 Sang Hyun Song1‡
Sang Hyun Song1‡ Dawool Han1,2‡
Dawool Han1,2‡ Kyeonghui Park1Jo Eun Um3
Kyeonghui Park1Jo Eun Um3 Seonghun Kim2,3†Minhee Ku4,5Jaemoon Yang4,5
Seonghun Kim2,3†Minhee Ku4,5Jaemoon Yang4,5 Tae-Hyun Yoo6
Tae-Hyun Yoo6 Jong In Yook1,2Nam Hee Kim1,2*
Jong In Yook1,2Nam Hee Kim1,2* Hyun Sil Kim1,2*
Hyun Sil Kim1,2*- 1Department of Oral Pathology, Yonsei University College of Dentistry, Seoul, Republic of Korea
- 2Oral Cancer Research Institute, Yonsei University College of Dentistry, Seoul, Republic of Korea
- 3R&D Center, MET Life Science, Seoul, Republic of Korea
- 4Department of Radiology, Yonsei University College of Medicine, Seoul, Republic of Korea
- 5Convergence Research Center for Systems Molecular Radiological Science, Yonsei University, Seoul, Republic of Korea
- 6Department of Internal Medicine, Yonsei University College of Medicine, Seoul, Republic of Korea
Background: Approximately 30% of diabetic patients develop diabetic nephropathy, a representative microvascular complication. Although the etiological mechanism has not yet been fully elucidated, renal tubular damage by hyperglycemia-induced expression of transforming growth factor-β (TGF-β) is known to be involved. Recently, a new type of cell death by iron metabolism called ferroptosis was reported to be involved in kidney damage in animal models of diabetic nephropathy, which could be induced by TGF-β. Bone morphogenetic protein-7 (BMP7) is a well-known antagonist of TGF-β inhibiting TGF-β-induced fibrosis in many organs. Further, BMP7 has been reported to play a role in the regeneration of pancreatic beta cells in diabetic animal models.
Methods: We used protein transduction domain (PTD)-fused BMP7 in micelles (mPTD-BMP7) for long-lasting in vivo effects and effective in vitro transduction and secretion.
Results: mPTD-BMP7 successfully accelerated the regeneration of diabetic pancreas and impeded progression to diabetic nephropathy. With the administration of mPTD-BMP7, clinical parameters and representative markers of pancreatic damage were alleviated in a mouse model of streptozotocin-induced diabetes. It not only inhibited the downstream genes of TGF-β but also attenuated ferroptosis in the kidney of the diabetic mouse and TGF-β-stimulated rat kidney tubular cells.
Conclusion: BMP7 impedes the progression of diabetic nephropathy by inhibiting the canonical TGF-β pathway, attenuating ferroptosis, and helping regenerate diabetic pancreas.
1 Introduction
Diabetic nephropathy (DN) is a major complication of diabetes and a causative factor of renal dysfunction; glomerulosclerosis, hypertrophy of basement membrane, extracellular matrix (ECM) accumulation, tubulointerstitial fibrosis, tubular cell damage, and cell death are well-known pathologic changes of DN. Approximately 30% of patients with diabetes develop DN; however, the biological mechanism of DN pathogenesis remains unclear. Previous studies demonstrated that changes in renal hemodynamics, hyperglycemia-induced oxidative stress, an inflammatory response, and enhanced renin-angiotensin-aldosterone system activity were involved in the pathogenesis of DN (1, 2). Recently, TGF-β1 has come to be viewed as the main regulator and therapeutic target of kidney fibrosis and tubular cell death, given its upregulation in patients with diabetes (3–7). Since the apoptosis or necroptosis of podocytes and renal tubular cells have been extensively studied (8), we focused on ferroptosis, which is emerging as a cause of cell death in many diseases including cancer and metabolic disease.
Ferroptosis is a newly proposed type of cell death characterized by iron-dependent lipid oxidation. The key players of ferroptosis are cysteine transporter and glutathione peroxidase 4 (GPX4); low intracellular cysteine levels result in reduced glutathione synthesis and the dysfunction of lipid oxidative damage repair, leading to ferroptosis (9, 10). Emerging evidence has revealed that ferroptosis is involved in acute kidney injury (AKI) and diabetic nephrology (11–13), including its pathogenesis. TGF-β1-stimulates tubular cells; erastin-stimulated tubular cells and streptozotocin (STZ)-induced diabetic mice show similar ferroptotic conditions in the kidney (14).
Multiple studies have reported that bone morphogenetic protein-7 (BMP7) effectively inhibits fibrosis involving various organs, such as retinopathy, liver fibrosis, and peritoneal fibrosis (15–17). In diverse renal disease models, BMP7 has been found to counteract fibrosis by suppressing the activation of multiple signaling pathways including the p38, p44/42 MAPK, and TGF-β pathways, as well as by reducing reactive oxygen species (ROS) formation (18–20). In an STZ-induced animal diabetes model, BMP7 was found to protect against diabetic kidney injury, counteracting TGF/SMADs signaling (20, 21). Further, several studies have identified the role of BMP7 in supporting β-cell regeneration and differentiation in adult and prenatal pancreas. For example, BMP7 plays a critical role during the transdifferentiation of ductal progenitor cells into exocrine beta cells. These results suggest the therapeutic potential of BMP7 for treating DN by protecting renal fibrosis as well as including pancreatic exocrine regeneration.
Since recombinant human BMP (rhBMP) undergoes rapid clearance and enzymatic degradation in vivo (22), we designed a micellar formulation of protein transduction domain (PTD)-BMP7 (mPTD-BMP7) to overcome the pharmacodynamic limitation of rhBMP7 in animal experimental models and to enhance endosomal transduction in vitro and in vivo (23, 24). We investigated whether mPTD-BMP7 ameliorated pancreatic damage in STZ-induced diabetes and attenuated diabetes-induced ferroptotic deterioration of the kidney.
2 Materials and methods
2.1 Cell culture and treatment of NRK-52E cells
Immortalized rat proximal tubular epithelial cells, NRK-52E ( CRL-5171, ATCC, VA, USA), were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (FBS) and 1% penicillin-streptomycin-amphotericin B. Subconfluent NRK-52E cells were starved in FBS-free condition for 24 hours; then, the medium was replaced with DMEM with 0.5% FBS. For the TGF-β1 group, TGF-β1 (10 ng/mL; R&D Systems, Minneapolis, MN, USA) was added to the culture, and erastin (Selleckchem) was used as the positive control for ferroptosis induction. Both groups were treated with mPTD-BMP7 (250 ng/mL). AGEs (advanced glycation end-products) were obtained through the non-enzymatic reaction of BSA (Sigma-Aldrich) and glycerolaldehyde as described by Valencia et al. (25). Briefly, sterile filtered 30% BSA solution was incubated with 70 mM glycolaldehyde dimer in sterile PBS without calcium chloride and magnesium chloride for 3 days at 37°C. After incubation, the AGEs were dialyzed against sterile PBS to remove remaining glycolaldehyde at 4°C using 10 kDa cut-off cassettes. Control BSA was prepared via the same process without reaction with glycolaldehyde dimer. Concentration of dialyzed BSA-AGEs was determined by BCA assay and extent of AGEs modification was confirmed by fluorescence and absorbance as described by Valencia et al. (25). After starvation in FBS-free condition for 24 hours, the medium was replaced with 0.5% FBS and 500 μg/mL BSA-AGEs or BSA-control and further incubated for 72 hours.
2.2 Animal experiments
Eight-week-old male C57BL/6 mice were injected with either the diluent (control; n=5) or streptozotocin (STZ; Sigma-Aldrich; 50 mg/kg; n=20) intraperitoneally for 5 days consecutively. To confirm diabetes mellitus (DM; fasting blood glucose >300 mg/dL), tail vein blood glucose was measured, and only 13 mice were diagnosed with DM. Five mice from the control group and seven from the DM group were treated with the vehicle (10 μg) intraperitoneally every 72 h. The other six mice from the DM group were treated with mPTD-BMP7 (10 μg) intraperitoneally every 72 h. After 16 weeks, the mice were anesthetized with Zoletil (10 mg/kg; Virbac, Carros, France), and their kidneys and pancreas were extracted. Body weight, kidney weight, and levels of blood glucose, HbA1c, serum bilirubin, serum creatinine, blood urea nitrogen (BUN), 24 h urinary albumin, and creatinine were determined at the time of sacrifice. Blood glucose level was measured using a glucometer, and HbA1c level was measured using a glycohemoglobin analyzer. Then, 24-h urinary albumin was assessed via the enzyme-linked immunosorbent assay (ELISA; Nephrat II, Exocell Inc., Philadelphia, PA, USA), and total bilirubin and 24-h urinary creatinine levels were analyzed using a Cobas 8000 C502 analyzer (Roche, Mannheim, Baden Württemberg, Germany). Insulin (NBP2-62853, Novus Biologicals, Littleton, CO, USA) and C-peptide (NBP2-82124, Novus Biologicals, Littleton, CO, USA) concentrations were determined by ELISA.
2.3 Quantitative real-time polymerase chain reaction
Total RNA was isolated from the cells, whole pancreas, and kidney samples using Trizol (Invitrogen), and the cDNA was synthesized using a reverse transcriptase premix product (AccuPower CycleScript RT PreMix, Bioneer). Transcript levels were detected using the 7300 Real-Time PCR system (Applied Biosystems) with TB Green® Premix Ex Taq TM II (Takara, Shiga, Japan). mRNA expression level was calculated using △CT values and the results are shown as relative expression normalized by that of the housekeeping genes (r18s, Actb, Rpl13a). The primer sequences used in this study are presented in Tables 1, 2.

Table 1 Primer sequences used in the qPCR analysis of the animal experiment.
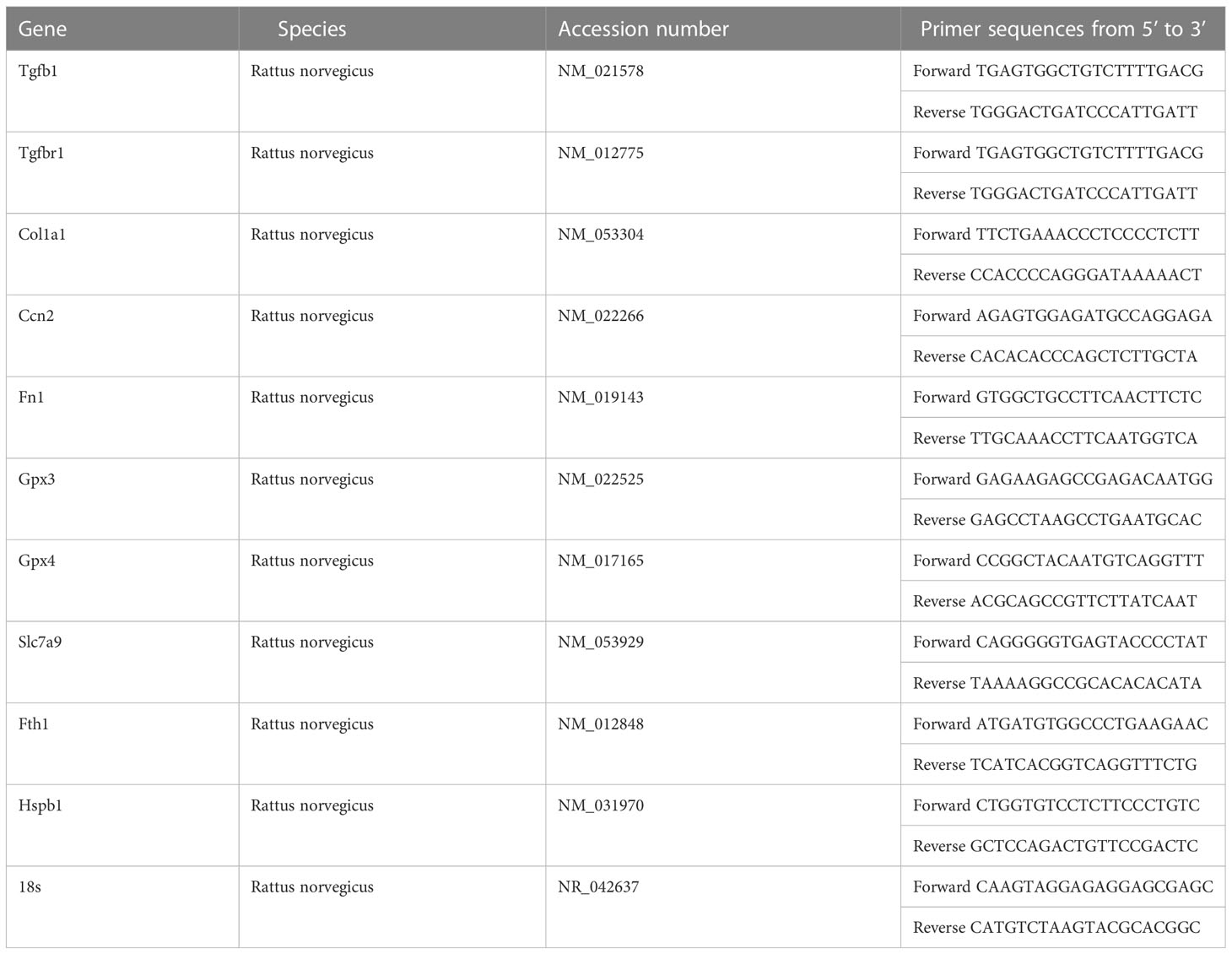
Table 2 Primer sequences used in qPCR analysis of the cell experiment.
2.4 Measurement of lipid peroxidation
The level of malondialdehyde (MDA) was measured using a lipid peroxidation assay kit (Abcam, Cambridge, United Kingdom). For live cell imaging, the Image-iT® lipid peroxidation kit (Thermo Fisher Scientific) was used. For the MDA assay, cultured cells were homogenized in MDA lysis buffer with 5% butylated hydroxytoluene, and the harvested kidney tissues were homogenized in distilled water with 5% butylated hydroxytoluene, followed by the addition of 2 N perchloric acid. After centrifugation of samples at 13,000 × g for 10 min at 4°C, the supernatant of each sample was collected, and MDA levels were measured using thiobarbituric acid, which reacts with MDA to produce a compound that has maximum absorbance at a wavelength of 532 nm. For the live cell analysis of lipid peroxidation, cells were seeded in four-well chambers (3 × 104 per well) and pretreated with mPTD-BMP7 for 16 h before TGF-β1 or erastin stimulation. The Image-iT® lipid peroxidation sensor was added into each well and incubated for 30 min at 37°C. The culture medium was removed, and samples were washed with phosphate-buffered saline (PBS). Images were acquired using an LSM700 confocal microscope (Carl Zeiss Vision, Hallbergmoos, Germany) under ×40 magnification. Fluorescence intensity was quantified using the publicly available ImageJ program.
2.5 Glutathione assay
Glutathione concentration (reduced form of glutathione only) was measured using a glutathione assay kit (Sigma-Aldrich). Cultured cells and harvested kidney tissues were lysed in glutathione buffer, and 5% sulfosalicylic acid was added later. The cells were centrifuged at 700 × g for 5 min and kidney samples were centrifuged at 8000 × g for 10 min; the supernatants were collected. NADPH-generating mix was added to the glutathione reaction buffer and incubated for 10 min at room temperature (RT) to generate NADPH. Then, 20 μL of supernatant and 160 μL of NADPH solution were mixed and incubated for 10 min at RT. Glutathione concentrations were determined by the reaction with 5,5’-dithio-bis-(2-nitrobenzoic acid) at a wavelength of 415 nm.
2.6 Immunohistochemistry
Serial paraffin sections were stained with hematoxylin and eosin (H&E) for routine morphological observation. For immunohistochemical (IHC) staining, tissue sections were deparaffinized with xylene, rehydrated in serially diluted ethyl alcohol, and immersed in 3% hydrogen peroxide. Antigen retrieval was carried out by microwave heating in 10-mM citric acid buffer (pH 6.0) for 20 min. Nonspecific binding of protein was blocked with 2.5% goat serum (MP-7451, Vector Laboratories) for 30 min at RT, followed by overnight incubation at 4°C with a primary antibody. After washing with PBS, the sections were incubated with a secondary antibody, horseradish peroxidase-conjugated goat anti-rabbit IgG (MP-7451, Vector Laboratories), for 30 min at RT. After washing with PBS, 3,3’ diaminobenzidine was added as the chromogen and the color developed was detected using Envision Detection Systemic of Agilent (K5007, Agilent). Hematoxylin was used for counter staining, followed by mounting. The primary antibodies used were as follows: anti-4-hydroxynonenal (4-HNE; NBP2-59353, Novus Biologicals), anti-MDA (NBP2-59367, Novus Biologicals), and anti-fibronectin (ab2413, abcam), anti-GPX4 (ab125066, abcam), anti-CTGF (ab209780, abcam), and anti-Collagen Ia (ab21286, abcam). Quantitative analysis of IHC and Sirius red staining was evaluated using five randomly taken images of renal cortex per subject. For each image, the positive signal was extracted through the color threshold process via the ImageJ program (open source software), and the final value was the average of multiplying the intensity and area of signal of five renal cortex images.
2.7 Statistical analysis
All data are representative of at least three independent experiments. Statistical differences were analyzed using paired t-test and Mann–Whitney test using Prism 5 (GraphPad, San Diego, CA). A P value < 0.05 was considered statistically significant.
3 Results
3.1 mPTD-BMP7 abrogates TGF-β1- and erastin-induced kidney tubular cell damage
To assess the previously known antagonizing impact of BMP7 on TGF-β1, the downstream genes of TGF-β1 were further evaluated. The expression levels of Tgfb1 and Tgfbr1 were significantly higher in the TGF-β1-stimulated NRK-52E cells that were stimulated for 96 h, followed by a significant increase in the expression of the downstream genes (Cola1, fibronectin, and Ccn2). The upregulation of fibrosis-related genes was alleviated by mPTD-BMP7 (Figure 1A). Next, we investigated the effect of mPTD-BMP7 on TGF-β1- and erastin-induced kidney tubular cell death. Cell viability was significantly reduced in TGF-β1- and erastin-treated tubular cell, but it was attenuated by mPTD-BMP7 (Figure 1B). Several key molecules of ferroptosis were evaluated to confirm whether TGF- β1 induces ferroptosis in renal tubular cell. Reduction of mRNA levels of glutathione peroxidase (Gpx3, Gpx4) and cysteine transporter (Slc7a9) were significantly attenuated by mPTD-BMP7 in TGF-β1-stimulated NRK-52E cells. Furthermore, the TGF-β1-induced changes in the mRNA levels of ferritin heavy chain (Fth1) and heat shock protein beta-1(Hspb1) were abrogated with mPTD-BMP7 treatment. NRK-52E cells stimulated with erastin, a ferroptosis inducer, showed similar findings as TGF-β1-stimulated cells (Figure 1C). Reduced glutathione (GSH) level was significantly reduced in TGF-β1 and erastin-stimulated conditions; however, the concentration increased with mPTD-BMP7 treatment. The increase in lipid peroxidation caused by TGF-β1 was attenuated by administration of mPTD-BMP7 at 2.5 μg/mL. In addition, mPTD-BMP7 abrogated lipid peroxidation in erastin-stimulated NRK-52E cells (Figures 1D, E). Taken together, these findings suggest that TGF- β1-induced tubular cell injury is involved in ferroptosis; further, mPTD-BMP7 is presumed to attenuate ferroptotic cell damage. When the same experiments were performed with advanced glycation end-products (AGEs), levels of which are elevated in hyperglycemic conditions, a similar tendency was observed as with erastin and TGF-β1, but in less degree (Figures 1B–E). To determine whether mPTD-BMP7 reduced lipid peroxidation of TGF-β1- and erastin-treated tubular cell, the level of lipid peroxidation was assed. Once oxidized, the fluorescence shift form a non-oxidative form (Texas red channel) to an oxidative form (FITC channel). The oxidative fluorescence intensity was increase in TGF-β1- and erastin-treated tubular cell. mPTD-BMP7 significantly reduced the process of red fluorescence to green fluorescence (Figures 1F, G). These results suggest that ferroptosis in renal tubular cell increased responding to highly elevated levels of TGF- β1, erastin and AGEs in diabetic condition.
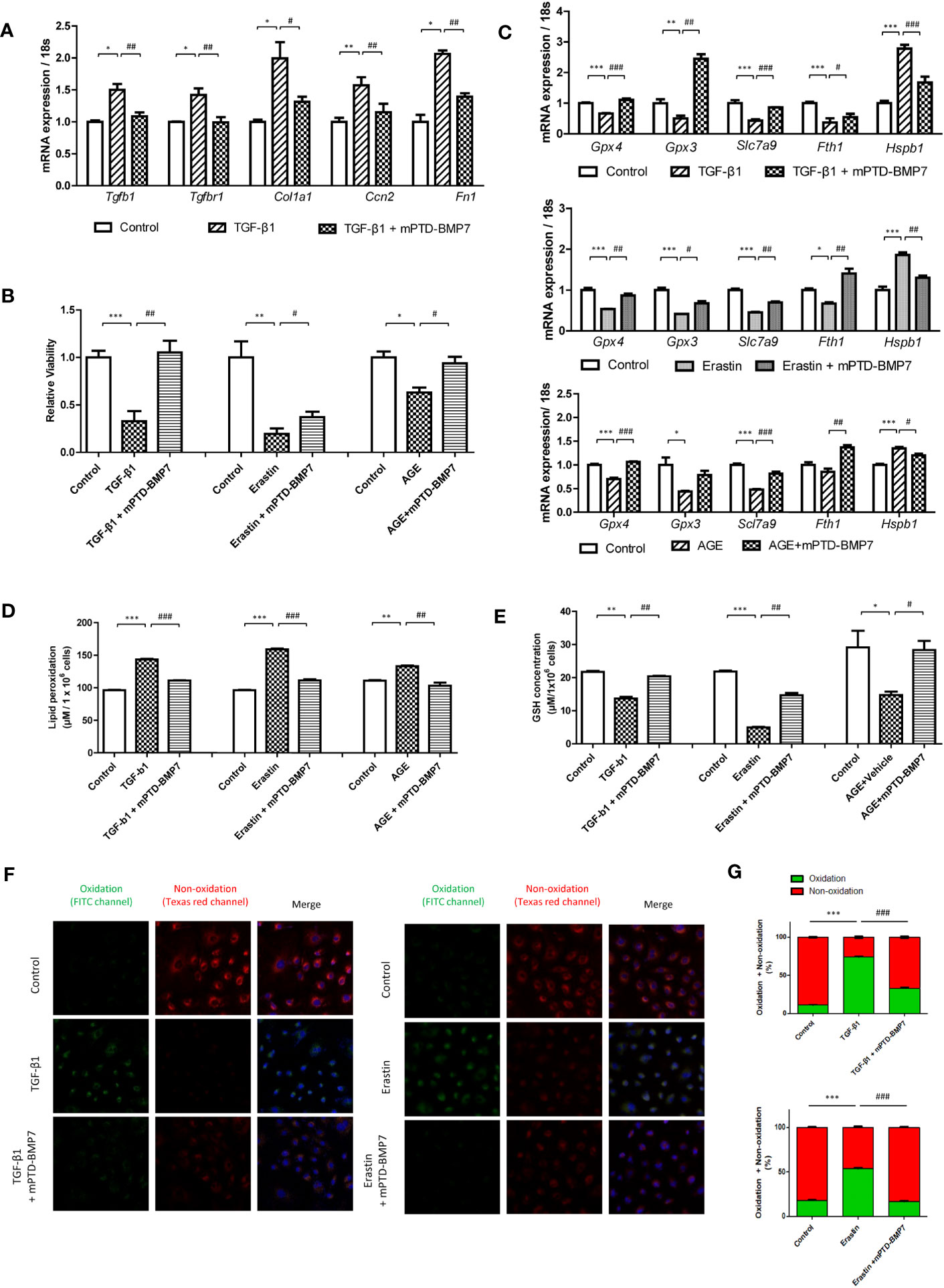
Figure 1 mPTD-BMP7 attenuates TGF-β1- or erastin- or AGEs-induced cellular damage in NRK-52E cells. (A) mPTD-BMP7 significantly ameliorates the expression of fibrosis-related downstream genes in transforming growth factor-beta1 (TGF-β1)-stimulated NRK-52E cells after 96 h. (B) Salvage from cell death by the administration of mPTD-BMP7 is observed on the JuLi real-time live cell analyzer (NanoEntek). NRK-52E cells were stimulated by TGF-β1 or erastin or AGEs. (C) The increase in the mRNA levels of ferroptosis-related genes in TGF-β1- or erastin- or AGEs-stimulated NRK-52E cells is halted after mPTD-BMP7 treatment. (D) Administration of mPTD-BMP7 significantly attenuates the increase in lipid peroxidation in TGF-β1- or erastin- or AGEs-stimulated NRK-52E. (E) Administration of mPTD-BMP7 led to significant recovery of reduced glutathione (GSH) levels in TGF-β1- or erastin- or AGEs-stimulated NRK-52E. (F) Increased lipid peroxidation assessed using Image-iT® in TGF-β1- or erastin-stimulated NRK-52E. (G) Quantitative analysis of image-iT® shows a significant decrease in lipid peroxidation after mPTD-BMP7 administration. (*P < 0.05; **P < 0.01; ***P < 0.001; versus control group. #P < 0.05; ##P < 0.01; ###P < 0.001; versus TGF-β1 or erastin or AGEs group).
3.2 mPTD-BMP7 attenuates pancreatic damage under diabetic conditions
To study the distribution of mPTD-BMP7, we covalently conjugated indocyanine green to mPTD-BMP7 and administered this compound to the mice via intra-peritoneal injection (Figure 2A). An ex vivo fluorescence analysis showed that a considerable amount of mPTD-BMP7 had accumulated in the pancreas at 6 h and persisted even at 72 h (Figure 2B). Based on the results of quantitative analysis of the ex vivo images of the pancreas and kidney, mPTD-BMP7 was administered twice a week to STZ-induced diabetic mice (Figure 2C). In the animal model of diabetes, the kidney/body weight, levels of blood glucose and serum bilirubin, and urine albumin-creatinine ratio (UACR) had significantly increased, these factors, especially UACR and bilirubin, showing overall improvements with mPTD-BMP7 treatment (P<0.05; Table 3). Fasting glucose (Figure 2D) and HbA1c levels (Table 3) showed little attenuation with mPTD-BMP7 treatment, whereas the concentrations of serum insulin and C-peptide showed significant reduction in diabetic mice (Figures 2E, F). To determine whether mPTD-BMP7 affected pancreas regeneration, we examined the qPCR result of a representative pancreas marker in diabetic pancreatic tissue. Among the pancreatic developmental markers, mRNA levels of pancreatic duodenal homeobox 1 (PDX1), which is a β-cell putative progenitor, adult pancreatic markers (Figure 2G, upper), and pancreatic developmental markers (Figure 2G, lower) severely decreased in the diabetic mice but improved significantly with mPTD-BMP7 treatment.
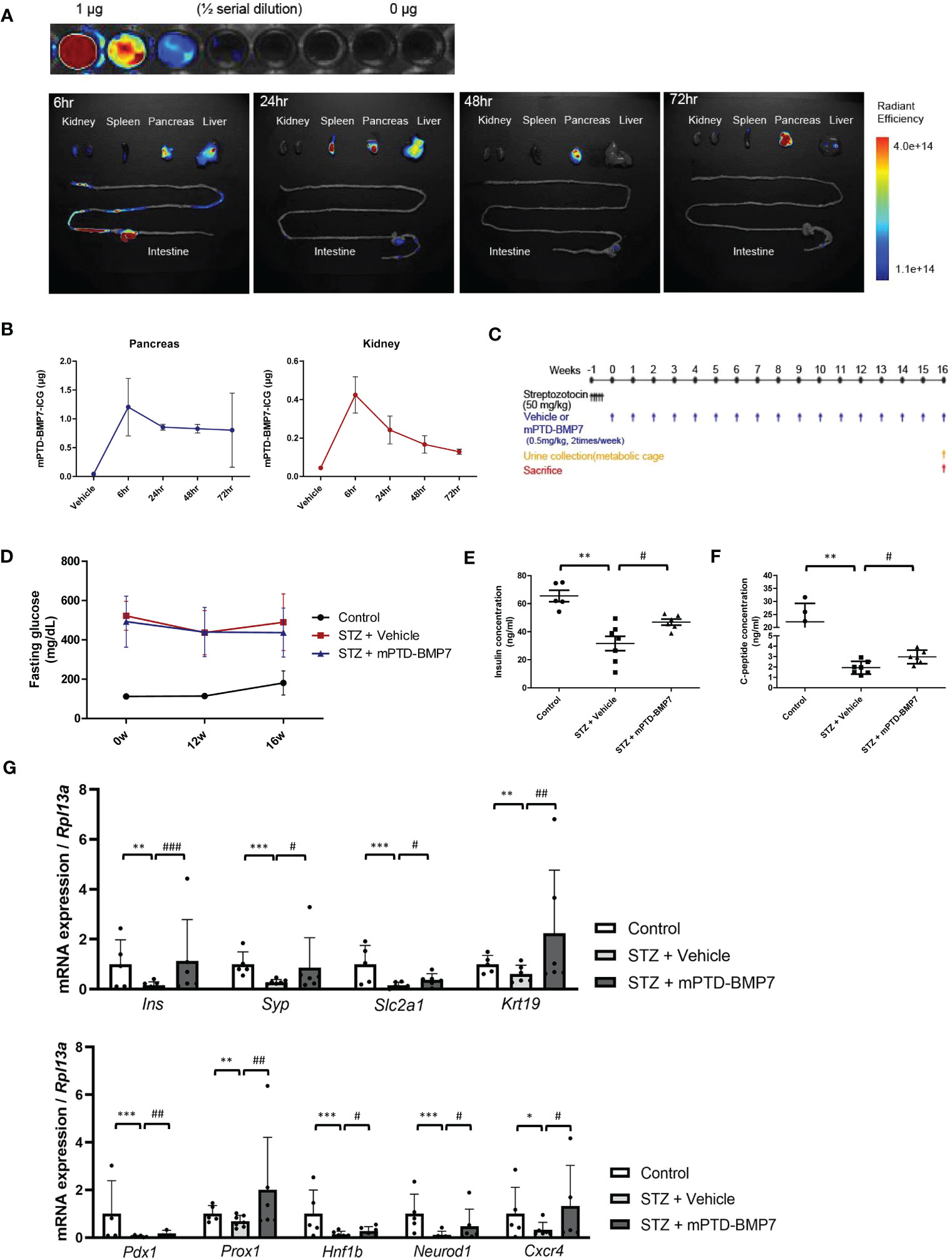
Figure 2 Effect of intraperitoneal administration of mPTD-BMP7 on pancreas function. (A) Ten micrograms of indocyanine green (ICG)-labeled mPTD-BMP7 was injected intraperitoneally, and the dissected major organs were assessed by ex vivo fluorescence imaging over time. (B) Quantitative analysis of the ex vivo fluorescence images of the pancreas and kidney. (C) Experimental schematics of streptozotocin (STZ)-induced diabetic mice. The diabetic condition was induced by an intraperitoneal injection of STZ (50 mg/kg) for 5 days consecutively, followed by an intraperitoneal injection of mPTD-BMP7 (0.5 mg/kg) twice per week. (D) Fasting glucose level was assessed over time for 16 weeks. Concentrations of insulin (E) and C-peptide (F) were measured in the extracted serum; these are reduced after mPTD-BMP7 administration. (G) mRNA levels of adult pancreatic markers (upper) and pancreatic developmental markers (lower) in STZ mice were attenuated after mPTD-BMP7 administration. (*P < 0.05; **P < 0.01; ***P < 0.001; versus control group. #P < 0.05; ##P < 0.01; ###P < 0.001; versus mPTD-BMP7 group).
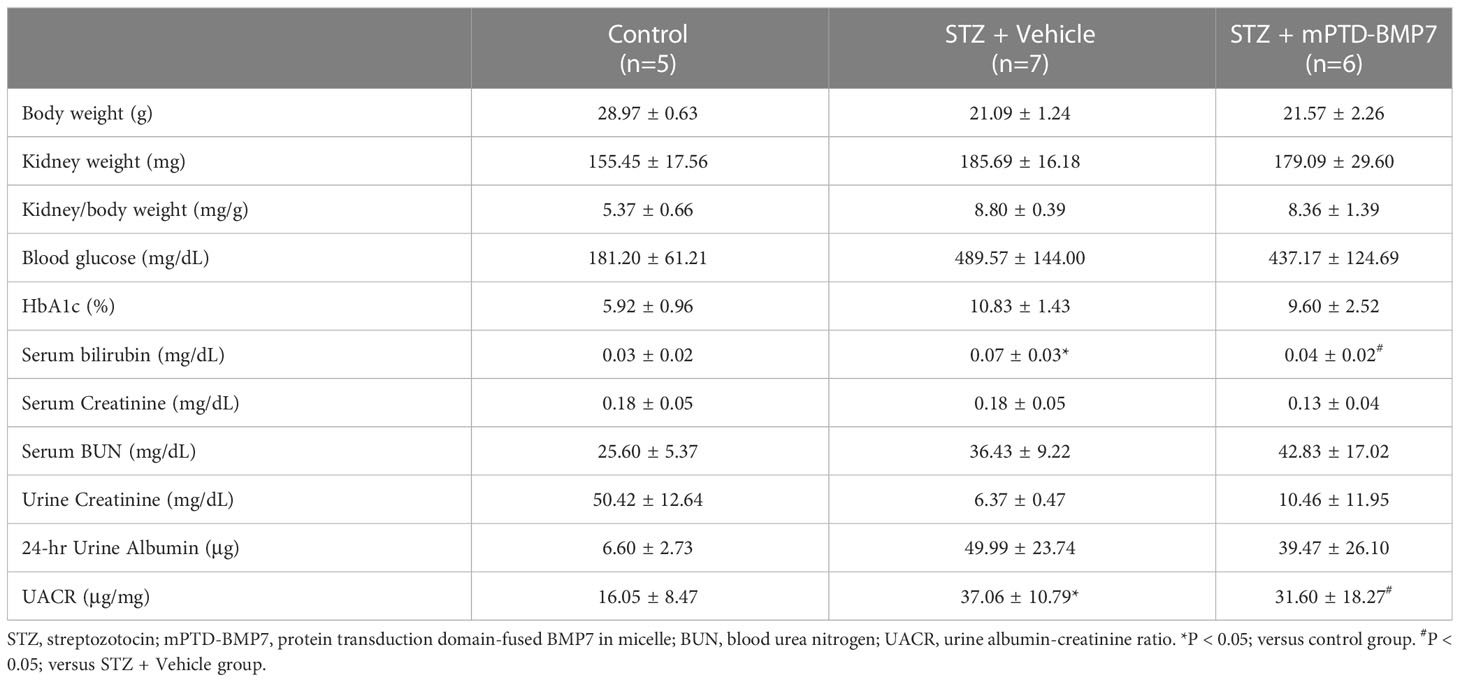
Table 3 Effect of mPTD-BMP7 assessed by clinical parameters in the animal models of diabetes.
3.3 mPTD-BMP7 ameliorates ferroptotic damage to the kidney in diabetic conditions
In the STZ-induced diabetic condition, renal tubular cells showed signs of cellular damage, but treatment with mPTD-BMP7 reduced damage. To confirm if ferroptotic damage had occurred in the kidneys of diabetic animals and was reduced by mPTD-BMP7 treatment, we evaluated key players in ferroptosis. mRNA levels of Gpx3, Gpx4, and Slc7a9 in diabetic kidney were significantly downregulated, but recovered after mPTD-BMP7 treatment (Figure 3A). Iron accumulation was confirmed in the diabetic kidney by Prussian blue staining and was prevented by mPTD-BMP7 (Figure 3B), consistent with significant change of mRNA expression levels of Fth1 and Hspb1 mRNA (Figure 3A). MDA and 4-HNE, the end products of lipid peroxidation, were significantly increased in the damaged tubular cells in diabetic kidney and markedly reduced by mPTD-BMP7 administration (Figures 3B, C). Additionally, reduced GSH concentration decreased in the diabetic kidney, but recovered after treatment with mPTD-BMP7 (Figure 3D). Based on these results, we confirmed that ferroptosis is a form of renal tubular damage induced by STZ and that mPTD-BMP7 can reduce this type of cellular damage.
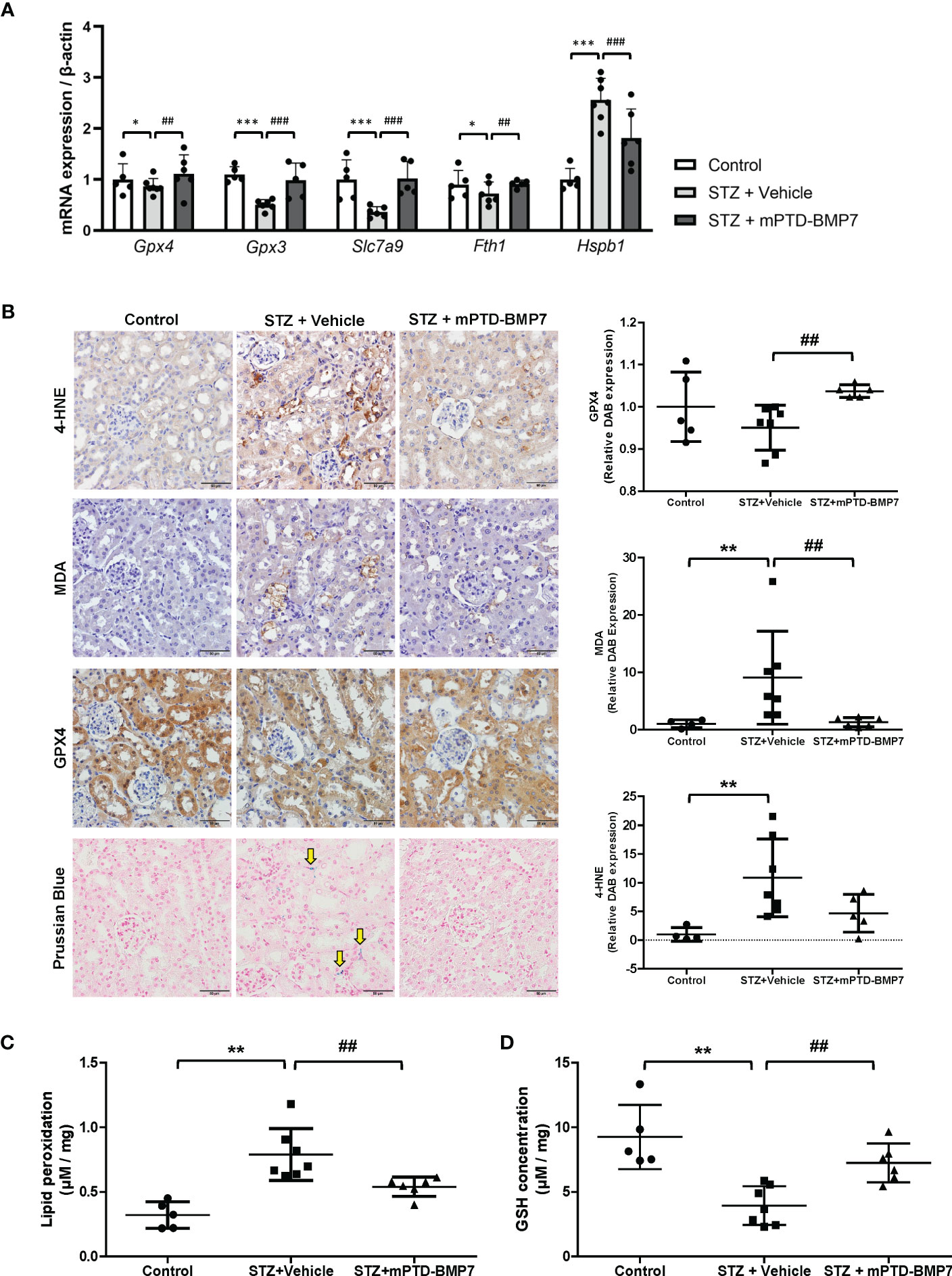
Figure 3 Effects of mPTD-BMP7 on ferroptosis in the kidneys of diabetic mice. (A) mRNA expression of ferroptosis-related genes significantly changed in the diabetic kidney, and the changes were abrogated by mPTD-BMP7. (B) Immunohistochemistry showed that damaged tubular cells exhibited increased level of 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA) in the vehicle group, and it was abrogated by mPTD-BMP7 administration. Prussian blue staining revealed iron deposition in the interstitium of diabetic kidney (yellow arrow), and it was attenuated by mPTD-BMP7. Original magnification: x400 (scale bar: 50μm) (C) Lipid oxidation, assessed via MDA levels, was increased in the kidneys of STZ mice, and reduced by treatment with mPTD-BMP7. (D) GSH concentration in the kidneys of STZ mice was decreased, and it was attenuated by treatment with mPTD-BMP7. (*P < 0.05; **P < 0.01; ***P < 0.001 versus control group. ##P < 0.01; ###P < 0.001; versus mPTD-BMP7 group).
3.4 Effect of mPTD-BMP7 on the progression of diabetic renal fibrosis
The STZ-induced diabetic kidney exhibited marked damage on renal cortical tubules, including tubular cell vacuolization, loss of nucleus, and structural destruction of tubule (Figure 4B). On the other hand, diabetic changes in the glomerulus, such as mesangial expansion and deposition of excessive extracellular matrix, were not marked. Most notably, tubulointersititial inflammation and fibrosis were observed in the STZ-treated group. However, administration of mPTD-BMP7 attenuated the cellular damage, tubulointerstitial inflammation and fibrosis (Figure 4B). The STZ-induced diabetic group exhibited a marked increase in Sirius red and Masson’s trichrome staining, as well as increased fibronectin, CTGF (connective tissue growth factor) and collagen type I-A expression on immunohistochemical staining, which are indications of fibrosis (Figure 4B). Also, the mRNA level of TGF-β1 was significantly increased in the diabetic kidney, and fibrosis-related downstream genes were upregulated. These upregulated transcriptions of TGF-β1 and fibrosis-related genes were hindered by mPTD-BMP7 (Figure 4A). Taken together, we confirmed that mPTD-BMP7 antagonized TGF-β1-induced fibrosis and ferroptotic damage in the STZ-induced diabetic nephropathy model (Figure 5).
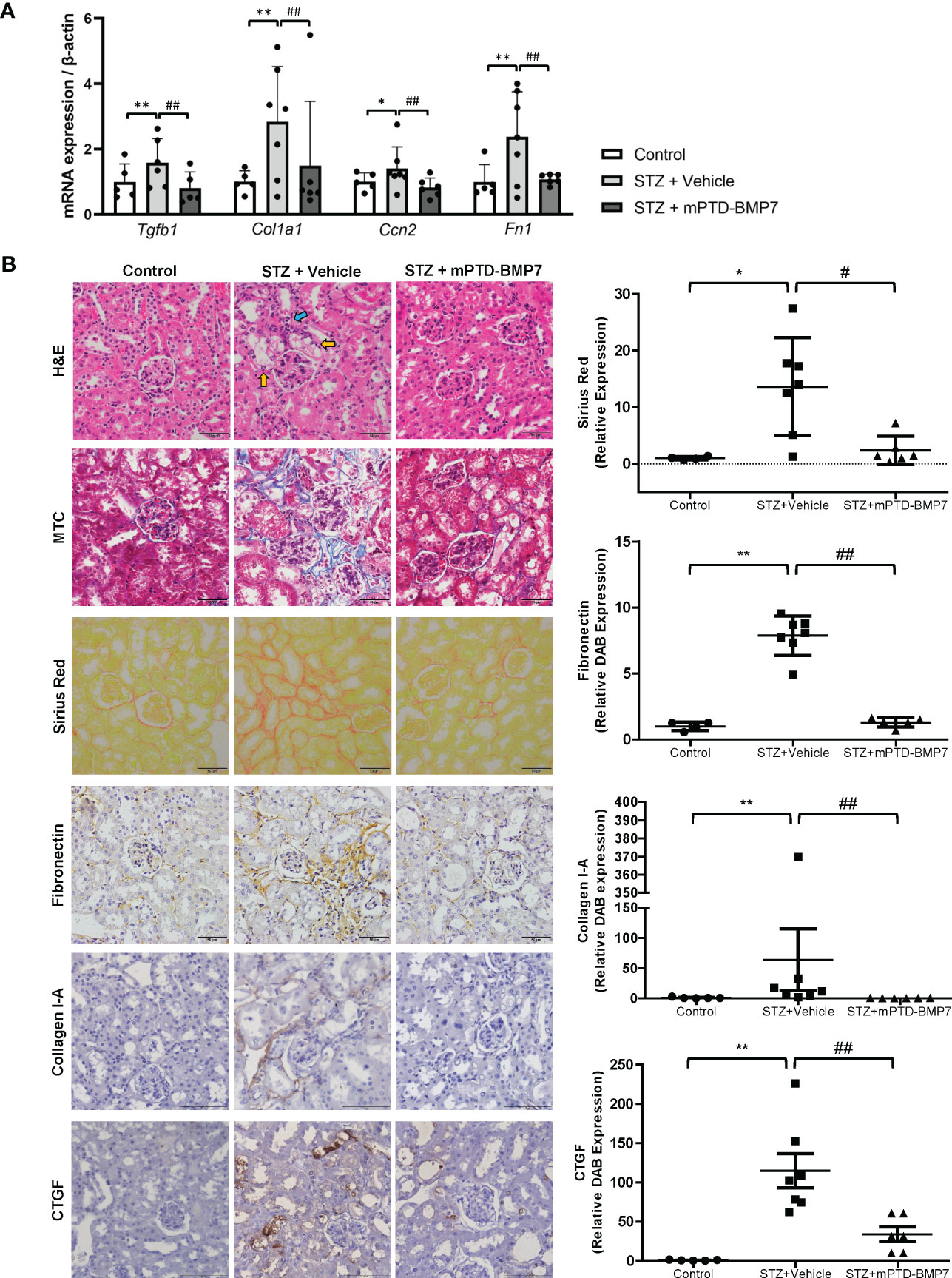
Figure 4 Effects of mPTD-BMP7 on fibrosis in the kidneys of diabetic mice. (A) Increased mRNA levels of fibrosis-related genes were attenuated by mPTD-BMP7 in the diabetic kidney. (B) Pathologic change in the diabetic kidney was observed in STZ-treated group, such as degeneration of tubular cells (yellow arrow) and tubulointerstitial inflammation (blue arrow) and fibrosis. It was abrogated by mPTD-BMP7 administration. Accumulation of collagen and fibronectin in the STZ-treated renal interstitium was detected in Masson’s trichrome (MTC), Sirius red staining and immunohistochemical staining of fibronectin and collagen type I-A, and it was reduced by mPTD-BMP2 administration. Original magnification: x400 (scale bar: 50μm). (*P < 0.05; **P < 0.01 versus control group. #P < 0.05; ##P < 0.01; versus mPTD-BMP7 group).
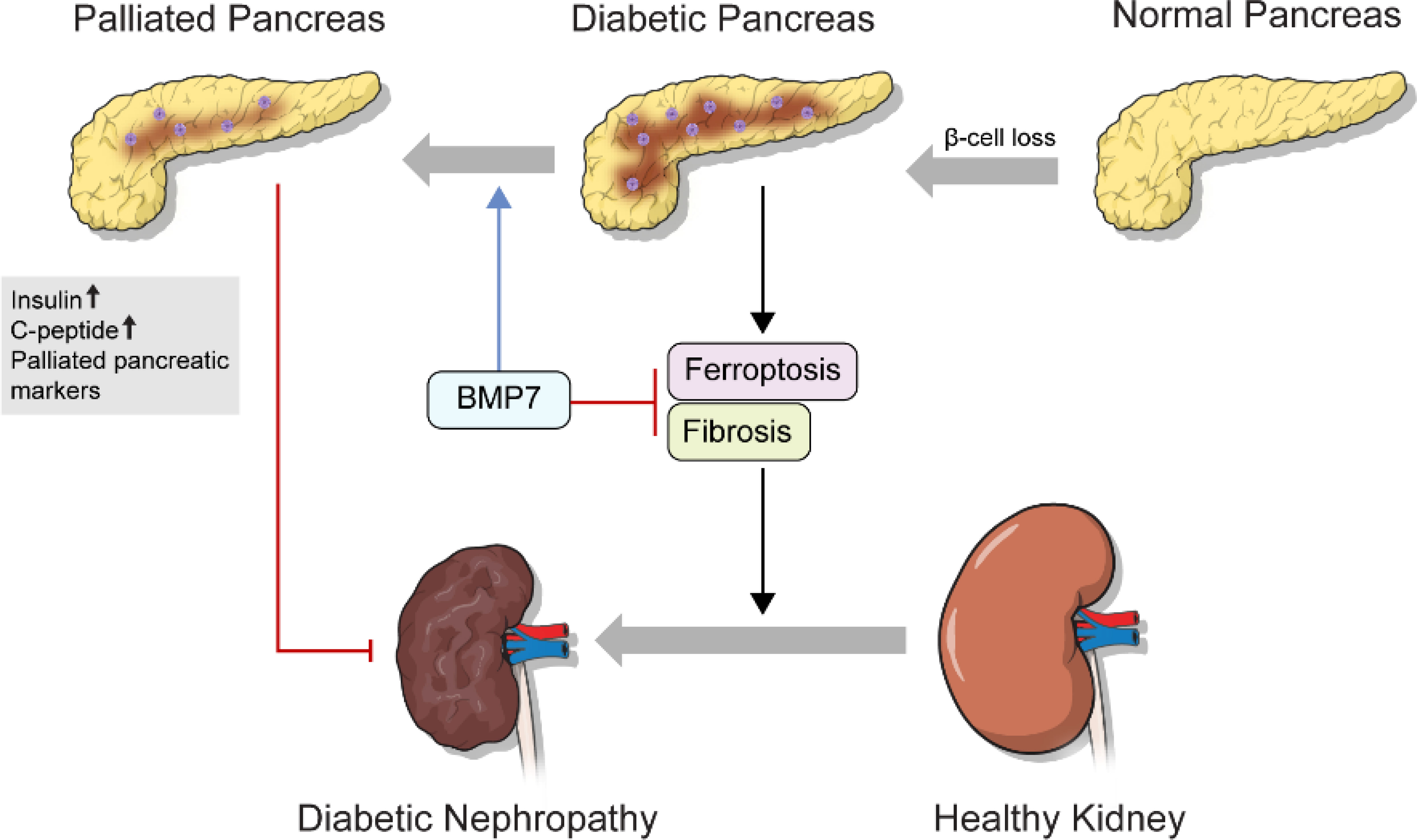
Figure 5 Schematic diagrams of the role of mPTD-BMP7 in the diabetic condition.
4 Discussion
The loss or dysfunction of β cells is involved in the pathogenesis of most types of diabetes. Many factors induce β cell loss or dysfunction, such as genetic mutation, extrinsic metabolic stress, autoimmune antibodies, inflammation, pancreatitis, pancreatic cancer, and Alzheimer’s disease (26–28). Insulin-producing β cells can be generated from non-pancreatic somatic cells, pancreatic exocrine cells, or pancreatic islet cells by transdifferentiation (29). Many studies have reported obtaining β cells from murine hepatocytes by the overexpression of pancreatic transcript factors that are important for β cells such as PDX1, NEUROD1, and neurogenin-3 (Ngn3) via adenoviral delivery in diabetic animal models (30–33). Efficient protocols of gene therapy have been proposed to convert α-cells into β cells, where adenoviral delivery of transgenic constructs such as PDX1 and MAFA genes via pancreatic duct has proven to expand β-cells in STZ-treated and nonobese diabetic mice (34). Further, acinar cells, as a prospective source of β cell generation, are abundant, in close proximity to, and share developmental origin with endocrine cells (29). Overexpression of pancreatic transcription factors such as PDX1, Ngn-3, and MafA or exposure to cytokines and growth factors enables efficient transdifferentiation into β cells in rodent models and rodent cell experiments (35–37). While the genetic manipulations are interesting, druggable therapeutics for pancreatic regeneration are limited currently. Recently, the β cell-like phenotype was induced by treatment with BMP7 in human nonendocrine pancreatic tissue (38). Further, BMP7 stimulated pancreatic progenitor cell proliferation by binding to ALK3, which is co-expressed with PDX1 in multipotent cells in exocrine pancreas (39). In our diabetic pancreas model, we found that reduced expression of many adult pancreatic markers (INS, SYP, Slc2a1, and KRT19) and pancreatic developmental markers (Pdx1, Prox1, Hnf1b, and Neurod1) in STZ-induced diabetic model was attenuated by the intraperitoneal administration of mPTD-BMP7, assuming pancreatic exocrine-endocrine conversion has partially occurred. Considering the irreversible damage to the insulin-producing beta cells of the pancreas by STZ, our study suggests the regenerative potential of mPTD-BMP7 in treating pancreatic beta cells.
Chronic kidney disease (CKD) caused by DM is diagnosed as DN, with gradual kidney dysfunction via nephron loss and characteristic pathological findings, such as increased mesangial substrate, tubular cell death, nodular lesions, and tubulointerstitial fibrosis (40, 41). Not only microvascular and glomerular injuries but also tubular atrophy and tubular epithelial cell death play a role in this condition. Hyperglycemia triggers the generation of free radicals and oxidative stress in the tubular cells, mediating proliferation, extracellular matrix deposition, and cell death (42). Since the proximal tubule requires a supply of high-energy phosphate compounds and relies on aerobic metabolism, reduced oxygen supply to the metabolically active tubular epithelial cells makes them vulnerable to diabetic ischemic injury (42, 43). Tubular cell injury and death are key findings of kidney damage in diseases such as unilateral ureteral obstruction, AKI, renal ischemia followed by reperfusion, and CKD of diabetic conditions (44–56). In our STZ model, diabetic kidney exhibited damaged renal tubules and tubulointerstitial fibrosis, but less pathologic change in the glomerulus. In addition, damage to the kidneys showed regional and early-to-mid stage destruction rather than diffuse and end-stage destruction. Our 16-week animal experiment may have been insufficient to induce chronic severe renal destruction; this was not considered to cause serious deterioration in serum and urinary functional parameters.
Many studies have focused on the modalities of apoptosis and necrosis in kidney disease; recent advances have enhanced our understanding of novel concepts of regulated necrosis, such as necroptosis, ferroptosis, pyroptosis, and mitochondrial permeability transition-dependent regulated necrosis (42). In an STZ-induced DN model, damaged tubular cells undergo various types of programmed cell death; apoptosis, autophagy, necroptosis and ferroptosis (8, 14, 57–59). Several recent studies have tried to identify the role of ferroptosis in this DN model (14, 60–62). Unlike autophagy and apoptosis, ferroptosis is a distinct type of regulated cell death characterized by iron and O2-dependent accumulation of lipid hydroperoxides to lethal levels. It was first proposed by Dixon et al. as a type of cell death triggered by the depletion of cellular amino acid cysteine or by inhibition of GPX4, causing iron and O2-dependent accumulation of lipid hydroperoxides to lethal levels (10). Disruption of the intracellular thiol antioxidant network progresses to non-apoptotic, iron-dependent, oxidative cell death, termed ferroptosis (9). In many cells, GSH synthesis is dependent on the continuous import of cysteine by the cell surface Cys2/glutamate antiporter system xc- (63, 64), the heterodimer of a membrane protein b0,+AT/SLC7A9, and its auxiliary subunit rBAT/SLC3A1, which is responsible for cysteine reabsorption, particularly in renal proximal tubules (65–68). Although Slc7a9 mutations have been reported to cause cystinuria, their functions in protecting proximal tubular cells against iron-induced oxidative damage (65, 66, 68) and ferroptosis are unclear. Slc7a9 showed a decreased expression in erastin- or TGF-β1-stimulated proximal tubular cells and in the kidneys of diabetic STZ mice. mPTD-BMP7 reversed this reduction in Slc7a9 expression. According to our results, Slc7a9 is assumed to be involved in ferroptosis as a novel cysteine importer in renal proximal tubular cells.
Among the factors that lead to the progression of DN, TGF-β1 is considered a master regulator of DN development in both type 1 and type 2 DM (4, 6, 7, 69). It is a pleiotropic cytokine involved in angiogenesis, immunomodulation, and ECM formation, stimulated by various factors including hyperglycemia and ROS in kidney cells (7). There is abundant evidence that TGF‐β plays a predominant role in kidney fibrosis, confirmed by the fact that suppression of the TGF‐β/Smad signaling pathway attenuates kidney fibrosis and injury (5). Recently, TGF‐β has been reported to induce ferroptosis by inhibiting GPX4 and cysteine transporters in hepatocellular carcinoma cells and renal tubular cells, enhancing lipid-oxidation (14, 70). Moreover, TGF‐β and ROS interact not only in their self-synthesis, but also in the downregulation of antioxidant enzymes such as glutaredoxin, catalase, superoxide dismutase, and GPX (71), the latter which has been proposed to alleviate the activation of the TGF‐β/SMAD signaling pathway, which is involved in the progression of myelofibrosis (72). In accordance with these findings, we demonstrated that Gpx4 was involved both in ferroptosis and fibrosis, and that its action was hindered by mPTD-BMP7.
Despite our results showing improvement of renal fibrosis and ferroptosis markers in tissue level and kidney functional parameters such as UACR by mPTD-BMP7, other parameters did not match the tissue status. Although a persistent decrease in eGFR (estimated glomerular filtration rate) or increase in UACR are the commonly accepted parameters in diagnosing diabetic nephropathy (73), GFR is not usually appropriate in laboratory animal model (74). However, other parameters such as BUN, serum creatinine, and urinary albumin secretion were not significantly improved. As seen in Figure 2A, the delivery of the mPTD-BMP7 was primarily concentrated in the pancreas and much less in kidney, suggesting that our form of BMP7 may not have been sufficient in kidney to significantly improve all renal functional parameters. Recently, we reported that interventional administration via renal artery in a large animal model revealed efficient delivery of mPTD-BMP7 into the kidney, resulting in inhibition of fibrotic progression of kidney (23). Therefore, peritoneal administration of mPTD-BMP7 in this study may have less therapeutic potential to the kidney.
Similarly, mPTD-BMP7 improves C-peptide and insulin in the STZ-induced mouse model, leaving blood glucose and HbA1c level. Considering the irreversible toxicity of STZ to beta cells and dietary control in mice, we believe that mPTD-BMP7 restores pancreatic damage from the STZ. Considering these together, BMP7 can be considered an alternative therapy against diabetes-induced damage to the pancreas and kidney. BMP7 helped prevent progression to DN via ferroptosis as well as via inhibiting the canonical TGF‐β signaling. Further pharmacologic and preclinical studies are required to identify clinical therapeutics for DM.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee of the Yonsei University.
Author contributions
SHS, DH performed experiments; KP, JU, and SK supported in vitro and in vivo experiments. MK and JY performed in vivo image analysis. T-HY, NK, JIY, and HK planned all experiments, analyzed the data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Research Foundation of Korea (NRF-2019R1A2C2084535, NRF-2021R1A2C3003496, NRF-2022R1A2C3004609) funded by the Korean government (MSIP).
Acknowledgments
We thank E. Tunkle for preparation of the manuscript.
Conflict of interest
T-HY, NK, HK, and JIY are the founders of MET Life Sciences Co., Ltd., and shareholders. Authors JU and SK were employed by MET Life Sciences Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sakuma H, Hagiwara S, Kantharidis P, Gohda T, Suzuki Y. Potential targeting of renal fibrosis in diabetic kidney disease using MicroRNAs. Front Pharmacol (2020) 11:587689. doi: 10.3389/fphar.2020.587689
2. Zhang Y, Jin, Kang X, Zhou R, Sun Y, Lian F, et al. Signaling pathways involved in diabetic renal fibrosis. Front Cell Dev Biol (2021) 9:696542. doi: 10.3389/fcell.2021.696542
3. Hathaway CK, Gasim AM, Grant R, Chang AS, Kim HS, Madden VJ, et al. Low TGFbeta1 expression prevents and high expression exacerbates diabetic nephropathy in mice. Proc Natl Acad Sci U S A (2015) 112(18):5815–20. doi: 10.1073/pnas.1504777112
4. Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes (1996) 45(4):522–30. doi: 10.2337/diab.45.4.522
5. Sun YB, Qu X, Caruana G, Li J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation (2016) 92(3):102–7. doi: 10.1016/j.diff.2016.05.008
6. Wang L, Wang HL, Liu TT, Lan HY. TGF-beta as a master regulator of diabetic nephropathy. Int J Mol Sci (2021) 22(15):7881. doi: 10.3390/ijms22157881
7. Zhao L, Zou Y, Liu F. Transforming growth factor-Beta1 in diabetic kidney disease. Front Cell Dev Biol (2020) 8:187. doi: 10.3389/fcell.2020.00187
8. Erekat NS. Programmed cell death in diabetic nephropathy: a review of apoptosis, autophagy, and necroptosis. Med Sci Monit (2022) 28:e937766. doi: 10.12659/msm.937766
10. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
11. Ni L, Yuan C, Wu X. Targeting ferroptosis in acute kidney injury. Cell Death Dis (2022) 13(2):182. doi: 10.1038/s41419-022-04628-9
12. Sha W, Hu F, Xi Y, Chu Y, Bu S. Mechanism of ferroptosis and its role in type 2 diabetes mellitus. J Diabetes Res (2021) 2021:9999612. doi: 10.1155/2021/9999612
13. Wang WJ, Jiang X, Gao CC, Chen ZW. Salusinbeta participates in high glucoseinduced HK2 cell ferroptosis in a Nrf2dependent manner. Mol Med Rep (2021) 24(3):674. doi: 10.3892/mmr.2021.12313
14. Kim S, Kang SW, Joo J, Han SH, Shin H, Nam BY, et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis (2021) 12(2):160. doi: 10.1038/s41419-021-03452-x
15. Yao H, Ge T, Zhang Y, Li M, Yang S, Li H, et al. BMP7 antagonizes proliferative vitreoretinopathy through retinal pigment epithelial fibrosis. Vivo vitro FASEB J (2019) 33(3):3212–24. doi: 10.1096/fj.201800858RR
16. Khan I, Agarwal P, Thangjam GS, Radhesh R, Rao SG, Kondaiah P. Role of TGF-beta and BMP7 in the pathogenesis of oral submucous fibrosis. Growth Factors (2011) 29(4):119–27. doi: 10.3109/08977194.2011.582839
17. Kinoshita K, Iimuro Y, Otogawa K, Saika S, Inagaki Y, Nakajima Y, et al. Adenovirus-mediated expression of BMP-7 suppresses the development of liver fibrosis in rats. Gut (2007) 56(5):706–14. doi: 10.1136/gut.2006.092460
18. Li RX, Yiu WH, Tang SC. Role of bone morphogenetic protein-7 in renal fibrosis. Front Physiol (2015) 6:114. doi: 10.3389/fphys.2015.00114
19. Isaka Y. Targeting TGF-beta signaling in kidney fibrosis. Int J Mol Sci (2018) 19(9):2532. doi: 10.3390/ijms19092532
20. Wang S, Chen Q, Simon TC, Strebeck F, Chaudhary L, Morrissey J, et al. Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy. Kidney Int (2003) 63(6):2037–49. doi: 10.1046/j.1523-1755.2003.00035.x
21. Peng W, Zhou X, Xu T, Mao Y, Zhang X, Liu H, et al. BMP-7 ameliorates partial epithelial-mesenchymal transition by restoring SnoN protein level via Smad1/5 pathway in diabetic kidney disease. Cell Death Dis (2022) 13(3):254. doi: 10.1038/s41419-022-04529-x
22. Lo KW, Ulery BD, Ashe KM, Laurencin CT. Studies of bone morphogenetic protein-based surgical repair. Adv Drug Delivery Rev (2012) 64(12):1277–91. doi: 10.1016/j.addr.2012.03.014
23. Kim S, Jeong CH, Song SH, Um JE, Kim HS, Yun JS, et al. Micellized protein transduction domain-bone morphogenetic protein-7 efficiently blocks renal fibrosis Via inhibition of transforming growth factor-Beta-Mediated epithelial-mesenchymal transition. Front Pharmacol (2020) 11:591275. doi: 10.3389/fphar.2020.591275
24. Kim S, Shin DH, Nam BY, Kang HY, Park J, Wu M, et al. Newly designed protein transduction domain (PTD)-mediated BMP-7 is a potential therapeutic for peritoneal fibrosis. J Cell Mol Med (2020) 24(22):13507–22. doi: 10.1111/jcmm.15992
25. Valencia JV, Weldon SC, Quinn D, Kiers GH, DeGroot J, TeKoppele JM, et al. Advanced glycation end product ligands for the receptor for advanced glycation end products: biochemical characterization and formation kinetics. Analytical Biochem (2004) 324(1):68–78. doi: 10.1016/j.ab.2003.09.013
26. Hart PA, Bellin MD, Andersen DK, Bradley D, Cruz-Monserrate Z, Forsmark CE, et al. Type 3c (pancreatogenic) diabetes mellitus secondary to chronic pancreatitis and pancreatic cancer. Lancet Gastroenterol Hepatol (2016) 1(3):226–37. doi: 10.1016/s2468-1253(16)30106-6
27. Mittal K, Mani RJ, Katare DP. Type 3 diabetes: cross talk between differentially regulated proteins of type 2 diabetes mellitus and alzheimer's disease. Sci Rep (2016) 6:25589. doi: 10.1038/srep25589
28. Wang HL, Wang L, Zhao CY, Lan HY. Role of TGF-beta signaling in beta cell proliferation and function in diabetes. Biomolecules (2022) 12(3):373. doi: 10.3390/biom12030373
29. Basile G, Kulkarni RN, Morgan NG. How, when, and where do human beta-cells regenerate? Curr Diabetes Rep (2019) 19(8):48. doi: 10.1007/s11892-019-1176-8
30. Ber I, Shternhall K, Perl S, Ohanuna Z, Goldberg I, Barshack I, et al. Functional, persistent, and extended liver to pancreas transdifferentiation. J Biol Chem (2003) 278(34):31950–7. doi: 10.1074/jbc.M303127200
31. Ferber S, Halkin A, Cohen H, Ber I, Einav Y, Goldberg I, et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med (2000) 6(5):568–72. doi: 10.1038/75050
32. Kojima H, Fujimiya M, Matsumura K, Younan P, Imaeda H, Maeda M, et al. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med (2003) 9(5):596–603. doi: 10.1038/nm867
33. Tang DQ, Shun L, Koya V, Sun Y, Wang Q, Wang H, et al. Genetically reprogrammed, liver-derived insulin-producing cells are glucose-responsive, but susceptible to autoimmune destruction in settings of murine model of type 1 diabetes. Am J Transl Res (2013) 5(2):184–99.
34. Xiao X, Guo P, Shiota C, Zhang T, Coudriet GM, Fischbach S, et al. Endogenous reprogramming of alpha cells into beta cells, induced by viral gene therapy, reverses autoimmune diabetes. Cell Stem Cell (2018) 22(1):78–90.e4. doi: 10.1016/j.stem.2017.11.020
35. Baeyens L, Lemper M, Leuckx G, De Groef S, Bonfanti P, Stange G, et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol (2014) 32(1):76–83. doi: 10.1038/nbt.2747
36. Rooman I, Bouwens L. Combined gastrin and epidermal growth factor treatment induces islet regeneration and restores normoglycaemia in C57Bl6/J mice treated with alloxan. Diabetologia (2004) 47(2):259–65. doi: 10.1007/s00125-003-1287-1
37. Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature (2008) 455(7213):627–32. doi: 10.1038/nature07314
38. Klein D, Alvarez-Cubela S, Lanzoni G, Vargas N, Prabakar KR, Boulina M, et al. BMP-7 induces adult human pancreatic exocrine-to-Endocrine conversion. Diabetes (2015) 64(12):4123–34. doi: 10.2337/db15-0688
39. Qadir MMF, Alvarez-Cubela S, Klein D, Lanzoni G, Garcia-Santana C, Montalvo A, et al. P2RY1/ALK3-expressing cells within the adult human exocrine pancreas are BMP-7 expandable and exhibit progenitor-like characteristics. Cell Rep (2018) 22(9):2408–20. doi: 10.1016/j.celrep.2018.02.006
40. Tervaert TW, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol (2010) 21(4):556–63. doi: 10.1681/ASN.2010010010
41. Yokoyama H, Okudaira M, Otani T, Sato A, Miura J, Takaike H, et al. Higher incidence of diabetic nephropathy in type 2 than in type 1 diabetes in early-onset diabetes in Japan. Kidney Int (2000) 58(1):302–11. doi: 10.1046/j.1523-1755.2000.00166.x
42. Priante G, Gianesello L, Ceol M, Del Prete D, Anglani F. Cell death in the kidney. Int J Mol Sci (2019) 20(14). doi: 10.3390/ijms20143598
43. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol (2002) 192(1):1–15. doi: 10.1002/jcp.10119
44. Jang HS, Padanilam BJ. Simultaneous deletion of bax and bak is required to prevent apoptosis and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol (2015) 309(6):F540–50. doi: 10.1152/ajprenal.00170.2015
45. Kennedy WA 2nd, Buttyan R, Garcia-Montes E, D'Agati V, Olsson CA, Sawczuk IS. Epidermal growth factor suppresses renal tubular apoptosis following ureteral obstruction. Urology (1997) 49(6):973–80. doi: 10.1016/s0090-4295(97)00101-5
46. Kennedy WA, Stenberg A, Lackgren G, Hensle TW, Sawczuk IS. Renal tubular apoptosis after partial ureteral obstruction. J Urol (1994) 152(2 Pt 2):658–64. doi: 10.1016/s0022-5347(17)32675-7
47. Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Renal Physiol (2002) 283(5):F861–75. doi: 10.1152/ajprenal.00362.2001
48. Nilsson L, Madsen K, Krag S, Frokiaer J, Jensen BL, Norregaard R. Disruption of cyclooxygenase type 2 exacerbates apoptosis and renal damage during obstructive nephropathy. Am J Physiol Renal Physiol (2015) 309(12):F1035–48. doi: 10.1152/ajprenal.00253.2015
49. Truong LD, Petrusevska G, Yang G, Gurpinar T, Shappell S, Lechago J, et al. Cell apoptosis and proliferation in experimental chronic obstructive uropathy. Kidney Int (1996) 50(1):200–7. doi: 10.1038/ki.1996.303
50. Hoste EA, Clermont G, Kersten A, Venkataraman R, Angus DC, De Bacquer D, et al. RIFLE criteria for acute kidney injury are associated with hospital mortality in critically ill patients: a cohort analysis. Crit Care (2006) 10(3):R73. doi: 10.1186/cc4915
52. Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol (2014) 25(12):2689–701. doi: 10.1681/ASN.2014030262
53. Price PM, Hodeify R. A possible mechanism of renal cell death after ischemia/reperfusion. Kidney Int (2012) 81(8):720–1. doi: 10.1038/ki.2011.495
54. Schumer M, Colombel MC, Sawczuk IS, Gobe G, Connor J, O'Toole KM, et al. Morphologic, biochemical, and molecular evidence of apoptosis during the reperfusion phase after brief periods of renal ischemia. Am J Pathol (1992) 140(4):831–8.
55. Khan S, Cleveland RP, Koch CJ, Schelling JR. Hypoxia induces renal tubular epithelial cell apoptosis in chronic renal disease. Lab Invest (1999) 79(9):1089–99.
56. Thomas GL, Yang B, Wagner BE, Savill J, El Nahas AM. Cellular apoptosis and proliferation in experimental renal fibrosis. Nephrol Dial Transplant (1998) 13(9):2216–26. doi: 10.1093/ndt/13.9.2216
57. Kumar D, Zimpelmann J, Robertson S, Burns KD. Tubular and interstitial cell apoptosis in the streptozotocin-diabetic rat kidney. Nephron Exp Nephrol (2004) 96(3):e77–88. doi: 10.1159/000076749
58. Guo M, Chen Q, Huang Y, Wu Q, Zeng Y, Tan X, et al. High glucose-induced kidney injury via activation of necroptosis in diabetic kidney disease. Oxid Med Cell Longev (2023) 2023:2713864. doi: 10.1155/2023/2713864
59. Ding Y, Choi ME. Autophagy in diabetic nephropathy. J Endocrinol (2015) 224(1):R15–30. doi: 10.1530/joe-14-0437
60. Wu Y, Chen Y. Research progress on ferroptosis in diabetic kidney disease. Front Endocrinol (Lausanne) (2022) 13:945976. doi: 10.3389/fendo.2022.945976
61. Wang Y, Bi R, Quan F, Cao Q, Lin Y, Yue C, et al. Ferroptosis involves in renal tubular cell death in diabetic nephropathy. Eur J Pharmacol (2020) 888:173574. doi: 10.1016/j.ejphar.2020.173574
62. Yang XD, Yang YY. Ferroptosis as a novel therapeutic target for diabetes and its complications. Front Endocrinol (Lausanne) (2022) 13:853822. doi: 10.3389/fendo.2022.853822
63. Bannai S, Tsukeda H, Okumura H. Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem Biophys Res Commun (1977) 74(4):1582–8. doi: 10.1016/0006-291x(77)90623-4
64. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem (1999) 274(17):11455–8. doi: 10.1074/jbc.274.17.11455
65. Correia MJ, Pimpao AB, Fernandes DGF, Morello J, Sequeira CO, Calado J, et al. Cysteine as a multifaceted player in kidney, the cysteine-related thiolome and its implications for precision medicine. Molecules (2022) 27(4):1416. doi: 10.3390/molecules27041416
66. Nagamori S, Wiriyasermkul P, Guarch ME, Okuyama H, Nakagomi S, Tadagaki K, et al. Novel cystine transporter in renal proximal tubule identified as a missing partner of cystinuria-related plasma membrane protein rBAT/SLC3A1. Proc Natl Acad Sci U S A (2016) 113(3):775–80. doi: 10.1073/pnas.1519959113
67. Okazaki Y. The role of ferric nitrilotriacetate in renal carcinogenesis and cell death: from animal models to clinical implications. Cancers (Basel) (2022) 14(6):1495. doi: 10.3390/cancers14061495
68. Chillaron J, Font-Llitjos M, Fort J, Zorzano A, Goldfarb DS, Nunes V, et al. Pathophysiology and treatment of cystinuria. Nat Rev Nephrol (2010) 6(7):424–34. doi: 10.1038/nrneph.2010.69
69. Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A (2000) 97(14):8015–20. doi: 10.1073/pnas.120055097
70. Kim DH, Kim WD, Kim SK, Moon DH, Lee SJ. TGF-beta1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis (2020) 11(5):406. doi: 10.1038/s41419-020-2618-6
71. Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic Biol Med (2010) 48(1):1–15. doi: 10.1016/j.freeradbiomed.2009.09.026
72. Li Z, Zhu Z, Liu Y, Liu Y, Zhao H. Function and regulation of GPX4 in the development and progression of fibrotic disease. J Cell Physiol (2022) 237(7):2808–24. doi: 10.1002/jcp.30780
73. Selby NM, Taal MW. An updated overview of diabetic nephropathy: diagnosis, prognosis, treatment goals and latest guidelines. Diabetes Obes Metab (2020) 22(S1):3–15. doi: 10.1111/dom.14007
Keywords: Bmp7, ferroptosis, TGF-β, fibrosis, diabetic nephropathy
Citation: Song SH, Han D, Park K, Um JE, Kim S, Ku M, Yang J, Yoo T-H, Yook JI, Kim NH and Kim HS (2023) Bone morphogenetic protein-7 attenuates pancreatic damage under diabetic conditions and prevents progression to diabetic nephropathy via inhibition of ferroptosis. Front. Endocrinol. 14:1172199. doi: 10.3389/fendo.2023.1172199
Received: 23 February 2023; Accepted: 04 May 2023;
Published: 24 May 2023.
Edited by:
Jia Fu, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Dong Zhou, University of Connecticut, United StatesXiangchen Gu, Shanghai Jiao Tong University, China
Copyright © 2023 Song, Han, Park, Um, Kim, Ku, Yang, Yoo, Yook, Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyun Sil Kim, S0hTQHl1aHMuYWM=; Nam Hee Kim, bWlnbzc3QHl1aHMuYWM=
†Present address: Seonghun Kim, Department of Neurosurgery, Duke University School of Medicine, Durham, NC, United States
‡These authors have contributed equally to this work and share first authorship