and preventing contractions. In addition, estrogen promotes the development of the fetal lungs, liver, and immune system. However, excessive estrogen production can lead to adverse outcomes, such as preeclampsia and gestational diabetes. Therefore, tight estrogen regulation is essential for optimum pregnancy outcomes (10).
Placenta acts as an endocrine organ during gestation, secretes several hormones essential for adapting maternal physiology, and transfers nutrients and gases to the growing fetus, thereby supporting fetal development (11). The trophoblast of the developing placenta produces hormones to ensure that the endometrium’s uterus lining can receive them for embryo implantation. As the gestation progress, the placental hormones control maternal physiology to supply adequate oxygen and nutrients to support fetus growth and development. Syncytiotrophoblasts are the cells that produce placental hormones in a tightly regulated manner. Several hormones regulate fetoplacental growth, including glucocorticoid, leptin, gonadotrophin, insulin, insulin growth factor, prostaglandin, and others.
Bisphenols can cross the placenta from maternal blood into the fetal compartment and are detectable in the amniotic fluid and cord blood, indicating exposure to developing fetuses (12). BPA exposure during a critical developmental window can affect trophoblast proliferation, migration, invasion, fusion, apoptosis, and placental morphology. In-vitro studies have shown that placental JEG-3 cells treated with low doses of BPA altered steroidogenic genes CYP11A1 and CYP19 (13). In addition, trophoblast cells (BeWo) increased β-hCG secretion and trophoblast cell fusion upon treatment with BPA (14). The exposure to low doses of BPA in pregnant mice from day 1-7 caused degenerative changes in the placenta's trophoblastic giant cells and spongiotrophoblast layers.
Moreover, the placenta's intervillous spaces were narrower in BPA-exposed mice (15). The pregnant CD-1 mice administered with BPA from gD 1 to 11 affected placental angiogenesis and caused necrosis and degeneration of giant cells in early pregnancy (16). In mice, exposure to BPA during preimplantation affects nutrient transport in embryo, preimplantation embryo development, and uterine receptivity (17).
Additionally, the progeny of mothers exposed to BPA was growth-restricted compared to the controls (18). Female rats orally administered with BPA showed reduced lumen diameter, acetylcholine-relaxation, and expressions of endothelial nitric oxide synthase 3 (NOS3), estrogen receptor α (ERα), and peroxisome proliferator-activated receptor ɣ (PPARɣ) in the uterine arteries (19). The low levels of BPA exposure have decreased the trophoblast invasion by altering the balance between the MMPs/TIMP in BeWo cells (20). Similar findings were reported in the in-vivo study conducted on pregnant mice exposed to BPA, which exhibited pre-eclampsia-like features like decreased trophoblast cell invasion, increased retention of smooth muscle cells, and a decreased vessel area at the junctional zone of the placenta (21). BPA exposure to the placenta is also reported to alter its metabolic state by altering the levels of GLUT1 expression in the villous explants (22) and placental HTR8/SVneo cells (23).
Increased expression of glucose transporters in the placenta promoted excess glucose transverse to the fetus, induce fetal hyperglycemia, and cause associated developmental defects. Pre- and post-conceptional exposure to bisphenols decreased the area occupied by a spongiotrophoblast relative to trophoblast giant cells within the junctional zone and markedly reduced placental serotonin (5-HT) concentrations. In addition, they lowered the 5-HT GC immunoreactivity (24). BPS-exposed placentas had lower E-cadherin expression, fewer binucleate cells, and higher glial cell missing-1 protein expression. Interestingly, BPA exposed placentas were unaffected, highlighting the intrinsic differences among bisphenol chemicals (25). This evidence suggests that exposure to BPA during pregnancy may adversely affect placental function and fetal development. Thus, due to EDC exposure, fetal development could be impaired due to altered placental response involving its growth, hormonal balance, gene expression, epigenetic modification, and angiogenic and invasive activities.
Placenta is the critical endocrine organ whose formation and development dictate the fate of a successful pregnancy. Placenta plays a vital role during gestation, including implantation of the fetus to the uterine wall, placentation, hormone secretion, uterine artery remodelling, gaseous exchange, transport of nutrients, removal of waste materials, and parturition. Any disruption or imbalances in hormonal production can adversely affect fetoplacental development. Data show that exposure to endocrine-disrupting chemicals like bisphenols A and their substitutions (BPS, BPF, TBBPA, BADGE) affect early development stages. Early developmental involves implantation, trophoblast invasion, and remodelling are modulated by BPA (16, 17, 26–32), BPS (33, 34), BPF (32, 34), and TBBPA (35) exposure. The trophoblast Syncytialization and nutrient exchange across the placenta are affected by BPA (22, 23), and BPS (36) exposure. Feto-placental growth outcomes such as intrauterine and fetal growth, & weight, ponderal index, femur length, abdominal circumference, head circumference, and ponderal index are altered by several EDC exposure, including BPA (23, 24, 37–41), BPS (24, 25, 39, 42), BPF (39), TBBPA (40, 43, 44), and BADGE (45).
2.1 Effects of maternal glucocorticoids on the fetoplacental growth and development
Endocrine stress elicits metabolic responses through the centrally coordinating neuroendocrine by activating the HPA-axis through hormonal signalling, leading to the systemic release of glucocorticoid (GC) from the adrenal (kidney) in humans (46). GCs act as maturation signal to sense EDC exposure and regulates intrauterine programming of adult physiological phenotype. Excess exposure to endogenous GCs activates maternal HPA by augmenting nutritional and stress response during pregnancy and increases fetal GC concentration. Fetal GC can be elevated independently of the mother to compensate reduced supply of nutrients and oxygen or by the changes in the placental activity of the 11HSD enzymes. The placenta produces an enzyme called 11-beta hydroxysteroid dehydrogenase type 2 (11β-HSD2), which inactivates cortisol in the placenta and prevents it from reaching the fetus.
Maternal GC affects the growth and development of the placenta by modulating surface areas for nutrient exchange, formation of fetal blood vessels, and expression of nutrient transporters across the placenta. Glucocorticoid receptor (GR) is ubiquitous in the cellular system and highly expressed in the placenta. Maternal GC has multiple ways of controlling fetoplacental growth, including its effects on the placental vascularization (47), expression of nutrient transporters in the placenta (48), cardio-metabolic maturation of the newborn heart (49), birth weight of the offspring (50), change in body fat deposition and body composition of the primate offspring (51), and others. Excess GC exposure during early fetal life in humans results in glucose intolerance later in life due to the reduced ability to secrete insulin at adult age (52). Placental 11β-HSD2 is a key GC metabolizing enzyme that controls fetal GC levels and mineralocorticoid receptors specificity in the kidney. In the kidney, the enzyme acts as a receptor-modulator of metabolically active GCs, such as cortisol (human) and cortisone (animal), to convert to their derivatives. In the physiological state, maternal GC levels are relatively higher than fetus but protected from excess GC exposure by the placental 11β-HSD2 enzymes. Inhibition or mutation of 11β-HSD2 is reported to lead to low birth weight (LBW) (53, 54). During human pregnancy, cortisol levels are elevated in the maternal circulation (55). However, the placental HSD11β protects the fetus from exposure to high levels of maternal GCs (56). The reduced placental activity and expression of HSD11β2 correlates with lower birth weight (57, 58), and dysregulated placental HSD11β2 expression is reported in intrauterine growth restriction (57, 59). Therefore, optimal placental HSD11β balance is critical for the organ growth of the fetus and its maturation.
2.2 Placental leptin and its roles in fetal growth
Leptin is a fat cell-produced hormone that regulates satiety, signalling, and energy balance (60). Leptin (16KDa) is the novel placenta-derived hormone in humans (61); it plays an endocrine role in controlling maternal satiety, energy metabolism, and fat metabolism and supports trophoblast proliferation and survival (62). Leptin produced by the placenta and adipose tissue has a similar size, charge, and reactivity, but their upstream regulations differ. The human placenta expressed a comparatively higher leptin mRNA and protein than rodents (63). Both placental and maternal circulatory leptin increases with gestation, indicating leptin’s role in maintaining pregnancy.
Expression of the leptin gene in placental vascular endothelial cells in contact with fetal blood first highlighted its potential role in placental angiogenesis. Leptin is co-localized with syncytiotrophoblasts on the maternal side in humans. In contrast, it is co-localized at both the maternal side and labyrinth of the cytotrophoblast of the developing fetus in rodents. The leptin receptor gene (OB-R) in mouse placenta has different variants, including signalling receptor (OB-Rb), transporter receptor (OB-Ra), and soluble receptor (OB-Re). The placental leptin content is regulated by changes in maternal hormones like progesterone and cortisol, which can affect leptin production in maternal adipose tissue in humans (64). A positive correlation exists among circulatory leptin, hCG, and estradiol concentration in pregnant women (65). Leptin and hCG act as a potential regulators of angiogenesis by controlling VEGF secretion at the maternal-fetal interface (66). The leptin receptor was upregulated by hypoxia in placental cells (67). Placental leptin is a biomarker of placental hypoxia in severe preeclampsia (68). Maternal fat deposition increases during pregnancy, but the mother’s BMI does not correlate with the gestational increase in circulatory leptin, indicating the contribution of leptin from the placenta, not from maternal adipose tissue (69). The leptin binding to soluble leptin receptors secreted by the placenta increased circulatory leptin and prevented leptin from binding to signalling receptors, leading to leptin resistance (70). The soluble receptor of leptin concentration was decreased rapidly in the later gestation, around 20-30 weeks (71), suggesting leptin’s role in the catabolic phase of pregnancy in regulating maternal appetite and fat metabolism. It was proposed that leptin could promote placental development by stimulating angiogenesis (72). Although leptin’s role in placental angiogenesis is unclear, in vitro, data showed that leptin stimulated tube formation independent of VEGF and upregulated expression of genes associated with angiogenesis in the placental cells (73). In addition to its angiogenic effects, placental leptin showed anti-inflammatory roles by producing excess in GDM and PE to counter the effects of proinflammatory cytokines (74). Leptin is essential in fetoplacental growth and development as cord blood leptin positively correlates with birth weight (75). Maternal hyperglycemia in insulin-dependent diabetes during pregnancy enforces the fetal pancreas to release insulin that increases fetal and placental leptin concentration in paracrine and induces fetal overgrowth and macrosomia (76). Maternal hyperglycaemia was associated with DNA methylation of the leptin gene, which could risk childhood obesity (77). Placental leptin gene expression was reduced due to BPA exposure (38). Leptin level was increased in BPA-exposed overweight mice in the non-fasting state, indicating the involvement of leptin resistance in these animals (78). BPS exposure potentiated the obesity induced by a high-fat diet and was linked with a higher fat mass, food intake, and hyperleptinemia (79).
Overall, the placenta regulates its growth by its leptin in an autocrine manner since cord blood leptin concentration correlates with placental size. Again, placental leptin executes paracrine regulation of maternal energy balance by increasing its concentration later in the pregnancy, thus sustaining fetal growth and development.
2.3 Maternal chorionic gonadotrophin and placental trophoblast functions
Chorionic gonadotrophin (hCG) is produced by the placenta. During early pregnancy, trophoblasts secrete hCG, as the first placental hormone (80). hCG promotes the production of ovarian progesterone and estrogen from the corpus luteum. Later, the placenta takes over the progesterone production as steroid biosynthesis occurs throughout the gestation (81). hCG is a glycoprotein with α and β subunits linked by a non-covalent bond. The α-subunit is encoded in chromosome 6 and is similar to other glycoproteins such as follicle-stimulating hormone (FSH), luteinizing hormone (LH), and thyroid-stimulating hormone (TSH). The β-subunit is encoded by chromosome 19 and is different for each hormone. hCG exists in different isoforms, such as classical hCG, hyperglycosylated hCG, and the free β unit of the hyperglycosylated hCG (82). The hCG levels peak during the first trimester, later decrease, and remain at basal levels throughout pregnancy (83). The biological functions of hCG include angiogenesis, vasculogenesis (84), and differentiation of cytotrophoblasts into syncytiotrophoblasts (85, 86). A retrospective cohort found that in the first trimester, low free β-hCG levels significantly increased the risk for preterm birth, LBW, intrauterine growth restriction (IUGR), and low APGAR score (87). Immunostaining of placental tissues reveals α-hCG staining rates decreased significantly in IUGR cases over the control (88).
In contrast, the high free β-hCG group had a significantly decreased risk of GDM and preterm birth. In the second trimester, low and high β-hCG had substantially higher risks of the most common adverse outcomes, i.e., spontaneous abortion, IUGR, and preterm birth (87). Moreover, free β-hCG was associated with an increased risk of spontaneous fetal loss (89). Serum β-hCG levels of the missed abortion were lower than those of the control group. Decreased hCG production in early pregnancy down-regulates the expression of the VEGF-MEK/ERK signal pathway, ultimately reducing the angiogenesis (90).
Moreover, studies also show a relationship between gestational trophoblastic disease and hCG. Thus, maternal hCGs involved in several gestational events during pregnancy while their abnormal levels are associated with adverse pregnancy outcomes. Higher exposure to BPA and phthalates was associated with a lower hCG in pregnancy, suggesting altered production or secretion of hCG by the placenta (91).
2.4 Insulin, placentation, and nutrient transport
The peptide hormones such as insulin, IGF1, and IGF2 perform mitogenic and metabolic effects by specifically binding to their surface receptors in target tissues. In normal physiology, insulin and IGFs bind to their receptor, such as IR or IGF1R, in a rapidly growing embryonic tissue (92), thereby regulating placental growth and transport, trophoblast invasion, and placental angiogenesis. Insulin (1ng/ml) stimulated angiogenesis by increased KDR expression in the extravillous HTR8/SVneo cells (93), which is required for vessel formation to promote angiogenesis in the placenta. Maternal diabetes could result in hypervascularization and excess angiogenesis in the placenta due to dysregulated insulin, growth factors, and excess pro-angiogenic effects of insulin in the endothelial cells (94). Placental IGF1 and IGF2 expression are regulated by compartment and gestational stages. For example, IGF2 was not detected at term but was highly expressed in the first trimester villous and extravillous cytotrophoblasts, indicating its significant roles in the early embryonic development involving the placentation (95–97). During the early trimester, these IGFs regulate the uterine-trophoblast invasion (98), migration (99), and placental LPL activity (100). Moreover, IGFs regulates FABP4 expression (101), transplacental glucose and amino acid transporters (102) via activation of mammalian target of rapamycin (mTOR) signalling (103), and increased basal membrane GLUT1 content and trans epithelial glucose transport (104). Several factors, including mTOR signalling, regulate the nutrient transport mechanism. In trophoblast cells, mTOR senses the nutrient signal for maternal supply and fetal demand. mTOR senses various maternal endocrine signals (such as insulin, leptin, and others) to regulate fetal growth by activating gene transcription and protein expression, resulting in modulated expression of nutrient transporters. The maternal hormones such as insulin, leptin, and IGF1 stimulate, while hypoxia and adiponectin downregulate the mTOR signalling (105). Protein restriction during pregnancy affects insulin signalling and glucose metabolism in the muscle tissue of the offspring by involving mTOR (106). Pregnancy complications associated with obesity and endocrine stress due to EDC exposure can affect the mTOR pathway, leading to altered placental nutrient transporter efficiencies and influencing the fetus and development.
2.5 Prostaglandin and placental trophoblast development
Prostaglandins (PGs) are lipid-derived hormone-like molecules that modulate several fetoplacental growth and development functions. Unlike hormones produced by endocrine glands released into the bloodstream, prostaglandins are produced by specific tissues at the site of action. Prostaglandins are produced from arachidonic acid, 20:4n-6 (ARA), by phospholipase A2, followed by cyclooxygenase (COX). A higher ARA level in the early placenta supports prostaglandin’s roles in early development, i.e., decidualization, vascularization, angiogenesis, organogenesis, and others (107–110). Moreover, ARA does not accumulate in the placenta near term since its derivative can trigger parturition. In contrast, the precursor of the vasorelaxant and anticoagulant PGE1 levels are increased at period, facilitating blood flow to support fetal growth spurt. Prostaglandin E2 (PGE2) is abundantly produced by the decidua that helps the migration of first-trimester extravillous trophoblast by increasing the intracellular calcium concentration (111). Upregulating PGE2 production and PGE2 receptor expression associated with increased levels of leukemia inhibitory factor favors EVT invasion during the placentation (112). Early stages of spiral artery remodeling mimic angiogenesis, are facilitated by several cytokine-producing cell types and therefore remain critical for early placentation that involves EVT (113, 114). PGE2 is involved in angiogenesis (115) by acting on various cell types that produce proangiogenic factors, such as VEGF, CXCL1, and bFGF, which act on target endothelial cells to promote the angiogenesis (116). Multiple signaling mediators by the various PGE2 receptors facilitate the PGE2-induced angiogenic response. PGE2 stimulated in vitro angiogenesis comparable with VEGF in trophoblast cells. However, PGE2-induced angiogenesis was inhibited by COX2 inhibitors. In contrast, VEGF-induced tube formation was the least affected (110), indicating that PGE2 may have an independent effect on angiogenesis in the placenta. Another PG, like PGF2α levels, increases during the window of implantation in the uterine lumen of humans and other mammals, suggesting its role in embryo implantation. PGF2α reported stimulating invasion and migration of the human trophoblast HTR8/SVneo cells (117). PGF2α increased decidual gelatinolytic activity in fetal membranes since its inhibition helps arrest the labor (118). Prostaglandins and their derivatives perform multiple roles associated with fetoplacental growth and development, including embryonic implantation, vascularization, angiogenesis, trophoblast invasion, migration, and labor.
3 Roles of placental steroid hormones in pregnancy
Placenta acts as the primary organ for steroid hormone synthesis during gestation. Placenta contains all the necessary enzymes for steroid biosynthesis. The cytochromes P450s (CYPs) and the hydroxysteroid dehydrogenases (HSD) are the enzymes in the placenta where hormonal biosynthesis takes place from the maternal cholesterol (119). The placenta converts cholesterol into pregnanolone (PREG) and finally to progesterone by a series of enzymatic reactions. In addition to the placenta, fetal tissues can convert PREG to dehydroepiandrosterone sulfate (DHEAS) and finally to 16α-OH-DHEAS. Hydroxylated DHEAS and DHEAS again enter the placenta and eventually transform into androstenedione and testosterone. In the placenta, the aromatase enzyme (CYP19A1) converts androstenedione and testosterone into estrogen [estrone (E1), estradiol (E2), or estriol (E3) (119). Progesterone predominantly mediates its actions by binding to the nuclear receptor (PR-A and PR-B) and membrane-bound receptor causing non-genomic actions.
High progesterone levels are correlated with the development of glucose abnormalities in pregnancy. Recent studies have shown that progesterone receptors knocked out in female mice (PR-/-) but not male had lower fasting glycemia than PR+/+ mice and showed higher insulin levels on glucose injection. Moreover, pancreatic islet cells were larger and secreted more insulin due to increased β-cell proliferation (120). Placental estriol, estradiol, and progesterone levels were lower in preeclampsia's placental tissues than in healthy pregnant women (121). In preeclampsia, a severe hypertensive disorder of pregnancy, the sirtuin1 expression was lower in serum and placenta. Progesterone alleviates preeclampsia-like symptoms mediated by SIRT1 deficiency (122). Progesterone prepares the uterus for pregnancy and prevents preterm births by inhibiting the contraction of uterine muscles. Vaginal progesterone was associated with a significant reduction in the risk of preterm birth (<33 weeks of gestation), spontaneous preterm birth, and admission to the neonatal intensive care unit in pregnancies with short cervix (123). Vaginal progesterone administration significantly decreased the risk of preterm birth, neonatal death, composite neonatal morbidity, and mortality in women with twin gestation and short cervix (124).
As mentioned, the enzyme aromatase converts androgen precursors from fetal and maternal adrenal glands into estrogen in the placenta. The classical estrogen receptors (ER-α & β) and the plasma-bound G protein-coupled ER (GPR30/GPER) are expressed in the uterine artery endothelial cells (UAECs) and smooth muscle of the uterus, mediating their actions through genomic or non-genomic pathways. Activation of ERs by estrogen enhances UAEC's nitric oxide production, thereby causing uterine vasodilation during pregnancy (125). Knockout of ER-α in the female mice decreased litter size and maternal nurturing behavior (126). Estrogen receptor-β is involved in implantation and parturition in the mature uterus (127). Insulin resistance associated with hyperestrogenemia occurs in GDM and PCOS. Estradiol can bind to insulin and may interfere with binding to its receptor, causing insulin resistance (128). Recent studies have shown a possible link between estradiol and glucose homeostasis. Cord blood estradiol in the GDM was significantly lower than in the control group.
Moreover, the cord blood estradiol concentrations were negatively correlated with birth weight (129). Estrogens are well known to modulate angiogenesis in both physiological and pathological conditions. Exogenous estrogen stimulates cell proliferation and angiogenesis in the pregnant ewe UAECs (130). Female ovariectomized ER knockout mice showed impaired angiogenesis, suggesting the role of ER in the angiogenesis (131). As estrogens promote angiogenesis and vasodilation, preeclampsia may be associated. Plasma 17β-estradiol levels were significantly reduced in severe PE subjects (132). Similar outcomes were found in patients with mild to severe PE (133). Placental tissue levels of estriol and estradiol were significantly lower in PE compared to healthy pregnant women (121). Overall data indicate the role of estrogen in the pathophysiology of preeclampsia, which could be caused due to aromatase deficiency in the placenta.
4 Endocrine modulating factors during pregnancy and gestational disease risks
Several studies mentioned in this section have reported a relationship between placental hormones and the development of IUGR. Low hCG concentrations during the late first trimester were associated with decreased fetal growth, birth weight, and small for gestational age (SGA) (134). A similar study was conducted on 9450 singleton pregnant women. SGA was associated with low maternal serum levels of pregnancy-associated plasma protein-A (PAPP-A), free β-hCG, and slow early fetal growth (135). β-hCG levels were significantly decreased in SGA cases compared to the control (136). Sex hormones also regulate IUGR. 17-β Estradiol (E2) is critical to a physiological pregnancy and synthesized by the conversion of androgens by the CYP19A1 gene. Maternal E2 level in plasma was significantly decreased in the IUGR. Although the placental expression of CYP19A1 was considerably higher than in control (137), serum levels of estriol (E3) were significantly lower in the IUGR (138). Moreover, the estrogen-related receptor gamma expression was reduced in the FGR placenta compared to the control (139).
Gestational diabetes mellitus is characterized by spontaneous hyperglycemia during pregnancy. The Hyperglycaemia and Adverse Pregnancy Outcome (HAPO) study found that maternal hyperglycemia independently increased the risk of preterm delivery, cesarean delivery, infants born large for gestational age, and neonatal hypoglycemia (140). Although GDM is a pregnancy disorder, it resolves following delivery but can have long-term health consequences on maternal and fetal life. Risk factors include obesity, diabetes, a sedentary lifestyle, genetics, and advanced maternal age. The primary pathophysiological condition in GDM is β-cell dysfunction. It is caused by an inability of β-cells to sense blood glucose concentration and release insulin adequately. Due to this condition, high blood glucose prevails, leading to hyperglycemia. Glucose is the primary energy source for both placenta and fetus development. As glucose is always required, the placenta expresses insulin-independent glucose transporter GLUT1. GLUT1 plays a role in implantation and angiogenesis. The expression of GLUT1 on the basal plasma membrane of the placenta is positively correlated with birth weight (141). However, the expression and function of GLUT1 are down-regulated in pre-eclampsia and IUGR (142).
Moreover, insulin-stimulated glucose uptake of the first-trimester trophoblast cells, HTR8/SVneo, is partially mediated via GLUT1 (93). Trophoblasts and endothelial cells of the placenta also express insulin receptors, which can be activated by insulin and alter the placental metabolism of nutrients (143). Higher hCG levels in early pregnancy are associated with a lower risk of GDM (144). In contrast, elevated hCG levels were especially predominant among women who developed GDM (145). The relationship between hCG and GDM was inconsistent and varied with the duration of pregnancy. Estradiol is an essential mediator of glucose homeostasis. In-vivo studies have shown that aromatase knockout mice (ArKO-/-) develop glucose intolerance and insulin resistance. Treatment of ArKO males with 17β-estradiol improved the glucose response (146). Cord blood estradiol was significantly lower in GDM than in control. Moreover, cord blood estradiol was negatively correlated with birth weight. Estrogen protects pancreatic β-cells from apoptosis and prevents insulin deficiency (147). A recent study has also shown a relationship between progesterone and GDM. Compared to controls, GDMs have significantly lower progesterone at weeks 10-14 (148).
Hyperglycemia in mothers can lead to macrosomia due to the excess glucose available to the fetus via the placental glucose transporter GLUT1. In addition to β-cell dysfunction, insulin resistance and several maternal hormones produced by adipose, liver, and placenta can develop GDM. Gestational hCG levels are associated with predictive risk with the development of GDM but inconsistently.
5 Endocrine-disrupting chemicals induce endocrine dysfunction and fetal programming of development and reproductive disease
Endocrine-disrupting chemicals interfere with the typical endocrine system and produce adverse effects. EDC was thought to exert its action primarily through nuclear hormone receptors such as ER, androgen receptors (AR), thyroid receptors (TR), and retinoid receptors. However, recent data suggest that EDC can also act on non-nuclear and orphan receptors. EDC can be synthetic or natural chemical compounds. Synthetic chemicals are used as plasticizers [bisphenols, phthalates], pharmaceutical agents [diethylstilbestrol (DES)], industrial solvents [polychlorinated biphenyls (PCBs), polybrominated biphenyls (PBBs), dioxins], agrochemicals [methoxychlor, chlorpyrifos, dichlorodiphenyltrichloroethane (DDT)], fungicides (vinclozolin)]. Natural chemicals include phytoestrogens such as genistein and coumesterol.
Humans are exposed to EDC by various routes such as ingestion, inhalation, and transdermal. EDCs have been detected in maternal blood, amniotic fluid, and cord blood (149). The EDC’s influence on maternal response may program several adult diseases, particularly during gestation. Figure 1 describes the life course exposure risk of EDCs on the diverse functional effects in modulating fetoplacental development that are mediated by maternal effects spanning from hormonal imbalance, epigenetic modification, dysregulated metabolic responses and risks of gonadal and reproductive functional deformities in the offspring.
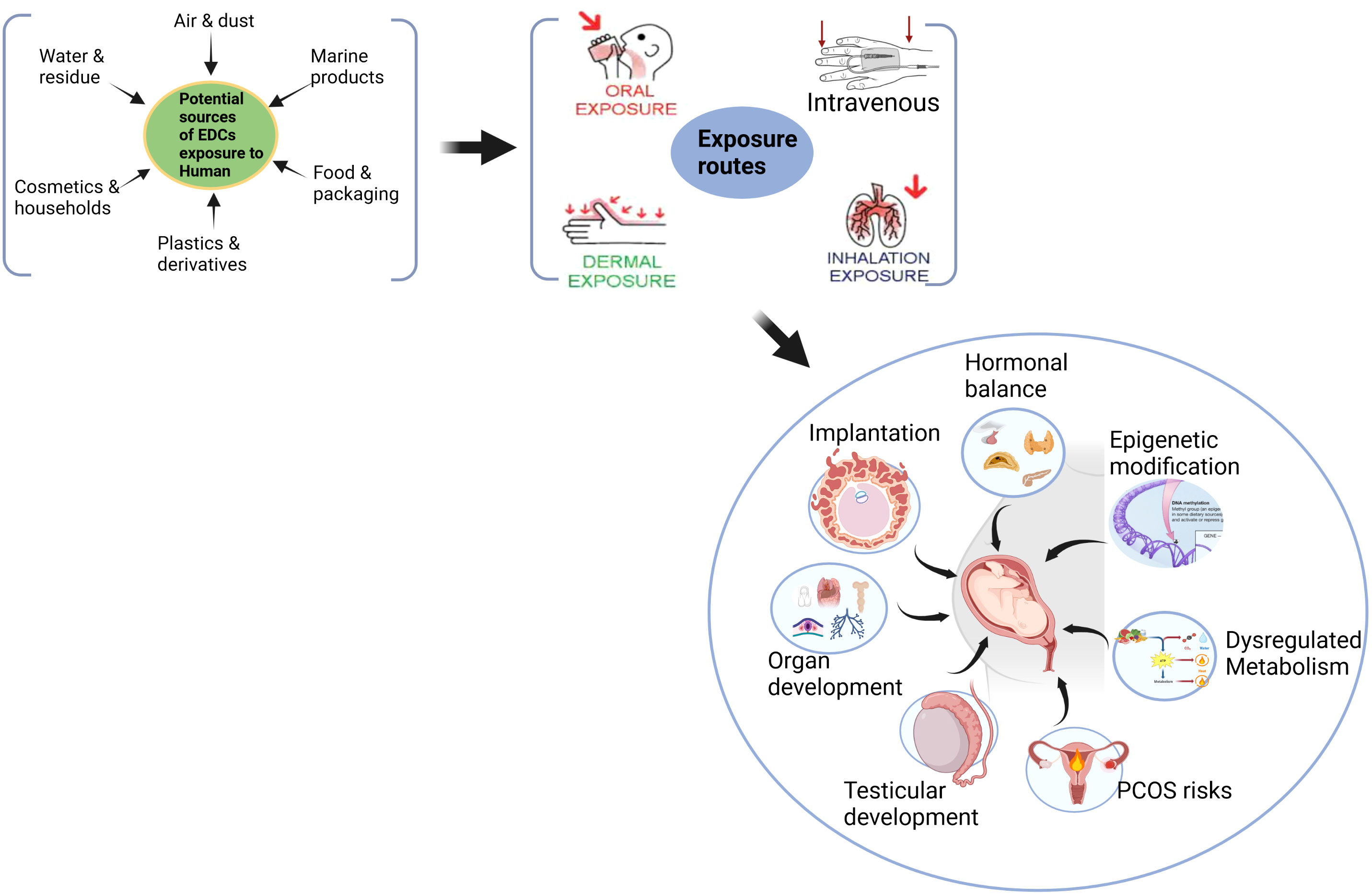
Figure 1 Maternal exposure risk of EDCs in modulating fetoplacental development that they might risk programming adult metabolic and reproductive diseases.
Adults can perform rapid first-pass metabolism to convert these into biologically inactive metabolites and rapidly clear them in the urine. Still, the unborn developing fetus and placenta do not have the enzymatic machinery to catabolize these compounds. As these chemicals cross the placenta, EDC threatens the unborn fetus. Placental trophoblast invasion, proliferation, and its several functions are affected due to exposure to bisphenol A (BPA) and their substitution chemicals (Figure 2). Research has shown that EDC exposure during the critical developmental window can have long-term health impacts on the fetus and its later adult life (2). Moreover, successful pregnancy depends on the physiological secretion of several hormones. Any changes in the hormonal milieu during development can cause pregnancy-related complications and diseases.
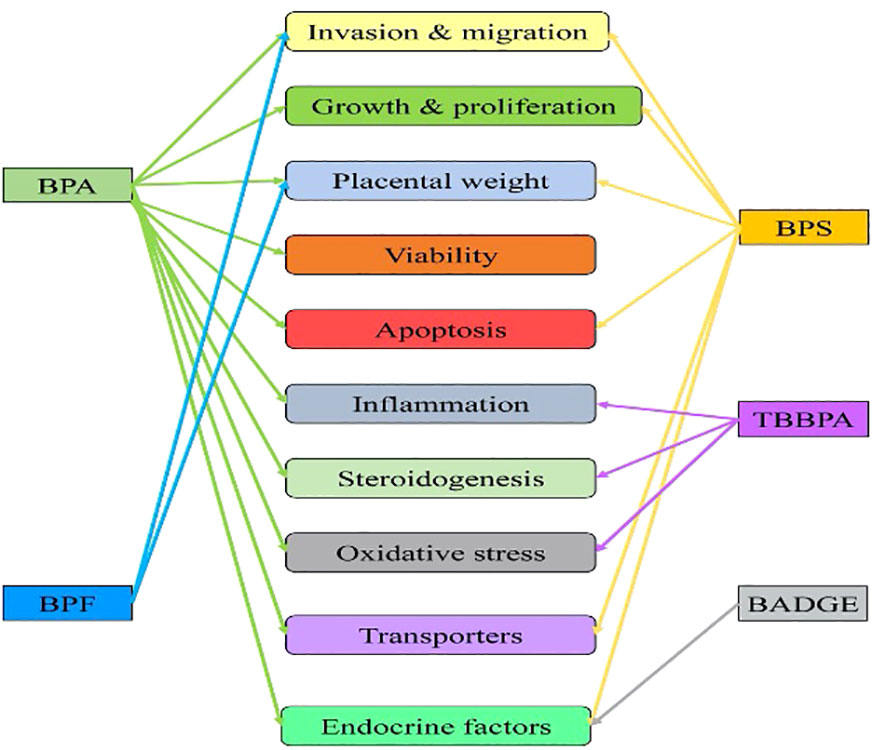
Figure 2 Influence of endocrine-disrupting chemicals (bisphenols) exposure on trophoblast invasion, development, and placental functions. Single-headed arrow indicates an independent study reported so far available from the PubMed database. BPA, Bisphenol A (23, 24, 26–28, 38); BPS, Bisphenol S (25, 33, 36, 42, 150, 151); BPF, Bisphenol F (34, 39); TBBPA, Tetrabromo Bisphenol A (35, 40, 43, 152, (44); BADGE, Bisphenol A diglycidyl ether (45).
Humans are constantly exposed to synthetic EDCs such as bisphenols and phthalates. The plastic-derived chemicals such as BPA and its substitute bisphenol S (BPS) are the major endocrine disruptors exposed ubiquitously since it mimics hormones and modulates the endocrine system, affecting the metabolic outcome. The food contaminated with packaging and chemical agents mixed with bisphenols is continuously exposed in micron levels through leaching, especially plastic-derived residual contaminants (153). Semisynthetic and plasticizer-laden food surfaces (154) are exposed to heat and change in pH, resulting in chemical hydrolysis of polyester bonds, releasing chemicals directly exposed to living systems (155). BPs are absorbed through the skin, and first-pass metabolized to their conjugates, resulting in a greater endocrine risk to human disease burden (156).
BPA is the most abundant in consumer products, including food and beverage containers, medical devices, toys, and thermal receipts. BPs are detected in human blood irrespective of gender. However, the findings on gender-specific exposure to BPA are inconsistent, as studies have shown that BPA concentrations were higher in men than women (157, 158). At the same time, others found equal concentrations of BPA in both males and females. The discrepancies in the data could be multifactorial, including but not limited to age, sample size, geographical location, and technique. The concentrations of BPA detected in the cord blood are closely related to levels in maternal blood, indicating that BPA readily crosses the placenta. Epidemiology and clinical evidence of EDCs exposure suggest a profound association between the exposure of BPA and its analogs during pregnancy and altered fetal and neonatal growth outcomes, including intrauterine growth, gestational length, birth weight, ponderal index, femur length, abdominal circumference, head circumference and others (Table 1).
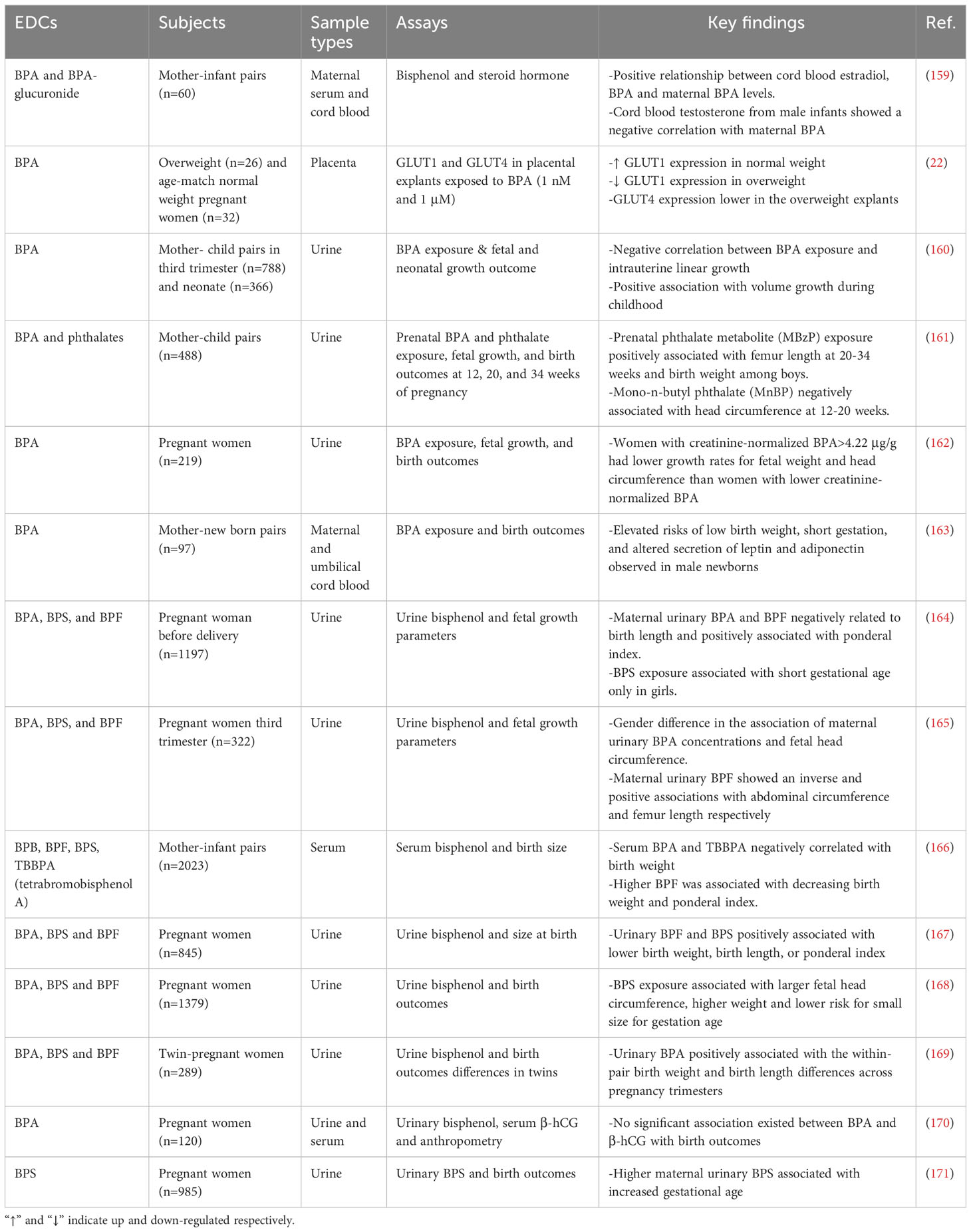
Table 1 Epidemiology and clinical evidence of bisphenol exposure on fetal and neonatal growth outcome.
Trans maternal exposure to BPA in non-obese diabetic mice increased the severity of insulitis and the incidence of diabetes in female offspring (172). Perinatal exposure to BPA alters the expression of genes involved in β-cell growth regulation, incrementing β-cell mass/area and β-cell proliferation during early life. Excess insulin production in early life may impair glucose metabolism in later life (173). Additionally, the metabolic changes caused by in-utero BPA exposure were found to be inheritable. i.e., maternal exposure to a low dose of BPA impaired insulin secretion induced islet inflammation and increased β-cell death in the offspring. Surprisingly, subsequent generations (174) and third-generation (175) inherited these changes, indicating that BPA-induced epigenetic modifications were inherited.
Although excess GC exposure can modulate fetus growth and development, little is known about how the GC system is vulnerable to BPs exposure, particularly during pregnancy. Data suggest that BPs significantly inhibited HSD11β2 activity in humans and rats, possibly by steroid binding site, as evidenced by docking analysis (176). It is well established that BPA exposure modulates cellular function by their estrogen agonist or androgen antagonist function. Still, their preference for binding GR or ER in the placenta is unknown. BPA may mediate its effects via a non-classical ER pathway without GC, where ER antagonists inhibit BPA-induced adipogenesis but not GR antagonists (177). BPA at lower concentrations increased the expression of both protein and mRNA of HSD11β2 in placental trophoblast cells (38). Our recent data showed that prenatal BPs exposure lowered HSD11β2 expression in the rat testis (178), indicating reduced protection against cortisol-mediated stress induced by bisphenols in these offspring.
BPA possesses pseudo-estrogenic and anti-androgenic effects that might interfere with the developmental programming of the male reproductive system in the offspring. Prenatal exposure to BPA and BPS at much lower concentrations than TDI, during gestational days 4 to 21, alters testis weights, histology, increased testicular inflammation, and oxidative stress in the adult offspring. Moreover, sperm DNA damage and DNA methylation were also increased in both BPA and BPS-exposed adult offspring (178). Exposure to BPA and its analogs bisphenol B (BPB), bisphenol F (BPF), and bisphenol S (BPS) during gestation resulted in decreased testosterone secretion, sperm production, and histological changes in the reproductive tissues of adult SD male rats (179). BPA exposure during pregnancy and lactation can also affect epididymal sperm count and sperm motility (180). CD-1 mice exposed to bisphenols, dysregulated serum estradiol-17β, testosterone, expression of steroidogenic enzymes, increased mRNA levels of DNA methyltransferases, and histone methyltransferases in F3 adult testis (181). Similar results were found with BPA and its analogs on female reproductive functions in mice. Early life exposure in mammals affects early oogenesis and follicle formation in the fetal ovary (182, 183). Bisphenol exposure in the F0 generation accelerated the onset of puberty and exhibited abnormal estrous cyclicity in F3 females (184). Gestation and lactation-specific BPA exposure decreased sex hormones and reduced the number of tertiary ovarian follicles in the female offspring (185). BPA exposure affected placentation and exhibited preeclampsia-like phenotype in pregnant mice (21). Exposure to BPA was also associated with a risk of preterm birth (186).
Growing evidence suggests that BPA and its substitute BPS have distinct action modes in different tissues. e.g., in-utero exposure to BPA altered endogenous long-chain fatty acid metabolism in the testis of adult offspring, thereby affecting sperm maturation and quality. However, no changes were observed in the testis of BPS-exposed offspring (187). The bisphenols interfere developmental programming of the reproductive system in the offspring (178) since fetuses do not equip with enzymatic machinery to catabolize these compounds (2). Thus, in-utero exposure to BPA during development can program the fetus for endocrine-related diseases in later life.
6 Endocrine-disrupting chemicals and endocrine risk of pre-pregnancy
Female infertility is a rapidly evolving endocrine disorder in pre-menopausal women, with an estimated prevalence of 5%-18% globally (188). Often it remained underdiagnosed and became one of the leading causes of female infertility. The risk of failed pregnancy can be associated with metabolic, reproductive, and endocrine features such as anovulation, hyperandrogenism, arrested folliculogenesis, infertility, polycystic ovaries, type 2 diabetes, and obesity (189). Dysregulated hypothalamic‐pituitary‐ovary (HPO) axis, followed by hyperandrogenism, increased serum luteinizing hormone, and metabolic disturbance, contribute to the complex etiology of PCOS (190). Despite studies, the pathophysiological cycle of hyperandrogenism, hyperinsulinemia, and anovulation in PCOS remains unclear. Studies pointed out that exposure to common household plastics containing chemicals could associate with risks of PCOs in humans.
BPA levels were significantly elevated in women with PCOs (191). Similarly, adolescent girls with PCOS had markedly increased serum BPA than controls, and BPA was correlated considerably with androgen (192). Thus, BPA may play a potential role in the etiology of PCOS, specifically a significant contributor to ovarian hyperandrogenism observed in women with PCOS. In addition to hormonal changes, BPA can exert its effects through epigenetic modifications. BPA can change DNA methylation or alter histone modifications without altering the nucleotide sequence of the genes. BPA altered stress-related promoter DNA methylation in placental cells (27). In-utero BPA exposure decreases the CpG islands in the promoter and intronic region of the HOXA10 gene, which is involved in uterine organogenesis (193). Prenatal or neonatal exposure to BPA causes an increase in levels of androgen-hyperandrogenism, one of the hallmarks of PCOS. Hyperandrogenism promotes epigenetic modifications of histone deacetylase 3 (HDAC3), peroxisome proliferator-activated receptor gamma 1 (PPARG1), and nuclear corepressor 1 (NCOR1) genes in granulosa cells of PCOS women (194). BPA exposure could dysregulate endocrine balance by changing the expression of several genes, such as CYP11A1, GnRH, AdipoQ ESR1, and StAR, whose disruption is commonly observed in the pathogenesis of PCOS (195).
The endocrine disruptor BPA can mimic estrogen to interact with its receptor. BPA exposure modulated steroidogenic enzymes' expression in the ovary, lowering aromatase expression and estrogen production in granulosa cells. BPA is reported to stimulate androgen synthesis in ovarian theca-interstitial cells. Moreover, BPA concentration was significantly higher in PCOS patients' follicular fluids. BPS is structurally similar to BPA, where sulphonyl groups substitute phenolic groups. In utero, BPS exposure also disrupted female reproductive development by changing the estrogen-responsive gene expression in the uterus and ovary (196). Whether exposure to bisphenols is causally related to developing PCOs risk is not established yet. However, BPA could be a causal factor for PCOS because it mimics the hormone estrogen's function (197). BPA (500 μg/kg/day) exposure causes irreversible alteration in the hypothalamic-pituitary-gonadal axis in rats, leading to anovulation and infertility (198).
Subcutaneously injected with BPA from postnatal days 1 to 10 resulted in the development of the PCOS morphology after 4 to 5 months. Serum testosterone and estradiol were elevated, with an irregular GnRH pulsatility (199). In a similar study, SD female offspring perinatally exposed to 1.2 mg/kg bw/day BPA from gD 6 till lactation exhibited increased body weight, altered patterns of estrous cyclicity, and decreased plasma LH levels (200). Prenatal exposure to BPA in CF-1 mice significantly reduced the days between the vaginal opening and the first oestrus (201). Similarly, ICR mice prenatally exposed to BPA induce early vaginal opening (202). Neonatal SD rats were subcutaneously exposed to BPA, which caused multiple cystic follicles forming in the ovary (203). Similarly, an increase in cystic ovaries and pathologies of the oviduct and uterus were observed in mice (204). BPA-exposed ovaries showed a decreased antral & corpus luteum and increased atretic & cystic follicles (205). Maternal BPA exposure increases antral follicles' size, elevates the incidence of ovarian cysts, and changes ovarian follicular dynamics (206).
BPA exposure affects steroidogenesis in the ovary by reduced E2 production and decreased aromatase (CYP19A1) expression in human granulosa cells (207). Prenatal exposure to BPA caused alterations in fetal ovarian steroidogenesis by increasing Cyp19 and 5α-reductase expression and miRNA expression of folliculogenesis in sheep (208). Moreover, it was shown that patients with PCOS have decreased mRNA expression of aromatase (CYP19A1) compared to the non-PCOS group. Hyperandrogenism in PCOS is due to reduced aromatase activity (209). Bisphenol A exposure can also modulate hormone levels. Lower E2 levels and higher testosterone were observed in all the BPA-treated groups compared to the control in rat ovarian theca-interstitial and granulosa cells (210). On the contrary, LH and E2 significantly increase in the BPA-treated Wistar rats (211).
BPA is reported to modulate the HPO axis and elevate serum LH. Still, their low-level exposure in utero to develop a PCOS-like phenotype in the offspring must be reported. Despite the broader presence of direct hormone-regulated animal models in studying human PCOS, the pathogenesis and treatment still need to be determined. Thus, investigating the PCOS-like phenotype in animals without direct hormonal intervention is expected to determine the underlying mechanism for better understanding the pathogenesis of PCOS. Moreover, evidence from animal models may clarify the causal roles of EDC exposure in determining the PCOS risk factors.
7 Maternal phthalate exposure, endocrine effects on fetal growth and reproductive development
In the food processing, pharmaceutical, and medical industries, Phthalates are used as plasticizers in daily use products such as cosmetics, personal care, detergents, toys, household products, solvents, sealants, and paints. Men and women are differentially exposed to cosmetics. A recent study of participants (n=9218) investigated the exposure to EDCs based on occupation and gender differences in personal care product use. Higher urinary concentration of EDCs (BPA and mono-n-butyl phthalate, MnBP) was observed in women compared to men since the latter were exposed to phthalates-containing cosmetics more frequently than men (212). Women are suspected of exposure to various EDCs, including abundant phthalates in cosmetics. Women can carry and transmit this risk exposure to a new life. Numerous forms of phthalate exist in our environment, in which DEHP (Di 2-ethyl hexyl phthalate) is prominently present in food items. Samples originating from meats, fats, and dairy products may contain DHEP at concentrations (≥300 μg/kg) and significantly contribute to exposure in epidemiological studies. The molecular weight of DEHP is 390.56 g/mol. The first step in the metabolism of DEHP is the hydrolysis of DEHP to mono (2-ethylhexyl) phthalate (MEHP) catalyzed by unspecific lipases. Upon oral exposure, phthalates are metabolized quickly into their monoester metabolites. These phthalate metabolites have been detected in urine, maternal blood, amniotic fluid, and cord blood. In mice, MEHP is metabolized into a wide range of secondary metabolites (diacids and keto acids), excreted as glucuronide conjugates in human urine, and unconjugated metabolites in rat urine. A single oral dose clinical study found that about 71% of the DEHP delivery was excreted after 24 h, and 4% was excreted in the following 20h (213). European Food Safety Authority (EFSA) reported that a 50 μg/kg/day dose could lead to testicular toxicity. In utero, DEHP exposure diminishes mineralocorticoid receptor expression in adult rat Leydig cells, affecting aldosterone-induced androgen formation and decreasing testosterone production. In-utero exposure to DEHP (100, 300, and 750 mg/kg/day) results in a 50% decrease in testosterone and aldosterone without changing corticosterone concentrations in male rats. DEHP was reported to enhance estrogenic activity at concentrations of 1.50 ppm in vitro and in vivo, suggesting its role in the transactivation of ER.
DEHP exposure decreased steroidogenic acute regulatory protein expression in pregnant mice and lowered fetal testicular 17α-hydroxylase and cytochrome P450 17A1 levels. Early trimester DEHP exposure is inversely associated with the anogenital distance of male newborns but not girls, indicating gender-specific effects on male genital development (214). In addition, the concentration of DEHP increases the chances of limb malformation in both litters and fetuses (215). The defects were specific to hind limbs and represented a spectrum of polydactyly phenotypes. In addition to the known effects of phthalate on fetal testicular functions, unexpected limb malformations and high rates of fetal death following exposure to 250 mg/kg DEHP was noted. However, further studies are needed to identify the mechanism responsible for DEHP-induced limb malformations and to determine the potential relevance of this finding concerning humans.
Due to their lipophilic nature, phthalates can act through nuclear membrane receptors and involves epigenetic modifications by altering the gene expression (216). Several animal studies and epidemiological data have been raised to define the risks of phthalates and their metabolites. Pregnant mice exposed to DEHP-induced fetal IUGR (217) lowered placental weight and reduced blood sinusoid area in the placental labyrinth layer in a stage-specific manner (218). Gestational DEHP exposure alters the expression of steroidogenic enzymes and circulatory steroid hormone levels in pregnant females (219). High doses of DEHP caused IUGR and induced fetal malformations (220). DEHP exposure also damaged DNA in the trophoblast (221) and altered the placental receptor expression (222).
Early exposure to phthalates can lead to metabolic, and reproductive diseases in later life. Gestational and lactation exposure to phthalates causes hyperglycemia, impaired glucose, and insulin tolerances, and altered insulin transduction pathways in adult male offspring (223). Gestational DEHP exposure results in impaired β-cell function by downregulating genes involved in their development and function by the increase in the global DNA methylation (224, 225). Recent data reported that maternal urinary phthalate metabolites are associated with DNA methylation profiling of the first-trimester placenta. The results showed 282 differentially methylated regions (DMRs) corresponding to 245 unique genes in the early human placenta for high compared to low total phthalate exposure. The phthalate metabolites in urine were related to changes in miRNA expression of the human placenta, where miR-142-3p, miR15a-5p, and miR-185 expression were associated between phthalate exposure and protein serine/threonine kinase activity (226). Phthalate exposure altered the first-trimester placental transcriptome and methylome connected with the placental growth factors (227). Maternal exposure to phthalates seems to promote the transgenerational inheritance of adult-onset disease risk factors in subsequent generations. Exposure of DHEP to F0 generation activated the PI3K/Akt/mTOR signaling in the germ cells of the adult mice testis in F1 and F2 generations (228). Prenatal DEHP exposure during gonads differentiation displayed symptoms similar to the human testicular dysgenesis syndrome in adult male offspring mice, which was causally associated with promoter silencing of genes involved in seminal vesicle secretory protein and antigens (229). Early and postnatal lactational DEHP exposure decreased testis weights and disturbed testis architecture by altering seminiferous tubule diameter and germinal epithelium height in adult offspring (230). Emerging epidemiological and clinical data on phthalates exposure as measured by their metabolites indicated pregnancy risks and skewed growth & development of the newborns. Maternal phthalates exposure was associated with fetal and neonatal growth parameters, including anogenital index, gestational age, birth length, birth weight, head circumference, and others (Table 2).
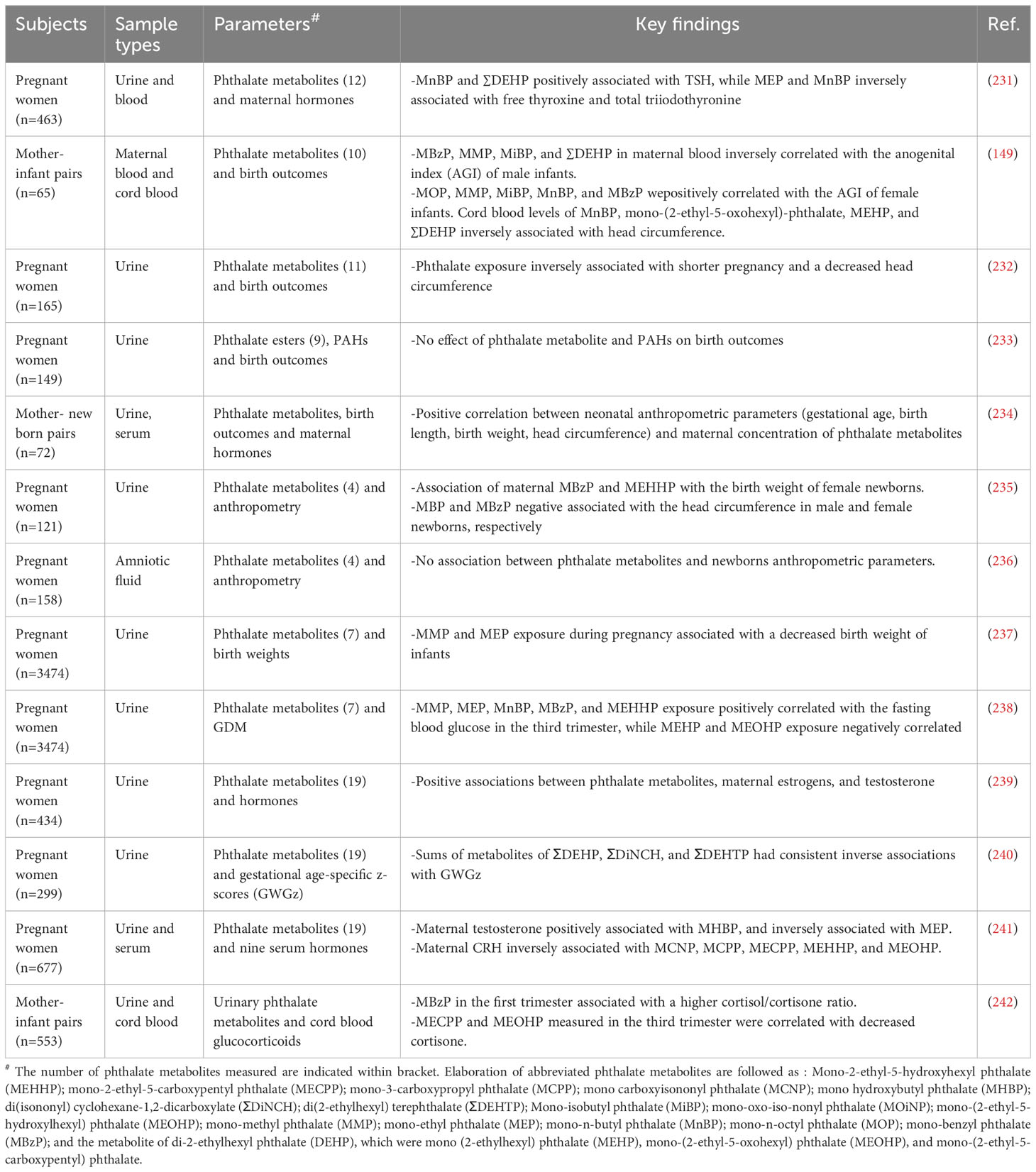
Table 2 Phthalates exposure, pregnancy risks and growth & development of the newborns: epidemiological and clinical data.
8 In utero phthalates exposure and testicular functions
Several in vivo studies show that phthalate exposure, irrespective of exposure stages can have long-term programming consequences in developing reproductive dysfunctions. Sprague Dawley dams were exposed to dibutyl phthalate (DBP) (2550 mg/kg) by oral gavage from gestational day 12 for 10 days, lowered the serum testosterone levels, caused fetal Leydig cell aggregation and lowered Lhcgr, Star, Insl3, and Nr5a1 levels in the male fetuses (243). Diisobutyl phthalate (DiBP) has structural and application properties similar to DBP and is used as a substitute for DBP. DiBP exposure to pregnant Wistar rats significantly reduced the male offspring’s anogenital distance and testicular production. The histology data showed Leydig cell hyperplasia, Sertoli cell vacuolization, and central location of gonocyte (244). In utero exposure to dipentyl phthalate (DPeP) from gD 14 to 21 decreased male offspring's serum testosterone and luteinizing hormone levels and increased the fetal Leydig cells by increasing proliferation (245). Gestational di-(2-ethylhexyl) phthalate (DEHP) exposure to SD rats significantly reduced serum testosterone levels, reduced fetal Leydig cell size, induced abnormal aggregation of these cells, and altered MDA levels in pup’s testis (246). Another synthetic phthalate plasticizer diisodecyl phthalate (DIDP), decreased serum testosterone, and induced abnormal aggregation of fetal Leydig cells. However, no effect on Leydig and Sertoli cell numbers was observed in the male pups (247). Diisopentyl phthalate (DiPeP) lowered mRNA levels of key steroidogenic proteins like Star, Cyp11a1, Cyp17a1, Ar, Esr1, Gper, and Insl3 in testis of prenatally exposed rats (248). In-utero exposure to diisononyl phthalate (DiNP) caused a reduction in fetal testosterone and Insl3 levels in male rats (249). Similar outcomes were observed with the exposure to unique phthalates such as bis (2-butoxyethyl) phthalate (BBOP) which contains oxygen atoms in the carbon backbone (250). In-vivo studies were conducted with mixtures of different phthalates to study their combined effect on testicular testosterone production. Co-administration of DBP and DEHP in pregnant SD rats reduced fetal testosterone levels in addition to reproductive malformations such as epididymal agenesis and reduced androgen-dependent organ weights (251). Similar findings were observed when co-administration of five phthalate esters i.e., benzylbutyl phthalate (BBP), di(n)butyl phthalate (DBP), diethylhexyl phthalate (DEHP), diisobutyl phthalate (DiBP) and dipentyl phthalate (DPP) in dams inhibited fetal testosterone production in the SD rats (252). In addition to testosterone reductions, phthalates impair sperm quality and induce inflammation of testicular Sertoli cells (253, 254). All these in vivo data indicate that phthalates lead to testicular dysgenesis syndrome irrespective of the type of the phthalate exposed (Figure 3).
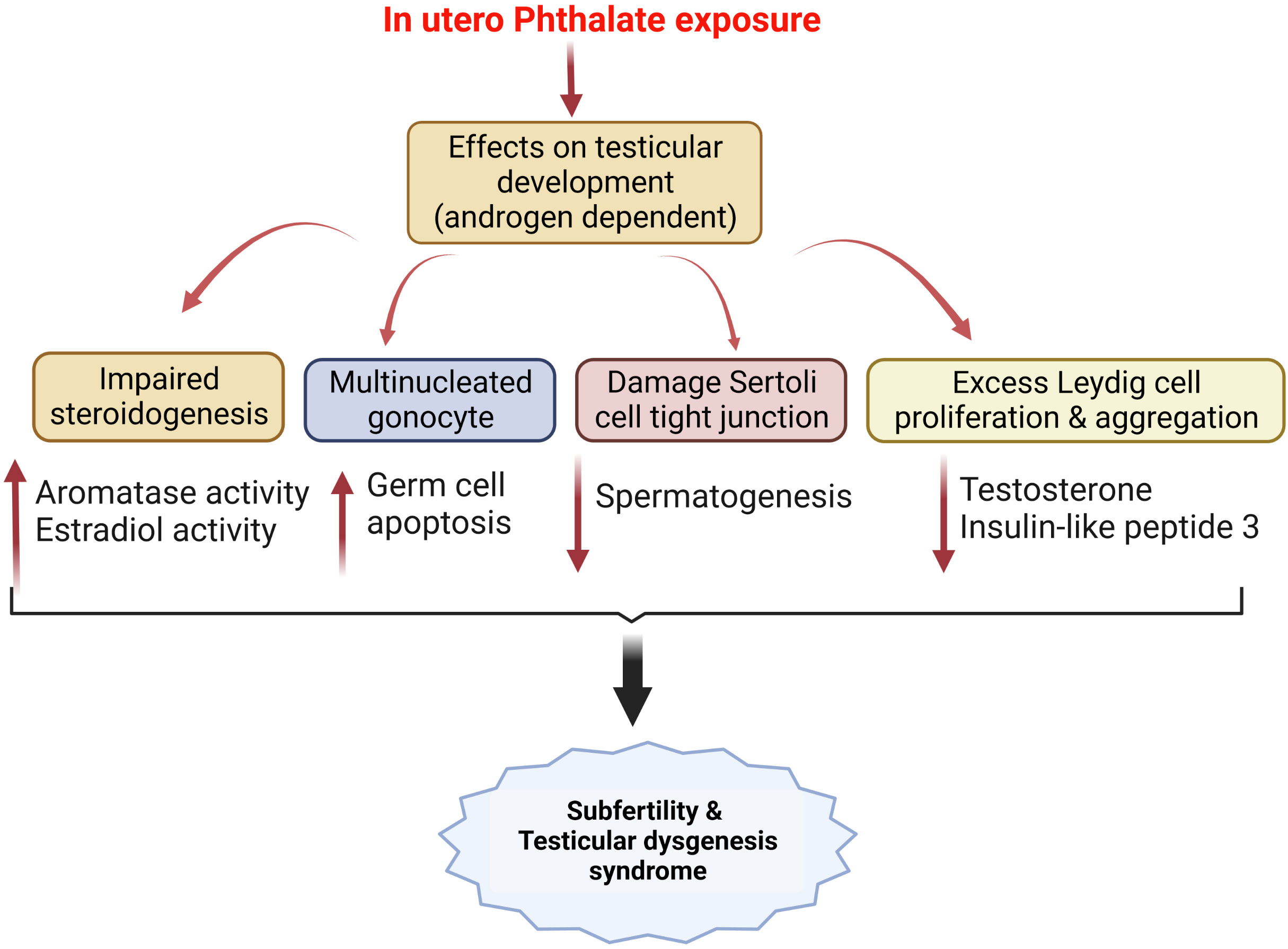
Figure 3 In utero phthalate exposure and its effects on testicular development modulates male fertility and results in testicular dysgenesis syndrome, as reported by several pre-clinical studies mentioned in the text.
9 Modulation of early development by epigenetic-DNA methylation: effects of endocrine disruptors
Epigenetic control of early development is susceptible to the factors associated with environmental and lifestyle practices since a set of genomic imprinters is programmed to execute their functions for early development. The window of fetoplacental development is plastic, capable of remodeling, and sensitive, during which changes in the maternal milieu by internal or external factors might significantly impact developmental programming. Epigenetic mechanisms involve multiple ways, including DNA methylation, histone modifications and microRNAs, and others. A dysregulated epigenetic-DNA methylation results in a change in fetal outcomes by regulating placental methylome and function (255). Maternal nutrition, stress, physical activity, hormones, and endocrine disruptors can modulate epigenetic control of fetal development during gestation. For example, maternal intake of alpha-linolenic acid during pregnancy and lactation causes changes in DNA methylation of the offspring’s liver (256), and maternal omega-3 deficiency increased 5-methylcytosine content in the mice’s placenta, indicating placenta’s sensitivity to epigenetic changes during development (257). Thus, a successful pregnancy requires controlled epigenetic modifications that lead to genome stability during developmental events, including organ formation from gamete production and fertilization to fetoplacental development and fetus outcome (258).
Unlike peptide hormones, steroid hormones are lipophilic that can cross cell membranes and signal through nuclear receptors, acting on transcription factors to turn on or off gene expression. Estrogen, a steroid hormone, binds to either membrane estrogen receptors (ER) or nuclear ERs. Endocrine disruptors like BPs are also known as synthetic estrogen. Activating a non-genomic pathway by binding estrogen to membrane ER causes activation of the PI3K-AKT signaling and downstream activation of gene transcription. Nuclear ER provides DNA-dependent regulation of gene expression mediated by binding of ER with coactivators or corepressors, which encode enzymes with histone acetyltransferase activity (259). RIZ1 is an ER coactivator, which represses transcription by methylation of lysine 9 of histone 3. Knockout of RIZ1 in female mice exhibit a decreased response to female sex hormones, reduced vaginal epithelial thickening in response to estrogen, and reduced litter sizes compared with their wild-type (260).
Although the role of epigenetics in successful pregnancy is well known, endocrine regulation of epigenetic processes is complex, and less data is available on the endocrine regulation of DNA methylation and histone modifications in response to endocrine disruptors like BPA. The sensitive window of early embryonic and placental development stages is more vulnerable as significant imprinting genes control these events. In utero, BPA exposure significantly altered the methylation levels of differentially methylated regions (DMR) imprinting control region and Igf2 DMR (261). Placental trophoblast cells respond to various exogenous agents by activating stress pathways, and epigenetic controls often regulate some of these responses. Data showed that alteration in DNA methylation profile in the first-trimester trophoblast genomic DNA correlated with CpG methylation of gene promoters associated with stress responses, including oxidative stress, inflammation, DNA damage, apoptosis, hypoxia, and the unfolded protein response (27).
Furthermore, the percentage of promoter methylation was lowered by BPA exposure compared with those of control cells. Normally, DNA methylation of gene promoters represses gene transcription to those that encode stress and toxicity pathways. However, stress response genes were de-repressed due to BPA exposure. In normal physiology, the trophoblast cortisol could protect the fetus by methylating genomic DNA to repress gene transcription of stress and toxicity mediators (262), which was unmethylated by the presence of BPA, indicating that activation of these pathways in response to BPA exposure by epigenetic modification. Recently we observed that gestational BPs exposure significantly raised plasma cortisol, body weight, and adiposity in 90d offspring (unpublished data). Thus, placental programming in DNA methylation due to BPA exposure could make the offspring susceptible to stress and anxiety behavior later in life.
BPA modulates gene expression through epigenetic modifications, such as DNA methylations, by either DNA hyper or hypo-methylation. The maternal exposure to BPA shifted the coat color of yellow Agouti mouse offspring toward yellow by decreasing CpG (cytosine-guanine dinucleotide) methylation in an intra-cisternal retrotransposon upstream of the Agouti gene (263). In-utero BPA exposure in pregnant CD-1 mice affects the expression of the homeobox (Hoxa10) gene by decreasing the promoter DNA methylation of CpG sites of the Hoxa10 gene in the reproductive tract of mice (193). In a recent study, the association between prenatal BPA exposure and DNA methylation in the placenta found that BPA causes differential hypo/hypermethylation of CpG sites in the BPA-exposed groups (264). Maternal BPA exposure caused promoter hypermethylation of three genes (CAPS2, TNFRSF25, and HKR1) in the cord blood (265). Perinatal BPA exposure results in abnormal DNA methylation in hepatic tissue, which precedes with the development of insulin resistance, indicating fetal reprogramming via epigenetic changes (266). Developmental exposure to BPA predisposed the offspring to progress steatohepatitis when fed a high-fat diet due to dysregulated gene expression in the liver (267). BPA exposure in utero induced preadipocyte differentiation by DNA hypermethylation of the CPT1A gene, indicating that BPA exposure induced obesity in the offspring by accumulating free fatty acids (268). Prenatal exposure to BPA altered the MEST promoter methylation, leading to increased risks of childhood obesity until about 6 years of age (269).
Further study confirmed that hypomethylation of the MEST promoter, a gene code for α/β hydrolase, is associated with obesity in mice exposed to BPA prenatally. Apart from being sex-specific, the tissue-specificity of prenatal exposure to BPA was also associated with the global methylation of long-interspersed nuclear element-1, a repetitive DNA element expressed in the liver (270). Maternal BPA exposure during gestation and lactation affected glucose homeostasis in the F2 offspring where hepatic glucokinase gene promoter was completely methylated in all CpG sites compared with unmethylated sites in controls of the exposed liver tissue, indicating BPA’s control on the metabolic fate of the offspring via epigenetic programming of metabolic gene expression (271). Furthermore, BPA-exposed mice exhibited preeclampsia-like features, including hypertension, disrupted angiogenesis biomarkers in circulation, and placenta’s involvement in the reprogramming of DNA methylation of WNT2/beta-catenin gene, indicating that exposure window of placental development is the most critical for the epigenetic targets, thereby, a key determinant for the progression of placental-disease preeclampsia (272).
Epigenetic changes are sensitive to environmental factors, including exposure to EDCs like phthalate. Phthalates can cross the placental barrier, and their metabolites have been detected in the placenta. During pregnancy, the placenta produces and releases diverse miRNAs into the maternal circulation, which can be modulated due to the impact of phthalate exposure. Studies have indicated that phthalates and their metabolites can alter miRNA profiles in placental tissue, suggesting a potential link between phthalate exposure, miRNA dysregulation, and adverse pregnancy outcomes. Urinary levels of 13 phthalates in pregnant women with uncomplicated term dichorionic, diamniotic twin pregnancies and the miRNA profile of placental EVs (EV-miRNAs) circulating in maternal blood were measured. The expression of miR-518e in the maternal blood was highest among women with high mono-benzyl phthalate Field urinary levels (273). A study on 179 women found associations between first-trimester phthalate levels and miRNA expression in the placenta. miR-142-3p, miR15a-5p, and miR-185 expressions were found to be associated with urine phthalate levels (226). Mono-(2-ethylhexyl) phthalate (MEHP) induces oxidative stress-responsive miR-17-5p, miR-155-5p, and miR-126-3p in HTR8/SVneo in a dose- and time-dependent manner by altering the expression of genes involved in oxidative stress (274). A clinical study conducted on 202 pregnant women between 22 and 29 gestational weeks measured 11 phthalate metabolites in the spot urine samples and global DNA methylation in the placental tissue of the fetal side at delivery. Mono-benzyl phthalate concentration was inversely associated with the placental methylation of Alu repeats.
Moreover, all phthalate biomarkers except for mono-carboxy-iso-octyl phthalate and mono (2-ethyl-5-hydroxyhexyl) (MEHHP) phthalate were associated with at least one differentially methylated region (275). A case-control study examined the associations of prenatal phthalate exposure, infant growth, and global DNA methylation in human placenta 119 subjects [55 fetal growth restriction (FGR) cases and 64 normal controls]. Concentrations of mono (2-ethyl-5-hydroxyhexyl) phthalate, mono (2-ethyl-5-oxohexyl) phthalate (MEOHP), and sum DEHP (molar sum of MEHP, MEHHP, and MEOHP) were significantly higher in FGR cases than those in normal controls. Placental LINE-1 methylation was positively associated with fetal birth weight and negatively associated with urinary phthalate metabolites concentrations (MEHHP and mean DEHP) (276). Another study conducted in 181 mother-newborn pairs (80 fetal growth restriction newborns, 101 normal newborns) measured third-trimester urinary phthalate metabolite concentrations and placental DNA methylation levels of IGF2 and found that urinary concentrations of MEHHP and MEOHP were inversely associated significantly with placental IGF2 DNA methylation (277). A recent study measured the effect of maternal phthalate exposure on the human placental DNA methylome, transcriptome and identified 39 genes with significantly altered methylation and gene expression in the high phthalate exposure group, with most of these relationships were inversely correlated (29 out of 39) (227).
Moreover, placental imprinting was assessed at birth in placental samples in 179 subjects and found a significant decrease in H19 methylation, which was associated with high levels of the sum of phthalate metabolites and metabolites of low molecular weight (LMW) phthalates. Σ phthalate and LMW phthalate concentrations were inversely associated with IGF2 differentially methylated regions methylation values (278). Placental exposure to phthalates may result in aberrant miRNA expression. Pregnant SD rats exposed to DEHP promoted the placental miR-155-5p expression, the cAMP/PKA inactivation, lipid metabolism and altered placental histopathology. Moreover, the knockdown of miR-155-5p abrogated DEHP-induced proliferative, migrative, and invasive inhibition in HTR8/SVneo cells (279). Phthalates (MEHP) have been reported to induce apoptosis by inducing miR-16, which alters the BCL-2/BAX ratio in the first trimer placental trophoblast HTR-8/SVneo cells (280).
Available data indicate that gestational phthalate exposure could modulate the placental gene expression related to its function or altered response to the fetus due to aberrant expression of miRNAs in the placenta.
10 Conclusion
EDCs exposure can significantly affect fetoplacental growth and pregnancy outcomes via different mechanisms. Several EDCs are estrogen-mimicking steroids, and those can quickly diffuse into the nucleus across the cell membrane and modulate the transcription of genes. These endocrine disruptors can mediate their effects by interfering endocrine function of the hypothalamic-pituitary glands, changing the secretion of gonadotropin-releasing hormone in the hypothalamus, promoting the proliferation of the pituitary, premature puberty, and resulting in infertility risks in both men and women. The epigenetic fingerprint of steroidogenesis and organogenesis in the uterus, follicles, implant, and placenta could risk the gonadal programming of the offspring. The most abundant EDCs include BPs, which report obesogenic effects, disrupting regular metabolic activity, making the body prone to overweight, and obesity. Being overweight and obese state impedes a successful pregnancy. In addition, prenatal BPs exposure may influence postnatal fetal HPA responsiveness due to alterations in cortisol levels and HSD11β activities.
Several epidemiological studies pointed out maternal exposure to BPs and Phthalates is associated with adverse outcomes on fetal growth and development, its life-long impact in the endocrine-linked growth process, and risks of reproductive organ deformities. However, data correlating of these EDC exposure with the early stage of puberty and sexual maturity and its follow-up with pre-pregnancy endocrine regulation is required for renewing better strategies in preventing EDCs-exposure induced harmful effects on pregnancy outcomes.
Author contributions
SB: writing, revision, and approval of the final version. SV: writing. AD: revision and approval of the last version. All authors have approved the final version for submission.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rolfo A, Nuzzo AM, De Amicis R, Moretti L, Bertoli S, Leone A. Fetal-maternal exposure to endocrine disruptors: correlation with diet intake and pregnancy outcomes. Nutrients (2020) 12:1744–63. doi: 10.3390/nu12061744
2. Basak S, Das MK, Duttaroy AK. Plastics derived endocrine-disrupting compounds and their effects on early development. Birth Defects Res (2020) 112:1308–25. doi: 10.1002/bdr2.1741
3. Gingrich J, Ticiani E, Veiga-Lopez A. Placenta disrupted: endocrine disrupting chemicals and pregnancy. Trends Endocrinol Metab (2020) 7:508–24. doi: 10.1016/j.tem.2020.03.003
4. Yang C, Song G, Lim W. A mechanism for the effect of endocrine disrupting chemicals on placentation. Chemosphere (2019) 231:326–36. doi: 10.1016/j.chemosphere.2019.05.133
5. Cariati F, D’Uonno N, Borrillo F, Iervolino S, Galdiero G, Tomaiuolo R. “Bisphenol a: an emerging threat to male fertility”. Reprod Biol Endocrinol (2019) 17:6. doi: 10.1186/s12958-018-0447-6
6. Qian Y, Shao H, Ying X, Huang W, Hua Y. The endocrine disruption of prenatal phthalate exposure in mother and offspring. Front Public Health (2020) 8:366. doi: 10.3389/fpubh.2020.00366
7. Urfer A, Turpin H, Dimitrova N, Borghini A, Plessen KJ, Morisod Harari M, et al. Consequences of prematurity on cortisol regulation and adjustment difficulties: A 9-year longitudinal study. Children (Basel Switzerland) (2021) 9:9–24. doi: 10.3390/children9010009
8. Korevaar TIM, Medici M, Visser TJ, Peeters RP. Thyroid disease in pregnancy: new insights in diagnosis and clinical management. Nat Rev Endocrinol (2017) 13:610–22. doi: 10.1038/nrendo.2017.93
9. Kumar P, Magon N. Hormones in pregnancy. Nigerian Med J (2012) 53:179–83. doi: 10.4103/0300-1652.107549
10. Pepe GJ, Albrecht ED. Actions of placental and fetal adrenal steroid hormones in primate pregnancy. Endocr Rev (1995) 16:608–48. doi: 10.1210/edrv-16-5-608
11. Duttaroy AK, Basak S. Endocrine factors and their effects on placentation. In: Early nutrition and lifestyle factors: effects on first trimester placenta. Cham: Springer International Publishing(2016). p. 91–100.
12. Balakrishnan B, Henare K, Thorstensen EB, Ponnampalam AP, Mitchell MD. Transfer of bisphenol A across the human placenta. Am J Obstet Gynecol (2010) 202:393 e1–7. doi: 10.1016/j.ajog.2010.01.025
13. Chu PW, Yang ZJ, Huang HH, Chang AA, Cheng YC, Wu GJ, et al. Low-dose bisphenol A activates the ERK signaling pathway and attenuates steroidogenic gene expression in human placental cells. Biol Reprod (2018) 98:250–8. doi: 10.1093/biolre/iox162
14. Narciso L, Ietta F, Romagnoli R, Paulesu L, Mantovani A, Tait S. Effects of Bisphenol A on endogenous retroviral envelopes expression and trophoblast fusion in BeWo cells. Reprod Toxicol (Elmsford N.Y.) (2019) 89:35–44. doi: 10.1016/j.reprotox.2019.07.001
15. Tachibana T, Wakimoto Y, Nakamuta N, Phichitraslip T, Wakitani S, Kusakabe K, et al. Effects of bisphenol A (BPA) on placentation and survival of the neonates in mice. J Reprod Dev (2007) 53:509–14. doi: 10.1262/jrd.18171
16. Tait S, Tassinari R, Maranghi F, Mantovani A. Bisphenol A affects placental layers morphology and angiogenesis during early pregnancy phase in mice. J Appl Toxicol JAT (2015) 35:1278–91. doi: 10.1002/jat.3176
17. Xiao S, Diao H, Smith MA, Song X, Ye X. Preimplantation exposure to bisphenol A (BPA) affects embryo transport, preimplantation embryo development, and uterine receptivity in mice. Reprod Toxicol (Elmsford N.Y.) (2011) 32:434–41. doi: 10.1016/j.reprotox.2011.08.010
18. Muller JE, Meyer N, Santamaria CG, Schumacher A, Luque EH, Zenclussen ML, et al. Bisphenol A exposure during early pregnancy impairs uterine spiral artery remodeling and provokes intrauterine growth restriction in mice. Sci Rep (2018) 8:9196. doi: 10.1038/s41598-018-27575-y
19. Barberio L, Paulesu L, Canesi L, Grasselli E, Mandalà M. Bisphenol a interferes with uterine artery features and impairs rat feto-placental growth. Int J Mol Sci (2021) 22:6912–24. doi: 10.3390/ijms22136912
20. Wang ZY, Lu J, Zhang YZ, Zhang M, Liu T, Qu XL. Effect of Bisphenol A on invasion ability of human trophoblastic cell line BeWo. Int J Clin Exp Pathol (2015) 8:14355–64.
21. Ye Y, Tang Y, Xiong Y, Feng L, Li X. Bisphenol A exposure alters placentation and causes preeclampsia-like features in pregnant mice involved in reprogramming of DNA methylation of WNT2. FASEB J (2019) 33:2732–42. doi: 10.1096/fj.201800934RRR
22. Ermini L, Nuzzo AM, Ietta F, Romagnoli R, Moretti L, Masturzo B, et al. Placental glucose transporters and response to bisphenol A in pregnancies from of normal and overweight mothers. Int J Mol Sci (2021) 22:6912–39. doi: 10.3390/ijms22126625
23. Benincasa L, Mandalà M, Paulesu L, Barberio L, Ietta F. Prenatal nutrition containing bisphenol A affects placenta glucose transfer: evidence in rats and human trophoblast. Nutrients (2020) 12:1375–88. doi: 10.3390/nu12051375
24. Mao J, Jain A, Denslow ND, Nouri MZ, Chen S, Wang T, et al. Bisphenol A and bisphenol S disruptions of the mouse placenta and potential effects on the placenta-brain axis. Proc Natl Acad Sci USA (2020) 117:4642–52. doi: 10.1073/pnas.1919563117
25. Gingrich J, Pu Y, Roberts J, Karthikraj R, Kannan K, Ehrhardt R, et al. Gestational bisphenol S impairs placental endocrine function and the fusogenic trophoblast signaling pathway. Arch Toxicol (2018) 92:1861–76. doi: 10.1007/s00204-018-2191-2
26. Spagnoletti A, Paulesu L, Mannelli C, Ermini L, Romagnoli R, Cintorino M, et al. Low concentrations of Bisphenol A and para-Nonylphenol affect extravillous pathway of human trophoblast cells. Mol Cell Endocrinol (2015) 412:56–64. doi: 10.1016/j.mce.2015.05.023
27. Basak S, Srinivas V, Duttaroy AK. Bisphenol-A impairs cellular function and alters DNA methylation of stress pathway genes in first trimester trophoblast cells. Reprod Toxicol (2018) 82:72–9. doi: 10.1016/j.reprotox.2018.10.009
28. Ponniah M, Billett EE, De Girolamo LA. Bisphenol A increases BeWo trophoblast survival in stress-induced paradigms through regulation of oxidative stress and apoptosis. Chem Res Toxicol (2015) 28:1693–703. doi: 10.1021/acs.chemrestox.5b00093
29. Ehrlich S, Williams PL, Missmer SA, Flaws JA, Berry KF, Calafat AM, et al. Urinary bisphenol A concentrations and implantation failure among women undergoing in vitro fertilization. Environ Health Perspect (2012) 120:978–83. doi: 10.1289/ehp.1104307
30. Berger RG, Foster WG, deCatanzaro D. Bisphenol-A exposure during the period of blastocyst implantation alters uterine morphology and perturbs measures of estrogen and progesterone receptor expression in mice. Reprod Toxicol (Elmsford N.Y.) (2010) 30:393–400. doi: 10.1016/j.reprotox.2010.06.006
31. Takai Y, Tsutsumi O, Ikezuki Y, Hiroi H, Osuga Y, Momoeda M, et al. Estrogen receptor-mediated effects of a xenoestrogen, bisphenol A, on preimplantation mouse embryos. Biochem Biophys Res Commun (2000) 270:918–21. doi: 10.1006/bbrc.2000.2548
32. Yang L, Baumann C, de la Fuente R, Viveiros MM. Bisphenol exposure disrupts cytoskeletal organization and development of pre-implantation embryos. Cells (2022) 11:3233. doi: 10.3390/cells11203233
33. Ticiani E, Pu Y, Gingrich J, Veiga-Lopez A. Bisphenol S impairs invasion and proliferation of extravillous trophoblasts cells by interfering with epidermal growth factor receptor signaling. Int J Mol Sci 23 (2022) 23:671–85. doi: 10.3390/ijms23020671
34. Fan H, Jiang L, Lee YL, Wong CKC, Ng EHY, Yeung WSB, et al. Bisphenol compounds regulate decidualized stromal cells in modulating trophoblastic spheroid outgrowth and invasion in vitro†. Biol Reprod (2020) 102:693–704. doi: 10.1093/biolre/ioz212
35. Park HR, Kamau PW, Korte C, Loch-Caruso R. Tetrabromobisphenol A activates inflammatory pathways in human first trimester extravillous trophoblasts in vitro. Reprod Toxicol (Elmsford N.Y.) (2014) 50:154–62. doi: 10.1016/j.reprotox.2014.10.005
36. Ticiani E, Gingrich J, Pu Y, Vettathu M, Davis J, Martin D, et al. and epidermal growth factor receptor signaling in human placental cytotrophoblasts. Environ Health Perspect (2021) 129:27005. doi: 10.1289/EHP7297
37. Huang H, Tan W, Wang CC, Leung LK. Bisphenol A induces corticotropin-releasing hormone expression in the placental cells JEG-3. Reprod Toxicol (Elmsford N.Y.) (2012) 34:317–22. doi: 10.1016/j.reprotox.2012.04.008
38. Rajakumar C, Guan H, Langlois D, Cernea M, Yang K. Bisphenol A disrupts gene expression in human placental trophoblast cells. Reprod Toxicol (2015) 53:39–44. doi: 10.1016/j.reprotox.2015.03.001
39. Liang J, Yang C, Liu T, Tan HJJ, Sheng Y, Wei L, et al. Prenatal exposure to bisphenols and risk of preterm birth: Findings from Guangxi Zhuang birth cohort in China. Ecotoxicology Environ Saf (2021) 228:112960. doi: 10.1016/j.ecoenv.2021.112960
40. Reed JM, Spinelli P, Falcone S, He M, Goeke CM, Susiarjo M. Evaluating the effects of BPA and TBBPA exposure on pregnancy loss and maternal-fetal immune cells in mice. Environ Health Perspect (2022) 130:37010. doi: 10.1289/EHP10640
41. Sugiura-Ogasawara M, Ozaki Y, Sonta S, Makino T, Suzumori K. Exposure to bisphenol A is associated with recurrent miscarriage. Hum Reprod (Oxford England) (2005) 20:2325–9. doi: 10.1093/humrep/deh888
42. Aker AM, Ferguson KK, Rosario ZY, Mukherjee B, Alshawabkeh AN, Calafat AM, et al. A repeated measures study of phenol, paraben and Triclocarban urinary biomarkers and circulating maternal hormones during gestation in the Puerto Rico PROTECT cohort. Environ Health Global Access Sci Source (2019) 18:28. doi: 10.1186/s12940-019-0459-5
43. Arita Y, Pressman M, Getahun D, Menon R, Peltier MR. Effect of Tetrabromobisphenol A on expression of biomarkers for inflammation and neurodevelopment by the placenta. Placenta (2018) 68:33–9. doi: 10.1016/j.placenta.2018.06.306
44. Shi L, Zhang B, Ying Y, Tang Y, Wang S, Zhu Y, et al. Halogen atoms determine the inhibitory potency of halogenated bisphenol A derivatives on human and rat placental 11β-hydroxysteroid dehydrogenase 2. Food Chem Toxicol (2023) 175:113739. doi: 10.1016/j.fct.2023.113739
45. Marqueño A, Pérez-Albaladejo E, Flores C, Moyano E, Porte C. Toxic effects of bisphenol A diglycidyl ether and derivatives in human placental cells. Environ pollut (Barking Essex (2019) 1987) 244:513–21. doi: 10.1016/j.envpol.2018.10.045
46. Herman JP, McKlveen JM, Ghosal S, Kopp B, Wulsin A, Makinson R, et al. Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr Physiol (2016) 6:603–21. doi: 10.1002/cphy.c150015
47. Hewitt DP, Mark PJ, Waddell BJ. Glucocorticoids prevent the normal increase in placental vascular endothelial growth factor expression and placental vascularity during late pregnancy in the rat. Endocrinology (2006) 147:5568–74. doi: 10.1210/en.2006-0825
48. Cowell W, Deyssenroth M, Chen J, Wright RJ. Maternal stress in relation to sex-specific expression of placental genes involved in nutrient transport, oxygen tension, immune response, and the glucocorticoid barrier. Placenta (2020) 96:19–26. doi: 10.1016/j.placenta.2020.05.004
49. Walejko JM, Antolic A, Koelmel JP, Garrett TJ, Edison AS, Keller-Wood M. Chronic maternal cortisol excess during late gestation leads to metabolic alterations in the newborn heart. Am J Physiol Endocrinol Metab (2019) 316:E546–e556. doi: 10.1152/ajpendo.00386.2018
50. Stoye DQ, Andrew R, Grobman WA, Adam EK, Wadhwa PD, Buss C, et al. Maternal glucocorticoid metabolism across pregnancy: A potential mechanism underlying fetal glucocorticoid exposure. J Clin Endocrinol Metab (2020) 105:e782–90. doi: 10.1210/clinem/dgz313
51. Kuo AH, Li J, Li C, Huber HF, Schwab M, Nathanielsz PW, et al. Prenatal steroid administration leads to adult pericardial and hepatic steatosis in male baboons. Int J Obes (2017) 41:1299–302. doi: 10.1038/ijo.2017.82
52. Riveline JP, Baz B, Nguewa JL, Vidal-Trecan T, Ibrahim F, Boudou P, et al. Exposure to glucocorticoids in the first part of fetal life is associated with insulin secretory defect in adult humans. J Clin Endocrinol Metab 105 (2020) 3:191–9. doi: 10.1210/clinem/dgz145
53. Kajantie E, Dunkel L, Turpeinen U, U.-H.k. Stenman PJ, Nuutila M, Andersson S. Placental 11β-hydroxysteroid dehydrogenase-2 and fetal cortisol/cortisone shuttle in small preterm infants. J Clin Endocrinol Metab (2003) 88:493–500. doi: 10.1210/jc.2002-021378
54. Holmes MC, Abrahamsen CT, French KL, Paterson JM, Mullins JJ, Seckl JR. The mother or the fetus? 11β-hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. J Neurosci (2006) 26:3840–4. doi: 10.1523/JNEUROSCI.4464-05.2006
55. Nolten WE, Rueckert PA. Elevated free cortisol index in pregnancy: possible regulatory mechanisms. Am J Obstet Gynecol (1981) 139:492–8. doi: 10.1016/0002-9378(81)90331-8
56. Yang K. Placental 11 beta-hydroxysteroid dehydrogenase: barrier to maternal glucocorticoids. Rev Reprod (1997) 2:129–32. doi: 10.1530/ror.0.0020129
57. McTernan CL, Draper N, Nicholson H, Chalder SM, Driver P, Hewison M, et al. Reduced placental 11beta-hydroxysteroid dehydrogenase type 2 mRNA levels in human pregnancies complicated by intrauterine growth restriction: an analysis of possible mechanisms. J Clin Endocrinol Metab (2001) 86:4979–83. doi: 10.1210/jcem.86.10.7893
58. Wyrwoll CS, Seckl JR, Holmes MC. Altered placental function of 11beta-hydroxysteroid dehydrogenase 2 knockout mice. Endocrinology (2009) 150:1287–93. doi: 10.1210/en.2008-1100
59. Aufdenblatten M, Baumann M, Raio L, Dick B, Frey BM, Schneider H, et al. Prematurity is related to high placental cortisol in preeclampsia. Pediatr Res (2009) 65:198–202. doi: 10.1203/PDR.0b013e31818d6c24
60. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature (1998) 395:763–70. doi: 10.1038/27376
61. Masuzaki H, Ogawa Y, Sagawa N, Hosoda K, Matsumoto T, Mise H, et al. Nonadipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans. Nat Med (1997) 3:1029–33. doi: 10.1038/nm0997-1029
62. Maymo JL, Perez AP, Gambino Y, Calvo JC, Sanchez-Margalet V, Varone CL. Review: Leptin gene expression in the placenta–regulation of a key hormone in trophoblast proliferation and survival. Placenta (2011) 32 Suppl 2:S146–53. doi: 10.1016/j.placenta.2011.01.004
63. Ashworth CJ, Hoggard N, Thomas L, Mercer JG, Wallace JM, Lea RG. Placental leptin. Rev Reprod (2000) 5:18–24. doi: 10.1530/ror.0.0050018
64. Henson MC, Castracane VD. Leptin in pregnancy. Biol Reprod (2000) 63:1219–28. doi: 10.1095/biolreprod63.5.1219
65. Islami D, Bischof P, Chardonnens D. Possible interactions between leptin, gonadotrophin-releasing hormone (GnRH-I and II) and human chorionic gonadotrophin (hCG). Eur J Obstet Gynecol Reprod Biol (2003) 110:169–75. doi: 10.1016/S0301-2115(03)00185-4
66. Islami D. Modulation of placental vascular endothelial growth factor by leptin and hCG. Mol Hum Reprod (2003) 9:395–8. doi: 10.1093/molehr/gag053
67. Klaffenbach D, Meissner U, Raake M, Fahlbusch F, Alejandre Alcazar MA, Allabauer I, et al. Upregulation of leptin-receptor in placental cells by hypoxia. Regul Peptides (2011) 167:156–62. doi: 10.1016/j.regpep.2010.12.007
68. Mise H, Sagawa N, Matsumoto T, Yura S, Nanno H, Itoh H, et al. Augmented placental production of leptin in preeclampsia: possible involvement of placental hypoxia. J Clin Endocrinol Metab (1998) 83:3225–9. doi: 10.1210/jc.83.9.3225
69. Henson MC. Leptin in pregnancy: an update. Biol Reprod (2006) 74:218–29. doi: 10.1095/biolreprod.105.045120
70. Gavrilova O, Barr V, Marcus-Samuels B, Reitman M. Hyperleptinemia of pregnancy associated with the appearance of a circulating form of the leptin receptor. J Biol Chem (1997) 272:30546–51. doi: 10.1074/jbc.272.48.30546
71. Lewandowski K, Horn R, O’Callaghan CJ, Dunlop D, Medley GF, O’Hare P, et al. Bound leptin, and soluble leptin receptor in normal and diabetic pregnancies. J Clin Endocrinol Metab (1999) 84:300–6. doi: 10.1210/jcem.84.1.5401
72. Sierra-Honigmann MR. Biological action of leptin as an angiogenic factor. Science (1998) 281:1683–6. doi: 10.1126/science.281.5383.1683
73. Basak S, Duttaroy AK. Leptin induces tube formation in first-trimester extravillous trophoblast cells. Eur J Obstet Gynecol Reprod Biol (2012) 164:24–9. doi: 10.1016/j.ejogrb.2012.05.033
74. Pérez-Pérez A, Toro A, Vilariño-García T, Maymó J, Guadix P, Dueñas JL, et al. Leptin action in normal and pathological pregnancies. J Cell Mol Med (2018) 22:716–27. doi: 10.1111/jcmm.13369
75. Stefaniak M, Dmoch-Gajzlerska E, Mazurkiewicz B, Gajzlerska-Majewska W. Maternal serum and cord blood leptin concentrations at delivery. PloS One (2019) 14:e0224863. doi: 10.1371/journal.pone.0224863
76. Lea RG, Howe D, Hannah LT, Bonneau O, Hunter L, Hoggard N. Placental leptin in normal, diabetic and fetal growth-retarded pregnancies. Mol Hum Reprod (2000) 6:763–9. doi: 10.1093/molehr/6.8.763
77. Gagné-Ouellet V, Breton E, Thibeault K, Fortin CA, Cardenas A, Guérin R, et al. Mediation analysis supports a causal relationship between maternal hyperglycemia and placental DNA methylation variations at the leptin gene locus and cord blood leptin levels. Int J Mol Sci (2020) 21:329–40. doi: 10.3390/ijms21010329
78. Garcia-Arevalo M, Alonso-Magdalena P, Servitja JM, Boronat-Belda T, Merino B, Villar-Pazos S, et al. Maternal exposure to bisphenol-A during pregnancy increases pancreatic beta-cell growth during early life in male mice offspring. Endocrinology (2016) 157:4158–71. doi: 10.1210/en.2016-1390
79. Ivry Del Moral L, Le Corre L, Poirier H, Niot I, Truntzer T, Merlin JF, et al. Obesogen effects after perinatal exposure of 4,4'-sulfonyldiphenol (Bisphenol S) in C57BL/6 mice. Toxicology (2016) 357-358:11–20. doi: 10.1016/j.tox.2016.05.023
80. Ogueh O, Clough A, Hancock M, Johnson MR. A longitudinal study of the control of renal and uterine hemodynamic changes of pregnancy. Hypertens Pregnancy (2011) 30:243–59. doi: 10.3109/10641955.2010.484079
81. Fournier T, Guibourdenche J, Evain-Brion D. Review: hCGs: different sources of production, different glycoforms and functions. Placenta (2015) 36 Suppl 1:S60–5. doi: 10.1016/j.placenta.2015.02.002
82. Cole LA. New discoveries on the biology and detection of human chorionic gonadotropin. Reprod Biol Endocrinol (2009) 7:1–37. doi: 10.1186/1477-7827-7-8
83. Braunstein GD, Rasor J, Adler D, Danzer H, Wade ME. Serum human chorionic gonadotropin levels throughout normal pregnancy. Am J Obstet Gynecol (1976) 126:678–81. doi: 10.1016/0002-9378(76)90518-4
84. Zygmunt M, Herr F, Münstedt K, Lang U, Liang OD. Angiogenesis and vasculogenesis in pregnancy. Eur J Obstet Gynecol Reprod Biol (2003) 110 Suppl 1:S10–8. doi: 10.1016/S0301-2115(03)00168-4
85. Shi QJ, Lei ZM, Rao CV, Lin J. Novel role of human chorionic gonadotropin in differentiation of human cytotrophoblasts. Endocrinology (1993) 132:1387–95. doi: 10.1210/endo.132.3.7679981
86. Weedon-Fekjaer MS, Duttaroy AK, Nebb HI. Liver X receptors mediate inhibition of hCG secretion in a human placental trophoblast cell line. Placenta (2005) 26:721–8. doi: 10.1016/j.placenta.2004.10.005
87. Sirikunalai P, Wanapirak C, Sirichotiyakul S, Tongprasert F, Srisupundit K, Luewan S, et al. Associations between maternal serum free beta human chorionic gonadotropin (β-hCG) levels and adverse pregnancy outcomes. J Obstet Gynaecol (2016) 36:178–82. doi: 10.3109/01443615.2015.1036400
88. Günyeli İ, ZergeroĞlu S, Danişman N, MollamahmutoĞlu L. The diagnostic significance of hCG and hPL via immunohistochemistry of placental tissues in pregnancies diagnosed with IUGR and IUD. J Obstet Gynaecol (2009) 29:521–5. doi: 10.1080/01443610902993376
89. Dugoff L, Hobbins JC, Malone FD, Porter TF, Luthy D, Comstock CH, et al. First-trimester maternal serum PAPP-A and free-beta subunit human chorionic gonadotropin concentrations and nuchal translucency are associated with obstetric complications: a population-based screening study (the FASTER Trial). Am J Obstet Gynecol (2004) 191:1446–51. doi: 10.1016/j.ajog.2004.06.052
90. Jing G, Yao J, Dang Y, Liang W, Xie L, Chen J, et al. The role of β-HCG and VEGF-MEK/ERK signaling pathway in villi angiogenesis in patients with missed abortion. Placenta (2021) 103:16–23. doi: 10.1016/j.placenta.2020.10.005
91. Derakhshan A, Shu H, Broeren MAC, Kortenkamp A, Lindh CH, Demeneix B, et al. Association of endocrine disrupting chemicals exposure with human chorionic gonadotropin concentrations in pregnancy. Environ Int (2023) 178:108091. doi: 10.1016/j.envint.2023.108091
92. Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol (1999) 19:3278–88. doi: 10.1128/MCB.19.5.3278
93. Basak S, Vilasagaram S, Naidu K, Duttaroy AK. Insulin-dependent, glucose transporter 1 mediated glucose uptake and tube formation in the human placental first trimester trophoblast cells. Mol Cell Biochem (2019) 451:91–106. doi: 10.1007/s11010-018-3396-7
94. Lassance L, Miedl H, Absenger M, Diaz-Perez F, Lang U, Desoye G, et al. Hyperinsulinemia stimulates angiogenesis of human fetoplacental endothelial cells: a possible role of insulin in placental hypervascularization in diabetes mellitus. J Clin Endocrinol Metab (2013) 98:E1438–47. doi: 10.1210/jc.2013-1210
95. Dalcik H, Yardimoglu M, Vural B, Dalcik C, Filiz S, Gonca S, et al. Expression of insulin-like growth factor in the placenta of intrauterine growth-retarded human fetuses. Acta Histochemica (2001) 103:195–207. doi: 10.1078/0065-1281-00580
96. Thomsen BM, Clausen HV, Larsen LG, Nurnberg L, Ottesen B, Thomsen HK. Patterns in expression of insulin-like growth factor-II and of proliferative activity in the normal human first and third trimester placenta demonstrated by non-isotopic in situ hybridization and immunohistochemical staining for MIB-1. Placenta (1997) 18:145–54. doi: 10.1016/S0143-4004(97)90086-2
97. Hill DJ, Clemmons DR, Riley SC, Bassett N, Challis JR. Immunohistochemical localization of insulin-like growth factors (IGFs) and IGF binding proteins -1, -2 and -3 in human placenta and fetal membranes. Placenta (1993) 14:1–12. doi: 10.1016/S0143-4004(05)80244-9
98. Hamilton GS, Lysiak JJ, Han VK, Lala PK. Autocrine-paracrine regulation of human trophoblast invasiveness by insulin-like growth factor (IGF)-II and IGF-binding protein (IGFBP)-1. Exp Cell Res (1998) 244:147–56. doi: 10.1006/excr.1998.4195
99. Irving JA, Lala PK. Functional role of cell surface integrins on human trophoblast cell migration: regulation by TGF-beta, IGF-II, and IGFBP-1. Exp Cell Res (1995) 217:419–27. doi: 10.1006/excr.1995.1105
100. Magnusson-Olsson AL, Hamark B, Ericsson A, Wennergren M, Jansson T, Powell TL. Gestational and hormonal regulation of human placental lipoprotein lipase. J Lipid Res (2006) 47:2551–61. doi: 10.1194/jlr.M600098-JLR200
101. Basak S, Sarkar A, Mathapati S, Duttaroy AK. Cellular growth and tube formation of HTR8/SVneo trophoblast: effects of exogenously added fatty acid-binding protein-4 and its inhibitor. Mol Cell Biochem (2018) 437:55–64. doi: 10.1007/s11010-017-3095-9
102. Kniss DA, Shubert PJ, Zimmerman PD, Landon MB, Gabbe SG. Insulinlike growth factors. Their regulation of glucose and amino acid transport in placental trophoblasts isolated from first-trimester chorionic villi. J Reprod Med (1994) 39:249–56.
103. Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, et al. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab (2013) 98:105–13. doi: 10.1210/jc.2012-2667
104. Baumann MU, Schneider H, Malek A, Palta V, Surbek DV, Sager R, et al. Regulation of human trophoblast GLUT1 glucose transporter by insulin-like growth factor I (IGF-I). PloS One (2014) 9:e106037. doi: 10.1371/journal.pone.0106037
105. Roos S, Powell TL, Jansson T. Placental mTOR links maternal nutrient availability to fetal growth. Biochem Soc Trans (2009) 37:295–8. doi: 10.1042/BST0370295
106. Mohammed S, Qadri S, Molangiri A, Basak S, Rajkumar H. Gestational low dietary protein induces intrauterine inflammation and alters the programming of adiposity & insulin sensitivity in the adult offspring. J Nutr Biochem (2023) 116:109330. doi: 10.1016/j.jnutbio.2023.109330
107. Basak S, Duttaroy AK. Maternal PUFAs, placental epigenetics, and their relevance to fetal growth and brain development. Reprod Sci (2023) 30:408–27. doi: 10.1007/s43032-022-00989-w
108. Duttaroy AK, Basak S. Maternal fatty acid metabolism in pregnancy and its consequences in the feto-placental development. Front Physiol (2022) 12:787848–66. doi: 10.3389/fphys.2021.787848
109. Basak S, Mallick R, Banerjee A, Pathak S, Duttaroy AK. Maternal supply of both arachidonic and docosahexaenoic acids is required for optimal neurodevelopment. Nutrients (2021) 13:2061–89. doi: 10.3390/nu13062061
110. Basak S, Duttaroy AK. Effects of fatty acids on angiogenic activity in the placental extravillious trophoblast cells. Prostaglandins Leukot Essent Fatty Acids (2013) 88:155–62. doi: 10.1016/j.plefa.2012.10.001
111. Nicola C, Lala PK, Chakraborty C. Prostaglandin E2-mediated migration of human trophoblast requires RAC1 and CDC42. Biol Reprod (2008) 78:976–82. doi: 10.1095/biolreprod.107.065433
112. Horita H, Kuroda E, Hachisuga T, Kashimura M, Yamashita U. Induction of prostaglandin E2 production by leukemia inhibitory factor promotes migration of first trimester extravillous trophoblast cell line, HTR-8/SVneo. Hum Reprod (2007) 22:1801–9. doi: 10.1093/humrep/dem125
113. Sonderegger S, Yap J, Menkhorst E, Weston G, Stanton PG, Dimitriadis E. Interleukin (IL)11 mediates protein secretion and modification in human extravillous trophoblasts. Hum Reprod (2011) 26:2841–9. doi: 10.1093/humrep/der259
114. Das MK, Basak S, Ahmed MS, Attramadal H, Duttaroy AK. Connective tissue growth factor induces tube formation and IL-8 production in first trimester human placental trophoblast cells. Eur J Obstet Gynecol Reprod Biol (2014) 181:183–8. doi: 10.1016/j.ejogrb.2014.07.045
115. Zhang Y, Daaka Y. PGE2 promotes angiogenesis through EP4 and PKA Cgamma pathway. Blood (2011) 118:5355–64. doi: 10.1182/blood-2011-04-350587
116. Trau HA, Brännström M, Curry TE Jr., Duffy DM. Prostaglandin E2 and vascular endothelial growth factor A mediate angiogenesis of human ovarian follicular endothelial cells. Hum Reprod (2016) 31:436–44. doi: 10.1093/humrep/dev320
117. Baryla M, Kaczynski P, Goryszewska E, Riley SC, Waclawik A. Prostaglandin F(2α) stimulates adhesion, migration, invasion and proliferation of the human trophoblast cell line HTR-8/SVneo. Placenta (2019) 77:19–29. doi: 10.1016/j.placenta.2019.01.020
118. Ulug U, Goldman S, Ben-Shlomo I, Shalev E. Matrix metalloproteinase (MMP)-2 and MMP-9 and their inhibitor, TIMP-1, in human term decidua and fetal membranes: the effect of prostaglandin F(2alpha) and indomethacin. Mol Hum Reprod (2001) 7:1187–93. doi: 10.1093/molehr/7.12.1187
119. Pasqualini JR. Enzymes involved in the formation and transformation of steroid hormones in the fetal and placental compartments. J Steroid Biochem Mol Biol (2005) 97:401–15. doi: 10.1016/j.jsbmb.2005.08.004
120. Picard F, Wanatabe M, Schoonjans K, Lydon J, O'Malley BW, Auwerx J. Progesterone receptor knockout mice have an improved glucose homeostasis secondary to beta -cell proliferation. Proc Natl Acad Sci USA (2002) 99:15644–8. doi: 10.1073/pnas.202612199
121. Açıkgöz Ş, Özmen Bayar Ü, Can M, Güven B, Mungan G, Doğan S, et al. Levels of oxidized LDL, estrogens, and progesterone in placenta tissues and serum paraoxonase activity in preeclampsia. Mediators Inflammation (2013) 2013:862982. doi: 10.1155/2013/862982
122. Pei J, Liu Z, Wang C, Chu N, Liu L, Tang Y, et al. Progesterone attenuates SIRT1-deficiency-mediated pre-eclampsia. Biomolecules (2022) 12:422–39. doi: 10.3390/biom12030422
123. Romero R, Conde-Agudelo A, Da Fonseca E, O'Brien JM, Cetingoz E, Creasy GW, et al. Vaginal progesterone for preventing preterm birth and adverse perinatal outcomes in singleton gestations with a short cervix: a meta-analysis of individual patient data. Am J Obstet Gynecol (2018) 218:161–80. doi: 10.1016/j.ajog.2017.11.576
124. Romero R, Conde-Agudelo A, El-Refaie W, Rode L, Brizot ML, Cetingoz E, et al. Vaginal progesterone decreases preterm birth and neonatal morbidity and mortality in women with a twin gestation and a short cervix: an updated meta-analysis of individual patient data. Ultrasound Obstet Gynecol (2017) 49:303–14. doi: 10.1002/uog.17397
125. Bai J, Qi QR, Li Y, Day R, Makhoul J, Magness RR, et al. Estrogen receptors and estrogen-induced uterine vasodilation in pregnancy. Int J Mol Sci (2020) 21:4349–98. doi: 10.3390/ijms21124349
126. Ribeiro AC, Musatov S, Shteyler A, Simanduyev S, Arrieta-Cruz I, Ogawa S, et al. siRNA silencing of estrogen receptor-α expression specifically in medial preoptic area neurons abolishes maternal care in female mice. Proc Natl Acad Sci USA (2012) 109:16324–9. doi: 10.1073/pnas.1214094109
127. Koehler KF, Helguero LA, Haldosén LA, Warner M, Gustafsson JA. Reflections on the discovery and significance of estrogen receptor beta. Endocr Rev (2005) 26:465–78. doi: 10.1210/er.2004-0027
128. Root-Bernstein R, Podufaly A, Dillon PF. Estradiol binds to insulin and insulin receptor decreasing insulin binding in vitro. Front Endocrinol (Lausanne) (2014) 5. doi: 10.3389/fendo.2014.00118
129. Qi X, Gong B, Yu J, Shen L, Jin W, Wu Z, et al. Decreased cord blood estradiol levels in related to mothers with gestational diabetes. Medicine (2017) 96:e6962. doi: 10.1097/MD.0000000000006962
130. Jobe SO, Ramadoss J, Koch JM, Jiang Y, Zheng J, Magness RR. Estradiol-17beta and its cytochrome P450- and catechol-O-methyltransferase-derived metabolites stimulate proliferation in uterine artery endothelial cells: role of estrogen receptor-alpha versus estrogen receptor-beta. Hypertension (Dallas Tex. (2010) 1979) 55:1005–11. doi: 10.1161/HYPERTENSIONAHA.109.146399
131. Johns A, Freay AD, Fraser W, Korach KS, Rubanyi GM. Disruption of estrogen receptor gene prevents 17 beta estradiol-induced angiogenesis in transgenic mice. Endocrinology (1996) 137:4511–3. doi: 10.1210/endo.137.10.8828515
132. Hertig A, Liere P, Chabbert-Buffet N, Fort J, Pianos A, Eychenne B, et al. Steroid profiling in preeclamptic women: evidence for aromatase deficiency. Am J Obstet Gynecol (2010) 203:477.e1–9. doi: 10.1016/j.ajog.2010.06.011
133. Jobe SO, Tyler CT, Magness RR. Aberrant synthesis, metabolism, and plasma accumulation of circulating estrogens and estrogen metabolites in preeclampsia implications for vascular dysfunction. Hypertension (Dallas Tex. (2013) 1979) 61:480–7. doi: 10.1161/HYPERTENSIONAHA.111.201624
134. Barjaktarovic M, Korevaar TI, Jaddoe VW, de Rijke YB, Visser TJ, Peeters RP, et al. Human chorionic gonadotropin (hCG) concentrations during the late first trimester are associated with fetal growth in a fetal sex-specific manner. Eur J Epidemiol (2017) 32:135–44. doi: 10.1007/s10654-016-0201-3
135. Kirkegaard I, Henriksen TB, Uldbjerg N. Early fetal growth, PAPP-A and free β-hCG in relation to risk of delivering a small-for-gestational age infant. Ultrasound Obstet Gynecol (2011) 37:341–7. doi: 10.1002/uog.8808
136. Pihl K, Larsen T, Krebs L, Christiansen M. First trimester maternal serum PAPP-A, β-hCG and ADAM12 in prediction of small-for-gestational-age fetuses. Prenatal Diagnosis (2008) 28:1131–5. doi: 10.1002/pd.2141
137. Anelli GM, Mandò C, Letizia T, Mazzocco MI, Novielli C, Lisso F, et al. Placental ESRRG-CYP19A1 expressions and circulating 17-beta estradiol in IUGR pregnancies. Front Pediatr (2019) 7:154. doi: 10.3389/fped.2019.00154
138. Ozer A, Tolun F, Aslan F, Hatırnaz S, Alkan F. The role of G protein-associated estrogen receptor (GPER) 1, corin, raftlin, and estrogen in etiopathogenesis of intrauterine growth retardation. J Maternal-fetal Neonatal Med (2021) 34:755–60. doi: 10.1080/14767058.2019.1615433
139. Zou Z, Harris LK, Forbes K, Heazell AEP. Placental expression of estrogen-related receptor gamma is reduced in fetal growth restriction pregnancies and is mediated by hypoxia†. Biol Reprod (2022) 107:846–57. doi: 10.1093/biolre/ioac108
140. Metzger BE, Lowe LP, Dyer AR, Trimble ER, Chaovarindr U, Coustan DR, et al. Hyperglycemia and adverse pregnancy outcomes. New Engl J Med (2008) 358:1991–2002. doi: 10.1056/NEJMoa0707943
141. Acosta O, Ramirez VI, Lager S, Gaccioli F, Dudley DJ, Powell TL, et al. Increased glucose and placental GLUT-1 in large infants of obese nondiabetic mothers. Am J Obstet Gynecol (2015) 212:227.e1–7. doi: 10.1016/j.ajog.2014.08.009
142. Lüscher BP, Marini C, Joerger-Messerli MS, Huang X, Hediger MA, Albrecht C, et al. Placental glucose transporter (GLUT)-1 is down-regulated in preeclampsia. Placenta (2017) 55:94–9. doi: 10.1016/j.placenta.2017.04.023
143. Hiden U, Maier A, Bilban M, Ghaffari-Tabrizi N, Wadsack C, Lang I, et al. Insulin control of placental gene expression shifts from mother to foetus over the course of pregnancy. Diabetologia (2006) 49:123–31. doi: 10.1007/s00125-005-0054-x
144. Liu Y, Guo F, Maraka S, Zhang Y, Zhang C, Korevaar TIM, et al. Associations between human chorionic gonadotropin, maternal free thyroxine, and gestational diabetes mellitus. Thyroid (2021) 31:1282–8. doi: 10.1089/thy.2020.0920
145. Merviel P, Müller F, Guibourdenche J, Berkane N, Gaudet R, Bréart G, et al. Correlations between serum assays of human chorionic gonadotrophin (hCG) and human placental lactogen (hPL) and pre-eclampsia or intrauterine growth restriction (IUGR) among nulliparas younger than 38 years. Eur J Obstet Gynecol Reprod Biol (2001) 95:59–67. doi: 10.1016/S0301-2115(00)00370-5
146. Takeda K, Toda K, Saibara T, Nakagawa M, Saika K, Onishi T, et al. Progressive development of insulin resistance phenotype in male mice with complete aromatase (CYP19) deficiency. J Endocrinol (2003) 176:237–46. doi: 10.1677/joe.0.1760237
147. Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, et al. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci USA (2006) 103:9232–7. doi: 10.1073/pnas.0602956103
148. Li M, Song Y, Rawal S, Hinkle SN, Zhu Y, Tekola-Ayele F, et al. Plasma prolactin and progesterone levels and the risk of gestational diabetes: A prospective and longitudinal study in a multiracial cohort. Front Endocrinol (Lausanne) (2020) 11:83. doi: 10.3389/fendo.2020.00083
149. Hwa HL, Peng FS, Ting TT, Chen HW, Chan HY, Yang DP, et al. Monitoring phthalates in maternal and cord blood: implications for prenatal exposure and birth outcomes. Environ Toxicol Chem (2022) 41:715–25. doi: 10.1002/etc.5280
150. Qin J, Ru S, Wang W, Hao L, Ru Y, Wang J, et al. Long-term bisphenol S exposure aggravates non-alcoholic fatty liver by regulating lipid metabolism and inducing endoplasmic reticulum stress response with activation of unfolded protein response in male zebrafish. Environ Pollut (Barking Essex (2020) 1987) 263:114535. doi: 10.1016/j.envpol.2020.114535
151. Darghouthi M, Rezg R, Boughmadi O, Mornagui B. Low-dose bisphenol S exposure induces hypospermatogenesis and mitochondrial dysfunction in rats: A possible implication of StAR protein. Reprod Toxicol (Elmsford N.Y.) (2022) 107:104–11. doi: 10.1016/j.reprotox.2021.11.007
152. Sheller-Miller S, Radnaa E, Arita Y, Getahun D, Jones RJ, Peltier MR, et al. Environmental pollutant induced cellular injury is reflected in exosomes from placental explants. Placenta (2020) 89:42–9. doi: 10.1016/j.placenta.2019.10.008
153. Nerín C, Aznar M, Carrizo D. Food contamination during food process. Trends Food Sci Technol (2016) 48:63–8. doi: 10.1016/j.tifs.2015.12.004
154. Wu LH, Zhang XM, Wang F, Gao CJ, Chen D, Palumbo JR, et al. Occurrence of bisphenol S in the environment and implications for human exposure: A short review. Sci Total Environ (2018) 615:87–98. doi: 10.1016/j.scitotenv.2017.09.194
155. Kubiak A, Biesaga M. Application of molecularly imprinted polymers for bisphenols extraction from food samples – A review. Crit Rev Analytical Chem (2020) 50:311–21. doi: 10.1080/10408347.2019.1626698
156. Cressey D. Bisphenol A goes through the skin. Nature (2010) 581:1038–9. doi: 10.1038/news.2010.581
157. Takeuchi T, Tsutsumi O. Serum bisphenol a concentrations showed gender differences, possibly linked to androgen levels. Biochem Biophys Res Commun (2002) 291:76–8. doi: 10.1006/bbrc.2002.6407
158. Kim YH, Kim CS, Park S, Han SY, Pyo MY, Yang M. Gender differences in the levels of bisphenol A metabolites in urine. Biochem Biophys Res Commun (2003) 312:441–8. doi: 10.1016/j.bbrc.2003.10.135
159. Gounden V, Warasally MZ, Magwai T, Naidoo R, Chuturgoon A. A pilot study: Relationship between Bisphenol A, Bisphenol A glucuronide and sex steroid hormone levels in cord blood in A South African population. Reprod Toxicol (2021) 100:83–9. doi: 10.1016/j.reprotox.2021.01.004
160. Lee YM, Hong YC, Ha M, Kim Y, Park H, Kim HS, et al. Prenatal Bisphenol-A exposure affects fetal length growth by maternal glutathione transferase polymorphisms, and neonatal exposure affects child volume growth by sex: From multiregional prospective birth cohort MOCEH study. Sci Total Environ (2018) 612:1433–41. doi: 10.1016/j.scitotenv.2017.08.317
161. Casas M, Valvi D, Ballesteros-Gomez A, Gascon M, Fernández MF, Garcia-Esteban R, et al. Exposure to bisphenol A and phthalates during pregnancy and ultrasound measures of fetal growth in the INMA-sabadell cohort. Environ Health Perspect (2016) 124:521–8. doi: 10.1289/ehp.1409190
162. Snijder CA, Heederik D, Pierik FH, Hofman A, Jaddoe VW, Koch HM, et al. Fetal growth and prenatal exposure to bisphenol A: the generation R study. Environ Health Perspect (2013) 121:393–8. doi: 10.1289/ehp.1205296
163. Chou WC, Chen JL, Lin CF, Chen YC, Shih FC, Chuang CY. Biomonitoring of bisphenol A concentrations in maternal and umbilical cord blood in regard to birth outcomes and adipokine expression: a birth cohort study in Taiwan. Environ Health (2011) 10:94. doi: 10.1186/1476-069X-10-94
164. Yang P, Lin BG, Zhou B, Cao WC, Chen PP, Deng YL, et al. Sex-specific associations of prenatal exposure to bisphenol A and its alternatives with fetal growth parameters and gestational age. Environ Int (2021) 146:106305. doi: 10.1016/j.envint.2020.106305
165. Zhou B, Yang P, Deng YL, Zeng Q, Lu WQ, Mei SR. Prenatal exposure to bisphenol a and its analogues (bisphenol F and S) and ultrasound parameters of fetal growth. Chemosphere (2020) 246:125805. doi: 10.1016/j.chemosphere.2019.125805
166. Liang J, Liu S, Liu T, Yang C, Wu Y, Jennifer Tan HJ, et al. Association of prenatal exposure to bisphenols and birth size in Zhuang ethnic newborns. Chemosphere (2020) 252:126422. doi: 10.1016/j.chemosphere.2020.126422
167. Hu J, Zhao H, Braun JM, Zheng T, Zhang B, Xia W, et al. Associations of trimester-specific exposure to bisphenols with size at birth: A Chinese prenatal cohort study. Environ Health Perspect (2019) 127:107001. doi: 10.1289/EHP4664
168. Sol CM, van Zwol-Janssens C, Philips EM, Asimakopoulos AG, Martinez-Moral MP, Kannan K, et al. Maternal bisphenol urine concentrations, fetal growth and adverse birth outcomes: A population-based prospective cohort. Environ Health Global Access Sci Source (2021) 20:60. doi: 10.1186/s12940-021-00747-6
169. Wang J, Mei H, Zhou AF, Huang LL, Cao ZQ, Hong AB, et al. The associations of birth outcome differences in twins with prenatal exposure to bisphenol A and its alternatives. Environ Res (2021) 200:111459. doi: 10.1016/j.envres.2021.111459
170. Amin MM, Ghasemi Z, Khoshhali M, Taheri E, Dehdashti B, Fatehizadeh A, et al. Association of maternal exposure to bisphenol A with her β-hCG level and neonatal anthropometric measures. Environ Sci Pollut Res Int (2021) 28:62809–15. doi: 10.1007/s11356-021-15094-9
171. Wan Y, Huo W, Xu S, Zheng T, Zhang B, Li Y, et al. Relationship between maternal exposure to bisphenol S and pregnancy duration. Environ Pollut (Barking Essex (2018) 1987) 238:717–24. doi: 10.1016/j.envpol.2018.03.057
172. Bodin J, Bølling AK, Becher R, Kuper F, Løvik M, Nygaard UC. Transmaternal bisphenol A exposure accelerates diabetes type 1 development in NOD mice. Toxicological Sci (2014) 137:311–23. doi: 10.1093/toxsci/kft242
173. Boronat-Belda T, Ferrero H, Al-Abdulla R, Quesada I, Gustafsson JA, Nadal Á, et al. Bisphenol-A exposure during pregnancy alters pancreatic β-cell division and mass in male mice offspring: A role for ERβ. Food Chem Toxicol (2020) 145:111681. doi: 10.1016/j.fct.2020.111681
174. Bansal A, Rashid C, Xin F, Li C, Polyak E, Duemler A, et al. Sex- and dose-specific effects of maternal bisphenol A exposure on pancreatic islets of first- and second-generation adult mice offspring. Environ Health Perspect (2017) 125:097022. doi: 10.1289/EHP1674
175. Bansal A, Li C, Xin F, Duemler A, Li W, Rashid C, et al. Transgenerational effects of maternal bisphenol: a exposure on offspring metabolic health. J Dev Origins Health Dis (2018) 10:164–75. doi: 10.1017/S2040174418000764
176. Zhang B, Wang S, Tang Y, Hu Z, Shi L, Lu J, et al. Direct inhibition of bisphenols on human and rat 11β-hydroxysteroid dehydrogenase 2: Structure-activity relationship and docking analysis. Ecotoxicol. Environ Saf (2023) 254:114715. doi: 10.1016/j.ecoenv.2023.114715
177. Boucher JG, Boudreau A, Atlas E. Bisphenol A induces differentiation of human preadipocytes in the absence of glucocorticoid and is inhibited by an estrogen-receptor antagonist. Nutr Diabetes (2014) 4:e102–2. doi: 10.1038/nutd.2013.43
178. Molangiri A, Varma S, Satyavani M, Kambham S, Duttaroy AK, Basak S. Prenatal exposure to bisphenol S and bisphenol A differentially affects male reproductive system in the adult offspring. Food Chem Toxicol (2022) 167:113292. doi: 10.1016/j.fct.2022.113292
179. Ullah A, Pirzada M, Jahan S, Ullah H, Razak S, Rauf N, et al. Prenatal BPA and its analogs BPB, BPF, and BPS exposure and reproductive axis function in the male offspring of Sprague Dawley rats. Hum Exp Toxicol (2019) 38:1344–65. doi: 10.1177/0960327119862335
180. Yang YJ, Hong YP, Chae SA. Reduction in semen quality after mixed exposure to bisphenol A and isobutylparaben in utero and during lactation periods. Hum Exp Toxicol (2016) 35:902–11. doi: 10.1177/0960327115608927
181. Shi M, Whorton AE, Sekulovski N, MacLean JA, Hayashi K. Prenatal exposure to bisphenol A, E, and S induces transgenerational effects on male reproductive functions in mice. Toxicological Sci (2019) 172:303–15. doi: 10.1093/toxsci/kfz207
182. Hunt PA, Lawson C, Gieske M, Murdoch B, Smith H, Marre A, et al. Bisphenol A alters early oogenesis and follicle formation in the fetal ovary of the rhesus monkey. Proc Natl Acad Sci USA (2012) 109:17525–30. doi: 10.1073/pnas.1207854109
183. Susiarjo M, Hassold TJ, Freeman E, Hunt PA. Bisphenol A exposure in utero disrupts early oogenesis in the mouse. PloS Genet (2007) 3:e5. doi: 10.1371/journal.pgen.0030005
184. Shi M, Whorton AE, Sekulovski N, MacLean JA, Hayashi K. Prenatal exposure to bisphenol A, E, and S induces transgenerational effects on female reproductive functions in mice. Toxicological Sci (2019) 170:320–9. doi: 10.1093/toxsci/kfz124
185. Ozkemahli G, Balci Ozyurt A, Erkekoglu P, Zeybek ND, Yersal N, Kocer-Gumusel B. The effects of prenatal and lactational bisphenol A and/or di(2-ethylhexyl) phthalate exposure on female reproductive system. Toxicol Mech Methods (2022) 32:597–605. doi: 10.1080/15376516.2022.2057265
186. Namat A, Xia W, Xiong C, Xu S, Wu C, Wang A, et al. Association of BPA exposure during pregnancy with risk of preterm birth and changes in gestational age: A meta-analysis and systematic review. Ecotoxicology Environ Saf (2021) 220:112400. doi: 10.1016/j.ecoenv.2021.112400
187. Varma S, Molangiri A, Kona SR, Ibrahim A, Duttaroy AK, Basak S. Fetal exposure to endocrine disrupting-bisphenol A (BPA) alters testicular fatty acid metabolism in the adult offspring: relevance to sperm maturation and quality. Int J Mol Sci (2023) 24:3769–87. doi: 10.3390/ijms24043769
188. The Lancet Regional Health – E. Polycystic ovary syndrome: What more can be done for patients? Lancet Regional Health – Europe (2022) 21:1–2. doi: 10.1016/j.lanepe.2022.100524
189. Dumesic DA, Oberfield SE, Stener-Victorin E, Marshall JC, Laven JS, Legro RS. Scientific statement on the diagnostic criteria, epidemiology, pathophysiology, and molecular genetics of polycystic ovary syndrome. Endocrine Rev (2015) 36:487–525. doi: 10.1210/er.2015-1018
190. Osuka S, Nakanishi N, Murase T, Nakamura T, Goto M, Iwase A, et al. Animal models of polycystic ovary syndrome: A review of hormone-induced rodent models focused on hypothalamus-pituitary-ovary axis and neuropeptides. Reprod Med Biol (2019) 18:151–60. doi: 10.1002/rmb2.12262
191. Kandaraki E, Chatzigeorgiou A, Livadas S, Palioura E, Economou F, Koutsilieris M, et al. Endocrine disruptors and polycystic ovary syndrome (PCOS): elevated serum levels of bisphenol A in women with PCOS. J Clin Endocrinol Metab (2011) 96:E480–4. doi: 10.1210/jc.2010-1658
192. Akın L, Kendirci M, Narin F, Kurtoglu S, Saraymen R, Kondolot M, et al. The endocrine disruptor bisphenol A may play a role in the aetiopathogenesis of polycystic ovary syndrome in adolescent girls. Acta Paediatrica (Oslo Norway (2015) 1992) 104:e171–7. doi: 10.1111/apa.12885
193. Bromer JG, Zhou Y, Taylor MB, Doherty L, Taylor HS. Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response. FASEB J (2010) 24:2273–80. doi: 10.1096/fj.09-140533
194. Qu F, Wang FF, Yin R, Ding GL, El-Prince M, Gao Q, et al. A molecular mechanism underlying ovarian dysfunction of polycystic ovary syndrome: hyperandrogenism induces epigenetic alterations in the granulosa cells. J Mol Med (Berlin Germany) (2012) 90:911–23. doi: 10.1007/s00109-012-0881-4
195. Mukhopadhyay R, Prabhu NB, Kabekkodu SP, Rai PS. Review on bisphenol A and the risk of polycystic ovarian syndrome: an insight from endocrine and gene expression. Environ Sci Pollut Res (2022) 29:32631–50. doi: 10.1007/s11356-022-19244-5
196. Hill CE, Sapouckey SA, Suvorov A, Vandenberg LN. Developmental exposures to bisphenol S, a BPA replacement, alter estrogen-responsiveness of the female reproductive tract: A pilot study. Cogent Med (2017) 4:1317690. doi: 10.1080/2331205X.2017.1317690
197. Rutkowska A, Rachoń D, Bisphenol A. (BPA) and its potential role in the pathogenesis of the polycystic ovary syndrome (PCOS). Gynecological Endocrinol (2014) 30:260–5. doi: 10.3109/09513590.2013.871517
198. Fernández M, Bourguignon N, Lux-Lantos V, Libertun C. Neonatal exposure to bisphenol a and reproductive and endocrine alterations resembling the polycystic ovarian syndrome in adult rats. Environ Health Perspect (2010) 118:1217–22. doi: 10.1289/ehp.0901257
199. Fernandez MO, Bourguignon NS, Arocena P, Rosa M, Libertun C, Lux-Lantos V. Neonatal exposure to bisphenol A alters the hypothalamic-pituitary-thyroid axis in female rats. Toxicol Lett (2018) 285:81–6. doi: 10.1016/j.toxlet.2017.12.029
200. Rubin BS, Murray MK, Damassa DA, King JC, Soto AM. Perinatal exposure to low doses of bisphenol A affects body weight, patterns of estrous cyclicity, and plasma LH levels. Environ Health Perspect (2001) 109:675–80. doi: 10.1289/ehp.01109675
201. Howdeshell KL, Hotchkiss AK, Thayer KA, Vandenbergh JG, vom Saal FS. Exposure to bisphenol A advances puberty. Nature (1999) 401:763–4. doi: 10.1038/44517
202. Honma S, Suzuki A, Buchanan DL, Katsu Y, Watanabe H, Iguchi T. Low dose effect of in utero exposure to bisphenol A and diethylstilbestrol on female mouse reproduction. Reprod Toxicol (Elmsford N.Y.) (2002) 16:117–22. doi: 10.1016/s0890-6238(02)00006-0
203. Kato H, Ota T, Furuhashi T, Ohta Y, Iguchi T. Changes in reproductive organs of female rats treated with bisphenol A during the neonatal period. Reprod Toxicol (Elmsford N.Y.) (2003) 17:283–8. doi: 10.1016/s0890-6238(03)00002-9
204. Newbold RR, Jefferson WN, Padilla-Banks E. Long-term adverse effects of neonatal exposure to bisphenol A on the murine female reproductive tract. Reprod Toxicol (Elmsford N.Y.) (2007) 24:253–8. doi: 10.1016/j.reprotox.2007.07.006
205. Ijaz S, Ullah A, Shaheen G, Jahan S. Exposure of BPA and its alternatives like BPB, BPF, and BPS impair subsequent reproductive potentials in adult female Sprague Dawley rats. Toxicol Mech Methods (2020) 30:60–72. doi: 10.1080/15376516.2019.1652873
206. Veiga-Lopez A, Beckett EM, Abi Salloum B, Ye W, Padmanabhan V. Developmental programming: prenatal BPA treatment disrupts timing of LH surge and ovarian follicular wave dynamics in adult sheep. Toxicol Appl Pharmacol (2014) 279:119–28. doi: 10.1016/j.taap.2014.05.016
207. Liu H, Jin H, Kim G, Bae J. A low dose of bisphenol A stimulates estradiol production by regulating β-catenin-FOXL2-CYP19A1 pathway in human ovarian granulosa cells. Biochem Biophys Res Commun (2021) 583:192–8. doi: 10.1016/j.bbrc.2021.10.070
208. Veiga-Lopez A, Luense LJ, Christenson LK, Padmanabhan V. Developmental programming: gestational bisphenol-A treatment alters trajectory of fetal ovarian gene expression. Endocrinology (2013) 154:1873–84. doi: 10.1210/en.2012-2129
209. Panghiyangani R, Soeharso P, Andrijono DA, Wiweko B, Kurniati M, Pujianto DA. CYP19A1 gene expression in patients with polycystic ovarian syndrome. J Hum Reprod Sci (2020) 13:100–3. doi: 10.4103/jhrs.JHRS_142_18
210. Zhou W, Liu J, Liao L, Han S, Liu J. Effect of bisphenol A on steroid hormone production in rat ovarian theca-interstitial and granulosa cells. Mol Cell Endocrinol (2008) 283:12–8. doi: 10.1016/j.mce.2007.10.010
211. Gámez JM, Penalba R, Cardoso N, Bernasconi PS, Carbone S, Ponzo O, et al. Exposure to a low dose of bisphenol A impairs pituitary-ovarian axis in prepubertal rats: effects on early folliculogenesis. Environ Toxicol Pharmacol (2015) 39:9–15. doi: 10.1016/j.etap.2014.10.015
212. Kim S, Min HS, Lee WJ, Choe SA. Occupational differences in personal care product use and urinary concentration of endocrine disrupting chemicals by gender. J Exposure Sci Environ Epidemiol (2023) 33:312–8. doi: 10.1038/s41370-022-00436-7
213. Koch HM, Bolt HM, Preuss R, Angerer J. New metabolites of di(2-ethylhexyl)phthalate (DEHP) in human urine and serum after single oral doses of deuterium-labelled DEHP. Arch Toxicol (2005) 79:367–76. doi: 10.1007/s00204-004-0642-4
214. Swan SH, Sathyanarayana S, Barrett ES, Janssen S, Liu F, Nguyen RH, et al. First trimester phthalate exposure and anogenital distance in newborns. Hum Reprod (2015) 30:963–72. doi: 10.1093/humrep/deu363
215. Ungewitter E, Rotgers E, Bantukul T, Kawakami Y, Kissling GE, Yao HH-C. From the cover: teratogenic effects of in utero exposure to di-(2-ethylhexyl)-phthalate (DEHP) in B6:129S4 mice. Toxicol Sci (2017) 157:8–19. doi: 10.1093/toxsci/kfx019
216. Hlisníková H, Petrovičová I, Kolena B, Šidlovská M, Sirotkin A. Effects and mechanisms of phthalates' Action on reproductive processes and reproductive health: A literature review. Int J Environ Res Public Health (2020) 17:6811–48. doi: 10.3390/ijerph17186811
217. Yu Z, Han Y, Shen R, Huang K, Xu YY, Wang QN, et al. Gestational di-(2-ethylhexyl) phthalate exposure causes fetal intrauterine growth restriction through disturbing placental thyroid hormone receptor signaling. Toxicol Lett (2018) 294:1–10. doi: 10.1016/j.toxlet.2018.05.013
218. Shen R, Zhao LL, Yu Z, Zhang C, Chen YH, Wang H, et al. Maternal di-(2-ethylhexyl) phthalate exposure during pregnancy causes fetal growth restriction in a stage-specific but gender-independent manner. Reprod Toxicol (Elmsford N.Y.) (2017) 67:117–24. doi: 10.1016/j.reprotox.2016.12.003
219. Saadeldin IM, Hussein MA, Suleiman AH, Abohassan MG, Ahmed MM, Moustafa AA, et al. Ameliorative effect of ginseng extract on phthalate and bisphenol A reprotoxicity during pregnancy in rats. Environ Sci Pollut Res Int (2018) 25:21205–15. doi: 10.1007/s11356-018-2299-1
220. Xu W, Wu H, Shang L. Gene expression in rat placenta after exposure to di(2-ethylhexyl) phthalate. Hum Exp Toxicol (2021) 40:504–14. doi: 10.1177/0960327120954259
221. Zhao S, Hong Y, Liang YY, Li XL, Shen JC, Sun CC, et al. Compartmentalized regulation of NAD(+) by Di (2-ethyl-hexyl) phthalate induces DNA damage in placental trophoblast. Redox Biol (2022) 55:102414. doi: 10.1016/j.redox.2022.102414
222. Zhang S, Sun C, Zhao S, Wang B, Wang H, Zhang J, et al. Exposure to DEHP or its metabolite MEHP promotes progesterone secretion and inhibits proliferation in mouse placenta or JEG-3 cells. Environ pollut (Barking Essex (2020) 1987) 257:113593. doi: 10.1016/j.envpol.2019.113593
223. Rajagopal G, Bhaskaran RS, Karundevi B. Maternal di-(2-ethylhexyl) phthalate exposure alters hepatic insulin signal transduction and glucoregulatory events in rat F(1) male offspring. J Appl Toxicol JAT (2019) 39:751–63. doi: 10.1002/jat.3764
224. Rajesh P, Balasubramanian K. Gestational exposure to di(2-ethylhexyl) phthalate (DEHP) impairs pancreatic β-cell function in F1 rat offspring. Toxicol Lett (2015) 232:46–57. doi: 10.1016/j.toxlet.2014.09.025
225. Rajesh P, Balasubramanian K. Phthalate exposure in utero causes epigenetic changes and impairs insulin signalling. J Endocrinol (2014) 223:47–66. doi: 10.1530/JOE-14-0111
226. LaRocca J, Binder AM, McElrath TF, Michels KB. First-trimester urine concentrations of phthalate metabolites and phenols and placenta miRNA expression in a cohort of U.S. Women. Environ Health Perspect (2016) 124:380–7. doi: 10.1289/ehp.1408409
227. Grindler NM, Vanderlinden L, Karthikraj R, Kannan K, Teal S, Polotsky AJ, et al. Exposure to phthalate, an endocrine disrupting chemical, alters the first trimester placental methylome and transcriptome in women. Sci Rep (2018) 8:6086. doi: 10.1038/s41598-018-24505-w
228. Zhang J, Yao Y, Pan J, Guo X, Han X, Zhou J, et al. Maternal exposure to Di-(2-ethylhexyl) phthalate (DEHP) activates the PI3K/Akt/mTOR signaling pathway in F1 and F2 generation adult mouse testis. Exp Cell Res (2020) 394:112151. doi: 10.1016/j.yexcr.2020.112151
229. Stenz L, Escoffier J, Rahban R, Nef S, Paoloni-Giacobino A. Testicular dysgenesis syndrome and long-lasting epigenetic silencing of mouse sperm genes involved in the reproductive system after prenatal exposure to DEHP. PloS One (2017) 12:e0170441. doi: 10.1371/journal.pone.0170441
230. Dorostghoal M, Moazedi AA, Zardkaf A. Long-term effects of maternal exposure to Di (2-ethylhexyl) Phthalate on sperm and testicular parameters in Wistar rats offspring. Iranian J Reprod Med (2012) 10:7–14.
231. Wu W, Ma ZL, Yang F, Wu P, Zhang DX, Zeng R, et al. Urinary phthalate metabolites in pregnant women: occurrences, related factors, and association with maternal hormones. Environ Sci Pollut Res Int (2022) 29:33372–82. doi: 10.1007/s11356-022-18590-8
232. Polańska K, Ligocka D, Sobala W, Hanke W. Effect of environmental phthalate exposure on pregnancy duration and birth outcomes. Int J Occup Med Environ Health (2016) 29:683–97. doi: 10.13075/ijomeh.1896.00691
233. Suzuki Y, Niwa M, Yoshinaga J, Mizumoto Y, Serizawa S, Shiraishi H. Prenatal exposure to phthalate esters and PAHs and birth outcomes. Environ Int (2010) 36:699–704. doi: 10.1016/j.envint.2010.05.003
234. Hlisníková H, Kolena B, Šidlovská M, Mlynček M, Petrovičová I. Urinary phthalate biomarkers during pregnancy, and maternal endocrine parameters in association with anthropometric parameters of newborns. Children (Basel Switzerland) (2022) 9:413–39. doi: 10.3390/children9030413
235. Darvishmotevalli M, Moradnia M, Hosseini R, Bina B, Feizi A, Ebrahimpour K, et al. Association between prenatal phthalate exposure and anthropometric measures of newborns in a sample of Iranian population. Environ Sci Pollut Res Int (2021) 28:50696–706. doi: 10.1007/s11356-021-14182-0
236. Golestanzadeh M, Goodarzi-Khoigani M, Shahrbanoo Daniali S, Ebrahimpour K, Zarean E, Yazdi M, et al. Association between phthalate metabolites in human amniotic fluid and offspring birth size: a sub-study of the PERSIAN birth cohort. Environ Sci Pollut Res Int (2022) 29:76970–82. doi: 10.1007/s11356-022-20839-1
237. Zhang YW, Gao H, Mao LJ, Tao XY, Ge X, Huang K, et al. Effects of the phthalate exposure during three gestation periods on birth weight and their gender differences: A birth cohort study in China. Sci Total Environ (2018) 613-614:1573–8. doi: 10.1016/j.scitotenv.2017.08.319
238. Zhang YW, Gao H, Huang K, Xu YY, Sheng J, Tao FB. [A cohort study on association between the first trimester phthalates exposure and fasting blood glucose level in the third trimester]. Zhonghua liu xing bing xue za zhi = Zhonghua liuxingbingxue zazhi (2017) 38:388–92. doi: 10.3760/cma.j.issn.0254-6450.2017.03.023
239. Pacyga DC, Gardiner JC, Flaws JA, Li Z, Calafat AM, Korrick SA, et al. Maternal phthalate and phthalate alternative metabolites and urinary biomarkers of estrogens and testosterones across pregnancy. Environ Int (2021) 155:106676. doi: 10.1016/j.envint.2021.106676
240. Pacyga DC, Patti MA, Papandonatos GD, Haggerty DK, Calafat AM, Gardiner JC, et al. Associations of individual and cumulative urinary phthalate and replacement biomarkers with gestational weight gain through late pregnancy. Sci Total Environ (2023) 855:158788. doi: 10.1016/j.scitotenv.2022.158788
241. Cathey AL, Watkins D, Rosario ZY, Vélez C, Alshawabkeh AN, Cordero JF, et al. Associations of phthalates and phthalate replacements with CRH and other hormones among pregnant women in Puerto Rico. J Endocrine Soc (2019) 3:1127–49. doi: 10.1210/js.2019-00010
242. Sun X, Li J, Jin S, Li Y, Liu W, Zhao H, et al. Associations between repeated measures of maternal urinary phthalate metabolites during pregnancy and cord blood glucocorticoids. Environ Int (2018) 121:471–9. doi: 10.1016/j.envint.2018.09.037
243. Ma L, Mo J, Chen Y, Li L, Xie L, Chen X, et al. In utero cadmium and dibutyl phthalate combination exposure worsens the defects of fetal testis in rats. Environ pollut (Barking Essex (2020) 1987) 265:114842. doi: 10.1016/j.envpol.2020.114842
244. Borch J, Axelstad M, Vinggaard AM, Dalgaard M. Diisobutyl phthalate has comparable anti-androgenic effects to di-n-butyl phthalate in fetal rat testis. Toxicol Lett (2006) 163:183–90. doi: 10.1016/j.toxlet.2005.10.020
245. Chen H, Xin X, Liu M, Ma F, Yu Y, Huang J, et al. In utero exposure to dipentyl phthalate disrupts fetal and adult Leydig cell development. Toxicol Appl Pharmacol (2021) 419:115514. doi: 10.1016/j.taap.2021.115514
246. Li Q, Zhu Q, Tian F, Li J, Shi L, Yu Y, et al. In utero di-(2-ethylhexyl) phthalate-induced testicular dysgenesis syndrome in male newborn rats is rescued by taxifolin through reducing oxidative stress. Toxicol Appl Pharmacol (2022) 456:116262. doi: 10.1016/j.taap.2022.116262
247. Zhang S, Chen X, Li X, Wang Y, Zhu Q, Huang T, et al. Effects of in utero exposure to diisodecyl phthalate on fetal testicular cells in rats. Toxicol Lett (2020) 330:23–9. doi: 10.1016/j.toxlet.2020.04.024
248. Curi TZ, Neubert da Silva G, Passoni MT, Lima Tolouei SE, Meldola H, Romano RM, et al. In utero and lactational exposure to diisopentyl phthalate induces fetal toxicity and antiandrogenic effects in rats. Toxicological Sci (2019) 171:347–58. doi: 10.1093/toxsci/kfz159
249. Earl Gray L Jr. Biologically relevant reductions in fetal testosterone and Insl3 induced by in utero exposure to high levels of di-isononyl phthalate (DINP) in male rats. Toxicol Appl Pharmacol (2023) 465:116454. doi: 10.1016/j.taap.2023.116454
250. Liu M, Chen H, Dai H, Zhou L, Wang Y, Xin X, et al. Effects of bis(2-butoxyethyl) phthalate exposure in utero on the development of fetal Leydig cells in rats. Toxicol Lett (2021) 351:65–77. doi: 10.1016/j.toxlet.2021.08.008
251. Howdeshell KL, Furr J, Lambright CR, Rider CV, Wilson VS, Gray LE Jr. Cumulative effects of dibutyl phthalate and diethylhexyl phthalate on male rat reproductive tract development: altered fetal steroid hormones and genes. Toxicological Sci (2007) 99:190–202. doi: 10.1093/toxsci/kfm069
252. Howdeshell KL, Wilson VS, Furr J, Lambright CR, Rider CV, Blystone CR, et al. A mixture of five phthalate esters inhibits fetal testicular testosterone production in the sprague-dawley rat in a cumulative, dose-additive manner. Toxicological Sci (2008) 105:153–65. doi: 10.1093/toxsci/kfn077
253. Dostalova P, Zatecka E, Ded L, Elzeinova F, Valaskova E, Kubatova A, et al. Gestational and pubertal exposure to low dose of di-(2-ethylhexyl) phthalate impairs sperm quality in adult mice. Reprod Toxicol (Elmsford N.Y.) (2020) 96:175–84. doi: 10.1016/j.reprotox.2020.06.014
254. Zhou Y, Ma T, Yan M, Meng X, Wu J, Ding J, et al. Exposure of DBP in gestation induces inflammation of testicular Sertoli cells in progeny by activating NLRP3 inflammasomes. Sci Total Environ (2020) 707:136139. doi: 10.1016/j.scitotenv.2019.136139
255. Xu X, Yekeen TA, Xiao Q, Wang Y, Lu F, Huo X. Placental IGF-1 and IGFBP-3 expression correlate with umbilical cord blood PAH and PBDE levels from prenatal exposure to electronic waste. Environ Pollut (Barking Essex (2013) 1987) 182:63–9. doi: 10.1016/j.envpol.2013.07.005
256. Niculescu MD, Lupu DS, Craciunescu CN. Perinatal manipulation of α-linolenic acid intake induces epigenetic changes in maternal and offspring livers. FASEB J (2013) 27:350–8. doi: 10.1096/fj.12-210724
257. Srinivas V, Molangiri A, Mallepogu A, Kona SR, Ibrahim A, Duttaroy AK, et al. Maternal n-3 PUFA deficiency alters uterine artery remodeling and placental epigenome in the mice. J Nutr Biochem (2021) 96:108784–96. doi: 10.1016/j.jnutbio.2021.108784
258. Zuccarello D, Sorrentino U, Brasson V, Marin L, Piccolo C, Capalbo A, et al. Epigenetics of pregnancy: looking beyond the DNA code. J Assist Reprod Genet (2022) 39:801–16. doi: 10.1007/s10815-022-02451-x
259. Leader JE, Wang C, Popov VM, Fu M, Pestell RG. Epigenetics and the estrogen receptor. Ann N Y Acad Sci (2006) 1089:73–87. doi: 10.1196/annals.1386.047
260. Carling T, Kim KC, Yang XH, Gu J, Zhang XK, Huang S. A histone methyltransferase is required for maximal response to female sex hormones. Mol Cell Biol (2004) 24:7032–42. doi: 10.1128/MCB.24.16.7032-7042.2004
261. Susiarjo M, Sasson I, Mesaros C, Bartolomei MS. Bisphenol a exposure disrupts genomic imprinting in the mouse. PloS Genet (2013) 9:e1003401. doi: 10.1371/journal.pgen.1003401
262. Sharma R, Frasch MG, Zelgert C, Zimmermann P, Fabre B, Wilson R, et al. Maternal–fetal stress and DNA methylation signatures in neonatal saliva: an epigenome-wide association study. Clin Epigenet (2022) 14:87. doi: 10.1186/s13148-022-01310-x
263. Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci USA (2007) 104:13056–61. doi: 10.1073/pnas.0703739104
264. Song X, Wang Z, Zhang Z, Miao M, Liu J, Luan M, et al. Differential methylation of genes in the human placenta associated with bisphenol A exposure. Environ Res (2021) 200:111389. doi: 10.1016/j.envres.2021.111389
265. Song X, Zhou X, Yang F, Liang H, Wang Z, Li R, et al. Association between prenatal bisphenol a exposure and promoter hypermethylation of CAPS2, TNFRSF25, and HKR1 genes in cord blood. Environ Res (2020) 190:109996. doi: 10.1016/j.envres.2020.109996
266. Ma Y, Xia W, Wang DQ, Wan YJ, Xu B, Chen X, et al. Hepatic DNA methylation modifications in early development of rats resulting from perinatal BPA exposure contribute to insulin resistance in adulthood. Diabetologia (2013) 56:2059–67. doi: 10.1007/s00125-013-2944-7
267. Strakovsky RS, Wang H, Engeseth NJ, Flaws JA, Helferich WG, Pan YX, et al. (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol Appl Pharmacol (2015) 284:101–12. doi: 10.1016/j.taap.2015.02.021
268. Onuzulu CD, Rotimi OA, Rotimi SO. Epigenetic modifications associated with in utero exposure to endocrine disrupting chemicals BPA, DDT and Pb. Rev Environ Health (2019) 34:309–25. doi: 10.1515/reveh-2018-0059
269. Junge KM, Leppert B, Jahreis S, Wissenbach DK, Feltens R, Grutzmann K, et al. MEST mediates the impact of prenatal bisphenol A exposure on long-term body weight development. Clin Epigenet (2018) 10:58. doi: 10.1186/s13148-018-0478-z
270. Faulk C, Kim JH, Anderson OS, Nahar MS, Jones TR, Sartor MA, et al. Detection of differential DNA methylation in repetitive DNA of mice and humans perinatally exposed to bisphenol A. Epigenetics (2016) 11:489–500. doi: 10.1080/15592294.2016.1183856
271. Li G, Chang H, Xia W, Mao Z, Li Y, Xu S. F0 maternal BPA exposure induced glucose intolerance of F2 generation through DNA methylation change in Gck. Toxicol Lett (2014) 228:192–9. doi: 10.1016/j.toxlet.2014.04.012
272. Ye Y, Tang Y, Xiong Y, Feng L, Li X. Bisphenol A exposure alters placentation and causes preeclampsia-like features in pregnant mice involved in reprogramming of DNA methylation of WNT2. FASEB J (2018) 2:2732–42. doi: 10.1096/fj.201800934RRR
273. Zhong J, Baccarelli AA, Mansur A, Adir M, Nahum R, Hauser R, et al. Maternal phthalate and personal care products exposure alters extracellular placental miRNA profile in twin pregnancies. Reprod Sci (2019) 26:289–94. doi: 10.1177/1933719118770550
274. Meruvu S, Zhang J, Choudhury M. Mono-(2-ethylhexyl) phthalate increases oxidative stress responsive miRNAs in first trimester placental cell line HTR8/SVneo. Chem Res Toxicol (2016) 29:430–5. doi: 10.1021/acs.chemrestox.6b00038
275. Jedynak P, Tost J, Calafat AM, Bourova-Flin E, Broséus L, Busato F, et al. Pregnancy exposure to phthalates and DNA methylation in male placenta - An epigenome-wide association study. Environ Int (2022) 160:107054. doi: 10.1016/j.envint.2021.107054
276. Zhao Y, Shi HJ, Xie CM, Chen J, Laue H, Zhang YH. Prenatal phthalate exposure, infant growth, and global DNA methylation of human placenta. Environ Mol Mutagenesis (2015) 56:286–92. doi: 10.1002/em.21916
277. Zhao Y, Chen J, Wang X, Song Q, Xu HH, Zhang YH. Third trimester phthalate exposure is associated with DNA methylation of growth-related genes in human placenta. Sci Rep (2016) 6:33449. doi: 10.1038/srep33449
278. LaRocca J, Binder AM, McElrath TF, Michels KB. The impact of first trimester phthalate and phenol exposure on IGF2/H19 genomic imprinting and birth outcomes. Environ Res (2014) 133:396–406. doi: 10.1016/j.envres.2014.04.032
279. Gu X, Liu H, Luo W, Wang X, Wang H, Li L. Di-2-ethylhexyl phthalate-induced miR-155-5p promoted lipid metabolism via inhibiting cAMP/PKA signaling pathway in human trophoblastic HTR-8/Svneo cells. Reprod Toxicol (Elmsford N.Y.) (2022) 114:22–31. doi: 10.1016/j.reprotox.2022.10.001
Keywords: in utero exposure, endocrine-disrupting chemicals, fetoplacental growth, bisphenol, glucocorticoids, leptin, bps
Citation: Basak S, Varma S and Duttaroy AK (2023) Modulation of fetoplacental growth, development and reproductive function by endocrine disrupters. Front. Endocrinol. 14:1215353. doi: 10.3389/fendo.2023.1215353
Received: 01 May 2023; Accepted: 14 September 2023;
Published: 03 October 2023.
Edited by:
Jayonta Bhattacharjee, Bangladesh Agricultural University, BangladeshReviewed by:
Maria De Falco, University of Naples Federico II, ItalyHuan Shen, Peking University, China
Copyright © 2023 Basak, Varma and Duttaroy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Asim K. Duttaroy, YS5rLmR1dHRhcm95QG1lZGlzaW4udWlvLm5v