
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 16 June 2023
Sec. Systems Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1177268
This article is part of the Research TopicGenetic variants and metabolic diseases, volume IIView all 13 articles
 Babu Kavitha1
Babu Kavitha1 Sampathkumar Ranganathan2
Sampathkumar Ranganathan2 Sundaramoorthy Gopi1
Sundaramoorthy Gopi1 Umashankar Vetrivel3,4
Umashankar Vetrivel3,4 Nagarajan Hemavathy3
Nagarajan Hemavathy3 Viswanathan Mohan5
Viswanathan Mohan5 Venkatesan Radha1*
Venkatesan Radha1*Background: HNF1A is an essential component of the transcription factor network that controls pancreatic β-cell differentiation, maintenance, and glucose stimulated insulin secretion (GSIS). A continuum of protein malfunction is caused by variations in the HNF1A gene, from severe loss-of-function (LOF) variants that cause the highly penetrant Maturity Onset Diabetes of the Young (MODY) to milder LOF variants that are far less penetrant but impart a population-wide risk of type 2 diabetes that is up to five times higher. Before classifying and reporting the discovered variations as relevant in clinical diagnosis, a critical review is required. Functional investigations offer substantial support for classifying a variant as pathogenic, or otherwise as advised by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) ACMG/AMP criteria for variant interpretation.
Objective: To determine the molecular basis for the variations in the HNF1A gene found in patients with monogenic diabetes in India.
Methods: We performed functional protein analyses such as transactivation, protein expression, DNA binding, nuclear localization, and glucose stimulated insulin secretion (GSIS) assay, along with structural prediction analysis for 14 HNF1A variants found in 20 patients with monogenic diabetes.
Results: Of the 14 variants, 4 (28.6%) were interpreted as pathogenic, 6 (42.8%) as likely pathogenic, 3 (21.4%) as variants of uncertain significance, and 1 (7.14%) as benign. Patients harboring the pathogenic/likely pathogenic variants were able to successfully switch from insulin to sulfonylureas (SU) making these variants clinically actionable.
Conclusion: Our findings are the first to show the need of using additive scores during molecular characterization for accurate pathogenicity evaluations of HNF1A variants in precision medicine.
The hepatocyte nuclear factor 1A (HNF1A)gene (MIM # 142410) encodes a crucial member of an auto-regulatory transcription circuit in mature and developing pancreas. Heterozygous mutations in HNF1A result in the most common form of MODY namely subtype HNF1A-MODY. Autosomal dominant inheritance, early onset, and progressive β-cell deterioration resulting in severe hyperglycemia define this type of monogenic diabetes (1–3). This kind of MODY has the highest prevalence and is more common than other subtypes, and it is more common in Europe, North America, and Asia (4–7).
Individuals with HNF1A MODY are likely to develop extra pancreatic symptoms such as glycosuria which will appear even before the onset of diabetes due to a low renal glucose threshold (8). This is mainly because HNF1A is expressed in tissues such as the kidney, liver, and small intestine, in addition to β-cells. The risk of micro- and macro-vascular problems in HNF1A-MODY is comparable to that of T1D and T2DM (9) and hence strict glucose management is required for these individuals. Patients harboring pathogenic variants in HNF1A gene are sensitive to low doses of sulfonylureas (10).
The HNF1A protein consists of three functional domains namely a dimerization domain (1 – 33 aa), a bipartite DNA-binding domain (homeo domain 100 –184 aa; POU domain 198 –281 aa), and a transactivation domain (282 –631 aa) (11, 12). It binds to DNA as a homodimer or with the structurally related transcription factor HNF1B as heterodimers (13, 14). To date, about 564 MODY-causing variants have been identified in the HNF1A gene (15, 16). These variations include missense, nonsense, frameshift, in-frame deletions/insertions/duplications, splice site, promoter region, and whole/partial gene deletions. Analyses of these variants have demonstrated that some of them render the protein unstable and poorly expressed (17, 18). Some of the variants affect either the DNA binding or transactivation ability of HNF1A. However, patients with the latter type of variants do not exhibit more severe phenotypes (19–21). Finally, a subgroup of variants exert a dominant-negative effect over the normal protein.
It is important that these candidate variants are subjected to rigorous evaluation of pathogenicity to avoid false annotation of causality, which would be an impediment to the translation of genomic research findings to clinical practice and precision medicine. False assignment of pathogenicity can also have severe consequences for patients, resulting in incorrect prognostic and therapeutic advice. Therefore, a comprehensive map is needed, linking mutation status, effect on protein function, and clinical effect that is genotype-function-phenotype. The recent American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) (ACMG-AMP) guidelines classification is based on five tier score system namely pathogenic (P), likely pathogenic (LP), variant of uncertain significance (VUS), likely benign (LB) and benign (B) (22). Our previous studies have shown that HNF1A -MODY is the most prevalent subtype in India (3) and we identified several variants which were of uncertain significance, Assessing the pathogenicity of these rare protein-coding genetic variants in HNF1A is very important in our patient cohort before assigning causality to these variants, as this may lead to change of treatment.
Functional investigation constitutes one of the strongest pieces of evidence for classifying a variant as pathogenic or benign (23). Each variant needs to be assessed by genomic, bioinformatic, structural, and functional lines of evidence for classifying them as pathogenic or benign. Hence, we hypothesized that functional evaluation would enhance the interpretation of the pathogenicity of HNF1A variants identified in individuals from families of Indian MODY subjects.
We investigated 14 HNF1A variants found in 20 unrelated individuals (11 females and 9 males) from 20 non-consanguineous Indian families. Patients were selected for MODY genetic screening based on the following criteria: a family history of diabetes in multiple generations; an early age at onset of diabetes (< 35 years); lack of obesity, ketosis, and beta cell autoimmunity with detectable endogenous insulin reserve as measured by C peptide which is one of the best biomarkers; and diabetes controllable without insulin for at least 2 years. The study was carried out in compliance with the Helsinki Declaration (2000); all study participants (or their guardians) provided written, informed consent, and the study was approved by the Madras Diabetes Research Foundation’s local institutional ethics committee.
Genomic DNA was isolated from whole blood using the standard protocol. Direct sequencing was carried out on an ABI 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA) using the Big Dye terminator V3.1 chemistry, and the sequences were compared with the public databases. Published primer sequences were used to amplify the DNA for HNF1A gene. In addition to the sequencing of patients, we also sequenced 100 normal glucose-tolerant subjects (fasting value <100 mg/dL and 2 hours value <140 mg/dL) to check for the presence or absence of variants in them.
All HNF1A variations were assessed using the ACMG guidelines, which classify variants as pathogenic (class 5), likely pathogenic (class 4), uncertain significance (class 3), likely benign (class 2), or benign (class 1). Criteria used for the classification of variants are listed in Supplementary Table 1. Public databases such as PubMed, the Human Gene Mutation Database, ClinVar, and LOVD were used and the genome aggregation database (GnomAD) was referred to for population frequency. Bioinformatic prediction tools such as SIFT, PolyPhen2, Mutation Taster, PROVEAN, CADD Score, i mutant 2.0, and Grantham scores were used to assess the pathogenicity (Supplementary Table 2).
Human HNF1A cDNA (NCBI Entrez Gene BC104910.1) (NM_000545.5) in pcDNA 3.1 His/C vector (Invitrogen Inc, Carlsbad, CA, USA), was used as a template for constructing individual HNF1A variants using the QuikChange Lightning Site-directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA), and all constructs were verified by Sanger sequencing. Transiently transfected HeLa and INS1 cells with WT, empty vector (pcDNA3.1), or variant HNF1A cDNA were used in functional studies, investigating HNF1A (i) transcriptional activity using a rat albumin (in HeLa cells) and HNF4A P2 (in INS1 cells) promoter-linked luciferase reporter assay system; (ii) DNA binding ability was analyzed using Episeeker DNA-protein binding assay kit (Abcam, ab117139) and a biotinylated oligonucleotide (Sigma Aldrich, St. Luis, MO, US) containing the HNF1A binding site in the rat albumin promoter; (iii) protein expression in whole cell lysates by immunoblotting;(iv) nuclear localization by indirect immunocytochemistry; and (v) the glucose-stimulated insulin secretion (GSIS) capacity of the variant HNF1A in INS1 β-cells were measured using insulin ELISA kit (Mercodia, Sweden). A detailed methodology is described in the Supplementary Material.
The human HNF1A protein sequence (P20823) was downloaded from the UniProt database. The Consurf server was used to obtain amino acid conservation scores within the orthologous protein family by comparing 150 homologous sequences. For the structure-based stability prediction, the available crystal structure of HNF1A in complex with DNA, PDB ID-1IC8 was remodeled with missing residues and was refined using Modeller10v. The refined Wild type (WT) HNF1A was considered for stability analysis of HNF1A and also the impact of mutants in the HNF1A-DNA complex. The structure of mutants was modeled with a WT-HNF1A template using Modeller10v, and the refined WT and MT HNF1A were subjected to molecular dynamics simulation studies using Gromacs2020 (10.1080/07391102.2021.1965030). Subsequently, PCA and FEL analyses were carried out to determine the near-native conformation, wherein the HNF1A-DNA interactions were analyzed using DNAproDB. A detailed methodology is given in the Supplementary Material.
The results of functional analyses of individual variants are presented as mean (in %) ± standard deviation (SD) and normalized to WT HNF1A activity (set as 100%), unless otherwise specified. Experiments were carried out on at least 3 independent occasions unless otherwise specified in the figure legends. Statistical differences between individual variants and WT function were analyzed using GraphPad Prism software (version 8.1.1, GraphPad Software, Inc. San Diego, CA, USA) and raw data (i.e., firefly/renilla ratios) and an unpaired 2-tailed t-test based on n=3. A p-value < 0.05 was considered statistically significant.
A total of 14 missense HNF1A variants identified in 20 clinical MODY patients were included in this study. All the patients were heterozygous for the variants. In three families, we were able to observe the segregation of variants in affected family members, but for other patients, family samples were not available. Pedigrees of the available families are shown in Supplementary Figure 1. All were negative for β-cell autoantibodies such as GAD and ZnT8 antibodies. The mean ± SD of biochemical parameters were as follows: age at onset of diabetes, 21 ± 6.5 years; Body Mass Index (BMI) - 23 ± 4 kg/m2; duration of diabetes, 9.9 ± 6.7 years; Fasting plasma glucose - 181 ± 64 mg/dL; post prandial plasma glucose - 277 ± 97 mg/dL; glycated hemoglobin (HbA1C)- 9.2 ± 2.4%; fasting C-peptide was 0.9 ± 0.4 pmol/L; stimulated C- peptide was 1.5 ± 0.6 pmol/L; total cholesterol - 169 ± 41 mg/dL; triglycerides - 137 ± 82 mg/dL; High Density Lipoprotein (HDL)- cholesterol - 39 ± 8.5 mg/dL and Low Density Lipoprotein (LDL)- cholesterol - 94 ± 36 mg/dL. Prior to functional genetic investigations, 11 patients were on insulin treatment; one patient was on insulin + metformin; four patients were on insulin + SU; one patient was on metformin alone and three patients were on SU treatment alone before the genetic investigation. Clinical and biochemical parameters are summarized in Table 1.
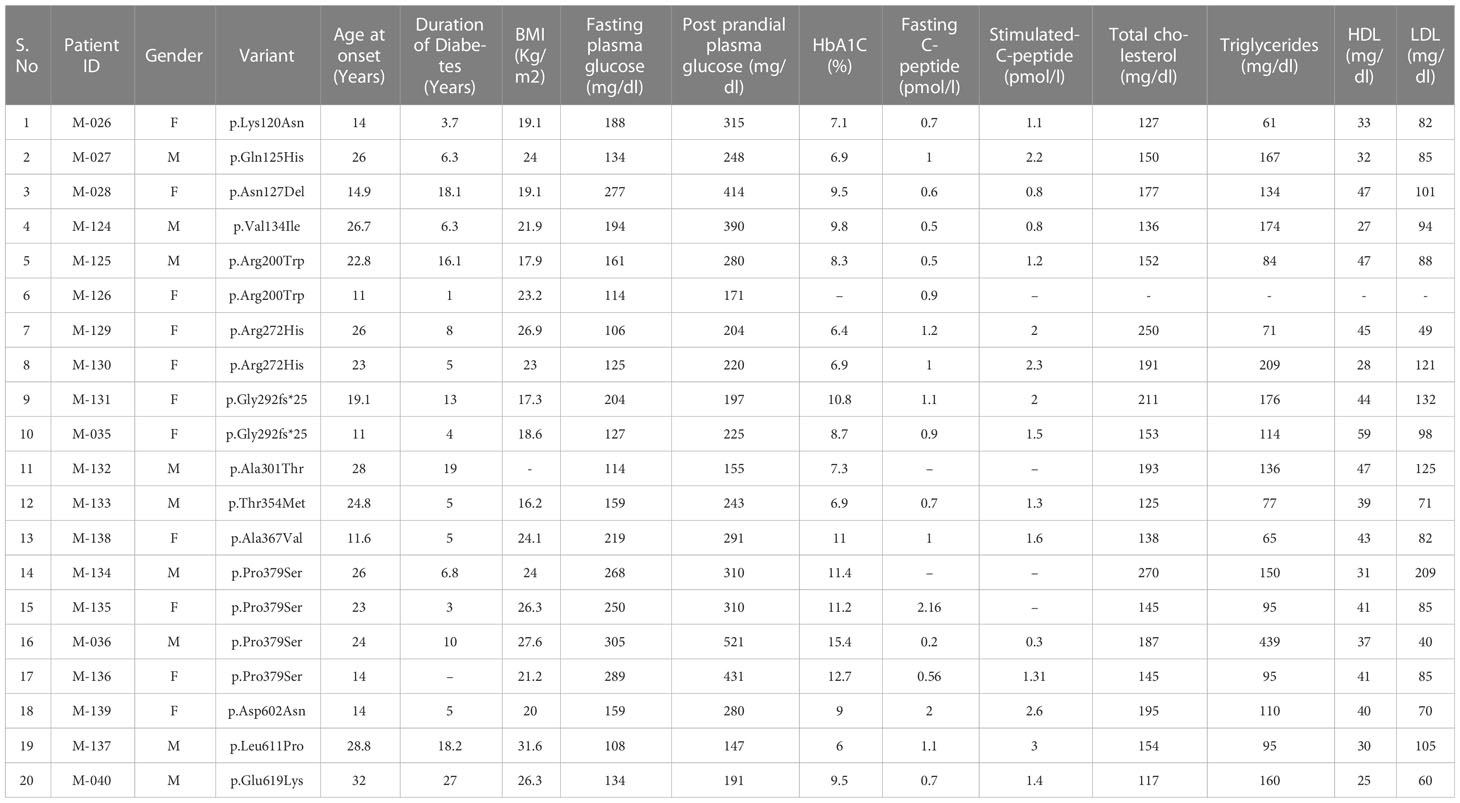
Table 1 Clinical and biochemical workup of subjects with HNF1A gene variants.
Among the 14 variants, four variants (p.Lys120Asn,p.Gln125His,p.Ala367Val,p.Asp602Asn) were novel and not reported in the literature, three variants were previously reported by us (3, 24), and the remaining seven variants were reported in other studies (20, 25–29). Of the 14 variants included in this study, six variants reside in DNA binding domain (91-281 a.a), specifically four variants were mapped to POUS domain (91-181 a.a), one variant was mapped to POUH domain (203-279 a.a) and one variant reside in the interface between the POUs and POUH domains of HNF1A protein. The other, eight variants were mapped to the transactivation domain (282- 631 a.a) of HNF1A protein (Supplementary Figure 2).
In HeLa cells compared to the WT HNF1A activity (set as 100%), the measured levels of transcriptional activity (TA) for five (p.Asn127*,p.Val134Ile,p.Arg200Trp and p.Gly292Fs*25)of the 14 variants were significantly lower (<40%) (Figure 1A, Table 2). Three variants (p.Lys120Asn,p.Pro379Ser, and p.Leu611Pro) had TA activity <50%, while two variants (p.Gln125His and p.Thr354Met) had TA activity of 53 and 62% respectively and reduction observed in all these variants were significant. Two variants p.Ala367Val (61%) and p.Asp602Asn (51%) showed a mildly reduced TA. Two other variants (p.Ala301Thr and p.Glu619Lys) demonstrated TA levels comparable to WT HNF1A levels (Figure 1A, Table 2). TA was consistently higher for all these variants when using HNF4A-P2 promoter in INS-1 cells (activity range 32%–137%) (Figure 1B, Table 2) versus rat albumin promoter in HeLa cells. This is most likely due to interference of endogenous HNF1A in INS-1 cells (2- to 4-fold higher basal promoter activity).
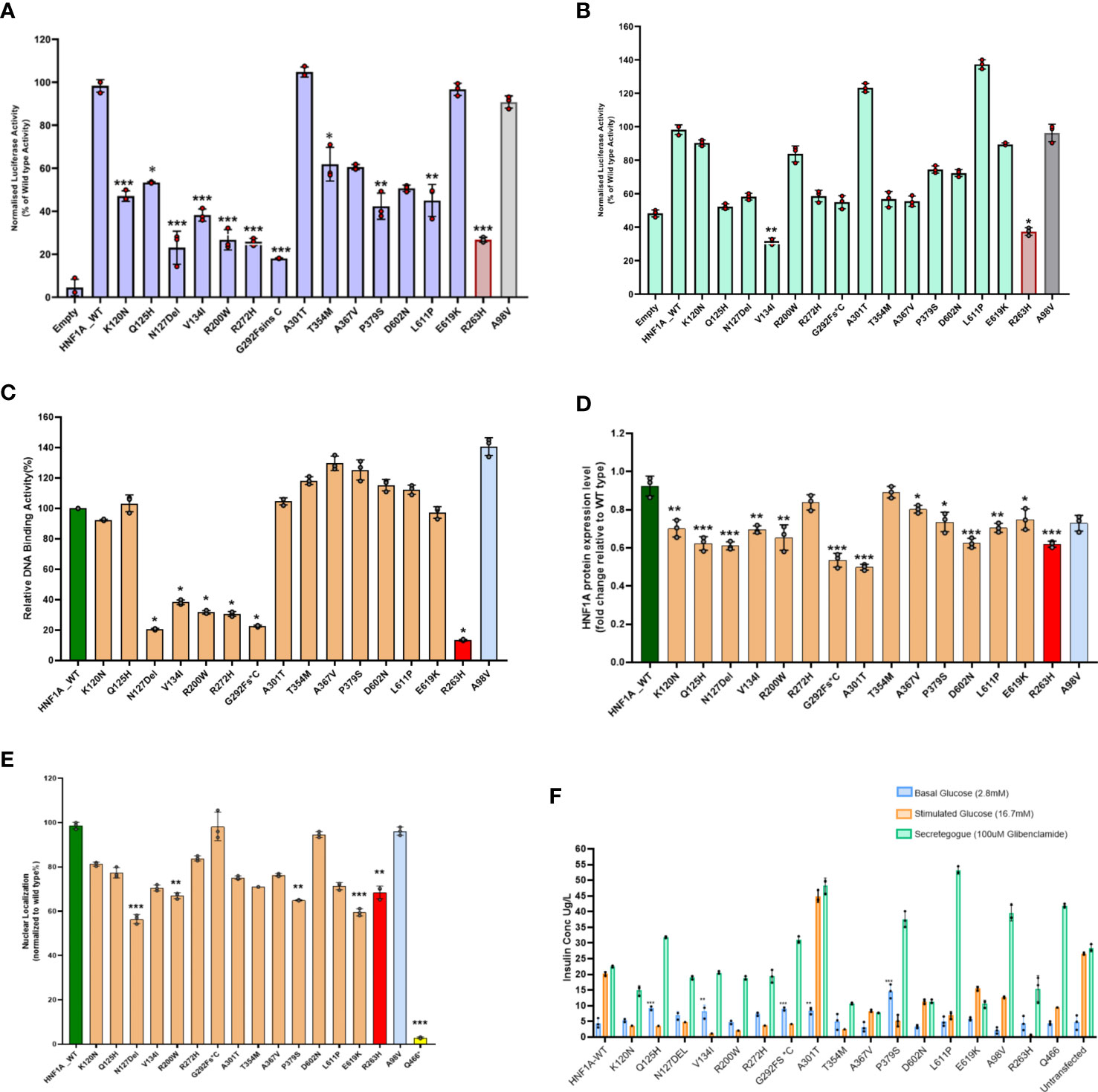
Figure 1 Summary of the data obtained from functional studies. (A) Transcriptional activity of the HNF1A protein variants in HeLa cells; (B) Transcriptional activity of the HNF1A protein variants in Ins1 cells using HNF4A P2 promoter; (C) Assessment of the DNA binding ability of the HNF1A protein variants; (D) Protein expression levels of the HNF1A protein variants in HeLa cells; (E) Nuclear localization of the HNF1A protein variants in HeLa cells; (F) Variant effect on Glucose Stimulated Insulin Secretion. Red bar indicates MODY 3 control variant; Grey bar indicates type 2 diabetes risk variant; Yellow bar indicates variant with poor nuclear translocation effect in HNF1A gene. Each bar represents the mean of three independent experiments (n=3) ± SD. P-values were obtained by un-paired student t-test. *** indicates p value <0.001; ** indicates p value <0.01; * indicates p value <0.05.
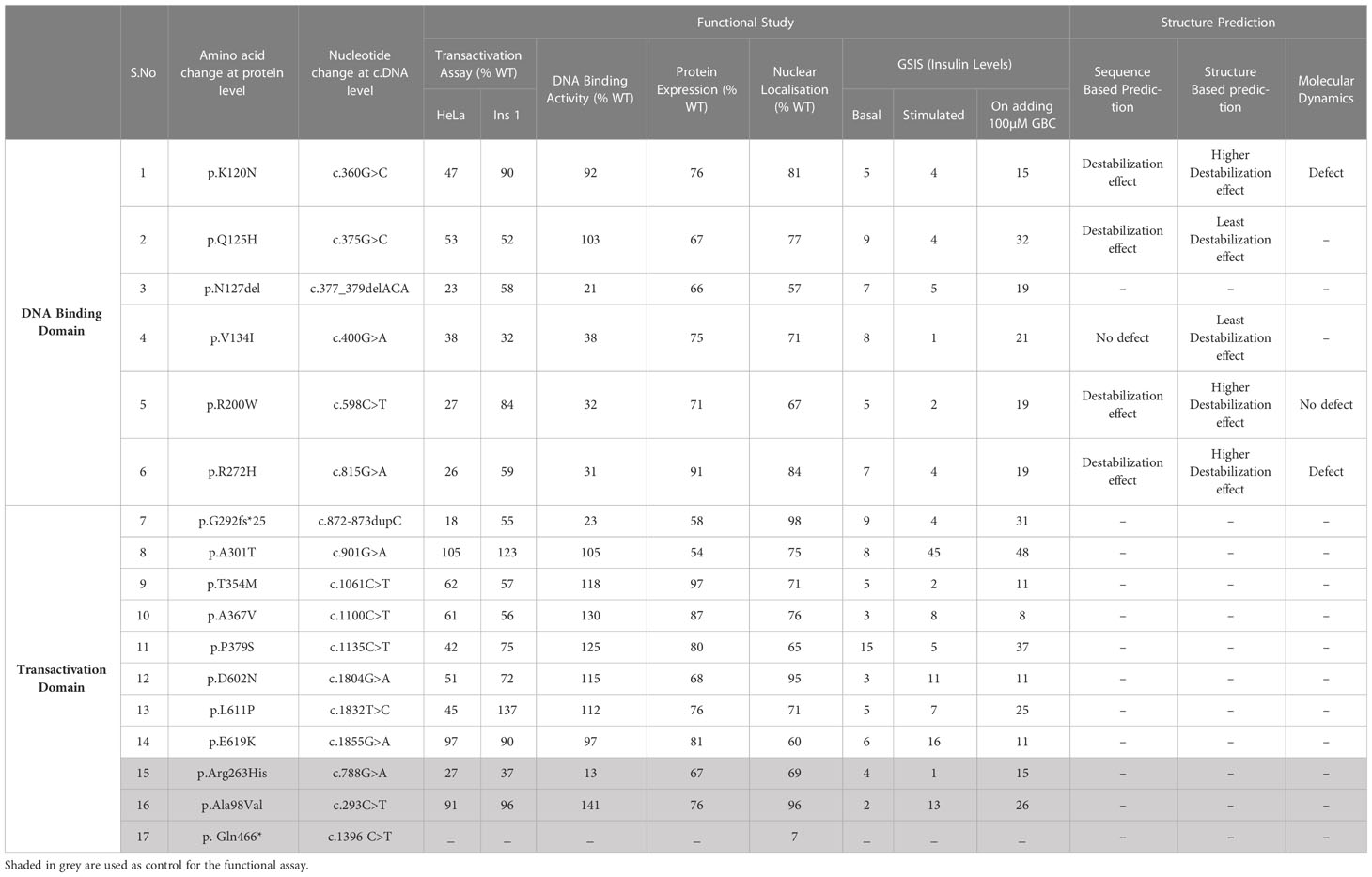
Table 2 Summary of the functional studies of the HNF1A variants identified in Indian MODY subjects.
Three variants (p.Asn127*, p.Arg200Trp and p.Arg272His) localized in the DBD and one variant (p.Gly292Fs*25) in TAD demonstrated severely reduced (<40%) activity. All other variants showed normal binding activity comparable to WT (Figure 1C, Table 2).
Two variants (p.Gly292Fs*25 and p.Ala301Thr) showed significantly reduced protein expression level (<60%); while four variants (p.Gln125His,p.Asn127*,p.Arg200Trp and p.Asp602Asn), demonstrated reduced expression level (61-75%) and were also significant (Figure 1D, Table 2).
All the 14 HNF1A variants were assessed for their ability to translocate to the nucleus of the cell in order to regulate their target gene expression. Only four variants showed reduced (~57-67%) nuclear translocation as assessed by indirect immunocytochemistry (Figure 1E, Table 2). Other variants showed normal nuclear translocation.
All 14 variants were also assessed for insulin secretion using GSIS. Under basal conditions (2.8mM glucose), these variants produced insulin in the range of 3-15µg/L of insulin and under stimulated conditions using 16.7mM glucose they produced 1-45µg/L of insulin. When they were treated with 100µM glibenclamide (GBC), the stimulated insulin secretion was enhanced ranging from 8-48µg/L in all the 14 variants tested (Figure 1F, Table 2).
Structural analysis was performed for variants found in DNA binding domain. These variants were mapped onto the crystal structure of HNF1A protein (PDB ID: 1IC8). Thereby, all the missense variants, namely p.Lys120Asn, p.Gln125His, p.Val134Ile, p.Arg200Trp, and p.Arg272His, were subjected to the following predictions such as sequence and structural-based stability prediction followed by molecular dynamics (MD).
Sequence-based stability study revealed that the HNF1A structure is destabilized by the variants p.Lys120Asn, p.Gln125His, p.Arg200Trp, and p.Arg272His, but not by the variant p.Val134Ile. The crystal structure of HNF1A in association with DNA (PDB ID-1IC8), was further modified with missing residues and refined using Modeller10v for the structure-based stability prediction (Figure 2A). According to structure-based prediction, the HNF1A variants p.Lys120Asn, p.Arg200Trp, and p.Arg272His were shown to have a larger destabilizing impact and more molecular flexibility than the other variants. Among these variants, the p.Arg200Trp variant has a higher destabilizing impact. Variants p.Gln125His and p.Val134Ile had the least destabilizing impact (Figures 2B–K). Since the three variants p.Lys120Asn, p.Arg200Trp and p.Arg272His, showed higher destabilizing effects they were chosen for the MD study.
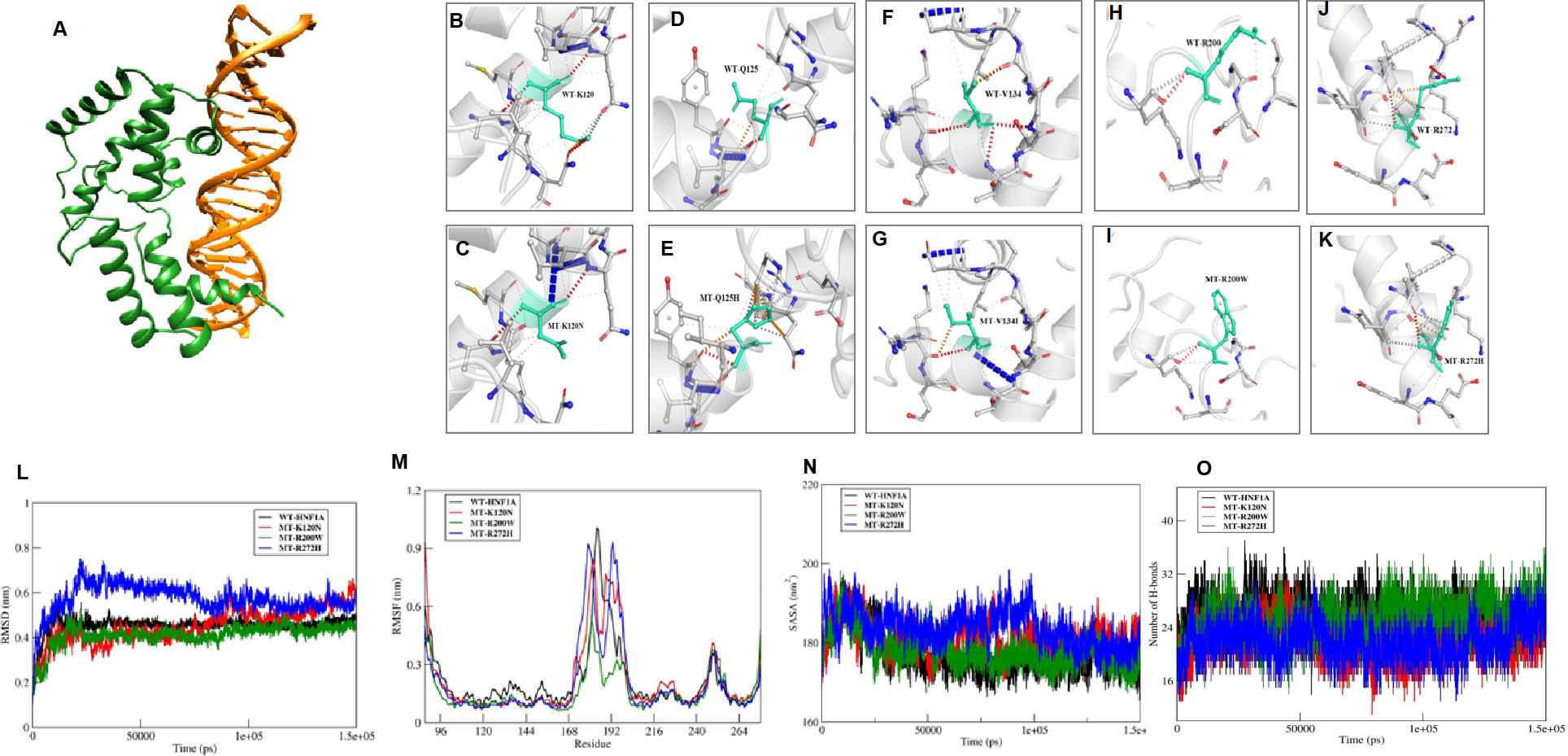
Figure 2 In Sillico structural prediction of wild type and variant HNF1A protein. (A) Remodelled and Refined Wild-type (WT) HNF1A-DNA complex; B-K)Prediction of Interatomic Interactions of the Wild and mutant forms of HNF1A variants, where the Wild-type and mutant residues are coloured in light-green and are also represented as sticks alongside the surrounding residues which are involved in any type of interactions; (L-O) Molecular dynamics simulation analysis of the wild and MT forms of HNF1A complexes (L) RMSD plot (M) RMSF plot (N) Solvent accessible surface area plot; (O) Number of inter hydrogen bonds maintained throughout the MD production run within HNF1A and DNA.
The WT-HNF1A template was used to simulate the structures of the mutants p.Lys120Asn, p.Arg200Trp, and p.Arg272His. The revised WT and MT HNF1A were then submitted to MD simulation investigations using Gromacs2020. When the complexes’ MD trajectories were compared to the WT, the variant p.Arg272His showed higher divergence than the variants p.Lys120Asn and p.Arg200Trp in the initial period of simulation. However, variant p.Lys120Asn showed more deviations than p.Arg272His during the last 20 ns of the root mean square deviation (RMSD) plot, a numerical measurement representing the difference between WT and variant protein structures (Figure 2L). The root mean square fluctuation (RMSF) plot, is a calculation of individual residue flexibility, or how much a particular residue moves (fluctuates) during a simulation (Figure 2M), and this showed that residues that interact with DNA were found to have larger deviations in all of the complexes; in particular, residues 179 and 180 of the p.Arg272His variant showed higher deviations of 0.9 nm and 192-193 of the p.Arg272His variant showed higher fluctuations of about 1 nm among the complexes. When compared to WT, the variants p.Lys120Asn and p.Arg272His lost their contact with DNA at the residue level, and their total interactions with DNA also decreased (Figures 2N, O). However, the variant p.Arg200Trp had an increased frequency of interactions with DNA and a greater accessible surface area of all buried solvents (Figures 2N, O). Particularly, the variant residue Trp200 interacts with the minor groove of DNA. From these results, it was revealed that variants p.Lys120Asn and p.Arg272His had lost their interaction with DNA resulting in structural defects.
Pathogenic HNF1A variants causing HNF1A-MODY are often characterized by significantly decreased TA, poor DNA binding, impaired nuclear targeting, and/or lower protein expression levels in the range of ~20-35% when compared to WT (100%) (19, 21, 30–33). In this study, the cut-off considerations were set at a slightly different level compared to the previous study by Althari et al. (31). Being a more distilled cohort of clinically proven MODY patients, the cut-off of TA<40% was used for pathogenic variants, and TA activity between 40-60% was used for likely pathogenic variants. In addition to this, DNA binding activity, GSIS, and clinical course were considered for ascribing pathogenic and likely pathogenic variants. Therefore, over and above the ACMG/AMP guidelines, the functional and clinical work such as the response to SU have been considered together to re-interpret the variants.
Variants p.Gly292Fs*25 and p.Asn127* were interpreted as pathogenic variants since they have low TA activity along with the reduced DNA binding activity and defect in insulin secretion.p.Arg272His was reinterpreted as a pathogenic variant from their initial interpretation. Seven variants (p.Lys120Asn, p.Gln125His, p.Val134Ile, p.Arg200Trp, p.Thr354Met, p.Pro379Ser, and p.Leu611Pro) were reclassified as likely pathogenic variants from VUS. Three variants (p.Ala367Val, p.Asp602Asn, and p.Glu619Lys) remained VUS after reinterpretation whereas variant p.Ala301Thr was reinterpreted as benign from VUS (Figure 3, Table 3).
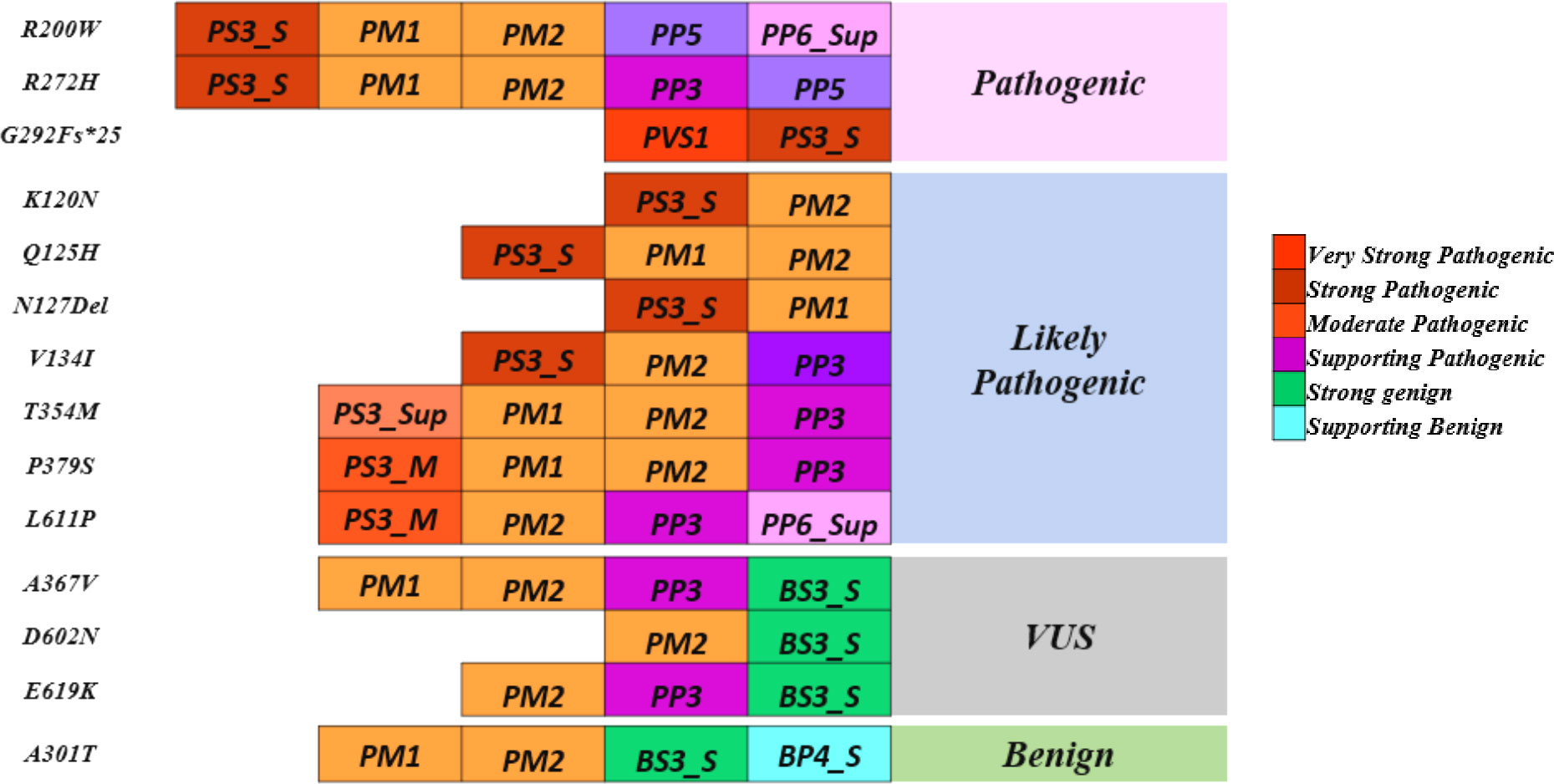
Figure 3 Reinterpretation of HNF1A variants using molecular characterization.
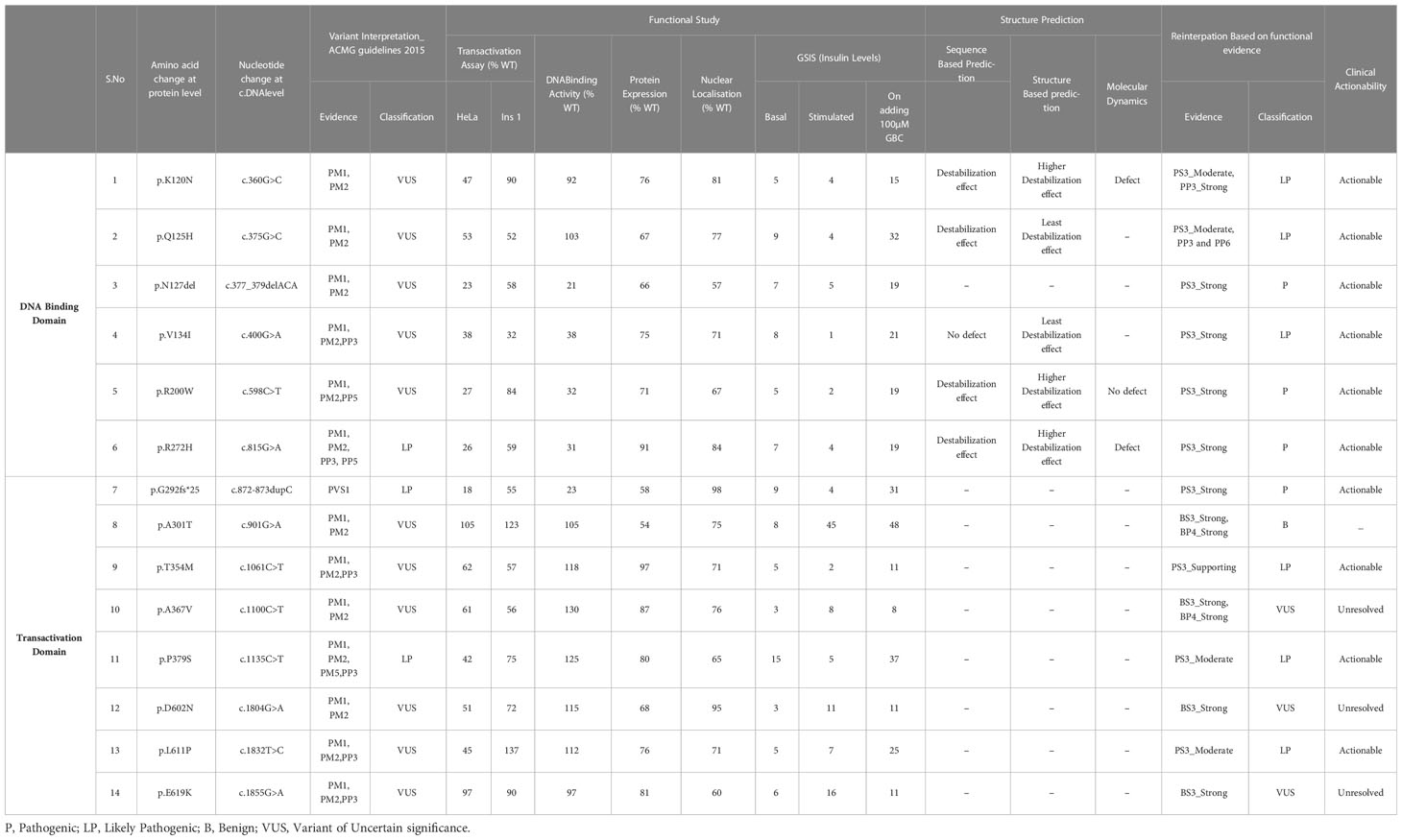
Table 3 Summary of re-interpretation of HNF1A gene variants and their clinical actionability, identified in Indian MODY patients based on molecular characterization.
Variants designated as pathogenic/likely pathogenic based on functional assessment were investigated for clinical actionability by collecting the follow-up details of the patients over a period of time. The patient (M-026) with variant p.Lys120Asn has been switched from insulin to two doses of SU (glimepiride) along with metformin per day. The patient M-027 with the mutation p.Gln125His (likely pathogenic variant) developed diabetes at the age of 25.7 years and had diabetes for 7 years. Before genetic testing, the patient was treated with insulin and oral hypoglycemic agents (OHA). As a result of genetic studies, the patient was transferred from insulin to two doses of gliclazide per day. His HbA1C levels dropped from 9.6% to 6.4% after his therapy was changed.
Patient M-028, who carries the pathogenic variant p.Asn127*, is diagnosed with diabetes at the age of 14.9 years, with a duration of 15.6 years (Figure 4). The patient was on OHA for around two years before being started on insulin. She is currently on insulin and SU therapy since her β cell reserve was low (CPF-0.6 and CPS-0.9) and she started to develop microvascular and macrovascular complications. Patient M-124 harboring the variant p.Val134Ile (Likely pathogenic variant) was diagnosed with diabetes at the age of 26.7 years with diabetes duration of 4 years. Based on functional evidence, patient M-124 with variant p.Val134Ile was transitioned from insulin to a single dose of glipizide per day.
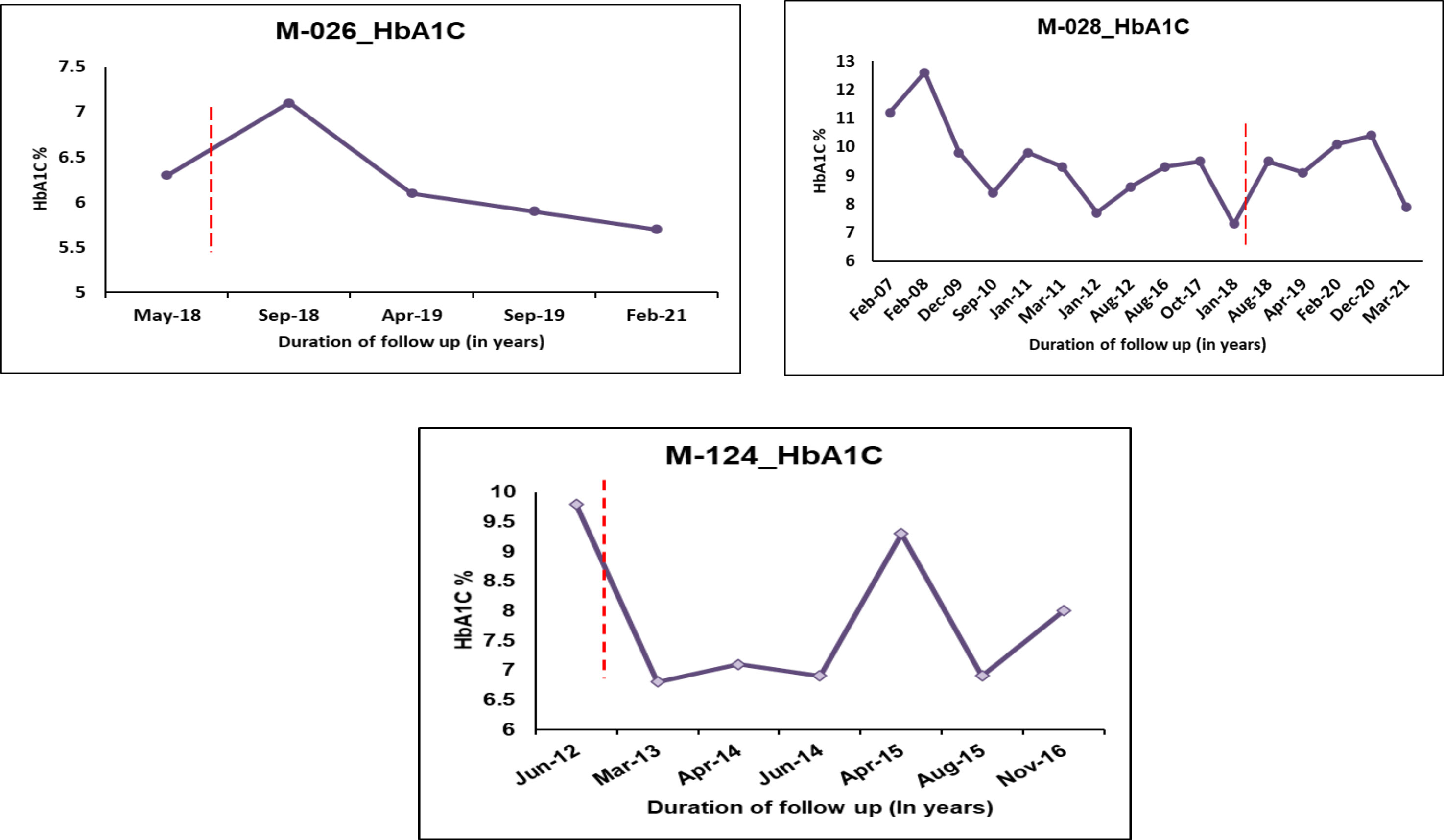
Figure 4 HbA1C Trajectories of few HNF1A MODY patients after change in treatment based on re-interpretation of the variants. Dotted lines indicate change of treatment.
Patient M-126 with the pathogenic variant p.Arg200Trp was switched from insulin to SU. It was advised to continue with SU for patient M-125 who had the same variant. Statins were given for patient M-125 in order to maintain a normal lipid profile. Previous studies have shown two other amino acid changes at the same codon such as p.Arg200Gly and p.Arg200Gln in multiple SU-sensitive HNF1A-MODY families (34, 35). The functional effects of these two variants, p.Arg200Gly and p.Arg200Gln, were however not mentioned. All of the patients, including the one from this study, who have the variation in this codon respond to SU. This suggests that the variation is pathogenic and clinically actionable. Patients with pathogenic variant (p.Arg200Trp, p.Arg272His and p.Gly292Fs*25) and likely pathogenic variant (p.Thr354Met and p.Leu611Pro) were also shifted from insulin to SU therapy.
The comprehension of disease mechanisms is improved by well-established functional investigations on variants, which also offer proof for the pathogenicity of the variants. Studies have demonstrated that functional studies help to clarify the interpretation of HNF1A-MODY variants, particularly in the absence of familial segregation or phenotypic data (32).
In this study, we have performed molecular characterization of 14 HNF1A variants identified in 20 unrelated individuals from 20 non-consanguineous families among Indian MODY subjects, where the majority of variants have not been reported. Normal transactivation activity of HNF1A protein, which depends on the capacity to bind target promoters (DNA) and on an adequate quantity of cellular (nuclear) protein, is necessary for normal HNF1A transcription factor function.
Because not all functional tests represent the underlying process and not all variants have the same effects on function (36), we aimed at improving the understanding and interpretation of these findings. Therefore, multiple assays were employed to fully examine the effects of a variant in order to come to a conclusion. These variants were examined utilizing in vitro functional pipelines, such as luciferase assays for transactivation, which measure the transcriptional activity of HNF1A variants, as well as assays of DNA binding activity, protein expression, and subcellular localization to determine the impact of the variants on the protein function. Additionally, a GSIS assay to examine the impact of these variants on insulin secretion was performed. A distinctive feature of this work is the in silico structural analyses to determine if it might identify the variants with functional defects. Since the crystal structure of HNF1A is available only for the DNA binding domain, structural investigations were carried out for the missense variants identified only in that region.
A multi-pronged approach using the ACMG guidelines, the functional and structural analyses have been considered together to re-classify these variants. In this work, we focused on the scoring systems and the criteria for re-interpreting the variants. PS3 was assigned when data from well-established in vitro functional studies supported a detrimental effect on the gene or gene product; PP3 was assigned when multiple lines of computational evidence and structural prediction supported a detrimental effect on the gene or gene product (conservation, evolutionary, etc.); and BS3 was assigned when well-established in vitro functional studies showed no detrimental effect on protein function. In addition, multiple levels of strength, such as strong, moderate, and supporting levels based on functional and structural data were applied to the scoring approaches employed in this study. Of the 14 variants considered in this study, 1 variant p.Arg272His was interpreted as likely pathogenic, and 11 variants were interpreted as VUS initially based on the ACMG/AMP guidelines. (Figure 3, Table 3).
According to previous studies on the effects of pathogenic HNF1A-MODY variants, pathogenic and MODY causal variants impair HNF1A activity, DNA binding, and localization (40% compared to WT HNF1A) (21, 32), whereas type 2 diabetes risk variants have an impact on HNF1A function ranging from 40% -60% compared to WT (30, 31, 33).
Based on the aforementioned cut-offs, many degrees of strength were assigned to each scoring criterion. PS3_Strong scoring criteria were assigned to variants that showed <40% activity than WT activity in at least two functional assays; PS3_Moderate was assigned to variants that showed activity between 40 and 60%; and PS3_Supporting was assigned to variants that showed activity less than 65%. PP3_Strong criterion was assigned when the variant showed defects in all the in silico structural prediction analysis. The variant meeting the BS3_Strong criterion had no negative effect on protein function in any of the functional experiments.
The p.Arg272His previously interpreted as likely pathogenic was re-interpreted as pathogenic based on the evidence PS3_Strong, PM1, PM2, PP5, and PP3_Strong. One variant p.Arg200Trp interpreted as VUS was re-interpreted as pathogenic based on the evidence PS3_Strong, PM1, PM2, PP3_Supporting, and PP5. Variant p.Gly292Fs*25 was interpreted as pathogenic based on the evidence PVS1 and PS3_Strong and variant p.Asn127* was interpreted as likely pathogenic based on the evidence PS3_Strong, PM1. Variants p.Lys120Asn and p.Gln125His interpreted as VUS was re-interpreted into likely pathogenic based on the evidence PS3_Moderate, PM2, PP3_Strong, and PS3_Moderate, PM2, PP3_Supporting, PP6 respectively. Variant p.Val134Ile was re-interpreted into likely pathogenic based on evidence PS3_Strong and PM2. Variant p.Thr354Met was re-interpreted as likely pathogenic based on PS3_Supporting, PM1, PM2, and PP3. Variant p.Pro379Ser was re-interpreted as likely pathogenic based on the evidence PS3_Moderate, PM1, PM2, and PP3. Variant p.Leu611Pro was re-interpreted as likely pathogenic based on the evidence PS3_Moderate, PM2, PP3, and PP6_Supporting. Variant p.Ala367Val remains VUS based on the evidence PM1, PM2, PP3, and BS3_Strong. Variants p.Asp602Asn and p.Glu619Lys remain VUS based on the evidence PM2, BS3_Strong and PM2, PP3, and BS3_Strong respectively. Variant p.Ala301Thr was re-interpreted as benign based on the evidence PM1, PM2, BS3_Strong, and BP4_Strong (Table 3). It is crucial to remember that functional evidence does not always associate a variant to disease outcome; in order to determine clinical actionability, the functional data must be assessed in combination with clinical data (30). It is important to be aware of the fact that both functional and longitudinal clinical follow up are important to establish the clinical actionability of the variants.
Clinical actionability is generally defined as clinically prescribed interventions that are effective for preventing or delaying clinical disease, lowering clinical burden, or improving clinical outcomes in an adult who has not previously received a diagnosis and are specific to the genetic disorder under consideration (37). Based on our results, 4 out of 14 (28.6%) variants were interpreted as pathogenic, 6 variants (42.8%) as likely pathogenic, 3 variants (21.4%) as variants of uncertain significance, and 1 variant (7.14%) as a benign variant. Patients with the ten P/LP variants were able to successfully switch from insulin to SU and sustain good glycemic control, thus making these variants clinically actionable (Table 3).
We performed 3D structural analysis to check whether in-silico analysis corroborated with functional investigations in identifying the pathogenic variants and also to have a structural understanding of the variant HNF1A proteins. Our in-silico analysis showed that variants p.Gln125His, p.Val134Ile have lesser structural defects while variants p.Lys120Asn and p.Arg272His have severe structural defects, and the variant p.Arg200Trp has moderate structural defects. In the case of the p.Val134Ile variant, we found differences between the functional and structural data. Although in-silico structural analysis showed that it has a lesser destabilizing effect despite being predicted to be a highly conserved structural residue, our functional data showed that variant p.Val134Ile has a defect in DNA binding thus down-regulating the target genes resulting in reduced insulin secretion (Table 2). Moreover, the patient follow-up also showed that the patient (M-124) responded well to treatment change to SU, making this variant a clinically actionable one (Figure 4).
Our study has a few limitations. Since we could not obtain family samples for many patients, we were unable to conduct family co-segregation studies. In some patients, we did not have adequate clinical data.
In summary, this paper exemplifies the importance of performing molecular characterization after genetic testing, since the understanding of the functional basis of genotypes helps in understanding the phenotype which could lead to changes in clinical treatment for monogenic disorders like MODY. Our findings are the first to show the need of using additive scores during molecular characterization for accurate pathogenicity evaluations of HNF1A variants in precision medicine. Furthermore, it is also one of the first to introduce structural understanding to functional implications. The study has led to the delineation of the VUS into pathogenic and disease-causing MODY variants, from non-pathogenic variants. Patients with most pathogenic HNF1A variants benefit from OHA treatment; hence, this would assist clinicians in determining the best course of action for patients. While the combination of functional and structural-based approaches may lead to increased certainty in variant–phenotype correlation in a research setting, a functional understanding of the variants helps in precision diagnosis and treatment in a monogenic disorder such as MODY.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by Institutional ethics committee, MDRF. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
VR and BK designed and implemented the functional study. BK analyzed the data and wrote the manuscript. SR designed and performed the structural analysis. UV and NH analyzed the structural data. SG performed segregation analysis. VM collected the clinical data and analyzed the manuscript. VR analyzed all data and corrected the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the Indian Council of Medical Research (ICMR), India, through the project Functional Studies on Variants of Pancreatic β-cell genes (HNF1A, HNF4A, ABCC8, and KCNJ11) in monogenic diabetes – an experimental approach with clinical translational potential; grant no: No. 5/4/5-2/Diab/2020-NCD-III awarded to VR.
The authors thank the patients and their parents for giving the blood samples for the study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1177268/full#supplementary-material
1. Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature (1996) 384(6608):455–8. doi: 10.1038/384455a0
2. Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab (2008) 4(4):200–13. doi: 10.1038/ncpendmet0778
3. Radha V, Ek J, Anuradha S, Hansen T, Pedersen O, Mohan V. Identification of novel variants in the hepatocyte nuclear factor-1alpha gene in south Indian patients with maturity onset diabetes of young. J Clin Endocrinol Metab (2009) 94(6):1959–65. doi: 10.1210/jc.2008-2371
4. Kavvoura FK, Owen KR. Maturity onset diabetes of the young: clinical characteristics, diagnosis and management. Pediatr Endocrinol Rev (2012) 10(2):234–42.
5. Radha V, Mohan V. Genetic basis of monogenic diabetes. Curr Sci (2017) 113:1277–86. doi: 10.18520/cs/v113/i07/1277-1286
6. Broome DT, Pantalone KM, Kashyap SR, Philipson LH. Approach to the patient with MODY-monogenic diabetes. J Clin Endocrinol Metab (2021) 106(1):237–50. doi: 10.1210/clinem/dgaa710
7. Hattersley AT, Greeley SAW, Polak M, Rubio-Cabezas O, Njølstad PR, Mlynarski W, et al. ISPAD clinical practice consensus guidelines 2018: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes (2018) 19:47–63. doi: 10.1111/pedi.12772
8. Pontoglio M, Prié D, Cheret C, Doyen A, Leroy C, Froguel P, et al. HNF1Alpha controls renal glucose reabsorption in mouse and man. EMBO Rep (2000) 1(4):359–65. doi: 10.1093/embo-reports/kvd071
9. Steele AM, Shields BM, Shepherd M, Ellard S, Hattersley AT, Pearson ER. Increased all-cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabetes Med (2010) 27(2):157–61. doi: 10.1111/j.1464-5491.2009.02913.x
10. Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet (2003) 362(9392):1275–81. doi: 10.1016/S0140-6736(03)14571-0
11. Baumhueter S, Mendel DB, Conley PB, Kuo CJ, Turk C, Graves MK, et al. HNF-1 shares three sequence motifs with the POU domain proteins and is identical to LF-B1 and APF. Genes Dev (1990) 4(3):372–9. doi: 10.1101/gad.4.3.372
12. Tronche F, Yaniv M. HNF1, a homeoprotein member of the hepatic transcription regulatory network. BioEssays (1992) 14(9):579–87. doi: 10.1002/bies.950140902
13. Mendel DB, Crabtree GR. HNF-1, a member of a novel class of dimerizing homeodomain proteins. J Biol Chem (1991) 266(2):677–80. doi: 10.1016/S0021-9258(17)35222-5
14. Galán M, García-Herrero CM, Azriel S, Gargallo M, Durán M, Gorgojo JJ, et al. Differential effects of HNF-1α mutations associated with familial young-onset diabetes on target gene regulation. Mol Med (2011) 17(3-4):256–65. doi: 10.2119/molmed.2010.00097
15. Cooper DN, Krawczak M. Human gene mutation database. Hum Genet (2021) 98(5):629. doi: 10.1007/s004390050272
16. Ellard S, Colclough K. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha (HNF1A) and 4 alpha (HNF4A) in maturity-onset diabetes of the young. Hum Mutat (2006) 27(9):854–69. doi: 10.1002/humu.20357
17. Vaxillaire M, Abderrahmani A, Boutin P, Bailleul B, Froguel P, Yaniv M, et al. Anatomy of a homeoprotein revealed by the analysis of human MODY3 mutations. J Biol Chem (1999) 274(50):35639–46. doi: 10.1074/jbc.274.50.35639
18. Valkovicova T, Skopkova M, Stanik J, Gasperikova D. Novel insights into genetics and clinics of the HNF1A-MODY. Endocr Regul (2019) 53(2):110–34. doi: 10.2478/enr-2019-0013
19. Bjørkhaug L, Sagen JV, Thorsby P, Søvik O, Molven A, Njølstad PR. Hepatocyte nuclear factor-1 alpha gene mutations and diabetes in Norway. J Clin Endocrinol Metab (2003) 88(2):920–31. doi: 10.1210/jc.2002-020945
20. Bellanné-Chantelot C, Carette C, Riveline JP, Valéro R, Gautier JF, Larger E, et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes (2008) 57(2):503–8. doi: 10.2337/db07-0859
21. Balamurugan K, Bjørkhaug L, Mahajan S, Kanthimathi S, Njølstad PR, Srinivasan N, et al. Structure-function studies of HNF1A (MODY3) gene mutations in south Indian patients with monogenic diabetes. Clin Genet (2016) 90(6):486–95. doi: 10.1111/cge.12757
22. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
23. Starita LM, Ahituv N, Dunham MJ, Kitzman JO, Roth FP, Seelig G, et al. Variant interpretation: functional assays to the rescue. Am J Hum Genet (2017) 101(3):315–25. doi: 10.1016/j.ajhg.2017.07.014
24. Mohan V, Radha V, Nguyen TT, Radha V, Nguyen TT, Stawiski EW, et al. Comprehensive genomic analysis identifies pathogenic variants in maturity-onset diabetes of the young (MODY) patients in south India. BMC Med Genet (2018) 19(1):22. doi: 10.1186/s12881-018-0528-6
25. Thomas H, Badenberg B, Bulman M, Lemm I, Lausen J, Kind L, et al. Evidence for haploinsufficiency of the human HNF1Alpha gene revealed by functional characterization of MODY3-associated mutations. Biol Chem (2002) 383(11):1691–700. doi: 10.1515/BC.2002.190
26. Chèvre JC, Hani EH, Boutin P, Vaxillaire M, Blanché H, Vionnet N, et al. Mutation screening in 18 Caucasian families suggest the existence of other MODY genes. Diabetologia (1998) 41(9):1017–23. doi: 10.1007/s001250051025
27. Kaisaki PJ, Menzel S, Lindner T, Oda N, Rjasanowski I, Sahm J, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in MODY and early-onset NIDDM: evidence for a mutational hotspot in exon 4. Diabetes (1997) 46(3):528–35. doi: 10.2337/diab.46.3.528
28. Dusátková P, Průhová S, Sumník Z, Kolousková S, Obermannová B, Cinek O, et al. HNF1A mutation presenting with fetal macrosomia and hypoglycemia in childhood prior to onset of overt diabetes. J Pediatr Endocrinol Metab (2011) 24(5-6):377–9. doi: 10.1515/jpem.2011.083
29. Elbein SC, Teng K, Yount P, Scroggin E. Linkage and molecular scanning analyses of MODY3/hepatocyte nuclear factor-1 alpha gene in typical familial type 2 diabetes: evidence for novel mutations in exons 8 and 10. J Clin Endocrinol Metab (1998) 83(6):2059–65. doi: 10.1210/jcem.83.6.4874
30. Najmi LA, Aukrust I, Flannick J, Molnes J, Burtt N, Molven A, et al. Functional investigations of HNF1A identify rare variants as risk factors for type 2 diabetes in the general population. Diabetes (2017) 66(2):335–46. doi: 10.2337/db16-0460
31. Althari S, Najmi LA, Bennett AJ, Aukrust I, Rundle JK, Colclough K, et al. Unsupervised clustering of missense variants in HNF1A using multidimensional functional data aids clinical interpretation. Am J Hum Genet (2020) 107(4):670–82. doi: 10.1016/j.ajhg.2020.08.016
32. Malikova J, Kaci A, Dusatkova P, Aukrust I, Torsvik J, Vesela K, et al. Functional analyses of HNF1A-MODY variants refine the interpretation of identified sequence variants. J Clin Endocrinol Metab (2020) 105(4):dgaa051. doi: 10.1210/clinem/dgaa051
33. SIGMA Type 2 Diabetes Consortium, Estrada K, Aukrust I, Burtt NP, Mercader JM, García-Ortiz H, et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA. Diabetes Consortium. (2014) 311(22):2305–14. doi: 10.1001/jama.2014.6511
34. Brnich SE, Rivera-Muñoz EA, Berg JS. Quantifying the potential of functional evidence to reclassify variants of uncertain significance in the categorical and Bayesian interpretation frameworks. Hum Mutat (2018) 39(11):1531–41. doi: 10.1002/humu.23609
35. Zubkova N, Burumkulova F, Plechanova M, Burtt NP, Mercader JM, García-Ortiz H, et al. High frequency of pathogenic and rare sequence variants in diabetes-related genes among Russian patients with diabetes in pregnancy. Acta Diabetol (2019) 56(4):413–20. doi: 10.1007/s00592-018-01282-6
36. Pruhova S, Ek J, Lebl J, Sumnik Z, Saudek F, Andel M, et al. Genetic epidemiology of MODY in the Czech republic: new mutations in the MODY genes HNF-4alpha, GCK and HNF-1alpha. Diabetologia (2003) 46(2):291–5. doi: 10.1007/s00125-002-1010-7
Keywords: Maturity Onset Diabetes of Young (MODY) subtype-3, acmg-amp guidelines, re-interpretation, pathogenic variants, functional characterization, structural analysis, ACMG-AMP guidelines
Citation: Kavitha B, Ranganathan S, Gopi S, Vetrivel U, Hemavathy N, Mohan V and Radha V (2023) Molecular characterization and re-interpretation of HNF1A variants identified in Indian MODY subjects towards precision medicine. Front. Endocrinol. 14:1177268. doi: 10.3389/fendo.2023.1177268
Received: 01 March 2023; Accepted: 31 May 2023;
Published: 16 June 2023.
Edited by:
Tarunveer Singh Ahluwalia, Steno Diabetes Center Copenhagen (SDCC), DenmarkReviewed by:
Amalia Sertedaki, National and Kapodistrian University of Athens, GreeceCopyright © 2023 Kavitha, Ranganathan, Gopi, Vetrivel, Hemavathy, Mohan and Radha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Venkatesan Radha, cmFkaGFydkB5YWhvby5jby5pbjs=; ZHJyYWRoYUBtZHJmLmlu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.