 Haiyan Zhu
Haiyan Zhu Xin Wang2
Xin Wang2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 06 June 2023
Sec. Cancer Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1195500
This article is part of the Research Topic Tumor Metabolism and Programmed Cell Death View all 5 articles
Clear cell renal cell carcinoma (ccRCC) is a malignancy that exhibits metabolic reprogramming as a result of genetic mutations. This reprogramming accommodates the energy and anabolic needs of the cancer cells, leading to changes in glucose, lipid, and bio-oxidative metabolism, and in some cases, the amino acid metabolism. Recent evidence suggests that ccRCC may be classified as a metabolic disease. The metabolic alterations provide potential targets for novel therapeutic interventions or biomarkers for monitoring tumor growth and prognosis. This literature review summarized recent discoveries of metabolic alterations in ccRCC, including changes in glucose, lipid, and amino acid metabolism. The development of metabolic drugs targeting these metabolic pathways was also discussed, such as HIF-2α inhibitors, fatty acid synthase (FAS) inhibitors, glutaminase (GLS) inhibitors, indoleamine 2,3-dioxygenase (IDO) inhibitors, and arginine depletion. Future trends in drug development are proposed, including the use of combination therapies and personalized medicine approaches. In conclusion, this review provides a comprehensive overview of the metabolic alterations in ccRCC and highlights the potential for developing new treatments for this disease.
Cancer is a disease characterized by uncontrolled cell growth, and to sustain this growth, cancer cells acquire large amounts of energy by altering their metabolic pathways (1, 2). Initially, it was thought that the enhanced glycolysis observed in tumor cells was inefficient in generating energy (3). However, it is now clear that cancer and metabolism are intricately linked, and metabolic reprogramming is a hallmark of cancer. This process involves genetic mutations that alter various metabolic processes, including glucose and lipid metabolism and oxidative phosphorylation (OXPHOS). In other words, cancer cells adapt their metabolism to meet their energy and building block needs (4). The increased glycolysis rate of cancer cells consumes most of the nutrients in the surrounding microenvironment. This metabolic restriction significantly promotes the formation of the tumor microenvironment and reduces the responsiveness of T cells (5).
Renal clear cell carcinoma (ccRCC) is a malignant tumor that arises from the renal tubular epithelium and accounts for 75% of all kidney cancer cases (6, 7). According to the American Cancer Society’s estimates for 2022, approximately 79,000 new cases of kidney cancer will be diagnosed in the United States with ccRCC being the most prevalent subtype (8). This malignant tumor has a poor prognosis, particularly when metastases occur. Surgical resection is often insufficient to manage the disease, and treatment decisions may involve targeted drug therapy, immunotherapy combined with targeted drug therapy, or dual immunotherapy combined (9). Evidence suggests that these therapies can extend the overall and progression-free survival of patients, but they are also associated with the occurrence of adverse events (10, 11).Currently, the International Metastatic Renal Cell Carcinoma Database score (IMDC score) is utilized to guide treatment decisions by providing prognostic stratification.
Metabolic reprogramming is a key feature of ccRCC, and altered gene expressions can lead to significant metabolic changes that fuel tumor growth (12). In particular, ccRCC is associated with abnormalities in glucose metabolism and OXPHOS, as well as unique metabolic alterations such as glutamine addiction (13). The Warburg effect, in which cancer cells preferentially use glycolysis rather than OXPHOS for energy production, is also commonly observed in ccRCC (14).
Understanding these metabolic changes and their impact on tumor growth is essential for developing new and effective treatments for ccRCC. In this article, we provided an overview of ccRCC and the metabolic alterations that occur in the disease, focusing on glucose metabolism, lipid metabolism, and OXPHOS. We also reviewed the pharmacological treatments that have been developed to target these metabolic modifications and discussed potential future directions for drug development in ccRCC.
Metabolic reprogramming provides tumors with a competitive advantage in acquiring survival resources (15). The Warburg effect is a prominent example of metabolic reprogramming in cancer cells, promoting cancer cell proliferation and creating an acidic environment that can facilitate cancer cell migration (16, 17). Glutamine metabolism and enhanced pentose phosphate pathway (PPP) promote macromolecular production and signal transmission in cancer cells (18). Furthermore, the hypoxic signaling pathway can activate neovascularization to facilitate tumor cell proliferation, even in the hypoxic environments surrounding the tumor cells (19). Despite these advantages, modulating the metabolic pathways of cancer cells can limit their metabolic dominance and prevent the development of ccRCC (4).
ccRCC is associated with changes in lipid and glucose metabolism, as evidenced by the presence of the translucent cytoplasm and significant lipid and glycogen accumulation observed in these cells (20).
Most cases of ccRCC arise from mutations in the Von Hippel-Lindau (VHL) allele, which results in altered tumor metabolism in 90% of patients with ccRCC (4). The VHL oncogene, located on the short arm of chromosome 3 encodes the Von Hippel-Lindau(VHL) tumor suppressor protein (pVHL) (21).
Under physiological conditions, pVHL ubiquitinates proline-containing residues in the oxygen-dependent degradation domains (ODDs) of hypoxia-inducible factors (HIFs), promoting their proteasomal degradation (22, 23). HIFs are transcription factors that regulate cellular adaptation to hypoxic environments (24). HIF-1α is the most commonly expressed HIF family member in cells and tissues (25), and its stability is negatively regulated by oxygen levels (26), and cellular metabolism. However, under the pathological hypoxic conditions, HIF-2α is less likely to degrade and instead forms a heterodimer HIF-1β, also known as the aromatic hydrocarbon receptor nuclear transporter (ARNT), that enters the nucleus and activates the transcription of numerous genes (27).
In ccRCC, mutations in VHL result in the accumulation of HIF-2α, which creates a state of “pseudo-hypoxia” (28) and induces metabolic changes, including angiogenesis, epithelial-mesenchymal transition, invasion, and metastatic spread (29). Recent research indicates that HIF-1α functions as a tumor suppressor, while HIF-2α acts as an oncogene in the biology of ccRCC (30).
Glucose metabolism is a multifaceted process of paramount importance for cellular energy production. The first and foremost step of glucose metabolism is glycolysis, which catalyzes the breakdown of glucose to yield pyruvate (31). Pyruvate is then subjected to aerobic oxidation via the tricarboxylic acid (TCA) cycle, leading to the generation of ATP (32), reduced nicotinamide adenine dinucleotide (NADH), and reduced flavin adenine dinucleotide (FADH2) or anaerobic fermentation to produce lactate and ATP (33). In addition, the pentose phosphate pathway (PPP) contributes to the production of glucose for lipid metabolism and nucleic acid synthesis by generating reduced nicotinamide adenine dinucleotide phosphate (NADPH) and ribose 5-phosphate (34).
In ccRCC, glucose metabolism undergoes significant alterations due to the pivotal role played by HIF-1α. HIF-1α stimulates lactate production and reduces pyruvate entry into the mitochondria, leading to diminished TCA cycle activity and ATP production (35). The Warburg effect, a metabolic reprogramming phenomenon observed in cancer cells, is characterized by decreased pyruvate entry into the mitochondria and increased lactate production, regardless of oxygen levels and ATP production efficiency (4). HIF-1α facilitates the expression of BCL2 Interacting Protein 3 (BNIP3), which decelerates mitochondrial metabolic activities (36) and inhibits the enzymatic activity of pyruvate dehydrogenase, thereby hindering the conversion of pyruvate to acetyl coenzyme A and promoting lactate production (37). Additionally, in ccRCC, HIF-1α upregulates the expression of glycolytic enzymes, such as hexokinase (HK), neuron-specific enolase (NSE), phosphoglycerate kinase (PGK), and pyruvate kinase (PK), further corroborating the Warburg effect (38, 39). Among these up-regulated key enzymes, HK2 is one of the key factors involved in the development of a variety of human cancers. Some literature shows that the high expression of HK2 is positively correlated with the advanced tumor, lymph node metastasis and the worst survival rate of renal cancer patients. The high expression of HK2 has been identified as an independent risk factor for RCC; It also shows a positive correlation with immune cell infiltration and prognosis in kidney cancer patients, playing an important role in the occurrence and development of cancer (40).
Glucose metabolism involves the pentose phosphate pathway (PPP), which is a source of NADPH and pentose phosphate (41). The PPP is upregulated in ccRCC to produce more NADPH, which is crucial for maintaining redox homeostasis and protecting against cellular damage caused by reactive oxygen species (ROS) (42). In ccRCC, the PPP is upregulated to produce more NADPH to counteract oxidative stress and lessen the harm caused by excess ROS to the tumor cells (43). Moreover, the pentose phosphate in the PPP also satisfies the high demand for 5-carbon sugars required for nucleotide biosynthesis (44).
In the TCA cycle of ccRCC, enzymes that replenish metabolic fluxes from other pathways are often downregulated (41). Citrate and cis-aconitate levels markedly increase in the TCA cycle of ccRCC, while malate and fumarate levels significantly decrease (13). These latter two decreases are linked to a reduction in succinate dehydrogenase (SDH), leading to a constant depletion of fumarate, and then malate (41), which contradicts the common perception that tumor tissues exhibit high levels of fumarate.
HIF-1α enhances glucose uptake by activating the glucose transporter protein (GLUT) gene and inhibits mitochondrial respiratory function by controlling the expression of microRNAs such as miR-210 (45). In contrast, HIF-2α in ccRCC primarily controls target genes related to glycolysis while also interact with several important oncogenes (Figure 1). Furthermore, it stimulates MYC and P53 activity and boosts the expression of cell cycle regulators (46), thus highlighting its critical role in the development of ccRCC (47). Notably, the selective antagonists of HIF-2α have demonstrated clinical responses in several clinical trials, particularly targeting xenograft tumor models in ccRCC (48).
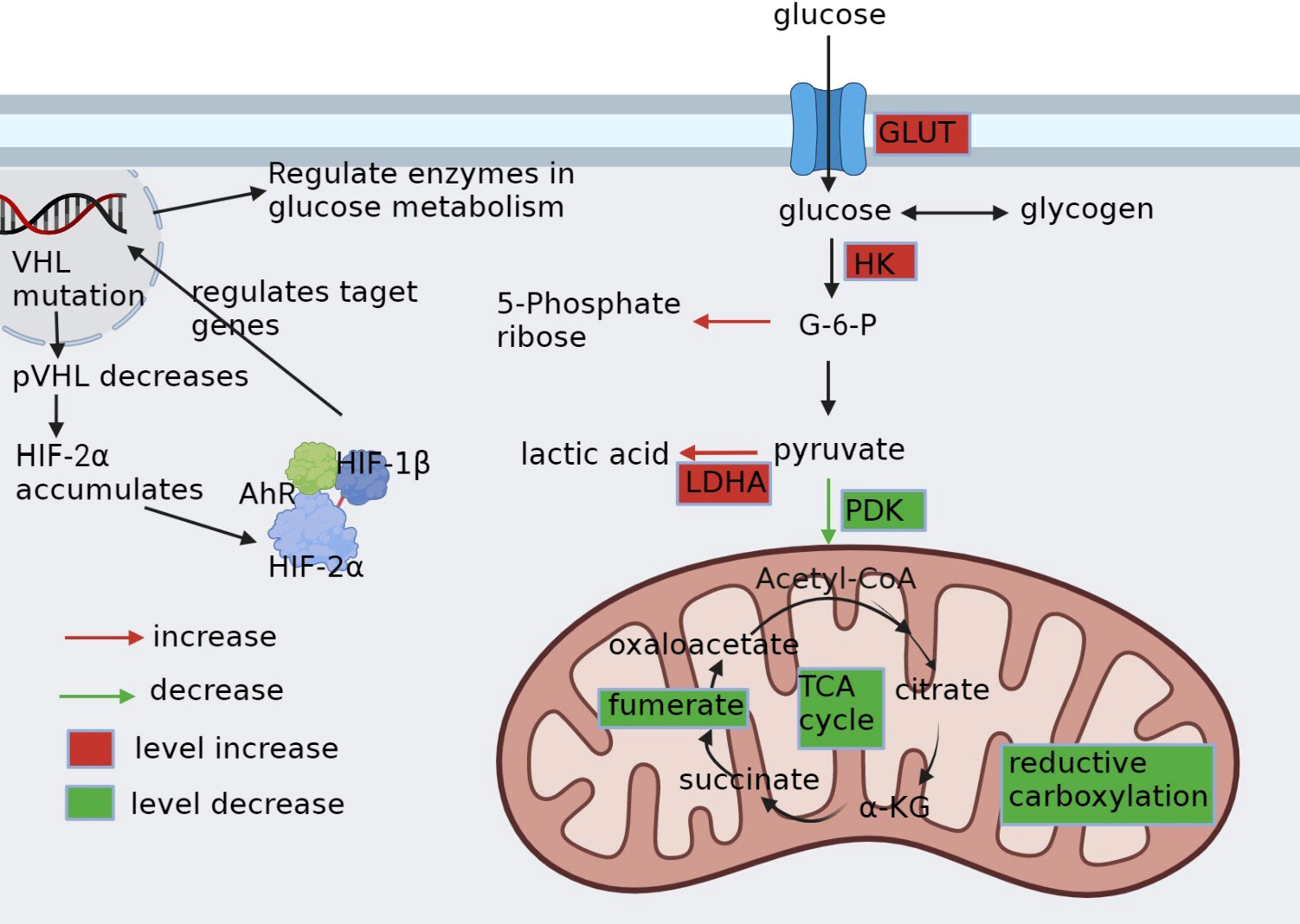
Figure 1 Glucose metabolic reprogramming in ccRCC. pVHL, the product of VHL gene mutation, is reduced. HIF-2α, which depends on pVHL for proteasomal degradation, accumulates intracellularly and binds to HIF-1β and AhR into the nucleus, as well as regulates the expression of carriers and enzyme genes related to glycolysis. It alters the whole glucose metabolism in cancer cells by upregulating glucose transporter protein GLUT, upregulating enzymes HK and LDHA related to glycolysis, downregulating enzyme PDK1 of aerobic glucose metabolism, promoting pentose phosphate pathway, and disrupting metabolites in the tricarboxylic acid cycle.
Recent research results indicate that the Warburg effect promotes tumor growth to a certain extent, but tumor growth is fundamentally dependent on functional mitochondria. In addition to having basic bioenergy functions, mitochondrial metabolism controls redox balance and coordinates cell death. Therefore, mitochondria are promising targets for developing new anticancer drugs (49).
In ccRCC, the TCA cycle undergoes changes. Firstly, there is a reduction in pyruvate entering the mitochondria, followed by changes in key enzymes such as fumarate hydratase (FH) and succinate dehydrogenase (SDH) in the TCA cycle, resulting in imbalanced metabolite content (50). Citrate and aconite levels are significantly higher, while succinate and malic acid saline levels are significantly lower (41). In addition, enzymes that supplement metabolic flux to the TCA cycle through other pathways are typically downregulated (50).
Oxidative phosphorylation (OXPHOS) is a major process in which human cells generate energy in the form of ATP. However, due to changes in the TCA cycle, ccRCC is associated with a decrease in OXPHOS activity (41). The VHL gene mutation in ccRCC leads to the accumulation of HIF-1α, which impairs the oxidation metabolism of glucose and suppresses pyruvate entry into the mitochondria, thereby reducing electron transfer efficiency (13, 51). The downregulation of complex-V and the repression of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) in ccRCC also contribute to the impaired OXPHOS (52). PGC-1α, a critical transcriptional coactivator of PPARγ, regulates mitochondrial biogenesis and respiration, and its inhibition leads to delayed respiration, reduced mitochondrial transcription factor A (TFAM) expression, and unfavorable prognosis of ccRCC (53).
In addition to HIF-1α, HIF-2α plays a role in the OXPHOS alterations in ccRCC. HIF-2α is associated with the upregulation of antioxidant genes, reducing reactive oxygen species (ROS), decreasing DNA damage from excess ROS, and promoting tumor cell survival (54). These changes in OXPHOS have implications for tumor growth and potential therapeutic targets, including the development of selective HIF-2α antagonists, which have demonstrated clinical efficacy in treating ccRCC (55). Therefore, understanding the mechanisms underlying the impaired OXPHOS in ccRCC could lead to the development of novel treatments for this aggressive cancer.
Mitochondria can promote malignant transformation through three main mechanisms, including (1) mitochondrial reactive oxygen species (ROS) support the accumulation of carcinogenic DNA changes and activation of carcinogenic pathways to some extent;(2) abnormal accumulation in specific mitochondrial metabolites, including fumarate, succinate, and 2-hydroxyglutarate (2HG); (3) functional deficits in MOMP or mitochondrial permeability transition (MPT) are usually necessary for the formation and survival of malignant precursors (49, 56).
Due to the high glycolysis rate of cancer cells and the gradual consumption of oxygen in the local environment, the ratio of NADH to NAD+in cancer cells increases, the redox imbalance, and the production of reactive oxygen species (ROS) increases (57). To prevent the accumulation of ROS, cells can resist oxidation through the thioredoxin system and glutathione system (58). In addition, when ROS levels increase, the main regulatory factor of antioxidant response, the nuclear factor erythroid 2-like 2 (NRF2), stabilizes because its negative regulatory factor, Kelch like ECH related protein 1 (KEAP1), is oxidized and loses its ability to chelate NRF2 in the cytosol for proteasome degradation (57). Excessive ROS production may exceed the antioxidant capacity of cancer cells, therefore cancer cells often upregulate their antioxidant defense mechanisms (59). In ccRCC, HIF-2 α Upregulation of antioxidant genes, reduction of reactive oxygen species (ROS), reduction of excessive ROS damage to DNA, and promotion of tumor cell survival (54).
Lipid metabolism is a functional process pivotal in energy storage and signal transduction (30). Dysregulation of lipid metabolism is a common abnormality in ccRCC (Figure 2), which is thought to contribute to the aggressive behavior of the tumor (60). In ccRCC, lipid synthesis and storage are upregulated, while utilization and oxidation are downregulated (61), resulting in the accumulation of cholesterol (62), fatty acids (13), and triglycerides (60). This lipid metabolism reprogramming promotes cell membrane synthesis and cell proliferation while inhibiting the β-oxidation of fatty acids (4). Specifically, increased cholesterol uptake by upregulated lipoprotein receptors, such as the very low-density lipoprotein receptor (VLDL-R) (63) and scavenger receptor B1 (SR-B1) (64), and the expression of acyl-coenzyme A:cholesterol acyltransferase (ACAT) contribute to the accumulation of cholesterol in ccRCC (62). In addition, HIF-2 α activating the expression of hypoxia induced lipid droplet associated protein (HILPDA) and selectively enriching polyunsaturated lipids (65).
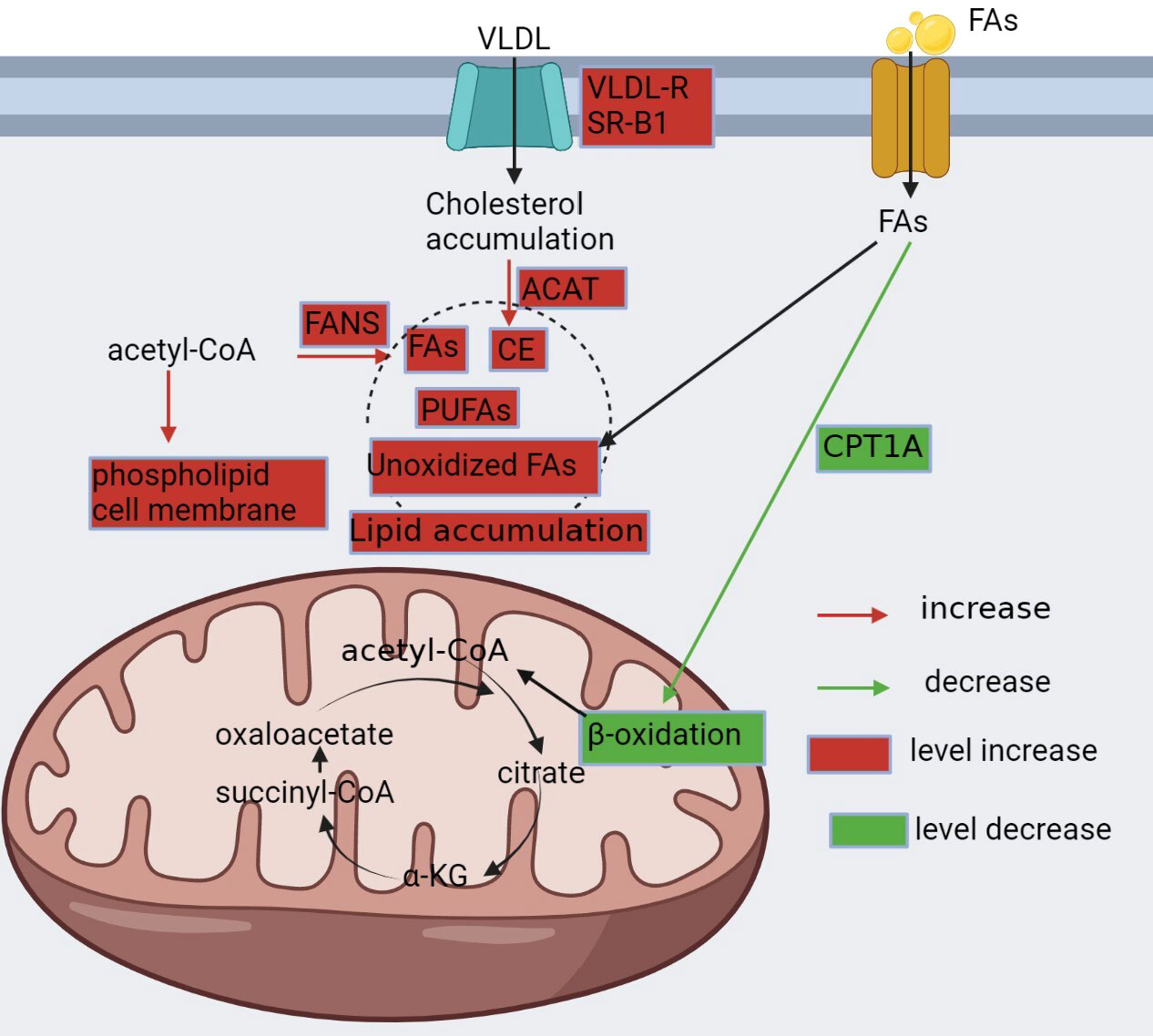
Figure 2 Reprogramming of fatty acid metabolism. In ccRCC, lipid metabolism is altered and characterized by increased synthesis, enhanced utilization, and decreased oxidation to meet the metabolic demands of cancer cells. Renal clear cells upregulate VLDLR and SR-B1 to promote cholesterol uptake, followed by upregulation of ACAT, which converts pure cholesterol into cholesteryl esters; downregulation of CPT1A, a key enzyme in fatty acid β-oxidation, which reduces fatty acid consumption; and upregulation of FASN, which promotes fatty acid synthesis. All of these contribute to the accumulation of lipids in cancer cells.
Furthermore, ccRCC exhibits fatty acid synthesis due to the upregulation of fatty acid synthase (FAS) (38). However, in contrast to other malignancies, ccRCC displays reduced fatty acid oxidation, which is primarily attributed to the inhibition of carnitine palmitoyltransferase 1A (CPT1A), the key enzyme for fatty acid oxidation (20). In addition, ccRCC shows an increase in the expression of fatty acid desaturase 1 (FADS1), a crucial enzyme in the metabolism of polyunsaturated fatty acids (PUFAs), leading to PUFA accumulation (60).
PUFA is one of the main targets of lipid peroxidation, and lipid peroxidation is a sign of ferroptosis. Studies have identified acyl-CoA synthetase long-chain family member 4 (ACSL4) as a key determinant of iron mortality sensitivity (66). After ACSL4 activation, lysophosphatidylcholine acyltransferase 3 (LPCAT3) is involved in ferrozotic signaling by inserting acyl groups in lysophospholipids, specifically phosphatidylcholine and phosphatidylethanolamine. It is important to note that iron death may also occur in a way that is not dependent on ACSL4. Pharmacologically induced ferroptosis in cancer cells is a promising anticancer strategy, although its role in tumors is still unclear (67).
Lipid metabolism is closely linked to glucose metabolism as glycerol and glucose metabolism are related through glycerol kinase and α-phosphoglycerol dehydrogenase, producing dihydroxyacetone phosphate (34). During glucose overload, acetyl coenzyme A, produced during glucose metabolism, and NADPH and H+ from the PPP are converted into fatty acids by FAS. Phospholipid and ketone body metabolism are also part of lipid metabolism (68). Notably, lipids not only provide energy but also function as signaling molecules that control cell growth and proliferation (30). Thus, targeting lipid metabolism has emerged as a potential therapeutic strategy for ccRCC. Identifying specific enzymes or signaling pathways involved in lipid metabolism could lead to the development of novel therapeutic approaches.
Glutamine is essential for maintaining the redox balance of normal cells by serving as a precursor for the production of α-ketoglutarate (α-KG) and glutathione, which are critical for intracellular redox homeostasis and the synthesis of other amino acids (18). Glutamine is transported into the cells via specialized transporter proteins, such as Solute Carrier Family 1 Member 5 (SLC1A5) (69), and is subsequently converted to glutamate by glutaminase (GLS) (70). In addition to participating in protein synthesis, glutamine also plays various synthetic and metabolic roles in cells, such as promoting the production of nucleotides, hexosamine units, and asparagine, participating in enhancing cellular oxidative stress defense, and consuming many essential amino acids (71). The activity of GLS is positively correlated with the rate of cell growth and malignancy, making it a potential target for anticancer therapies. Inhibition of GLS activity or expression prevent tumor growth (72). The altered cellular metabolism in ccRCC increases glutamine addiction and subsequent cellular pathways (73). The increased demand for glutamine is necessary to support the fast growth and proliferation of malignant cells (73). In addition, high levels of glutamic acid can damage the input of cystine, and can also lead to ROS imbalance and T cell dysfunction, thus forming a tumor microenvironment conducive to tumor survival (5).
In ccRCC, the upregulation of Solute Carrier Family 7 Member 5 (SLC7A5), an amino acid transporter that facilitates glutamine entry into cells, is controlled by HIF-2α, which is a hallmark of this malignancy (74). Once inside the cells, glutamine is converted to glutamate by GLS and further metabolized to α-KG by glutamate dehydrogenase (GDH), serving as the primary mechanism for carbon delivery to the TCA cycle that is essential for cellular lifespan (75). Additionally, the reductive carboxylation of glutamate produces isocitrate, which generates acetyl coenzyme A for lipid synthesis (18, 41).
Elevated glutamine levels in ccRCC are associated with enhanced glutamate production, which is a crucial mechanism for neutralizing ROS (76). Glutamine also promotes glycolysis, cell proliferation, and immortalization while decreasing cancer cell death by inhibiting the expression of thioredoxin-interacting protein[]. Therefore, the altered glutamine metabolism in ccRCC highlights its potential as a therapeutic target for treating this malignancy. Figure 3 illustrates the glutamine metabolism pathway.
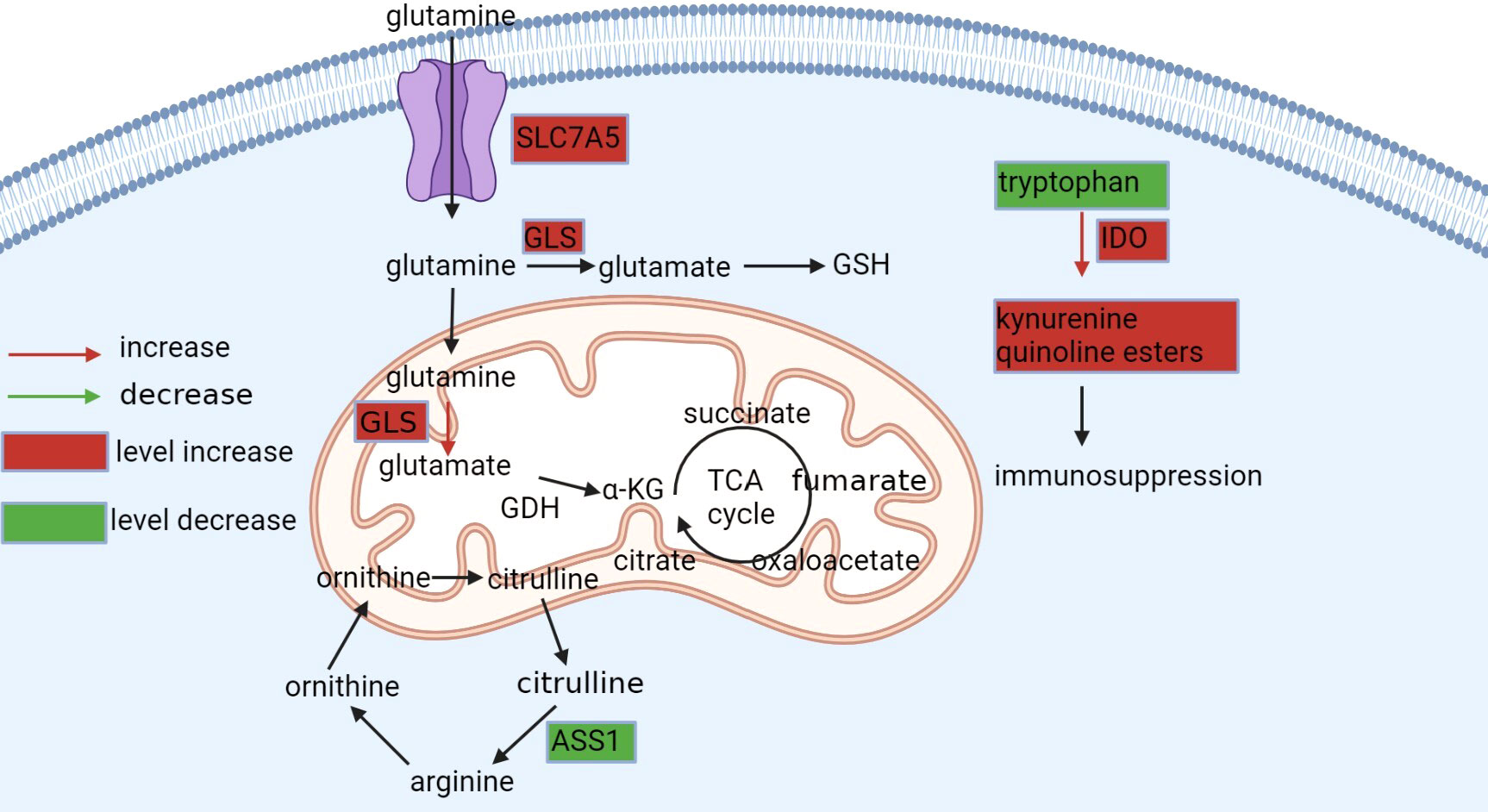
Figure 3 Metabolism of glutamine, arginine, and tryptophan. Glutamine is available intracellularly for various anabolic pathways. Tumor cells exhibit glutamine dependence in ccRCC, upregulating SLC7A5 glutamine transporter protein and GLS enzymes to take up more glutamine and promote its conversion to glutamate, which maintains the redox state in vivo or to α-KG, an alternative substrate for the tricarboxylic acid cycle and synthesis of lipids. Tryptophan is degraded in the kynurenine pathway to produce immunosuppressive products, while in ccRCC, this process is upregulated by IOD, leading to the activation of the entire pathway. Arginine is synthesized from citrulline via ASS1, but AAS1 is not expressed or downregulated in ccRCC and cannot synthesize this amino acid and can only be taken up from the blood.
Tryptophan plays a vital role in promoting T cell-mediated immune response against tumors. However, the excessive oxidation of tryptophan to kynurenine pathway metabolites leads to T-cell disfunction, allowing tumors to evade immune surveillance. Moreover, kynurenine pathway inhibit T-cell activation (77). In tumor-draining lymph nodes, overactive indoleamine 2,3-dioxygenase (IDO) promotes dendritic cells to directly suppress and inhibit T cells, thus impairing antigen responses and recognition (78).
In ccRCC, the dysregulation of immune checkpoint molecules has been linked to elevated indoleamine 2,3-dioxygenase (IDO) expression, which results in decreased levels of tryptophan and activation of the kynurenine pathway. This metabolic reprogramming promotes tumor cell proliferation by hindering the effectiveness of IFN-α therapy and inducing immunosuppression (79, 80). Furthermore, IDO overexpression is strongly associated with cancer metastasis. In vitro experiments utilizing lung cancer cells demonstrate that IDO overexpression increases cell viability, while IDO knockdown decreases cell viability. In animal models, injecting human lung cancer cells that overexpress IDO into mice increases metastases in the brain, liver, and bone (81) (Figure 3). These findings indicate that IDO may serve as a potential therapeutic target for ccRCC and other malignancies, highlighting the need for further research in this area.
In ccRCC, alterations in arginine metabolism can be observed, including changes in arginine transporters and enzymes such as arginase and arginine succinate synthase 1 (ASS1). Tumor cells often display a downregulation or absence of ASS1, the enzyme responsible for synthesizing arginine from citrulline. This leads to a dependence on exogenous arginine for cancer cell survival, which has been confirmed by proteomic profiling of biopsy samples from patients with ccRCC (Figure 3). Therefore, targeting arginine metabolism may be a viable strategy to inhibit cancer growth by depriving cancer cells of arginine[]. Moreover, studies have identified arginine deprivation as a promising therapeutic approach to induce selective cytotoxicity in ASS1-deficient tumors (82). Therefore, a better understanding of the altered arginine metabolism in ccRCC can provide insights into potential therapeutic targets for treating this cancer (83).
ccRCC is frequently associated with genetic mutations that lead to hypoxic alterations, with the most commonly mutated gene being VHL (84). VHL mutations lead to the intracellular accumulation of HIF-α, which in turn upregulates the expression of vascular endothelial growth factors (VEGFs) (19).
Previously, the main therapeutic approach for ccRCC was to target angiogenesis using VEGF receptor (VEGFR) or VEGF inhibitors like sunitinib (85). However, these inhibitors have limited efficacy and can cause adverse effects such as vascular toxicity and off-target effects (86).
As ccRCC is characterized by metabolic reprogramming, targeting specific enzymes or proteins implicated in dysregulated metabolic pathways has shown promise in developing drugs that can selectively kill tumor cells with minimal adverse effects on normal cells (87). Among these metabolic changes, the most classic is the increase in glycolysis. In ccRCC, we can use glycolysis inhibitors to inhibit tumor cells, following the treatment methods of hepatocellular carcinoma (15). Unfortunately, although early clinical studies have shown that targeting the glycolysis pathway as a treatment method to inhibit cancer progression is effective, there have been no clinical trials to confirm it (88), so there is no detailed explanation in this article. Table 1 and Figure 4 provide an overview of the specific enzymes and proteins targeted in this manner. Focusing on these dysregulated metabolic pathways offers the potential to develop more effective and better-tolerated treatment options for patients with ccRCC (41, 87).
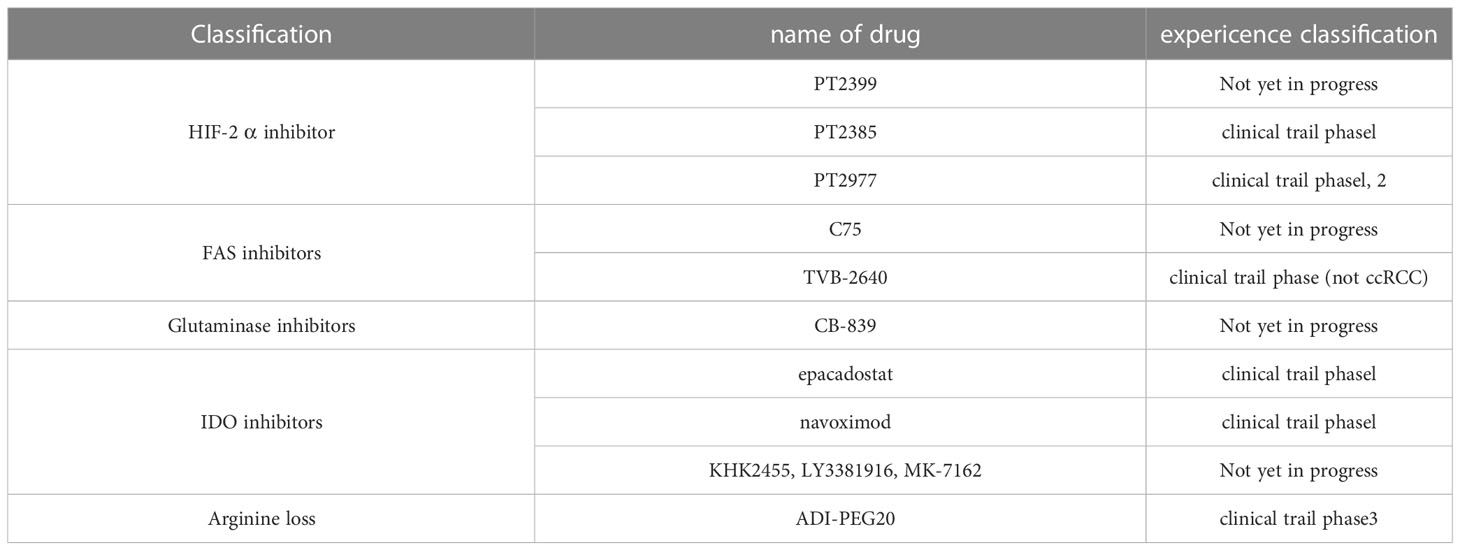
Table 1 Metabolic drug for ccRCC.
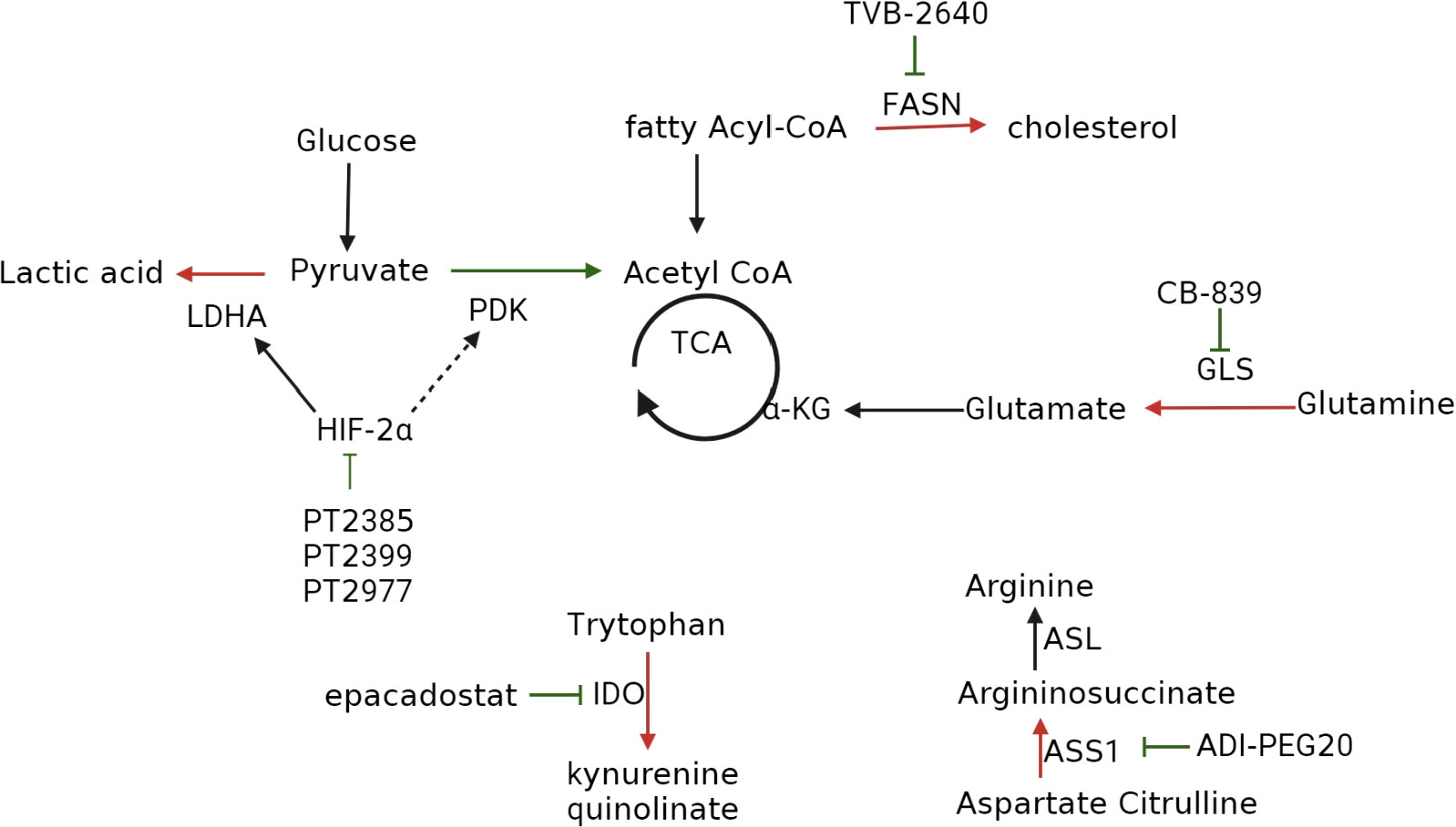
Figure 4 Inhibitors that target metabolic reprogramming in ccRCC. Glycolysis can be inhibited with HIF-2α inhibitors such as PT2385, PT2399, and PT2977; FAS can be suppressed with FAS inhibitor TVB-2640; tryptophan metabolism can be inhibited with IDO inhibitors epacadostat, navoximod, KHK2455, LY3381916, MK-7162; glutamine metabolism can be inhibited using the GLS inhibitor CB-839; extracellular arginine can be depleted using ADI-PEG20.
Targeting the HIF-2α pathway represents a promising approach for treating ccRCC, as HIF-2α is a key downstream effector of the VHL tumor suppressor protein, which is frequently mutated in ccRCC (47, 89). HIF-2α promotes tumorigenesis and metastasis by regulating angiogenesis, cell proliferation, and metabolism, making it an attractive therapeutic target.
Although the HIF-2α pathway was once considered “undruggable” (90), recent technological advancements have revealed a structural defect in HIF-2α, specifically PAS-B, leading to the development of first-generation HIF-2α antagonists, including PT2399 and PT2385. These antagonists induce a conformational change in PAS-B, thereby impeding the formation of the HIF-2α/HIF-1β heterodimer, and represent a promising avenue for the treatment of ccRCC (91).
PT2399 has exhibited superior activity to sunitinib and has been effective against sunitinib-resistant tumors. However, its long-term use can lead to drug resistance through mutations in the HIF-2α binding site or HIF-1β second site inhibition mutations (92). Moreover, PT2399 does not affect the expression of HIF-2α target genes, which can prevent the segregation of HIF-2α and maintain the expression of the HIF-2β gene. In contrast, PT2385 has demonstrated encouraging results in a phase I trial, exhibiting good tolerance with no dose-limited toxicity or discontinuation of treatment due to adverse events. Remarkably, 2%, 12%, and 52% of patients achieved complete, partial, and stable remissions, respectively (48, 93).
Second-generation HIF-2α inhibitors, such as PT2977 (MK-6482, belzutifan), have been developed to overcome some of the limitations of first-generation compounds (94). PT2977 binds to HIF-2α at a site adjacent to the PAS-B domain and induces a conformational change that prevents HIF-2α from binding to its target genes. PT2977 has demonstrated excellent pharmacological properties, including low lipophilicity, high oral bioavailability, and a favorable safety profile. In a phase II clinical trial, patients with ccRCC who received 120 mg of PT2977 orally per day had an objective response rate of 49%. The main adverse events reported were grade 1 and grade 2, which can be managed with appropriate measures (95, 96).
In summary, selective HIF-2α antagonists a promising therapeutic strategy for treating ccRCC. Second-generation inhibitors such as PT2977 have shown improved efficacy and safety compared with first-generation compounds, highlighting their potential as a new class of anticancer agents.
ccRCC is characterized by specific genetic mutations that dysregulate cellular processes, including lipid metabolism. In particular, upregulation of FAS expression in ccRCC increases fatty acid levels that fuel the cancer cells and post-translationally modify proteins. Fatty acids also play a crucial role in maintaining redox homeostasis and energy levels, which are vital for tumor cell growth and survival (97). Given the importance of lipid metabolism in ccRCC, limiting the production of fatty acids is an effective strategy for treating this type of cancer.
Studies have shown a positive correlation between FAS expression and tumor aggressiveness and a negative correlation with prognosis in ccRCC (98). Hence, the use of FAS inhibitors could be a promising treatment strategy for ccRCC. Preclinical experiments have demonstrated the inhibitory effect of C75, a FAS inhibitor, on the aggressiveness and proliferation of ccRCC (99).
TVB-2640 is a noval FAS inhibitor that has shown great promise in clinical studies (100). In a phase I clinical trial, TVB-2640 effectively reduced fatty acid production in patients with non-small cell lung cancer. In subsequent clinical trials for breast and ovarian cancer, TVB-2640 exhibited favorable clinical activity and safety, with no gastrointestinal or serum toxicity observed (101, 102). Adverse events were mainly mild and included skin and eye effects, which were manageable with appropriate measures. TVB-2640 is currently being evaluated in multiple ongoing clinical trials for various types of cancer, including ccRCC.
Overall, these findings suggest that FAS inhibitors such as TVB-2640 represent a promising therapeutic approach for ccRCC, and ongoing clinical trials will provide further insight into their effectiveness and safety.
Glutamine plays a critical role in energy production, maintenance of redox stability, and macromolecule synthesis in cancer cells, making it an attractive target for clinical cancer treatment. In ccRCC, GLS functions as a compensatory mechanism to restore the TCA cycle and stimulate cell proliferation to a lesser extent (4). CB-839 is a GLS inhibitor that has shown promising results in preclinical studies. When combined with Everolimus, an mTOR inhibitor commonly used in the treatment of ccRCC, it has been shown to enhance antitumor activity in animal models. While there is a lack of clinical trials exploring this potential treatment strategy for ccRCC, the preclinical data suggests that it could be an effective approach (103).
Therefore, it is important to conduct clinical trials to explore the effectiveness and safety of combining CB-839 with Everolimus for the treatment of ccRCC. Such trials will provide valuable insights into the potential of this treatment strategy and whether it can be used as an effective therapeutic approach for ccRCC patients.
IDO is an enzyme that plays a role in the catabolism of tryptophan via the kynurenine pathway. By depleting tryptophan and activating T cells, IDO inhibits the immunosuppressive effect in the local tumor microenvironment and suppresses antitumor T cells, thereby promoting tumor metastasis (80). Therefore, IDO has emerged as a potential therapeutic target for cancer treatment.
Epacadostat, a selective IDO-targeted inhibitor, has shown promising results in preclinical trials by improving the lysis of tumor antigen-specific T cells (104). However, its efficacy in clinical trials has been disappointing, with some studies reporting adverse effects such as toxicity and lack of efficacy. Although phase I clinical trials have demonstrated promising antitumor activity against various advanced solid tumors with the combination of Epacadostat and the PD-1 inhibitor pembrolizumab, further investigation is necessary to determine its optimal use and potential limitations (93, 105).
Navoximod is another IDO inhibitor. It is well tolerated at a dose of 800 mg BID, with rapid absorption and moderate bioavailability. Despite its promising pharmacokinetic profile, Navoximod as a monotherapy has demonstrated only modest efficacy against tumors (106, 107). However, a combination therapy of Navoximod and Atezolizumab has demonstrated acceptable safety and observed antitumor activity. Nonetheless, it remains uncertain whether adding Navoximod to Atezolizumab confers any additional benefits to patients (108).
To fully activate the host immune system, a combined regimen that involves one or more specific immunotherapeutic agents may be required, as IDO inhibitors have demonstrated limited efficacy as monotherapies (106). Several other IDO inhibitors, such as KHK2455, LY3381916, and MK-7162, are undergoing clinical trials to assess their safety, tolerability, and antitumor activity (78). These agents have different mechanisms of action and pharmacokinetic profiles, which may result in improved clinical outcomes when combined with other immunotherapeutic drugs. Further investigations are needed to fully elucidate the optimal combinations and dosing regimens for these agents in different cancer types and patient populations.
Arginine dystrophy is a metabolic alteration in some tumors where cells become addicted to extracellular arginine supply due to the absence or low levels of ASS1 (109, 110). Arginine is essential for metabolic pathways, including nitric oxide synthesis and protein biosynthesis (111). In ccRCC, the use of polyethylene glycol form of arginine deaminase (ADI-PEG20) can lower circulating arginine levels by catabolizing arginine to citrulline, thereby limiting tumor growth. However, this therapeutic approach may be limited by the re-expression of ASS1 (4).
Clinical trials have demonstrated the safety, tolerability, and clinical efficacy of ADI-PEG20 in reversing medication resistance in patients with arginine-dystrophic tumors (112). Furthermore, promising results have been reported in clinical studies evaluating ADI-PEG20 as a potential therapy for various cancer types, such as non-small cell lung cancer, acute myeloid leukemia, and uveal melanoma (113, 114).
Considering the potential of arginine depletion as a therapeutic strategy, further studies are warranted to optimize its clinical use in combination with other cancer therapies and investigate the mechanisms of resistance to this approach.
Clear cell renal cell carcinoma (ccRCC) is characterized by significant metabolic reprogramming, leading to altered energy requirements and redox homeostasis. Cancer cells predominantly utilize anaerobic glycolysis and HIF-driven lactate metabolism, while efficient TCA cycling is downregulated. NADPH synthesis in the PPP is upregulated to protect against ROS and nucleotide damage. The high lactate content of the tumor microenvironment promotes immunosuppression and migration, while tryptophan breakdown leads to increased synthesis of immunosuppressive kynurenine metabolites. Lipid synthesis and utilization are elevated, whereas lipid oxidation is suppressed in ccRCC. However, glutamine absorption is increased to produce fatty acids and counteract oxidative stress and ROS. Further research is needed to fully understand the complex metabolic alterations in ccRCC and develop effective therapeutic strategies targeting these pathways.
The development of drugs that specifically target abnormal metabolism represents a promising avenue for the treatment of ccRCC, which currently has limited therapeutic options in clinical practice. Targeting enhanced or altered metabolic pathways that selectively affect proliferating cancer cells while sparing normal cells is a feasible strategy for the development of novel therapies for ccRCC, given its unique metabolic properties.
However, there are potential drawbacks to targeting metabolic pathways, including the possibility of affecting other rapidly proliferating cells, and the effectiveness of antimetabolic drugs is related to cancer cell mutation patterns.
Nonetheless, the benefits of developing antitumor cell metabolism drugs outweigh the drawbacks. Anti-metabolites may offer a more favorable safety profile for ccRCC compared with traditional anti-angiogenic medications, potentially reducing cardiovascular side effects. Furthermore, metabolomics can aid in the discovery of targeted treatments for ccRCC. With the anticipated emergence of new techniques in novel metabolism, we expect to see an increase in therapeutic options for treating ccRCC in the near future.
HZ wrote the manuscript. XW and SL made a revised version. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
2. Vazquez A, Kamphorst JJ, Markert EK, Schug ZT, Tardito S, Gottlieb E. Cancer metabolism at a glance. J Cell Sci (2016) 129:3367–73. doi: 10.1242/jcs.181016
3. Warburg O. On the origin of cancer cells. Science (1956) 123:309–14. doi: 10.1126/science.123.3191.309
4. Wettersten HI, Aboud OA, Lara PN Jr., Weiss RH. Metabolic reprogramming in clear cell renal cell carcinoma. Nat Rev Nephrol (2017) 13:410–9. doi: 10.1038/nrneph.2017.59
5. Cassim S, Pouyssegur J. Tumor microenvironment: a metabolic player that shapes the immune response. Int J Mol Sci (2019) 21:2–3. doi: 10.3390/ijms21010157
6. Hu SL, Chang A, Perazella MA, Okusa MD, Jaimes EA, Weiss RH. The nephrologist's tumor: basic biology and management of renal cell carcinoma. J Am Soc Nephrol (2016) 27:2227–37. doi: 10.1681/ASN.2015121335
7. Szymanski Ł., Helbrecht I, Fiedorowicz M, Matak D, Bartnik E, Golik P, et al. Cancer stem cells in renal carcinoma. Postepy Biochem (2019) 65:95–102. doi: 10.18388/pb.2019_250
8. Siddiqi A, Rani M, Bansal P, Rizvi MMA. Renal cell carcinoma management: a step to nano-chemoprevention. Life Sci (2022) 308:120922. doi: 10.1016/j.lfs.2022.120922
9. Barata PC, Rini BI. Treatment of renal cell carcinoma: current status and future directions. CA Cancer J Clin (2017) 67:507–24. doi: 10.3322/caac.21411
10. Motzer RJ, Powles T, Burotto M, Escudier B, Bourlon MT, Shah AY, et al. Nivolumab plus cabozantinib versus sunitinib in first-line treatment for advanced renal cell carcinoma (CheckMate 9ER): long-term follow-up results from an open-label, randomised, phase 3 trial. Lancet Oncol (2022) 23:888–98. doi: 10.1016/S1470-2045(22)00290-X
11. Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med (2019) 380:1116–27. doi: 10.1056/NEJMoa1816714
12. Jonasch E, Walker CL, Rathmell WK. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat Rev Nephrol (2021) 17:245–61. doi: 10.1038/s41581-020-00359-2
13. Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, et al. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell (2016) 29:104–16. doi: 10.1016/j.ccell.2015.12.004
14. Fu Q, Xu L, Wang Y, Jiang Q, Liu Z, Zhang J, et al. Tumor-associated macrophage-derived interleukin-23 interlinks kidney cancer glutamine addiction with immune evasion. Eur Urol (2019) 75:752–63. doi: 10.1016/j.eururo.2018.09.030
15. Cassim S, Raymond VA, Dehbidi-Assadzadeh L, Lapierre P, Bilodeau M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle (2018) 17:903–16. doi: 10.1080/15384101.2018.1460023
16. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci (2016) 41:211–8. doi: 10.1016/j.tibs.2015.12.001
17. Cassim S, Raymond VA, Lacoste B, Lapierre P, Bilodeau M. Metabolite profiling identifies a signature of tumorigenicity in hepatocellular carcinoma. Oncotarget (2018) 9:26868–83. doi: 10.18632/oncotarget.25525
18. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest (2013) 123:3678–84. doi: 10.1172/JCI69600
19. Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer (2011) 12:9–22. doi: 10.1038/nrc3183
20. Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat Commun (2017) 8:1769. doi: 10.1038/s41467-017-01965-8
21. Linehan WM, Schmidt LS, Crooks DR, Wei D, Srinivasan R, Lang M, et al. The metabolic basis of kidney cancer. Cancer Discovery (2019) 9:1006–21. doi: 10.1158/2159-8290.CD-18-1354
22. Chitrakar A, Budda SA, Henderson JG, Axtell RC, Zenewicz LA. E3 ubiquitin ligase Von hippel-lindau protein promotes Th17 differentiation. J Immunol (2020) 205:1009–23. doi: 10.4049/jimmunol.2000243
23. Chappell JC, Payne LB, Rathmell WK. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J Clin Invest (2019) 129:442–51. doi: 10.1172/JCI120855
24. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol (2020) 21:268–83. doi: 10.1038/s41580-020-0227-y
25. Al Tameemi W, Dale TP, Al-Jumaily RMK, Forsyth NR. Hypoxia-modified cancer cell metabolism. Front Cell Dev Biol (2019) 7:4. doi: 10.3389/fcell.2019.00004
26. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest (2013) 123:3664–71. doi: 10.1172/JCI67230
27. Choudhry H, Harris AL. Advances in hypoxia-inducible factor biology. Cell Metab (2018) 27:281–98. doi: 10.1016/j.cmet.2017.10.005
28. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von hippel-lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292:468–72. doi: 10.1126/science.1059796
29. Motzer RJ, Banchereau R, Hamidi H, Powles T, McDermott D, Atkins MB, et al. Molecular subsets in renal cancer determine outcome to checkpoint and angiogenesis blockade. Cancer Cell (2020) 38:803–817.e4. doi: 10.1016/j.ccell.2020.10.011
30. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
31. Adeva-Andany MM, Pérez-Felpete N, Fernández-Fernández C, Donapetry-García C, Pazos-García C. Liver glucose metabolism in humans. Biosci Rep (2016) 36:8. doi: 10.1042/BSR20160385
33. Han HS, Kang G, Kim JS, Choi BH, Koo SH. Regulation of glucose metabolism from a liver-centric perspective. Exp Mol Med (2016) 48:e218. doi: 10.1038/emm.2015.122
34. Wanders RJ, Vreken P, Ferdinandusse S, Jansen GA, Waterham HR, van Roermund CW, et al. Peroxisomal fatty acid alpha- and beta-oxidation in humans: enzymology, peroxisomal metabolite transporters and peroxisomal diseases. Biochem Soc Trans (2001) 29:250–67. doi: 10.1042/bst0290250
35. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
36. Mylonis I, Simos G, Paraskeva E. Hypoxia-inducible factors and the regulation of lipid metabolism. Cells (2019) 8(3):214. doi: 10.3390/cells8030214
37. Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate metabolism in human lung tumors. Cell (2017) 171:358–371.e9. doi: 10.1016/j.cell.2017.09.019
38. Xie H, Simon MC. Oxygen availability and metabolic reprogramming in cancer. J Biol Chem (2017) 292:16825–32. doi: 10.1074/jbc.R117.799973
39. Nakazawa MS, Keith B, Simon MC. Oxygen availability and metabolic adaptations. Nat Rev Cancer (2016) 16:663–73. doi: 10.1038/nrc.2016.84
40. Liu C, Li H, Huang H, Zheng P, Li Z. The correlation of HK2 gene expression with the occurrence, immune cell infiltration, and prognosis of renal cell carcinoma. Dis Markers (2022) 2022:1452861. doi: 10.1155/2022/1452861
41. Chakraborty S, Balan M, Sabarwal A, Choueiri TK, Pal S. Metabolic reprogramming in renal cancer: events of a metabolic disease. Biochim Biophys Acta Rev Cancer (2021) 1876:188559. doi: 10.1016/j.bbcan.2021.188559
42. Nogueira V, Hay N. Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin Cancer Res (2013) 19:4309–14. doi: 10.1158/1078-0432.CCR-12-1424
43. Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J (2009) 418:29–37. doi: 10.1042/BJ20081258
44. Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell (2014) 5:592–602. doi: 10.1007/s13238-014-0082-8
45. Ivan M, Huang X. miR-210: fine-tuning the hypoxic response. Adv Exp Med Biol (2014) 772:205–27. doi: 10.1007/978-1-4614-5915-6_10
46. Schödel J, Grampp S, Maher ER, Moch H, Ratcliffe PJ, Russo P, et al. Hypoxia, hypoxia-inducible transcription factors, and renal cancer. Eur Urol (2016) 69:646–57. doi: 10.1016/j.eururo.2015.08.007
47. Kondo K, Kim WY, Lechpammer M, Kaelin WG Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PloS Biol (2003) 1:E83. doi:10.1371/journal.pbio.0000083
48. Courtney KD, Ma Y, Diaz de Leon A, Christie A, Xie Z, Woolford L, et al. HIF-2 complex dissociation, target inhibition, and acquired resistance with PT2385, a first-in-Class HIF-2 inhibitor, in patients with clear cell renal cell carcinoma. Clin Cancer Res (2020) 26:793–803. doi: 10.1158/1078-0432.CCR-19-1459
49. Cassim S, Vučetić M, Ždralević M, Pouyssegur J. Warburg and beyond: the power of mitochondrial metabolism to collaborate or replace fermentative glycolysis in cancer. Cancers (Basel) (2020) 12:1119. doi: 10.3390/cancers12051119
50. Massari F, Ciccarese C, Santoni M, Brunelli M, Piva F, Modena A, et al. Metabolic alterations in renal cell carcinoma. Cancer Treat Rev (2015) 41:767–76. doi: 10.1016/j.ctrv.2015.07.002
51. Wettersten HI, Hakimi AA, Morin D, Bianchi C, Johnstone ME, Donohoe DR, et al. Grade-dependent metabolic reprogramming in kidney cancer revealed by combined proteomics and metabolomics analysis. Cancer Res (2015) 75:2541–52. doi: 10.1158/0008-5472.CAN-14-1703
52. Simonnet H, Alazard N, Pfeiffer K, Gallou C, Béroud C, Demont J, et al. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis (2002) 23:759–68. doi: 10.1093/carcin/23.5.759
53. LaGory EL, Wu C, Taniguchi CM, Ding CC, Chi JT, von Eyben R, et al. Suppression of PGC-1α is critical for reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep (2015) 12:116–27. doi: 10.1016/j.celrep.2015.06.006
54. Yu Y, Yu Q, Zhang X. Allosteric inhibition of HIF-2α as a novel therapy for clear cell renal cell carcinoma. Drug Discovery Today (2019) 24:2332–40. doi: 10.1016/j.drudis.2019.09.008
55. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med (2018) 24:1036–46. doi: 10.1038/s41591-018-0052-4
56. Porporato PE, Filigheddu N, Pedro JMB, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res (2018) 28:265–80. doi: 10.1038/cr.2017.155
57. Zhu J, Thompson CB. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol (2019) 20:436–50. doi: 10.1038/s41580-019-0123-5
58. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Sci 368 (2020), 5. doi: 10.1126/science.aaw5473
59. Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature (2015) 527:186–91. doi: 10.1038/nature15726
60. Heravi G, Yazdanpanah O, Podgorski I, Matherly LH, Liu W. Lipid metabolism reprogramming in renal cell carcinoma. Cancer Metastasis Rev (2022) 41:17–31. doi: 10.1007/s10555-021-09996-w
61. Ganti S, Taylor SL, Abu Aboud O, Yang J, Evans C, Osier MV, et al. Kidney tumor biomarkers revealed by simultaneous multiple matrix metabolomics analysis. Cancer Res (2012) 72:3471–9. doi: 10.1158/0008-5472.CAN-11-3105
62. Gebhard RL, Clayman RV, Prigge WF, Figenshau R, Staley NA, Reesey C, et al. Abnormal cholesterol metabolism in renal clear cell carcinoma. J Lipid Res (1987) 28:1177–84. doi: 10.1016/S0022-2275(20)38606-5
63. Saito K, Arai E, Maekawa K, Ishikawa M, Fujimoto H, Taguchi R, et al. Lipidomic signatures and associated transcriptomic profiles of clear cell renal cell carcinoma. Sci Rep (2016) 6:28932. doi: 10.1038/srep28932
64. Wu G, Wang Q, Xu Y, Li J, Zhang H, Qi G, et al. Targeting the transcription factor receptor LXR to treat clear cell renal cell carcinoma: agonist or inverse agonist? Cell Death Dis (2019) 10:416. doi: 10.1038/s41419-019-1654-6
65. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun (2019) 10:1617. doi: 10.1038/s41467-019-09277-9
66. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13:81–90. doi: 10.1038/nchembio.2238
67. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy (2021) 17:2054–81. doi: 10.1080/15548627.2020.1810918
68. Huang C, Freter C. Lipid metabolism, apoptosis and cancer therapy. Int J Mol Sci (2015) 16:924–49. doi: 10.3390/ijms16010924
69. Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, et al. A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab (2020) 31:267–283.e12. doi: 10.1016/j.cmet.2019.11.020
70. Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest (2021) 131:e140100. doi: 10.1172/JCI140100
71. Pavlova NN, Zhu J, Thompson CB. The hallmarks of cancer metabolism: still emerging. Cell Metab (2022) 34:355–77. doi: 10.1016/j.cmet.2022.01.007
72. Lobo C, Ruiz-Bellido MA, Aledo JC, Márquez J, Núñez De Castro I, Alonso FJ. Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem J 348 Pt (2000) 2:257–61. doi: 10.1042/bj3480257
73. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci (2010) 35:427–33. doi: 10.1016/j.tibs.2010.05.003
74. Onishi Y, Hiraiwa M, Kamada H, Iezaki T, Yamada T, Kaneda K, et al. Hypoxia affects Slc7a5 expression through HIF-2α in differentiated neuronal cells. FEBS Open Bio (2019) 9:241–7. doi: 10.1002/2211-5463.12559
75. Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or akt signaling. Cancer Res (2009) 69:7986–93. doi: 10.1158/0008-5472.CAN-09-2266
76. Wettersten HI. Reprogramming of metabolism in kidney cancer. Semin Nephrol (2020) 40:2–13. doi: 10.1016/j.semnephrol.2019.12.002
77. Fiore A, Murray PJ. Tryptophan and indole metabolism in immune regulation. Curr Opin Immunol (2021) 70:7–14. doi: 10.1016/j.coi.2020.12.001
78. Platten M, Nollen EAA, Röhrig UF, Fallarino F, Opitz CA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discovery (2019) 18:379–401. doi: 10.1038/s41573-019-0016-5
79. Riesenberg R, Weiler C, Spring O, Eder M, Buchner A, Popp T, et al. Expression of indoleamine 2,3-dioxygenase in tumor endothelial cells correlates with long-term survival of patients with renal cell carcinoma. Clin Cancer Res (2007) 13:6993–7002. doi: 10.1158/1078-0432.CCR-07-0942
80. Trott JF, Kim J, Abu Aboud O, Wettersten H, Stewart B, Berryhill G, et al. Inhibiting tryptophan metabolism enhances interferon therapy in kidney cancer. Oncotarget (2016) 7:66540–57. doi: 10.18632/oncotarget.11658
81. Tang D, Yue L, Yao R, Zhou L, Yang Y, Lu L, et al. P53 prevent tumor invasion and metastasis by down-regulating IDO in lung cancer. Oncotarget (2017) 8:54548–57. doi: 10.18632/oncotarget.17408
82. Yao S, Janku F, Subbiah V, Stewart J, Patel SP, Kaseb A, et al. Phase 1 trial of ADI-PEG20 plus cisplatin in patients with pretreated metastatic melanoma or other advanced solid malignancies. Br J Cancer (2021) 124:1533–9. doi: 10.1038/s41416-020-01230-8
83. Perroud B, Ishimaru T, Borowsky AD, Weiss RH. Grade-dependent proteomics characterization of kidney cancer. Mol Cell Proteomics (2009) 8:971–85. doi: 10.1074/mcp.M800252-MCP200
84. Hsieh JJ, Le VH, Oyama T, Ricketts CJ, Ho TH, Cheng EH, et al. HIF, and epigenetic deregulation in clear cell renal cell carcinoma. J Clin Oncol (2018) 36:Jco2018792549. doi: 10.1200/JCO.2018.79.2549
85. Roskoski R Jr. Vascular endothelial growth factor (VEGF) and VEGF receptor inhibitors in the treatment of renal cell carcinomas. Pharmacol Res (2017) 120:116–32. doi: 10.1016/j.phrs.2017.03.010
86. Keefe D, Bowen J, Gibson R, Tan T, Okera M, Stringer A. Noncardiac vascular toxicities of vascular endothelial growth factor inhibitors in advanced cancer: a review. Oncologist (2011) 16:432–44. doi: 10.1634/theoncologist.2010-0271
87. DeBerardinis RJ, Keshari KR. Metabolic analysis as a driver for discovery, diagnosis, and therapy. Cell (2022) 185:2678–89. doi: 10.1016/j.cell.2022.06.029
88. Abdel-Wahab AF, Mahmoud W, Al-Harizy RM. Targeting glucose metabolism to suppress cancer progression: prospective of anti-glycolytic cancer therapy. Pharmacol Res (2019) 150:104511. doi: 10.1016/j.phrs.2019.104511
89. Kaelin WG Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell (2008) 30:393–402. doi: 10.1016/j.molcel.2008.04.009
90. Koehler AN. A complex task? direct modulation of transcription factors with small molecules. Curr Opin Chem Biol (2010) 14:331–40. doi: 10.1016/j.cbpa.2010.03.022
91. Scheuermann TH, Li Q, Ma HW, Key J, Zhang L, Chen R, et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol (2013) 9:271–6. doi: 10.1038/nchembio.1185
92. Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature (2016) 539:112–7. doi: 10.1038/nature19796
93. Courtney KD, Infante JR, Lam ET, Figlin RA, Rini BI, Brugarolas J, et al. Phase I dose-escalation trial of PT2385, a first-in-Class hypoxia-inducible factor-2α antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J Clin Oncol (2018) 36:867–74. doi: 10.1200/JCO.2017.74.2627
94. Xu R, Wang K, Rizzi JP, Huang H, Grina JA, Schlachter ST, et al. 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile (PT2977), a hypoxia-inducible factor 2α (HIF-2α) inhibitor for the treatment of clear cell renal cell carcinoma. J Med Chem (2019) 62:6876–93. doi: 10.1021/acs.jmedchem.9b00719
95. Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, et al. Belzutifan for renal cell carcinoma in von hippel-lindau disease. N Engl J Med (2021) 385:2036–46. doi: 10.1056/NEJMoa2103425
96. Choueiri TK, Bauer TM, Papadopoulos KP, Plimack ER, Merchan JR, McDermott DF, et al. Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis. Nat Med (2021) 27:802–5. doi: 10.1038/s41591-021-01324-7
97. Angeles TS, Hudkins RL. Recent advances in targeting the fatty acid biosynthetic pathway using fatty acid synthase inhibitors. Expert Opin Drug Discovery (2016) 11:1187–99. doi: 10.1080/17460441.2016.1245286
98. Horiguchi A, Asano T, Asano T, Ito K, Sumitomo M, Hayakawa M. Fatty acid synthase over expression is an indicator of tumor aggressiveness and poor prognosis in renal cell carcinoma. J Urol (2008) 180:1137–40. doi: 10.1016/j.juro.2008.04.135
99. Horiguchi A, Asano T, Asano T, Ito K, Sumitomo M, Hayakawa M. Pharmacological inhibitor of fatty acid synthase suppresses growth and invasiveness of renal cancer cells. J Urol (2008) 180:729–36. doi: 10.1016/j.juro.2008.03.186
100. Wang J, Lin W, Li R, Cheng H, Sun S, Shao F, et al. The deubiquitinase USP13 maintains cancer cell stemness by promoting FASN stability in small cell lung cancer. Front Oncol (2022) 12:899987. doi: 10.3389/fonc.2022.899987
101. Falchook G, Infante J, Arkenau HT, Patel MR, Dean E, Borazanci E, et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine (2021) 34:100797. doi: 10.1016/j.eclinm.2021.100797
102. Franz A, Coscia F, Shen C, Charaoui L, Mann M, Sander C. Molecular response to PARP1 inhibition in ovarian cancer cells as determined by mass spectrometry based proteomics. J Ovarian Res (2021) 14:140. doi: 10.1186/s13048-021-00886-x
103. Emberley E, Pan A, Chen J, Dang R, Gross M, Huang T, et al. The glutaminase inhibitor telaglenastat enhances the antitumor activity of signal transduction inhibitors everolimus and cabozantinib in models of renal cell carcinoma. PloS One (2021) 16:e0259241. doi: 10.1371/journal.pone.0259241
104. Jochems C, Fantini M, Fernando RI, Kwilas AR, Donahue RN, Lepone LM, et al. The IDO1 selective inhibitor epacadostat enhances dendritic cell immunogenicity and lytic ability of tumor antigen-specific T cells. Oncotarget (2016) 7:37762–72. doi: 10.18632/oncotarget.9326
105. Naing A, Powderly JD, Nemunaitis JJ, Luke JJ, Mansfield AS, Messersmith WA, et al. Exploring the safety, effect on the tumor microenvironment, and efficacy of itacitinib in combination with epacadostat or parsaclisib in advanced solid tumors: a phase I study. J Immunother Cancer (2022) 10(3):e004223. doi: 10.1136/jitc-2021-004223
106. Nayak-Kapoor A, Hao Z, Sadek R, Dobbins R, Marshall L, Vahanian NN, et al. Phase ia study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) in patients with recurrent advanced solid tumors. J Immunother Cancer (2018) 6:61. doi: 10.1186/s40425-018-0351-9
107. Ma S, Suchomel J, Yanez E, Yost E, Liang X, Zhu R, et al. Investigation of the absolute bioavailability and human mass balance of navoximod, a novel IDO1 inhibitor. Br J Clin Pharmacol (2019) 85:1751–60. doi: 10.1111/bcp.13961
108. Jung KH, LoRusso P, Burris H, Gordon M, Bang YJ, Hellmann MD, et al. Phase I study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) administered with PD-L1 inhibitor (Atezolizumab) in advanced solid tumors. Clin Cancer Res (2019) 25:3220–8. doi: 10.1158/1078-0432.CCR-18-2740
109. Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T, et al. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer (2010) 126:2762–72. doi: 10.1002/ijc.25202
110. Haines RJ, Pendleton LC, Eichler DC. Argininosuccinate synthase: at the center of arginine metabolism. Int J Biochem Mol Biol (2011) 2:8–23.
111. Wu G, Morris SM Jr. Arginine metabolism: nitric oxide and beyond. Biochem J (1998) 336:1–17. doi: 10.1042/bj3360001
112. Hall PE, Lewis R, Syed N, Shaffer R, Evanson J, Ellis S, et al. A phase I study of pegylated arginine deiminase (Pegargiminase), cisplatin, and pemetrexed in argininosuccinate synthetase 1-deficient recurrent high-grade glioma. Clin Cancer Res (2019) 25:2708–16. doi: 10.1158/1078-0432.CCR-18-3729
113. Tomlinson BK, Thomson JA, Bomalaski JS, Diaz M, Akande T, Mahaffey N, et al. Phase I trial of arginine deprivation therapy with ADI-PEG 20 plus docetaxel in patients with advanced malignant solid tumors. Clin Cancer Res (2015) 21:2480–6. doi: 10.1158/1078-0432.CCR-14-2610
Keywords: ccRCC, cancer, metabolism reprogramming, metabolism, glucose, lipids ccRCC, lipids
Citation: Zhu H, Wang X, Lu S and Ou K (2023) Metabolic reprogramming of clear cell renal cell carcinoma. Front. Endocrinol. 14:1195500. doi: 10.3389/fendo.2023.1195500
Received: 28 March 2023; Accepted: 16 May 2023;
Published: 06 June 2023.
Edited by:
Hongming Miao, Army Medical University, ChinaCopyright © 2023 Zhu, Wang, Lu and Ou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kongbo Ou, T3Vrb25nYm9AMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.