 Catarina I. Gonçalves
Catarina I. Gonçalves Josianne N. Carriço
Josianne N. Carriço Omneya M. Omar
Omneya M. Omar Ebtesam Abdalla
Ebtesam Abdalla Manuel C. Lemos
Manuel C. Lemos- 1CICS-UBI, Health Sciences Research Centre, University of Beira Interior, Covilhã, Portugal
- 2Department of Pediatrics, Faculty of Medicine, Alexandria University, Alexandria, Egypt
- 3Department of Human Genetics, Medical Research Institute, Alexandria University, Alexandria, Egypt
The HDR syndrome is a rare autosomal dominant disorder characterised by Hypoparathyroidism, Deafness, and Renal dysplasia, and is caused by inactivating heterozygous germline mutations in the GATA3 gene. We report an 11-year-old girl with HDR syndrome caused by a heterozygous mutation located at the splice acceptor site of exon 5 of the GATA3 gene (NM_001002295.2: c.925-1G>T). Functional studies using a minigene assay showed that this splice site mutation abolished the normal splicing of the GATA3 pre-mRNA and led to the use of a cryptic splice acceptor site, resulting in the loss of the first seven nucleotides (TCTGCAG) of exon 5 in the GATA3 mRNA. These findings increase the understanding of the mechanisms by which GATA3 splicing mutations can cause HDR syndrome.
1 Introduction
The HDR syndrome (OMIM 146255), also known as Barakat syndrome, is a rare autosomal dominant disorder characterised by Hypoparathyroidism (H), Deafness (D), and Renal dysplasia (R), and is caused by germline mutations of the GATA3 gene (1, 2).
The primary hypoparathyroidism manifests as low serum concentrations of parathyroid hormone (PTH) leading to symptomatic or asymptomatic hypocalcemia (3). The deafness is usually bilateral, sensorineural, and more evident at higher frequencies (4, 5). The renal abnormalities can manifest as renal aplasia or hypoplasia, vesicoureteral reflux, and renal cysts that may cause compression and deformities leading to renal failure (6).
The clinical expression of each component of the disorder can vary widely (2). Although the hearing loss is commonly diagnosed during childhood, the hypocalcemia and renal abnormalities often stay asymptomatic and undiagnosed for several years, particularly when there is no family history to alert to this diagnosis (7). The type of underlying mutation may influence the severity and age of onset of each HDR feature (2).
The GATA3 gene is located on chromosome 10p14, comprises six exons and encodes a 444 amino acid protein. The GATA3 protein is a dual zinc-finger transcription factor that is expressed in the developing parathyroid, inner ear, and kidneys (1, 2). In the year 2000, heterozygous loss-of-function mutations of GATA3 were found to be responsible for the HDR syndrome (8). Since then, mutations in GATA3 have been reported in at least 124 kindreds, consisting of 40% frameshift deletions or insertions, 23% missense mutations, 14% nonsense mutations, 6% splice site mutations, 1% inframe deletions or insertions, 15% whole-gene deletions, and 1% whole-gene duplications (2).
We present the clinical and genetic characteristics of a patient with HDR syndrome, and the functional characterization of a splice site mutation in the GATA3 gene.
2 Materials and methods
2.1 Clinical studies
The patient is an 11-year-old girl, born to non-consanguineous Egyptian parents with unremarkable family histories. Since early life, she suffered several episodes of convulsions and tetany. The first episode occurred at the age of 14 days, during which hypocalcaemia was confirmed. This was attributed to vitamin D deficiency and treated accordingly. Since then, she suffered multiple episodes of convulsions with frequent hospital visits to receive intravenous calcium. At the age of 3 years, an audiogram revealed bilateral severe sensorineural hearing impairment. Upon current admission, physical examination revealed spasm of hands and feet. She had normal facial appearance, no dysmorphic features, and no skeletal abnormalities. She was wearing hearing aids. Systemic examination was unremarkable including cardiac examination. Laboratory assessment revealed low total serum calcium 5.2 mg/dL (ref: 8.8-10.8), low PTH concentration 9.1 pg/mL (ref: 9-52), and high serum phosphorus 10 mg/dL (ref: 4-7). An abdominal ultrasound showed a simple cyst (1.5 x 1.6 cm) with thin wall and clear content in the mid-zone of the left kidney with normal right kidney and normal cortical echogenicity of the kidneys. Magnetic Resonance Imaging (MRI) revealed absent basal ganglia calcification.
2.2 Genomic deoxyribonucleic acid sequencing
The genetic studies were approved by the Institutional Ethics Committees of both the Faculty of Health Sciences, University of Beira Interior (Ref: CE-FCS-2013-017) and the Medical Research Institute, University of Alexandria (Ref: IORG0008812). Written informed consent was obtained from the patient’s legal guardian. DNA was extracted from peripheral blood leucocytes of the patient and her unaffected mother (the unaffected father was unavailable for the study) using previously described methods (9). The patient was screened for mutations in GATA3 by polymerase chain reaction (PCR) amplification of the six coding exons and exon–intron boundaries, and bidirectional sequencing using a CEQ DTCS sequencing kit (Beckman Coulter, Fullerton, CA, USA) and an automated capillary DNA sequencer (GenomeLab TM GeXP, Genetic Analysis System, Beckman Coulter). Primer sequences were designed using Primer3Plus (10) (available upon request). The GATA3 variant was analyzed by Franklin (Genoox Ltd, https://franklin.genoox.com/) and classified according to American College of Medical Genetics and Genomics (ACMG) criteria (11). The nomenclature of the variant was based on the GATA3 cDNA reference sequence (GenBank accession number NM_001002295.2).
2.3 In silico prediction
To predict the effect of the splice site variant, we applied the bioinformatic program NNSplice (http://fruitfly.org/seq_tools/splice.html) that uses machine learning to predict potential splice sites, with a score ranging from 0 (low) to 1 (high) (12).
2.4 In vitro functional studies
To assess the effect of this splice site variant on the messenger ribonucleic acid (mRNA) of GATA3, we used a minigene technique (13). Using the patient genomic DNA as template, the wild-type and mutant alleles of the GATA3 exon 5, along with 384 base-pairs (bp) of 5’ and 423 bp of 3’ intronic flanking sequences were amplified by PCR using iProof High-Fidelity DNA Polymerase (Bio-Rad Laboratories, Hercules, CA, USA) with the following oligonucleotides: forward 5’-CTGACTGACATATGCTGAAAGCCCAGTTCCAAAA-3’, and reverse 5’-TCAGTCAGAGATCTCCCTGCCACACATTACAATTC-3’. Both oligonucleotides carry NdeI and BglII restriction enzyme sites at the 5’ end, respectively. These restrictions enzymes were used to clone the PCR product into the pcAT7-Glo1 plasmid (a kind gift from Dr. Kristen W. Lynch, Perelman School of Medicine, University of Pennsylvania, USA). COS-7 cells were cultured in a 6-well plate, in Dulbecco’s modified Eagle medium, with 4.5 g/L glucose, L-glutamine, sodium pyruvate, 1.5 g/L NaHCO3 (PAN-Biotech GmbH, Aidenbach, Germany), and supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in 5% CO2. When a confluence of 60-70% (or 0.3 X 106 cells per well) was achieved, the cultured cells were transfected with 5 µg of the minigene plasmid DNA using Xfect transfection reagent (Takara Bio USA, San Jose, CA, USA), according to the manufacturer’s protocol. Total RNA was harvested from transfected cells, after 24 h, using QIAshredder spin columns (QIAGEN, Hilden, Germany) and RNeasy Mini kit (QIAGEN, Hilden, Germany) following the manufacturer’s recommendations. Reverse transcription reactions were performed with 600 ng of the total extracted RNAs using the RevertAid First Strand cDNA Synthesis kit (Thermo Fisher Scientific Baltics UAB, Vilnius, Lithuania). PCR amplifications were performed with the generated cDNAs as template, using the oligonucleotides: Act 5’-TTCGGCTTCTGGCGTGTGACCGGCGGCTCTAGC-3’ and ActT7R 5’-CACAGTCGAGGCTGATCAGCGG-3’. The amplified products were then sequenced with the CEQ DTCS sequencing kit (Beckman Coulter, Fullerton, CA, USA) and an automated capillary DNA sequencer (GenomeLab TM GeXP, Genetic Analysis System, Beckman Coulter).
3 Results
DNA sequencing of the GATA3 gene in the patient revealed a heterozygous variant in the splice acceptor site of exon 5 (NM_001002295.2: c.925-1G>T) (Figure 1A). The variant was absent in the Genome Aggregation Database (gnomAD) (14). The variant was not found in her unaffected mother. Her unaffected father was unavailable for the study.
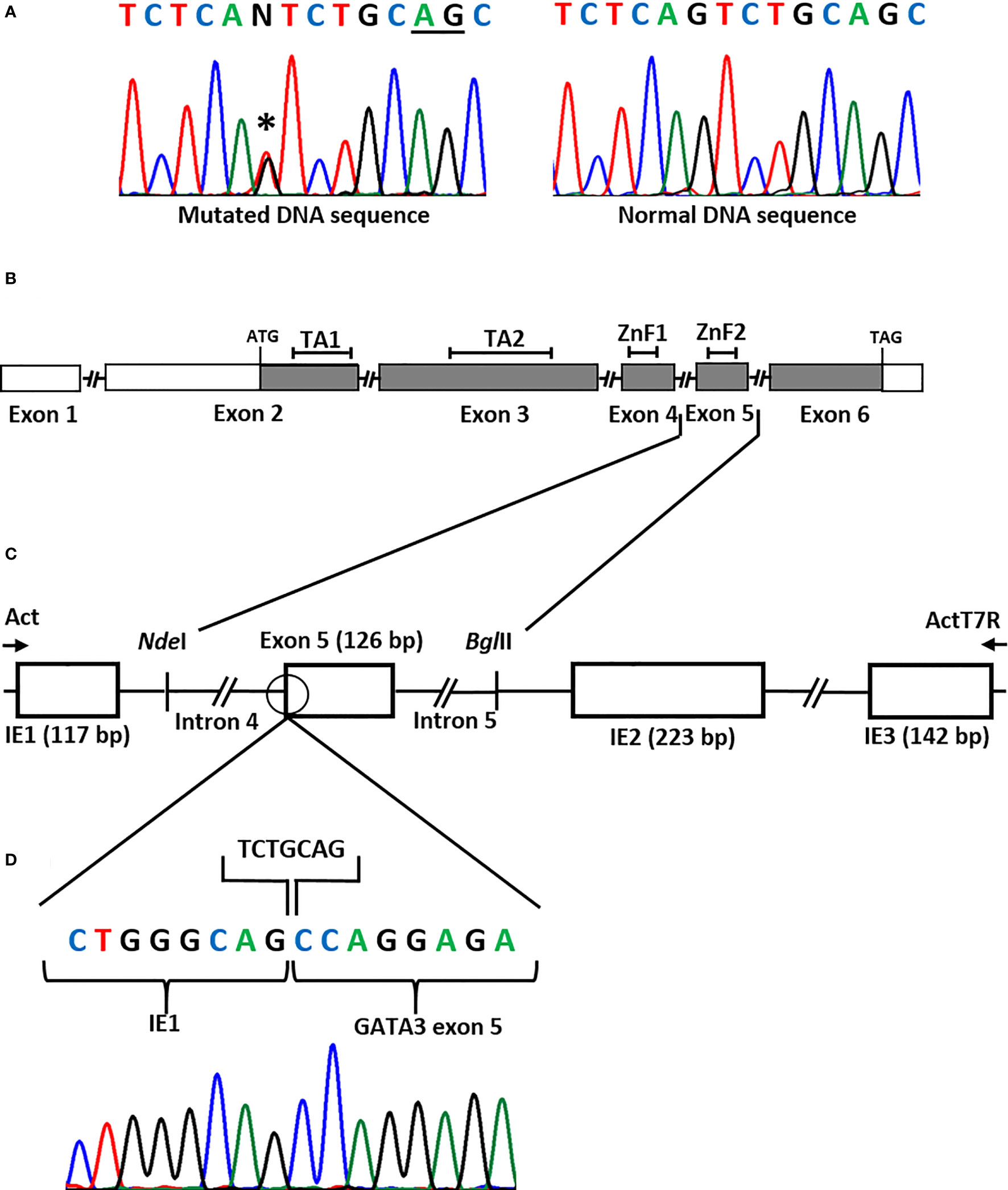
Figure 1 (A) DNA sequence of the GATA3 exon 5 splice acceptor site showing a heterozygous splice site mutation (NM_001002295.2: c.925-1G>T) (asterisk) in the patient. Underlined nucleotides (AG) represent the cryptic splice acceptor site used by the spliceosome. (B) Representation of the GATA3 gene. Boxes represent the exons, filled boxes represent the coding regions, open boxes represent non-coding regions, connecting lines represent the introns (not drawn to scale). TA1, Transactivating domain 1; TA2, Transactivating domain 2; ZnF1, Zinc Finger 1; ZnF2, Zinc Finger 2. The ATG (translation start) codon is in exon 2 and the TAG (stop) codon is in exon 6. (C) Minigene assay to assess splicing. The patient had a G>T substitution in the splice acceptor site (open circle) in intron 4 of the GATA3 gene. To check its effect on the splicing of the transcript, the wild-type and the mutant sequences of exon 5, together with the flanking introns (introns 4 and 5), were cloned into the NdelI and BglII restrictions sites of the pcAT7-Glo1 vector, between intrinsic exons IE1 and IE2. The constructs were transfected into COS-7 cells and RNA was extracted. Reverse Transcription (RT)-PCR using Act and ActT7R flanking primers (arrows) amplified the spliced products. Bp, base-pair. (D) DNA sequence of the spliced product showing the loss of seven nucleotides (TCTGCAG) at the beginning of exon 5 due to the use of an alternative splice acceptor site (TCTGCAG).
The bioinformatic program NNSplice indicated that the normal splice acceptor site of exon 5 had a score of 0.89 and that the next best potential splice site was located seven nucleotides downstream, with a score of 0.93.
The functional studies using the minigene technique showed that the splice site variant abolished the normal splicing of the GATA3 pre-mRNA and that a cryptic splice acceptor site in exon 5 was used instead. This resulted in the loss of the first seven nucleotides (TCTGCAG) of exon 5 in the GATA3 mRNA (Figures 1B–D).
According to the available evidence, the variant fulfilled the ACMG criteria for “Pathogenic” (criteria PVS1, PS3, PM2).
4 Discussion
Our study of an Egyptian girl with HDR syndrome identified a GATA3 mutation in the splice acceptor site of exon 5 (c.925-1G>T). We demonstrated that the loss of this splice site leads to the use of a cryptic splice acceptor site, located seven nucleotides downstream, that presents a surrounding splice junction sequence similar to the splice junction consensus sequence (N-Y12-14NYAG) (15). This leads to a frameshift that is predicted to produce a missense peptide with a termination at codon 355, resulting in the loss of the GATA3 ZnF2 domain.
The way by which the splicing machinery acts in identifying and removing introns is a central and a conserved step of gene expression in all eukaryotes. RNA splicing depends on the recognition of nucleotide sequences located at the exon-intron boundaries, which include the highly conserved AG and GT dinucleotides at the splice acceptor and donor sites, respectively (16). Mutations that change splicing consensus sequences are often associated with diseases (17, 18). These mutations may result in abnormal splicing through exon skipping, intron retention, or activation of cryptic splice sites (19). Cryptic splice sites have similar splicing consensus sequences, but are not normally used in RNA splicing and are only activated when the authentic splice site is lost as the result of a mutation (20). The consequences of splice site mutations can sometimes be predicted using bioinformatic programs (12), but ultimately, functional studies are needed to confirm what happens at the cellular level. Cell-based analysis of minigene splicing is widely used to investigate the effects of sequence variants on RNA splicing (13). This is usually carried out by cloning the relevant exons of the gene into a plasmid containing endogenously expressed exons and analyzing the transcribed RNA (13).
About 6% of GATA3 mutations associated with HDR are splice site mutations and it is interesting to note that these are located exclusively in introns 4 and 5 (2). Only four splice site mutations have been studied for their functional consequences. These consist of c.924 + 4_924 + 19del (21) and c.924 + 5G>C (22) that result in skipping of exon 4, and c.1051-1G>T (23) and c.1051-2A>G (24) that result in the use of an alternative splice acceptor site in exon 6.
The mutation found in our patient (c.925-1G>T) was also identified in an Italian patient with hearing loss (25), but no clinical details or functional studies for this patient were presented. Thus, our study is the first to demonstrate the mechanisms by which this mutation disrupts the function of GATA3.
Our patient presented the full triad of the syndrome since early age, which is in agreement with the type of mutation. A review of 177 reported HDR patients showed that the average age of diagnosis of hypoparathyroidism, deafness, and renal defects, was 15.3, 7.5, and 14.0 years, respectively (2). However, GATA3 protein-truncating mutations (frameshift, nonsense, and splice site), which are likely to have a more severe effect than missense mutations, were associated with an earlier expression of the disorder (2). Our patient had no family history of HDR. Therefore, she likely represents a sporadic case caused by a de novo mutation, as occurs in approximately half of HDR patients (2).
In conclusion, our results increase the understanding of the mechanisms by which GATA3 splicing mutations can cause HDR syndrome.
Data availability statement
The datasets presented in this study can be found in an online repository. The name of the repository and ID number(s) can be found below: LOVD - 0000436803 (phenotype), 0000325519 (DNA seq), and 0000927946 (variant), available at https://databases.lovd.nl/shared/individuals/00435323.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Ethics Committees of the Faculty of Health Sciences, University of Beira Interior (Ref: CE-FCS-2013-017) Institutional Ethics Committees of the Medical Research Institute, University of Alexandria (Ref: IORG0008812). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
CIG and JNC performed the genetic and functional studies, and wrote the first draft of the manuscript. OMO and EA diagnosed the patient and collected the clinical data. MCL conceived and supervised the study. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Portuguese Foundation for Science and Technology (FCT) (project grant UIDB/00709/2020), and by Programa Operacional Regional do Centro (project grant CENTRO-08-5864-FSE-000039).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Barakat AJ, Raygada M, Rennert OM. Barakat syndrome revisited. Am J Med Genet A (2018) 176:1341–8. doi: 10.1002/ajmg.a.38693
2. Lemos MC, Thakker RV. Hypoparathyroidism, deafness, and renal dysplasia syndrome: 20 Years after the identification of the first GATA3 mutations. Hum Mutat (2020) 41:1341–50. doi: 10.1002/humu.24052
3. Bilezikian JP. Hypoparathyroidism. J Clin Endocrinol Metab (2020) 105:1722–36. doi: 10.1210/clinem/dgaa113
4. van Looij MAJ, Meijers-Heijboer H, Beetz R, Thakker RV, Christie PT, Feenstra LW, et al. Characteristics of hearing loss in HDR (hypoparathyroidism, sensorineural deafness, renal dysplasia) syndrome. Audiol Neurootol (2006) 11:373–9. doi: 10.1159/000095899
5. Wang L, Lin Q-F, Wang H-Y, Guan J, Lan L, Xie L-Y, et al. Clinical auditory phenotypes associated with GATA3 gene mutations in familial hypoparathyroidism-deafness-renal dysplasia syndrome. Chin Med J (Engl) (2017) 130:703–9. doi: 10.4103/0366-6999.201600
6. Upadhyay J, Steenkamp DW, Milunsky JM. The syndrome of hypoparathyroidism, deafness, and renal anomalies. Endocr Pract (2013) 19:1035–42. doi: 10.4158/EP13050.RA
7. Horta M, Lino C, Lemos MC. Hypoparathyroidism, deafness and renal dysplasia (HDR) syndrome and GATA3. QJM (2017) 110:837–8. doi: 10.1093/qjmed/hcx176
8. Van Esch H, Groenen P, Nesbit MA, Schuffenhauer S, Lichtner P, Vanderlinden G, et al. GATA3 haplo-insufficiency causes human HDR syndrome. Nature (2000) 406:419–22. doi: 10.1038/35019088
9. Lemos MC, Regateiro FJ. N-acetyltransferase genotypes in the Portuguese population. Pharmacogenetics (1998) 8:561–4. doi: 10.1097/00008571-199812000-00013
10. Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res (2007) 35:W71–4. doi: 10.1093/nar/gkm306
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol (1997) 4:311–23. doi: 10.1089/cmb.1997.4.311
13. Smith SA, Lynch KW. Cell-based splicing of minigenes. Methods Mol Biol (2014) 1126:243–55. doi: 10.1007/978-1-62703-980-2_18
14. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
15. Vaz-Drago R, Custódio N, Carmo-Fonseca M. Deep intronic mutations and human disease. Hum Genet (2017) 136:1093–111. doi: 10.1007/s00439-017-1809-4
16. De Conti L, Baralle M, Buratti E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip Rev RNA (2013) 4:49–60. doi: 10.1002/wrna.1140
17. Wang G-S, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet (2007) 8:749–61. doi: 10.1038/nrg2164
18. Abramowicz A, Gos M. Correction to: Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet (2019) 60:231. doi: 10.1007/s13353-019-00493-z
19. Buratti E, Chivers M, Královicová J, Romano M, Baralle M, Krainer AR, et al. Aberrant 5’ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res (2007) 35:4250–63. doi: 10.1093/nar/gkm402
20. Roca X, Sachidanandam R, Krainer AR. Intrinsic differences between authentic and cryptic 5’ splice sites. Nucleic Acids Res (2003) 31:6321–33. doi: 10.1093/nar/gkg830
21. Belge H, Dahan K, Cambier J-F, Benoit V, Morelle J, Bloch J, et al. Clinical and mutational spectrum of hypoparathyroidism, deafness and renal dysplasia syndrome. Nephrol Dial Transplant (2017) 32:830–7. doi: 10.1093/ndt/gfw271
22. Matsuo K, Kobayashi A, Tanahashi Y, Maruyama S, Niitsu Y, Katsuta H, et al. Novel splice site mutation in GATA3 in a patient with HDR syndrome. Clin Pediatr Endocrinol (2017) 26:271–3. doi: 10.1297/cpe.26.271
23. Nesbit MA, Bowl MR, Harding B, Ali A, Ayala A, Crowe C, et al. Characterization of GATA3 mutations in the hypoparathyroidism, deafness, and renal dysplasia (HDR) syndrome. J Biol Chem (2004) 279:22624–34. doi: 10.1074/jbc.M401797200
24. Ali A, Christie PT, Grigorieva IV, Harding B, Van Esch H, Ahmed SF, et al. Functional characterization of GATA3 mutations causing the hypoparathyroidism-deafness-renal (HDR) dysplasia syndrome: insight into mechanisms of DNA binding by the GATA3 transcription factor. Hum Mol Genet (2007) 16:265–75. doi: 10.1093/hmg/ddl454
Keywords: HDR syndrome, hypoparathyroidism, deafness, renal dysplasia, GATA3, splice site mutation, cryptic splice site
Citation: Gonçalves CI, Carriço JN, Omar OM, Abdalla E and Lemos MC (2023) Hypoparathyroidism, deafness and renal dysplasia syndrome caused by a GATA3 splice site mutation leading to the activation of a cryptic splice site. Front. Endocrinol. 14:1207425. doi: 10.3389/fendo.2023.1207425
Received: 17 April 2023; Accepted: 13 July 2023;
Published: 04 August 2023.
Edited by:
Anatoly Tiulpakov, Research Centre for Medical Genetics, RussiaReviewed by:
Hao Zhang, Shanghai Jiao Tong University, ChinaAndrew Mallett, Townsville University Hospital, Australia
Copyright © 2023 Gonçalves, Carriço, Omar, Abdalla and Lemos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel C. Lemos, bWNsZW1vc0BmY3NhdWRlLnViaS5wdA==
†ORCID: Manuel C. Lemos, orcid.org/0000-0001-9326-8900
‡These authors have contributed equally to this work and share first authorship