 Anne Gulbins
Anne Gulbins Gina-Eva Görtz
Gina-Eva Görtz Erich Gulbins
Erich Gulbins Anja Eckstein
Anja Eckstein- 1Department of Ophthalmology, University Hospital Essen, University of Duisburg-Essen, Essen, Germany
- 2Institute of Molecular Biology, University Hospital Essen, University of Duisburg-Essen, Essen, Germany
Graves’ disease (GD) is caused by an autoimmune formation of autoantibodies and autoreactive T-cells against the thyroid stimulating hormone receptor (TSHR). The autoimmune reaction does not only lead to overstimulation of the thyroid gland, but very often also to an immune reaction against antigens within the orbital tissue leading to thyroid eye disease, which is characterized by activation of orbital fibroblasts, orbital generation of adipocytes and myofibroblasts and increased hyaluronan production in the orbit. Thyroid eye disease is the most common extra-thyroidal manifestation of the autoimmune Graves’ disease. Several studies indicate an important role of sphingolipids, in particular the acid sphingomyelinase/ceramide system and sphingosine 1-phosphate in thyroid eye disease. Here, we discuss how the biophysical properties of sphingolipids contribute to cell signaling, in particular in the context of thyroid eye disease. We further review the role of the acid sphingomyelinase/ceramide system in autoimmune diseases and its function in T lymphocytes to provide some novel hypotheses for the pathogenesis of thyroid eye disease and potentially allowing the development of novel treatments.
1 Introduction
Sphingolipids have been shown to be important in several autoimmune diseases such as multiple sclerosis and arthritis (1–4). Both, the de novo synthesis of sphingolipids and the sphingomyelinase-pathway are implied in the pathogenesis of auto-immune disorders (1–4). However, at present only limited information is available on the role of sphingolipids in Graves’ disease (GD) and thyroid eye disease. GD is an autoimmune disorder, which is caused by activation of the immune system against the endogenous thyroid stimulating hormone receptor (TSHR), with the formation of auto-antibodies that trigger alterations and the clinical symptoms affecting the thyroid, the eye orbit and other tissues. Here, we summarize the current knowledge about the role of sphingolipids in autoimmune diseases, in particular GD, present an overview about the pathophysiology of GD and thyroid eye disease and discuss hypotheses how to combine the two areas of research. The current knowledge on thyroid eye disease is briefly summarized in Figure 1 (please refer for an extended presentation to the chapters below). The pathways of ceramide synthesis and sphingomyelinase-activities are given in Figure 2.
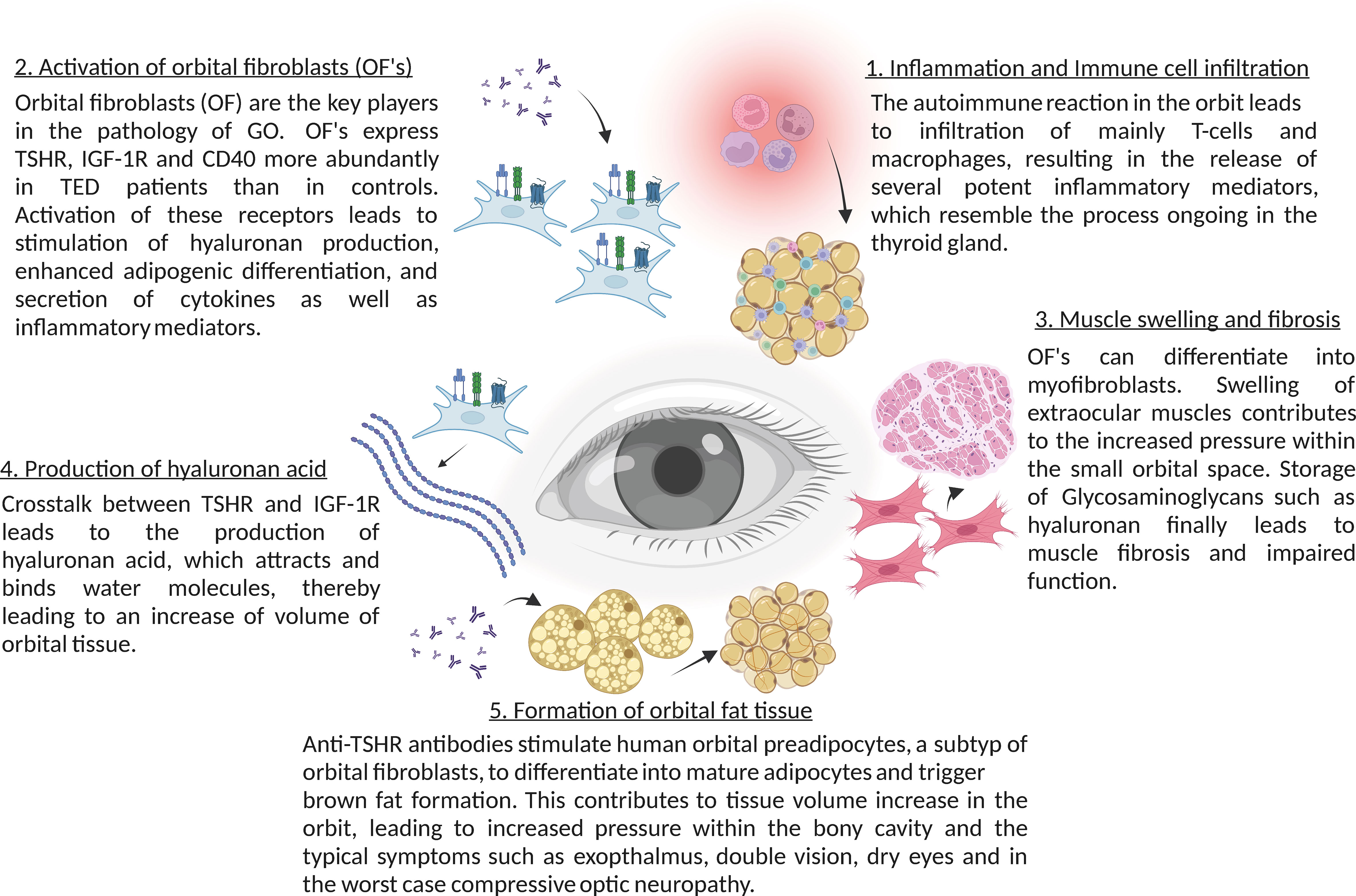
Figure 1 Summary of the main molecular mechanisms in thyroid eye disease (TED).
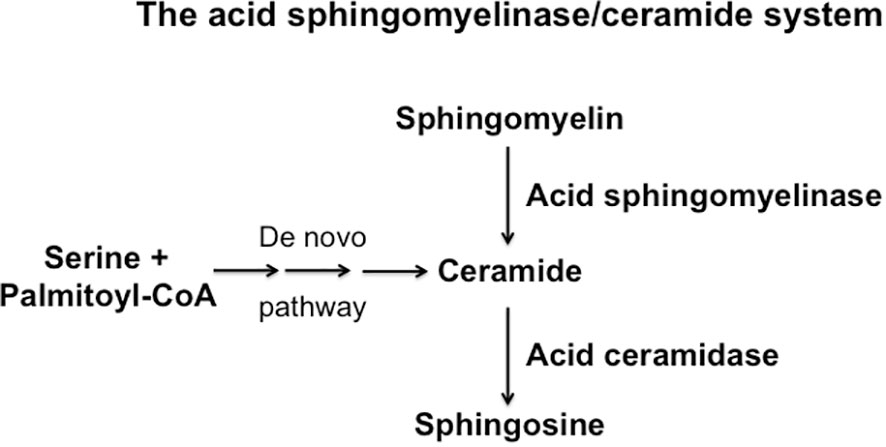
Figure 2 Schematic overview over the synthesis and consumption of ceramide, sphingomelin and sphingosine.
2 Sphingolipids and ceramide
Sphingolipids belong to the major components of the biological membranes. They are composed of the very hydrophobic ceramide moiety and a hydrophilic headgroup, for instance phosphorylcholine for the abundant membrane lipid sphingomyelin. Sphingolipids are critically involved in the structure of cellular membranes (5, 6). However, they are not only structural molecules of cellular membranes, but also involved in cellular signal transduction, cell stimulation, stress and death. Sphingomyelin molecules interact with each other and together with cholesterol molecules that stabilize the bulky sphingomyelin aggregates, they contribute to the organization of cellular membranes and the formation of small distinct membrane domains (5–7). These very small domains, with a diameter of approximately 20 nm (8), were named rafts (7), which may serve to sort receptor molecules, although this function still needs to be formally proved in vivo.
The hydrolysis of sphingomyelin results in the formation of ceramide, which is an amide ester of D-erythro-sphingosine and a fatty acid containing 2-32 (or even longer) carbon atoms in the acyl chain (5, 6). The hydrolysis of ceramide is mediated by acid, neutral or alkaline sphingomyelinases. However, ceramide can be also generated by the de novo synthase pathway, a retrograde production from sphingosine via ceramidases and upon hydrolysis of complex glycosylated lipids or dephosphorylation of ceramide 1-phosphate (5, 9–11). The generation of ceramide from sphingomyelin in small rafts has a dramatic effect on the biophysical properties of these small membrane domains: Ceramide molecules are very hydrophobic and thereby these molecules have the tendency to spontaneously self-associate to small microdomains that further fuse to large ceramide-enriched membrane domains, also named platforms (5, 6, 12, 13). A variety of studies has shown the formation of such ceramide-enriched membrane domains in particular upon application of stress and stimulation of certain receptors, for instance upon infection of mammalian cells with pathogens such as Pseudomonas aeruginosa, Neisseriae gonorrhoeae, Rhinovirus or SARS-CoV-2, stimulation of receptors such as CD95, CD40 or DR5 or application of stress stimuli such as platelet activating factor, irradiation, UV-light or Cu2+ application (14–26). Although ceramide-enriched membrane domains do not seem to have specific signaling functions for all of these diverse stimuli, they are central for cell activation, because they serve to trap, cluster and organize receptor and intracellular signaling molecules in the cell membrane, such as CD95, CD40, DR5, β1-integrin or NADPH-oxidase (12, 19, 20, 23, 25, 27). The reorganization process of receptors and signaling molecules results in the spatial and temporal organization of the signalosome generated by a specific receptor or stress stimulus. This re-organization process allows receptors to associate with down-stream signaling molecules, to exclude inhibitory molecules, to concentrate a high number of receptor molecules in a small area of the plasma membrane and thereby to greatly amplify the primary signals finally permitting a specific receptor to transmit its signal into the cell (27). This mechanism is, in particular, mediated by the acid sphingomyelinase, which resides in lysosomes. Upon cellular activation, the acid sphingomyelinase is often exposed onto the cell surface by fusion of secretory lysosomes with the plasma membrane (12, 28, 29). This fusion process of secretory lysosomes with the plasma membrane is mediated by a syntaxin 4-regulated transport of intracellular vesicles (28), although many details of this process are still unknown. The acid sphingomyelinase generates ceramide on the cell surface, for instance after irradiation or CD95 stimulation (12, 29), but it is also possible that ceramide is already generated in secretory lysosomes and exposed together with the acid sphingomyelinase. These processes are certainly not exclusive and a local surface acid sphingomyelinase activity may lead to further formation of ceramide and thereby control the strength and the duration of signals generated by receptor clustering.
In addition to re-organizing the cell membrane and to form membrane platforms, ceramide has been also shown to directly regulate several molecules, i.e. cathepsin D (30), phospholipase A2 (31), kinase suppressor of Ras (32), ceramide-activated protein serine-threonine phosphatases [CAPP] (33), protein kinase C isoforms (34, 35), phosphatase 2A (36), Lc3B (37) and phospholipase D (38). Studies by Schneider-Schaulies et al. demonstrated an important impact of ceramide on the cytoskeleton, in particular F-actin, ezrin and moesin, and migratory properties of lymphocytes (39), although it is unknown whether this is mediated by a direct interaction of membrane ceramide with cytoskeletal proteins or via an indirect effect of ceramide. Further, it has been demonstrated that ceramide regulates the activity of ion channels, such as the potassium channel Kv1.3 and calcium release-activated calcium channels (40–43), but it is again unknown whether this effect is direct or mediated via re-organization and/or lipid-protein interactions of the ion channels in ceramide-enriched membrane domains.
3 Thyroid eye disease
3.1 Clinics
Graves’ disease (GD) is an autoimmune disorder that is caused by autoantibodies against the thyroid stimulating hormone receptor (TSHR). These anti-TSHR-antibodies are often stimulating antibodies of the TSHR and therefore lead to an overstimulation of the thyroid gland (44). TSHR-stimulating autoantibodies bind to the TSHR at a similar extracellular site as the thyroid stimulating hormone (TSH) itself and cause deregulated TSHR hyper-activation (44) resulting in Graves’ disease (45).
The continuous stimulation of TSHR leads to uncontrolled production of thyroid hormones T3 and T4 in the thyroid. To date, TSHR hyper-activation is treated by anti-thyroid drugs correcting the “clinically evident” consequences. This involves pharmacological downstream suppression of the enzyme thyroperoxidase to impair thyroid hormone synthesis and thyroid hormone release from the thyroid. About half of the patients go into remission over time due to spontaneous decay of anti-TSHR antibodies, the other half suffers from chronic relapsing course due to persistence of the immune response (46). In these patients the thyroid will be treated by thyroidectomy or radioiodine therapy. In some of these patients anti-TSHR antibodies persist particularly after radioiodine therapy, and therefore extrathyroidal manifestations can also occur later in the course of these cases with Graves’ disease (47). The most common extrathyroidal manifestation is thyroid eye disease, also known as Graves’ Orbitopathy (TED or GO) (46, 48, 49). Acropachy and pretiabial myxedema are much less common. About half of Graves’ disease patients develop thyroid eye disease and 3 to 5% develop a severe course of disease threatening their vision (46).
3.2 Molecular mechanisms of thyroid eye disease
The TSHR belongs to the glycoprotein hormone receptors, a subfamily of class A G-protein-coupled receptors. The interplay between TSH and TSHR constitutes the pituitary-thyroid axis, which tightly regulates growth, development and metabolism. There have been numerous publications showing the relation between anti-TSHR antibodies level and activity and thyroid eye disease (e.g. 50–56).
Anti-TSHR antibodies do not only bind to follicular epithelial cells of the thyroid gland, but also to retro-orbital fibroblasts, which express TSHR (57). Therefore, autoimmune anti-TSHR autoantibodies activate the TSHR in the orbit causing Thyroid eye disease, a potentially quality of life-reducing and sight-threatening disease.
Binding of anti-TSHR antibodies to the TSHR results in direct activation of target cells, but also immediate stimulation of hyaluronan secretion, one of the central patho-mechanisms of thyroid eye disease, since hyaluronic acid binds water molecules, which leads to increase of volume of orbital tissues (58).
Anti-TSHR antibodies also stimulate human orbital preadipocytes to differentiate into mature adipocytes via phosphoinositide 3-kinase activation, which also contributes to the tissue volume increase in the orbit (59, 60). These two pathomechanisms lead clinically to swollen extraocular muscles and exophthalmos. Since the volume increases occur in a bony-limited orbit, it can lead to more or less compression of the orbit depending on the strength of the orbital ligaments, impairment of orbital venous and lymphatic outflow and tissue hypoxia (61, 62).
In addition to the stimulation of TSHR in fibroblasts and adipocytes, the autoimmune reaction in the orbit also leads to infiltration with macrophages and T cells into orbital tissues, resulting in the release of several potent inflammatory mediators. In the initial phase of the disease an increased activity of Th1 lymphocytes has been shown, which leads to the production and release of typical cytokines, such as IL-1β, IL-2, TNF-α and IFN-γ, enhancing the proliferation of orbital fibroblasts and production of glycosaminoglycans (63). These cytokines further attract infiltration of immune cells T and B cells, monocytes, and neutrophils via cytokines like CCL2, CCL5 and CCL20, which migrate into the orbital tissue and enhance the inflammatory process (64, 65). During the ongoing inflammatory process Th2 lymphocytes get activated as well and further release cytokines, such as IL-4, IL-5, IL-10 and IL-13 (66). In the later stages of the disease tissue expansion, remodeling and fibrosis dominate the autoimmune disorder (67). In addition, an impaired suppressor function of Treg lymphocytes in patients with thyroid eye disease has been suggested (65). Tregs are important for tolerance to self-antigens and for modulating the autoimmune system by the release of anti-inflammatory cytokines (68). Patients with thyroid eye disease show a significant increase in Th17-cells in the serum, contributing to the inflammatory infiltration by mediating pro-inflammatory responses (69). This suggests a dysregulation of the Th17/Treg balance towards lower Tregs and higher Th17 levels in thyroid eye disease patients.
In addition, sphingolipids are known to be involved in the control of development, differentiation, activation, proliferation and attraction of lymphocytes (70–72).
These changes are found to varying degrees in human tissue and also in the thyroid eye disease animal model. This high variability of autoimmune-driven inflammation in in vivo and in vitro models is also reflected by the clinical manifestation, which varies greatly among patients depending on their age and risk factors.
In the past, therapeutic efforts focused on anti-inflammatory therapy with the aim to stop the inflammatory reaction in the orbit and the autoimmune reaction in general (73). In 2017, the results of a phase 2 study were published, in which for the first time patients with thyroid eye disease were treated with an antibody that blocks the IGF1R (74). The results mark a turning point of thyroid eye disease treatment, because for the first time, exophthalmos (80%) and motility impairment (approx. 50%) could be efficiently remedied (75). However, first case reports for the IGF-1R antibody therapy show recurrences after this therapy, since the autoimmune reaction does not seem to be influenced by this therapy (76). Therefore there is a strong need to further elucidate the autoimmune reaction in thyroid eye disease to develop causative treatments. Sphingolipids may play an important role here.
4 Sphingolipids in thyroid eye disease
The role of sphingolipids in thyroid eye disease is largely unknown. Studies by our group demonstrated that fibroblasts in the orbit of patients with thyroid eye disease exhibit an altered phenotype with the expression of CD40, which is a co-stimulatory receptor usually expressed on the surface of immune cells such as B-lymphocytes or antigen-presenting cells (for review see e.g. 77). Activated T lymphocytes express the ligand of CD40, i.e. CD154. Binding of CD154 to its cognate receptor CD40 results in B lymphocyte activation, in particular immunoglobulin class switching from IgM to IgG (77). The T cell mediated help is a central step in immune stimulation in general and also plays an important role in the pathogenesis of autoimmune disorders (77). Moreover, expression of CD40 is not restricted to B-lymphocytes and antigen presenting cells, but it is also present on fibroblasts of the orbit in patients with thyroid eye disease (78–80). Here, the expression of CD40 in the diseased orbit creates a link to previous studies demonstrating that cellular stimulation via CD40 results in a rapid activation of the acid sphingomyelinase, a translocation of the acid sphingomyelinase onto the cell surface and the generation of surface ceramide with the subsequent formation of large ceramide-enriched membrane platforms that cluster and signal CD40 (19). Molecular studies demonstrated that the transmembranous domain of CD40 mediates clustering of CD40 (81). In these studies, the transmembranous domain of CD40 was exchanged with the transmembranous domain of CD45, which does not cluster in ceramide-enriched membrane domains (81). This chimeric fusion protein did not cluster in ceramide-enriched membrane domains anymore and did not signal under physiological conditions, while forced cross-linking and clustering of this chimeric molecule using crosslinked antibodies reconstituted CD40-typical signaling (81). This establishes a central role of ceramide-mediated clustering of CD40 for signaling of CD40.
Consistent with the hypothesis that CD40 clustering also mediates activation of orbital fibroblasts in thyroid eye disease, we observed an activation of the acid sphingomyelinase and a release of ceramide upon activation of CD40 in thyroid eye disease-derived fibroblasts (80).
The generation of ceramide also allows increased formation of sphingosine 1-phosphate and it was demonstrated that activation of fibroblasts isolated from the orbit of patients with thyroid eye disease also shows a higher stimulation of sphingosine kinase 1 (80), which converts sphingosine to sphingosine 1-phosphate (82). CD40 stimulation of these fibroblasts resulted in a further increased formation of sphingosine and a release of sphingosine 1-phosphate (80). Sphingosine 1-phosphate is known to be central in the regulation of T lymphocyte emigration into tissues (82). Several studies (72, 83, 84) demonstrated that sphingosine 1-phosphate is important in key aspects of thyroid eye disease, including the induction of inflammation, increased adipogenesis in the orbit and the induction of orbital fibrosis (72, 83, 84). These studies demonstrated increased expression of sphingosine 1-phosphate receptors 1, 2 and 3 in the orbit from patients with thyroid eye disease (72, 83, 84). In vitro data showed that adipocytes responded with increased differentiation upon stimulation with sphingosine 1-phosphate, which was prevented by an inhibitor of the sphingosine 1-phosphate receptor 1 (72). Further, orbital fibroblasts from patients with Graves’ orbitopathy exhibited an increased production of reactive oxygen species, which was also blocked by an inhibitor of sphingosine 1-phosphate receptors (83). It remains to be determined whether the increased production of reactive oxygen species also mediates a constitutive activation of the acid sphingomyelinase, which is regulated by redox mechanisms (85, 86). Such a stimulation would also result in the formation of ceramide-enriched membrane platforms that serve to cluster NADPH-oxidase and thereby further promote the formation of reactive oxygen species (86). Such a vicious cycle, once initiated by the inflammation-triggered alteration of orbital fibroblasts may continuously drive the autoimmune disease, even after down-regulating antigen expression. It is also tempting to speculate that cigarette smoking, one of the most important risks for thyroid eye disease (87), drives such a reactive oxygen species – acid sphingomyelinase – ceramide cycle and thereby promotes the disease. In accordance, Ko et al. demonstrated increased expression levels of sphingosine 1-phosphate 1 receptor mRNA and of sphingosine 1-phosphate in orbital fibroblasts from patients with GD, in particular after stimulation with cigarette smoke extract (84). The changes of sphingosine 1-phosphate correlated with increased formation of collagen Iα, fibronectin, and α-smooth muscle actin, as well as IL-1β–induced expression of metalloproteases MMP-1, MMP-2, MMP-9, and TIMP-1, events that were prevented by inhibition of sphingosine 1-phosphate receptors (83).
Our own studies demonstrated that stimulation of fibroblasts isolated from the orbit of patients with Graves’ orbitopathy via CD40 attracted T lymphocytes, which was blocked by a sphingosine kinase inhibitor (80). Thus, alterations of fibroblasts in the orbit of patients with Graves’ orbitopathy may drive the influx of T lymphocytes into the orbit via the generation of sphingosine 1-phosphate.
Collectively, these studies indicate that sphingosine 1-phosphate seems to be important for several aspects of thyroid eye disease, in particular adipogenesis and activation of local fibroblasts. At present, the role of ceramide and sphingosine in thyroid eye disease requires definition.
5 Sphingolipids in autoimmune lymphocyte activation
The state of the current knowledge on the role of sphingolipids in thyroid eye disease is presented above. However, there is good evidence to postulate an important role of the acid sphingomyelinase in autoimmune disorders. Thus, it was shown that the acid sphingomyelinase plays an important role in multiple sclerosis (1, 2, 4), the most important autoimmune disorder of the central nervous system and the leading cause of neurological disability among young adults in the Western world (88, 89). Multiple sclerosis is characterized by focal inflammation in the central nervous system eventually leading to local demyelination, loss of neurons, glia scars and neurological symptoms, such as motor and sensory deficits or impairment of vision. As in thyroid eye disease the autoimmune process is initiated by infiltration of CD4+ and CD8+ T lymphocytes as well as macrophages (88, 90–92), which then orchestrate and execute central nervous inflammation and damage. Studies on the role of the acid sphingomyelinase in multiple sclerosis demonstrated that genetic deficiency of acid sphingomyelinase prevented many aspects of multiple sclerosis, including blood-brain barrier disruption, influx of immune cells into the central nervous system and, thereby, neuroinflammation (1, 2). Acid sphingomyelinase-deficient mice were almost completely protected against the induction of an experimental multiple sclerosis, i.e. experimental autoimmune encephalomyelitis (1, 2). Experimental autoimmune encephalomyelitis was also inhibited by treatment of the mice with functional inhibitors of the acid sphingomyelinase, such as amitriptyline or sertraline (2). These drugs interfere with binding of the acid sphingomyelinase to membranes and displace the acid sphingomyelinase from intralysosomal membranes resulting in proteolytic degradation of the acid sphingomyelinase (93–95). These drugs are often antidepressants (94, 95).
Deficiency or pharmacological blockade of the acid sphingomyelinase also prevented the induction of another autoimmune disorder, i.e. autoimmune arthritis (3). These studies revealed an induction of severe arthritis upon autoimmune immunization, which was prevented by genetic deficiency or pharmacological inhibition of the acid sphingomyelinase (3). In summary, the data clearly demonstrate that the acid sphingomyelinase plays a pivotal role in autoimmune disorders, although the molecular mechanisms of the involvement of the acid sphingomyelinase are still unknown. It is therefore tempting to speculate that the acid sphingomyelinase also plays a role in GD, although this needs to be tested.
Sphingomyelinases have been shown to regulate several aspects of lymphocyte biology, although a comprehensive picture is missing.
Several studies have shown that deletion of the acid sphingomyelinase results in an increased formation of regulatory T lymphocytes that are able to down-regulate or even inhibit immune responses (96, 97). Acid sphingomyelinase-deficient mice have higher number of regulatory T cells compared to littermate control mice. In vitro stimulation of acid sphingomyelinase-deficient T-lymphocytes resulted in higher number of Foxp3+ -induced regulatory T cells, compared with control T-cells (97). This effect might be mediated by Akt and Rictor, since acid sphingomyelinase-deficient induced regulatory T cells demonstrated reduced phosphorylation of Akt and reduced expression of Rictor, a protein that complexes with mTor2 and has been shown to be involved in regulation of cell growth and proliferation (96).
The generation of regulatory T cells in mice lacking the acid sphingomyelinase might explain, at least in part, the inhibition of autoimmune responses in acid sphingomyelinase-deficient mice or upon treatment with pharmacological inhibitors of the acid sphingomyelinase.
A recent study using T cell-specific acid sphingomyelinase- or acid ceramidase-deficient mice revealed that T cell-specific ablation of acid sphingomyelinase resulted in reduced ceramide levels in T cells and an impairment of T cell responses, whereas T cell specific deletion of the acid ceramidase elevated T cell activation (98). Decreased ceramide concentrations promoted differentiation of CD4+ regulatory T cells, but also negatively interfered with cytotoxic activity of CD8+ T cells, which was improved by elevated ceramide concentrations (98). The studies also linked the T cell receptor (TCR/CD3) complex to ceramide-enriched membrane domains, since ceramide co-localized with the TCR/CD3 complex forming an immune synapse in stimulated T cells, an event that seems to regulate the strength of TCR/CD3 signaling (99).
Several studies imply that also neutral sphingomyelinases are important in T cell activation (100, 101). These studies demonstrated that inhibition of neutral sphingomyelinases interfered with lymph node homing and adhesion of T cells to activated endothelial cells (99, 100). In accordance, pharmacological or genetic ablation of neutral sphingomyelinase 2 interfered with T cell polarization, suggesting that neutral sphingomyelinases are involved in T cell recruitment and migration (99, 100). Besides regulation of T cell polarization and migration, neutral sphingomyelinase-2 is also critically involved in fine-tuning of signaling generated via the TCR/CD3 (99, 100). Thus, deletion of neutral sphingomyelinase-2 resulted in hyper-responsivity of T cells upon stimulation via CD3/CD28 (99, 100). This effect seems to be mediated by an increased metabolic activity with an accumulation of ATP in mitochondria and higher basal glycolytic activity in lymphocytes upon downregulation of neutral sphingomyelinase-2 (101). Whether down-regulation of neutral sphingomyelinase-2 results in an altered immune response in vivo remains to be determined.
6 Perspective
The role of sphingolipids in thyroid eye disease, in particular sphingomyelin, ceramide, sphingosine and sphingosine 1-phosphate and of the enzymes involved in the metabolism of these sphingolipids such as acid sphingomyelinase, acid ceramidase and sphingosine kinases requires definition and only a few data are available. An overview is presented in Figure 3. However, already these few data clearly indicate that this pathway and in particular the acid sphingomyelinase and ceramide have a very important role in the immune-pathogenesis of autoimmune disorders such as multiple sclerosis, immune arthritis and possibly also thyroid eye disease. Further, sphingolipids, in particular ceramide and sphingosine 1-phosphate seem to be critically involved in regulating and mediating the local alterations of the orbit in thyroid eye disease. Therefore, it might be very interesting to test the development of thyroid eye disease in animal models and to investigate the impact of a genetic deficiency or a pharmacological inhibition of the acid sphingomyelinase on thyroid eye disease. These studies may also serve to develop novel treatments of thyroid eye disease based on inhibition of the acid sphingomyelinase and/or neutralization of ceramide and sphingosine 1-phosphate.

Figure 3 Possible function of sphingolipids in thyroid eye disease (TED).
Author contributions
The authors wrote the manuscript together and all authors contributed to writing and editing.
Funding
The manuscript was supported by DFG grant Gu 335/35-2 to EG.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Becker KA, Halmer R, Davies L, Henry BD, Ziobro-Henry R, Decker Y, et al. Blockade of experimental multiple sclerosis by inhibition of the acid sphingomyelinase/ceramide system. Neurosignals (2017) 25:88–97. doi: 10.1159/000484621
2. Walter S, Gulbins E, Halmer R, Jahromi NH, Becker KA, Schottek A, et al. Pharmacological inhibition of acid sphingomyelinase ameliorates experimental autoimmune encephalomyelitis. Neurosignals (2019) 27:20–31. doi: 10.33594/000000183
3. Beckmann N, Becker KA, Walter S, Becker JU, Kramer M, Hessler G, et al. Regulation of arthritis severity by the acid sphingomyelinase. Cell Physiol Biochem (2017) 43:1460–71. doi: 10.1159/000481968
4. Schiffmann S, Ferreiros N, Birod K, Eberle M, Schreiber Y, Pfeilschifter W, et al. Ceramide synthase 6 plays a critical role in the development of experimental autoimmune encephalomyelitis. J Immunol (2012) 188:5723–33. doi: 10.4049/jimmunol.1103109
5. Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol (1998) 14:111–36. doi: 10.1146/annurev.cellbio.14.1.111
6. Kolesnick RN, Goni FM, Alonso A. Compartmentalization of ceramide signaling: Physical foundations and biological effects. J Cell Physiol (2000) 184:285–300. doi: 10.1002/1097-4652(200009)184:3<285::AID-JCP2>3.0.CO;2-3
7. Simons K, Ikonen E. Functional rafts in cell membranes. Nature (1997) 387:569–72. doi: 10.1038/42408
8. Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, Polyakova S, et al. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature (2009) 457:1159–62. doi: 10.1038/nature07596
9. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat Rev Mol Cell Biol (2008) 9:139–50. doi: 10.1038/nrm2329
10. Okino N, He X, Gatt S, Sandhoff K, Ito M, Schuchman EH. The reverse activity of human acid ceramidase. J Biol Chem (2003) 278:29948–53. doi: 10.1074/jbc.M303310200
11. Ishibashi Y, Nakasone T, Kiyohara M, Horibata Y, Sakaguchi K, Hijikata A, et al. A novel endoglycoceramidase hydrolyzes oligogalactosylceramides to produce galactooligosaccharides and ceramides. J Biol Chem (2007) 282:11386–96. doi: 10.1074/jbc.M608445200
12. Grassmè H, Jekle A, Riehle A, Schwarz H, Berger J, Sandhoff K, et al. CD95 signaling via ceramide-rich membrane rafts. J Biol Chem (2001) 276:20589–96. doi: 10.1074/jbc.M101207200
13. Gulbins E, Kolesnick R. Raft ceramide in molecular medicine. Oncogene (2003) 22:7070–7. doi: 10.1038/sj.onc.1207146
14. Grassmè H, Jendrossek V, Riehle A, von Kurthy G, Berger J, Schwarz H, et al. Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat Med (2003) 9:322–30. doi: 10.1038/nm823
15. Grassmè H, Gulbins E, Brenner B, Ferlinz K, Sandhoff K, Harzer K, et al. Acidic sphingomyelinase mediates entry of N. gonorrhoeae into nonphagocytic cells. Cell (1997) 91:605–15. doi: 10.1016/S0092-8674(00)80448-1
16. Hauck CR, Grassmè H, Bock J, Jendrossek V, Ferlinz K, Meyer TF, et al. Acid sphingomyelinase is involved in CEACAM receptor-mediated phagocytosis of neisseria gonorrhoeae. FEBS Lett (2000) 478:260–6. doi: 10.1016/S0014-5793(00)01851-2
17. Cremesti A, Paris F, Grassmè H, Holler N, Tschopp J, Fuks Z, et al. Ceramide enables fas to cap and kill. J Biol Chem (2001) 276:23954–61. doi: 10.1074/jbc.M101866200
18. Grassmè H, Schwarz H, Gulbins E. Molecular mechanisms of ceramide-mediated CD95 clustering. Biochem Biophys Res Commun (2001) 284:1016–30. doi: 10.1006/bbrc.2001.5045
19. Grassmè H, Jendrossek V, Bock J, Riehle A, Gulbins E. Ceramide-rich membrane rafts mediate CD40 clustering. J Immunol (2002) 168:298–307. doi: 10.4049/jimmunol.168.1.298
20. Dumitru CA, Gulbins E. TRAIL activates acid sphingomyelinase via a redox mechanism and releases ceramide to trigger apoptosis. Oncogene (2006) 25:5612–25. doi: 10.1038/sj.onc.1209568
21. Göggel R, Winoto-Morbach S, Vielhaber G, Imai Y, Lindner K, Brade L, et al. PAF-mediated pulmonary edema: A new role for acid sphingomyelinase and ceramide. Nat Med (2004) 10:155–60. doi: 10.1038/nm977
22. Grassmè H, Riehle A, Wilker B, Gulbins. E. Rhinoviruses infect human epithelial cells via ceramide-enriched membrane platforms. J Biol Chem (2005) 280:26256–62. doi: 10.1074/jbc.M500835200
23. Rotolo JA, Zhang J, Donepudi M, Lee H, Fuks Z, Kolesnick R. Caspase-dependent and -independent activation of acid sphingomyelinase signaling. J Biol Chem (2005) 280:26425–34. doi: 10.1074/jbc.M414569200
24. Lang PA, Schenck M, Nicolay JP, Becker JU, Kempe DS, Lupescu A, et al. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat Med (2007) 13:164–70. doi: 10.1038/nm1539
25. Grassmé H, Henry B, Ziobro R, Becker KA, Riethmüller J, Gardner A, et al. β1-integrin accumulates in cystic fibrosis luminal airway epithelial membranes and decreases sphingosine promoting bacterial infections. Cell Host Microbes (2017) 21:707–18. doi: 10.1016/j.chom.2017.05.001
26. Carpinteiro A, Edwards MJ, Hoffmann M, Kochs G, Gripp B, Weigang S, et al. Pharmacological inhibition of acid sphingomyelinase prevents uptake of SARS-CoV-2 by epithelial cells. Cell Rep Med (2020) 1:100142. doi: 10.1016/j.xcrm.2020.100142
27. Jin S, Zhou F, Katirai F, Li PL. Lipid raft redox signaling: Molecular mechanisms in health and disease. Antioxid Redox Signal (2011) 15:1043–83. doi: 10.1089/ars.2010.3619
28. Perrotta C, Bizzozero L, Cazzato D, Morlacchi S, Assi E, Simbari F, et al. Syntaxin 4 is required for acid sphingomyelinase activity and apoptotic function. J Biol Chem (2010) 285:40240–51. doi: 10.1074/jbc.M110.139287
29. Ferranti CS, Cheng J, Thompson C, Zhang J, Rotolo JA, Buddaseth S, et al. Fusion of lysosomes to plasma membrane initiates radiation-induced apoptosis. J Cell Biol (2020) 219:e201903176. doi: 10.1083/jcb.201903176
30. Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R, et al. Cathepsin d targeted by acid sphingomyelinase-derived ceramide. EMBO J (1999) 18:5252–63. doi: 10.1093/emboj/18.19.5252
31. Huwiler A, Johansen B, Skarstad A, Pfeilschifter J. Ceramide binds to the CaLB domain of cytosolic phospholipase A2 and facilitates its membrane docking and arachidonic acid release. FASEB J (2001) 15:7–9. doi: 10.1096/fj.00-0370fje
32. Zhang Y, Yao B, Delikat S, Bayoumy S, Lin XH, Basu S, et al. Kinase suppressor of ras is ceramide-activated protein kinase. Cell (1997) 89:63–72. doi: 10.1016/S0092-8674(00)80183-X
33. Dobrowsky RT, Hannun YA. Ceramide-activated protein phosphatase: Partial purification and relationship to protein phosphatase 2A. Adv Lipid Res (1993) 25:91–104.
34. Muller G, Ayoub M, Storz P, Rennecke J, Fabbro D, Pfizenmaier K. PKC zeta is a molecular switch in signal transduction of TNF-alpha, bifunctionally regulated by ceramide and arachidonic acid. EMBO J (1995) 14:1961–9. doi: 10.1002/j.1460-2075.1995.tb07188.x
35. Huwiler A, Fabbro D, Pfeilschifter J. Selective ceramide binding to protein kinase c-alpha and -delta isoenzymes in renal mesangial cells. Biochemistry (1998) 37:14556–62. doi: 10.1021/bi981401i
36. Avota E, Gulbins E, Schneider-Schaulies S. Ceramide generation is essential for enhancement of viral uptake in dendretic cells. PloS Pathog (2009) 5:e1000623.
37. Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol (2012) 8:831–8. doi: 10.1038/nchembio.1059
38. Chalfant CE, Szulc Z, Roddy P, Bielawska A, Hannun YA. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J Lipid Res (2004) 45:496–506. doi: 10.1194/jlr.M300347-JLR200
39. Gassert E, Avota E, Harms H, Krohne G, Gulbins E, Schneider-Schaulies S. Induction of membrane ceramides: A novel strategy to interfere with T lymphocyte cytoskeletal reorganisation in viral immunosuppression. PloS Pathog (2009) 5(10):e1000623. doi: 10.1371/journal.ppat.1000623
40. Szabo I, Gulbins E, Apfel H, Zhang X, Barth P, Busch AE, et al. Tyrosine phosphorylation-dependent suppression of a voltage-gated k+ channel in T lymphocytes upon fas stimulation. J Biol Chem (1996) 271:20465–9. doi: 10.1074/jbc.271.34.20465
41. Gulbins E, Szabo I, Baltzer K, Lang. F. Ceramide-induced inhibition of T lymphocyte voltage-gated potassium channel is mediated by tyrosine kinases. Proc Natl Acad Sci USA (1997) 94:7661–6. doi: 1073/pnas.94.14.7661
42. Lepple-Wienhues A, Belka C, Laun T, Walter B, Wieland U, Welz M, et al. Stimulation of CD95 (Fas) blocks T lymphocyte calcium channels through sphingomyelinase and sphingolipids. Proc Natl Acad Sci USA (1999) 96:13795–800. doi: 10.1073/pnas.96.24.13795
43. Bock J, Szabo I, Gamper N, Adams C, Gulbins E. Ceramide inhibits the potassium channel Kv1.3 by the formation of membrane platforms. Biochem Biophys Res Commun (2003) 305:890–7. doi: 10.1016/S0006-291X(03)00763-0
44. Rapoport B, Chazenbalk GD, Jaume JC, McLachlan SM. The thyrotropin (TSH) receptor: Interaction with TSH and autoantibodies. Endocrine Rev (1998) 19:673–716. doi: 10.1210/edrv.19.6.0352
45. Davies TF, Andersen S, Latif R, Nagayama Y, Barbesino G, Brito M, et al. Graves' disease. Nat Rev Dis Primers (2020) 6:52. doi: 10.1038/s41572-020-0184-y
46. Wiersinga WM, Bartalena L. Epidemiology and prevention of graves' ophthalmopathy. Thyroid (2002) 12:855–60. doi: 10.1089/105072502761016476
47. Laurberg P, Wallin G, Tallstedt L, Abraham-Nordling M, Lundell G, Tørring O. TSH-receptor autoimmunity in graves' disease after therapy with anti-thyroid drugs, surgery, or radioiodine: A 5-year prospective randomized study. Eur J Endocrinol (2008) 158:69–75. doi: 10.1530/EJE-07-0450
48. Eckstein AK, Johnson KTM, Thanos M, Esser J, Ludgate M. Current insights into the pathogenesis of graves’ orbitopathy. Hormone Metab Res (2009) 41(6):456–64. doi: 10.1055/s-0029-1220935
49. Dumont JE, Lamy F, Roger P, Maenhaut C. Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiol Rev (1992) 72:667–97. doi: 10.1152/physrev.1992.72.3.667
50. Gerding MN, van der Meer JW, Broenink M, Bakker O, Wiersinga WM, Prummel MF. Association of thyrotrophin receptor antibodies with the clinical features of graves' ophthalmopathy. Clin Endocrinol (2000) 52:267–71. doi: 10.1046/j.1365-2265.2000.00959.x
51. Goh SY, Ho SC, Seah LL, Fong KS, Khoo DH. Thyroid autoantibody profiles in ophthalmic dominant and thyroid dominant graves’ disease differ and suggest ophthalmopathy is a multiantigenic disease. Clin Endocrinol (2004) 60:600–7. doi: 10.1111/j.1365-2265.2004.02033.x
52. Khoo DH, Eng PH, Ho SC, Fok AC. Differences in the levels of TSH-binding inhibitor immunoglobulins in goitrous and agoitrous autoimmune thyroiditis after twelve months of l-thyroxine therapy. Clin Endocrinol (Oxf) (1999) 51:73–9. doi: 10.1046/j.1365-2265.1999.00740.x
53. Lytton SD, Ponto KA, Kanitz M, Matheis N, Kohn LD, Kahaly GJ. A novel thyroid stimulating immunoglobulin bioassay is a functional indicator of activity and severity of graves' orbitopathy. J Clin Endocrinol Metab (2010) 95:2123–31. doi: 10.1210/jc.2009-2470
54. Eckstein AK, Plicht M, Lax H, Hirche H, Quadbeck B, Mann K, et al. Clinical results of anti-inflammatory therapy in graves' ophthalmopathy and association with thyroidal autoantibodies. Clin Endocrinol (2004) 61:612–8. doi: 10.1111/j.1365-2265.2004.02143.x
55. Eckstein AK, Plicht M, Lax H, Neuhäuser M, Mann K, Lederbogen S, et al. Thyrotropin receptor autoantibodies are independent risk factors for graves' ophthalmopathy and help to predict severity and outcome of the disease. J Clin Endocrinol Metab (2006) 91:3464–70. doi: 10.1210/jc.2005-2813
56. Stöhr M, Oeverhaus M, Lytton SD, Horstmann M, Zwanziger D, Möller L, et al. Predicting the relapse of hyperthyroidism in treated graves' disease with orbitopathy by serial measurements of TSH-receptor autoantibodies. Horm Metab Res (2021) 53:235–44. doi: 10.1055/a-1373-5523
57. Endo T, Kobayashi T. Expression of functional TSH receptor in white adipose tissues of Hyt/Hyt mice induces lipolysis in vivo. am. J Physiology: Endocrinol Metab (2012) 302:E1569–75. doi: 10.1152/ajpendo.00572.2011
58. Krieger CC, Neumann S, Place RF, Marcus-Samuels B, Gershengorn MC. Bidirectional TSH and IGF-1 receptor cross talk mediates stimulation of hyaluronan secretion by graves' disease immunoglobins. J Clin Endocrinol Metab (2015) 100:1071–7. doi: 10.1210/jc.2014-3566
59. Kumar S, Coenen MJ, Scherer PE, Bahn RS. Evidence for enhanced adipogenesis in the orbits of patients with graves' ophthalmopathy. J Clin Endocrinol Metab (2004) 89:930–5. doi: 10.1210/jc.2003-031427
60. Kumar S, Nadeem S, Stan MN, Coenen M, Bahn RS. A stimulatory TSH receptor antibody enhances adipogenesis via phosphoinositide 3-kinase activation in orbital preadipocytes from patients with graves’ ophthalmopathy. J Mol Endocrinol (2011) 46:155–63. doi: 10.1530/JME-11-0006
61. Görtz GE, Horstmann M, Aniol B, Reyes BD, Fandrey J, Eckstein A, et al. Hypoxia-dependent HIF-1 activation impacts on tissue remodeling in graves' ophthalmopathy-implications for smoking. J Clin Endocrinol Metab (2016) 101:4834–42. doi: 10.1210/jc.2016-1279
62. Görtz GE, Philipp S, Bruderek K, Jesenek C, Horstmann M, Henning Y, et al. Macrophage-orbital fibroblast interaction and hypoxia promote inflammation and adipogenesis in graves' orbitopathy. Endocrinology (2022) 164:bqac203. doi: 10.1210/endocr/bqac203
63. Wakelkamp IM, Bakker O, Baldeschi L, Wiersinga WM, Prummel MF. TSH-r expression and cytokine profile in orbital tissue of active vs. inactive graves' ophthalmopathy patients. Clin Endocrinol- (Oxf). (2003) 58:280–7. doi: 10.1046/j.1365-2265.2003.01708.x
64. Tanda ML, Piantanida E, Lai A, Lombardi V, Dalle Mule I, Liparulo L, et al. Thyroid autoimmunity and environment. Horm Metab Res (2009) 41:436–42. doi: 10.1055/s-0029-1215568
65. Łacheta D, Miśkiewicz P, Głuszko A, Nowicka G, Struga M, Kantor I, et al. Immunological aspects of graves' ophthalmopathy. Biomed Res Int (2019) 2019:7453260. doi: 10.1155/2019/7453260
66. Mikoś H, Mikoś M, Obara-Moszyńska M, Niedziela M. The role of the immune system and cytokines involved in the pathogenesis of autoimmune thyroid disease (AITD). Endokrynol Pol (2014) 65:150–5. doi: 10.5603/EP.2014.0021
67. Wick G, Grundtman C, Mayerl C, Wimpissinger TF, Feichtinger J, Zelger B, et al. The immunology of fibrosis. Annu Rev Immunol (2013) 31:107–35. doi: 10.1146/annurev-immunol-032712-095937
68. Weaver CT, Hatton RD. Interplay between the TH17 and TReg cell lineages: A (co-)evolutionary perspective. Nat Rev Immunol (2009) 9:883–9. doi: 10.1038/nri2660
69. Fang S, Zhang S, Huang Y, Wu Y, Lu Y, Zhong S, et al. Evidence for associations between Th1/Th17 “Hybrid” phenotype and altered lipometabolism in very severe graves orbitopathy. J Clin Endocrinol Metab (2020) 105:dgaa124. doi: 10.1210/clinem/dgaa124
70. Kim SE, Lee JH, Chae MK, Lee EJ, Yoon JS. The role of sphingosine-1-phosphate in adipogenesis of graves' orbitopathy. Invest Ophthalmol Vis Sci (2016) 57:301–11. doi: 10.1167/iovs.15-17863
71. Ko J, Kim JY, Chae MK, Lee EJ, Yoon JS. Sphingosine-1-phosphate mediates fibrosis in orbital fibroblasts in graves' orbitopathy. J Mol Endocrinol (2021) 66(4):313–23. doi: 10.1530/JME-21-0057
72. Seo Y, Chae MK, Han SA, Lee EJ, Lee JH, Yoon JS. Sphingosine-1-phosphate is involved in inflammatory reactions in patients with graves’ orbitopathy. Inflammation Res (2017) 66(6):535–45. doi: 10.1007/s00011-017-1037-3
73. Eckstein A, Philipp S, Goertz G, Banga JP, Berchner-Pfannschmidt U. Lessons from mouse models of graves' disease. Endocrine (2020) 68:265–70. doi: 10.1007/s12020-020-02311-7
74. Bartalena L, Kahaly GJ, Baldeschi L, Dayan CM, Eckstein A, Marcocci C, et al. The 2021 European group on graves' orbitopathy (EUGOGO) clinical practice guidelines for the medical management of graves' orbitopathy. Eur J Endocrinol (2021) 185:G43–67. doi: 10.1530/EJE-21-0479
75. Smith TJ, Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, Tang RA, et al. Teprotumumab for thyroid-associated ophthalmopathy. N Engl J Med (2017) 376:1748–61. doi: 10.1056/NEJMoa1614949
76. Winn BJ, Kersten RC. Teprotumumab interpreting the clinical trials in the context of thyroid eye disease pathogenesis and current therapies. Ophthalmology (2021) 128:1627–51. doi: 10.1016/j.ophtha.2021.04.024
77. Görtz GE, Moshkelgosha S, Jesenek C, Edelmann B, Horstmann M, Banga JP, et al. Pathogenic phenotype of adipogenesis and hyaluronan in orbital fibroblasts from female graves' orbitopathy mouse model. Endocrinology (2016) 157:3771–8. doi: 10.1210/en.2016-1304
78. Plöhn S, Edelmann B, Japtok L, He X, Hose M, Hansen W, et al. CD40 enhances sphingolipids in orbital fibroblasts: Potential role of sphingosine-1-phosphate in inflammatory T-cell migration in graves' orbitopathy. Invest Ophthalmol Vis Sci (2018) 59:5391–7. doi: 10.1167/iovs.18-25466
79. Bock J, Gulbins E. The transmembranous domain of CD40 determines CD40 partitioning into lipid rafts. FEBS-Lett (2002) 534:169–74. doi: 10.1016/S0014-5793(02)03784-5
80. Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol (2012) 22:50–60. doi: 10.1016/j.tcb.2011.09.003
81. Kim SE, Lee JH, Chae MK, Lee EJ, Yoon JS. The role of sphingosine-1-phosphate in adipogenesis of graves’ orbitopathy. Invest Ophthalmol Vis Sci (2016) 57:301–11. doi: 10.1167/iovs.15-17863
82. Ko J, Chae MK, Lee JH, Lee EJ, Yoon JS. Sphingosine-1-phosphate mediates fibrosis in orbital fibroblasts in graves’ orbitopathy. Invest Ophthalmol Vis Sci (2017) 58:2544–53. doi: 10.1167/iovs.16-20684
83. Qiu H, Edmunds T, Baker-Malcolm J, Karey KP, Estes S, Schwarz C, et al. Activation of human acid sphingomyelinase through modification or deletion of c-terminal cysteine. J Biol Chem (2003) 278:32744–52. doi: 10.1074/jbc.M303022200
84. Zhang AY, Yi F, Jin S, Xia M, Chen QZ, Gulbins E, et al. Acid sphingomyelinase and its redox amplification in formation of lipid raft redox signaling platforms in endothelial cells. Antioxid Redox Signal (2007) 9:817–28. doi: 10.1089/ars.2007.1509
85. Prummel MF, Wiersinga WM. Smoking and risk of graves' disease. JAMA (1993) 269:479–82. doi: 10.1001/jama.1993.03500040045034
86. Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. New Engl J Med (2000) 343:938–52. doi: 10.1056/NEJM200009283431307
87. Warnke C, Kieseier BC, Hartung HP. Biotherapeutics for the treatment of multiple sclerosis: Hopes and hazards. J Neural Transm (2013) 120 Suppl 1:S55–60. doi: 10.1007/s00702-013-1055-4
88. Traugott U, Reinherz EL, Raine CS. Multiple sclerosis: Distribution of T cell subsets within active chronic lesions. Science (1983) 219:308–10. doi: 10.1126/science.6217550
89. Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol (1986) 19:578–87. doi: 10.1002/ana.410190610
90. Prineas JW, Wright RG. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis. Lab Invest (1978) 38:409–21.
91. Engelhardt B, Ransohoff RM. Capture, crawl, cross: The T cell code to breach the blood-brain barriers. Trends Immunol (2012) 33:579–89. doi: 10.1016/j.it.2012.07.004
92. Kawakami N, Bartholomaus I, Pesic M, Mues M. An autoimmunity odyssey: How autoreactive T cells infiltrate into the CNS. Immunol Rev (2012) 248:140–55. doi: 10.1111/j.1600-065X.2012.01133.x
93. Hurwitz R, Ferlinz K, Sandhoff K. The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol Chem Hoppe Seyler (1994) 375:447–50. doi: 10.1515/bchm3.1994.375.7.447
94. Kornhuber J, Tripal P, Reichel M, Terfloth L, Bleich S, Wiltfang J, et al. Identification of new functional inhibitors of acid sphingomyelinase using a structure-property-activity relation model. J Med Chem (2008) 51:219–37. doi: 10.1021/jm070524a
95. Gulbins E, Palmada M, Reichel M, Lüth A, Böhmer C, Amato D, et al. Acid sphingomyelinase/ceramide system mediates effects of antidepressant drugs. Nat Med (2013) 19:934–8. doi: 10.1038/nm.3214
96. Zhou Y, Salker MS, Walker B, Münzer P, Borst O, Gawaz M, et al. Acid sphingomyelinase (ASM) is a negative regulator of regulatory T cell (Treg) development. Cell Physiol Biochem (2016) 39:985–95. doi: 10.1159/000447806
97. Hollmann C, Werner S, Avota E, Reuter D, Japtok L, Kleuser B, et al. Inhibition of acid sphingomyelinase allows for selective targeting of CD4+ conventional versus Foxp3+ regulatory T cells. J Immunol (2016) 197:3130–41. doi: 10.4049/jimmunol.1600691
98. Hose M, Günther A, Naser E, Schumacher F, Schönberger T, Falkenstein J, et al. Cell-intrinsic ceramides determine T cell function during melanoma progression. Elife (2022) 11:e83073. doi: 10.7554/eLife.83073
99. Collenburg L, Schneider-Schaulies S, Avota E. The neutral sphingomyelinase 2 in T cell receptor signaling and polarity. Biol Chem (2018) 399:1147–55. doi: 10.1515/hsz-2017-0280
100. Collenburg L, Beyersdorf N, Wiese T, Arenz C, Saied EM, Becker-Flegler KA, et al. The activity of the neutral sphingomyelinase is important in T cell recruitment and directional migration. Front Immunol (2017) 8:1007. doi: 10.3389/fimmu.2017.01007
Keywords: thyroid eye disease (TED), Graves’ disease, sphingolipids, ceramide, acid sphingomyelinase 2
Citation: Gulbins A, Görtz G-E, Gulbins E and Eckstein A (2023) Sphingolipids in thyroid eye disease. Front. Endocrinol. 14:1170884. doi: 10.3389/fendo.2023.1170884
Received: 21 February 2023; Accepted: 22 March 2023;
Published: 04 April 2023.
Edited by:
Cao Li, Beijing Tiantan Hospital, Capital Medical University, ChinaReviewed by:
Yang Zhang, University of Houston, United StatesPin-Lan Li, Virginia Commonwealth University, United States
Copyright © 2023 Gulbins, Görtz, Gulbins and Eckstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anja Eckstein, YW5qYS5lY2tzdGVpbkB1ay1lc3Nlbi5kZQ==; Erich Gulbins, ZXJpY2guZ3VsYmluc0B1ay1lc3Nlbi5kZQ==
†These authors share first authorship