 Keying Zhang
Keying Zhang Zhangning Fu
Zhangning Fu Yifan Zhang
Yifan Zhang Xiangmei Chen
Xiangmei Chen Guangyan Cai
Guangyan Cai Quan Hong
Quan Hong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 17 July 2023
Sec. Diabetes: Molecular Mechanisms
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1173933
This article is part of the Research TopicA Year in Review: Discussions in Diabetes: Molecular MechanismsView all 8 articles
Diabetic nephropathy (DN) is one of the most common complications of diabetes, and its main manifestations are progressive proteinuria and abnormal renal function, which eventually develops end stage renal disease (ESRD). The pathogenesis of DN is complex and involves many signaling pathways and molecules, including metabolic disorders, genetic factors, oxidative stress, inflammation, and microcirculatory abnormalities strategies. With the development of medical experimental techniques, such as single-cell transcriptome sequencing and single-cell proteomics, the pathological alterations caused by kidney cell interactions have attracted more and more attention. Here, we reviewed the characteristics and related mechanisms of crosstalk among kidney cells podocytes, endothelial cells, mesangial cells, pericytes, and immune cells during the development and progression of DN and highlighted its potential therapeutic effects
Diabetic nephropathy (DN) is one of the most frequent and serious microvascular complications of diabetes (1, 2). According to the World Health Organization, the worldwide prevalence of diabetes was estimated to rise from 2.8% in 2000 to 4.4% in 2030. The number of people with diabetes would rise from 171 million to 366 million during the three decades (3). It has been estimated that more than 40% of patients with diabetes would develop chronic kidney disease (CKD), and eventually develop end stage renal disease (ESRD) (4).
Mitochondrial dysfunction leads to microvascular dysfunction (5). Subsequently, the crosstalk with mesangial cells and podocytes further promotes the development of DN (6). Greka et al. also found that there is a close relationship between the decrease in the number of podocytes and the changes in podocyte shape (mainly manifested as podocyte foot process effacement). Podocytes surrounding the glomerular vascular wall, provide an anatomical location for crosstalk between podocytes and endothelial cells. Studies have also found that endothelial cells and podocytes of the filtration barrier can directly interact with Glomerular Basement Membrane (GBM) in a hyperglycemic environment, or regulate with mesangial cells through crosstalk to promote the progression of DN (6).
Renal tubulointerstitial lesions also play a very important role in the development of DN, for their ability to predict the prognosis of renal disease (7). Renal tubulointerstitial injury appears in the early stage of DN and precedes glomerular lesions (8). Interstitial fibrosis in DN is closely related to the injury of renal tubular epithelial cells. Tubular epithelial cells (TECs), the most important cellular component of the renal interstitium, have the function of reabsorption and excretion. Targeted inhibition of renal tubule SGLT2 protects renal function in patients with DN (9). The hyperglycemic and hypoxic environment of DN promotes renal interstitial vascular endothelial cells and pericytes to differentiate into fibroblasts. The dysfunction of endothelial cells aggravates renal tubular epithelial cell apoptosis and tubular atrophy. Damaged renal tubular epithelial cells further drive inflammatory cells into the tubulointerstitial. Moreover, renal tubular cells can interact with pericytes and infiltrating inflammatory cells, participating in the progression of DN (10–12).
Therefore, the crosstalk among kidney cells and interaction with immune cells play an important role in the occurrence and progression of DN. Many reports have confirmed the molecular mode of crosstalk, but there is a lack of systematic understanding. This paper summarizes the molecular mechanism of the interaction among different cells, hoping to provide new ideas for further research on the pathogenesis of DN and drug development.
A systematized narrative review was performed to identify the crosstalk of DKD. This review uses the literature study. The PubMed was searched to relevant articles. The keywords are crosstalk, diabetic nephropathy, intercellular communication, cellular crosstalk. A filter was placed to include only articles’ IF≥5. In addition, the reference lists of eligible studies were hand searched.
The inclusion criteria were:
1. Primary literature. The references must be authentic, complete, easy to search and verify, and avoid wrong citations.
2. The article focuses on the study of diabetic nephropathy caused by type 2 diabetes mellitus, especially the molecular mechanisms among renal cells.
3. The references mainly concentrate on basic research and pathogenesis of diabetic nephropathy.
The exclusion criteria were:
1. Secondary literature.
2. The references only focus on the drug research and treatment of diabetic nephropathy without considering the underlying mechanism.
3. Only the articles of single gene sequencing or simple experimental design were completed.
Podocytes and endothelial cells maintain capillary homeostasis by crosstalk in the normal physiological process. In the pathological condition of hyperglycemia, hemodynamic abnormality promotes the injury of podocytes and endothelial cells. The activation of intracellular signaling molecules leads to the secretion dysregulation of vascular growth factors such as vascular endothelial growth factor (VEGF) and TGF-β, which are involved in the regulation of abnormal angiogenesis, and eventually albuminuria (13).
The VEGF family mainly consists of three members. They are polypeptides that belong to the cystine-knot super-family of signaling proteins. A variety of subtypes are formed due to different exon shears, which constitute the vascular endothelial growth factor subtype family. Among them, VEGF-A plays a major role in vasculogenesis (14), and is therefore considered as the angiogenic factor. VEGF-A overexpression can cause glomerulomegaly, mesangial proliferation, podocyte effacement and lead to albuminuria (15). VEGF-B is a crucial factor in promoting abnormal lipid metabolism (16). VEGF-B is related to the promotion of lipid accumulation and lipotoxicity in podocyte. VEGF-C can disrupt the glomerular filtration barrier (17). Vascular endothelial growth factor C is involved in angiogenesis and lymphogenesis. Some research showed that amelioration of intrarenal inflammation and fibrosis is associated with attenuated lymphatic proliferation in the kidney (18).
The VEGF signaling pathway is one of the most important pathways involved in podocyte-endothelial cell interaction. In the early stage of DN, the VEGF signaling pathway is activated in the glomerulus, which leads to cell dysfunction and abnormal angiogenesis, eventually promoting glomerular cell hypertrophy and albuminuria (19).
In the kidney, VEGF-A is mainly expressed on glomerular podocytes. VEGF R2 on the surface of endothelial cells is phosphorylated after binding with VEGF and increases intracellular calcium ion level through PI3K/Akt signaling pathway. After binding with calmodulin enhances eNOS activity and increases NO production (20–22). On the pathological conditions of hyperglycemia, excessive production of VEGF-A produced by podocytes can induce abnormal angiogenesis of endothelial cells, leading to immature capillaries in the glomerulus (14). Overactivation of VEGFR2 also increases the production of ROS, especially superoxide (O2-), aggravating endothelial injury, and leading to the occurrence and progression of the renal microvascular disease (23, 24). Increased expression levels of VEGF-A in the blood lead to changes in GBM and a decrease in the glycocalyx of endothelial cells (25), which further promotes the increase of vascular permeability and causes leakage. Although the activation of the VEGF/VEGFR signaling pathway in the early stage of DN leads to the formation of new blood vessels and glomerular injury, excessive loss of podocytes in the late stage leads to the weakening of VEGF signaling, which further leads to vascular thinning and renal fibrosis, aggravating the development of DN (26). Both loss and overexpression of VEGF lead to glomerular abnormality; loss of VEGF-A prevents glomerular angiogenesis and the development of a glomerular filtration barrier (27). Overexpression of VEGF-A in podocytes is similar in different renal diseases (28). For example, there is no difference between glomerulopathy caused by overexpression of VEGF164 and glomerulopathy caused by early diabetes nephropathy (15, 29).
Nowadays, increased use of glucose-lowering agents and glycemic control have not resulted in a reduced prevalence of DN. More and more pathogeneses are considered to drive the development of DN (30). Thus, we looked for new mechanisms. In muscle, VEGF-B has been shown to control lipid accumulation through regulation of endothelial fatty acid (FA) transcytosis, and it may thus be a potential target in treating type 2 diabetes (T2DM) (31) VEGF-B was overexpressed in podocytes, while VEGFR1 was mainly found to be expressed on endothelial cells. The article showed reducing VEGF-B signaling could ameliorate glomerular lipotoxicity and, as a consequence, the progression of DN. Glomerular VEGF-B levels were upregulated in subjects with DN, suggesting that anti-VEGF-B treatment may be useful as a therapeutic strategy to treat DN in humans (32).
VEGF-C can protect endothelial cells from the influence of VEGF-A reduction on cell permeability and plays a role in protecting endothelial cells (33). Additionally, apart from blood glucose, increased levels of advanced glycation end products (AGEs) can also promote VEGF expression in podocytes, thereby increasing the oxidative stress response of endothelial cells and podocytes (34, 35).
TGF-β is a multifunctional dipeptide. It is a family of factors that promote cell growth and differentiation, consisting of more than 30 proteins with similar structures (36). TGF-β1 is one of the fibrogenic factors secreted by glomerular endothelial cells, glomerular mesangial cells, and renal tubule epithelial cells.
Unlike the VEGF signaling pathway necessary to maintain the glomerular filtration barrier, in a high glucose environment, the over-activation of the TGF- β signaling pathway is harmful to mesangial cells, podocytes, and endothelial cells. TGF- β 1 can induce the occurrence of epithelial-mesenchymal transition (EMT) (37) and the accumulation of extracellular matrix (ECM) in renal tubular epithelial cells, which is closely related to the progression of renal interstitial fibrosis in DN. Inhibiting the TGF- β 1 signaling pathway can reduce EMT and fibrosis of DN (38).
In addition, in the diabetes mouse model, TGF- β signaling pathway can induce dedifferentiation or apoptosis of endothelial cells and podocytes (39). TGF-β1 secreted by endothelial cells binds to TGF-βR1 of podocytes, which can activate downstream Smad signals, including Smad2, Smad3, and Smad7 (40). Activation of Smad2/3 promotes the secretion of ECM, leading to glomerular sclerosis and fibrosis, while Smad7 induces apoptosis of podocytes by blocking the activity of NF-kB. Meanwhile, Smad7 can directly activate caspase3. Cysteingl aspartate specific protease(Caspase)is an important cytokine in the molecular mechanism of cell apoptosis. Smad7 and p38MAPK induce podocyte apoptosis by activating caspase3 (41). Similarly, in high glucose condition, the endothelial cell secretes TGF- β to promote apoptosis and ultimately aggravates renal microvascular disease (42).
Angiotensin (Ang 1/Ang 2) plays an important role in maintaining endothelial integrity and participates in the pathophysiological process of DN. Ang-1 is mainly produced by podocytes and can promote angiogenesis and microvascular growth by binding to the Tie2 receptor expressed by glomerular endothelial cells, reducing the permeability of endothelial cells and regulating the VEGF signaling (43, 44).
Ang2 is mainly produced by GECs (Glomerular Endothelial Cells) and inhibits Ang1 through competitive binding to the Tie-2 receptor (45). Studies have demonstrated that Ang-2 expression is up-regulated in the glomerulus of diabetic nephropathy patients, which antagonists Ang-1 induced Tie 2 activation, and thus inhibits the anti-apoptotic effect of Ang-1 on endothelial cells (46). In pathological conditions of DN, decreased glomerular VEGF-A expression is accompanied by increased Ang-2/Ang-1 ratio, resulting in increased apoptosis of endothelial cells (47).
Endothelin (ET-1) is a vasoconstrictive peptide mainly produced by GECs.ET-1 binds to two subtypes of receptors, the endothelin A receptor (ETAR) and the endothelin B receptor (ETBR) (48). Studies about animal models of diabetes have shown the expression of ET-1 in glomeruli was increased by 5 times compared with the receptor, suggesting that ET-1 plays an important role in DN (49). In the pathological conditions of diabetes, ET-1 is associated with vasoconstriction, kidney injury, mesangial hyperplasia, glomerulosclerosis, fibrosis, and inflammation (50). ETAR is mainly expressed in podocytes of the glomerulus. ET-1 combined with ETAR to promote vasoconstriction, cell proliferation, fibrosis, podocyte injury, and inflammatory response. ETBR is mainly distributed in glomerular endothelial cells and renal tubular epithelial cells. ET-1 combined with ETBR can promote vascular dilation, anti-proliferation, and anti-fibrosis, and play a protective role.
ET-1 can also be secreted by podocytes. In DN progression, TGF-β signaling is activated in podocytes, followed by increased secretion of ET-1. Binding to ETAR in GECs mediates mitochondrial oxidative stress and adjacent endothelial cell dysfunction (35, 51, 52). Selective blocking of ETAR has been demonstrated to reduce the expression of chemokines and cytokines, as well as decrease the secretion of various mediators of renal fibrosis. It can prevent podocyte loss, albuminuria, and glomerulosclerosis (53).
ET-1 seems to be a key mediator in podocytes-to-ECs and ECs-to-podocytes communications promoting cell injury in several renal pathologies including DN. Endothelin A (ETA) receptor antagonist—Atrasentan entered the field of vision as a new drug, and has got some achievements in DN. The SONAR (The Study of Diabetic Nephropathy with Atrasentan) (54) is the first trial to evaluate the long-term effectiveness of ETA receptor antagonists on albuminuria DN. The results showed that Atrasentan reduced the risk of progression to CKD (including DN) by 35% compared with placebo and was safe and well tolerated.
GECs directly contact and interact with mesangial cells in structure. In the high glycemic environment, the crosstalks between GEC and GMC (Glomerulus Mesangial Cells) induce the inflammatory responses of the kidney and glomerulosclerosis (55, 56), which damage the integrity of the glomerular tissue and cause renal dysfunction.
PDGFB is mainly expressed in GECs, while PDGFR-β is mainly expressed in MCs. PDGFB/PDGFR is the main medium in the crosstalk between GEC and MCs (57). During glomerular development, the interaction of GEC and MCs promotes mesangial cell maturation and maintains through the PDGF-B/PDGFR signaling pathway (58). The PDGFB/PDGFR signaling pathway is activated in the glomeruli of diabetic mice, which promotes the progression of DN, and the mechanism may be related to oxidative stress and mesangial expansion (59). It has been demonstrated that GECs paracrine PDGFB to regulate mesangial cell proliferation, and the inhibition of PDGFB and its receptor can inhibit mesangial proliferation in diabetic rats (51). The expression of PDGFB in the kidney of diabetic nephropathy patients is significantly increased (60). The non-expression of PDGFB and its receptor PDGFR-β can reduce the accumulation of extracellular matrix and mesangial cells proliferation (61). In addition to mesangial cells, renal vascular smooth muscle cells (SMC) are also the target of PDGF-B signaling, which accelerates the development or neovasculature of blood vessels by promoting the proliferation of SMC (62).
ET-1 may be involved in the occurrence of diabetic vascular diseases through mesangial cell proliferation, promoting fibrosis and inflammation.ET-1, one of the most effective vasoconstrictors and a growth factor for mesangial cells (48, 63), is positively associated with increased albumin excretion in patients with diabetic nephropathy. ET-1 is secreted by endothelial cells, binds to ETAR and ETBR on mesangial cells, and exerts its effect. For example, mesangial proliferation and ECM accumulation are accelerated by up-regulating the expression of ECM-related genes (64). Some studies have shown that HG activates RhoA/ROCK signaling pathway in mesangial cells and promotes the progression of DN. ET-1 mediates ETAR activation in mesangial cells and enhances the expression of ECM-related genes (65, 66). Studies have shown that high glucose activates the RhoA/ROCK pathway in mesangial cells and promotes the progression of DN, which also depends on the secretion of ET-1. On the other hand, the combination of ET-1 with ETBR can inhibit the NF-κB signaling pathway and reduce the secretion of ET-1 by endothelial cells. This negative feedback reduces the inflammatory responses of endothelial cells (63).
In the early stage of DN, renal tubules are damaged to varying degrees, and tubular epithelial cell damage can lead to abnormal endothelial cell function. The crosstalk between the two kinds of cells induces inflammatory responses and promotes the occurrence and progress of DN.
As the first barrier of the glomerular filtration membrane, GECs come into directly contact with substances in the circulating blood and are more vulnerable to damage by inflammatory factors (67). Except for functioning in the interaction between podocytes and endothelial cells, VEGF/VEGFR signaling pathway also plays an important role in the interaction between epithelial cells and endothelial cells in the renal tubulointerstitial. In the diabetic environment, tubule epithelial cells (TECs) are susceptible to hemodynamic changes in metabolic disorders, resulting in the secretion of multiple inflammatory mediators, leading to interstitial inflammation. Urinary albumin in DN patients activates TECs to produce pro-inflammatory factors such as CRP, IL, TNF-α, NF-κB, and ROS, which can lead to GEC injury, apoptosis, and EndMT. Stimulated by inflammatory factors, the glomerular vascular network suffered apoptosis and necrosis, and the structure and function of GECs were destroyed. The injured GEC reduced the blood supply to the renal tubules, resulting in increased TEC injury (68). The interaction aggravates the progress of DN.
In addition to playing a role in the interaction between podocytes and endothelial cells, VEGF/VEGFR signaling also plays an important role in the interaction between epithelial cells and endothelial cells in the renal tubulointerstitial. In the renal tubulointerstitial, VEGF-A can also be synthesized and secreted by renal tubule epithelial cells (TECs) and subsequently combines with VEGFR of endothelial cells (GECs) to regulate the structure and function of GECs (69). VEGF/VEGFR activation in early DN leads to neovascularization. However, in the late stage of DN, the aggravation of TEC injury and deletion in late DN is accompanied by decreased production of VEGF-A, decreased endothelial cell angiogenesis and permeability of endothelial cells, and endothelial function destruction (70). Subsequently, eNOS deficiency, NO reduction, interstitial ischemia, and hypoxia occur, leading to TEC damage, decreasing VEGF-A synthesis, and aggravating a vicious cycle in disease progression (71).
The abnormalities of podocytes and mesangial cells play a key role in the development of DN. Recent researches suggest that crosstalks between the two types of cells may be mediated by exosomes. Exosomes are spherical extracellular vesicles (ECVs) with a phospholipid bilayer membrane structure that are 40-100 nm in diameter and contain a variety of molecules, such as proteins, lipids, DNA, miRNA, and LncRNA. Normally, exosomes can be secreted by kidney cells, such as podocytes and mesangial cells. In recent years, a large number of data have proved that exosomes are involved in the glomerular, renal tubule, and tubulointerstitial lesions, which are related to the pathological changes and prognosis of DN (72, 73). In the high glucose condition, exosomes released by GMCs may affect podocyte function by carrying TGF-β1, participating in the pathological process of DN, and eventually leading to albuminuria (74, 75).
Podocyte and Endothelial cell:In the pathological condition of high glucose, the abnormal renal expression of VEGF, angiopoietins, TGF- β, and endothelin-1 in early Diabetic nephropathy (DN) induces endothelial cell dysfunction and contributes to the disappearance of podocyte foot processes.
Endothelial cell and Mesanginal cell:PDGFB secreted by GECs binds to PDGFR-βR on mesangial cells, which contributes to the development of mesangial cells. ET-1 is a growth factor of mesangial cells. ET-1 may be involved in the pathogenesis of diabetic vascular diseases through mesangial cell proliferation, promoting fibrosis and promoting inflammation.
Podocyte and Macrophage:Macrophage is the main immune cell that causes kidney injury in DN. Infiltration, recruitment, and activation of macrophages can lead to the generation and release of many inflammatory factors, pro-fibrotic factors, and anti-angiogenic factors, such as TNF-α, ROS, IL-1, IL-6, TGF-β, and VEGF, which can interact with podocytes.
The single-cell sequencing study by Fu et al. provided direct evidence that macrophages were the main infiltrating immune cells in the kidney tissue of diabetic mice, and M1 macrophages were more significant in early DN (76). Experimental evidence also confirmed that there were different degrees of immune cell infiltration in renal tissues of DN patients, which was related to DN staging (77). All these phenomena suggest that in the diabetic environment, macrophages interact with cytokines, causing renal function damage and accelerating the progression of DN (78).
Toll-like receptor (TLRs) protein is an important receptor that can regulate immune response and inflammatory diseases. TLR4 expression was increased in macrophages of diabetic nephropathy patients, while TLR2 expression was not changed (79). The expression level of TLR4 was directly related to the infiltration of tubulointerstitial macrophages and was inversely proportional to the glomerular filtration rate. In the TLR4 knockout diabetes mice, the infiltration of interstitial macrophages, proteinuria, and progress of renal function was significantly improved. Itsmechanism was confirmed by cell experiments: high glucose induces TLR4 expression by activating PKC in human proximal tubule epithelial cells, and the activation of IkB/NF-κB leads to up-regulated expression of IL-6 and CCL-2 and leads to the inflammatory response (79). Therefore, TLR4 mediates the action of macrophages and renal tubular epithelial cells to increase the tubulointerstitial inflammatory response and promote diabetic kidney injury.
TEC-derived exosome (EVe) activates the inflammatory phenotype of macrophages, inducing the expression and release of proinflammatory cytokines, and inducing the release of macrophage-derived exosome (EVm). EVm can also induce the apoptosis of lipotoxic TECs, thus forming a vicious cycle, and promoting kidney inflammation and damage in DN (80). Mechanism studies have shown that Eve contains high levels of miR-19b-3p. It can activate the NF-κB signaling pathway by targeting the inhibition of SOCS-1, leading to the phenotype polarization of M1 macrophages, further increasing the expression of MCP-1, IL-1β, and other renal inflammatory cytokines, and promoting the occurrence of renal tubulointerstitial inflammation in diabetic nephropathy (81). Vascular lesions caused by diabetes lead to renal ischemia and hypoxia. Li et al. confirmed that renal tubular epithelial cells in an anoxic environment could release mirNA-23A-rich exosomes, which could be absorbed by macrophages and transformed into pro-inflammatory phenotypes through the HIF-1α pathway, promoting tubulointerstitial inflammation (82). These studies confirmed that microRNAs in exosomes mediate interactions between renal tubular epithelial cells and macrophages in DN tubulointerstitial inflammation.
Infiltration, recruitment and activation of macrophages can lead to the generation and release of many inflammatory, pro-fibrotic and anti-angiogenic factors. For example, TNF-α, ROS, IL-1, IL-6, TGF-β and VEGF can interact with podocytes (83).In addition, cytokines act on podocytes, the major components of the filtration barrier, through multiple signaling pathways, such as p38 MAPK, NF-κB, Toll-like receptors, or proteins, ultimately damage renal cell and aggravate the progression of the disease.
Pericytes are a class of undifferentiated cells. It is generally believed that mesenchymal cells adjacent to microvascular endothelial cells are pericytes (84). In the kidney, pericytes are highly specialized cells that make up 30% of the total tissue and are involved in the regulation of glomerular ultrafiltration. It plays an important role in the pathophysiological activities of microvascular. PDGFR-α and -β were expressed only in pericytes and myofibroblasts. Inhibition of PDGF signaling by imatinib or neutralizing PDGFR antibodies can reduce macrophage infiltration and fibrosis (85). Hu et al. showed that C1q tumor necrosis factor-associated protein-3 could reduce mesangial cell proliferation and extracellular matrix accumulation induced by high glucose, and inhibit pericytes to differentiate into mesangial cells (86). This evidence suggests that the interaction between pericytes and other cells also plays a significant role in the progression of DN.
Endothelial cell and Renal tubular epithelial cell:The crosstalk between endothelial cells and renal tubular epithelial cell plays an important role in the occurrence and development of DN. There are many signaling pathways between GECs and TECs, in which crosstalk plays a vast role. The abnormal secretion of VEGF and inflammatory factors(such as CRP、IL-6、TNF-αand so on) promote injury to GECs during the progress of DN.
Tubular epithelial cells and macrophages:Macrophage infiltration around renal tubular epithelial cells (TECs) is a hallmark of DN. In the diabetic environment, macrophages interact with tubule epithelial cells through cytokines, causing renal function damage and accelerating the progression of DN.
Pericyte and other cells:There are many common signaling pathways among podocytes, mesangial cell endothelial cells, and pericytes, in which crosstalk plays a vast role, Such as C1q(Complement 1q).
Recently, new therapeutics including sodium-glucose transport protein 2 (SGLT2) inhibitors, endothelin antagonists, glucagon like peptide-1 (GLP-1) agonists, and mineralocorticoid receptor antagonists (MRA), have provided additional treatment options for patients with DN.
Recently, SGLT2 inhibitors have emerged as a new class of drugs, to control blood sugar by increasing the excretion of sugar in the urine. Ipragliflozin, was injected into streptozotocin induced diabetic mice to reduce the blood glucose level of diabetic mice. The experimental results showed that it alleviated the damage of diabetic endothelial function, improved the phosphorylation of eNOS, and reduced the expression of inflammatory molecules (87).
Furthermore, Glucagon-like peptide-1 (GLP-1) is an intestinal hormone. It has the properties of increasing insulin secretion and inhibiting glucagon secretion after eating. It plays an important role in maintaining blood glucose homeostasis (88). The inhibition of oxidative stress, inflammation, fibrosis, and induction of natriuresis have been mainly implicated as mechanisms underlying the attenuation of DN by GLP-1 receptor agonists.GLP-1 receptor agonists have a renal protective effect in addition to hypoglycemic effects (89).
Dipeptidyl peptidase-4 (DPP-4) inhibitors reduce the levels of blood glucose by increasing the half-life of short-lived endogenous incretins, such as GLP-1 and glucose-dependent insulinotropic polypeptide (90). DPP-4inhibitors (insulin-based therapy), a new class of hypoglycemic agents for clinical practice, their role in diabetic nephropathy, with a particular focus on renal protection and alternative markers of cardiovascular disease (91). Trials such as SAVOR-TIMI 53 (92) and CARMELINA (93) showed a possible reduction in albuminuria and improvement in the histological changes of kidney in patients receiving DPP-4 inhibitors.
In addition to GLP-1 and SGLT2 discussed earlier, there is a new class of drugs called MRAs (Mineralocorticoid receptor antagonists).MRAs are drugs that inhibit the effect of aldosterone on mineral mineralocorticoid receptor (MR), such as fenelidone (94). MRAs block MR-mediated sodium reabsorption and MR Overactivation in renal tissues. In patients with diabetic nephropathy, MRA reduces the upregulation of pro-inflammatory mediators, including TGF-β, PDGF,CCL2 and so on (95). In preclinical studies, the protective effect of MRAs in animal models of diabetes nephropathy was reported in 2001 (96). Mineralocorticoid receptor-mediated inflammation has been proposed to be partially caused by injury, oxidative stress or cell apoptosis (95).
Glomerular endothelial cells, podocytes and renal tubular epithelial cells play important roles in the pathophysiological progression of DN. Many studies have proved that crosstalk among them, as well as neighboring pericytes and immune cells, which is related to the progression of diabetic nephropathy (As shown in the Figures 1, 2). The cell damage caused by this crosstalk is oxidative stress; macrophage infiltration, inflammatory response, the increasing secretion of TGF-b; abnormal angiogenesis caused by increased secretion of VEGF. There is a table (As shown in the Table 1) with important cellular crosstalks that contribute to understand the progression of diabetic nephropathy.
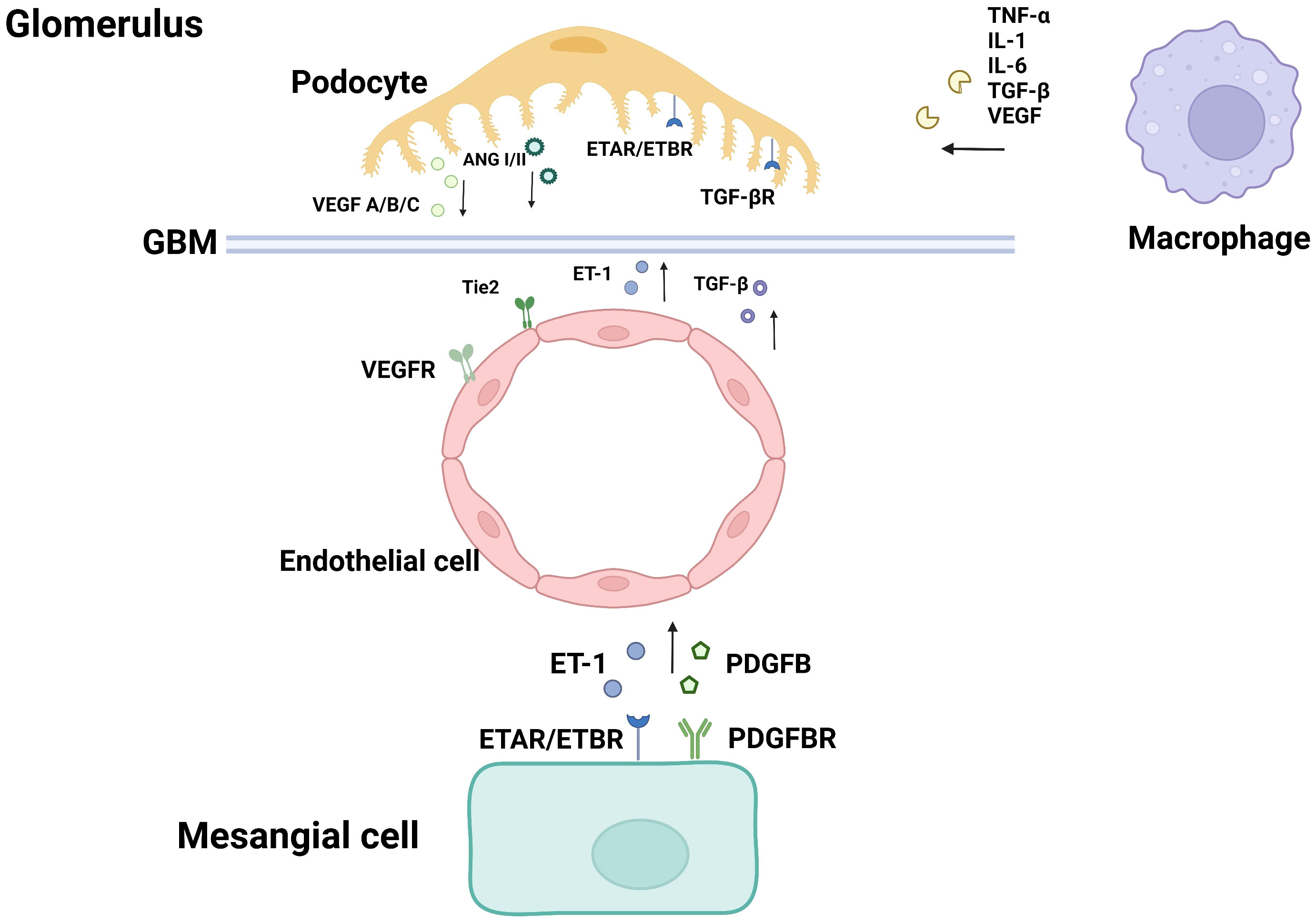
Figure 1 Cellular crosstalk in the glomerulus.
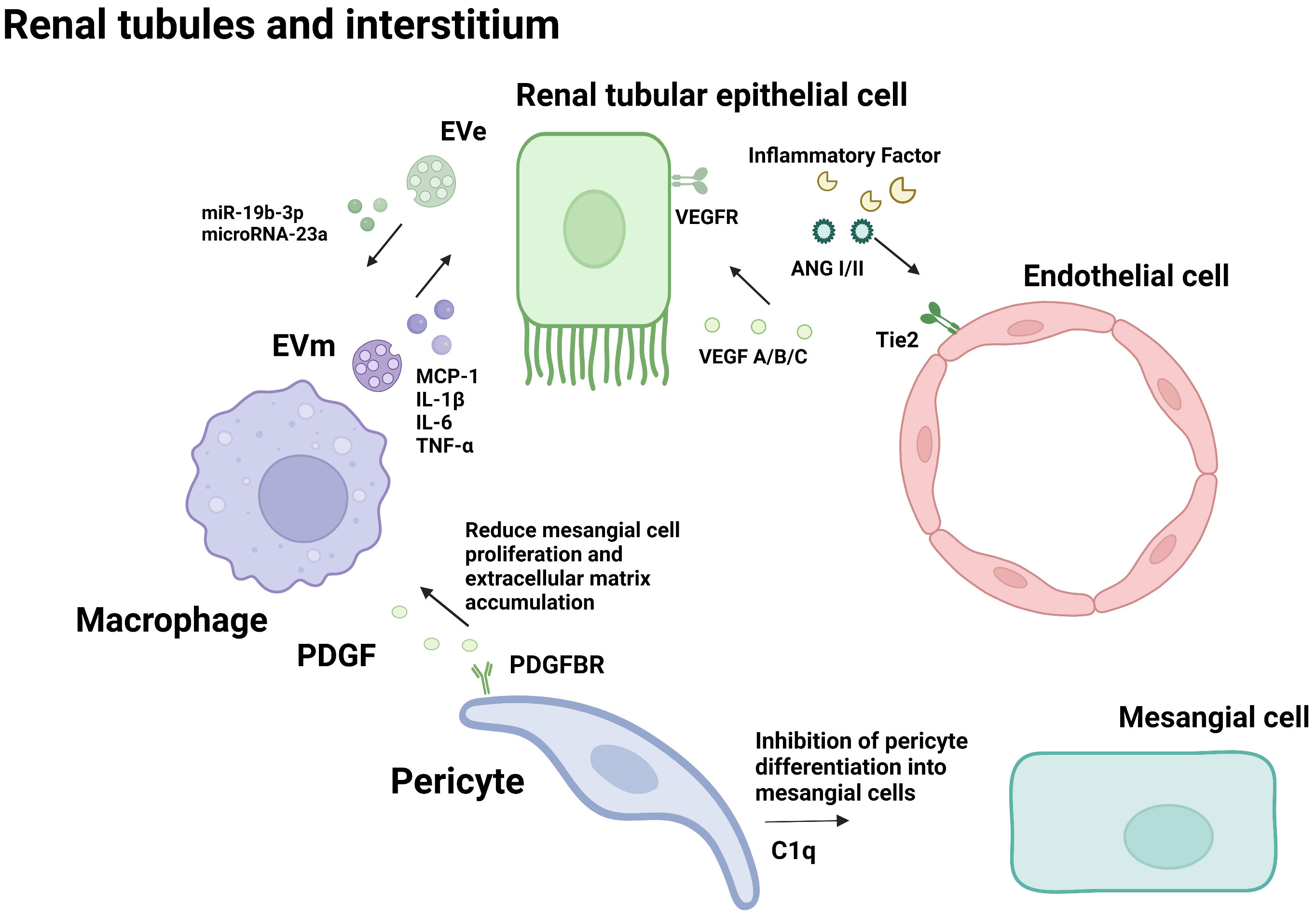
Figure 2 Cellular crosstalk in the renal tubules and interstitium.
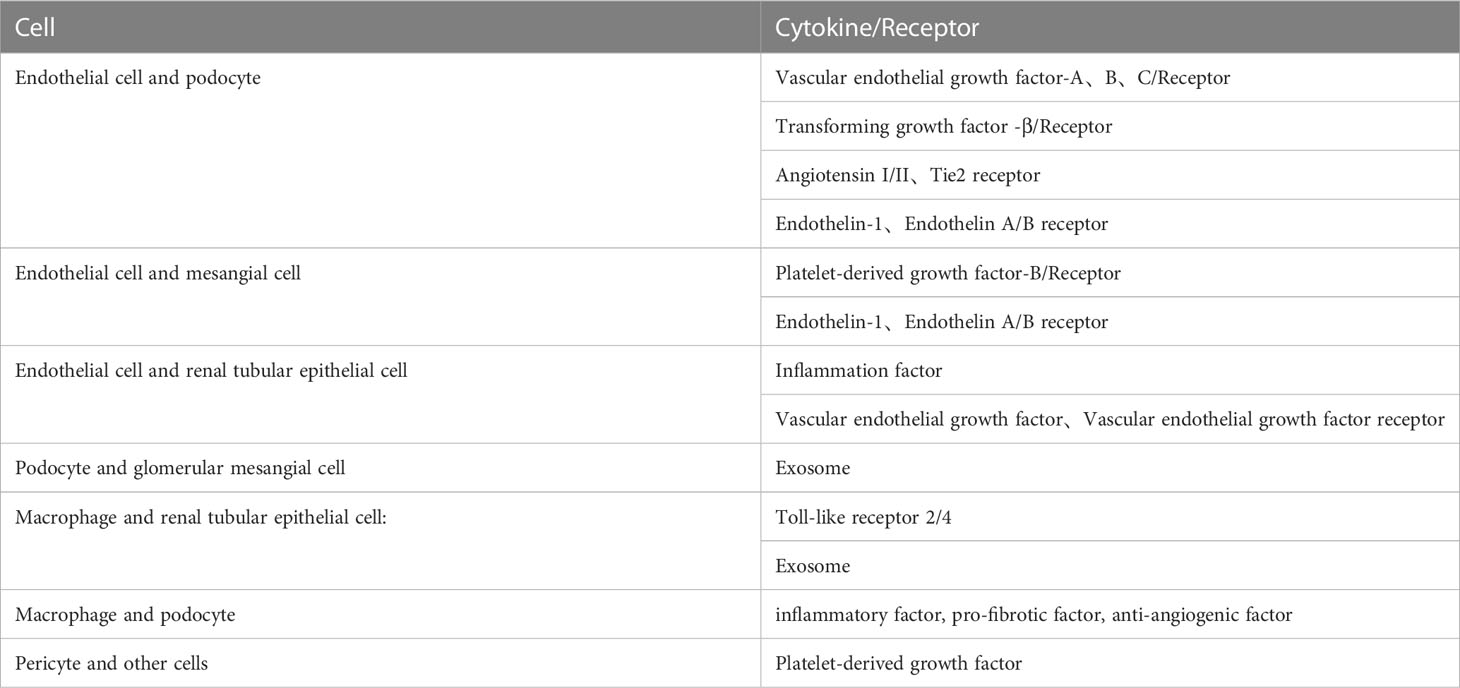
Table 1 The full search words about crosstalk between renal cells and other cells in the DN.
At present, many studies need to be further clarified on the role of cell crosstalk in the occurrence and development of DN. For example, the mechanism of VEGF and TGF-β. The VEGF-A and VEGF-C crosstalks are controversial. VEGF-A is a critical mediator of angiogenesis and vasculogenesis and is involved in the formation of glomerular filtration barriers. Human kidney biopsies showed high VEGF-A expression at early stages of DN, and lower VEGF-A expression in patients with more advanced stage of DN because loss of podocyte (14). Studies suggest that in the diabetic nephropathy, the disruption of podocyte function after VEGF-A depletion arises from endothelial cell dysfunction. Excessive VEGF-A production results in neovascularization leading to pathologic microangiopathy. In the later stages, the reduction of VEGF signaling may occur due to the loss of podocytes, contributing to vascular rarefication and renal fibrosis. VEGF-C is mainly involved in lymphangiogenesis and binds to VEGFR-2, VEGFR-3.Some studies showed that renal inflammation and fibrosis were improved in diabetic mice by down-regulating VEGF-C and VEGFR-3 expression (97). However, in DN, a chronic proinflammatory state, the overgrown lymph eventually becomes incomplete and dysfunctional due to chronic upregulation of VEGF-C (98). Its detailed and systematic mechanisms still need further study.
In addition, in DN, early studies have shown that that crosstalk between TGF-β and hormones has a complex mechanism in DN. The data on diabetic nephropathy and TGF signaling pathway appear to be controversial. The results suggest that sex, sex hormones and diabetic conditions influence differences in expression of TGF-β1, its receptor and bone morphogenetic protein 7 (BMP7). Complex crosstalk between sex hormones, sex-dependent expression pattern and profibrotic signals for the precise course of DN development (92). So, we still have a lot to figure out about crosstalk between cells.
The interaction of glomerular endothelial cells, podocytes, renal tubular epithelial cells, and other cells is closely related to the progression of DN. The pathogenesis of DN is numerous and complex, and it remains unknown. The interaction among cells is the key factor that promotes DN progression. The new treatment plan to improve the damage to kidney cells and maintain the normal crosstalk among cells may become a new strategy for the prevention and treatment of DN in the future. Targeted drug research on crosstalk among cells to maintain cell function may be a new perspective, which will provide a brand-new strategy for the prevention and treatment of DN.
KZ, YZ, and ZF oversaw the preparation of the manuscript and wrote the first draft. XC, GC, and QH design this topic and give guidance. All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China (NO.82204744, 82270758 and 82070741) and National KeyResearch and Development Project (2018YFE0126600).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fonseca VA, Bennett MV, Booker B, Cefalu WT, Clark N, Deeb LC, et al. American Diabetes association statement on emergency and disaster preparedness - a report of the disaster response task force. Diabetes Care (2007) 30(9):2395–8.
2. Warram JH, Gearin G, Laffel L, Krolewski AS. Effect of duration of type I diabetes on the prevalence of stages of diabetic nephropathy defined by urinary albumin/creatinine ratio. J Am Soc Nephrol (1996) 7(6):930–7. doi: 10.1681/ASN.V76930
3. Rathmann W, Giani G. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030 - response to wild et al. Diabetes Care (2004) 27(10):2568–9. doi: 10.2337/diacare.27.10.2568
4. Gheith O, Farouk N, Nampoory N, Halim MA, Al-Otaibi T. Diabetic nephropathy: world wide difference of prevalence and risk factors. J Nephropharmacol (2016) 5(1):49–56.
5. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell (2013) 154(3):651–63. doi: 10.1016/j.cell.2013.06.037
6. Fu J, Lee K, Chuang PY, Liu Z, He JC. Glomerular endothelial cell injury and cross talk in diabetic nephropathy. Am J Physiology-Renal Physiol (2015) 308(4):F287–97. doi: 10.1152/ajprenal.00533.2014
7. Mise K, Hoshino J, Ueno T, Hazue R, Sumida K, Hiramatsu R, et al. Clinical and pathological predictors of estimated GFR decline in patients with type 2 diabetes and overt proteinuric diabetic nephropathy. Diabetes-Metab Res Rev (2015) 31(6):572–81. doi: 10.1002/dmrr.2633
8. Levin A, Rocco M. KDOQI clinical practice guideline and clinical practice recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis (2007) 50(3):471–530. doi: 10.1053/j.ajkd.2006.03.010
9. Defronzo RA, Norton L, Abdul-Ghani M. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat Rev Nephrol (2017) 13(1):11–26. doi: 10.1038/nrneph.2016.170
10. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med (2010) 16(5):535–U27. doi: 10.1038/nm.2144
11. Liu B-C, Tang T-T, Lv L-L, Lan HY. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int (2018) 93(3):568–79. doi: 10.1016/j.kint.2017.09.033
12. Lipphardt M, Song JW, Matsumoto K, Dadafarin S, Dihazi H, Müller G, et al. The third path of tubulointerstitial fibrosis: aberrant endothelial secretome. Kidney Int (2017) 92(3):558–68. doi: 10.1016/j.kint.2017.02.033
13. Forbes JM, Fukami K, Cooper ME. Diabetic nephropathy: where hemodynamics meets metabolism. Exp Clin Endocrinol Diabetes (2007) 115(2):69–84. doi: 10.1055/s-2007-949721
14. Tufro A, Veron D. VEGF and podocytes in diabetic nephropathy. Semin Nephrol (2012) 32(4):385–93. doi: 10.1016/j.semnephrol.2012.06.010
15. Veron D, Reidy KJ, Bertuccio C, Teichman J, Villegas G, Jimenez J, et al. Overexpression of VEGF-a in podocytes of adult mice causes glomerular disease. Kidney Int (2010) 77(11):989–99. doi: 10.1038/ki.2010.64
16. Karpanen T, Bry M, Ollila HM, Seppänen-Laakso T, Liimatta E, Leskinen H, et al. Overexpression of vascular endothelial growth factor-b in mouse heart alters cardiac lipid metabolism and induces myocardial hypertrophy. Circ Res (2008) 103(9):1018–26. doi: 10.1161/CIRCRESAHA.108.178459
17. Shen Y, Chen W, Han L, Bian Q, Fan J, Cao Z, et al. VEGF-b antibody and interleukin-22 fusion protein ameliorates diabetic nephropathy through inhibiting lipid accumulation and inflammatory responses. Acta Pharm Sin B (2021) 11(1):127–42. doi: 10.1016/j.apsb.2020.07.002
18. Stevens M, Neal CR, Salmon AHJ, Bates DO, Harper SJ, Oltean S, et al. VEGF-A(165) b protects against proteinuria in a mouse model with progressive depletion of all endogenous VEGF-a splice isoforms from the kidney. J Physiol (2017) 595(19):6281–98. doi: 10.1113/JP274481
19. Dei Cas A, Gnudi L. VEGF and angiopoietins in diabetic glomerulopathy: how far for a new treatment? Metabolism-Clinical Exp (2012) 61(12):1666–73. doi: 10.1016/j.metabol.2012.04.004
20. Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest (1997) 100(12):3131–9. doi: 10.1172/JCI119868
21. Tsurumi Y, Murohara T, Krasinski K, Chen D, Witzenbichler B, Kearney M, et al. Reciprocal relation between VEGF and NO in the regulation of endothelial integrity. Nat Med (1997) 3(8):879–86. doi: 10.1038/nm0897-879
22. Gao N, Ding M, Zheng JZ, Zhang Z, Leonard SS, Liu KJ, et al. Vanadate-induced expression of hypoxia-inducible factor 1 alpha and vascular endothelial growth factor through phosphatidylinositol 3-kinase/Akt pathway and reactive oxygen species. J Biol Chem (2002) 277(35):31963–71. doi: 10.1074/jbc.M200082200
23. Daehn IS. Glomerular endothelial cell stress and cross-talk with podocytes in early diabetic nephropathy. Front Med (2018) 5:76. doi: 10.3389/fmed.2018.00076
24. Sison K, Eremina V, Baelde H, Wang M, Quaggin SE. Glomerular structure and function require paracrine, not autocrine, VEGF VEGFR-2 signaling. J Am Soc Nephrol (2011) 22(7):1390–0. doi: 10.1681/ASN.2010030295
25. Xia Y, Li Y, Fu BM. Differential effects of vascular endothelial growth factor on glycocalyx of endothelial and tumor cells and potential targets for tumor metastasis. APL Bioeng (2022) 6(1):016101. doi: 10.1063/5.0064381
26. Yang J, Liu Z. Mechanistic pathogenesis of endothelial dysfunction in diabetic nephropathy and retinopathy. Front Endocrinol (2022) 12(1):13. doi: 10.3389/fendo.2022.816400
27. Ingber DE, Folkman J. Mechanochemical switching between growth and differentiation during fibroblast growth factor-stimulated angiogenesis in vitro: role of extracellular matrix. J Cell Biol (1989) 109(1):317–30. doi: 10.1083/jcb.109.1.317
28. Eremina V, Sood M, Haigh J, Nagy A, Quaggin SE. Glomerular-specific alterations of VEGF-a expression lead to distinct congenital and acquired renal diseases. J Clin Invest (2003) 111(5):707–16. doi: 10.1172/JCI17423
29. Veron D, Reidy K, Marlier A, Bertuccio C, Villegas G, Jimenez J, et al. Induction of podocyte VEGF164 overexpression at different stages of development causes congenital nephrosis or steroid-resistant nephrotic syndrome. Am J Pathol (2010) 177(5):2225–33. doi: 10.2353/ajpath.2010.091146
30. De Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic nephropathy in the united states. Jama (2011) 305(24):2532–9. doi: 10.1001/jama.2011.861
31. Hagberg CE, Mehlem A, Falkevall A, Muhl L, Fam BC, Ortsäter H, et al. Targeting VEGF-b as a novel treatment for insulin resistance and type 2 diabetes. Nature (2012) 490(7420):426–30. doi: 10.1038/nature11464
32. Falkevall A, Mehlem A, Palombo I, Sahlgren BH, Ebarasi L, He L, et al. Reducing VEGF-b signaling ameliorates renal lipotoxicity and protects against diabetic nephropathy. Cell Metab (2017) 25(3):713–26. doi: 10.1016/j.cmet.2017.01.004
33. Mahtal N, Lenoir O, Tharaux P-L. Glomerular endothelial cell crosstalk with podocytes in diabetic nephropathy. Front Med (2021), 8. doi: 10.3389/fmed.2021.659013
34. Wendt TM, Tanji N, Guo JC, Kislinger TR, Qu W, Lu Y, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol (2003) 162(4):1123–37. doi: 10.1016/S0002-9440(10)63909-0
35. Tsuchida K, Makita Z, Yamagishi S, Atsumi T, Miyoshi H, Obara S, et al. Suppression of transforming growth factor beta and vascular endothelial growth factor in diabetic nephropathy in rats by a novel advanced glycation end product inhibitor, OPB-9195. Diabetologia (1999) 42(5):579–88. doi: 10.1007/s001250051198
36. Gil CL, Hooker E, Larrivee B. Diabetic nephropathy, endothelial damage, and podocyte-endothelial crosstalk. Kidney Med (2021) 3(1):105–15. doi: 10.1016/j.xkme.2020.10.005
37. Ma J, Zhang L, Hao J, Li N, Tang J, Hao L, et al. Up-regulation of microRNA-93 inhibits TGF-beta 1-induced EMT and renal fibrogenesis by down-regulation of Orai1. J Pharmacol Sci (2018) 136(4):218–27. doi: 10.1016/j.jphs.2017.12.010
38. Wang Y, Le Y, Xue J-Y, et al. Let-7d miRNA prevents TGF-beta 1-induced EMT and renal fibrogenesis through regulation of HMGA2 expression. Biochem Biophys Res Commun (2016) 479(4):676–82. doi: 10.1016/j.bbrc.2016.09.154
39. Ghayur A, Margetts PJ. Transforming growth factor-beta and the glomerular filtration barrier. Kidney Res Clin Pract (2013) 32(1):3–10. doi: 10.1016/j.krcp.2013.01.003
40. Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol (2012) 74:299–323. doi: 10.1146/annurev-physiol-020911-153238
41. Schiffer M, Bitzer M, Roberts ISD, Kopp JB, Dijke ten P, Mundel P, et al. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest (2001) 108(6):807–16. doi: 10.1172/JCI200112367
42. Mcginn S, Poronnik P, King M, Gallery EDM, Pollock CA. High glucose and endothelial cell growth: novel effects independent of autocrine TGF-beta 1 and hyperosmolarity. Am J Physiol Cell Physiol (2003) 284(6):C1374–86. doi: 10.1152/ajpcell.00466.2002
43. Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med (2000) 6(4):460–3. doi: 10.1038/74725
44. Gale NW, Yancopoulos GD. Growth factors acting via endothelial cell-specific receptor tyrosine kinases: VEGFs, angiopoietins, and ephrins in vascular development. Genes Dev (1999) 13(9):1055–66. doi: 10.1101/gad.13.9.1055
45. Qi H, Casalena G, Shi S, Yu L, Ebefors K, Sun Y, et al. Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic nephropathy susceptibility. Diabetes (2017) 66(3):763–78. doi: 10.2337/db16-0695
46. Davis B, Cas AD, Long DA, White KE, Hayward A, Ku CH, et al. Podocyte-specific expression of angiopoietin-2 causes proteinuria and apoptosis of glomerular endothelia. J Am Soc Nephrol (2007) 18(8):2320–9. doi: 10.1681/ASN.2006101093
47. Blank U, Seto ML, Adams DC, Wojchowski DM, Karolak MJ, Oxburgh L, et al. An in vivo reporter of BMP signaling in organogenesis reveals targets in the developing kidney. BMC Dev Biol (2008) 8:86. doi: 10.1186/1471-213X-8-86
48. Zanatta CM, Veronese FV, Loreto MDS, Sortica DA, Carpio VN, Eldeweiss Albano MI, et al. Endothelin-1 and endothelin a receptor immunoreactivity is increased in patients with diabetic nephropathy. Renal Failure (2012) 34(3):308–15. doi: 10.3109/0886022X.2011.647301
49. Zhang Q, Xiao X, Li M, Li W, Yu M, Zhang H, et al. Gene expression profiling in glomeruli of diabetic nephropathy rat. Exp Biol Med (2012) 237(8):903–11. doi: 10.1258/ebm.2012.012032
50. Anguiano L, Riera M, Pascual J, Soler MJ. Endothelin blockade in diabetic nephropathy. J Clin Med (2015) 4(6):1171–92. doi: 10.3390/jcm4061171
51. Daehn I, Casalena G, Zhang T, Shi S, Fenninger F, Barasch N, et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest (2014) 124(4):1608–21. doi: 10.1172/JCI71195
52. Van De Lest NA, Bakker AE, Dijkstra KL, Zandbergen M, Heemskerk SAC, Wolterbeek R, et al. Endothelial endothelin receptor a expression is associated with podocyte injury and oxidative stress in patients with focal segmental glomerulosclerosis. Kidney Int Rep (2021) 6(7):1939–48. doi: 10.1016/j.ekir.2021.04.013
53. Ferland-Mccollough D, Slater S, Richard J, Reni C, Mangialardi G. Pericytes, an overlooked player in vascular pathobiology. Pharmacol Ther (2017) 171:30–42. doi: 10.1016/j.pharmthera.2016.11.008
54. Heerspink HJL, Parving HH, Andress DL, Bakris G, Correa-Rotter R, Hou F-F, et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): a double-blind, randomised, placebo-controlled trial. Lancet (2019) 393(10184):1937–47. doi: 10.1016/S0140-6736(19)30772-X
55. Sasser JM, Sullivan JC, Hobbs JL, Yamamoto T, Pollock DM, Carmines PK, et al. Endothelin a receptor blockade reduces diabetic renal injury via an anti-inflammatory mechanism. J Am Soc Nephrol (2007) 18(1):143–54. doi: 10.1681/ASN.2006030208
56. Murphy M, Docherty NG, Griffin B, Howlin J, McArdle E, McMahon R, et al. IHG-1 amplifies TGF-beta 1 signaling and is increased in renal fibrosis. J Am Soc Nephrol (2008) 19(9):1672–80. doi: 10.1681/ASN.2007101080
57. Lindahl P, Hellstrom M, Kalen M, Karlsson L, Pekny M, Pekna M, et al. Paracrine PDGF-B/PDGF-R beta signaling controls mesangial cell development in kidney glomeruli. Development (1998) 125(17):3313–22. doi: 10.1242/dev.125.17.3313
58. Uehara G, Suzuki D, Toyoda M, Umezono T, Sakai H. Glomerular expression of platelet-derived growth factor (PDGF)-a, -b chain and PDGF receptor-alpha, -beta in human diabetic nephropathy. Clin Exp Nephrol (2004) 8(1):36–42. doi: 10.1007/s10157-003-0265-8
59. Sohn E, Kim J, Kim C-S, Jo K, Lee YM, Kim JS, et al. Root of polygonum cuspidatum extract reduces progression of diabetes-induced mesangial cell dysfunction via inhibition of platelet-derived growth factor-BB (PDGF-BB) and interaction with its receptor in streptozotocin-induced diabetic rats. BMC Complement Altern Med (2014) 14:477. doi: 10.1186/1472-6882-14-477
60. Fagerudd JA, Groop PH, Honkanen E, Teppo AM, Grönhagen-Riska C. Urinary excretion of TGF-beta 1, PDGF-BB and fibronectin in insulin-dependent diabetes mellitus patients. Kidney Int Suppl (1997) 63:S195–7. doi: 10.1038/ki.1997.508
61. Alan S, Şalva E, Yılmaz İ, Akbuğa J. The effectiveness of chitosan-mediated silencing of PDGF-b and PDGFR-β in the mesangial proliferative glomerulonephritis therapy. Exp Mol Pathol (2019) 110:104280. doi: 10.1016/j.yexmp.2019.104280
62. Betsholtz C, Raines EW. Platelet-derived growth factor: a key regulator of connective tissue cells in embryogenesis and pathogenesis. Kidney Int (1997) 51(5):1361–9. doi: 10.1038/ki.1997.186
63. Zou H-H, Wang L, Zheng X-X, Turan SÖ, Akbuğa J. Endothelial cells secreted endothelin-1 augments diabetic nephropathy via inducing extracellular matrix accumulation of mesangial cells in ETBR-/- mice. Aging-Us (2019) 11(6):1804–20. doi: 10.18632/aging.101875
64. Yang C, Ling H, Zhang M, Yang Z, Wang X, Zeng F, et al. Oxidative stress mediates chemical hypoxia-induced injury and inflammation by activating NF-kappa b-COX-2 pathway in HaCaT cells. Mol Cells (2011) 31(6):531–8. doi: 10.1007/s10059-011-1025-3
65. Peng F, Wu D, Gao B, Ingram AJ, Zhang B, Chorneyko K, et al. RhoA/Rho-kinase contribute to the pathogenesis of diabetic renal disease. Diabetes (2008) 57(6):1683–92. doi: 10.2337/db07-1149
66. Kolavennu V, Zeng L, Peng H, Wang Y, Danesh FR. Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes (2008) 57(3):714–23. doi: 10.2337/db07-1241
67. Chen S-J, Lv L-L, Liu B-C, Tang R-N. Crosstalk between tubular epithelial cells and glomerular endothelial cells in diabetic nephropathy. Cell Proliferation (2020) 53(3):e12763. doi: 10.1111/cpr.12763
68. Ling L, Tan Z, Zhang C, Gui S, Cui Y, Hu Y, et al. CircRNAs in exosomes from high glucose-treated glomerular endothelial cells activate mesangial cells. Am J Trans Res (2019) 11(8):4667–82.
69. Pan L, Lian W, Zhang X, Han S, Cao C, Li X, et al. Human circular RNA-0054633 regulates high glucose-induced vascular endothelial cell dysfunction through the microRNA-218/roundabout 1 and microRNA-218/heme oxygenase-1 axes. Int J Mol Med (2018) 42(1):597–606. doi: 10.3892/ijmm.2018.3625
70. Dimke H, Sparks MA, Thomson BR, Frische S, Coffman TM, Quaggin SE, et al. Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J Am Soc Nephrol (2015) 26(5):1027–38. doi: 10.1681/ASN.2014010060
71. Pierce RW, Shabanova V, Canarie M, Pinto M, Silva YS, Bhandari V, et al. Angiopoietin level trajectories in toddlers with severe sepsis and septic shock and their effect on capillary endothelium. Shock (2019) 51(3):298–305. doi: 10.1097/SHK.0000000000001172
72. Rani S, O’brien K, Kelleher FC, Corcoran C, Germano S, Radomski MW, et al. Isolation of exosomes for subsequent mRNA, MicroRNA, and protein profiling. Methods Mol Biol (2011) 784:181–95. doi: 10.1007/978-1-61779-289-2_13
73. Zhang W, Zhou X, Yao Q, Liu Y, Zhang H, Dong Z, et al. HIF-1-mediated production of exosomes during hypoxia is protective in renal tubular cells. Am J Physiology-Renal Physiol (2017) 313(4):F906–13. doi: 10.1152/ajprenal.00178.2017
74. Wu X-M, Gao Y-B, Cui F-Q, Zhang N. Exosomes from high glucose-treated glomerular endothelial cells activate mesangial cells to promote renal fibrosis. Biol Open (2016) 5(4):484–91. doi: 10.1242/bio.015990
75. Wu X, Gao Y, Xu L, Dang W, Yan H, Zou D, et al. Exosomes from high glucose-treated glomerular endothelial cells trigger the epithelial-mesenchymal transition and dysfunction of podocytes. Sci Rep (2017) 7:9371. doi: 10.1038/s41598-017-09907-6
76. Fu J, Akat KM, Sun Z, Zhang W, Schlondorff D, Liu Z, et al. Single-cell RNA profiling of glomerular cells shows dynamic changes in experimental diabetic nephropathy. J Am Soc Nephrol (2019) 30(4):533–45. doi: 10.1681/ASN.2018090896
77. Yao X, Shen H, Cao F, He H, Li B, Zhang H, et al. Bioinformatics analysis reveals crosstalk among platelets, immune cells, and the glomerulus that may play an important role in the development of diabetic nephropathy. Front Med (Lausanne) (2021) 8:657918. doi: 10.3389/fmed.2021.657918
78. Pietropaolo M, Barinas-Mitchell E, Pietropaolo SL, Kuller LH, Trucco M. Evidence of islet cell autoimmunity in elderly patients with type 2 diabetes. Diabetes (2000) 49(1):32–8. doi: 10.2337/diabetes.49.1.32
79. Lin M, Yiu WH, Wu HJ, Chan LYY, Leung JCK, Au WS, et al. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J Am Soc Nephrol (2012) 23(1):86–102. doi: 10.1681/ASN.2010111210
80. Shimada M, Ishimoto T, Lee PY, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, et al. Toll-like receptor 3 ligands induce CD80 expression in human podocytes via an NF-kappa b-dependent pathway. Nephrol Dialysis Transplant (2012) 27(1):81–9. doi: 10.1093/ndt/gfr271
81. Krausgruber T, Fortelny N, Fife-Gernedl V, Senekowitsch M, Schuster LC, Lercher A, et al. Structural cells are key regulators of organ-specific immune responses. Nature (2020) 583(7815):296–+. doi: 10.1038/s41586-020-2424-4
82. Li ZL, Lv LL, Tang TT, Wang B, Feng Y, Zhou L-T, et al. HIF-1alpha inducing exosomal microRNA-23a expression mediates the cross-talk be tween tubular epithelial cells and macrophages in tubulointerstitial inflammation. Kidney Int (2019) 95(2):388–404. doi: 10.1016/j.kint.2018.09.013
83. A/L B Vasanth Rao VR, Tan SH, Candasamy M, Bhattamisra SK. Diabetic nephropathy: an update on pathogenesis and drug development. Diabetes Metab syndrome (2019) 13(1):754–62. doi: 10.1016/j.dsx.2018.11.054
84. Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res (2005) 97(6):512–23. doi: 10.1161/01.RES.0000182903.16652.d7
85. Chen Y-T, Chang F-C, Wu C-F, Chou Y-H, Hsu H-L, Chiang W-C, et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int (2011) 80(11):1170–81. doi: 10.1038/ki.2011.208
86. Hu T-Y, Li L-M, Pan Y-Z. CTRP3 inhibits high glucose-induced human glomerular mesangial cell dysfunction. J Cell Biochem (2019) 120(4):5729–36. doi: 10.1002/jcb.27859
87. Salim HM, Fukuda D, Yagi S, Soeki T, Shimabukuro M, Sata M, et al. Glycemic control with ipragliflozin, a novel selective SGLT2 inhibitor, ameliorated endothelial dysfunction in streptozotocin-induced diabetic mouse. Front Cardiovasc Med (2016) 3:43. doi: 10.3389/fcvm.2016.00043
88. Ahrén B. Glucagon-like peptide-1 receptor agonists for type 2 diabetes: a rational drug development. J Diabetes Investig (2019) 10(2):196–201. doi: 10.1111/jdi.12911
89. Kawanami D, Takashi Y. GLP-1 receptor agonists in diabetic nephropathy: from clinical outcomes to mechanisms. Front Pharmacol (2020) 11:967. doi: 10.3389/fphar.2020.00967
90. Panchapakesan U, Pollock CA. DPP-4 inhibitors-renoprotection in diabetic nephropathy? Diabetes (2014) 63(6):1829–30. doi: 10.2337/db14-0366
91. Panchapakesan U, Mather A, Pollock C. Role of GLP-1 and DPP-4 in diabetic nephropathy and cardiovascular disease. Clin Sci (Lond) (2013) 124(1):17–26. doi: 10.1042/CS20120167
92. Ziller N, Kotolloshi R, Esmaeili M, Liebisch M, Mrowka R, Baniahmad A, et al. Sex differences in diabetes- and TGF-β1-Induced renal damage. Cells (2020) 9(10):2236. doi: 10.3390/cells9102236
93. Rosenstock J, Kahn SE, Johansen OE, Zinman B, Espeland MA, Woerle HJ, et al. Effect of linagliptin vs glimepiride on major adverse cardiovascular outcomes in patients with type 2 diabetes: the CAROLINA randomized clinical trial. Jama (2019) 322(12):1155–66. doi: 10.1001/jama.2019.13772
94. Santos R, Oudit GY, Verano-Braga T, Canta G, Steckelings UM, Bader M, et al. The renin-angiotensin system: going beyond the classical paradigms. Am J Physiol Heart Circ Physiol (2019) 316(5):H958–h970. doi: 10.1152/ajpheart.00723.2018
95. Barrera-Chimal J, Lima-Posada I, Bakris GL, Jaisser F. Mineralocorticoid receptor antagonists in diabetic nephropathy - mechanistic and therapeutic effects. Nat Rev Nephrol (2022) 18(1):56–70. doi: 10.1038/s41581-021-00490-8
96. Miric G, Dallemagne C, Endre Z, Margolin S, Taylor SM, Brown L, et al. Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br J Pharmacol (2001) 133(5):687–94. doi: 10.1038/sj.bjp.0704131
97. Kim Y, Hwang SD, Lim JH, Kim MY, Kim EN, Choi BS, et al. Attenuated lymphatic proliferation ameliorates diabetic nephropathy and high-fat diet-induced renal lipotoxicity. Sci Rep (2019) 9(1):1994. doi: 10.1038/s41598-018-38250-7
98. Matsui K, Nagy-Bojarsky K, Laakkonen P, Krieger S, Mechtler K, Uchida S, et al. Lymphatic microvessels in the rat remnant kidney model of renal fibrosis: aminopeptidase p and podoplanin are discriminatory markers for endothelial cells of blood and lymphatic vessels. J Am Soc Nephrol (2003) 14(8):1981–9. doi: 10.1097/01.ASN.0000076078.50889.43
Keywords: crosstalk, diabetic kidney disease, diabetic nephropathy, intercellular communication, cellular crosstalk
Citation: Zhang K, Fu Z, Zhang Y, Chen X, Cai G and Hong Q (2023) The role of cellular crosstalk in the progression of diabetic nephropathy. Front. Endocrinol. 14:1173933. doi: 10.3389/fendo.2023.1173933
Received: 25 February 2023; Accepted: 26 June 2023;
Published: 17 July 2023.
Edited by:
Marcia Hiriart, Universidad Nacional Autonoma de Mexico, MexicoReviewed by:
Akira Sugawara, Tohoku University, JapanCopyright © 2023 Zhang, Fu, Zhang, Chen, Cai and Hong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quan Hong, aG9uZ3F1YW5AMzAxaG9zcGl0YWwuY29tLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.