
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 10 August 2023
Sec. Pediatric Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1209577
This article is part of the Research Topic Thyroid Nodules and Tumors in Childhood View all 10 articles
 Walter Maria Sarli1,2
Walter Maria Sarli1,2 Silvia Ricci1,2
Silvia Ricci1,2 Lorenzo Lodi1,2Federica Cavone3
Lorenzo Lodi1,2Federica Cavone3 Lucia Pacillo4,5,6Carmela Giancotta4Graziamaria Ubertini7
Lucia Pacillo4,5,6Carmela Giancotta4Graziamaria Ubertini7 Giampiero Baroncelli3Caterina Cancrini4,5,6Chiara Azzari1,2
Giampiero Baroncelli3Caterina Cancrini4,5,6Chiara Azzari1,2 Stefano Stagi1,8*
Stefano Stagi1,8*Introduction: The chromosome 22q11.2 deletion syndrome comprises phenotypically similar diseases characterized by abnormal development of the third and fourth branchial arches, resulting in variable combinations of congenital heart defects, dysmorphisms, hypocalcemia, palatal dysfunction, developmental or neuropsychiatric disorders, and impairment of the immune system due to thymic dysfunction. Other genetic syndromes, often called DiGeorge-like, share clinical and immunological features with 22q11.2 deletion syndrome. This syndrome has been rarely associated with malignancies, mainly hematological but also hepatic, renal, and cerebral. Rarely, malignancies in the head and neck region have been described, although no aggregate of data on the development of thyroid neoplasms in patients with this clinical phenotype has been conducted so far.
Materials and methods: To characterize this possible association, a multicenter survey was made. Thus, we present a case series of five pediatric patients with 22q11.2 deletion syndrome or DiGeorge-like syndrome who were occasionally found with confirmed or highly suspected neoplasms of the thyroid gland during their follow-up. In three cases, malignancies were histologically confirmed, but their outcome was good due to an early recognition of suspicious nodules and precocious surgery.
Conclusions: This study underlines for clinicians the higher risk of neoplasms in the head and neck district for patients affected by these syndromes. It also emphasizes the importance of a prolonged clinical and ultrasound follow-up for patients with this clinical and immunological phenotype.
The chromosome 22q11.2 deletion syndrome (22q11.2DS) is caused by a hemizygous deletion that is de novo in more than 90% of cases (1). In addition, 22q11.2DS encompasses several phenotypically similar diseases that share abnormal development of the third and fourth branchial arches, resulting in variable combinations of congenital heart defects, dysmorphisms, hypocalcemia, palatal dysfunction, developmental delay, neuropsychiatric disorders, and impaired immune function due to thymic hypoplasia or aplasia (2).
The most famous of these diseases is DiGeorge syndrome (DGS) that is usually caused by the loss of the TBX1 gene (MIM_188400), located in the 22q11 region, which is an important transcription factor necessary for the development of the thyroid, parathyroids, palate, teeth, thymus, and heart (2). Sometimes, DGS is not caused by 22q11.2 deletion but TBX1 pathogenic mutations (3). Nevertheless, there are many other genetic syndromes, often called DiGeorge-like, with clinical features like DGS but other chromosomal deletions, such as 10p13 (MIM_266500), 17p13.1 (MIM_613776), 16p11.2 (MIM_611913), and 4q34.1q35.2 (4–7), or others within the context of CHARGE (MIM_214800) or Opitz G/BBB syndrome (MIM_300000) (8, 9). However, in 6%–17% of patients, a genetic cause remains unknown (10).
Thyroid anomalies are frequently discovered in 22q11.2DS. Approximately half of the patients have structural anomalies such as thyroid hypoplasia, absent isthmus, and abnormal extension probably due to TBX1 haploinsufficiency (11–13). Thyroid dysfunction can also be caused by autoimmunity. It is estimated that over 8% of patients with 22q11.2DS will develop autoimmunity with age (14). Recently, we described 73 children with 22q11.2DS, followed up for 9.51 ± 5.72 years. Totally, 21.9% developed autoimmune thyroid disease (ATD) before the age of 18, including 20.5% Hashimoto’s thyroiditis (HT) and 1.4% Graves’ disease (GD) (15). Among 22q11.2DS patients who developed ATD, one case of thyroid cancer in one adolescent developing GD was described, strongly recommending periodic screening with ultrasound scan in these patients (15).
It is known that many congenitally inherited syndromes are associated with a higher risk of malignancy (16, 17) with the development of malignancies, mainly hematological but also hepatic, renal, and cerebral (8, 18). Malignant tumors in the head and neck region, as well as in other districts, have rarely been described (19–21). The risk of malignancy in 22q11.2DS is moderately increased when compared to the healthy general pediatric population with a reported frequency of 1% in the 22q11.2DS population (20). A recent Finnish nationwide register-based cohort study by Wahrmann et al. (22) described a 2.1% rate of malignancy in 98 patients with 22q11.2DS.
Very few cases of patients with 22q11.2DS and thyroid neoplasms, benign or malign, have been described so far (8, 14, 19). On the other hand, to the best of our knowledge, no cases of thyroid neoplasms have been reported in patients with DiGeorge-like syndromes.
To characterize this possible association, a multicenter survey was made. Thus, we present a case series of five pediatric patients with 22q11.2DS or DGS-like who were occasionally found with confirmed or highly suspected neoplasms of the thyroid gland during their follow-up.
We performed a retrospective multicenter study of patients with 22q11.2DS or DGS-like who presented with thyroid or parathyroid neoplasms before the age of 18 between 4 January 1985 and 6 July 2022. The study recruited patients with a confirmed genetic diagnosis of 22q11.2DS or with a DGS-like, based on a highly evocative clinical and laboratory phenotype, from the Paediatric Immunology Division and Auxo-endocrinology Division of Meyer Children’s Hospital in Florence (University of Florence), Santa Chiara’s Hospital in Pisa (University of Pisa), and Bambino Gesù Children Hospital in Rome (University of Roma Tor Vergata), Italy. The Tobias and the European Society of Immunodeficiencies (ESID) criteria were initially used to evaluate patients’ susceptibility to genetic analysis for 22q11.2DS (23, 24). All patients were monitored annually or biannually up to the age of 18 through clinical assessments and thyroid ultrasounds and blood tests to measure thyroid function and autoantibodies. A total of three patients with 22q11.2DS and confirmed thyroid cancers were extensively described below.
The study was conducted according to the Declaration of Helsinki II. Written informed consent from the patients’ parents or legal guardians was acquired before data collection. Patient data were retrospectively retrieved from the clinical records and anonymously collected in an Excel® spreadsheet. A specific approval by the local ethical committee was not required because all analyses included in this study were performed as part of routine clinical activity according to Good Clinical Practice.
Clinical, ultrasound, and histological data were collected from the clinical records of all patients during the follow-up. Ultrasound scans were performed with a linear multifrequency transducer. An extended immunologic phenotype was performed. All values were evaluated by standard methods and compared with age-matched normal values.
Between 4 January 1985 and 6 July 2022, a total of 275 patients with 22q11.2DS (260 patients) and DGS-like (15 patients) were consecutively observed in the three centers for multidisciplinal follow-up and periodic thyroid ultrasound scans. During follow-up, thyroid malignancies were found by ultrasound scans in three patients with 22q11.2DS (two boys and one girl) with a median age of 10.30 years ± 1.48 years at cancer diagnosis. None of them was previously exposed to radiation (Figure 1). None of them had a family history of thyroid or parathyroid carcinoma or adenoma. Two patients had autoimmune hypothyroidism and hyperthyroidism, respectively, at the time the nodules were discovered. The mean maximum diameter of confirmed malignant nodules was 23.16 ± 15.18 mm. According to the classification of the American Thyroid Association (ATA), all three nodules showed ultrasound characteristics of a high risk of malignancy (25). The clinical, ultrasound, and immunophenotypic characteristics of the patients are described in Figure 2 and Tables 1, 2.

Figure 1 Graphical representation of the main clinical features of the patients. *Confirmed thyroid cancers.
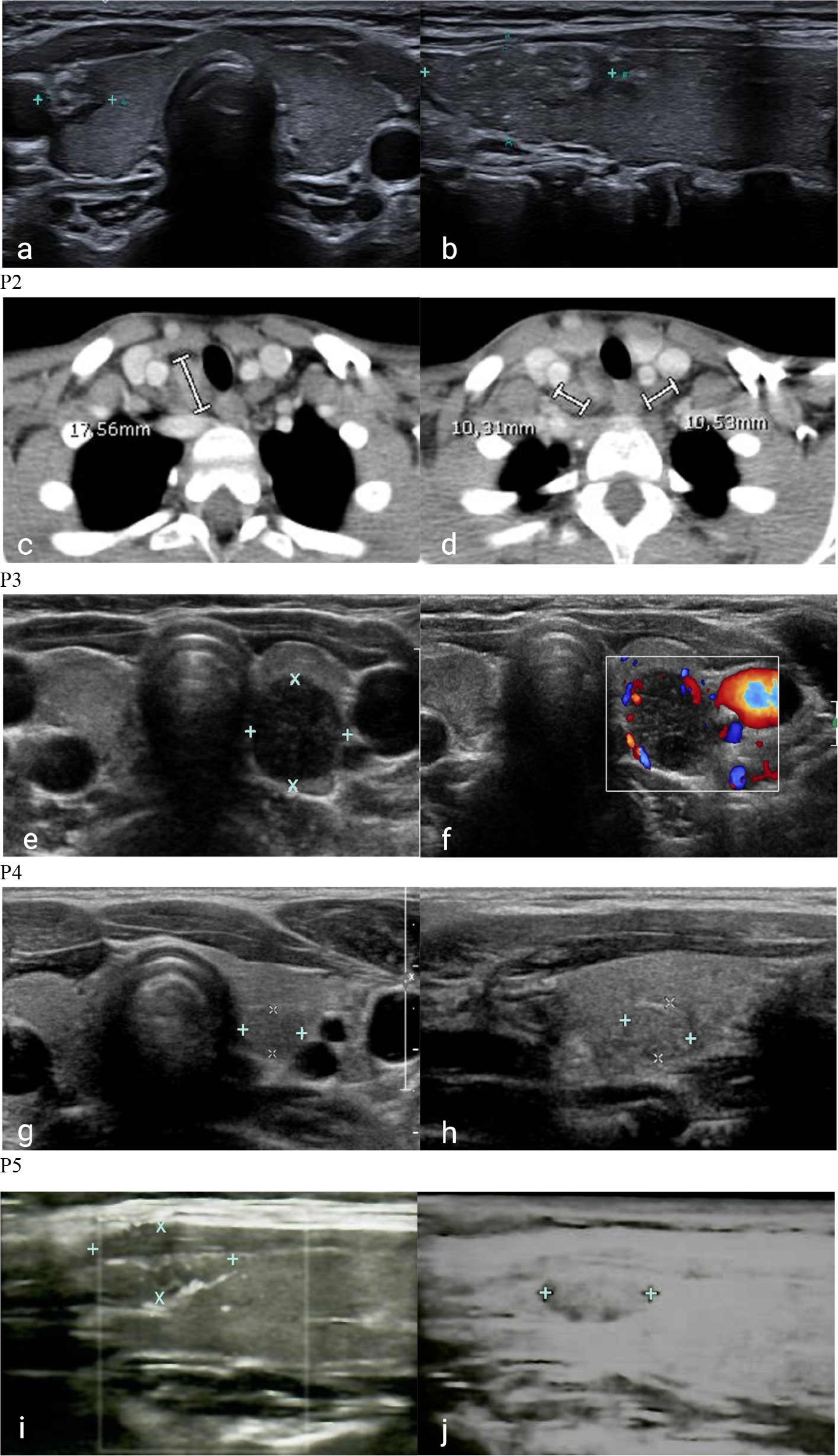
Figure 2 Cervical ultrasonography and computerized tomography of patients with 22q11DS and DiGeorge-like syndrome. P1. Inhomogeneous hypoechoic nodule in the right lower lobe in coronal (A) and sagittal (B) ultrasound projections. P2. Metastatic involvement of right lateral cervical lymph nodes (C) and normal lymph nodes (D) in coronal CT. P3. Inhomogeneous nodule with hypoechogenic areas (E) and peripheral vascular overflow (F) in the left lobe in coronal ultrasound projections. P4. Roundish hypoechoic nodule in the posterior region of the left lobe in close proximity to the carotid artery in coronal (G) and sagittal (H) ultrasound projections. P5. Iso-hypoechoic nodule in the upper third of the right lobe with blurred margins, and intra- and perilesional vascularity in sagittal ultrasound projections in sagittal ultrasound projection: first identification (I) and 1-year follow-up (J).
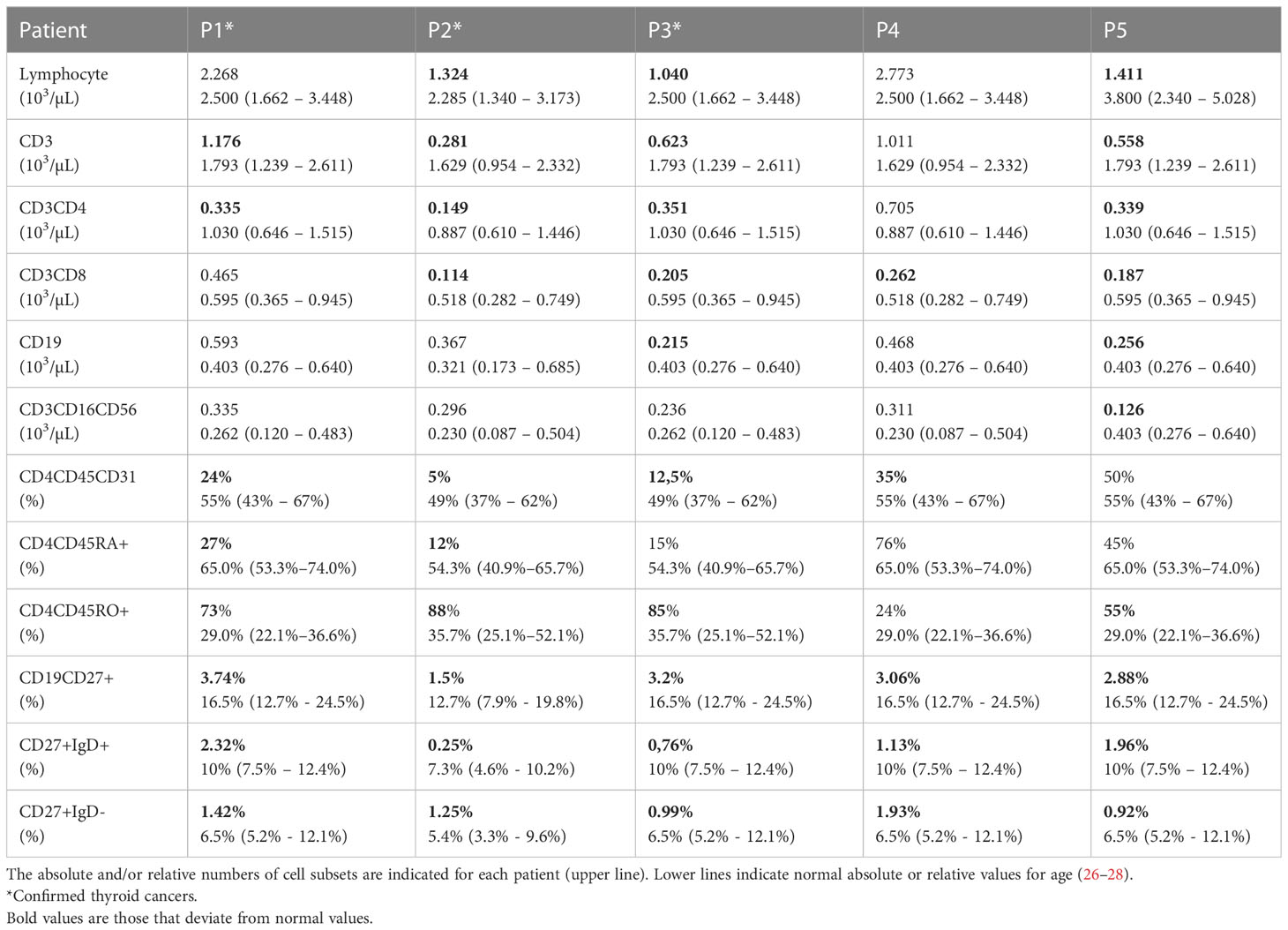
Table 1 Lymphocyte subsets of the patients.
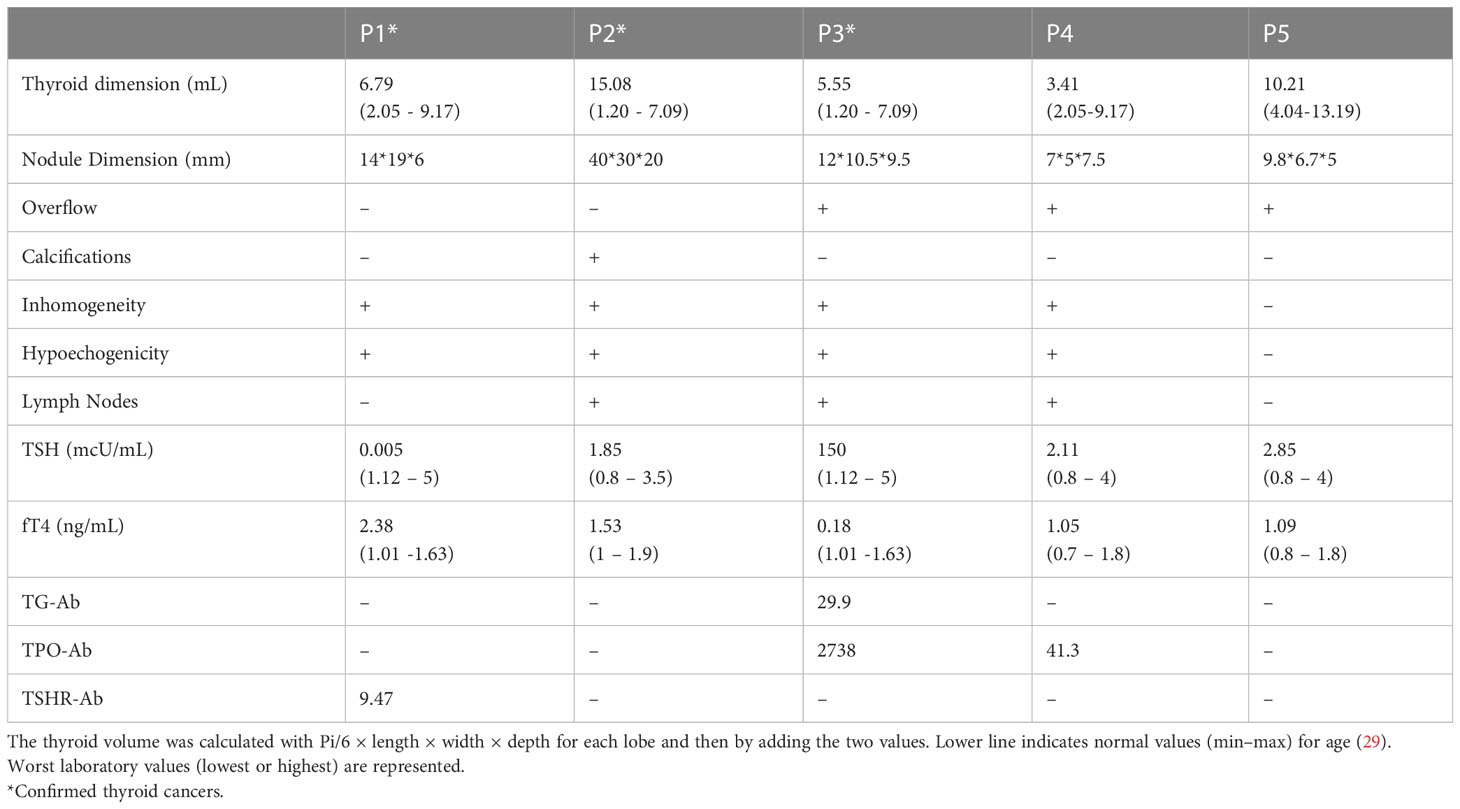
Table 2 Ultrasound features and laboratory details of patients with 22q11.2DS and DiGeorge-like syndrome and thyroid neoplasms.
P1 is a 12-year-old boy with 22q11.2DS diagnosed through comparative genomic hybridization array (CGH-array) and fluorescence in situ hybridization (FISH) at age 9 for a history of speech delay, palatal insufficiency, and submucosal cleft palate. Initial cardiologic evaluation identified pervious foramen ovale (PFO) and aortic root ectasia. During follow-up, his growth was good, and he did not complain of relevant infections. At age 11, routine blood tests for thyroid function and autoimmunity showed decreased thyroid-stimulating hormone (TSH) levels and significantly elevated TSH receptor (TSHR) antibodies. Ultrasound scans revealed diffuse thyroid gland overflow, leading to a diagnosis of Graves’ disease (GD). Treatment with Tapazole was initiated, resulting in good disease control without side effects. However, subsequent ultrasound scans performed after 6 and 9 months revealed diffuse inhomogeneity and a hypoechogenic nodule in the right lower lobe (14 × 19 × 6 mm), indicative of a potential thyroid abnormality. Fine-needle aspirate biopsy (FNAB) was conducted, identifying TIR3b cytology, and total thyroidectomy was performed. Histologic examination confirmed the presence of follicular carcinoma, which was classified as T1N0M0 according to TNM staging. Due to the early diagnosis, the patient did not require adjuvant radiometabolic therapy and is currently disease-free with substitutive therapy.
P2, a 24-year-old man, underwent surgery for intestinal occlusion due to malrotation at 6 days of age. The diagnosis of 22q11.2DS was prompted by dysmorphic features and an interventricular defect and confirmed at 1 month of age through FISH. During the follow-up, multiple thyroid ultrasound scans were performed, all yielding negative results. At the age of 9, a routine ultrasound scan revealed a solid nodular formation completely occupying the right thyroid lobe (40 × 30 × 20 mm). The nodule exhibited hyperechogenic spots in the peripheral zone and inhomogeneous hypoechogenicity in the central zone. Additionally, a right cervical lymphadenopathy was identified, with hyperechogenic spots. FNAB identified a TIR4 phenotype; thus, total thyroidectomy with cervical right lymphadenectomy was performed. Histologic examination revealed papillary thyroid carcinoma infiltrating the peri-thyroid lax tissues, which was classified as T3N1bMx due to metastatic involvement of four right recurrent lymph nodes (NR1, NR3, NR4) and three right lateral cervical lymph nodes (N1, N2). After 5 months, recurrence of disease was observed at the right lateral cervical site (right mandibular angle), necessitating additional surgery and radiometabolic therapy. P2 is currently disease-free and continues to receive care from an adult immunology center.
P3 is an 18-year-old woman with 22q11.2DS diagnosed through FISH at the age of 12 months for dysmorphic features and neurodevelopmental delay. Recurrent respiratory infections were observed since early childhood. At age 3, she was diagnosed with ATD based on increased TSH levels and positive autoantibodies. Substitution therapy with levothyroxine was initiated. Of note, familiar history was positive for autoimmune diseases (aunt with ATD and grandmother with Sjögren’s syndrome). During the follow-up, multiple thyroid ultrasound scans were performed. At the age of 10, a routine ultrasound scan revealed an inhomogeneous nodule (12 × 10.5 × 9.5 mm), with hypoechogenic areas, peripheral vascular overflow, and a suspected lymph node (long axis 10.5 mm). The first FNAB identified a TIR1 cytology, but 5 months later, the second FNAB identified a TIR3B phenotype. Consequently, hemithyroidectomy was performed. Histologic examination identified a papillary thyroid carcinoma Warthin-like variant (pTIb). Histology also showed lymphocyte infiltration as for Hashimoto thyroiditis, while no signs of metastases were found in lymph nodes. She subsequently underwent total thyroidectomy and then radiometabolic therapy with 131-I for ablation of residual thyroid tissue. Later, she remained stably disease-free with substitutive therapy.
Of note, it is worth mentioning that two additional patients with suspected thyroid malignant nodular lesions were identified during their follow-up. The first patient is a boy with 22q11.2DS who is under close surveillance with serial ultrasounds due to the presence of a slightly growing suspicious thyroid nodule that cannot be biopsied due to its proximity to the carotid artery (P4) while awaiting a more radical intervention in case of worsening ultrasound findings. The other patient is a girl with DGS-like who underwent FNAB and right lobectomy for a suspected malignant nodular lesion, which was subsequently identified as Hurtle cell adenoma (P5). Their clinical, ultrasound, and immunophenotypic characteristics are described for comparison and completeness alongside those of patients P1, P2, and P3 in Figure 2 and Tables 1, 2.
Our results seem to suggest that patients with 22q11.2DS and probably DGS-like can develop a broad range of thyroid neoplasms with a higher degree of malignancy. It is known to date that the prevalence of pediatric thyroid diseases of surgical interest, particularly thyroid cancer, is a rare entity and significantly lower than that of adults (30). Epidemiological surveillance data described 1.2 cases per 100,000 patients under the age of 20 but 0.4 cases per 100,000 in patients aged under 15 (31). However, the incidence of pediatric thyroid nodules and cancer appears to be steadily increasing and associated with a worse prognosis (32), far higher than the expected value in the adult population (5%) (33). In clinical practice, the management of pediatric thyroid nodules largely follows the adult thyroid guidelines (25), with some peculiarities related to pediatric age.
Although limited, our data appear to indicate that 22q11.2DS patients have a significantly higher prevalence of thyroid cancers, confirming the suggestions of the literature (34, 35) and strongly recommending periodic screening also with thyroid ultrasound scan in these patients. In fact, considering only malignant thyroid neoplasms confirmed by histological investigation through a long follow-up and periodic routine thyroid ultrasound scans, a total of three cases of malignant thyroid nodules (P1, P2, and P3) were identified out of a total of 260 patients with 22q11.2DS (1.15%).
Although not specifically intended for drawing conclusion on the thyroid cancer rate, we chose to mention in this case series two patients with 22q11.2DS and DGS-like phenotype who exhibited suspicious thyroid nodules during their follow-up to raise awareness among physicians about the potential risk associated with these conditions.
Indeed, the incidence of thyroid malignancy in patients with 22q11.2DS may be underestimated. This is exemplified by the case of P4, who presents a thyroid nodule with suspicious features of malignancy but cannot be biopsied. Patients with DGS-like phenotype may also have an increased risk of developing thyroid neoplasms, but due to the lack of specific literature on neoplasms in DGS-like patients, this association may have been overlooked. It is important to note that many DGS-like patients remain undiagnosed and thus do not receive comprehensive immunologic and endocrinologic follow-up. However, given their shared clinical and immunophenotypic features with 22q11.2DS patients, some centers adopt a similar follow-up approach for both groups. Our findings highlight the importance of regular ultrasound monitoring in DGS-like patients, as exemplified by the detection of a suspicious nodule in P5, although not confirmed as malignant.
To date, less is known about why patients with clinical phenotypes including 22q11.2DS, and probably DGS-like too, may have an increased risk of malignancies. One of the reasons could lay on the T-cell defects that are common in 22q11.2DS and in DGS-like too (10, 36, 37). As widely described for HIV infection, T lymphopenia or reduced natural killer cells, as well as their dysfunction, may predispose to poor surveillance against neoplastic cells. T-cell dysfunction can also lead to B-cell hyperproliferation and overactivation resulting in a higher risk of developing lymphomas (38, 39). In fact, cases of B lymphoma in patients with 22q11.2DS and severe T-cell impairment have been reported in the literature in the past (40, 41). It is unclear whether the few thyroid cancers described to date in patients with 22q11.2DS were associated with T lymphopenia (34, 42). Generally, there is limited understanding regarding the relationship between the development of thyroid adenomas or carcinomas and lymphopenia. A recent work by Rabold et al. (43) found that T-cell lymphopenia in patients with thyroid cancer indicates an aggressive tumor behavior and might badly influence the outcome of the disease. Lymphopenia could play a role also for persistent infections by oncogenic viruses. As proof for this, there is increasing evidence in the literature on the association between viral infections and thyroid cancer development. Recent work by Moghoofei et al. (44) described the detection of Epstein Barr virus (EBV) DNA in 71.9% of 57 thyroid tumor specimens and thus demonstrated that persistent EBV infection could be associated with an increased risk of thyroid cancer. This evidence was confirmed by a systematic literature review conducted in 2020 by Mostafaei et al. (45) who demonstrated the potential pathogenetic association with several viral infections, not only EBV.
All patients (P1, P2, and P3) with 22q11.2DS who developed thyroid cancer had T lymphopenia, especially in the CD3CD4+ and CD3CD8+ compartments at immunophenotype analysis. Variable degrees of T lymphopenia were also found in P4 and P5 as described in Table 1.
Probably, this may have played a role in cancer development, but we cannot assume that T lymphopenia alone was their only risk factor because there are numerous inborn errors of immunity with profound T-cell deficiency in which an increased risk of thyroid neoplasms is not reported in the literature.
Thus, likely in addition to lymphopenia, there could be other reasons related to the syndrome that we do not fully understand to date but that could contribute to the risk of developing thyroid cancers.
Since allelic losses of chromosome 22q arm were described by molecular analysis of well-differentiated thyroid cancer specimens (46), authors have tried to emphasize the role of this chromosomal region loss in thyroid cancer predisposition. For instance, atypical deletions could involve the tumor suppressor gene SMARCB1 (MIM_609322), which is associated with rhabdoid tumors, or DGCR8 gene (MIM_609030), which has been found to result in aberrant levels of miRNA leading to an increased susceptibility to malignancies (47, 48). However, if the reason was related only to this, many more cases of thyroid tumors among patients with 22q11.2DS would have been described so far.
Moreover, this pathogenesis cannot explain why patients with DGS-like, who do not have 22q deletion, may eventually share an increased risk.
Since, in 22q11.2DS, the development of the third and fourth branchial arches from which the thyroid gland derives is impaired (2), an embryological role in predisposition to thyroid cancer cannot be ruled out. Recently indeed, a model of fetal carcinogenesis of thyroid tumors has been proposed (49). According to this model, within a differentiated thyroid gland, there would exist three different types of fetal thyroid cells that could give rise to thyroid cancer cells. Finally, it is necessary to consider thyroid autoimmunity. It is well known that patients with 22q11.2DS have an increased risk of autoimmunity, both thyroid and non-thyroid (2). According to Shugar et al. (50), there is a higher rate of hypothyroidism and hyperthyroidism in children with 22q11.2DS than in the general population. However, as demonstrated in our previous work, through a long ultrasound and endocrinological follow-up, thyroid autoimmunity could be detected in up to 21.9% of patients with 22q11.2DS (15). Moreover, sometimes also ultrasound changes could precede the rise of thyroid autoantibodies or hormone impairment.
A recent study by Montin et al. (51) in patients with 22q11.2DS showed that certain immunologic features, including reduced numbers of recent thymic emigrants (RTEs), reduced naive T cells, and reduced B memory switched, are associated with the development of hematologic autoimmunity. This was also demonstrated in a study by Ricci et al. (52). Much evidence suggests that inflammation induced by thyroid autoimmunity may promote tumor development, although the precise mechanism has not been identified. Nevertheless, the presence of a proinflammatory environment, variable expression of transcriptional regulators, and increased TSH values together could promote tumor development or growth (53).
Two patients (P1 and P3) developed thyroid autoimmunity before tumor, although with different temporal latencies. Both had predisposing conditions for autoimmunity as explained above. In fact, both had reduced numbers of circulating naive T cells, RTEs, and switched B memory. However, these alterations were also present in P2 who never presented thyroid autoimmunity.
Although, in the case of P3, the temporal latency from the onset of autoimmunity suggests that autoimmunity may have contributed to cancer development, we cannot assume the same for P1. In fact, in this case, it is not possible to determine with certainty, given the short temporal latency, whether it was the autoimmunity that induced the tumor or the tumor at an early stage that fostered the autoimmunity.
Beyond the various hypotheses presented and depicted in the summary in Figure 3, with this work, we draw attention to the fact that prolonged follow-up protocols that include annual or biannual thyroid ultrasound scans not only allow recognition of autoimmune thyroid pathologies before autoantibodies or clinical symptoms become positive but also allow an earlier identification of thyroid nodules, particularly in adolescents with 22q11DS. In fact, all of our cases, both confirmed malignant and suspected, occurred in the age range of 9–15 years. We therefore suggest increasing clinicians’ attention and thyroid ultrasound follow-up in patients within this age group. Early ultrasonographic detection of a thyroid nodule is indeed important because it provides early identification of suspicious features of malignancy, early referral of patients to FNAB, and eventually surgery. Moreover, in case of malignant lesions, it offers the possibility of lowering the risk of metastasis, the need for radiometabolic therapy, and unfavorable outcomes. In addition, it also enables a detailed tracking of any morphological changes in suspicious nodules that cannot undergo early surgery because of a difficult location, as demonstrated for P4. Among the described patients, three patients developed malignant thyroid nodules, and only one of them had a recurrence of disease that required new surgery for lymphadenectomy. In any case, none of these patients presented distant metastases. All of them, reflecting the importance of an early diagnosis, had an excellent prognosis and all of them are steadily disease-free.
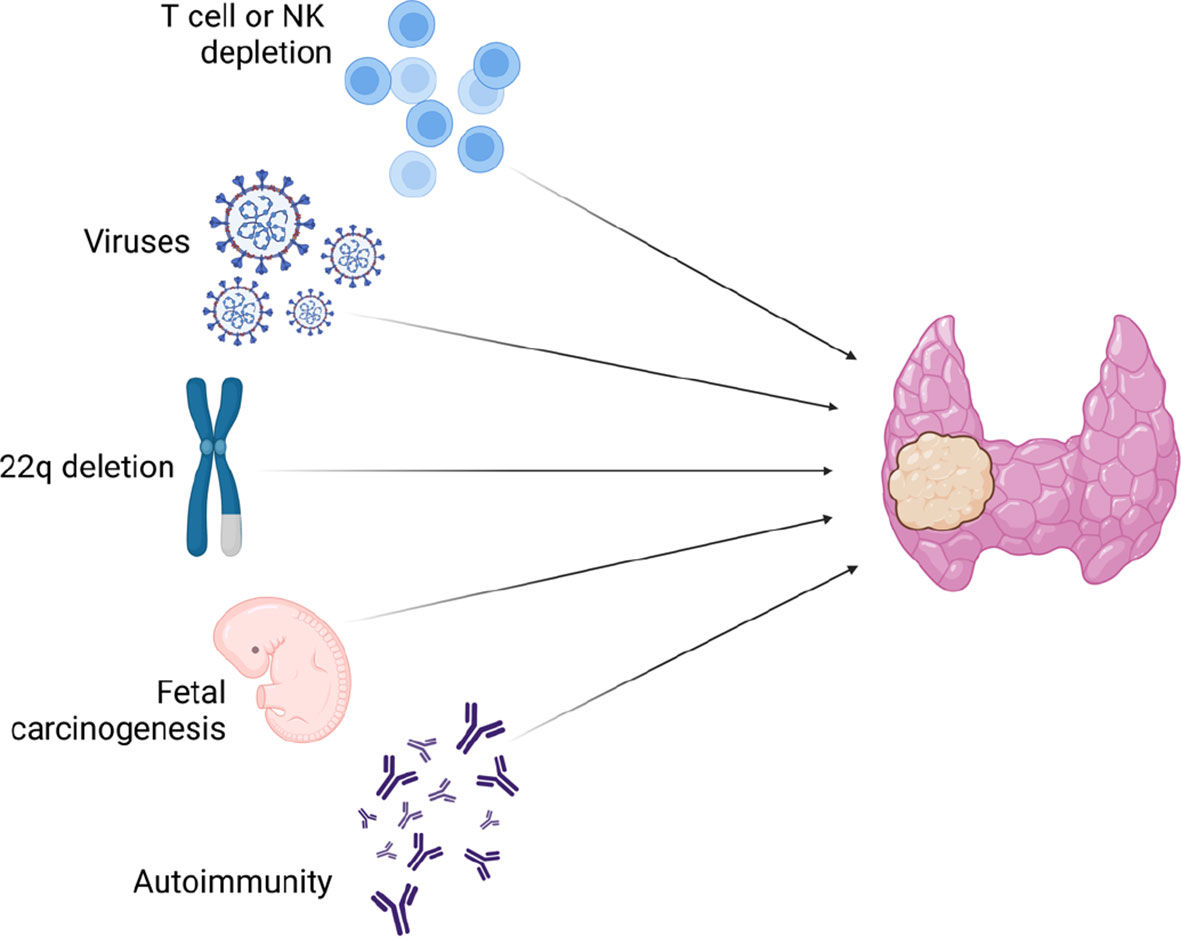
Figure 3 Etiologic hypotheses for increased risk of thyroid neoplasms in 22q11.2DS and DiGeorge-like syndrome patients.
To the best of our knowledge, this work represents the first aggregate of data on the development of thyroid neoplasms in patients with this clinical phenotype. Our data suggest a significantly increased prevalence of neoplasms among patients with 22q11.2DS and highlight how the thyroid may be one of the preferred sites for the development of neoplasms in these patients. Our data also demonstrate that the risk of developing thyroid neoplasms could also be shared by patients with DiGeorge-like syndromes. Thus, in patients with this clinical and immunological phenotype, ultrasound scans of thyroid and parathyroid glands should be performed at the time of diagnosis and then periodically, mostly in adolescence, to ensure early detection and more proper and precocious treatment of neoplasms. A long clinical and ultrasound follow-up for these patients, especially in the presence of more pronounced immunologic changes, is recommended since the risk of neoplasms could persist lifelong.
Anyway, our data are limited to obtain significant conclusions regarding the effective increase of the thyroid cancer risk for 22q11.2DS and DGS-like patients, and more extensive studies will be necessary to be conclusive. A large registry of studies enrolling individuals with 22q11.2DS, regardless of reason or age at diagnosis, would be useful to define the exact risk, especially given the variability in reported cancer types, and begin to understand the precise mechanisms behind.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
All authors participated in the drafting of the article unanimously. All authors read and approved the final article.
GB is the Representative of ERN-BOND, and this work is generated within the European Reference Network for Rare Bone Diseases.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kyritsi EM, Kanaka-Gantenbein C. Autoimmune thyroid disease in specific genetic syndromes in childhood and adolescence. Front Endocrinol (Lausanne) (2020) 11:543. doi: 10.3389/fendo.2020.00543
2. McLean-Tooke A, Spickett GP, Gennery AR. Immunodeficiency and autoimmunity in 22q11.2 deletion syndrome. Scandinavian J Immunol (2007) 66:1–7. doi: 10.1111/j.1365-3083.2007.01949.x
3. Ogata T, Niihori T, Tanaka N, Kawai M, Nagashima T, Funayama R, et al. TBX1 mutation identified by exome sequencing in a Japanese family with 22q11.2 deletion syndrome-like craniofacial features and hypocalcemia. PloS One (2014) 9:e91598. doi: 10.1371/journal.pone.0091598
4. Cuturilo G, Menten B, Krstic A, Drakulic D, Jovanovic I, Parezanovic V, et al. 4q34.1-q35.2 deletion in a boy with phenotype resembling 22q11.2 deletion syndrome. Eur J OF Pediatr (2011) 170:1465–70. doi: 10.1007/s00431-011-1533-3
5. Pignata C, D'Agostino A, Finelli P, Fiore M, Scotese I, Cosentini E, et al. Progressive deficiencies in blood T cells associated with a 10p12-13 interstitial deletion. Clin Immunol Immunopathol (1996) 80:9–15. doi: 10.1006/clin.1996.0088
6. Greenberg F, Elder FF, Haffner P, Northrup H, Ledbetter DH. Cytogenetic findings in a prospective series of patients with DiGeorge anomaly. Am J Hum Genet (1988) 43:605–11.
7. Ballif BC, Hornor SA, Jenkins E, Madan-Khetarpal S, Surti U, Jackson KE, et al. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2-p12.2. Nat Genet (2007) 39:1071–3. doi: 10.1038/ng2107
8. McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, Lynch D, Sullivan K, et al. Autosomal dominant ‘Opitz’ GBBB syndrome due to a 22q11.2 deletion. Am J Med Genet (1995) 59:103–13. doi: 10.1002/ajmg.1320590122
9. Devriendt K, Fryns JP, Mortier G, van Thienen MN, Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet (1998) 35:789–90. doi: 10.1136/jmg.35.9.789-a
10. Alberio AMQ, Legitimo A, Bertini V, Baroncelli GI, Costagliola G, Valetto A, et al. Clinical, immunological, and genetic findings in a cohort of patients with the diGeorge phenotype without 22q11.2 deletion. J Clin Med (2022) 11:2025. doi: 10.3390/jcm11072025
11. Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet (2001) 27:286–91. doi: 10.1038/85845
12. Stagi S, Lapi E, Gambineri E, Salti R, Genuardi M, Colarusso G, et al. Thyroid function and morphology in subjects with microdeletion of chromosome 22q11 (del(22)(q11)). Clin Endocrinol (2010) 72:839–44. doi: 10.1111/j.1365-2265.2009.03736.x
13. De Almeida JR, James AL, Papsin BC, Weksburg R, Clark H, Blaser S, et al. Thyroid gland and carotid artery anomalies in 22qll.2 deletion syndromes. Laryngoscope (2009) 119.
14. Jawad AF, McDonald-McGinn DM, Zackai E, Sullivan KE. Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J Pediatr (2001) 139:715–23. doi: 10.1067/mpd.2001.118534
15. Ricci S, Sarli WM, Lodi L, Canessa C, Lippi F, Azzari C, et al. Characterization of autoimmune thyroid disease in a cohort of 73 paediatric patients affected by 22q11.2 deletion syndrome: longitudinal single-centre study. Genes (Basel) (2022) 13:1552. doi: 10.3390/genes13091552
16. Powell JE, Kelly AM, Parkes SE, Cole TR, Mann JR. Cancer and congenital abnormalities in Asian children: a population-based study from the West Midlands. Br J Cancer (1995) 72:1563–9. doi: 10.1038/bjc.1995.549
17. Mann JR, Dodd HE, Draper GJ, Waterhouse JA, Birch JM, Cartwright RA, et al. Congenital abnormalities in children with cancer and their relatives: results from a case-control study (IRESCC). Br J Cancer (1993) 68:357–63. doi: 10.1038/bjc.1993.340
18. Marom T, Roth Y, Goldfarb A, Cinamon U. Head and neck manifestations of 22q11.2 deletion syndromes. Eur Arch Otorhinolaryngol (2012) 269:381–7. doi: 10.1007/s00405-011-1745-1
19. Adams JW, Malicki D, Levy M, Crawford JR. Coincident pineocytoma and probable brainstem glioma in a child with 22q11.2 deletion syndrome. BMJ Case Rep (2022) 15:e249232. doi: 10.1136/bcr-2022-249232
20. Scattone A, Caruso G, Marzullo A, Piscitelli D, Gentile M, Bonadonna L, et al. Neoplastic disease and deletion 22q11.2: a multicentric study and report of two cases. Pediatr Pathol Mol Med (2003) 22:323–41. doi: 10.1080/pdp.22.4.323.341
21. Ferreira E, Mohaghegh M, Venkat S, Drachenberg D, Battistuzzi S, Ji S. A case of an adult wilms tumour in a patient with velocardiofacial syndrome. Urology (2020) 137:e8–9. doi: 10.1016/j.urology.2019.12.019
22. Wahrmann S, Kainulainen L, Kytö V, Lempainen J. Childhood manifestations of 22q11.2 deletion syndrome: A Finnish nationwide register-based cohort study. Acta Paediatrica (2023) 112:1312–8. doi: 10.1111/apa.16737
23. Tobias ES, Morrison N, Whiteford ML, Tolmie JL. Towards earlier diagnosis of 22q11 deletions. Arch Dis Child (1999) 81:513–4. doi: 10.1136/adc.81.6.513
24. ESID - European Society for Immunodeficiencies. Available at: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria.
25. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American thyroid association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: the american thyroid association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid (2016) 26:1–133. doi: 10.1089/thy.2015.0020
26. Tosato F, Bucciol G, Pantano G, Putti MC, Sanzari MC, Basso G, Plebani M, et al. Lymphocytes subsets reference values in childhood. Cytomet Part A (2015) 87:81–5. doi: 10.1002/cyto.a.22520
27. Garcia-Prat M, Álvarez-Sierra D, Aguiló-Cucurull A, Salgado-Perandrés S, Briongos-Sebastian S, Franco-Jarava C, et al. Extended immunophenotyping reference values in a healthy pediatric population. Cytomet B Clin Cytom (2019) 96:223–33. doi: 10.1002/cyto.b.21728
28. Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for B cell subpopulations from infancy to adulthood. Clin Exp Immunol (2010) 162:271–9. doi: 10.1111/j.1365-2249.2010.04206.x
29. Aydıner Ö., Karakoç Aydıner E, Akpınar İ., Turan S, Bereket A. Normative data of thyroid volume-ultrasonographic evaluation of 422 subjects aged 0-55 years. J Clin Res Pediatr Endocrinol (2015) 7:98–101. doi: 10.4274/jcrpe.1818
30. Hodax JK, Bowerman K, Quintos JB. Benign thyroid nodules in pediatric patients: determining best practices for repeat ultrasound evaluations. J Pediatr Endocrinol Metab (2019) 32:895–901. doi: 10.1515/jpem-2018-0476
31. SEER*Explorer application. Available at: https://seer.cancer.gov/statistics-network/explorer/application.html.
32. Spinelli C, Ghionzoli M, Oreglio C, Sanna B, De Napoli L, Morganti R, et al. Increased trend of thyroid cancer in childhood over the last 30 years in EU countries: a call for the pediatric surgeon. Eur J Pediatr (2022) 181:3907–13. doi: 10.1007/s00431-022-04596-4
33. Rossi ED, Pantanowitz L, Hornick JL. A worldwide journey of thyroid cancer incidence centred on tumour histology. Lancet Diabetes Endocrinol (2021) 9:193–4. doi: 10.1016/S2213-8587(21)00049-8
34. McDonald-McGinn DM, Reilly A, Wallgren-Pettersson C, Hoyme HE, Yang SP, Adam M, et al. Malignancy in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Am J Med Genet A (2006) 140:906–9. doi: 10.1002/ajmg.a.31199
35. Lambert MP, Arulselvan A, Schott A, Markham SJ, Crowley TB, Zackai EH, et al. The 22q11.2 deletion syndrome: Cancer predisposition, platelet abnormalities and cytopenias. Am J Med Genet A (2018) 176:2121–7. doi: 10.1002/ajmg.a.38474
36. Davies EG. Immunodeficiency in diGeorge syndrome and options for treating cases with complete athymia. Front Immunol (2013) 4:322. doi: 10.3389/fimmu.2013.00322
37. Cirillo E, Giardino G, Gallo V, Galasso G, Romano R, D'Assante R, et al. DiGeorge-like syndrome in a child with a 3p12.3 deletion involving MIR4273 gene born to a mother with gestational diabetes mellitus. Am J Med Genet A (2017) 173:1913–8. doi: 10.1002/ajmg.a.38242
38. Riaz IB, Faridi W, Patnaik MM, Abraham RS. A systematic review on predisposition to lymphoid (B and T cell) neoplasias in patients with primary immunodeficiencies and immune dysregulatory disorders (Inborn errors of immunity). Front Immunol (2019) 10:777. doi: 10.3389/fimmu.2019.00777
39. Pai S-Y, Lurain K, Yarchoan R. How immunodeficiency can lead to malignancy. Hematology (2021) 2021:287–95. doi: 10.1182/hematology.2021000261
40. Hong R, Shen V, Rooney C, Hughes DP, Smith C, Comoli P, et al. Correction of DiGeorge anomaly with EBV-induced lymphoma by transplantation of organ-cultured thymus and Epstein-Barr-specific cytotoxic T lymphocytes. Clin Immunol (2001) 98:54–61. doi: 10.1006/clim.2000.4948
41. Ramos JT, López-Laso E, Ruiz-Contreras J, Giancaspro E, Madero S. B cell non-Hodgkin’s lymphoma in a girl with the DiGeorge anomaly. Arch Dis Child (1999) 81:444–5. doi: 10.1136/adc.81.5.444
42. Shah A, Sinnott B. Newly diagnosed hypoparathyroidism as the initial presentation of diGeorge syndrome in a 26-year-old man. AACE Clin Case Rep (2022) 8:181–2. doi: 10.1016/j.aace.2022.02.001
43. Rabold K, Gielen PR, Kers-Rebel ED, Netea MG, Smit JWA, Adema GJ, et al. T-cell lymphopenia in patients with advanced thyroid carcinoma is associated with poor prognosis. Oncologist (2019) 24:e106–10. doi: 10.1634/theoncologist.2018-0422
44. Moghoofei M, Mostafaei S, Nesaei A, Etemadi A, Sadri Nahand J, Mirzaei H, et al. Epstein-Barr virus and thyroid cancer: The role of viral expressed proteins. J Cell Physiol (2019) 234:3790–9. doi: 10.1002/jcp.27144
45. Mostafaei S, Keshavarz M, Sadri Nahand J, Farhadi Hassankiadeh R, Moradinazar M, Nouri M, et al. Viral infections and risk of thyroid cancer: A systematic review and empirical bayesian meta-analysis. Pathol - Res Pract (2020) 216:152855. doi: 10.1016/j.prp.2020.152855
46. Kitamura Y, Shimizu K, Ito K, Tanaka S, Emi M. Allelotyping of follicular thyroid carcinoma: frequent allelic losses in chromosome arms 7q, 11p, and 22q. J Clin Endocrinol Metab (2001) 86:4268–72. doi: 10.1210/jcem.86.9.7853
47. Bosse KR, Shukla AR, Pawel B, Chikwava KR, Santi M, Tooke L, et al. Malignant rhabdoid tumor of the bladder and ganglioglioma in a 14 year-old male with a germline 22q11.2 deletion. Cancer Genet (2014) 207:415–9. doi: 10.1016/j.cancergen.2014.05.007
48. Sellier C, Hwang VJ, Dandekar R, Durbin-Johnson B, Charlet-Berguerand N, Ander BP, et al. Decreased DGCR8 expression and miRNA dysregulation in individuals with 22q11.2 deletion syndrome. PloS One (2014) 9:e103884. doi: 10.1371/journal.pone.0103884
49. Takano T. Fetal cell carcinogenesis of the thyroid: a modified theory based on recent evidence. Endocr J (2014) 61:311–20. doi: 10.1507/endocrj.EJ13-0517
50. Shugar AL, Shapiro JM, Cytrynbaum C, Hedges S, Weksberg R, Fishman L, et al. An increased prevalence of thyroid disease in children with 22q11.2 deletion syndrome. Am J Med Genetics Part A (2015) 167:1560–4. doi: 10.1002/ajmg.a.37064
51. Montin D, Marolda A, Licciardi F, Robasto F, Di Cesare S, Ricotti E, et al. Immunophenotype anomalies predict the development of autoimmune cytopenia in 22q11.2 deletion syndrome. J Allergy Clin Immunol: In Pract (2019) 7:2369–76. doi: 10.1016/j.jaip.2019.03.014
52. Ricci S, Masini M, Valleriani C, Casini A, Cortimiglia M, Grisotto L, et al. Reduced frequency of peripheral CD4 + CD45RA + CD31 + cells and autoimmunity phenomena in patients affected by Del22q11 syndrome. Clin Immunol (2018) 188:81–4. doi: 10.1016/j.clim.2017.12.011
Keywords: thyroid cancer, thyroid nodules, 22q11 deletion syndrome, DiGeorge, genetic syndrome
Citation: Sarli WM, Ricci S, Lodi L, Cavone F, Pacillo L, Giancotta C, Ubertini G, Baroncelli G, Cancrini C, Azzari C and Stagi S (2023) Risk of thyroid neoplasms in patients with 22q11.2 deletion and DiGeorge-like syndromes: an insight for follow-up. Front. Endocrinol. 14:1209577. doi: 10.3389/fendo.2023.1209577
Received: 20 April 2023; Accepted: 14 July 2023;
Published: 10 August 2023.
Edited by:
Maria Cristina Vigone, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Gerdi Tuli, Regina Margherita Hospital, ItalyCopyright © 2023 Sarli, Ricci, Lodi, Cavone, Pacillo, Giancotta, Ubertini, Baroncelli, Cancrini, Azzari and Stagi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Stagi, c3RlZmFuby5zdGFnaUB5YWhvby5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.