 Inah Camaya
Inah Camaya Sheila Donnelly
Sheila Donnelly- School of Life Sciences, Faculty of Science, University of Technology Sydney, Ultimo, NSW, Australia
Diabetes is the fastest growing chronic disease globally, with prevalence increasing at a faster rate than heart disease and cancer. While the disease presents clinically as chronic hyperglycaemia, two distinct subtypes have been recognised. Type 1 diabetes (T1D) is characterised as an autoimmune disease in which the insulin-producing pancreatic β-cells are destroyed, and type 2 diabetes (T2D) arises due to metabolic insufficiency, in which inadequate amounts of insulin are produced, and/or the actions of insulin are diminished. It is now apparent that pro-inflammatory responses cause a loss of functional β-cell mass, and this is the common underlying mechanism of both T1D and T2D. Macrophages are the central immune cells in the pathogenesis of both diseases and play a major role in the initiation and perpetuation of the proinflammatory responses that compromise β-cell function. Furthermore, it is the crosstalk between macrophages and β-cells that orchestrates the inflammatory response and ensuing β-cell dysfunction/destruction. Conversely, this crosstalk can induce immune tolerance and preservation of β-cell mass and function. Thus, specifically targeting the intercellular communication between macrophages and β-cells offers a unique strategy to prevent/halt the islet inflammatory events underpinning T1D and T2D. Due to their potent ability to regulate mammalian immune responses, parasitic worms (helminths), and their excretory/secretory products, have been examined for their potential as therapeutic agents for both T1D and T2D. This research has yielded positive results in disease prevention, both clinically and in animal models. However, the focus of research has been on the modulation of immune cells and their effectors. This approach has ignored the direct effects of helminths and their products on β-cells, and the modulation of signal exchange between macrophages and β-cells. This review explores how the alterations to macrophages induced by helminths, and their products, influence the crosstalk with β-cells to promote their function and survival. In addition, the evidence that parasite-derived products interact directly with endocrine cells to influence their communication with macrophages to prevent β-cell death and enhance function is discussed. This new paradigm of two-way metabolic conversations between endocrine cells and macrophages opens new avenues for the treatment of immune-mediated metabolic disease.
1 Introduction
While the term ‘diabetes’ is defined as an individual’s inability to regulate blood glucose concentrations with resultant chronic hyperglycemia, traditionally two major clinically distinct subtypes have been characterized. Type 1 diabetes (T1D) results from the complete autoimmune mediated destruction of the insulin producing beta (β) cells within the pancreatic islets (1). In contrast, type 2 diabetes (T2D) arises because the β-cell population cannot satisfy insulin demand and/or peripheral tissues are resistant to the actions of insulin (2). Although T1D and T2D have been clinically classified as separate disease entities with distinct pathogeneses, there is now increasing evidence that they share disease sequalae. Loss of β-cell function and mass is the common underlying mechanism driving the progression of both conditions, and β-cell death/dysfunction is caused by pro-inflammatory responses largely initiated and perpetuated by macrophages (3).
The incidence of T1D and T2D have been exponentially increasing in recent decades, with the global prevalence predicted to reach almost 600 million cases by 2035 (4). Such rapid increases in disease prevalence cannot be attributable to genetic modifications, and instead suggest the removal of a protective environmental factor or introduction of a predisposing agent (5, 6). Initially, epidemiological studies established a robust inverse relationship between the incidence of multiple autoimmune/inflammatory diseases, and the prevalence of endemic helminth infections (7). Subsequently, compelling results from several human and animal studies have corroborated a protective effect of helminthic infection (or their excretory/secretory [ES] molecules) against the development of both TID and T2D (8). It has been broadly proposed that this positive impact on disease outcome is mediated by the potent ability of helminths to regulate pro-inflammatory host immune responses.
Interestingly, macrophages have been identified as the key players in both the modulation of host responses during helminth infections (9), and the initiation and perpetuation of pro-inflammatory responses during diabetes development. Over the years, the central role of macrophages in β-cell differentiation and homeostasis has been well demonstrated (10). However, more recently macrophages have emerged as central players in the initiation of autoimmune insulitis (immune cell infiltration of the islets) in T1D, and as the dominant immune cell population causing intra-islet inflammation in T2D (11, 12). Macrophages are highly dynamic and adopt distinct phenotypes and functions in response to cues received from adjacent cells and the surrounding microenvironment (13, 14). Further, macrophages are among the first immune cells to traffic to the islets during the development of T1D and T2D (11, 12). Thus, it is not surprising that macrophages can, and do, communicate intimately with β-cells, and vice versa. This intercell crosstalk determines if the islet micro-environment becomes pro- or anti-inflammatory, thereby promoting or mitigating the development of diabetes, respectively (15, 16).
This review discusses the evidence suggesting that modulation of the host immune responses by helminths alters the interplay between macrophages and β-cells to prevent β-cell dysfunction and death, and therefore prevent the development of both T1D and T2D. Helminth infection has also been shown to alter host metabolism (17), suggesting an additional effect on cells with endocrine function, such as β-cells. Therefore, this review also explores the biological changes mediated by helminths, and their ES products, to determine if they may directly alter the communication between β-cells and macrophages, to enhance β-cell survival and function, thereby preventing the development of T1D and T2D.
2 β-cell dysfunction and death underpin T1D and T2D
In T1D, β-cells are lost due to a sustained process of autoimmune destruction driven by pro-inflammatory immune cells. This destructive process progresses over several years such that at diagnosis approximately only 10-20% of β-cell mass remains (1). Within the autoimmune islet environment, dying β-cells are phagocytosed by antigen presenting cells (macrophages and dendritic cells), and autoantigens are processed and presented to autoreactive Th1 and Th17 CD4+ cells. Subsequently, autoreactive cytotoxic CD8+ T cells undergo activation and clonal expansion, and traffic to the pancreas where they infiltrate the islets and destroy β-cells (18, 19). During insulitis, infiltrating immune cells, such as macrophages and T cells, secrete pro-inflammatory cytokines (notably IL-1β, TNF, and IFNγ), which further promotes β-cell apoptosis (20, 21) (Figure 1). These pro-inflammatory macrophages are pivotal to T1D development as their deletion attenuates disease development (22).
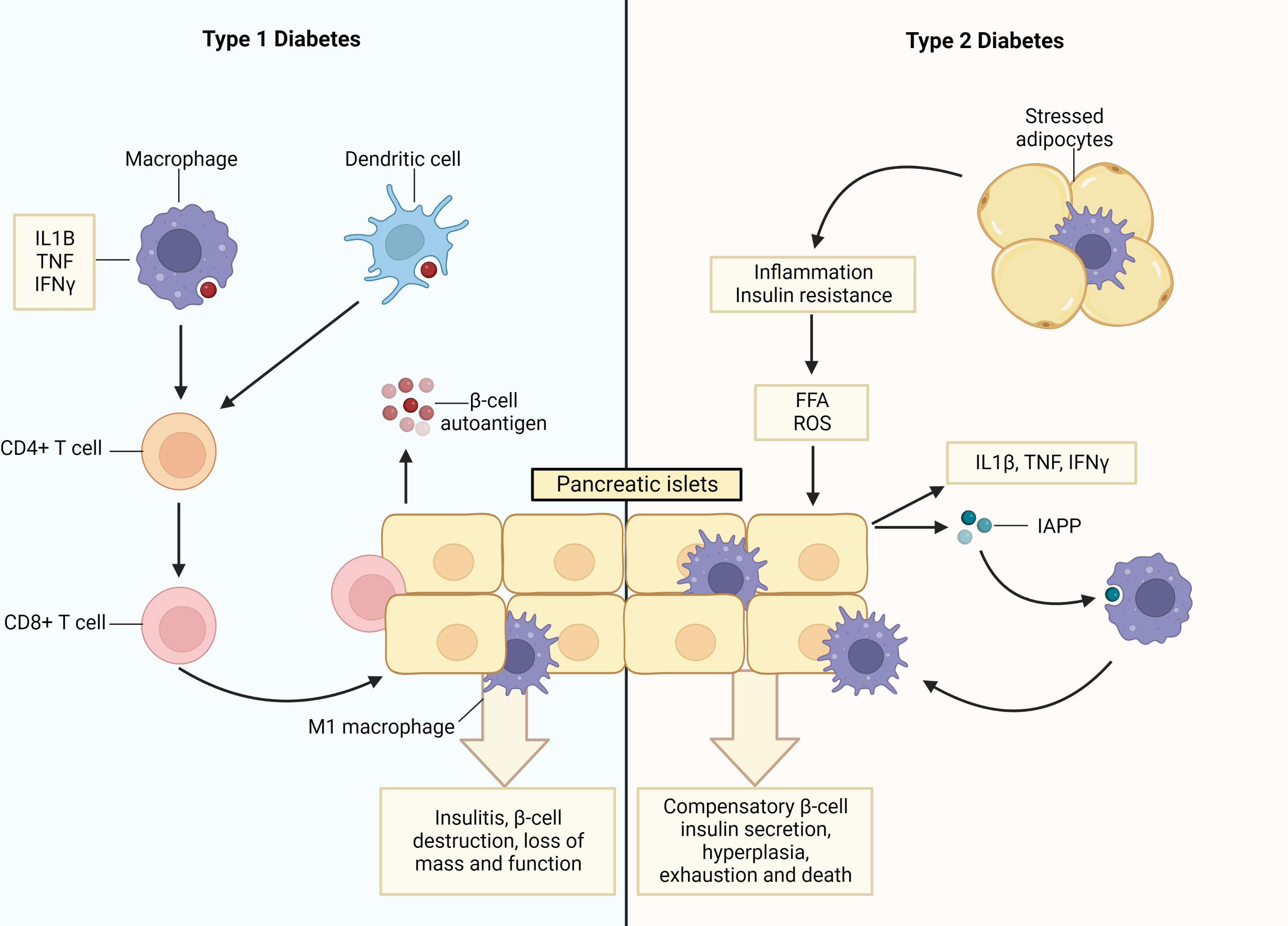
Figure 1 Pancreatic islet inflammation and β-cell death underpin type 1 and type 2 diabetes. In Type 1 diabetes (T1D) apoptotic β-cells are a source of auto- and neo-antigens resulting in the activation of antigen presenting cells (APCs; macrophages and dendritic cells) after their phagocytosis. This initiates inflammation and the priming of autoreactive Th1 and Th17 CD4+ cells, which, in turn, prime autoreactive cytotoxic CD8+ T cells that infiltrate the islets (termed insulitis) and destroy the β-cells. During insulitis, infiltrating macrophages and T cells secrete pro-inflammatory cytokines (IL1β, TNF and IFNγ), which increase rates of β-cell apoptosis. Additionally, stressed β-cells upregulate expression levels of MHC I/II and co-stimulatory molecules enabling them to function as APCs and also induce the activation and clonal expansion of autoreactive T-cell populations. In type 2 diabetes (T2D), there are increased levels of free fatty acids (FFAs) and inflammation. This induces β-cell endoplasmic reticulum (ER) stress and secretion of pro-inflammatory cytokines, akin to the intra-islet events observed in T1D, as well as islet amyloid polypeptide (IAPP). In turn, macrophages are recruited into the islet and islet resident macrophages become activated, both of which exhibit pro-inflammatory phenotypes. This process is exacerbated by stressed adipocytes that trigger inflammation in peripheral organs and initiate insulin resistance. Consequently, β-cells undergo compensatory hyperplasia and increased insulin secretion, which ultimately leads to β-cell exhaustion, death and T2D. Created with BioRender.com.
While the events initiating β-cell loss are different, T2D is also ultimately an inflammatory syndrome of the islets. Indeed, conditions of overnutrition trigger inflammation and insulin resistance, along with elevated levels of inflammatory factors, such as circulating glucose and free fatty acids (FFA) (23). This induces β-cell endoplasmic reticulum (ER) stress and β-cell secretion of pro-inflammatory cytokines/chemokines (such as IL-1β, TNF, and IFNγ) as well as islet amyloid polypeptide (IAPP). In turn, this pro-inflammatory milieu leads to the recruitment of macrophages to the islet, akin to insulitis development in T1D, in addition to the activation of islet resident macrophages. These macrophage populations exhibit a pro-inflammatory M1-like phenotype and release the same inflammatory cytokines/chemokines characteristic of T1D development (24, 25). This inflammatory sequalae is exacerbated by stressed and/or necrotic adipocytes, which similarly cause the recruitment of pro-inflammatory macrophages, and reductions in anti-inflammatory macrophage and regulatory T cell populations within the adipose tissue. This, in turn, triggers inflammation in organs targeted by insulin and initiates insulin resistance. To compensate for these adverse metabolic events, β-cells produce and secrete increased amounts of insulin, resulting in β-cell hyperplasia, stress, exhaustion, and ultimately death (26–28). Collectively, these processes lead to the perpetuation and maintenance of a pro-inflammatory environment within the islet, ultimately culminating in β-cell dysfunction, death and T2D (2) (Figure 1).
3 β-cells are active participants in their own destruction
The destructive consequences of insulitis on β-cells led to the long-held paradigm that β-cells were passive victims of the detrimental pro-inflammatory islet environments characteristic of T1D and T2D. However, this notion has been challenged by more recent evidence that β-cells are active participants in their own demise (1, 29).
The neonatal phase of pancreatic remodeling is characterized by waves of β-cell proliferation, apoptosis and neogenesis. While this is intended as a physiological phenomenon, the process can lead to the generation of β-cell autoantigens, and the stimulation of autoreactive T-cells, which play a major role in the β-cell destruction that leads to diabetes progression. However, the frequencies of these autoreactive T-cell populations in the peripheral blood of T1D patients is comparable to those observed in healthy individuals. This suggests that the activation of insulitis, which underpins the initiation and progression of diabetes, requires the immune tolerance to β-cell autoantigens to be broken (1, 30).
Analyses of human insulitis lesions have suggested that signals released from stressed β-cells precedes, and putatively initiates, the development of insulitis (31). This sequence of events is recapitulated in a humanized mouse model of diabetes, in which the presence of autoreactive T-cells alone was insufficient to induce disease, which was triggered only when β-cells were stressed by the addition of the diabetogenic agent, streptozotocin (STZ) (32). Furthermore, immunosuppressive therapies that solely target T-cells fail to provide long-term protection against T1D as they do not typically address the underlying loss of β-cell immune tolerance (33). Thus, it has now been proposed that activation of the cellular stress response in β-cells due to their metabolic activities promotes cell death pathways, and participates in the initiation and amplification of inflammation and the active destruction of β-cells in T1D (34). Likewise, β-cells undergo metabolic stress in T2D due to overnutrition and inflammation that causes β-cell compensatory insulin secretion, followed by exhaustion and ultimately death (2).
Beta cells rapidly respond to the minute-to-minute fluctuations in blood glucose levels by tightly regulating insulin secretion. Such high metabolic demand to produce and secrete insulin can render the β-cells susceptible to exceeding ER protein folding capacity, in turn leading to accumulation of misfolded proteins and ER stress (35). This can dysregulate several processes, such as inhibition of β-cell function, induction of apoptosis and activation of cytosolic post-translational modification (PTM) enzymes (36), which can generate a group of neoantigens called hybrid insulin peptides (HIPs) by covalently linking insulin peptides to β-cell granule peptides, such as insulin c-peptide and IAPP. These HIPs contribute to autoimmune responses and ultimately β-cell destruction, as they are recognized by autoreactive CD4+ T cells in both mouse models and human patients of T1D (37, 38). Beta cells from diabetic patients also express increased levels of MHC-I and MHC-II molecules (HLA-I and HLA-II, respectively, in humans), the latter being conventionally expressed only by antigen presenting cells. This enables β-cells to present peptides (notably autoantigens) to CD8+ and CD4+ T cells, respectively (39, 40). Moreover, immune cells have direct access to islets through the dense network of islet vasculature, which can be particularly detrimental in the presence of activated pro-inflammatory immune cells and cytokines that induce β-cell stress and apoptosis (34, 41).
These β-cell characteristics make them vulnerable to destruction, and this is exacerbated in the pathogenic conditions of T1D and T2D, in which there are increased levels of pro-inflammatory cytokines, reactive oxygen species (ROS) and nitric oxide (NO), which are especially detrimental since β-cells have limited antioxidant defense capabilities (42, 43), and are highly sensitive to cytokine mediated damage. The central pro-inflammatory cytokines, IL-1β, TNF and IFNγ, have been shown to inhibit β-cell function and activate apoptotic pathways in β-cell lines, human islets, and rodent models (34, 44). In turn, this inflammatory milieu stimulates the β-cells themselves to secrete pro-inflammatory cytokines and chemokines, thereby actively contributing to their own destruction (45). Indeed, β-cells of diabetic animals and within the islets of T2D patients were observed to produce IL-1β (46). This is corroborated by in vitro studies, wherein cultured human islets exposed to high glucose were observed to secrete IL-1β, with production increasing due to autocrine feedback (47).
Furthermore, β-cells produce several chemokines: (i) C-C ligand 5 (CCL5), also named RANTES, (ii) C-X-C motif chemokine ligand 10 (CXCL10), also called IP-10, and (iii) C-C ligand 2 (CCL2), also termed monotype chemoattractant protein or MCP1 (34). CXCL10 and CCL5 are secreted by murine islets, β-cell lines, and cultured human islets after exposure to IL-1β, TNF, and IFNγ. Both chemokines attract activated immune cells to the islets (48, 49), and CXCL10 has been shown to exert direct toxicity on the β-cells (50). Similarly, CCL2 is produced by human and murine islets in response to IL-1β and can be induced in vivo by environmental triggers (such as viral infections), causing inflammation and macrophage recruitment (51–53).
Collectively, these β-cell vulnerabilities, along with the signals they release under conditions of stress within the islet microenvironment, can create a self-perpetuating cycle of β-cell destruction, which is exacerbated in conjunction with pro-inflammatory immune cells (Figure 2).
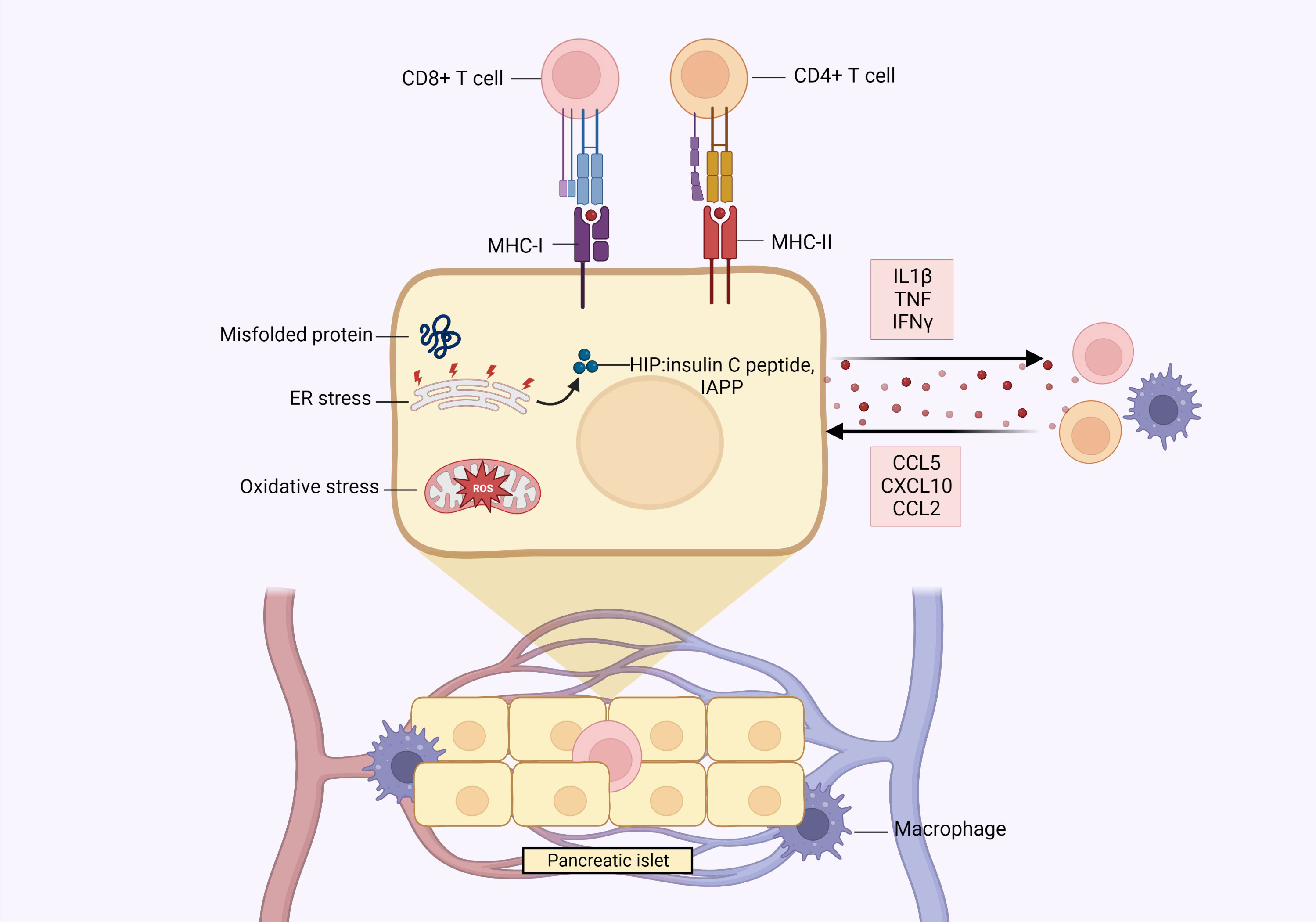
Figure 2 Pancreatic β-cells play an active role in their own destruction. The high metabolic demand of insulin production/secretion can render β-cells vulnerable to exceeding endoplasmic reticulum (ER) protein folding capacity, which can lead to accumulation of misfolded proteins and ER stress. This results in dysregulation of β-cell function, induction of apoptosis and generation of hybrid insulin peptides (HIPs), such as insulin c-peptide and islet amyloid polypeptide (IAPP), which can be recognized by autoreactive immune cells, thereby contributing to β-cell destruction. Beta cells also express MHC-I and MHC-II, the latter conventionally only being expressed by bonafide antigen presenting cells to present peptides to CD8+ and CD4+ T cells, respectively. The dense network of islet vasculature also allows immune cells direct access to the islets, which can be especially detrimental under diabetic conditions wherein immune cells generally exhibit pro-inflammatory activities. In a diabetic environment, there are elevated levels of reactive oxygen species (ROS) and pro-inflammatory cytokines, to which β-cells have suboptimal defense mechanisms due to their limited production of antioxidant enzymes and sensitivity to cytokine damage. In response to this inflammation, β-cells themselves secrete pro-inflammatory cytokines (IL1β, TNF, IFNγ) and chemokines (CCL5, CXCL10, CCL2), which contribute to β-cell destruction through self-toxicity or activation/recruitment of immune cells, such as macrophages. Created with BioRender.com.
4 Crosstalk between macrophages and β-cells predetermines the fate of β-cells
Macrophages constitute a heterogeneous and dynamic immune cell population whose phenotype and function are highly dependent on the tissue microenvironment. Macrophages can derive from embryonic hematopoietic precursors and inhabit specific tissues, in which they actively participate in maintaining homeostasis under physiological conditions. Following pathogen infection, tissue damage, release of inflammatory mediators, or metabolic cues, circulating monocytes are recruited in different tissues where they differentiate into resident macrophages. Here, stimuli in the surrounding micro-environment induce macrophages to adopt pro-inflammatory or anti-inflammatory phenotypes and functions, thereby mediating inflammatory/autoimmune or homeostatic/reparative responses, respectively (22, 54).
Macrophages are among the first cells to traffic to the pancreas under both physiological and diabetogenic conditions. This is because macrophages play central roles during neonatal islet remodeling, which is characterized by waves of β-cell differentiation, proliferation, and apoptosis, due to their potent phagocytic abilities. After taking up residence in the islets, macrophages remain in intimate contact with the β-cells, where they act as exquisite sensors of the function and viability of β-cells (15). Through a two-way exchange of signals, such as cytokines and chemokines, hormones and growth factors, insulin-containing vesicles and exosomes, and metabolites, macrophages are highly responsive to β-cell activities, and the composition of their secretions is modulated accordingly to promote β-cell survival and metabolic activity (under physiological cues). On the other hand, macrophage/β-cell crosstalk can drive β-cell dysfunction and apoptosis when physiological equilibrium is disturbed by the inflammatory signals present during the development of T1D and T2D (16) (Figure 3).
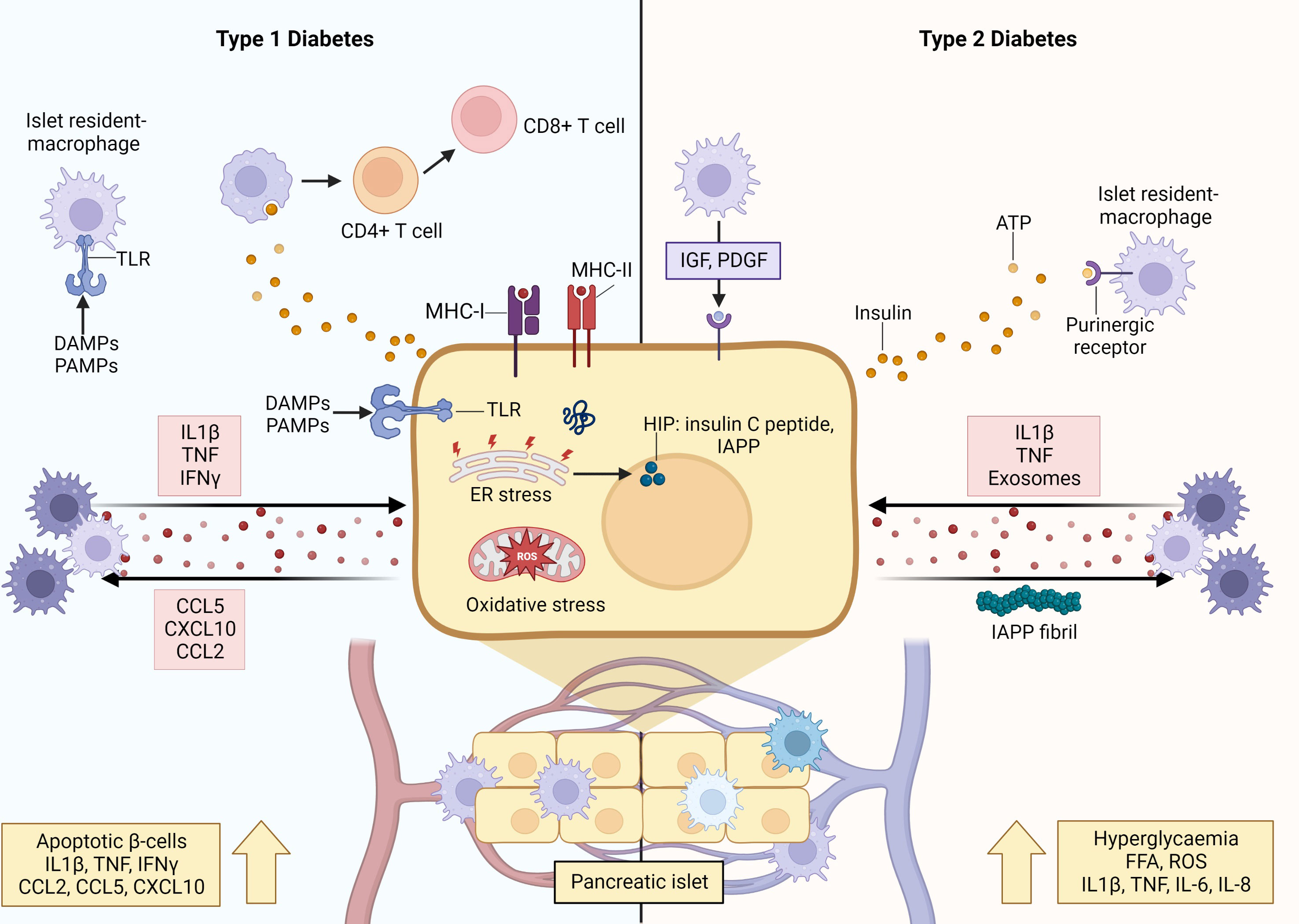
Figure 3 Crosstalk between macrophages and β-cells orchestrates inflammation and β-cell destruction in type 1 and type 2 diabetes. When physiological homeostasis is disrupted by inflammatory signals, communication between β-cells and macrophages drive β-cell dysfunction and apoptosis. In the initiating stages of T1D, resident islet macrophages exhibit a pro-inflammatory M1-like phenotype characterized by secretion of pro-inflammatory cytokines, upregulated MHC-II expression, and increased antigen presentation capacity. The β-cells also secrete pro-inflammatory cytokines and chemokines that recruit monocyte-derived macrophages with pro-inflammatory phenotype into the islet. Furthermore, β-cells also secrete insulin vesicles containing immunogenic peptides that are taken up by macrophages for presentation to autoreactive T cells, and express MHC-II, allowing them to present autoantigens as well. Together, these interactions reinforce and amplify β-cell apoptosis and destruction underlying the irreversible loss of β-cell mass leading to T1D. During T2D, hyperglycaemia and overnutrition cause β-cell hyperactivation and hyperplasia leading to functional changes and increased metabolic rates (ER and oxidative stress) within the β-cells. These changes are sensed by resident islet macrophages via uptake of insulin-containing vesicles, or through overactivation of purinergic receptors by ATP. Furthermore, β-cells secrete islet amyloid polypeptide (IAPP), which can aggregate to form plaques and fibrils that skew macrophages to a destructive pro-inflammatory M1-like phenotype. Islet macrophages do not purely exhibit M1 features and can display anti-inflammatory M2 activity, wherein they exhibit increased expression levels of reparative factors, such as IGF-1, that promote β-cell survival and proliferation. Created with BioRender.com .
In the context of T1D, the populations of resident islet macrophages in both animal models and humans display an pro-inflammatory phenotype, characterized by the secretion of cytokines (such as IL-1β and TNF), upregulated expression of MHC-II and increased antigen presentation capacity, and decreased phagocytosis of apoptotic β-cells (55–57). Decreased uptake of dying β-cells by macrophages and/or increased rates of β-cell apoptosis leads to an accumulation of dying β-cells, and their progression to necrosis, during the intended physiological process of neonatal islet remodeling. Also, after the phagocytosis of apoptotic β-cells, macrophages secrete increased levels of pro-inflammatory IL-12 and decreased levels of IL-10, which contradicts the expected anti-inflammatory pro-resolving response (58–60). Aside from this cytokine profile, intra-islet macrophages secrete NO and chemokines, and express chemokine receptors (notably CCR5, CXCR3 and CCR8) which further recruits and activates other pro-inflammatory immune cells (61, 62). These phenomena modulate the crosstalk between β-cells and macrophages to trigger the initiation of disease.
Furthermore, monocyte-derived macrophages are among the first, and most abundant, immune cells to traffic into the islet in both murine models and human patients (11, 63), and it is hypothesized that this initial influx is induced by increased levels of CCL2. Indeed, CCL2 overexpression in murine β-cells promoted monocyte recruitment to islets, infiltration, and β-cell destruction (53). Moreover, macrophages express C-C chemokine receptor 2 (CCR2) to which CCL2 can bind, thereby inducing macrophage secretion of the pro-inflammatory mediators, IL-1β, TNF, IL-12, and CXCL10. These islet infiltrating macrophages are predominantly characterized as a pro-inflammatory phenotype and release potent inflammatory mediators (such as IL-1β and CCL2) that promote β-cell apoptosis (64). Simultaneously, signals from apoptotic β-cells (IL-1β, CCL5, CXCL10, CCL2) amplify/maintain this pro-inflammatory phenotype (65).
This inflammatory environment can be exacerbated by priming factors that mediate crosstalk between macrophages and β-cells. For instance, β-cells and islet resident macrophages express toll-like receptors (TLRs), which are activated by pathogen activated molecular patterns (PAMPs) and damage associated molecular patterns (DAMPs) released due to viral infection and other tissue damaging agents. This stimulates the secretion of pro-inflammatory cytokines (such as IL-1β and TNF) that can reinforce and amplify a pro-inflammatory function/phenotype of islet resident macrophages and activate apoptotic pathways within β-cells (66). As aforementioned, stressed and/or apoptotic β-cells release pro-inflammatory signals, which are endocytosed by macrophages, thereby perpetuating the cycle of inflammation. Furthermore, insulin vesicles secreted by β-cells contain immunogenic peptides, which can also be taken up by resident M1-like macrophages for processing and presentation to autoreactive T cells. Aside from the uptake of secreted vesicles, intracellular cargo from apoptotic β-cells can be transferred to macrophages during phagocytosis (64). Additionally, under the pro-inflammatory conditions β-cells begin to express MHC-II, which enables them to present autoantigens (notably insulin neoantigens) to antigen presenting cells (40). Ultimately, the activities of β-cells and macrophages culminate in the activation and clonal expansion of autoreactive T cells, which drive the accelerated and irreversible loss of β-cell mass leading to T1D.
Similarly, under T2D conditions the crosstalk between macrophages and β-cells plays a major initiating role in establishing β-cell dysfunction, and a pro-inflammatory macrophage phenotype. In T2D, overnutrition and transient hyperglycemia induce β-cell hyperactivation and hyperplasia to satisfy the increased insulin demand (23). Aside from increased insulin secretory rates, β-cells undergo multiple functional changes induced by the nutrient rich environment and increased metabolic rates, including the production of pro-inflammatory mediators, such as NO and ROS (67). As islet macrophages constantly probe their microenvironment for signals, they sense these fluctuations in β-cell activity, via uptake of insulin-containing vesicles or through overactivation of their purinergic receptors by ATP. This can lead to desensitization and downregulation of homeostatic cues, as supported by reports of decreased purinergic receptor gene expression in T2D macrophages (68). Islet resident macrophages also contribute to islet vascular remodeling that supports compensatory hyperinsulinemia (69). While this may be advantageous in the early phases of compensatory prediabetes, this advantage is nullified by the chronic background of inflammation in which β-cell hyperactivation and dysfunction are amplified, ultimately driving β-cell exhaustion and loss.
Moreover, in T2D patients, β-cells secrete IAPP along with insulin (70). The former can aggregate to form plaques and fibrils, which skew islet macrophages to a pro-inflammatory phenotype and induce IL-1β secretion in circulating monocyte-derived macrophages to facilitate their recruitment into the islet (71). Additionally, this inflammatory dialogue is amplified by increased levels of FFAs (i.e. overnutrition) and pro-inflammatory cytokines (IL-1β, TNF, IL-6, and IL-8) (72, 73). Collectively, this environment induces β-cell dysfunction and eventual death (due to the actions of inflammatory mediators and metabolic exhaustion) (15). In addition, pro-inflammatory M1-like macrophages in T2D mice were found to secrete exosomes containing the microRNA, miR-212-5p, that can be taken up by β-cells, in turn inhibiting SIRT2-mediated Akt activation and subsequently impairment of glucose stimulated insulin secretion (74).
Importantly, islet macrophages do not exclusively display an M1-like phenotype, expressing both pro-inflammatory (CD68) and anti-inflammatory (CD163) markers (75). Furthermore, it has been observed that in animal models of T2D, islet macrophages undergo local expansion and specific transcriptional changes, with two distinct subsets identified: intra-islet (CD11c+) and peri-islet (CD11c-) macrophages. The former was found to inhibit β-cell function putatively through increased uptake of β-cell secreted insulin vesicles in a cell-to-cell contact dependent manner. On the other hand, both macrophage populations promoted compensatory β-cell proliferation (76). It has also been reported that in response to increased β-cell death in induced murine models of diabetes (STZ alone or in combination with a high fat diet) and in a genetic mouse model of T2D, islet macrophages exhibited increased expression levels of insulin like growth factor 1 (IGF-1), concomitant with decreased expression of IL-6 and TNF, a profile that is associated with a reparative phenotype (77). This functional activity is supported by a multitude of studies demonstrating that anti-inflammatory M2-like macrophages are induced in various models of pancreatic injury and inflammation and operate to promote β-cell survival, proliferation, and maintenance of functional β-cell mass, through the release of protective factors, such as IGF-1, epidermal growth factor (EGF), transforming growth factor (TGF), platelet-derived growth factor (PDGF), and Wnt3a (78).
Collectively, these observations demonstrate that macrophages exhibit distinct expression profiles and associated functional activities that are highly specialized in response to the islet microenvironment. As a result, the crosstalk between β-cells and macrophages plays a central role in both the maintenance of islet homeostasis and the initiation/perpetuation of inflammation. Accordingly, the macrophage / β-cell crosstalk can dictate autoimmunity and β-cell destruction, or conversely, immune tolerance and preservation of β-cell mass and function. Manipulating this metabolic conversation offers a unique and new strategy to directly affect β-cell survival and preservation of β-cell mass, and thus prevent the islet inflammatory events underpinning T1D and T2D.
5 Using the immune regulatory mechanisms of helminths to positively modulate macrophage and β-cell crosstalk for diabetes prevention
The inverse correlation between the prevalence of infection with helminths and the incidence of immune-mediated metabolic disease has been explained by the ‘old friends hypothesis’. This proposes that the coexistence of helminths and their human hosts over millennia has enabled them to potently regulate the mammalian immune system and skew responses to be anti-inflammatory/tolerogenic/reparative (79–81). This phenomenon affords mutual benefit by promoting longevity and tissue integrity for helminth and host, respectively.
Consequently, the elimination of helminths from human populations, due to enhanced sanitation practices, has also removed their regulatory influence on host immune responses. The result of this is the aberrant activation of inflammatory pathways, thereby increasing the incidence of immune-mediated diseases, such as T1D and T2D. Given these observations, live helminth infection, or the administration of their ES products, are being actively investigated for therapeutic potential (82–84).
5.1 Helminths are potent modulators of macrophage phenotype and function
To have sufficient time to mature and reproduce, thereby completing their lifecycle, helminths must establish chronic infections within their mammalian hosts. Accordingly, helminths have developed elegant mechanisms to educate the host’s immune system to tolerate their presence, and therefore support their prolonged survival. Thus, all helminth parasites typically drive the immune response of their hosts towards a predominantly anti-inflammatory phenotype and suppress the development of a pro-inflammatory immune response (85). Reflecting this bias in immune cell activation, helminth infection is commonly associated with an increase in Th2 cytokines, such as IL-4 and IL-13, and a reduction in Th1 cytokines, such as IL-12 and IFNγ (85–87).
Within the immune response to helminth infection, macrophages have emerged as the dominant innate immune cells playing a central role in controlling the development of the adaptive immune response, and the pathological outcomes of infection. During parasite infection, macrophages primarily display an anti-inflammatory M2-like phenotype, as characterized by the expression of specific effector molecules, such as arginase-1 (Arg1), Ym1 and resistin-like molecules (RELM). The primary functional role for these cells is the mediation of tissue repair mechanisms (9). Arg1 metabolizes L-arginine into polyamines, urea and L-orthinine, which contributes to collagen synthesis, fibrosis, and wound healing (88, 89). This consumption of L-arginine can also inhibit NO synthesis, as it is the same substrate required by inducible nitric oxide synthase (iNOS) (90), thereby regulating inflammatory activity. Similarly, Ym1 and RELMα are both associated with wound healing, with the former also directly regulating tissue repair (91–93), and the latter mediating collagen deposition and vascular stability (94, 95). In addition to these characteristic markers, helminth induced M2-like macrophages secrete growth factors, such as IGF1, vascular endothelial growth factor (VEGF) and PDGF, which orchestrate collagen deposition, angiogenesis and the recruitment, activation, and proliferation of reparative cells, such as fibroblasts and endothelial cells (9, 96). Besides this functional activity, the M2-like macrophages also regulate excessive inflammation by dampening pro-inflammatory macrophages (9). Thus, as a combined effect, the modulation of macrophage phenotypes during helminth infections acts to minimize/prevent excessive inflammatory immune responses within the host and to repair tissue damage caused by the migratory and feeding activities of the parasite (85).
The modulation of macrophage activities must be mediated by the molecules that are actively secreted by the parasites, and generally referred to as ES products. The ES products from all parasites are heterogenous mixes of proteins, glycoproteins and lipids, which mimic the modulation of immune responses induced by parasite infection per se, thus supporting the notion that these ES molecules hold considerable immune modulatory power (97). Accordingly, the ES products of several parasites have been mined to characterize the individual constituent molecules with a capacity to interact with, and modulate, the phenotype/function of host macrophages (Table 1).
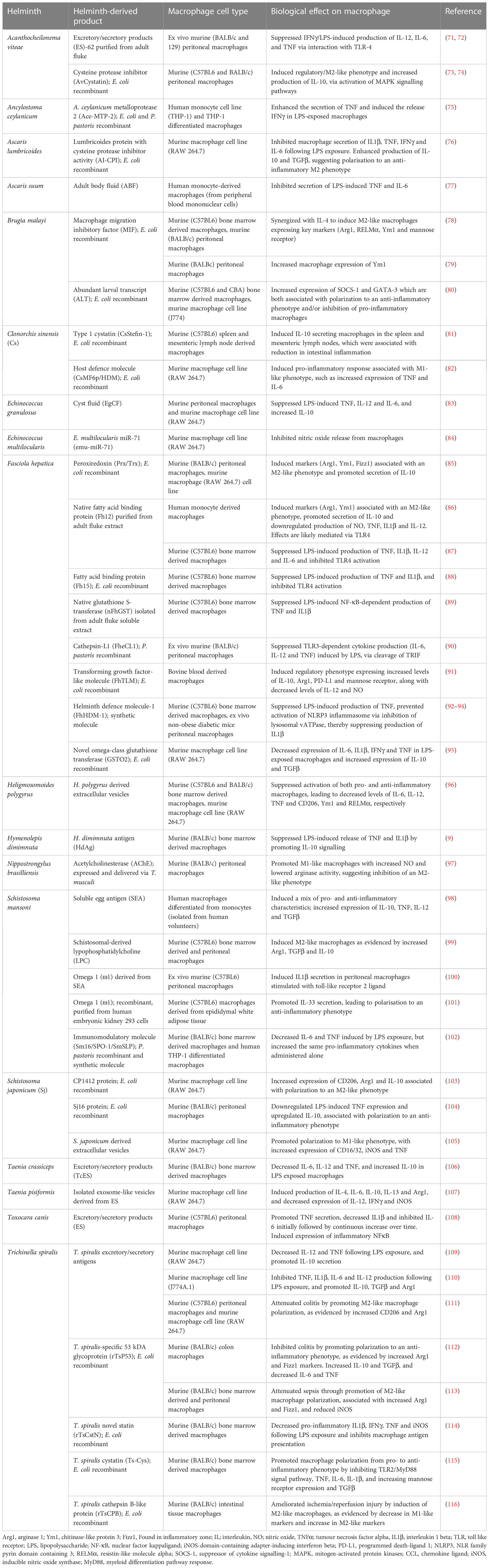
Table 1 Helminth-derived products and molecules that modulate macrophage phenotype and function.
5.2 Regulation of macrophage phenotypes by helminths prevents the development of T1D and T2D
The possibility that macrophages mediate the beneficial effect of helminths in diabetes emerged from experimental studies using mouse models in which the depletion of T-cells, or the genetic ablation of T-cell signaling molecules, failed to impact the protection from disease elicited by infection with Litomosoides sigmodontis, Heligmosomoides polygyrus, or Schistosoma mansoni (98, 99). Further analysis of immune cell populations within the infected non-diabetic animals showed an increase in the expression levels of M2 macrophage markers in the pancreatic lymph nodes. This finding indicated a central role for M2 macrophages in disease protection (100). Subsequent studies using the ES products of Fasciola hepatica and Taenia crassiceps confirmed the association between helminth-mediated protection and the recruitment of M2-like macrophages (101, 102). A functional role for macrophages was confirmed by depletion studies, in which the removal of macrophage populations, by the administration of clodronate-liposomes reversed the protective effect of the T. crassiceps ES products (102). Albeit, such macrophage ablation studies must be interpreted with caution, as in a recent study, the anti-inflammatory effects of clodronate liposomes in arthritis models were attributed to the modulation of neutrophil effector functions after phagocytosis of liposomes (103).
Analysis of the individual constituents of the F. hepatica ES products identified a single protein (FhHDM-1), which mimicked the protective effect of the ES products in preventing both insulitis and hyperglycemia in the non-obese diabetic (NOD) mouse model of T1D (104). While the reduction in disease progression was correlated to the modulation of macrophage activity, in contrast to the administration of the ES products, FhHDM-1-mediated protection was associated with the reduced ability of macrophages to respond to pro-inflammatory ligands, suggesting the regulation of an M1-like phenotype, and indicating multiple mechanisms mediating an overall switch from a predominance of pro-inflammatory to anti-inflammatory macrophages (104).
Helminth-mediated macrophage modulation is also advantageous in the prevention of T2D. Indeed, infection with various helminths, such as S. mansoni (105), Strongyloides venezuelensis, L. sigmodontis (106), Nippostrongylus brasiliensis (107), Trichinella spiralis (108), and H. polygyrus (109, 110), have been shown to exert beneficial effects (such as improvement of glucose tolerance and insulin sensitivity) in various T2D mouse models (Table 2). The mechanism of action has been putatively attributed to the activation/recruitment of M2-like macrophage populations within the adipose tissue. These M2-like populations oppose the obesity-induced elevation of pro-inflammatory macrophages and promote an anti-inflammatory environment akin to that observed in lean adipose tissue. Of these helminths, a wide array of molecules derived from S. mansoni (105, 111–113) and L. sigmodontis (106) similarly provided protection from T2D through the same mechanism (Table 2). This helminth-mediated reduction of inflammation in adipocyte tissue and subsequent improvements in glucose homeostasis could also have positive effects on the β-cells, by reducing FFAs levels and hyperglycemia that contribute to β-cell stress and exhaustion (8).
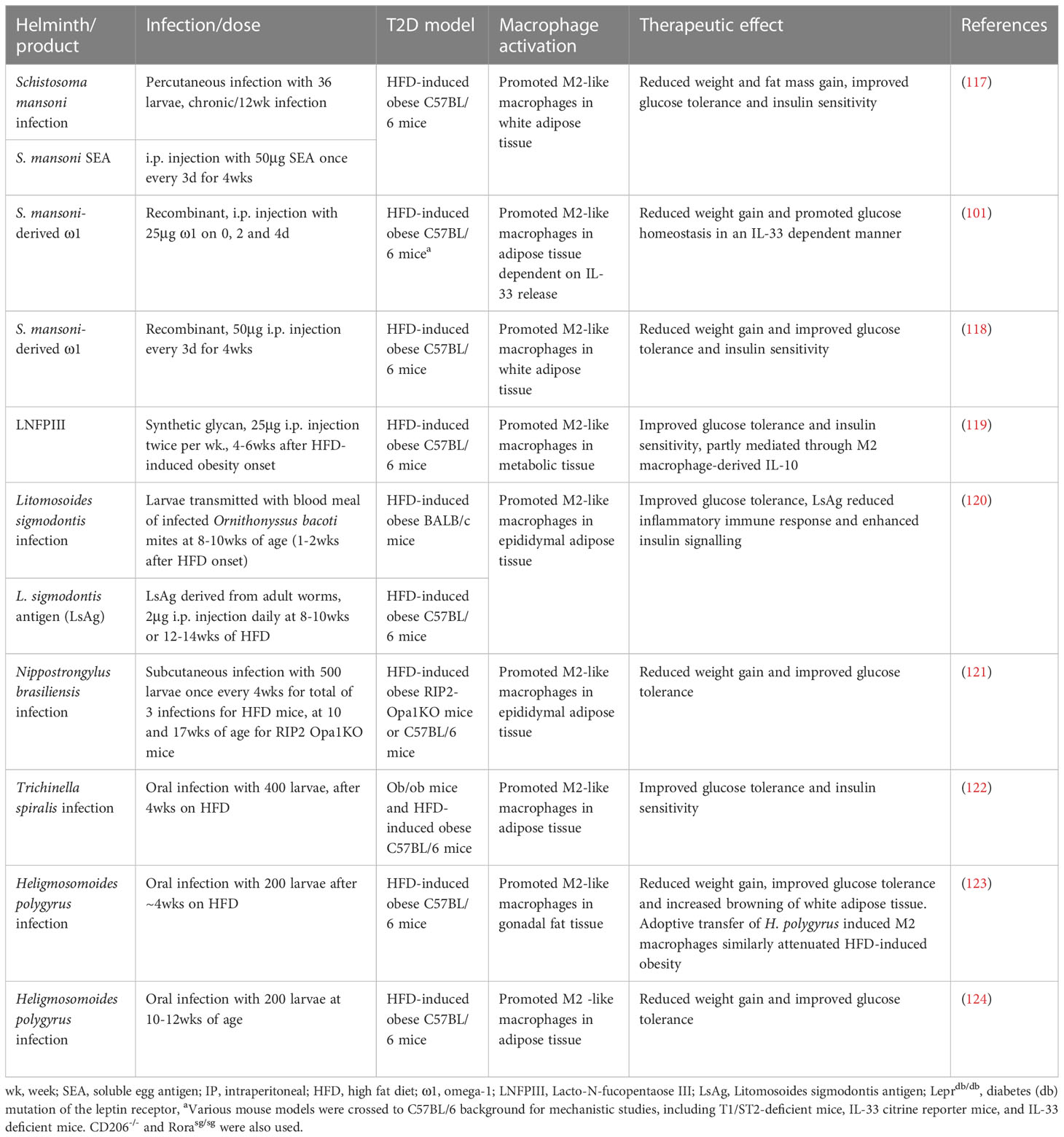
Table 2 Helminth infection and helminth-derived products exert protective effects against obesity and type 2 diabetes.
5.3 Helminth-mediated changes to macrophages may alter the crosstalk with β-cells
While the aforementioned studies clearly demonstrate a critical role for macrophages in the protective mechanisms of helminths against diabetes development, most reports simply concluded that the beneficial effects were attributable to a switch in the predominant inflammatory phenotype of macrophage. However, we propose that the changes to macrophages elicited by helminths, and their ES products, are also exerting a protective effect through communication with β-cells. Indeed, the M2-like macrophages induced by helminth infection exhibited increased expression levels of IGF1 (114). Additionally, ablation of IGF1 signaling in macrophages has been shown to disrupt endocrine IGF1-mediated signaling, resulting in a significant increase in insulin resistance in mice (114). Although a specific link to β-cell function was not explored in these studies, it has been reported that macrophages are the primary source of IGF1 in pancreatic islets, and that these cells function to enhance insulin secretion from β-cells (77). Furthermore, other growth factors (such as TGFβ1 and EGF) secreted by M2-like macrophages have been shown to enhance the survival and proliferation of β-cells through the increased expression levels of SMAD7 (115).
Supporting the beneficial effect of M2-anti-inflammatory macrophages in diabetes prevention, is the helminth-mediated regulation of pro-inflammatory macrophages. Under diabetic conditions, phagocytosis of apoptotic β-cells by macrophages generates ROS, leading to inflammasome activation, and the secretion of pro-inflammatory cytokines (notably IL-1 β), all hallmarks of an M1-like phenotype (60). As mentioned above, these macrophages have deleterious effects on β-cells by impairing glucose-stimulated insulin secretion, inducing apoptosis, and causing β-cell dedifferentiation. Further crosstalk between pro-inflammatory macrophages and pathogenic β-cells work in concert to drive inflammation and disease. Indeed, macrophages within the islets of NOD mice have been found to exhibit M1-like features (10), and depletion of these macrophages prevented T1D onset (116). Furthermore, typically, M1-like macrophages exhibit decreased phagocytic ability, as observed in diabetes-prone NOD mice (125) and humans (126), which could initiate and exacerbate inflammation because apoptotic β-cells are not efficiently cleared. Helminths can hinder this deleterious activity of pro-inflammatory macrophages (through inhibition of the inflammasome and/or secretion of pro-inflammatory cytokines (127)) or skew macrophages towards an anti-inflammatory M2-like phenotype. Consequently, the destructive pathway of macrophage/β-cell communication would be inhibited, the perpetuation of β-cell destruction prevented, and anti-inflammatory pathways conducive with preservation of β-cell function and mass, and disease prevention, would prevail.
6 Helminths modulate whole body metabolism independently of immune regulation
In recent years, it has become evident that infection with helminth parasites triggers a remodelling of the systemic metabolism of the host, which is demonstrated by altered production of pancreatic hormones, namely incretins and adipokines (128). Additionally, there is a shift in metabolic pathways in both infected tissue and in organs beyond the location of the parasite (17). The current theoretical framework suggests that this metabolic shift is required to direct the host immune status, and eventually impacts cells and organs that are not directly involved in the immune response (17, 129). This rewiring of the host’s metabolism commonly manifests as a significant increase in insulin sensitivity in non-diabetic helminth infected humans and mice (114, 117).
6.1 Helminth derived molecules interact with endocrine cells
We have recently shown that the F. hepatica FhHDM-1, which was previously shown to prevent T1D via modulation of macrophage activity, can also interact directly with pancreatic β-cells to modulate their activities (120, 121). After interaction through a currently unknown binding partner, FhHDM-1 activates the PI3K/Akt signaling pathway in β-cells. This consequently promotes β-cell survival and function, without enhancing proliferation, and inhibits pro-inflammatory cytokine induced apoptosis in both a NOD-derived (diabetogenic) β-cell line and in human islets (121).
Due to the focus on the contributory role for immune modulation in the protective effect mediated by helminths, there are limited additional studies specifically investigating the interactions of helminths, and their ES products, with β-cells. However, there is evidence supporting this premise. As mentioned previously, pancreatic β-cells express TLRs, which can modulate β-cell viability, influence insulin homeostasis (122), and contribute to T1D initiation upon interaction with high-mobility group box 1 (HMGB1) (123). Since the molecules secreted by helminths can interact with TLRs, these receptors may allow direct helminth regulation of β-cells (124). Furthermore, given the diversity of helminth molecules (including unique glycoproteins) it is plausible that helminth-derived molecules can directly modulate metabolic processes through glycan activation of metabolic cell receptors and C-type lectins (119). For example, the glycan, lacto-N-fucopentaose III (LNFPIII), which is the predominant polylactosamine sugar found on secreted egg antigens of S. mansoni exerted a similar dual effect as FhHDM-1. Under pre-T2D conditions (obesity), LNFPIII improved glucose tolerance and insulin sensitivity, by modulating pro-inflammatory responses and by directly inhibiting lipogenesis in hepatocytes, thereby ameliorating hepatic steatosis (112). Similarly, the T2 RNase ω1 secreted from the same helminth, promoted metabolic homeostasis in a murine model of diet induced T2D. In this case, ω1 was found to enhance adipocyte expression and secretion of IL-33, likely through interaction with the mannose receptor (111). Several other studies have reported similar direct helminth modulation of non-immune, metabolically active cells (8, 107, 118) (Table 3), predominantly adipocytes and hepatocytes, which communicate with, and influence the function of, β-cells. Indeed, adipocytes secrete several adipokines, such as leptin, adipsin and adiponectin, that contribute to regulation of β-cell insulin secretion and proliferation (132). Furthermore, extracellular vesicles (EVs) secreted by healthy adipocytes have been shown to promote normal β-cell physiology and provided protection from palmitate or pro-inflammatory cytokine induced death. Conversely, EVs derived from obese adipocytes led to β-cell dysfunction and death. Similar crosstalk has been observed between β-cells and hepatocytes (133). Hepatocytes secrete EVs and proliferative factors, such as IGF1, that contribute to compensatory β-cell hyperplasia in conditions of obesity/T2D. Moreover, under this inflammatory environment, pancreatic β-cell dysfunction leads to impaired insulin secretion that normally modulates glucagon release, thus leading to elevated glucagon levels. In turn, this exacerbates the increased glucose output from the liver associated with insulin resistance and induces production of kisspeptin1 in hepatocytes that can further impair β-cell insulin secretion (132). Therefore, helminth-mediated changes to adipocytes and hepatocytes would be expected to have significant knock-on effects to the β-cells.
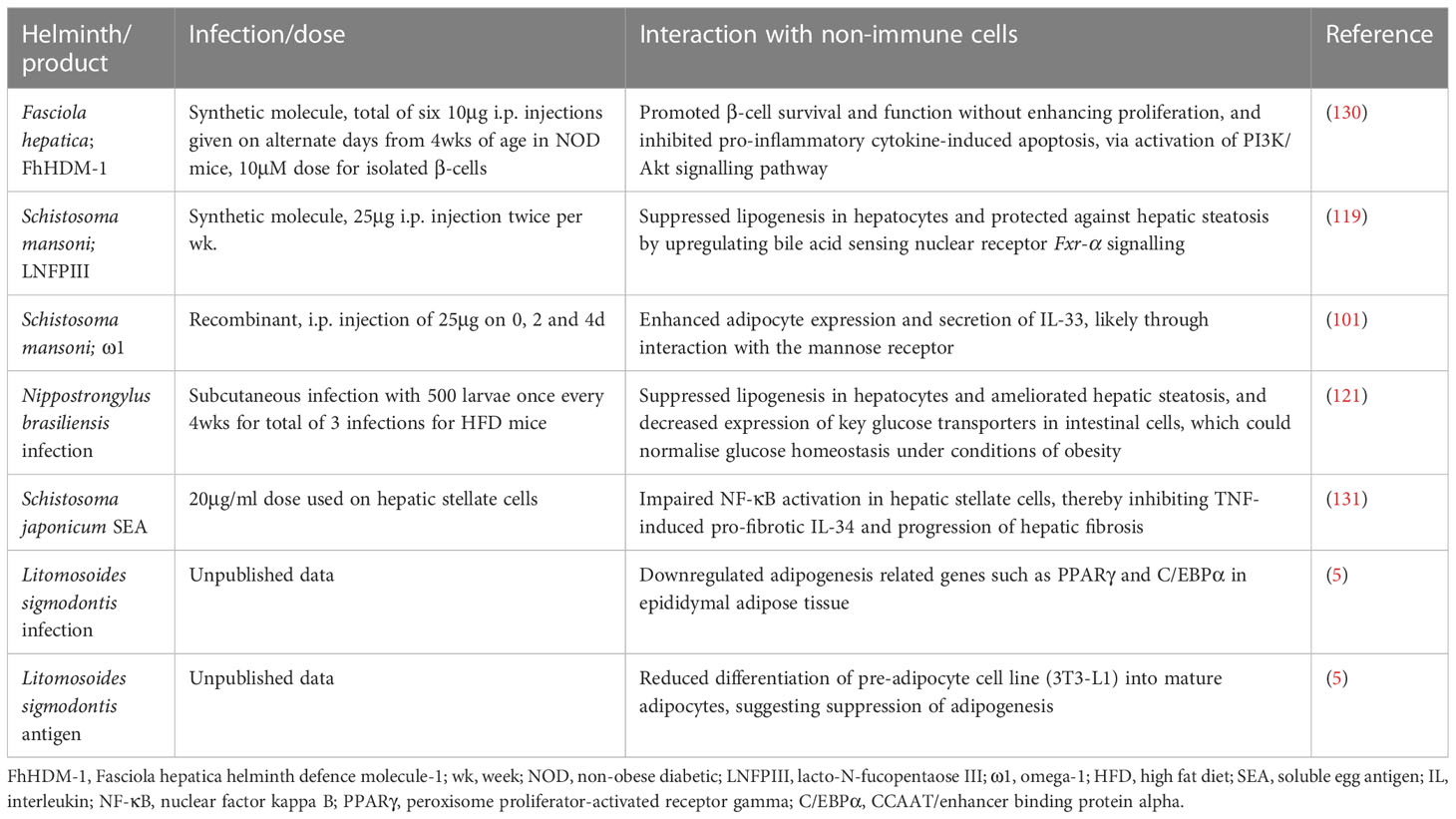
Table 3 Helminths and helminth derived products directly affect endocrine cells.
6.2 Helminths alter the gut-islet axis
Extensive analyses of the human microbiome have revealed that the gut microbiota is deeply interconnected with diabetes, with alterations to the populations of species in the gut impacting several metabolic effects and immune response processes (134, 135). It is widely acknowledged that gastrointestinal factors, such as glucagon like peptide (GLP)-1 and gastric inhibitory polypeptide (GIP), interact directly with β-cells to induce proliferation, and enhance resistance to apoptosis, thereby increasing/maintaining β-cell mass (136, 137). In addition, short chain fatty acids (SCFA) released by gut microbiota modulate the function and survival of β-cells (132). Thus, any changes to the bacteria populating the gut will subsequently alter the metabolic activity of β-cells within the pancreas.
As the gut mucosa hosts the largest population of macrophages in the human body (138), it is not surprising that SCFA released by gut microbiota also influences their functional phenotypes. For example, it has been reported that an increased abundance in Bacteroides fragilis, Lactobacillus spp. and Clostridia class induce the polarization of anti-inflammatory M2-like macrophages (139, 140). In contrast, the presence of Enterococcus faecalis polarizes colonic macrophages towards a pro-inflammatory phenotype (141). However, these effects are not confined to the intestinal microenvironment, as it has been shown that butyrate produced by gut microbiota induces the expression of mouse β-defensin 14 in pancreatic islets, which in turn drives the activation of regulatory macrophages and prevents the development of T1D in a mouse model (130).
Several studies of human and murine helminth infection have demonstrated that the presence of the parasites alters both the abundance and diversity of gut microbiomes (142, 143). More recently it has been demonstrated that these changes mediate a beneficial effect in models of T2D diabetes. Infection of mice with either S. venezuelensis (144) or Nippostrongylus brasiliensis (145) resulted in significant compositional changes in gut microbiota, most notably by increasing Lactobacillus spp., a bacterial species which has been assessed as a beneficial probiotic for diabetes (131). In both cases, these changes in gut microbiota resulted in improved insulin signaling and sensitivity. Although a specific effect on β-cells was not assessed in these studies, increased expression of M2 macrophage markers were found in adipose tissue, liver, and gut.
7 Concluding remarks
T1D and T2D are characterised by a progressive dysfunction and loss of the insulin-producing β-cells in the pancreatic islets leading to insulin deficiency. Despite this knowledge, none of the currently prescribed anti-diabetic agents effectively target the maintenance of functional β-cell mass. In T1D, the focus remains on blocking autoreactive immune cells and their mediators. However, diabetes reversal is not achieved, protection of residual β-cell mass is short-term, and global immune suppression is induced (146–148). Due to the scarcity of organ donors, significant β-cell loss during islet preparation, and allogenic and recurrent autoimmune reactions, which mediate β-cell death post-transplant, islet transplantation is not a realistic cure for T1D. In T2D, therapeutic options, such as dipeptidyl peptidase-4 (DPP-4) inhibitors, incretin-based drugs, and GLP-1 analogues, target β-cell function, but not all patients are responsive and approximately 50% ultimately require daily insulin injections due to β-cell exhaustion and death (149). Clearly, alternate, more effective strategies, are urgently required. By regulating the activities of islet macrophages and β-cells (and other endocrine cells), helminth parasites shape their crosstalk (Figure 4). This offers a unique opportunity to exploit helminths’ mechanisms for survival in their mammalian hosts to establish an environment that preserves β-cell mass and function and thus offers the potential as a cure for both T1D and T2D.
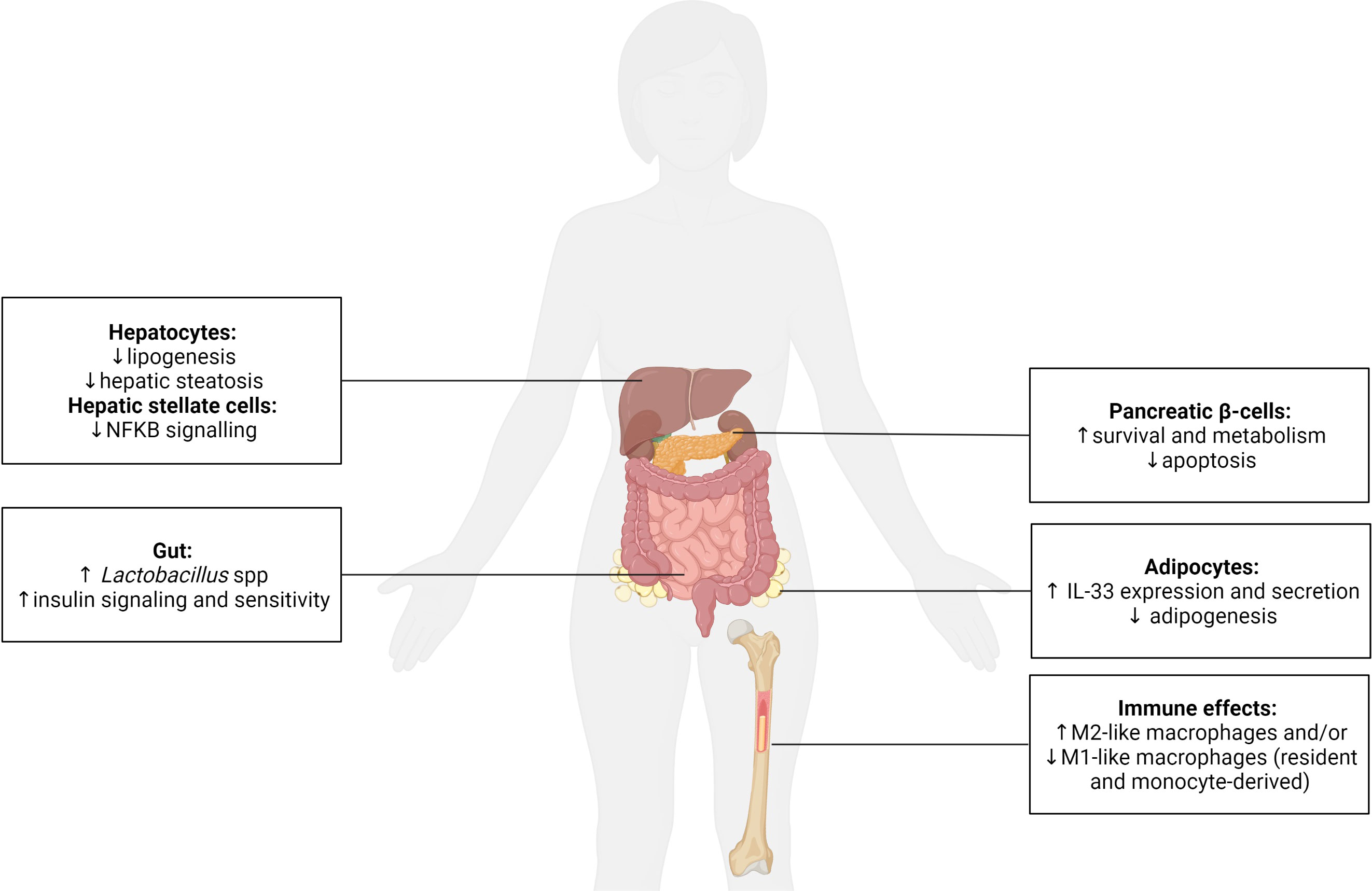
Figure 4 The systemic effect of helminths and their secreted molecules on the function of metabolic and immune cells. Helminths are recognized as potent immune modulators, typically promoting the polarization of macrophages (resident and monocyte-derived) to an anti-inflammatory/regulatory M2-like phenotype and/or inhibiting a pro-inflammatory M1-like phenotype. This immune regulation has been shown to be beneficial for type 1 and type 2 diabetes. Helminths also positively impact endocrine cells and have been demonstrated to (i) enhance pancreatic β-cell survival and metabolism, (ii) promote adipocyte expression of anti-inflammatory IL-33 and inhibit adipogenesis, (iii) reduce lipogenesis and prevent hepatic steatosis within hepatocytes, and decrease pro-inflammatory nuclear factor kappa (κ) B signaling in hepatic stellate cells, and (iv) increase the presence of probiotic Lactobacillus spp. in the gut, which promotes insulin signaling/sensitivity. Created with BioRender.com.
Author contributions
IC, BO’B and SD conceived and planned the review. IC conducted the literature search, designed and wrote the first draft of the manuscript. BO’B and SD contributed to the writing and editing of the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by a National Health and Medical Research Council project grant (APP1087341), and a JDRF Beta Cell Regeneration Innovative grant (1-INO-2019-785-S-B).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Roep BO, Thomaidou S, van Tienhoven R, Zaldumbide A. Type 1 diabetes mellitus as a disease of the β-Cell (Do not blame the immune system?). Nat Rev Endocrinol (2021) 17(3):150–61. doi: 10.1038/s41574-020-00443-4
2. Hudish LI, Reusch JE, Sussel L. β Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J Clin Invest (2019) 129(10):4001–8. doi: 10.1172/jci129188
3. Ikegami H, Babaya N, Noso S. β-Cell failure in diabetes: common susceptibility and mechanisms shared between type 1 and type 2 diabetes. J Diabetes Investig (2021) 12(9):1526–39. doi: 10.1111/jdi.13576
4. Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract (2014) 103(2):137–49. doi: 10.1016/j.diabres.2013.11.002
5. Traversi D, Rabbone I, Scaioli G, Vallini C, Carletto G, Racca I, et al. Risk factors for type 1 diabetes, including environmental, behavioural and gut microbial factors: A case-control study. Sci Rep (2020) 10(1):17566. doi: 10.1038/s41598-020-74678-6
6. Tremblay J, Hamet P. Environmental and genetic contributions to diabetes. Metabolism (2019) 100s:153952. doi: 10.1016/j.metabol.2019.153952
7. Zaccone P, Fehervari Z, Phillips JM, Dunne DW, Cooke A. Parasitic worms and inflammatory diseases. Parasite Immunol (2006) 28(10):515–23. doi: 10.1111/j.1365-3024.2006.00879.x
8. Berbudi A, Ajendra J, Wardani AP, Hoerauf A, Hübner MP. Parasitic helminths and their beneficial impact on type 1 and type 2 diabetes. Diabetes Metab Res Rev (2016) 32(3):238–50. doi: 10.1002/dmrr.2673
9. Coakley G, Harris NL. Interactions between macrophages and helminths. Parasite Immunol (2020) 42(7):e12717. doi: 10.1111/pim.12717
10. Calderon B, Carrero JA, Ferris ST, Sojka DK, Moore L, Epelman S, et al. The pancreas anatomy conditions the origin and properties of resident macrophages. J Exp Med (2015) 212(10):1497–512. doi: 10.1084/jem.20150496
11. Damond N, Engler S, Zanotelli VRT, Schapiro D, Wasserfall CH, Kusmartseva I, et al. A map of human type 1 diabetes progression by imaging mass cytometry. Cell Metab (2019) 29(3):755–68.e5. doi: 10.1016/j.cmet.2018.11.014
12. Orliaguet L, Dalmas E, Drareni K, Venteclef N, Alzaid F. Mechanisms of macrophage polarization in insulin signaling and sensitivity. Front Endocrinol (Lausanne) (2020) 11:62. doi: 10.3389/fendo.2020.00062
13. Loke P, Lin JD. Redefining inflammatory macrophage phenotypes across stages and tissues by single-cell transcriptomics. Sci Immunol (2022) 7(70):eabo4652. doi: 10.1126/sciimmunol.abo4652
14. Gordon S, Plüddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol (2017) 15(1):53. doi: 10.1186/s12915-017-0392-4
15. Cosentino C, Regazzi R. Crosstalk between macrophages and pancreatic β-Cells in islet development, homeostasis and disease. Int J Mol Sci (2021) 22(4):1765. doi: 10.3390/ijms22041765
16. Zinselmeyer BH, Vomund AN, Saunders BT, Johnson MW, Carrero JA, Unanue ER. The resident macrophages in murine pancreatic islets are constantly probing their local environment, capturing beta cell granules and blood particles. Diabetologia (2018) 61(6):1374–83. doi: 10.1007/s00125-018-4592-4
17. Kokova D, Verhoeven A, Perina EA, Ivanov VV, Heijink M, Yazdanbakhsh M, et al. Metabolic homeostasis in chronic helminth infection is sustained by organ-specific metabolic rewiring. ACS Infect Dis (2021) 7(4):906–16. doi: 10.1021/acsinfecdis.1c00026
18. Trudeau JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT. Neonatal beta-cell apoptosis: A trigger for autoimmune diabetes? Diabetes (2000) 49(1):1–7. doi: 10.2337/diabetes.49.1.1
19. Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological β Cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med (2003) 198(10):1527–37. doi: 10.1084/jem.20030966
20. Eizirik DL, Mandrup-Poulsen T. A choice of death – the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia (2001) 44(12):2115–33. doi: 10.1007/s001250100021
21. Cnop M, Welsh N, Jonas J-C, Joürns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes (2005) 54(suppl_2):S97–S107. doi: 10.2337/diabetes.54.suppl_2.S97
22. Yang S, Zhao M, Jia S. Macrophage: key player in the pathogenesis of autoimmune diseases. Front Immunol (2023) 14:1080310. doi: 10.3389/fimmu.2023.1080310
23. Caputo T, Gilardi F, Desvergne B. From chronic overnutrition to metaflammation and insulin resistance: adipose tissue and liver contributions. FEBS Lett (2017) 591(19):3061–88. doi: 10.1002/1873-3468.12742
24. Cuenco J, Dalmas E. Islet inflammation and β Cell dysfunction in type 2 diabetes. Handb Exp Pharmacol (2022) 274:227–51. doi: 10.1007/164_2021_571
25. Gao D, Jiao J, Wang Z, Huang X, Ni X, Fang S, et al. The roles of cell-cell and organ-organ crosstalk in the type 2 diabetes mellitus associated inflammatory microenvironment. Cytokine Growth Factor Rev (2022) 66:15–25. doi: 10.1016/j.cytogfr.2022.04.002
26. Winer S, Winer D. The adaptive immune system as a fundamental regulator of adipose tissue inflammation and insulin resistance. Immunol Cell Biol (2012) 90:755–62. doi: 10.1038/icb.2011.110
27. Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes (2008) 57(12):3239–46. doi: 10.2337/db08-0872
28. Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol (2011) 11(11):738–49. doi: 10.1038/nri3071
29. Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG, Gamble DR. In situ characterization of autoimmune phenomena and expression of hla molecules in the pancreas in diabetic insulitis. N Engl J Med (1985) 313(6):353–60. doi: 10.1056/nejm198508083130604
30. Erdem N, Montero E, Roep BO. Breaking and restoring immune tolerance to pancreatic beta-cells in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes (2021) 28(4):397–403. doi: 10.1097/med.0000000000000646
31. Piganelli JD, Mamula MJ, James EA. The role of β Cell stress and neo-epitopes in the immunopathology of type 1 diabetes. Front Endocrinol (Lausanne) (2020) 11:624590. doi: 10.3389/fendo.2020.624590
32. Tan S, Li Y, Xia J, Jin CH, Hu Z, Duinkerken G, et al. Type 1 diabetes induction in humanized mice. Proc Natl Acad Sci USA (2017) 114(41):10954–9. doi: 10.1073/pnas.1710415114
33. Kolb H, von Herrath M. Immunotherapy for type 1 diabetes: why do current protocols not halt the underlying disease process? Cell Metab (2017) 25(2):233–41. doi: 10.1016/j.cmet.2016.10.009
34. Toren E, Burnette KS, Banerjee RR, Hunter CS. Tse HM. Partners in crime: beta-cells and autoimmune responses complicit in type 1 diabetes pathogenesis. Front Immunol (2021) 12:756548. doi: 10.3389/fimmu.2021.756548
35. Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, et al. Ire1α Induces thioredoxin-interacting protein to activate the nlrp3 inflammasome and promote programmed cell death under irremediable er stress. Cell Metab (2012) 16(2):250–64. doi: 10.1016/j.cmet.2012.07.007
36. Marré ML, James EA, Piganelli JD. β Cell er stress and the implications for immunogenicity in type 1 diabetes. Front Cell Dev Biol (2015) 3:67. doi: 10.3389/fcell.2015.00067
37. Baker RL, Rihanek M, Hohenstein AC, Nakayama M, Michels A, Gottlieb PA, et al. Hybrid insulin peptides are autoantigens in type 1 diabetes. Diabetes (2019) 68(9):1830–40. doi: 10.2337/db19-0128
38. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic cd4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science (2016) 351(6274):711–4. doi: 10.1126/science.aad2791
39. Sano H, Imagawa A. Pancreatic β-cells express major histocompatibility complex class ii: do diabetic β-cells have the capacity of antigen-presenting cells? J Diabetes Investig (2020) 11(2):281–3. doi: 10.1111/jdi.13155
40. Zhao Y, Scott NA, Quah HS, Krishnamurthy B, Bond F, Loudovaris T, et al. Mouse pancreatic beta cells express mhc class ii and stimulate cd4(+) T cells to proliferate. Eur J Immunol (2015) 45(9):2494–503. doi: 10.1002/eji.201445378
41. Mallone R, Eizirik DL. Presumption of innocence for beta cells: why are they vulnerable autoimmune targets in type 1 diabetes? Diabetologia (2020) 63(10):1999–2006. doi: 10.1007/s00125-020-05176-7
42. Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes (1997) 46(11):1733–42. doi: 10.2337/diab.46.11.1733
43. Miki A, Ricordi C, Sakuma Y, Yamamoto T, Misawa R, Mita A, et al. Divergent antioxidant capacity of human islet cell subsets: A potential cause of beta-cell vulnerability in diabetes and islet transplantation. PloS One (2018) 13(5):e0196570. doi: 10.1371/journal.pone.0196570
44. Wang C, Guan Y, Yang J. Cytokines in the progression of pancreatic β-cell dysfunction. Int J Endocrinol (2010) 2010:515136. doi: 10.1155/2010/515136
45. Donath MY, Böni-Schnetzler M, Ellingsgaard H, Halban PA, Ehses JA. Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol Metab (2010) 21(5):261–7. doi: 10.1016/j.tem.2009.12.010
46. Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, et al. Glucose-induced beta cell production of il-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest (2002) 110(6):851–60. doi: 10.1172/jci15318
47. Böni-Schnetzler M, Thorne J, Parnaud G, Marselli L, Ehses JA, Kerr-Conte J, et al. Increased interleukin (Il)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of il-1beta in human islets by glucose and autostimulation. J Clin Endocrinol Metab (2008) 93(10):4065–74. doi: 10.1210/jc.2008-0396
48. Frigerio S, Junt T, Lu B, Gerard C, Zumsteg U, Holländer GA, et al. Beta cells are responsible for cxcr3-mediated T-cell infiltration in insulitis. Nat Med (2002) 8(12):1414–20. doi: 10.1038/nm1202-792
49. Carvalho-Pinto C, García MI, Gómez L, Ballesteros A, Zaballos A, Flores JM, et al. Leukocyte attraction through the ccr5 receptor controls progress from insulitis to diabetes in non-obese diabetic mice. Eur J Immunol (2004) 34(2):548–57. doi: 10.1002/eji.200324285
50. Schulthess FT, Paroni F, Sauter NS, Shu L, Ribaux P, Haataja L, et al. Cxcl10 impairs beta cell function and viability in diabetes through tlr4 signaling. Cell Metab (2009) 9(2):125–39. doi: 10.1016/j.cmet.2009.01.003
51. Chen MC, Proost P, Gysemans C, Mathieu C, Eizirik DL. Monocyte chemoattractant protein-1 is expressed in pancreatic islets from prediabetic nod mice and in interleukin-1 beta-exposed human and rat islet cells. Diabetologia (2001) 44(3):325–32. doi: 10.1007/s001250051622
52. Sarkar SA, Lee CE, Victorino F, Nguyen TT, Walters JA, Burrack A, et al. Expression and regulation of chemokines in murine and human type 1 diabetes. Diabetes (2012) 61(2):436–46. doi: 10.2337/db11-0853
53. Martin AP, Rankin S, Pitchford S, Charo IF, Furtado GC, Lira SA. Increased expression of ccl2 in insulin-producing cells of transgenic mice promotes mobilization of myeloid cells from the bone marrow, marked insulitis, and diabetes. Diabetes (2008) 57(11):3025–33. doi: 10.2337/db08-0625
54. Ruytinx P, Proost P, Van Damme J, Struyf S. Chemokine-induced macrophage polarization in inflammatory conditions. Front Immunol (2018) 9:1930. doi: 10.3389/fimmu.2018.01930
55. Ferreira RC, Guo H, Coulson RM, Smyth DJ, Pekalski ML, Burren OS, et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes (2014) 63(7):2538–50. doi: 10.2337/db13-1777
56. Bradshaw EM, Raddassi K, Elyaman W, Orban T, Gottlieb PA, Kent SC, et al. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing th17 cells. J Immunol (2009) 183(7):4432–9. doi: 10.4049/jimmunol.0900576
57. Sen P, Bhattacharyya S, Wallet M, Wong CP, Poligone B, Sen M, et al. Nf-kappa B hyperactivation has differential effects on the apc function of nonobese diabetic mouse macrophages. J Immunol (2003) 170(4):1770–80. doi: 10.4049/jimmunol.170.4.1770
58. Henson PM, Bratton DL. Antiinflammatory effects of apoptotic cells. J Clin Invest (2013) 123(7):2773–4. doi: 10.1172/jci69344
59. Gordon S, Plüddemann A. Macrophage clearance of apoptotic cells: A critical assessment. Front Immunol (2018) 9:127. doi: 10.3389/fimmu.2018.00127
60. Ward MG, Li G, Hao M. Apoptotic β-cells induce macrophage reprogramming under diabetic conditions. J Biol Chem (2018) 293(42):16160–73. doi: 10.1074/jbc.RA118.004565
61. Alleva DG, Pavlovich RP, Grant C, Kaser SB, Beller DI. Aberrant macrophage cytokine production is a conserved feature among autoimmune-prone mouse strains: elevated interleukin (Il)-12 and an imbalance in tumor necrosis factor-alpha and il-10 define a unique cytokine profile in macrophages from young nonobese diabetic mice. Diabetes (2000) 49(7):1106–15. doi: 10.2337/diabetes.49.7.1106
62. Cantor J, Haskins K. Recruitment and activation of macrophages by pathogenic cd4 T cells in type 1 diabetes: evidence for involvement of ccr8 and ccl1. J Immunol (2007) 179(9):5760–7. doi: 10.4049/jimmunol.179.9.5760
63. Hutchings P, Rosen H, O'Reilly L, Simpson E, Gordon S, Cooke A. Transfer of diabetes in mice prevented by blockade of adhesion-promoting receptor on macrophages. Nature (1990) 348(6302):639–42. doi: 10.1038/348639a0
64. Zirpel H, Roep BO. Islet-resident dendritic cells and macrophages in type 1 diabetes: in search of bigfoot's print. Front Endocrinol (Lausanne) (2021) 12:666795. doi: 10.3389/fendo.2021.666795
65. Denroche HC, Nackiewicz D, Verchere CB. When beta cells talk back. Diabetologia (2018) 61(1):39–42. doi: 10.1007/s00125-017-4443-8
66. Chittezhath M, Wai CMM, Tay VSY, Chua M, Langley SR, Ali Y. Tlr4 signals through islet macrophages to alter cytokine secretion during diabetes. J Endocrinol (2020) 247(1):87. doi: 10.1530/joe-20-0131
67. Dludla PV, Mabhida SE, Ziqubu K, Nkambule BB, Mazibuko-Mbeje SE, Hanser S, et al. Pancreatic β-cell dysfunction in type 2 diabetes: implications of inflammation and oxidative stress. World J Diabetes (2023) 14(3):130–46. doi: 10.4239/wjd.v14.i3.130
68. Weitz JR, Jacques-Silva C, Qadir MMF, Umland O, Pereira E, Qureshi F, et al. Secretory functions of macrophages in the human pancreatic islet are regulated by endogenous purinergic signaling. Diabetes (2020) 69(6):1206–18. doi: 10.2337/db19-0687
69. Chittezhath M, Gunaseelan D, Zheng X, Hasan R, Tay VSY, Lim ST, et al. Islet macrophages are associated with islet vascular remodeling and compensatory hyperinsulinemia during diabetes. Am J Physiol Endocrinol Metab (2019) 317(6):E1108–e20. doi: 10.1152/ajpendo.00248.2019
70. Kanatsuka A, Kou S, Makino H. Iapp/amylin and β-cell failure: implication of the risk factors of type 2 diabetes. Diabetol Int (2018) 9(3):143–57. doi: 10.1007/s13340-018-0347-1
71. Westwell-Roper CY, Ehses JA, Verchere CB. Resident macrophages mediate islet amyloid polypeptide-induced islet il-1β Production and β-cell dysfunction. Diabetes (2014) 63(5):1698–711. doi: 10.2337/db13-0863
72. Böni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, Prazak R, et al. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology (2009) 150(12):5218–29. doi: 10.1210/en.2009-0543
73. Hung HC, Tsai SF, Chou HW, Tsai MJ, Hsu PL, Kuo YM. Dietary fatty acids differentially affect secretion of pro-inflammatory cytokines in human thp-1 monocytes. Sci Rep (2023) 13(1):5511. doi: 10.1038/s41598-023-32710-5
74. Qian B, Yang Y, Tang N, Wang J, Sun P, Yang N, et al. M1 macrophage-derived exosomes impair beta cell insulin secretion via mir-212-5p by targeting sirt2 and inhibiting akt/gsk-3β/β-catenin pathway in mice. Diabetologia (2021) 64(9):2037–51. doi: 10.1007/s00125-021-05489-1
75. Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes (2007) 56(9):2356–70. doi: 10.2337/db06-1650
76. Ying W, Lee YS, Dong Y, Seidman JS, Yang M, Isaac R, et al. Expansion of islet-resident macrophages leads to inflammation affecting β Cell proliferation and function in obesity. Cell Metab (2019) 29(2):457–74.e5. doi: 10.1016/j.cmet.2018.12.003
77. Nackiewicz D, Dan M, Speck M, Chow SZ, Chen YC, Pospisilik JA, et al. Islet macrophages shift to a reparative state following pancreatic beta-cell death and are a major source of islet insulin-like growth factor-1. iScience (2020) 23(1):100775. doi: 10.1016/j.isci.2019.100775
78. Jensen DM, Hendricks KV, Mason AT, Tessem JS. Good cop, bad cop: the opposing effects of macrophage activation state on maintaining or damaging functional β-cell mass. Metabolites (2020) 10(12):485. doi: 10.3390/metabo10120485
79. Rook GAW. 99th dahlem conference on infection, inflammation and chronic inflammatory disorders: darwinian medicine and the ‘Hygiene’ or ‘Old friends’ Hypothesis. Clin Exp Immunol (2010) 160(1):70–9. doi: 10.1111/j.1365-2249.2010.04133.x
80. Rook GA. Regulation of the immune system by biodiversity from the natural environment: an ecosystem service essential to health. Proc Natl Acad Sci (2013) 110(46):18360–7. doi: 10.1073/pnas.1313731110
81. Allen JE, Maizels RM. Diversity and dialogue in immunity to helminths. Nat Rev Immunol (2011) 11(6):375–88. doi: 10.1038/nri2992
82. Smallwood TB, Giacomin PR, Loukas A, Mulvenna JP, Clark RJ, Miles JJ. Helminth immunomodulation in autoimmune disease. Front Immunol (2017) 8:453. doi: 10.3389/fimmu.2017.00453
83. Ryan SM, Eichenberger RM, Ruscher R, Giacomin PR, Loukas A. Harnessing helminth-driven immunoregulation in the search for novel therapeutic modalities. PloS Pathog (2020) 16(5):e1008508. doi: 10.1371/journal.ppat.1008508
84. Venkatakrishnan A, Sarafian JT, Jirků-Pomajbíková K, Parker W. Socio-medical studies of individuals self-treating with helminths provide insight into clinical trial design for assessing helminth therapy. Parasitol Int (2022) 87:102488. doi: 10.1016/j.parint.2021.102488
85. Maizels RM, Balic A, Gomez-Escobar N, Nair M, Taylor MD, Allen JE. Helminth parasites–masters of regulation. Immunol Rev (2004) 201:89–116. doi: 10.1111/j.0105-2896.2004.00191.x
86. Vacca F, Le Gros G. Tissue-specific immunity in helminth infections. Mucosal Immunol (2022) 15(6):1212–23. doi: 10.1038/s41385-022-00531-w
87. Wiedemann M, Voehringer D. Immunomodulation and immune escape strategies of gastrointestinal helminths and schistosomes. Front Immunol (2020) 11:572865. doi: 10.3389/fimmu.2020.572865
88. Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol (2004) 4(8):583–94. doi: 10.1038/nri1412
89. Morris SM Jr. Arginine metabolism: boundaries of our knowledge. J Nutr (2007) 137(6 Suppl 2):1602s–9s. doi: 10.1093/jn/137.6.1602S
90. Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol (2014) 5:532. doi: 10.3389/fimmu.2014.00532
91. Chen F, Liu Z, Wu W, Rozo C, Bowdridge S, Millman A, et al. An essential role for th2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med (2012) 18(2):260–6. doi: 10.1038/nm.2628
92. Sutherland TE, Logan N, Rückerl D, Humbles AA, Allan SM, Papayannopoulos V, et al. Chitinase-like proteins promote il-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat Immunol (2014) 15(12):1116–25. doi: 10.1038/ni.3023
93. Sutherland TE, Rückerl D, Logan N, Duncan S, Wynn TA, Allen JE. Ym1 induces relmα and rescues il-4rα Deficiency in lung repair during nematode infection. PloS Pathog (2018) 14(11):e1007423. doi: 10.1371/journal.ppat.1007423
94. Liu T, Jin H, Ullenbruch M, Hu B, Hashimoto N, Moore B, et al. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of il-4/il-13 and mediation via stat-6. J Immunol (2004) 173(5):3425–31. doi: 10.4049/jimmunol.173.5.3425
95. Knipper JA, Willenborg S, Brinckmann J, Bloch W, Maaß T, Wagener R, et al. Interleukin-4 receptor α Signaling in myeloid cells controls collagen fibril assembly in skin repair. Immunity (2015) 43(4):803–16. doi: 10.1016/j.immuni.2015.09.005
96. Motran CC, Silvane L, Chiapello LS, Theumer MG, Ambrosio LF, Volpini X, et al. Helminth infections: recognition and modulation of the immune response by innate immune cells. Front Immunol (2018) 9:664. doi: 10.3389/fimmu.2018.00664
97. Harnett W. Secretory products of helminth parasites as immunomodulators. Mol Biochem Parasitol (2014) 195(2):130–6. doi: 10.1016/j.molbiopara.2014.03.007
98. Liu Q, Sundar K, Mishra PK, Mousavi G, Liu Z, Gaydo A, et al. Helminth infection can reduce insulitis and type 1 diabetes through cd25- and il-10-independent mechanisms. Infect Immun (2009) 77(12):5347–58. doi: 10.1128/iai.01170-08
99. Osada Y, Yamada S, Nabeshima A, Yamagishi Y, Ishiwata K, Nakae S, et al. Heligmosomoides polygyrus infection reduces severity of type 1 diabetes induced by multiple low-dose streptozotocin in mice via stat6- and il-10-independent mechanisms. Exp Parasitol (2013) 135(2):388–96. doi: 10.1016/j.exppara.2013.08.003
100. Osada Y, Fujiyama T, Kamimura N, Kaji T, Nakae S, Sudo K, et al. Dual genetic absence of stat6 and il-10 does not abrogate anti-hyperglycemic effects of schistosoma mansoni in streptozotocin-treated diabetic mice. Exp Parasitol (2017) 177:1–12. doi: 10.1016/j.exppara.2017.03.008
101. Lund ME, O'Brien BA, Hutchinson AT, Robinson MW, Simpson AM, Dalton JP, et al. Secreted proteins from the helminth fasciola hepatica inhibit the initiation of autoreactive T cell responses and prevent diabetes in the nod mouse. PloS One (2014) 9(1):e86289. doi: 10.1371/journal.pone.0086289
102. Espinoza-Jiménez A, De Haro R, Terrazas LI. Taenia crassiceps antigens control experimental type 1 diabetes by inducing alternatively activated macrophages. Mediators Inflamm (2017) 2017:8074329. doi: 10.1155/2017/8074329
103. Culemann S, Knab K, Euler M, Wegner A, Garibagaoglu H, Ackermann J, et al. Stunning of neutrophils accounts for the anti-inflammatory effects of clodronate liposomes. J Exp Med (2023) 220(6):e20220525. doi: 10.1084/jem.20220525
104. Lund ME, Greer J, Dixit A, Alvarado R, McCauley-Winter P, To J, et al. A parasite-derived 68-mer peptide ameliorates autoimmune disease in murine models of type 1 diabetes and multiple sclerosis. Sci Rep (2016) 6:37789. doi: 10.1038/srep37789
105. Hussaarts L, García-Tardón N, van Beek L, Heemskerk MM, Haeberlein S, van der Zon GC, et al. Chronic helminth infection and helminth-derived egg antigens promote adipose tissue M2 macrophages and improve insulin sensitivity in obese mice. FASEB J (2015) 29(7):3027–39. doi: 10.1096/fj.14-266239
106. Berbudi A, Surendar J, Ajendra J, Gondorf F, Schmidt D, Neumann AL, et al. Filarial infection or antigen administration improves glucose tolerance in diet-induced obese mice. J Innate Immun (2016) 8(6):601–16. doi: 10.1159/000448401
107. Yang Z, Grinchuk V, Smith A, Qin B, Bohl JA, Sun R, et al. Parasitic nematode-induced modulation of body weight and associated metabolic dysfunction in mouse models of obesity. Infect Immun (2013) 81(6):1905–14. doi: 10.1128/iai.00053-13
108. Okada H, Ikeda T, Kajita K, Mori I, Hanamoto T, Fujioka K, et al. Effect of nematode trichinella infection on glucose tolerance and status of macrophage in obese mice. Endocr J (2013) 60(11):1241–9. doi: 10.1507/endocrj.ej13-0312
109. Su CW, Chen CY, Li Y, Long SR, Massey W, Kumar DV, et al. Helminth infection protects against high fat diet-induced obesity via induction of alternatively activated macrophages. Sci Rep (2018) 8(1):4607. doi: 10.1038/s41598-018-22920-7
110. Obi S, Shimokawa C, Katsuura M, Olia A, Imai T, Suzue K, et al. Il-33 is essential to prevent high-fat diet-induced obesity in mice infected with an intestinal helminth. Parasite Immunol (2020) 42(9):e12700. doi: 10.1111/pim.12700
111. Hams E, Bermingham R, Wurlod FA, Hogan AE, O'Shea D, Preston RJ, et al. The helminth T2 rnase Ω1 promotes metabolic homeostasis in an il-33- and group 2 innate lymphoid cell-dependent mechanism. FASEB J (2016) 30(2):824–35. doi: 10.1096/fj.15-277822
112. Bhargava P, Li C, Stanya KJ, Jacobi D, Dai L, Liu S, et al. Immunomodulatory glycan lnfpiii alleviates hepatosteatosis and insulin resistance through direct and indirect control of metabolic pathways. Nat Med (2012) 18(11):1665–72. doi: 10.1038/nm.2962
113. van der Zande HJP, Gonzalez MA, de Ruiter K, Wilbers RHP, García-Tardón N, van Huizen M, et al. The helminth glycoprotein omega-1 improves metabolic homeostasis in obese mice through type 2 immunity-independent inhibition of food intake. FASEB J (2021) 35(2):e21331. doi: 10.1096/fj.202001973R
114. Spadaro O, Camell CD, Bosurgi L, Nguyen KY, Youm YH, Rothlin CV, et al. Igf1 shapes macrophage activation in response to immunometabolic challenge. Cell Rep (2017) 19(2):225–34. doi: 10.1016/j.celrep.2017.03.046
115. Xiao X, Gaffar I, Guo P, Wiersch J, Fischbach S, Peirish L, et al. M2 macrophages promote beta-cell proliferation by up-regulation of smad7. Proc Natl Acad Sci (2014) 111(13):E1211–E20. doi: 10.1073/pnas.1321347111
116. Carrero JA, McCarthy DP, Ferris ST, Wan X, Hu H, Zinselmeyer BH, et al. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of nod mice. Proc Natl Acad Sci USA (2017) 114(48):E10418–e27. doi: 10.1073/pnas.1713543114
117. Mishra PK, Palma M, Bleich D, Loke P, Gause WC. Systemic impact of intestinal helminth infections. Mucosal Immunol (2014) 7(4):753–62. doi: 10.1038/mi.2014.23
118. Chen L, Yu Y, Liu E, Duan L, Zhu D, Chen J, et al. Schistosoma japonicum soluble egg antigen inhibits tnf-α-induced il-34 expression in hepatic stellate cells. Parasitol Res (2019) 118(2):551–7. doi: 10.1007/s00436-018-6165-3
119. van der Zande HJP, Zawistowska-Deniziak A, Guigas B. Immune regulation of metabolic homeostasis by helminths and their molecules. Trends Parasitol (2019) 35(10):795–808. doi: 10.1016/j.pt.2019.07.014
120. Alvarado R, O'Brien B, Tanaka A, Dalton JP, Donnelly S. A parasitic helminth-derived peptide that targets the macrophage lysosome is a novel therapeutic option for autoimmune disease. Immunobiology (2015) 220(2):262–9. doi: 10.1016/j.imbio.2014.11.008
121. Camaya I, Mok TY, Lund M, To J, Braidy N, Robinson MW, et al. The parasite-derived peptide FhHDM-1 activates the PI3K/Akt pathway to prevent cytokine-induced apoptosis of β-cells. J Mol Med (Berl) (2021) 99(11):1605–21. doi: 10.1007/s00109-021-02122-x
122. Garay-Malpartida HM, Mourão RF, Mantovani M, Santos IA, Sogayar MC, Goldberg AC. Toll-like receptor 4 (Tlr4) expression in human and murine pancreatic beta-cells affects cell viability and insulin homeostasis. BMC Immunol (2011) 12(1):18. doi: 10.1186/1471-2172-12-18
123. Li M, Song L, Gao X, Chang W, Qin X. Toll-like receptor 4 on islet β Cells senses expression changes in high-mobility group box 1 and contributes to the initiation of type 1 diabetes. Exp Mol Med (2012) 44(4):260–7. doi: 10.3858/emm.2012.44.4.021
124. Surendar J, Indulekha K, Hoerauf A, Hübner MP. Immunomodulation by helminths: similar impact on type 1 and type 2 diabetes? Parasite Immunol (2017) 39(5). doi: 10.1111/pim.12401
125. O'Brien BA, Huang Y, Geng X, Dutz JP, Finegood DT. Phagocytosis of apoptotic cells by macrophages from nod mice is reduced. Diabetes (2002) 51(8):2481–8. doi: 10.2337/diabetes.51.8.2481
126. Lecube A, Pachón G, Petriz J, Hernández C, Simó R. Phagocytic activity is impaired in type 2 diabetes mellitus and increases after metabolic improvement. PloS One (2011) 6(8):e23366. doi: 10.1371/journal.pone.0023366
127. Alvarado R, To J, Lund ME, Pinar A, Mansell A, Robinson MW, et al. The immune modulatory peptide fhhdm-1 secreted by the helminth fasciola hepatica prevents nlrp3 inflammasome activation by inhibiting endolysosomal acidification in macrophages. FASEB J (2017) 31(1):85–95. doi: 10.1096/fj.201500093R
128. Rajamanickam A, Munisankar S, Thiruvengadam K, Menon PA, Dolla C, Nutman TB, et al. Impact of helminth infection on metabolic and immune homeostasis in non-diabetic obesity. Front Immunol (2020) 11:2195. doi: 10.3389/fimmu.2020.02195
129. Kokova D, Mayboroda OA. Twenty years on: metabolomics in helminth research. Trends Parasitol (2019) 35(4):282–8. doi: 10.1016/j.pt.2019.01.012
130. Miani M, Le Naour J, Waeckel-Enée E, Verma SC, Straube M, Emond P, et al. Gut microbiota-stimulated innate lymphoid cells support β-defensin 14 expression in pancreatic endocrine cells, preventing autoimmune diabetes. Cell Metab (2018) 28(4):557–72.e6. doi: 10.1016/j.cmet.2018.06.012
131. Pintarič M, Langerholc T. Probiotic mechanisms affecting glucose homeostasis: A scoping review. Life (Basel) (2022) 12(8):1187. doi: 10.3390/life12081187
132. Fernández-Millán E, Guillén C. Multi-organ crosstalk with endocrine pancreas: A focus on how gut microbiota shapes pancreatic beta-cells. Biomolecules (2022) 12(1):104. doi: 10.3390/biom12010104
133. Gesmundo I, Pardini B, Gargantini E, Gamba G, Birolo G, Fanciulli A, et al. Adipocyte-derived extracellular vesicles regulate survival and function of pancreatic β cells. JCI Insight (2021) 6(5):e141962. doi: 10.1172/jci.insight.141962
134. Li WZ, Stirling K, Yang JJ, Zhang L. Gut microbiota and diabetes: from correlation to causality and mechanism. World J Diabetes (2020) 11(7):293–308. doi: 10.4239/wjd.v11.i7.293
135. Yuan X, Wang R, Han B, Sun C, Chen R, Wei H, et al. Functional and metabolic alterations of gut microbiota in children with new-onset type 1 diabetes. Nat Commun (2022) 13(1):6356. doi: 10.1038/s41467-022-33656-4
136. Mayendraraj A, Rosenkilde MM, Gasbjerg LS. Glp-1 and gip receptor signaling in beta cells - a review of receptor interactions and co-stimulation. Peptides (2022) 151:170749. doi: 10.1016/j.peptides.2022.170749
137. Marzook A, Tomas A, Jones B. The interplay of glucagon-like peptide-1 receptor trafficking and signalling in pancreatic beta cells. Front Endocrinol (Lausanne) (2021) 12:678055. doi: 10.3389/fendo.2021.678055
138. Amamou A, O'Mahony C, Leboutte M, Savoye G, Ghosh S, Marion-Letellier R. Gut microbiota, macrophages and diet: an intriguing new triangle in intestinal fibrosis. Microorganisms (2022) 10(3):490. doi: 10.3390/microorganisms10030490
139. Ivanov II, Honda K. Intestinal commensal microbes as immune modulators. Cell Host Microbe (2012) 12(4):496–508. doi: 10.1016/j.chom.2012.09.009
140. Jang SE, Han MJ, Kim SY, Kim DH. Lactobacillus plantarum clp-0611 ameliorates colitis in mice by polarizing M1 to M2-like macrophages. Int Immunopharmacol (2014) 21(1):186–92. doi: 10.1016/j.intimp.2014.04.021
141. Yang Y, Wang X, Huycke T, Moore DR, Lightfoot SA, Huycke MM. Colon macrophages polarized by commensal bacteria cause colitis and cancer through the bystander effect. Transl Oncol (2013) 6(5):596–606. doi: 10.1593/tlo.13412
142. Llinás-Caballero K, Caraballo L. Helminths and bacterial microbiota: the interactions of two of humans' "old friends". Int J Mol Sci (2022) 23(21):13358. doi: 10.3390/ijms232113358
143. Loke P, Lim YA. Helminths and the microbiota: parts of the hygiene hypothesis. Parasite Immunol (2015) 37(6):314–23. doi: 10.1111/pim.12193
144. Pace F, Carvalho BM, Zanotto TM, Santos A, Guadagnini D, Silva KLC, et al. Helminth infection in mice improves insulin sensitivity via modulation of gut microbiota and fatty acid metabolism. Pharmacol Res (2018) 132:33–46. doi: 10.1016/j.phrs.2018.04.008
145. Khudhair Z, Alhallaf R, Eichenberger RM, Whan J, Kupz A, Field M, et al. Gastrointestinal helminth infection improves insulin sensitivity, decreases systemic inflammation, and alters the composition of gut microbiota in distinct mouse models of type 2 diabetes. Front Endocrinol (Lausanne) (2020) 11:606530. doi: 10.3389/fendo.2020.606530
146. Quattrin T, Haller MJ, Steck AK, Felner EI, Li Y, Xia Y, et al. Golimumab and beta-cell function in youth with new-onset type 1 diabetes. N Engl J Med (2020) 383(21):2007–17. doi: 10.1056/NEJMoa2006136
147. von Herrath M, Bain SC, Bode B, Clausen JO, Coppieters K, Gaysina L, et al. Anti-interleukin-21 antibody and liraglutide for the preservation of β-cell function in adults with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol (2021) 9(4):212–24. doi: 10.1016/s2213-8587(21)00019-x
148. Bluestone JA, Buckner JH, Herold KC. Immunotherapy: building a bridge to a cure for type 1 diabetes. Science (2021) 373(6554):510–6. doi: 10.1126/science.abh1654
Keywords: helminth, diabetes, macrophage, β-cells, Fasciola hepatica, FhHDM-1
Citation: Camaya I, O’Brien B and Donnelly S (2023) How do parasitic worms prevent diabetes? An exploration of their influence on macrophage and β-cell crosstalk. Front. Endocrinol. 14:1205219. doi: 10.3389/fendo.2023.1205219
Received: 13 April 2023; Accepted: 10 July 2023;
Published: 26 July 2023.
Edited by:
Pingping Han, The University of Queensland, AustraliaReviewed by:
Bruno Guigas, Leiden University, NetherlandsSamson Mukaratirwa, Ross University School of Veterinary Medicine, Saint Kitts and Nevis
Copyright © 2023 Camaya, O’Brien and Donnelly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheila Donnelly, U2hlaWxhLkRvbm5lbGx5QHV0cy5lZHUuYXU=