 Karly E. Masse
Karly E. Masse Van B. Lu
Van B. Lu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 25 July 2023
Sec. Gut Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1169624
This article is part of the Research Topic The Cross-Talk Between Gut Microbiota and Endogenous Metabolites in Endocrine Diseases, volume II View all 7 articles
The gastrointestinal tract hosts the largest ecosystem of microorganisms in the body. The metabolism of ingested nutrients by gut bacteria produces novel chemical mediators that can influence chemosensory cells lining the gastrointestinal tract. Specifically, hormone-releasing enteroendocrine cells which express a host of receptors activated by these bacterial metabolites. This review will focus on the activation mechanisms of glucagon-like peptide-1 releasing enteroendocrine cells by the three main bacterial metabolites produced in the gut: short-chain fatty acids, secondary bile acids and indoles. Given the importance of enteroendocrine cells in regulating glucose homeostasis and food intake, we will also discuss therapies based on these bacterial metabolites used in the treatment of metabolic diseases such as diabetes and obesity. Elucidating the mechanisms gut bacteria can influence cellular function in the host will advance our understanding of this fundamental symbiotic relationship and unlock the potential of harnessing these pathways to improve human health.
The gut microbiota encompasses all the microorganisms (bacteria, fungi, archaea, viruses) that have colonized the gastrointestinal tract of host animals. The gut microbiota is composed of trillions of microbes, with current estimates suggesting the collective genome of gut bacteria outnumbers the host human genome over 1000:1 (1). Bacteria residing in the gut predominantly belong to the phyla Bacillota (also known as Firmicutes) and Bacteriodota (aka Bacteroidetes); however, bacteria belonging to Actinomycetota (aka Actinobacteria), Pseudomonadota (aka Proteobacteria), or Verrucomicrobiota (aka Verrucomicrobia) are also represented (2). This complex and diverse environment of microorganisms contributes to a symbiotic relationship with the host, assisting in host physiological functions such as nutrient and energy metabolism, maintenance of intestinal barrier integrity, and immune protection (3–5). Changes in gut microbiota populations have been associated with a multitude of human disease states, including the metabolic diseases Type 2 diabetes mellitus (T2DM) and obesity (Figure 1). Reduced bacterial diversity and richness have been reported in human and animal models of obesity and diabetes (6–8). Dysbiosis of host-gut microbiota equilibrium may precede metabolic disease as similar shifts in intestinal gut bacteria composition can disrupt nutrient and energy metabolism (9). Due to the global health burden of metabolic diseases, there is great interest in developing novel therapeutic approaches including targeting mechanisms involving the gut microbiota.
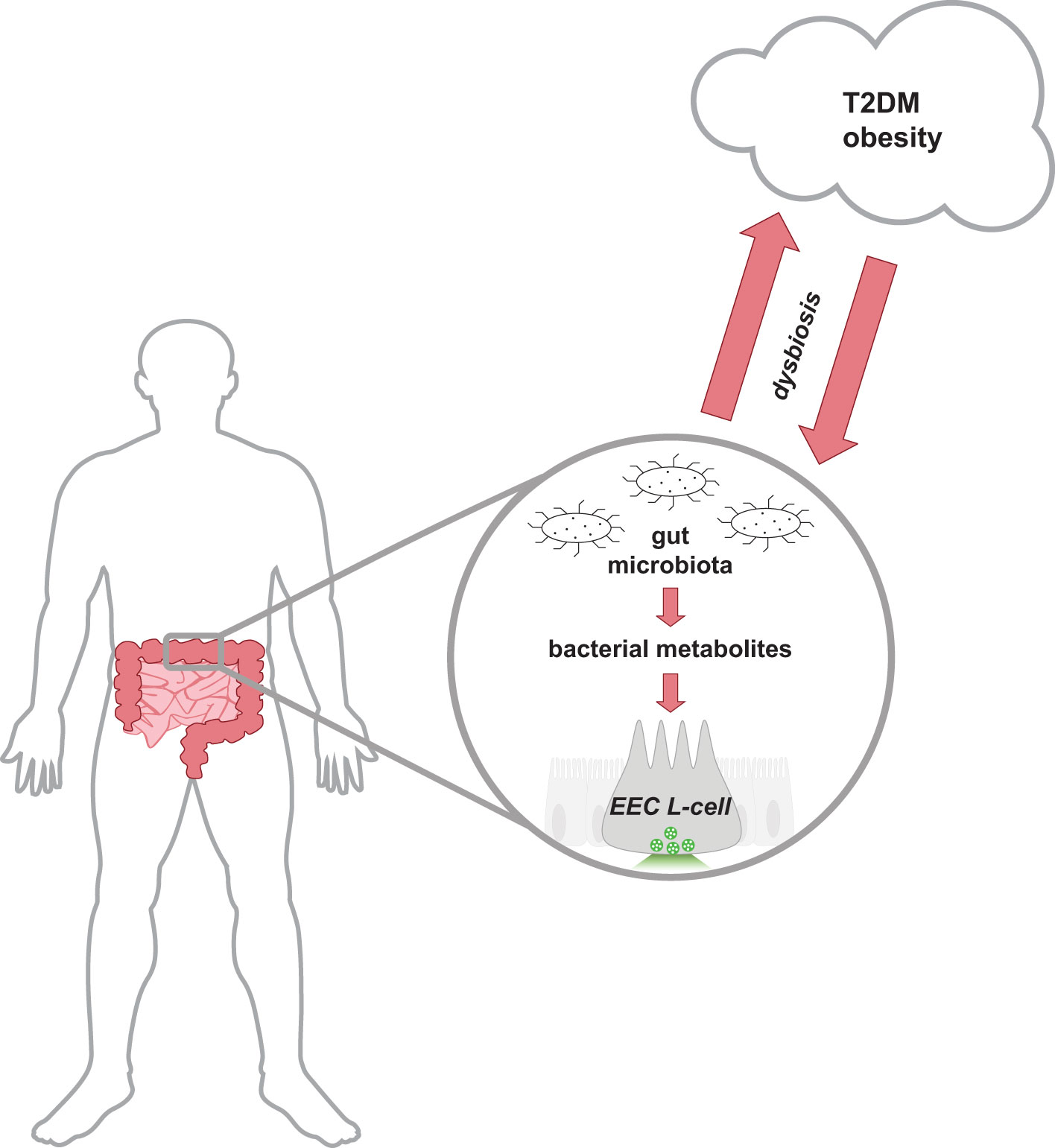
Figure 1 Schematic representation of the bidirectional relationship between the host-gut microbiota equilibrium and metabolic health. Cells lining the distal gastrointestinal tract are in direct contact with bacterial metabolites produced by the gut microbiota, and thus can contribute to host health. In a healthy state, the gut microbiota produces metabolites that activate receptors on distal EECs to mediate insulinotropic effects by the release of secretory vesicles containing GLP-1. Gut dysbiosis alters the intestinal composition and metabolites produced and is associated with the development of T2DM and obese-related diseases. Research has proposed that the dysregulation of metabolism in metabolic diseases releases molecules that can reduce the abundance of intestinal bacteria and alter the function of the ecosystem. EEC, enteroendocrine cell; GLP-1, glucagon-like peptide-1; T2DM, type 2 diabetes mellitus.
Diet is a key factor in metabolic health and can influence the progression of metabolic disease. It can also regulate gut microbiota health as resident gut bacteria metabolize host-digested macronutrients to produce an additional class of active biomolecules. For instance, complex carbohydrates undergo bacterial fermentation to produce short-chain fatty acids (SCFAs) (10–12) and the amino acid tryptophan is further metabolized by gut bacteria to produce indole and other indole-derivatives (13, 14). Cholesterol-derived bile acids released from hepatocytes are also modified by gut bacteria to improve solubility and facilitate recycling of bile acids in the distal colon (15). These bacterial metabolites themselves may mediate the effects of gut microbiota on host health as changes in the levels of SCFAs, indoles, and secondary bile acids are associated with metabolic disease (16–18) and restoration of levels can attenuate disease progression and severity (19–24). Although many studies have carefully identified and quantified the levels of bacterial metabolites produced in humans (10, 25–27), the signaling pathways mediating the cross-talk between microbiota-derived metabolites and host physiology has yet to be fully elucidated.
A specialized population of intestinal epithelial cells called enteroendocrine cells (EECs) are strategically positioned to mediate the effects of bacterial metabolites on host health. EECs have an open-type morphology that spans the intestinal epithelial cell layer. The apical cell side faces the luminal interface with microvilli-like structures that are exposed to nutrients and bacterial metabolites. EECs also express several different types of nutrient-sensitive receptors (28) that facilitate their role as intestinal chemosensors. The basolateral cell side of EECs connects the release of hormones to the intestinal circulatory system. The gut hormone released can exert effects on host physiology thereby providing a mechanistic link between bacterial metabolism of nutrients and host health. The gut hormone glucagon-like peptide-1 (GLP-1), secreted from a subset of EECs called L-cells, is of interest in the context of metabolic disease because of GLP-1’s anorexigenic and hypoglycemic properties (29). GLP-1 mimetics have been used for the treatment of obesity and T2DM (30–35). Furthermore, the mechanism of improved metabolic status following bariatric surgery has been attributed to enhanced GLP-1 release (36–38). EEC L-cells also secrete peptide-YY (PYY), a gut hormone involved in appetite regulation (39, 40). Interestingly, the distal small intestine and colon harbor the greatest density of PYY and GLP-1 releasing L-cells (41), paralleling the distribution of gut bacteria (42). Thus, studying bacterial metabolite sensing in EEC L-cells can advance our understanding of the mechanisms by which gut microbiota regulate host metabolic health. It can also provide a novel therapeutic avenue for the treatment and management of metabolic disease.
The focus of this review will be gut microbiota-derived metabolites that are most abundant in the human colon, specifically SCFAs, secondary bile acids and indoles, and how each bacterial metabolite modulate EEC L-cell function (43–47). We will detail the signaling pathways that are recruited in EEC L-cells following exposure to each bacterial metabolite. In addition, we will describe how the levels of these bacterial metabolites are altered during metabolic disease and discuss therapeutic approaches that target these bacterial metabolite signaling pathways.
SCFAs are monocarboxylic acids of 1-5 carbon chain lengths and are the most abundant bacterial metabolite produced in the gut (11, 12). The majority of bacterial SCFAs synthesized (>95%) include acetate (C2), propionate (C3), and butyrate (C4) in a molar ratio of approximately 3:1:1, respectively (10, 48). Production of SCFAs is most abundant in the caecum and ascending limb of the colon in humans (>100 mM) (10). In humans, undigested fiber passes through the small intestine largely unabsorbed before entry into the colon and metabolism by both Gram-negative and Gram-positive bacteria. The production of the smaller chained SCFAs acetate and propionate are favored by Bacteroidota, whereas Bacillota primarily produce butyrate as a metabolic product (49). Bacterial fermentation of indigestible carbohydrates yields the majority of SCFAs produced, but a small fraction of SCFAs produced (1%) stem from bacterial metabolism of dietary amino acids (50). Notably, the liver can generate significant levels of acetate (~1 mM) during bouts of chronic alcohol consumption that can enter circulation and impact gastrointestinal function (51).
Following production, SCFAs are almost exclusively taken up by colonocytes via H+-dependent or sodium-dependent monocarboxylate transporters (MCTs and SMCTs, respectively; Figure 2) (52). Colonic absorption of SCFAs accounts for approximately 5-10% of the body’s total energy requirement, with butyrate acting as the predominant source of energy (53). The absorbed butyrate is largely utilized by colonocytes for energy, and the remaining absorbed SCFAs are transported through portal blood circulation back to the liver where SCFAs are primarily converted to glucose stores (4). Therefore, the levels of SCFAs that reach systemic circulation are much lower in concentration than the levels measured in the colon (10, 54).
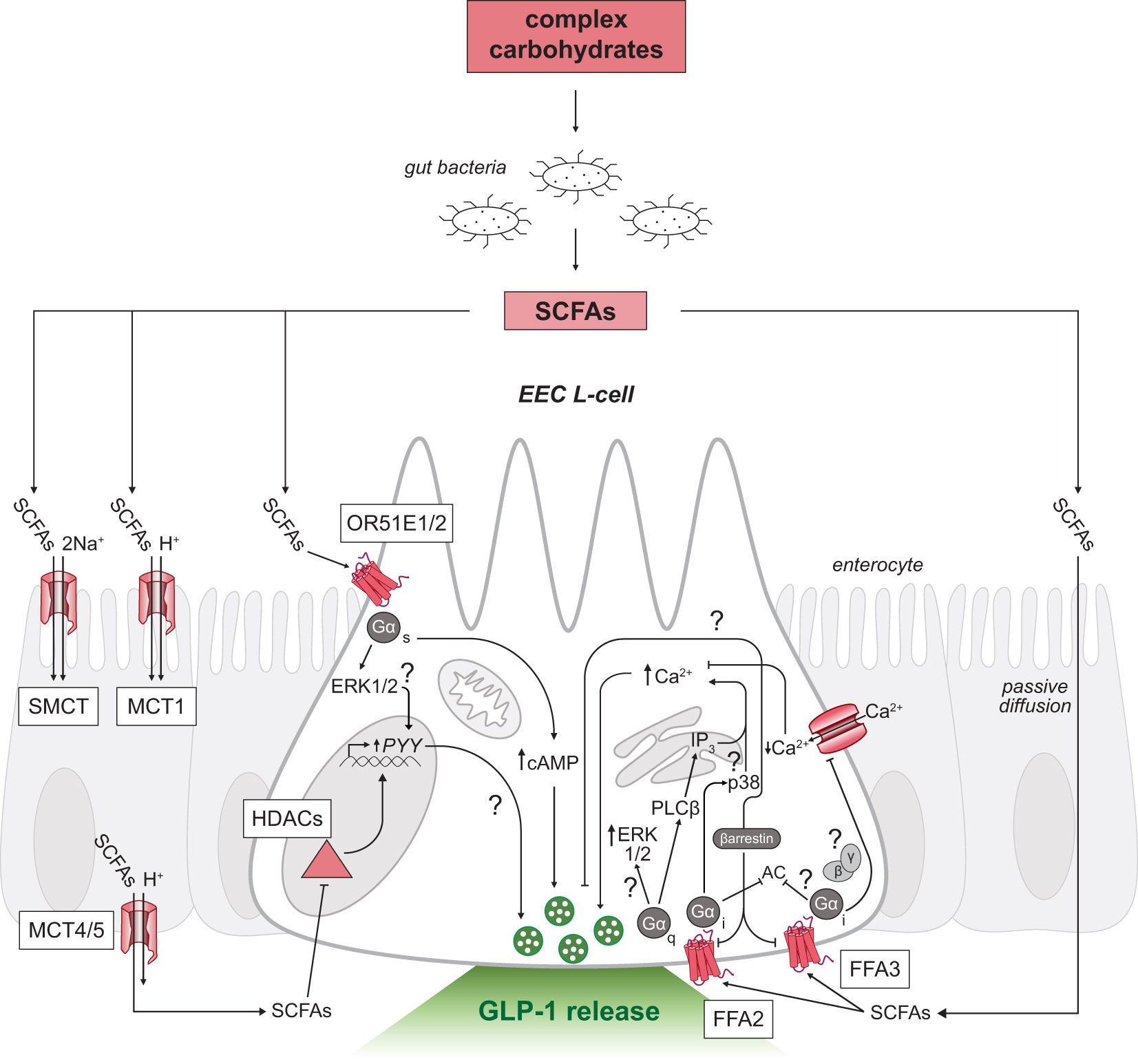
Figure 2 SCFA-triggered intracellular signaling mechanisms in GLP-1 releasing EEC L-cells. Schematic of an L-cell (white) surrounded by enterocytes (grey). Complex carbohydrates are substrates for resident gut bacteria to produce SCFAs in the distal gastrointestinal tract. SCFAs can signal through multiple receptors on both the apical (top) and basolateral membranes (bottom) of L-cells. Uptake of SCFAs by SMCT and MCT across the intestinal epithelium to the basolateral side shown in enterocytes. SCFAs inhibit HDACs or activate G-protein coupled receptors FFA2, FFA3 and OR51E1/2 in L-cells. Unresolved mechanisms are marked with a question mark. EEC, enteroendocrine cell; SCFAs, short-chain fatty acids; FFA2/3, free fatty acid receptor 2 or 3; OR51E1/2, olfactory receptor subfamily 5E1 or 2; GLP-1, glucagon-like peptide-1; Ca2+, calcium ions; SMCT, sodium-dependent monocarboxylate transporter; MCT, H+-dependent monocarboxylate transporter; Na+, sodium ions; HDACs, histone deacetylases; AC, adenylyl cyclase; cAMP, cyclic adenosine monophosphate; PLCβ, phospholipase C beta; ERK1/2, extracellular signal-regulated kinases; IP3, inositol triphosphate.
Bile acids are the primary metabolic end products of cholesterol catabolism (55, 56) and account for the majority of cholesterol turnover in humans. Hydroxylation and modification of cholesterol in the liver generates the primary bile acids, cholic acid (CA) and chenodeoxycholic acid (CDCA) in humans (57), and CA and muricholic acid (MCA) in rodents (58). Most primary bile acids are conjugated with glycine or taurine, to increase solubility properties, and are stored in the gallbladder (56). Conjugated bile acids comprise the majority of secreted bile; however, phospholipids, cholesterol, exogenous drugs, and environmental toxins contribute a small component (59). Following the consumption of fat, the gut hormone cholecystokinin is released which stimulates the contraction of the gallbladder to release bile acids into the proximal small intestine. Bile acids act as powerful detergent molecules, forming solubilizing micelles that promote the digestion and absorption of dietary lipids and fat-soluble vitamins (59). The total levels of bile acids in the enterohepatic circulation, or bile acid pool, remains consistent due to highly efficient (95%) reabsorption of bile acids in the small intestine (55). Conjugated primary bile acids are actively reabsorbed in the distal ileum via the apical sodium-dependent bile acid transporter (ASBT; also known as the ileal sodium-dependent bile acid transporter, IBAT; Figure 3), whereas unconjugated bile acids can passively diffuse through enterocytes. The reabsorbed bile acids are shuttled across enterocytes to the basolateral membrane and are recycled back to the liver through portal blood circulation. Conjugated bile acids are taken up by hepatocytes via the sodium taurocholate cotransporting polypeptide (NTCP) while unconjugated bile acids are taken up by the organic anion transporting polypeptide (OATP), which is also responsible for uptake of bilirubin (55, 59). The bile acid pool is tightly regulated through the coordination between synthesis, reabsorption, and excretion of bile acids by the liver.
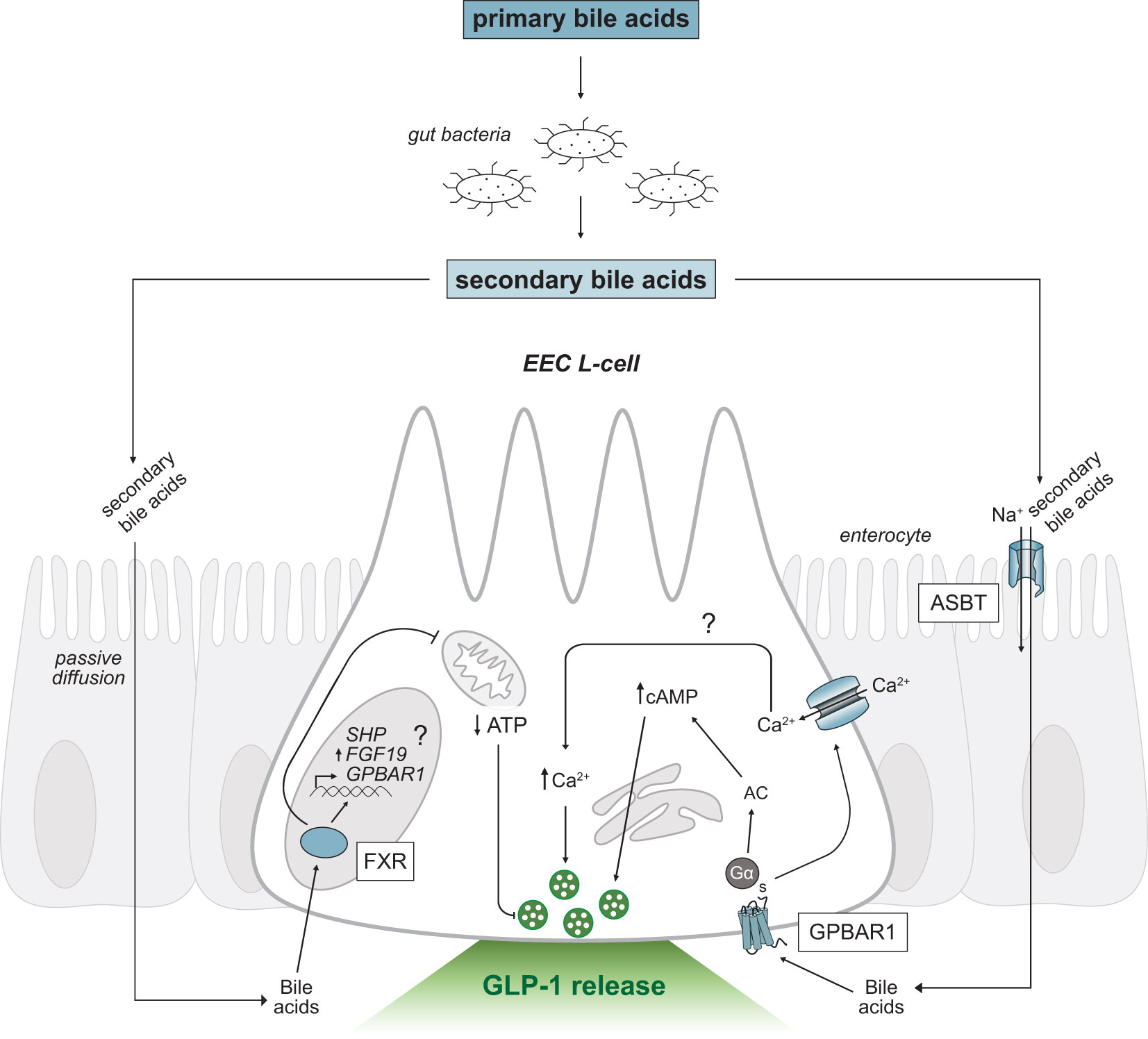
Figure 3 Secondary bile acid-triggered intracellular signaling mechanisms in GLP-1 releasing EEC L-cells. Schematic as in Figure 2. Primary bile acids are converted to secondary bile acids by intestinal gut bacteria. Secondary bile acids passively diffuse or are transported across the intestinal epithelium via ASBT and activate the G-protein coupled receptor, GPBAR1 and nuclear receptor, FXR. Activation of GPBAR1 by secondary bile acids results in GLP-1 secretion. FXR regulates SHP and FGF19 expression and may regulate GPBAR1 gene transcription (marked by a question mark). EEC, enteroendocrine cell; GPBAR1, G-protein coupled bile acid receptor; FXR, Farnesoid-X receptor; GLP-1, glucagon-like peptide-1; ASBT, apical sodium-dependent bile acid transporter; Ca2+, calcium ions; AC, adenylyl cyclase; cAMP, cyclic adenosine monophosphate; ATP, adenosine triphosphate; SHP, short heterodimer protein; FGF19, fibroblast growth factor 19; Na+, sodium ions.
The remaining bile acids that escape absorption in the small intestine (5%) act as substrates for anaerobic metabolism in the colon (15, 60) or are excreted with feces. Bile salt hydrolase (BSH), produced by intestinal bacteria, converts conjugated primary bile acids to secondary bile acids through a series of biotransformation reactions, thus increasing the diversity of bile acids. In humans, deoxycholic acid (DCA) and lithocholic acid (LCA) are the predominant secondary bile acids produced (15, 57), whereas in rodents the predominant secondary bile acids generated are DCA and ω-MCA (58). At the phyla level, bacterial populations encoding BSH, such as Bacillota, Bacteroidota and Actinomycetota, have been shown to play an important role in the production of secondary bile acids (61). Other human intestinal archaea species, Methanobrevibacter smithii and Methanosphera stadmanae, also encode for BSH and can contribute to the production of secondary bile acids (62). Other bile acid transformations catalyzed by bacterial enzymes include the actions of hydroxysteroid dehydrogenases (HSDs), which alters the hydrophobicity and toxicity of bile acids (15).
Tryptophan is an essential aromatic amino acid that must be consumed as the body lacks the enzymes necessary to synthesize tryptophan. Following protein digestion, most of the liberated tryptophan is absorbed in the small intestine and endogenously metabolized: up to 95% of ingested tryptophan is converted to kynurenic acid or nicotinamide adenine dinucleotide (NAD+) (63, 64) via the kynurenine pathway, and 1-2% of ingested tryptophan is converted to serotonin via tryptophan hydroxylase 1 activity (65, 66). The remaining ingested tryptophan that escapes absorption (4-6%) enters the colon and is metabolized by intestinal bacteria (13). More than 85 different Gram-positive and Gram-negative bacterial species express tryptophanase (67), the enzyme that catalyzes the hydrolytic β-elimination of tryptophan to indole, pyruvate, and ammonia (68). Indole production also depends on the tryptophan-specific transporter, TnaB, expressed in bacteria to facilitate tryptophan uptake. Other transporters such as AroP and Mtr permeases may also facilitate bacterial uptake of tryptophan (69). Indole is the most abundant bacterial metabolite of tryptophan degradation produced with the average physiological concentration between 0.25-1.1 mM in human feces (14). Bacterial metabolism of tryptophan can also give rise to other indole-moiety containing derivatives. Indole-3-acetic acid (IAA) is an intermediate formed during a series of decarboxylation reactions from indole-3-pyruvic acid (IPyA). IAA can be further catabolized to indole-3-aldehyde (IAld) and 3-methylindole (skatole). Alternatively, bacterial enzymes catalyze reduction and dehydration reactions to produce indole-3-propionic acid (IPA). Physiological levels of IPA range between 1-10 µM in human serum (70). Another bacterial transformation of tryptophan can occur through the actions of tryptophan decarboxylases to produce tryptamine (71). Bacteria also express decarboxylases to convert indole to tryptamine (72).
Following production, indole and other metabolic derivatives can passively diffuse through the plasma membrane to exert intracellular effects on intestinal epithelial cells (Figure 4). Metabolites may also enter enterohepatic circulation and undergo further oxidative metabolism by cytochrome P450 (CYP450) or detoxification by enzymes in the liver (73). For instance, indole undergoes sulfation in the liver to produce indoxyl sulfate, a uremic toxin which accumulates during renal insufficiency inducing fibrosis in damaged proximal tubule cells (74). Alternatively, indole may be reabsorbed passively or actively across bacterial membranes and activate a variety of bacterial processes (75–77).
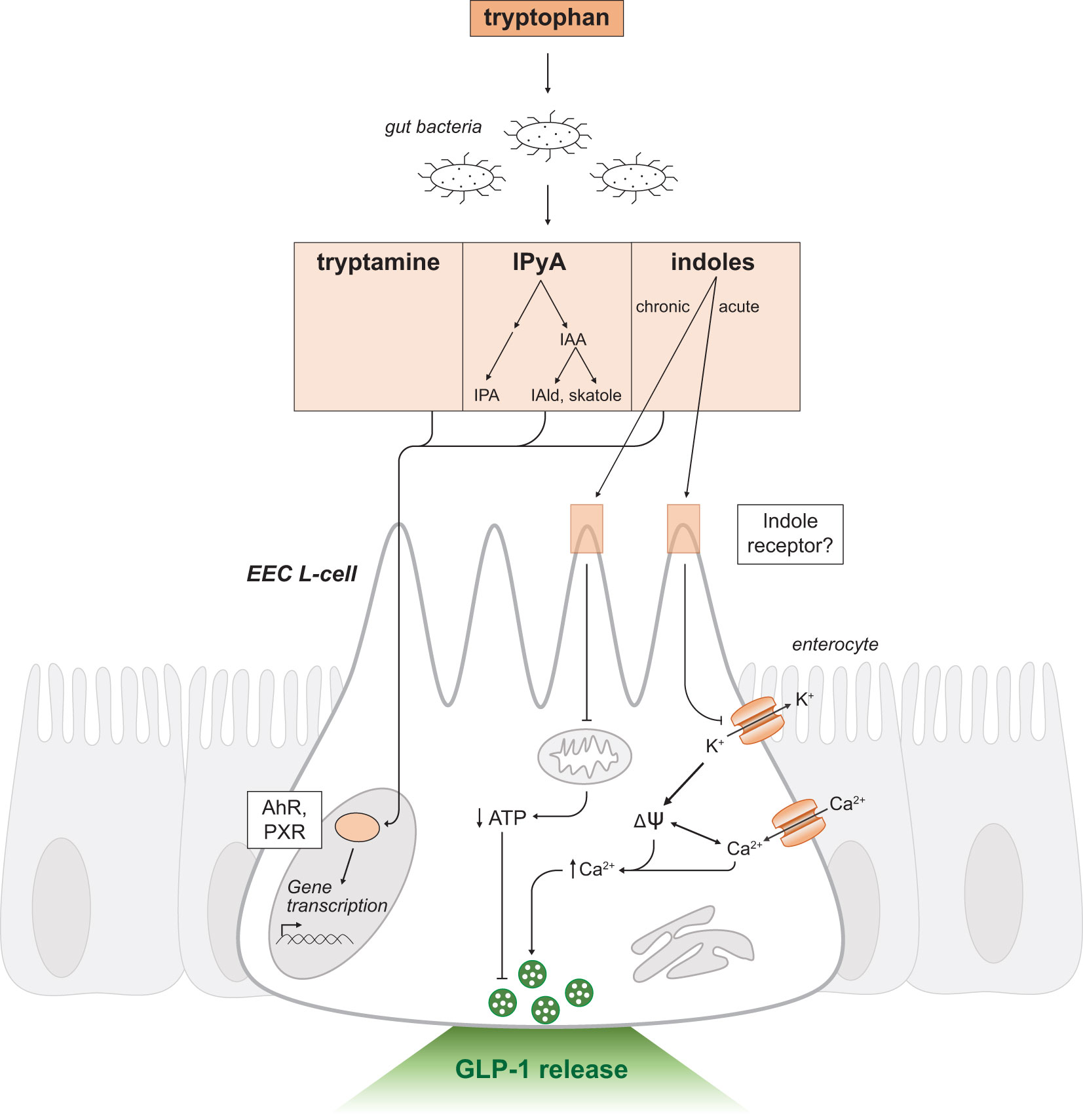
Figure 4 Indole-triggered intracellular signaling mechanisms in GLP-1 releasing EEC L-cells. Schematic as in Figure 2. Tryptophan is a substrate for resident gut bacteria to produce the metabolites tryptamine, IPyA and indoles. IPyA is a precursor to the metabolites IAA, IAld, skatole and IPA. Indole and other metabolites passively diffuse across the intestinal epithelium and activate the transcription factors AhR and PXR to regulate gene transcription. Indole may mediate effects through a yet to be determined receptor. There are dual and opposing effects on GLP-1 release by acute and chronic exposure to indoles. Acute indole stimulates release of secretory vesicles containing GLP-1 by a voltage-gated potassium channel blockade. Chronic indole exposure suppresses mitochondrial activity to produce ATP. IPyA, indole-3-pyruvic acid; IAA, indole-3-acetic acid; IAld, indole-3-aldehyde; IPA, indole-3-propionic acid; EEC, enteroendocrine cell; AhR, Aryl hydrocarbon receptor; PXR, Pregnane-X-receptor; ψ, membrane potential; K+, potassium ions; Ca2+, calcium ions; ATP, adenosine triphosphate.
Multiple intracellular signaling pathways have been implicated in gut hormone secretion with concurrent recruitment of several different pathways suggested to be necessary to stimulate release from EECs (78, 79). This section examines the various signaling pathways activated by bacterial metabolites highlighted in this review, and their known effects on GLP-1 release from EEC L-cells.
In addition to acting as a local energy source for colonocytes, SCFAs may signal through cell surface G-protein coupled receptors (GPCRs) that activate a series of intracellular effector molecules to produce various physiological responses (Figure 2). SCFAs activate several GPCRs including: the free fatty acid receptor 2 and 3 (FFA2 and FFA3) (80, 81), the olfactory receptor subfamily 51E1 and 51E2 (OR51E1 in human/Olfr558 in mouse; OR51E2 in human/Olfr78 in mouse) (82), and the hydroxycarboxylic acid receptor, HCAR2 (also known as the G-protein coupled receptor 109A (GPR109A) or the niacin receptor) (83). SCFAs can also affect gene expression through the inhibition of histone deacetylases (HDACs) (84). EECs express HDACs and SCFA-responsive GPCRs (85–89), except HCAR2 which is primarily localized in adipose and immune tissue (83).
The most potent endogenous ligands of FFA2 identified thus far are acetate and propionate (80, 81). To a weaker extent, butyrate can also stimulate the receptor. Ffar2 expression increases along the longitudinal axis of the gastrointestinal tract, with highest expression in the distal ileum and colon (90). Ffar2 expression was also found in leukocytes below the epithelial layer using an Ffar2-reporter mouse (88) and in the colonic epithelial layer of rats (90) and humans (89) by immunohistochemistry analysis.
Activation of FFA2 couples intracellularly through the G-protein families, Gαq and Gαi (91–93). Recruitment of the Gαq-coupled FFA2 signaling pathway in intestinal murine L-cells triggered SCFA-mediated GLP-1 secretion by promoting activity of phospholipase C-dependent production of inositol triphosphate (IP3), thereby increasing mobilization of calcium from intracellular stores (88, 92). GLP-1 release was found to be stimulatory in the presence of SCFAs with murine cell lines (94, 95) and murine primary colonic cultures, an effect attributed to FFA2- and FFA3-dependent mechanisms (92, 95). Compound 1, a selective FFA2 agonist, stimulated GLP-1 secretion by FFA2 (96) and this effect was lost in the presence of a Gαq inhibitor, FR900359 (91), thereby supporting Gαq-mediated FFA2 signaling mechanisms. Furthermore, pertussis toxin, a Gαi-protein uncoupler was shown to not be involved in SCFA-triggered GLP-1 release nor was the Gαi-biased FFA2 ligand, AZ1729 (91).
Recruitment of Gαi-coupled FFA2 remains unclear. Canonical Gαi-signaling mechanisms decrease cyclic adenosine monophosphate (cAMP) production by inhibiting adenylyl cyclase and thus, hormone secretion. However, in duodenal STC-1 and primary colonic cultures, propionate promoted GLP-1 secretion via Gαi-coupled FFA2 activation and downstream phosphorylation of a class of mitogen-activated protein kinases, p38 (97). This study suggests a spatial discrimination between the pleiotropic actions of the FFA2 receptor as Gαq-mediated FFA2 signaling occurred at the cell membrane and Gαi signaling was internalized, thus diversifying the downstream effector molecules activated in EECs (97). A possible convergence of downstream signaling pathways involving the phosphorylation of extracellular signal-regulated kinases (ERK) is possible as both Gαq- and Gαi-coupled signaling pathways can activate this effector molecule (94). Future research into how Gαi-coupled FFA2 is affected in other in vitro and in vivo L-cell models is warranted to confirm signaling through p38.
In addition, FFA2 is postulated to recruit β-arrestin, a protein involved in the downregulation of GPCRs (98). FFA2 has been shown to employ β-arrestin dependent signaling for transcriptional regulation of proinflammatory cytokine expression in vitro (99). In murine and human overexpression studies, agonist stimulation of FFA2 supported β-arrestin recruitment (94) suggesting a possible signaling pathway mediated in L-cells. FFA2 may have the capacity to employ different effector molecules depending on the spatial and temporal gradients of the receptor and needs of the host. Further studies are needed to investigate the functional selectivity of FFA2 under different metabolic conditions.
FFA3 is another free fatty acid receptor responsive to SCFAs that preferentially binds to the SCFAs propionate, butyrate and valerate (80, 81). Mouse Ffar3 expression paralleled the expression of glucagon (Gcg, the gene that encodes for GLP-1), with high transcript levels found in the distal small intestine and colon (92). EEC expression of Ffar3 was confirmed in a reporter mouse model (88). Additional Ffar3 expression was described in enteric neurons and vagal afferent neurons that innervate the gastrointestinal tract (100). However, other studies using in situ hybridization failed to detect Ffar3 expression in the nodose ganglion, but rather found expression in sympathetic ganglia innervating the intestines (101). Interestingly, in epithelial cells of the human colonic mucosa, Tazoe et al. (2009), demonstrated co-localization of FFA3 with PYY, the gut hormone co-localized with GLP-1. Co-localization was not observed between FFA3 and serotonin, a marker of another EEC population the enterochromaffin cells (102).
FFA3 exclusively recruits G-proteins of the Gαi family (93), but the cellular mechanism of FFA3 signaling has yet to be fully demonstrated. A study found that the selective FFA3 agonist, AR420626, promoted GLP-1 release from primary colonic cultures (88) and perfused intact colons (103). However, FFA3 activation in sympathetic neurons has been shown to inhibit voltage-gated calcium channels through a Gβγ-mediated mechanism (101), thus inhibiting neurotransmitter release. Similarly, β-arrestin may also be involved in FFA3 activation. In monocytes, FFA3 activation increased intracellular calcium signaling and recruited β-arrestin 2 (104), though, the involvement of β-arrestin in an EEC system has yet to be determined. Indeed, SCFA-triggered FFA3 signaling warrants further investigation.
It is possible that other SCFA-responsive receptors could be involved in the outcome of GLP-1 secretion in L-cells. Originally discovered in olfactory epithelium, the olfactory receptor subfamily 51E1/2 (OR51E1/2; Olfr558/Olfr78 in mouse) are other receptors responsive to SCFAs (82). Acetate and propionate, but not butyrate, are potent endogenous ligands for these receptors (87, 105). Expression of Olfr78 is localized in murine EECs of the colon, especially PYY-positive cells (87) and serotonin-producing enterochromaffin cells (106), though the function of the receptor remains unclear. Expression of the OR51E1 in a human L-cell line NCI-H716 was demonstrated, as well as stimulated GLP-1 secretion following selective receptor activation (107). The mechanism of enhanced GLP-1 secretion involved an increase in intracellular cAMP and phosphorylated ERK (107). Moreover, OR51E1 knockdown reduced GLP-1 secretion, supporting the receptor’s role in mediating the effects of SCFAs on EEC L-cells (107).
In addition to activating cell membrane receptors, SCFAs exert genomic effects by the inhibition of HDACs. Activation of HDACs modifies chromatin structure by removing an acetyl group from histone proteins which reduces DNA accessibility to transcriptional activity. HDAC activity has been implicated in gut development (108) and immune tissue regulation (85, 109). In the colon, HDACs are inhibited by both butyrate and propionate (85, 86), though butyrate is the most effective inhibitor of HDACs (110, 111). This is supported by previous studies suggesting that HDAC inhibition by butyrate induces expression of many genes in various tissues and cell lines (109, 112). Understanding HDAC-mediated changes in expression is physiologically relevant as diets high in fiber results in chronic elevation of SCFA levels, which can lead to lasting changes in gut function. For instance, colonic Gcg expression was increased in rats on a fiber-rich diet compared with animals on a chow-fed diet (113). SCFAs have also increased the number of L-cells in the intestinal epithelium and increased endogenous secretion of GLP-1 in both mouse and human organoids in vitro (114). As further support, there is a reduction of GLP-1 releasing L-cells in germ-free mice lacking intestinal microbiota (115, 116). However, in cell line models of human EECs, GCG expression minimally changed and PYY expression dramatically increased following prolonged exposure to butyrate (117). The long-lasting effects of SCFAs on EEC L-cells suggest they may be key regulators of metabolic health and a promising dietary intervention for the treatment and management of T2DM and obesity.
Bile acids have a functional role in lipid digestion and absorption, but also act as signaling molecules to cells lining the gastrointestinal tract. Bile acids exclusively activate two main receptors in L-cells, the cell surface G-protein coupled bile acid receptor 1 (GPBAR1, also called the membrane-type bile acid receptor, M-BAR, or the Takeda G-protein coupled receptor 5, TGR5) (118) and the nuclear transcription factor, Farnesoid-X receptor (FXR) (119–121)(Figure 3). Interestingly, primary bile acids preferentially activate FXR (CDCA>CA>LCA>DCA), whereas secondary bile acids are more potent endogenous ligands for GPBAR1 activation (LCA>DCA>CDCA>CA) (119). Bile acids also activate the nuclear receptors, pregnane-X receptor (PXR) (122), vitamin D receptor (VDR) (123), constitutive androstane receptor (CAR) (124), liver-X receptor (LXR) (125), and G-protein coupled sphingosine-1-phosphate receptor 2 (SIPR2) (126); however, these receptors are more selective for other endogenous and xenobiotic ligands such as steroid hormones and oxysterols.
Bile acids exert non-genomic effects through the activation of the membrane receptor, GPBAR1. Expression analysis data localizes GPBAR1 to brown adipose tissue, skeletal muscle, spleen, immune cells, gallbladder and the intestine (118, 127, 128). In the intestine, GPBAR1 is highly expressed in the ileum and colon of EECs (129, 130) and in the enteric ganglia and nerve fiber plexuses (131). GPBAR1 is localized on the basolateral face of L-cells, suggesting a mechanism is required for bile acids to be absorbed before activating the receptor (129). The transporter responsible for transporting conjugated bile acids across the epithelial layer in the small intestine is ASBT. Transport of bile acids across the epithelial layer is critical for gut hormone secretion from EECs as blocking ASBT in the terminal ileum reduced GLP-1 release (129, 132). An alternative mechanism to transport bile acids across the epithelium is required in the colon due to very low ASBT expression. Resident gut bacteria can improve bile acid permeability and potency by converting primary bile acids to secondary bile acids (132). Secondary bile acids, specifically LCA and the taurine conjugate TLCA, are the most potent stimulants of GPBAR1 activation (127, 133, 134). Multiple studies have identified bile acids as a robust trigger of GLP-1 release (28, 47, 129, 130) and bile acid-triggered GLP-1 release was diminished in a GPBAR1 knockout model (129, 133–135). GPBAR1 stimulates Gαs-protein coupling and increases intracellular cAMP levels through activation of adenylyl cyclase (47, 78, 129, 130). Activation of GPBAR1 in L-cells also increases membrane electrical activity via increased calcium current through L-type voltage-gated calcium channels (28). Bile acids can indirectly alter GLP-1 release by modulating L-cell differentiation. GPBAR1 agonists enhanced the number of GLP-1 producing L-cells in the intestinal epithelium (135).
The functional role of FXR activation is well documented (120, 136, 137). FXR regulates a multitude of genes involved in bile acid, lipid, and glucose metabolism (15). Expression of FXR is most abundant in the liver and intestine (119–121), with the highest expression levels found in the terminal ileum of EEC L-cells (45). FXR expression has also been identified in immune cells, adipose tissue, and skeletal muscle (138). FXR is a primary bile acid sensor, preferentially binding to CDCA in humans and CA to a weaker extent (119). In mice, CA is the primary ligand for FXR as mice lack CDCA (121). In the terminal ileum, activation of FXR induces expression of target genes including the small heterodimer partner (SHP) and fibroblast growth factor 19, FGF19 (Fgf15 in mice). FGF19 is released from enterocytes, transported to the liver via enterohepatic circulation, and binds to the tyrosine kinase receptor fibroblast growth factor receptor 4 (FGFR4) expressed in hepatocytes (139). Together, SHP and FGF19 suppress CYP7A1, a key gene involved in de novo biosynthesis of bile acids (140). The microbial ecosystem is speculated to play an important role in regulating expression of ileal FXR target genes. Under conditions of reduced gut microbiota, either germ-free or antibiotic-treated mice, elevated levels of the taurine conjugated β-muricholic acid (TβMCA) bile acid were detected (141). TβMCA acts as an FXR antagonist, resulting in reduced expression of Fgf15 and increased Cyp7a1 expression. Similarly, Li et al., (2013) found that reduced BSH activity diminished synthesis of secondary bile acids and inhibited FXR-induced signaling. Interestingly, the inhibition of intestinal FXR signaling altered bile acid composition in mice (142, 143) and decreased the incidence of obesity.
Activation of intestinal FXR is inhibitory to GLP-1 release in L-cells (45, 46). Trabelsi et al., (2015) determined FXR activation decreased glucose-stimulated GLP-1 secretion by blocking glycolysis, and thus glucose production in both mice and human intestinal L-cells. Similarly, Niss et al., (2020) found that inhibition of GLP-1 release by FXR is not only attributed to a downregulation in glycolysis, but also reduced glucose transport.
Tryptophan, the substrate for bacterial metabolism, can directly affect EEC L-cell function. In distal regions of the gastrointestinal tract, the bioavailability of digested peptides or amino acids is low as the bulk of protein digestion and absorption occurs in the small intestine before reaching the colon. Thus, the exposure of colonic EEC L-cells to tryptophan is limited. However, GLP-1 releasing L-cells in the proximal small intestine have been described (41, 144–146) and can respond to the presence of tryptophan. In vitro, enhanced GLP-1 release was observed in various EEC L-cell models exposed to tryptophan (147–149). However, contrary in vivo studies have reported a lack of stimulated GLP-1 release by intraluminal tryptophan in a perfused small intestine (150). Several G-protein coupled receptors have been implicated in EEC-sensing of tryptophan including the extracellular calcium-sensing receptor (CaSR) (151) and G-protein receptor 142 (GPR142) (149). The signaling mechanisms in EEC L-cells downstream of GPR142 activation is thought to be similar to pathways elucidated in other secretory cell types such as pancreatic β-cells (152). Both Gαq and Gαs-proteins are thought to be recruited to increase intracellular IP3 and cAMP levels, respectively (152, 153). Wang et al. (2016) also demonstrated that GPR142 activation led to an increase in inositol monophosphate accumulation, thus promoting the phosphorylation of ERK. The signaling mechanisms of CaSR in EEC L-cells have not been fully characterized, but in other duodenal EEC populations activated CaSR couples to Gαq-protein and downstream effectors PKC and IP3 receptors (154).
Indoles have been shown to alter EEC L-cell function. Acute application of indole increased GLP-1 secretion by increasing calcium mobilization in L-cells (44, 155) (Figure 4). The mechanism of action involved inhibition of voltage-gated potassium channels, thereby causing membrane depolarization and increased mobilization of calcium (44). However, chronic indole exposure reduced GLP-1 secretion by suppressing mitochondrial adenosine triphosphate (ATP) production, thus demonstrating dual and opposing effects of indole (44). The receptor responsible for mediating the effects of indole on GLP-1 release was not identified in this study. Also, the possible actions of indoles on tryptophan-sensitive receptors CaSR and GPR142 remain to be determined.
Another regulator of indole signaling is the aryl hydrocarbon receptor (AhR). Indole, tryptamine, skatole, IAA and other indole-derivatives are ligands for AhR (71, 156). AhR is a basic helix-loop-helix (bHLH) transcription factor (157) primarily expressed in host immune cells and its activation has been shown to mediate lipid and fatty acid metabolism and intestinal homeostasis (158). Inactive AhR forms a complex with heat shock protein 90 (Hsp90), the Hsp90 chaperone p23 (P23) and X-associated protein 2 (XAP2). Ligand binding induces a conformational change and translocation of the receptor complex to the nucleus (71). Within the nucleus, gene expression is activated through binding of the AhR nuclear translocator (ARNT) protein and cis-acting AhR response elements (AhREs) in target gene promoters (157). Interestingly, indoles, IAA and IPA also activate PXR (159). As mentioned, PXR is a nuclear receptor with DNA-binding and ligand-binding domains. Activation of PXR by several products of bacterial metabolism, including secondary bile acids and indoles, suggests a convergence of gut microbiota sensing pathways. Further investigation is warranted to understand the interactions between the different activating ligands and identify common downstream effectors. These studies will provide novel insights into the mechanisms underlying gut microbiota-host interactions.
Intestinal gut composition is an important determinant of health and many studies have attributed the pathogenesis of obesity and T2DM to an altered microbial ecosystem, particularly reduced bacterial diversity (160–163). Indeed, the dysregulation of nutrient metabolism, energy homeostasis, and appetite (164), all of which occur in obese-related diseases, are associated with a colonic shift in the relative abundance of three major phyla, Bacillota, Bacteroidota, and Verrucomicrobiota (164–167). In earlier studies, obesity and insulin resistance were associated with an increased abundance of Bacillota and concomitant decrease of Bacteroidota in both animal (7, 168), and human studies (6, 167, 169). However, recent reports found a reduction of the Bacillota population in obese subjects, whereas Bacteroidota significantly increased (16, 170–173). Some studies have even reported no change in the abundance of the two main microbial phyla (174–177). So, the exact changes in Bacillota and Bacteroidota during metabolic disease remains unresolved and we may need to consider other patient factors such as sex (178) and diet (179). Akkermansia¸ and its main species, Akkermansia muciniphilia (A. muciniphilia), is an abundant intestinal acetate- and butyrate-producing microbe from the phylum Verrucomicrobiota (180–182) and is gaining interest for its protective role against T2DM and obesity (183). The presence of A. muciniphilia in the gut is correlated with a healthy intestine and the decline in enrichment of A. muciniphilia has been linked to impairments in insulin sensitivity (165) and obese-related diseases (183–185). A. muciniphilia improves insulin sensitivity and glucose tolerance through various anti-inflammatory and energy mechanisms (186–189).
Surgical and pharmacological interventions that improve metabolic health also alter gut microbiota populations. Patients undergoing bariatric surgery, commonly Roux-en-Y-gastric bypass (RYGB) and vertical sleeve gastrectomy (VSG), often achieve sustained weight loss and T2DM resolution (190–192). A partial restoration of healthy intestinal microbiota composition was observed six months post-bariatric surgery in morbidly-obese female participants (193).
The manipulation of gut microbiota populations may be an approach to exploit in next generation therapeutics for metabolic disease. Interestingly, fecal microbiota transplantation from an individual with a healthy gut to an individual with metabolic syndrome resulted in significantly improved insulin sensitivity, accompanied by an altered microbial composition (163). Consistent with this report, the obese phenotype in mice was found to be transmissible by transplanting the gut microbiota of conventional obese mice to normal weight germ-free mice (167, 194). Furthermore, administration of A. muciniphilia was safe, and improved several metabolic parameters including increased insulin activity, a reduction in insulinemia, and decreased weight status in obese, insulin-resistant patients (195). However, A. muciniphilia was not linked to the improved glucose homeostasis pre- or post-bariatric surgery (185). Despite these discrepancies, manipulation of gut microbiota populations or administration of the metabolites produced by beneficial gut bacteria represent a promising therapeutic approach for improving metabolic health.
SCFAs in the gastrointestinal tract improve host gut health by increasing mucus production (196) and maintaining the intestinal gut barrier (197). SCFAs also promote crosstalk along the gut-brain axis (198) and are heavily involved in glucose and lipid metabolism (199). For example, SCFAs have been shown to stimulate the secretion of GLP-1 from intestinal L-cells (92, 95, 200) which promotes insulin release post-prandially. Similarly, butyrate attenuates insulin resistance of mice on a high-fat diet (HFD) by promoting energy expenditure (199).
Increased production of SCFAs by gut bacteria has been linked to reduced risk of obese-related chronic diseases (201). A shift in microbiome composition away from SCFA-producing bacterial species has been linked to prediabetes (173), a key step in the progression of diabetes (202). This may reflect diets consisting of high-fat and low-fiber content (203). Indeed, studies have shown that consumption of fiber- and ω-3 fatty acid-rich diets have increased levels of SCFA-producing bacteria (204, 205). The beneficial role of SCFAs was further supported by studies demonstrating increased dietary fiber intake reduced the risk of developing metabolic diseases, such as T2DM (206–210). Mechanisms proposed include direct effects on insulin sensitivity and energy expenditure (4, 199, 201, 211), improved glucose tolerance (49, 212, 213) or increased GLP-1 levels (212). Even direct delivery of SCFAs was beneficial as acute rectal infusions of sodium acetate enhanced PYY release in overweight human subjects and modulated whole-body metabolism (19–21). An increase in the abundance of acetate-producing bacteria was also found following SCFA administration (49). Therefore, supplementation of SCFAs may help to reverse gut dysbiosis as well as promote host metabolic health.
Epigenetic regulation, including HDAC-mediated mechanisms, has been linked to the development of T2DM in multiple organ systems (214). SCFAs are important regulators of gene expression through their actions as potent HDAC inhibitors (113). Reduced butyrate production led to a concomitant increase in colonic HDAC activity in a non-obese diabetic model and were associated with the increase in reactive oxidative species and alterations of colonic permeability (215). Butyrate supplementation suppressed HDAC activity in the liver of mice, leading to decreased gluconeogenesis and improved glucose homeostasis (216). This suggests that butyrate supplementation may induce epigenetic modifications that supports a healthy gut microbiota and metabolic health.
SCFAs may be important regulators of metabolic function, particularly in colonic stimulation of EECs and inhibition of HDAC activity. Harnessing the crosstalk mechanisms between resident gut bacteria and host by SCFAs may be a successful therapeutic strategy for the management of metabolic diseases in humans.
In an initial study linking cholesterol metabolism to diabetes, diabetic patients had an elevated bile acid pool size and greater excretion of bile acids in fecal samples (17). Furthermore, obese, diabetic db/db mice produced more bile acids leading to a larger total bile acid pool size (217), supporting the observation in humans.
The administration of bile acid sequestrants promoted insulin sensitivity in T2DM patients (218–220) and diabetic mice (221), likely by enhancing de novo synthesis of bile acids (222). Supporting studies have shown that in diabetic rats, intestinal sequestration of the bile acid pool improved insulin sensitivity and the mechanism involved may be mediated by GPBAR1 activation (223). Similarly, Trabelsi et al., (2015) found upon treatment with bile acid sequestrants to FXR-deficient cells, glucose tolerance improved by a GLP-1 mediated release mechanism. Direct targeting of GPBAR1 also produces positive metabolic health outcomes. In HFD-fed mice, overexpression of GPBAR1 increased secretion of GLP-1 induced insulin release, an effect that was lost in GPBAR1 deficient mice (134). Administration of oleanolic acid, the endogenous GPBAR1 agonist, ameliorated insulin sensitivity in mice upon HFD-feeding (224) and application of a selective GPBAR1 agonist, INT-777, enhanced GLP-1 secretion (133). The administration of taurocholate, the taurine conjugate of cholic acid, augmented GLP-1 release from L-cells and enhanced insulin release in humans (225). Other studies have shown that the administration of tauroursodeoxycholic acid, the taurine conjugated secondary bile acid of ursodeoxycholic acid, improved insulin sensitivity of obese humans (22) and obese mice (23). Furthermore, dual activation of FXR and GPBAR1 promotes GLP-1 release, thereby ameliorating insulin resistance (226). Thus, the receptors activated by bile acids and their metabolites present a powerful means to regulate glucose metabolism. However, intense adverse events have limited use of this approach in the clinic (227).
Dysregulation of energy utilization is often associated with obesity-related diseases (228). Activation of intestinal FXR promotes the secretion of FGF19, which could be exploited therapeutically as patients with obesity and T2DM have lower FGF19 levels (229). Administration of FGF19 to HFD-fed mice increased energy expenditure and reversed weight gain, thereby improving insulin sensitivity (230) and aided in T2DM resolution of patients following bariatric surgery (231). Interestingly, administration of a gut-biased FXR agonist protected against diet-induced weight gain while simultaneously enhancing energy expenditure (232).
Both RYGB and VSG enhanced the bile acid pool in rodents (233, 234) and humans (235, 236). Studies have shown that HFD-fed mice subjected to a bile diversion procedure increased the abundance of circulating bile acids (141, 237, 238) and improved glucose homeostasis (142). This was confirmed by the loss of significant weight reduction in FXR knockout mice on a HFD following VSG (239). However, increased abundance of bile acids has been reported in obese individuals with T2DM (240). Further, Jahansouz et al., (241) found a hypocaloric diet, mimicking weight loss, reduced the abundance of unconjugated bile acids. Bile acid signaling is involved in energy expenditure and glucose control following bariatric surgery in obese mice (239, 242) but further study is needed to define the role of bile acid signaling after bariatric surgery in humans.
Amino acid metabolism has been linked to metabolic health for decades; however, the involvement of metabolic products from bacterial amino acid metabolism in host metabolic health has recently become of interest. Fecal concentrations of indole, IAA and tryptamine were significantly reduced in mice fed a HFD compared with chow-fed mice (18, 243). A corresponding reduction in the concentration of indole-derivatives was reported in the feces of clinically obese (BMI>30) or T2DM human participants. Similar reductions in serum levels of indole-derivatives were reported in obese participants compared with non-obese controls (244). Following RYGB bariatric surgery, there was a significant improvement in glucose tolerance in T2DM subjects which coincided with an increase in IPA and tryptamine levels (245). Other retrospective studies have demonstrated that higher IPA levels were associated with lower risk of developing T2DM (70, 245).
Classical studies investigating the benefits of tryptophan consumption on metabolic disease progression may be attributed to bacterial-derived tryptophan metabolites. Ingesting tryptophan-enriched diets lowered the risk of developing obesity and T2DM in humans (246) and suppressed hyperglycemia and weight gain in animal models (247–249). Some of the mechanisms proposed for tryptophan-suppressed hyperglycemia include reduced insulin production and protection of pancreatic β-cells in diabetic rats (247) or inhibition of gluconeogenesis in rats and guinea pigs (248). The gut bacteria-derived tryptophan metabolite IPA was also associated with improved β-cell function (250) and rats fed an IPA-rich diet had significantly reduced fasting glucose levels (24). Furthermore, lower serum IPA levels were the most relevant indicators of early-onset T2DM (251) and body weight changes in obese rats (252). Recent studies have evaluated serum IPA levels as a risk biomarker for developing T2DM (250) or obesity (245) in humans. However, increased levels of tryptophan in the blood has been proposed as a predictor of increased risk of developing T2DM (253). Perhaps, this discrepancy between tryptophan and indolic metabolites in predicting metabolic disease states is due to the diverging metabolic pathways involved in the degradation of tryptophan. Tryptophan metabolites derived from the kynurenine pathway and IAA were found to be positively associated with T2DM risk (254). The kynurenine pathway exclusively involves host metabolism of tryptophan and excess kynurenine-metabolites have been associated with neurotoxicity and inflammation (255), while IAA is an intermediate of bacterial tryptophan metabolism and may be indicative of disrupted indole biosynthesis. As previously mentioned, the exclusive microbial-derived metabolite IPA has antidiabetic properties (24, 250). Therefore, the diversion of tryptophan metabolism to bacterial production of indoles or IPA has proven benefits in enhancing a healthy metabolic state.
Indole has been previously studied as an intercellular signaling molecule within the gut microbiota ecosystem. The capacity of indoles to stimulate GLP-1 releasing L-cells (44) has recently become of interest as a potential therapeutic target to regulate metabolic dysfunction (250). Indole-stimulated GLP-1 release can trigger the systemic effects of GLP-1 including enhanced insulin secretion, reduction in appetite and slowing of gastric emptying (29). Another possible mechanism by which indoles may regulate metabolic function could be through the regulation of intestinal microbial populations. Indoles directly affect bacterial functions associated with protection and host colonization (256). Since the relative abundance of several intestinal bacteria is associated with metabolic disease states, the role of indoles in determining the composition of the gut microbiota may contribute to the beneficial outcomes of bacterial indole production.
Indoles and other indole-derived metabolites produced by gut bacteria have significant physiological effects which may be exploited in future therapeutics. However, the risks associated with production of toxic by-products such as indoxyl sulfate will have to be carefully considered. Engineering enterobacteria that favor production of non-toxic indolic metabolites or the development of synthetic indole analogues that bypass first-pass metabolism are potential approaches to explore in designing future gut microbiota-based therapies. Herbal medicines may provide a source of inspiration to design structural analogues of indoles to treat metabolic disease. Indole alkaloids, which are bioactive compounds isolated from plants, have been found to inhibit dipeptidyl peptidase IV (DPP-IV) (257), the enzyme responsible for the inactivation of GLP-1. Therefore, indole alkaloids increase the half-life of the gut hormone responsible for enhancing insulin release post-prandially. Pharmacological strategies based on plant extracts containing indole alkaloids are effective in treating diabetic rats (258, 259) but their effectiveness and safety in human clinical trials remain to be determined. The direct effects of indole alkaloids on GLP-1 release are unknown, raising the possibility of these plant-based therapies improving glucose homeostasis through multiple synergistic pathways. Novel therapeutic approaches could also exploit indole signaling pathways. Recent studies have shown that targeting AhR by indoles may be able to ameliorate diabetes. Supplementation of indole-3-carbinol (I3C), an endogenous ligand of AhR, increased expression of AhR in the intestine (260) and promoted weight loss of HFD-fed obese mice and improved glucose tolerance (261). The involvement of AhR signaling in reducing proinflammatory responses (262, 263) could promote additional improvements in the treatment of metabolic diseases.
We have focused our review on the main metabolites produced by gut bacteria, namely short-chain fatty acids, secondary bile acids and indoles. However, other bacterial metabolites have also been implicated in host metabolic health. Bacterial fermentation of other dietary amino acids can give rise to several active metabolic compounds. For instance, p-Cresol is produced following tyrosine or phenylalanine degradation by gut bacteria. Serum concentration of p-Cresol is negatively correlated with T2DM and administration of p-Cresol reduced body weight, improved glucose homeostasis and β-cell function in HFD-fed mice (264). Despite these promising results, p-Cresol as a therapy for metabolic disease is limited as oral routes of administration are contraindicated due to sulfation by host cells to the nephrotoxic metabolite p-Cresol sulfate (264, 265). In addition, p-Cresol itself is a volatile compound that induces detrimental neurological, liver and respiratory effects at high concentrations (266). Imidazole propionate (IMP) is another amino acid-derived metabolite, produced by gut bacterial metabolism of histidine. Human subjects with prediabetes or T2DM have increased serum IMP levels compared with healthy individuals and administration of IMP to mice impairs glucose tolerance and disrupts insulin receptor signaling pathways (267, 268). Similarly, inosine, a purine metabolite involved in nucleotides and nucleic acids, is positively correlated with T2DM risk (269). Both IMP and inosine may be potential biomarkers to identify metabolite changes by the gut microbiota or may be exploited to uncover future treatments. More work is warranted to reconcile the changes that occur to the microbial composition and the mechanism by which these metabolites act to increase metabolic disease risk.
The gut microbiota has emerged as a pivotal regulator of GLP-1 releasing L-cells. Various approaches to treat obese-related diseases have been of interest for decades, although novel therapeutic strategies are urgently needed to treat a growing patient population. Approaches such as combining existing therapies in order to further enhance weight loss with fewer side effects or targeting the gut microbiota are currently in use. Altering gut bacterial populations with pre- or probiotics is a popular strategy to exploit this important relationship and restore deficiencies of nutrient and energy homeostasis observed in obesity-associated diseases. More work is needed to understand the precise cellular mechanisms that govern the bidirectional communication between the gut microbiota and EECs. Targeting common downstream effectors of converging signaling pathways recruited following bacterial metabolite receptor activation may be a feasible strategy as well. Due to the increasing incidence of metabolic disease, understanding the symbiotic relationship between gut bacteria and host cellular function, will provide greater clarity for the development of novel therapeutic strategies for the treatment of obese-related diseases.
KM and VL wrote and reviewed the manuscript. Both authors contributed to the article and approved the submitted version.
Work in the authors’ laboratories is supported by a grant from the Natural Sciences and Engineering Research Council of Canada (NSERC, RGPIN-2022-04187).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tierney BT, Yang Z, Luber JM, Beaudin M, Wibowo MC, Baek C, et al. The landscape of genetic content in the gut and oral human microbiome. Cell Host Microbe (2019) 26:283–95.e8. doi: 10.1016/j.chom.2019.07.008
2. Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet J-P, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol (2009) 11:2574–84. doi: 10.1111/j.1462-2920.2009.01982.x
3. Gensollen T, Iyer SS, Kasper DL, Blumberg RS. How colonization by microbiota in early life shapes the immune system. Science (2016) 352:539–44. doi: 10.1126/science.aad9378
4. den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res (2013) 54:2325. doi: 10.1194/JLR.R036012
5. Natividad JMM, Verdu EF. Modulation of intestinal barrier by intestinal microbiota: pathological and therapeutic implications. Pharmacol Res (2013) 69:42–51. doi: 10.1016/j.phrs.2012.10.007
6. Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA (2005) 102:11070. doi: 10.1073/PNAS.0504978102
7. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nat (2006) 444:1027–31. doi: 10.1038/nature05414
8. Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes (2012) 3:279–88. doi: 10.4161/gmic.19625
9. Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA (2004) 101:15718–23. doi: 10.1073/pnas.0407076101
10. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut (1987) 28:1221–7. doi: 10.1136/gut.28.10.1221
11. Miller TL, Wolin MJ. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl Environ Microbiol (1996) 62:1589–92. doi: 10.1128/aem.62.5.1589-1592.1996
12. Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett (2009) 294:1–8. doi: 10.1111/j.1574-6968.2009.01514.x
13. Yokoyama MT, Carlson JR. Microbial metabolites of tryptophan in the intestinal tract with special reference to skatole. Am J Clin Nutr (1979) 32:173–8. doi: 10.1093/ajcn/32.1.173
14. Karlin DA, Mastromarino AJ, Jones RD, Stroehlein JR, Lorentz O. Fecal skatole and indole and breath methane and hydrogen in patients with large bowel polyps or cancer. J Cancer Res Clin Oncol (1985) 109:135–41. doi: 10.1007/BF00391888
15. Ridlon JM, Kang D-J, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res (2006) 47:241–59. doi: 10.1194/jlr.R500013-JLR200
16. Schwiertz A, Taras D, Schäfer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obes (Silver Spring) (2010) 18:190–5. doi: 10.1038/oby.2009.167
17. Bennion LJ, Grundy SM. Effects of diabetes mellitus on cholesterol metabolism in man. N Engl J Med (1977) 296:1365–71. doi: 10.1056/NEJM197706162962401
18. Natividad JM, Agus A, Planchais J, Lamas B, Jarry AC, Martin R, et al. Impaired aryl hydrocarbon receptor ligand production by the gut microbiota is a key factor in metabolic syndrome. Cell Metab (2018) 28:737–749.e4. doi: 10.1016/j.cmet.2018.07.001
19. Canfora EE, van der Beek CM, Jocken JWE, Goossens GH, Holst JJ, Olde Damink SWM, et al. Colonic infusions of short-chain fatty acid mixtures promote energy metabolism in overweight/obese men: a randomized crossover trial. Sci Rep (2017) 7:2360. doi: 10.1038/s41598-017-02546-x
20. Freeland KR, Wolever TMS. Acute effects of intravenous and rectal acetate on glucagon-like peptide-1, peptide YY, ghrelin, adiponectin and tumour necrosis factor-alpha. Br J Nutr (2010) 103:460–6. doi: 10.1017/S0007114509991863
21. van der Beek CM, Canfora EE, Lenaerts K, Troost FJ, Damink SWMO, Holst JJ, et al. Distal, not proximal, colonic acetate infusions promote fat oxidation and improve metabolic markers in overweight/obese men. Clin Sci (Lond) (2016) 130:2073–82. doi: 10.1042/CS20160263
22. Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, et al. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes (2010) 59:1899–905. doi: 10.2337/db10-0308
23. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science (2006) 313:1137–40. doi: 10.1126/science.1128294
24. Abildgaard A, Elfving B, Hokland M, Wegener G, Lund S. The microbial metabolite indole-3-propionic acid improves glucose metabolism in rats, but does not affect behaviour. Arch Physiol Biochem (2018) 124:306–12. doi: 10.1080/13813455.2017.1398262
25. Zheng X, Xie G, Zhao A, Zhao L, Yao C, Chiu NHL, et al. The footprints of gut microbial-mammalian co-metabolism. J Proteome Res (2011) 10:5512–22. doi: 10.1021/pr2007945
26. Alnouti Y, Csanaky IL, Klaassen CD. Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. J Chromatogr B Analyt Technol BioMed Life Sci (2008) 873:209–17. doi: 10.1016/j.jchromb.2008.08.018
27. Chen MX, Wang S-Y, Kuo C-H, Tsai I-L. Metabolome analysis for investigating host-gut microbiota interactions. J Formos Med Assoc (2019) 118 Suppl 1:S10–22. doi: 10.1016/j.jfma.2018.09.007
28. Goldspink DA, Lu VB, Billing LJ, Larraufie P, Tolhurst G, Gribble FM, et al. Mechanistic insights into the detection of free fatty and bile acids by ileal glucagon-like peptide-1 secreting cells. Mol Metab (2018) 7:90–101. doi: 10.1016/j.molmet.2017.11.005
29. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology (2007) 132:2131–57. doi: 10.1053/j.gastro.2007.03.054
30. Gutzwiller JP, Drewe J, Göke B, Schmidt H, Rohrer B, Lareida J, et al. Glucagon-like peptide-1 promotes satiety and reduces food intake in patients with diabetes mellitus type 2. Am J Physiol (1999) 276:R1541–4. doi: 10.1152/ajpregu.1999.276.5.R1541
31. Marre M, Shaw J, Brändle M, Bebakar WMW, Kamaruddin NA, Strand J, et al. Liraglutide, a once-daily human GLP-1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with Type 2 diabetes (LEAD-1 SU). Diabetes Med (2009) 26:268–78. doi: 10.1111/j.1464-5491.2009.02666.x
32. Bays H, Pi-Sunyer X, Hemmingsson JU, Claudius B, Jensen CB, Van Gaal L. Liraglutide 3.0 mg for weight management: weight-loss dependent and independent effects. Curr Med Res Opin (2017) 33:225–9. doi: 10.1080/03007995.2016.1251892
33. Garber A, Henry R, Ratner R, Garcia-Hernandez PA, Rodriguez-Pattzi H, Olvera-Alvarez I, et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet (2009) 373:473–81. doi: 10.1016/S0140-6736(08)61246-5
34. Nauck M, Frid A, Hermansen K, Shah NS, Tankova T, Mitha IH, et al. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care (2009) 32:84–90. doi: 10.2337/dc08-1355
35. Zinman B, Gerich J, Buse JB, Lewin A, Schwartz S, Raskin P, et al. Efficacy and safety of the human glucagon-like peptide-1 analog liraglutide in combination with metformin and thiazolidinedione in patients with type 2 diabetes (LEAD-4 Met+TZD). Diabetes Care (2009) 32:1224–30. doi: 10.2337/dc08-2124
36. Miras AD, le Roux CW. Mechanisms underlying weight loss after bariatric surgery. Nat Rev Gastroenterol Hepatol (2013) 10:575–84. doi: 10.1038/nrgastro.2013.119
37. le Roux CW, Welbourn R, Werling M, Osborne A, Kokkinos A, Laurenius A, et al. Gut hormones as mediators of appetite and weight loss after Roux-en-Y gastric bypass. Ann Surg (2007) 246:780–5. doi: 10.1097/SLA.0b013e3180caa3e3
38. Larraufie P, Roberts GP, McGavigan AK, Kay RG, Li J, Leiter A, et al. Important role of the GLP-1 axis for glucose homeostasis after bariatric surgery. Cell Rep (2019) 26:1399–1408.e6. doi: 10.1016/j.celrep.2019.01.047
39. Halatchev IG, Ellacott KLJ, Fan W, Cone RD. Peptide YY3-36 inhibits food intake in mice through a melanocortin-4 receptor-independent mechanism. Endocrinology (2004) 145:2585–90. doi: 10.1210/en.2003-1754
40. Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature (2002) 418:650–4. doi: 10.1038/nature00887
41. Suzuki K, Iwasaki K, Murata Y, Harada N, Yamane S, Hamasaki A, et al. Distribution and hormonal characterization of primary murine L cells throughout the gastrointestinal tract. J Diabetes Investig (2018) 9:25–32. doi: 10.1111/jdi.12681
42. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PloS Biol (2016) 14:e1002533. doi: 10.1371/journal.pbio.1002533
43. Pathak P, Liu H, Boehme S, Xie C, Krausz KW, Gonzalez F, et al. Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. J Biol Chem (2017) 292:11055–69. doi: 10.1074/jbc.M117.784322
44. Chimerel C, Emery E, Summers DK, Keyser U, Gribble FM, Reimann F. Bacterial metabolite indole modulates incretin secretion from intestinal enteroendocrine L cells. Cell Rep (2014) 9:1202–8. doi: 10.1016/j.celrep.2014.10.032
45. Trabelsi M-S, Daoudi M, Prawitt J, Ducastel S, Touche V, Sayin SI, et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat Commun (2015) 6:7629. doi: 10.1038/ncomms8629
46. Niss K, Jakobsson ME, Westergaard D, Belling KG, Olsen JV, Brunak S. Effects of active farnesoid X receptor on GLUTag enteroendocrine L cells. Mol Cell Endocrinol (2020) 517:110923. doi: 10.1016/j.mce.2020.110923
47. Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun (2005) 329:386–90. doi: 10.1016/j.bbrc.2005.01.139
48. Fernandes J, Su W, Rahat-Rozenbloom S, Wolever TMS, Comelli EM. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr Diabetes (2014) 4:e121. doi: 10.1038/nutd.2014.23
49. Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science (2018) 359:1151–6. doi: 10.1126/science.aao5774
50. Smith EA, Macfarlane GT. Dissimilatory amino Acid metabolism in human colonic bacteria. Anaerobe (1997) 3:327–37. doi: 10.1006/anae.1997.0121
51. Nuutinen H, Lindros K, Hekali P, Salaspuro M. Elevated blood acetate as indicator of fast ethanol elimination in chronic alcoholics. Alcohol (1985) 2:623–6. doi: 10.1016/0741-8329(85)90090-4
52. Vijay N, Morris ME. Role of monocarboxylate transporters in drug delivery to the brain. Curr Pharm Des (2014) 20:1487–98. doi: 10.2174/13816128113199990462
53. Bergman EN. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev (1990) 70:567–90. doi: 10.1152/physrev.1990.70.2.567
54. Boets E, Gomand SV, Deroover L, Preston T, Vermeulen K, De Preter V, et al. Systemic availability and metabolism of colonic-derived short-chain fatty acids in healthy subjects: a stable isotope study. J Physiol (2017) 595:541–55. doi: 10.1113/JP272613
55. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem (2003) 72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712
56. Chiang JYL. Bile acids: regulation of synthesis. J Lipid Res (2009) 50:1955–66. doi: 10.1194/jlr.R900010-JLR200
57. Ellis E, Goodwin B, Abrahamsson A, Liddle C, Mode A, Rudling M, et al. Bile acid synthesis in primary cultures of rat and human hepatocytes. Hepatology (1998) 27:615–20. doi: 10.1002/hep.510270241
58. Botham KM, Boyd GS. The metabolism of chenodeoxycholic acid to beta-muricholic acid in rat liver. Eur J Biochem (1983) 134:191–6. doi: 10.1111/j.1432-1033.1983.tb07550.x
59. Boyer JL. Bile formation and secretion. Compr Physiol (2013) 3:1035–78. doi: 10.1002/cphy.c120027
60. Funabashi M, Grove TL, Wang M, Varma Y, McFadden ME, Brown LC, et al. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature (2020) 582:566–70. doi: 10.1038/s41586-020-2396-4
61. Jones BV, Begley M, Hill C, Gahan CGM, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA (2008) 105:13580–5. doi: 10.1073/pnas.0804437105
62. Ridlon JM, Harris SC, Bhowmik S, Kang D-J, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes (2016) 7:22–39. doi: 10.1080/19490976.2015.1127483
63. Nikiforov A, Kulikova V, Ziegler M. The human NAD metabolome: Functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol (2015) 50:284–97. doi: 10.3109/10409238.2015.1028612
64. Liu L, Su X, Quinn WJ, Hui S, Krukenberg K, Frederick DW, et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab (2018) 27:1067–1080.e5. doi: 10.1016/j.cmet.2018.03.018
65. Agus A, Planchais J, Sokol H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe (2018) 23:716–24. doi: 10.1016/j.chom.2018.05.003
66. Gostner JM, Geisler S, Stonig M, Mair L, Sperner-Unterweger B, Fuchs D. Tryptophan metabolism and related pathways in psychoneuroimmunology: the impact of nutrition and lifestyle. Neuropsychobiology (2020) 79:89–99. doi: 10.1159/000496293
67. Lee J-H, Lee J. Indole as an intercellular signal in microbial communities. FEMS Microbiol Rev (2010) 34:426–44. doi: 10.1111/j.1574-6976.2009.00204.x
68. Watanabe T, Snell EE. Reversibility of the tryptophanase reaction: synthesis of tryptophan from indole, pyruvate, and ammonia. Proc Natl Acad Sci USA (1972) 69:1086–90. doi: 10.1073/pnas.69.5.1086
69. Li G, Young KD. Indole production by the tryptophanase TnaA in Escherichia coli is determined by the amount of exogenous tryptophan. Microbiol (Reading) (2013) 159:402–10. doi: 10.1099/mic.0.064139-0
70. Tuomainen M, Lindström J, Lehtonen M, Auriola S, Pihlajamäki J, Peltonen M, et al. Associations of serum indolepropionic acid, a gut microbiota metabolite, with type 2 diabetes and low-grade inflammation in high-risk individuals. Nutr Diabetes (2018) 8:35. doi: 10.1038/s41387-018-0046-9
71. Kim CH. Immune regulation by microbiome metabolites. Immunology (2018) 154:220–9. doi: 10.1111/imm.12930
72. Williams BB, Van Benschoten AH, Cimermancic P, Donia MS, Zimmermann M, Taketani M, et al. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe (2014) 16:495–503. doi: 10.1016/j.chom.2014.09.001
73. Banoglu E, Jha GG, King RS. Hepatic microsomal metabolism of indole to indoxyl, a precursor of indoxyl sulfate. Eur J Drug Metab Pharmacokinet (2001) 26:235–40. doi: 10.1007/BF03226377
74. Miyazaki T, Ise M, Hirata M, Endo K, Ito Y, Seo H, et al. Indoxyl sulfate stimulates renal synthesis of transforming growth factor-beta 1 and progression of renal failure. Kidney Int Suppl (1997) 63:S211–4.
75. Piñero-Fernandez S, Chimerel C, Keyser UF, Summers DK. Indole transport across Escherichia coli membranes. J Bacteriol (2011) 193:1793–8. doi: 10.1128/JB.01477-10
76. Yanofsky C, Horn V, Gollnick P. Physiological studies of tryptophan transport and tryptophanase operon induction in Escherichia coli. J Bacteriol (1991) 173:6009–17. doi: 10.1128/jb.173.19.6009-6017.1991
77. Kawamura-Sato K, Shibayama K, Horii T, Iimuma Y, Arakawa Y, Ohta M. Role of multiple efflux pumps in Escherichia coli in indole expulsion. FEMS Microbiol Lett (1999) 179:345–52. doi: 10.1111/j.1574-6968.1999.tb08748.x
78. Hauge M, Ekberg JP, Engelstoft MS, Timshel P, Madsen AN, Schwartz TW. Gq and Gs signaling acting in synergy to control GLP-1 secretion. Mol Cell Endocrinol (2017) 449:64–73. doi: 10.1016/j.mce.2016.11.024
79. Hauge M, Vestmar MA, Husted AS, Ekberg JP, Wright MJ, Di Salvo J, et al. GPR40 (FFAR1) - Combined Gs and Gq signaling in vitro is associated with robust incretin secretagogue action ex vivo and in vivo. Mol Metab (2015) 4:3–14. doi: 10.1016/j.molmet.2014.10.002
80. Le Poul E, Loison C, Struyf S, Springael J-Y, Lannoy V, Decobecq M-E, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem (2003) 278:25481–9. doi: 10.1074/jbc.M301403200
81. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem (2003) 278:11312–9. doi: 10.1074/jbc.M211609200
82. Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA (2013) 110:4410–5. doi: 10.1073/pnas.1215927110
83. Feingold KR, Moser A, Shigenaga JK, Grunfeld C. Inflammation stimulates niacin receptor (GPR109A/HCA2) expression in adipose tissue and macrophages. J Lipid Res (2014) 55:2501–8. doi: 10.1194/jlr.M050955
84. Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell (1978) 14:105–13. doi: 10.1016/0092-8674(78)90305-7
85. Li M, van Esch BCAM, Henricks PAJ, Folkerts G, Garssen J. The anti-inflammatory effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor α-stimulated endothelial cells via activation of GPR41/43 and inhibition of HDACs. Front Pharmacol (2018) 9:533. doi: 10.3389/fphar.2018.00533
86. Silva LG, Ferguson BS, Avila AS, Faciola AP. Sodium propionate and sodium butyrate effects on histone deacetylase (HDAC) activity, histone acetylation, and inflammatory gene expression in bovine mammary epithelial cells. J Anim Sci (2018) 96:5244–52. doi: 10.1093/jas/sky373
87. Fleischer J, Bumbalo R, Bautze V, Strotmann J, Breer H. Expression of odorant receptor Olfr78 in enteroendocrine cells of the colon. Cell Tissue Res (2015) 361:697–710. doi: 10.1007/s00441-015-2165-0
88. Nøhr MK, Pedersen MH, Gille A, Egerod KL, Engelstoft MS, Husted AS, et al. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology (2013) 154:3552–64. doi: 10.1210/en.2013-1142
89. Karaki S-I, Tazoe H, Hayashi H, Kashiwabara H, Tooyama K, Suzuki Y, et al. Expression of the short-chain fatty acid receptor, GPR43, in the human colon. J Mol Histol (2008) 39:135–42. doi: 10.1007/s10735-007-9145-y
90. Karaki S, Mitsui R, Hayashi H, Kato I, Sugiya H, Iwanaga T, et al. Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell Tissue Res (2006) 324:353–60. doi: 10.1007/s00441-005-0140-x
91. Bolognini D, Moss CE, Nilsson K, Petersson AU, Donnelly I, Sergeev E, et al. A novel allosteric activator of free fatty acid 2 receptor displays unique gi-functional bias. J Biol Chem (2016) 291:18915–31. doi: 10.1074/jbc.M116.736157
92. Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes (2012) 61:364–71. doi: 10.2337/db11-1019
93. Avet C, Mancini A, Breton B, Le Gouill C, Hauser AS, Normand C, et al. Effector membrane translocation biosensors reveal G protein and βarrestin coupling profiles of 100 therapeutically relevant GPCRs. Elife (2022) 11:e74101. doi: 10.7554/eLife.74101
94. Hudson BD, Due-Hansen ME, Christiansen E, Hansen AM, Mackenzie AE, Murdoch H, et al. Defining the molecular basis for the first potent and selective orthosteric agonists of the FFA2 free fatty acid receptor. J Biol Chem (2013) 288:17296–312. doi: 10.1074/jbc.M113.455337
95. Psichas A, Sleeth ML, Murphy KG, Brooks L, Bewick GA, Hanyaloglu AC, et al. The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int J Obes (Lond) (2015) 39:424–9. doi: 10.1038/ijo.2014.153
96. Forbes S, Stafford S, Coope G, Heffron H, Real K, Newman R, et al. Selective FFA2 Agonism Appears to Act via Intestinal PYY to Reduce Transit and Food Intake but Does Not Improve Glucose Tolerance in Mouse Models. Diabetes (2015) 64:3763–71. doi: 10.2337/db15-0481
97. Caengprasath N, Gonzalez-Abuin N, Shchepinova M, Ma Y, Inoue A, Tate EW, et al. Internalization-dependent free fatty acid receptor 2 signaling is essential for propionate-induced anorectic gut hormone release. iScience (2020) 23:101449. doi: 10.1016/j.isci.2020.101449
98. DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol (2007) 69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749
99. Lee SU, In HJ, Kwon MS, Park B, Jo M, Kim M-O, et al. β-Arrestin 2 mediates G protein-coupled receptor 43 signals to nuclear factor-κB. Biol Pharm Bull (2013) 36:1754–9. doi: 10.1248/bpb.b13-00312
100. Nøhr MK, Egerod KL, Christiansen SH, Gille A, Offermanns S, Schwartz TW, et al. Expression of the short chain fatty acid receptor GPR41/FFAR3 in autonomic and somatic sensory ganglia. Neuroscience (2015) 290:126–37. doi: 10.1016/j.neuroscience.2015.01.040
101. Won Y-J, Lu VB, Puhl HL, Ikeda SR. β-Hydroxybutyrate modulates N-type calcium channels in rat sympathetic neurons by acting as an agonist for the G-protein-coupled receptor FFA3. J Neurosci (2013) 33:19314–25. doi: 10.1523/JNEUROSCI.3102-13.2013
102. Tazoe H, Otomo Y, Karaki S-I, Kato I, Fukami Y, Terasaki M, et al. Expression of short-chain fatty acid receptor GPR41 in the human colon. BioMed Res (2009) 30:149–56. doi: 10.2220/biomedres.30.149
103. Christiansen CB, Gabe MBN, Svendsen B, Dragsted LO, Rosenkilde MM, Holst JJ. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am J Physiol Gastrointest Liver Physiol (2018) 315:G53–65. doi: 10.1152/ajpgi.00346.2017
104. Ang Z, Xiong D, Wu M, Ding JL. FFAR2-FFAR3 receptor heteromerization modulates short-chain fatty acid sensing. FASEB J (2018) 32:289–303. doi: 10.1096/fj.201700252RR
105. Nishida A, Miyamoto J, Shimizu H, Kimura I. Gut microbial short-chain fatty acids-mediated olfactory receptor 78 stimulation promotes anorexigenic gut hormone peptide YY secretion in mice. Biochem Biophys Res Commun (2021) 557:48–54. doi: 10.1016/j.bbrc.2021.03.167
106. Priori D, Colombo M, Clavenzani P, Jansman AJM, Lallès J-P, Trevisi P, et al. The olfactory receptor OR51E1 is present along the gastrointestinal tract of pigs, co-localizes with enteroendocrine cells and is modulated by intestinal microbiota. PloS One (2015) 10:e0129501. doi: 10.1371/journal.pone.0129501
107. Han YE, Kang CW, Oh JH, Park SH, Ku CR, Cho YH, et al. Olfactory receptor OR51E1 mediates GLP-1 secretion in human and rodent enteroendocrine L cells. J Endocr Soc (2018) 2:1251–8. doi: 10.1210/js.2018-00165
108. Zimberlin CD, Lancini C, Sno R, Rosekrans SL, McLean CM, Vlaming H, et al. HDAC1 and HDAC2 collectively regulate intestinal stem cell homeostasis. FASEB J (2015) 29:2070–80. doi: 10.1096/fj.14-257931
109. Aoyama M, Kotani J, Usami M. Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating GPR-41/GPR-43 pathways. Nutrition (2010) 26:653–61. doi: 10.1016/j.nut.2009.07.006
110. Cousens LS, Gallwitz D, Alberts BM. Different accessibilities in chromatin to histone acetylase. J Biol Chem (1979) 254:1716–23. doi: 10.1016/S0021-9258(17)37831-6
111. Yuille S, Reichardt N, Panda S, Dunbar H, Mulder IE. Human gut bacteria as potent class I histone deacetylase inhibitors in vitro through production of butyric acid and valeric acid. PloS One (2018) 13:e0201073. doi: 10.1371/journal.pone.0201073
112. Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr (2003) 133:2485S–93S. doi: 10.1093/jn/133.7.2485S
113. Zhou J, Martin RJ, Tulley RT, Raggio AM, McCutcheon KL, Shen L, et al. Dietary resistant starch upregulates total GLP-1 and PYY in a sustained day-long manner through fermentation in rodents. Am J Physiol Endocrinol Metab (2008) 295:E1160–6. doi: 10.1152/ajpendo.90637.2008
114. Petersen N, Reimann F, Bartfeld S, Farin HF, Ringnalda FC, Vries RGJ, et al. Generation of L cells in mouse and human small intestine organoids. Diabetes (2014) 63:410–20. doi: 10.2337/db13-0991
115. Arora T, Akrami R, Pais R, Bergqvist L, Johansson BR, Schwartz TW, et al. Microbial regulation of the L cell transcriptome. Sci Rep (2018) 8:1207. doi: 10.1038/s41598-017-18079-2
116. Wichmann A, Allahyar A, Greiner TU, Plovier H, Lundén GÖ, Larsson T, et al. Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe (2013) 14:582–90. doi: 10.1016/j.chom.2013.09.012
117. Larraufie P, Martin-Gallausiaux C, Lapaque N, Dore J, Gribble FM, Reimann F, et al. SCFAs strongly stimulate PYY production in human enteroendocrine cells. Sci Rep (2018) 8:74. doi: 10.1038/s41598-017-18259-0
118. Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, et al. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun (2002) 298:714–9. doi: 10.1016/s0006-291x(02)02550-0
119. Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science (1999) 284:1365–8. doi: 10.1126/science.284.5418.1365
120. Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science (1999) 284:1362–5. doi: 10.1126/science.284.5418.1362
121. Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell (1999) 3:543–53. doi: 10.1016/s1097-2765(00)80348-2
122. Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA (2001) 98:3369–74. doi: 10.1073/pnas.051551698
123. Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, et al. Vitamin D receptor as an intestinal bile acid sensor. Science (2002) 296:1313–6. doi: 10.1126/science.1070477
124. Moore LB, Maglich JM, McKee DD, Wisely B, Willson TM, Kliewer SA, et al. Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol Endocrinol (2002) 16:977–86. doi: 10.1210/mend.16.5.0828
125. De Marino S, Carino A, Masullo D, Finamore C, Marchianò S, Cipriani S, et al. Hyodeoxycholic acid derivatives as liver X receptor α and G-protein-coupled bile acid receptor agonists. Sci Rep (2017) 7:43290. doi: 10.1038/srep43290
126. Nagahashi M, Takabe K, Liu R, Peng K, Wang X, Wang Y, et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology (2015) 61:1216–26. doi: 10.1002/hep.27592
127. Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem (2003) 278:9435–40. doi: 10.1074/jbc.M209706200
128. Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature (2006) 439:484–9. doi: 10.1038/nature04330
129. Brighton CA, Rievaj J, Kuhre RE, Glass LL, Schoonjans K, Holst JJ, et al. Bile acids trigger GLP-1 release predominantly by accessing basolaterally located G protein-coupled bile acid receptors. Endocrinology (2015) 156:3961–70. doi: 10.1210/en.2015-1321
130. Parker HE, Wallis K, le Roux CW, Wong KY, Reimann F, Gribble FM. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br J Pharmacol (2012) 165:414–23. doi: 10.1111/j.1476-5381.2011.01561.x
131. Poole DP, Godfrey C, Cattaruzza F, Cottrell GS, Kirkland JG, Pelayo JC, et al. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil (2010) 22:814–25. doi: 10.1111/j.1365-2982.2010.01487.x
132. Christiansen CB, Trammell SAJ, Wewer Albrechtsen NJ, Schoonjans K, Albrechtsen R, Gillum MP, et al. Bile acids drive colonic secretion of glucagon-like-peptide 1 and peptide-YY in rodents. Am J Physiol Gastrointest Liver Physiol (2019) 316:G574–84. doi: 10.1152/ajpgi.00010.2019
133. Harach T, Pols TWH, Nomura M, Maida A, Watanabe M, Auwerx J, et al. TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Sci Rep (2012) 2:430. doi: 10.1038/srep00430
134. Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab (2009) 10:167–77. doi: 10.1016/j.cmet.2009.08.001
135. Lund ML, Sorrentino G, Egerod KL, Kroone C, Mortensen B, Knop FK, et al. L-cell differentiation is induced by bile acids through GPBAR1 and paracrine GLP-1 and serotonin signaling. Diabetes (2020) 69:614–23. doi: 10.2337/db19-0764
136. Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell (1995) 81:687–93. doi: 10.1016/0092-8674(95)90530-8
137. Grober J, Zaghini I, Fujii H, Jones SA, Kliewer SA, Willson TM, et al. Identification of a bile acid-responsive element in the human ileal bile acid-binding protein gene. Involvement of the farnesoid X receptor/9-cis-retinoic acid receptor heterodimer. J Biol Chem (1999) 274:29749–54. doi: 10.1074/jbc.274.42.29749
138. Fiorucci S, Distrutti E. The pharmacology of bile acids and their receptors. Handb Exp Pharmacol (2019) 256:3–18. doi: 10.1007/164_2019_238
139. Xie MH, Holcomb I, Deuel B, Dowd P, Huang A, Vagts A, et al. FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine (1999) 11:729–35. doi: 10.1006/cyto.1999.0485
140. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab (2005) 2:217–25. doi: 10.1016/j.cmet.2005.09.001
141. Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H-U, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab (2013) 17:225–35. doi: 10.1016/j.cmet.2013.01.003
142. Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, et al. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun (2013) 4:2384. doi: 10.1038/ncomms3384
143. Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun (2015) 6:10166. doi: 10.1038/ncomms10166
144. Habib AM, Richards P, Cairns LS, Rogers GJ, Bannon CAM, Parker HE, et al. Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology (2012) 153:3054–65. doi: 10.1210/en.2011-2170
145. Glass LL, Calero-Nieto FJ, Jawaid W, Larraufie P, Kay RG, Göttgens B, et al. Single-cell RNA-sequencing reveals a distinct population of proglucagon-expressing cells specific to the mouse upper small intestine. Mol Metab (2017) 6:1296–303. doi: 10.1016/j.molmet.2017.07.014
146. Sun EW, de Fontgalland D, Rabbitt P, Hollington P, Sposato L, Due SL, et al. Mechanisms controlling glucose-induced GLP-1 secretion in human small intestine. Diabetes (2017) 66:2144–9. doi: 10.2337/db17-0058
147. Osuga Y, Harada K, Tsuboi T. Identification of a regulatory pathway of L-phenylalanine-induced GLP-1 secretion in the enteroendocrine L cells. Biochem Biophys Res Commun (2022) 588:118–24. doi: 10.1016/j.bbrc.2021.12.043
148. Pais R, Gribble FM, Reimann F. Signalling pathways involved in the detection of peptones by murine small intestinal enteroendocrine L-cells. Peptides (NY) (2016) 77:9–15. doi: 10.1016/j.peptides.2015.07.019
149. Lin HV, Efanov AM, Fang X, Beavers LS, Wang X, Wang J, et al. GPR142 controls tryptophan-induced insulin and incretin hormone secretion to improve glucose metabolism. PloS One (2016) 11:e0157298. doi: 10.1371/journal.pone.0157298
150. Modvig IM, Kuhre RE, Jepsen SL, Xu SFS, Engelstoft MS, Egerod KL, et al. Amino acids differ in their capacity to stimulate GLP-1 release from the perfused rat small intestine and stimulate secretion by different sensing mechanisms. Am J Physiol Endocrinol Metab (2021) 320:E874–85. doi: 10.1152/ajpendo.00026.2021
151. Mace OJ, Schindler M, Patel S. The regulation of K- and L-cell activity by GLUT2 and the calcium-sensing receptor CasR in rat small intestine. J Physiol (2012) 590:2917–36. doi: 10.1113/jphysiol.2011.223800
152. Al-Amily IM, Dunér P, Groop L, Salehi A. The functional impact of G protein-coupled receptor 142 (Gpr142) on pancreatic β-cell in rodent. Pflugers Arch (2019) 471:633–45. doi: 10.1007/s00424-019-02262-7
153. Wang J, Carrillo JJ, Lin HV. GPR142 agonists stimulate glucose-dependent insulin secretion via Gq-dependent signaling. PloS One (2016) 11:e0154452. doi: 10.1371/journal.pone.0154452
154. Zhao X, Xian Y, Wang C, Ding L, Meng X, Zhu W, et al. Calcium-sensing receptor-mediated L-tryptophan-induced secretion of cholecystokinin and glucose-dependent insulinotropic peptide in swine duodenum. J Vet Sci (2018) 19:179–87. doi: 10.4142/jvs.2018.19.2.179
155. Phuah P, Cheng S, Norton M, Roberts A, Otsubo E, Gribble F, et al. Microbial tryptophan metabolites modulate L-cell induced GLP-1 secretion to improve glucose homeostasis. Endocrine Abstracts (2021) 77:OC3.5. doi: 10.1530/ENDOABS.77.OC3.5
156. Scott SA, Fu J, Chang PV. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc Natl Acad Sci USA (2020) 117:19376–87. doi: 10.1073/pnas.2000047117
157. Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol (1995) 35:307–40. doi: 10.1146/annurev.pa.35.040195.001515
158. Krishnan S, Ding Y, Saedi N, Choi M, Sridharan GV, Sherr DH, et al. Gut microbiota-derived tryptophan metabolites modulate inflammatory response in hepatocytes and macrophages. Cell Rep (2018) 23:1099–111. doi: 10.1016/j.celrep.2018.03.109
159. Koutsounas I, Theocharis S, Patsouris E, Giaginis C. Pregnane X receptor (PXR) at the crossroads of human metabolism and disease. Curr Drug Metab (2013) 14:341–50. doi: 10.2174/1389200211314030009
160. Krajmalnik-Brown R, Ilhan Z-E, Kang D-W, DiBaise JK. Effects of gut microbes on nutrient absorption and energy regulation. Nutr Clin Pract (2012) 27:201–14. doi: 10.1177/0884533611436116
161. Chassaing B, Raja SM, Lewis JD, Srinivasan S, Gewirtz AT. Colonic microbiota encroachment correlates with dysglycemia in humans. Cell Mol Gastroenterol Hepatol (2017) 4:205–21. doi: 10.1016/j.jcmgh.2017.04.001
162. Org E, Blum Y, Kasela S, Mehrabian M, Kuusisto J, Kangas AJ, et al. Relationships between gut microbiota, plasma metabolites, and metabolic syndrome traits in the METSIM cohort. Genome Biol (2017) 18:70. doi: 10.1186/s13059-017-1194-2
163. Kootte RS, Levin E, Salojärvi J, Smits LP, Hartstra AV, Udayappan SD, et al. Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab (2017) 26:611–619.e6. doi: 10.1016/j.cmet.2017.09.008
164. Torres-Fuentes C, Schellekens H, Dinan TG, Cryan JF. The microbiota-gut-brain axis in obesity. Lancet Gastroenterol Hepatol (2017) 2:747–56. doi: 10.1016/S2468-1253(17)30147-4
165. Zhang J, Ni Y, Qian L, Fang Q, Zheng T, Zhang M, et al. Decreased abundance of akkermansia muciniphila leads to the impairment of insulin secretion and glucose homeostasis in lean type 2 diabetes. Adv Sci (Weinh) (2021) 8:e2100536. doi: 10.1002/advs.202100536
166. Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe (2008) 3:213–23. doi: 10.1016/j.chom.2008.02.015
167. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature (2006) 444:1022–3. doi: 10.1038/4441022a
168. Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science (2005) 307:1915–20. doi: 10.1126/science.1104816
169. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature (2012) 490:55–60. doi: 10.1038/nature11450
170. Larsen N, Vogensen FK, van den Berg FWJ, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PloS One (2010) 5:e9085. doi: 10.1371/journal.pone.0009085
171. Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA (2009) 106:2365–70. doi: 10.1073/pnas.0812600106
172. Duan M, Wang Y, Zhang Q, Zou R, Guo M, Zheng H. Characteristics of gut microbiota in people with obesity. PloS One (2021) 16:e0255446. doi: 10.1371/journal.pone.0255446
173. Zhang X, Shen D, Fang Z, Jie Z, Qiu X, Zhang C, et al. Human gut microbiota changes reveal the progression of glucose intolerance. PloS One (2013) 8:e71108. doi: 10.1371/journal.pone.0071108
174. Chambers ES, Viardot A, Psichas A, Morrison DJ, Murphy KG, Zac-Varghese SEK, et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut (2015) 64:1744–54. doi: 10.1136/gutjnl-2014-307913
175. Mai V, McCrary QM, Sinha R, Glei M. Associations between dietary habits and body mass index with gut microbiota composition and fecal water genotoxicity: an observational study in African American and Caucasian American volunteers. Nutr J (2009) 8:49. doi: 10.1186/1475-2891-8-49
176. Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone AM, Louis P, et al. Human colonic microbiota associated with diet, obesity and weight loss. Int J Obes (Lond) (2008) 32:1720–4. doi: 10.1038/ijo.2008.155
177. Jumpertz R, Le DS, Turnbaugh PJ, Trinidad C, Bogardus C, Gordon JI, et al. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am J Clin Nutr (2011) 94:58–65. doi: 10.3945/ajcn.110.010132
178. Markle JGM, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science (2013) 339:1084–8. doi: 10.1126/science.1233521
179. Yun E-J, Imdad S, Jang J, Park J, So B, Kim J-H, et al. Diet is a stronger covariate than exercise in determining gut microbial richness and diversity. Nutrients (2022) 14(12):2507. doi: 10.3390/nu14122507
180. Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol (2004) 54:1469–76. doi: 10.1099/ijs.0.02873-0
181. Belzer C, de Vos WM. Microbes inside–from diversity to function: the case of Akkermansia. ISME J (2012) 6:1449–58. doi: 10.1038/ismej.2012.6
182. Huang K, Wang MM, Kulinich A, Yao HL, Ma HY, Martínez JER, et al. Biochemical characterisation of the neuraminidase pool of the human gut symbiont Akkermansia muciniphila. Carbohydr Res (2015) 415:60–5. doi: 10.1016/j.carres.2015.08.001
183. Zhou Q, Zhang Y, Wang X, Yang R, Zhu X, Zhang Y, et al. Gut bacteria Akkermansia is associated with reduced risk of obesity: evidence from the American Gut Project. Nutr Metab (Lond) (2020) 17:90. doi: 10.1186/s12986-020-00516-1
184. Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature (2013) 498:99–103. doi: 10.1038/nature12198
185. Dao MC, Belda E, Prifti E, Everard A, Kayser BD, Bouillot J-L, et al. Akkermansia muciniphila abundance is lower in severe obesity, but its increased level after bariatric surgery is not associated with metabolic health improvement. Am J Physiol Endocrinol Metab (2019) 317:E446–59. doi: 10.1152/ajpendo.00140.2019
186. Loizou S, Lekakis I, Chrousos GP, Moutsatsou P. Beta-sitosterol exhibits anti-inflammatory activity in human aortic endothelial cells. Mol Nutr Food Res (2010) 54:551–8. doi: 10.1002/mnfr.200900012
187. Remely M, Hippe B, Zanner J, Aumueller E, Brath H, Haslberger AG. Gut Microbiota of Obese, Type 2 Diabetic Individuals is Enriched in Faecalibacterium prausnitzii, Akkermansia muciniphila and Peptostreptococcus anaerobius after Weight Loss. Endocr Metab Immune Disord Drug Targets (2016) 16:99–106. doi: 10.2174/1871530316666160831093813
188. Zhao S, Liu W, Wang J, Shi J, Sun Y, Wang W, et al. Akkermansia muciniphila improves metabolic profiles by reducing inflammation in chow diet-fed mice. J Mol Endocrinol (2017) 58:1–14. doi: 10.1530/JME-16-0054
189. Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med (2012) 18:59–68. doi: 10.1016/j.molmed.2011.07.010
190. Kadera BE, Lum K, Grant J, Pryor AD, Portenier DD, DeMaria EJ. Remission of type 2 diabetes after Roux-en-Y gastric bypass is associated with greater weight loss. Surg Obes Relat Dis (2009) 5:305–9. doi: 10.1016/j.soard.2009.02.007
191. Courcoulas AP, Christian NJ, Belle SH, Berk PD, Flum DR, Garcia L, et al. Weight change and health outcomes at 3 years after bariatric surgery among individuals with severe obesity. JAMA (2013) 310:2416–25. doi: 10.1001/jama.2013.280928
192. Schauer PR, Bhatt DL, Kirwan JP, Wolski K, Aminian A, Brethauer SA, et al. Bariatric surgery versus intensive medical therapy for diabetes - 5-year outcomes. N Engl J Med (2017) 376:641–51. doi: 10.1056/NEJMoa1600869
193. Koffert J, Lahti L, Nylund L, Salminen S, Hannukainen JC, Salminen P, et al. Nuutila P. Partial restoration of normal intestinal microbiota in morbidly obese women six months after bariatric surgery. PeerJ (2020) 8:e10442. doi: 10.7717/peerj.10442
194. Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science (2010) 328:228–31. doi: 10.1126/science.1179721
195. Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S, et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med (2019) 25:1096–103. doi: 10.1038/s41591-019-0495-2
196. Gaudier E, Rival M, Buisine M-P, Robineau I, Hoebler C. Butyrate enemas upregulate Muc genes expression but decrease adherent mucus thickness in mice colon. Physiol Res (2009) 58:111–9. doi: 10.33549/physiolres.931271
197. Peng L, Li Z-R, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr (2009) 139:1619–25. doi: 10.3945/jn.109.104638
198. Dalile B, Van Oudenhove L, Vervliet B, Verbeke K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat Rev Gastroenterol Hepatol (2019) 16:461–78. doi: 10.1038/s41575-019-0157-3
199. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes (2009) 58:1509–17. doi: 10.2337/db08-1637
200. Yadav H, Lee J-H, Lloyd J, Walter P, Rane SG. Beneficial metabolic effects of a probiotic via butyrate-induced GLP-1 hormone secretion. J Biol Chem (2013) 288:25088–97. doi: 10.1074/jbc.M113.452516
201. Lu Y, Fan C, Li P, Lu Y, Chang X, Qi K. Short chain fatty acids prevent high-fat-diet-induced obesity in mice by regulating G protein-coupled receptors and gut microbiota. Sci Rep (2016) 6:37589. doi: 10.1038/srep37589
202. Sanna S, van Zuydam NR, Mahajan A, Kurilshikov A, Vich Vila A, Võsa U, et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat Genet (2019) 51:600–5. doi: 10.1038/s41588-019-0350-x
203. Pastorino S, Richards M, Pierce M, Ambrosini GL. A high-fat, high-glycaemic index, low-fibre dietary pattern is prospectively associated with type 2 diabetes in a British birth cohort. Br J Nutr (2016) 115:1632–42. doi: 10.1017/S0007114516000672
204. Gutiérrez-Díaz I, Fernández-Navarro T, Sánchez B, Margolles A, González S. Mediterranean diet and faecal microbiota: a transversal study. Food Funct (2016) 7:2347–56. doi: 10.1039/c6fo00105j
205. Gutiérrez-Díaz I, Fernández-Navarro T, Salazar N, Bartolomé B, Moreno-Arribas MV, de Andres-Galiana EJ, et al. Adherence to a mediterranean diet influences the fecal metabolic profile of microbial-derived phenolics in a spanish cohort of middle-age and older people. J Agric Food Chem (2017) 65:586–95. doi: 10.1021/acs.jafc.6b04408
206. Li YJ, Chen X, Kwan TK, Loh YW, Singer J, Liu Y, et al. Dietary Fiber Protects against Diabetic Nephropathy through Short-Chain Fatty Acid-Mediated Activation of G Protein-Coupled Receptors GPR43 and GPR109A. J Am Soc Nephrol (2020) 31:1267–81. doi: 10.1681/ASN.2019101029
207. Meyer KA, Kushi LH, Jacobs DR, Slavin J, Sellers TA, Folsom AR. Carbohydrates, dietary fiber, and incident type 2 diabetes in older women. Am J Clin Nutr (2000) 71:921–30. doi: 10.1093/ajcn/71.4.921
208. Fung TT, Hu FB, Pereira MA, Liu S, Stampfer MJ, Colditz GA, et al. Whole-grain intake and the risk of type 2 diabetes: a prospective study in men. Am J Clin Nutr (2002) 76:535–40. doi: 10.1093/ajcn/76.3.535
209. van Dam RM, Hu FB, Rosenberg L, Krishnan S, Palmer JR. Dietary calcium and magnesium, major food sources, and risk of type 2 diabetes in U.S. black women. Diabetes Care (2006) 29:2238–43. doi: 10.2337/dc06-1014
210. Montonen J, Knekt P, Järvinen R, Aromaa A, Reunanen A. Whole-grain and fiber intake and the incidence of type 2 diabetes. Am J Clin Nutr (2003) 77:622–9. doi: 10.1093/ajcn/77.3.622
211. Anastasovska J, Arora T, Sanchez Canon GJ, Parkinson JRC, Touhy K, Gibson GR, et al. Fermentable carbohydrate alters hypothalamic neuronal activity and protects against the obesogenic environment. Obes (Silver Spring) (2012) 20:1016–23. doi: 10.1038/oby.2012.6
212. Roshanravan N, Mahdavi R, Alizadeh E, Jafarabadi MA, Hedayati M, Ghavami A, et al. Effect of butyrate and inulin supplementation on glycemic status, lipid profile and glucagon-like peptide 1 level in patients with type 2 diabetes: a randomized double-blind, placebo-controlled trial. Horm Metab Res (2017) 49:886–91. doi: 10.1055/s-0043-119089
213. Perraudeau F, McMurdie P, Bullard J, Cheng A, Cutcliffe C, Deo A, et al. Improvements to postprandial glucose control in subjects with type 2 diabetes: a multicenter, double blind, randomized placebo-controlled trial of a novel probiotic formulation. BMJ Open Diabetes Res Care (2020) 8(1):e001319. doi: 10.1136/bmjdrc-2020-001319
214. Ling C, Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab (2019) 29:1028–44. doi: 10.1016/j.cmet.2019.03.009
215. Noureldein MH, Bitar S, Youssef N, Azar S, Eid AA. Butyrate modulates diabetes-linked gut dysbiosis: epigenetic and mechanistic modifications. J Mol Endocrinol (2020) 64:29–42. doi: 10.1530/JME-19-0132
216. Khan S, Jena G. Sodium butyrate reduces insulin-resistance, fat accumulation and dyslipidemia in type-2 diabetic rat: A comparative study with metformin. Chem Biol Interact (2016) 254:124–34. doi: 10.1016/j.cbi.2016.06.007
217. Herrema H, Meissner M, van Dijk TH, Brufau G, Boverhof R, Oosterveer MH, et al. Bile salt sequestration induces hepatic de novo lipogenesis through farnesoid X receptor- and liver X receptor alpha-controlled metabolic pathways in mice. Hepatology (2010) 51:806–16. doi: 10.1002/hep.23408
218. Schwartz SL, Lai Y-L, Xu J, Abby SL, Misir S, Jones MR, et al. The effect of colesevelam hydrochloride on insulin sensitivity and secretion in patients with type 2 diabetes: a pilot study. Metab Syndr Relat Disord (2010) 8:179–88. doi: 10.1089/met.2009.0049
219. Hansen M, Sonne DP, Mikkelsen KH, Gluud LL, Vilsbøll T, Knop FK. Bile acid sequestrants for glycemic control in patients with type 2 diabetes: A systematic review with meta-analysis of randomized controlled trials. J Diabetes Complications (2017) 31:918–27. doi: 10.1016/j.jdiacomp.2017.01.011
220. Brufau G, Stellaard F, Prado K, Bloks VW, Jonkers E, Boverhof R, et al. Improved glycemic control with colesevelam treatment in patients with type 2 diabetes is not directly associated with changes in bile acid metabolism. Hepatology (2010) 52:1455–64. doi: 10.1002/hep.23831
221. Kobayashi M, Ikegami H, Fujisawa T, Nojima K, Kawabata Y, Noso S, et al. Prevention and treatment of obesity, insulin resistance, and diabetes by bile acid-binding resin. Diabetes (2007) 56:239–47. doi: 10.2337/db06-0353
222. Einarsson K, Ericsson S, Ewerth S, Reihnér E, Rudling M, Ståhlberg D, et al. Bile acid sequestrants: mechanisms of action on bile acid and cholesterol metabolism. Eur J Clin Pharmacol (1991) 40(Suppl 1):S53–8. doi: 10.1007/BF03216291
223. Shang Q, Saumoy M, Holst JJ, Salen G, Xu G. Colesevelam improves insulin resistance in a diet-induced obesity (F-DIO) rat model by increasing the release of GLP-1. Am J Physiol Gastrointest Liver Physiol (2010) 298(3): G419–24. doi: 10.1152/ajpgi.00362.2009
224. Sato H, Genet C, Strehle A, Thomas C, Lobstein A, Wagner A, et al. Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem Biophys Res Commun (2007) 362:793–8. doi: 10.1016/j.bbrc.2007.06.130
225. Adrian TE, Gariballa S, Parekh KA, Thomas SA, Saadi H, Al Kaabi J, et al. Rectal taurocholate increases L cell and insulin secretion, and decreases blood glucose and food intake in obese type 2 diabetic volunteers. Diabetologia (2012) 55:2343–7. doi: 10.1007/s00125-012-2593-2
226. Pathak P, Xie C, Nichols RG, Ferrell JM, Boehme S, Krausz KW, et al. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology (2018) 68:1574–88. doi: 10.1002/hep.29857
227. Zhang F, Xiao X, Li Y, Wu H, Deng X, Jiang Y, et al. Therapeutic opportunities of GPBAR1 in cholestatic diseases. Front Pharmacol (2021) 12:805269. doi: 10.3389/fphar.2021.805269
228. Villegas R, Shu XO, Yang G, Matthews CE, Li H, Cai H, et al. Energy balance and type 2 diabetes: a report from the Shanghai Women’s Health Study. Nutr Metab Cardiovasc Dis (2009) 19:190–7. doi: 10.1016/j.numecd.2008.06.003
229. Mráz M, Lacinová Z, Kaválková P, Haluzíková D, Trachta P, Drápalová J, et al. Serum concentrations of fibroblast growth factor 19 in patients with obesity and type 2 diabetes mellitus: the influence of acute hyperinsulinemia, very-low calorie diet and PPAR-α agonist treatment. Physiol Res (2011) 60:627–36. doi: 10.33549/physiolres.932099
230. Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology (2004) 145:2594–603. doi: 10.1210/en.2003-1671
231. Martinez de la Escalera L, Kyrou I, Vrbikova J, Hainer V, Sramkova P, Fried M, et al. Impact of gut hormone FGF-19 on type-2 diabetes and mitochondrial recovery in a prospective study of obese diabetic women undergoing bariatric surgery. BMC Med (2017) 15:34. doi: 10.1186/s12916-017-0797-5
232. Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med (2015) 21:159–65. doi: 10.1038/nm.3760
233. Flynn CR, Albaugh VL, Cai S, Cheung-Flynn J, Williams PE, Brucker RM, et al. Bile diversion to the distal small intestine has comparable metabolic benefits to bariatric surgery. Nat Commun (2015) 6:7715. doi: 10.1038/ncomms8715
234. Pournaras DJ, Glicksman C, Vincent RP, Kuganolipava S, Alaghband-Zadeh J, Mahon D, et al. The role of bile after Roux-en-Y gastric bypass in promoting weight loss and improving glycaemic control. Endocrinology (2012) 153:3613–9. doi: 10.1210/en.2011-2145
235. Albaugh VL, Flynn CR, Cai S, Xiao Y, Tamboli RA, Abumrad NN. Early increases in bile acids post Roux-en-Y gastric bypass are driven by insulin-sensitizing, secondary bile acids. J Clin Endocrinol Metab (2015) 100:E1225–33. doi: 10.1210/jc.2015-2467
236. Patti M-E, Houten SM, Bianco AC, Bernier R, Larsen PR, Holst JJ, et al. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obes (Silver Spring) (2009) 17:1671–7. doi: 10.1038/oby.2009.102
237. Kohli R, Kirby M, Setchell KDR, Jha P, Klustaitis K, Woollett LA, et al. Intestinal adaptation after ileal interposition surgery increases bile acid recycling and protects against obesity-related comorbidities. Am J Physiol Gastrointest Liver Physiol (2010) 299:G652–60. doi: 10.1152/ajpgi.00221.2010
238. Kohli R, Setchell KD, Kirby M, Myronovych A, Ryan KK, Ibrahim SH, et al. A surgical model in male obese rats uncovers protective effects of bile acids post-bariatric surgery. Endocrinology (2013) 154:2341–51. doi: 10.1210/en.2012-2069
239. Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature (2014) 509:183–8. doi: 10.1038/nature13135
240. Vincent RP, Omar S, Ghozlan S, Taylor DR, Cross G, Sherwood RA, et al. Higher circulating bile acid concentrations in obese patients with type 2 diabetes. Ann Clin Biochem (2013) 50:360–4. doi: 10.1177/0004563212473450
241. Jahansouz C, Xu H, Hertzel AV, Serrot FJ, Kvalheim N, Cole A, et al. Bile acids increase independently from hypocaloric restriction after bariatric surgery. Ann Surg (2016) 264:1022–8. doi: 10.1097/SLA.0000000000001552
242. McGavigan AK, Garibay D, Henseler ZM, Chen J, Bettaieb A, Haj FG, et al. TGR5 contributes to glucoregulatory improvements after vertical sleeve gastrectomy in mice. Gut (2017) 66:226–34. doi: 10.1136/gutjnl-2015-309871
243. Liu Y, Yang K, Jia Y, Shi J, Tong Z, Fang D, et al. Gut microbiome alterations in high-fat-diet-fed mice are associated with antibiotic tolerance. Nat Microbiol (2021) 6:874–84. doi: 10.1038/s41564-021-00912-0
244. Cussotto S, Delgado I, Anesi A, Dexpert S, Aubert A, Beau C, et al. Tryptophan metabolic pathways are altered in obesity and are associated with systemic inflammation. Front Immunol (2020) 11:557. doi: 10.3389/fimmu.2020.00557
245. Jennis M, Cavanaugh CR, Leo GC, Mabus JR, Lenhard J, Hornby PJ. Microbiota-derived tryptophan indoles increase after gastric bypass surgery and reduce intestinal permeability in vitro and in vivo. Neurogastroenterol Motil (2018) 30(2):e13178. doi: 10.1111/nmo.13178
246. Wang W, Wang X, Liu L, Liu Z, Han T, Sun C, et al. Dietary tryptophan and the risk of obesity and type 2 diabetes: Total effect and mediation effect of sleep duration. Obes (Silver Spring) (2022) 30:515–23. doi: 10.1002/oby.23343
247. Inubushi T, Kamemura N, Oda M, Sakurai J, Nakaya Y, Harada N, et al. L-tryptophan suppresses rise in blood glucose and preserves insulin secretion in type-2 diabetes mellitus rats. J Nutr Sci Vitaminol (Tokyo) (2012) 58:415–22. doi: 10.3177/jnsv.58.415
248. Smith SA, Elliott KR, Pogson CI. Inhibition of hepatic gluconeogenesis by tryptophan metabolites in rats and guinea pigs. Biochem Pharmacol (1979) 28:2145–8. doi: 10.1016/0006-2952(79)90196-5
249. Gartner SN, Aidney F, Klockars A, Prosser C, Carpenter EA, Isgrove K, et al. Intragastric preloads of l-tryptophan reduce ingestive behavior via oxytocinergic neural mechanisms in male mice. Appetite (2018) 125:278–86. doi: 10.1016/j.appet.2018.02.015
250. de Mello VD, Paananen J, Lindström J, Lankinen MA, Shi L, Kuusisto J, et al. Indolepropionic acid and novel lipid metabolites are associated with a lower risk of type 2 diabetes in the Finnish Diabetes Prevention Study. Sci Rep (2017) 7:46337. doi: 10.1038/srep46337
251. Mercer KE, Yeruva L, Pack L, Graham JL, Stanhope KL, Chintapalli SV, et al. Xenometabolite signatures in the UC Davis type 2 diabetes mellitus rat model revealed using a metabolomics platform enriched with microbe-derived metabolites. Am J Physiology-Gastrointestinal Liver Physiol (2020) 319:G157–69. doi: 10.1152/ajpgi.00105.2020
252. Konopelski P, Konop M, Gawrys-Kopczynska M, Podsadni P, Szczepanska A, Ufnal M. Indole-3-propionic acid, a tryptophan-derived bacterial metabolite, reduces weight gain in rats. Nutrients (2019) 11:591. doi: 10.3390/nu11030591
253. Chen T, Zheng X, Ma X, Bao Y, Ni Y, Hu C, et al. Tryptophan predicts the risk for future type 2 diabetes. PloS One (2016) 11:e0162192. doi: 10.1371/journal.pone.0162192
254. Qi Q, Li J, Yu B, Moon J-Y, Chai JC, Merino J, et al. Host and gut microbial tryptophan metabolism and type 2 diabetes: an integrative analysis of host genetics, diet, gut microbiome and circulating metabolites in cohort studies. Gut (2022) 71:1095–105. doi: 10.1136/gutjnl-2021-324053
255. Chen L-M, Bao C-H, Wu Y, Liang S-H, Wang D, Wu L-Y, et al. Tryptophan-kynurenine metabolism: a link between the gut and brain for depression in inflammatory bowel disease. J Neuroinflamm (2021) 18:135. doi: 10.1186/s12974-021-02175-2
256. Di Martino P, Fursy R, Bret L, Sundararaju B, Phillips RS. Indole can act as an extracellular signal to regulate biofilm formation of Escherichia coli and other indole-producing bacteria. Can J Microbiol (2003) 49:443–9. doi: 10.1139/w03-056
257. Shukla A, Srinivasan BP. 16,17-Dihydro-17b-hydroxy isomitraphylline alkaloid as an inhibitor of DPP-IV, and its effect on incretin hormone and β-cell proliferation in diabetic rat. Eur J Pharm Sci (2012) 47:512–9. doi: 10.1016/j.ejps.2012.07.012
258. Azmi MB, Qureshi SA. Methanolic root extract of rauwolfia serpentina benth improves the glycemic, antiatherogenic, and cardioprotective indices in alloxan-induced diabetic mice. Adv Pharmacol Sci (2012) 2012:376429. doi: 10.1155/2012/376429
259. Qureshi SA, Nawaz A, Udani SK, Azmi B. Hypoglycaemic and hypolipidemic activities of rauwolfia serpentina in alloxan-induced diabetic rats. Int J Pharmacol (2009) 5:323–6. doi: 10.3923/ijp.2009.323.326
260. Liu W-C, Chen P-H, Chen L-W. Supplementation of endogenous Ahr ligands reverses insulin resistance and associated inflammation in an insulin-dependent diabetic mouse model. J Nutr Biochem (2020) 83:108384. doi: 10.1016/j.jnutbio.2020.108384
261. Chang H-P, Wang M-L, Chan M-H, Chiu Y-S, Chen Y-H. Antiobesity activities of indole-3-carbinol in high-fat-diet-induced obese mice. Nutrition (2011) 27:463–70. doi: 10.1016/j.nut.2010.09.006
262. Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao C-C, Ardura-Fabregat A, et al. Microglial control of astrocytes in response to microbial metabolites. Nature (2018) 557:724–8. doi: 10.1038/s41586-018-0119-x
263. Shinde R, Hezaveh K, Halaby MJ, Kloetgen A, Chakravarthy A, da Silva Medina T, et al. Apoptotic cell–induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat Immunol (2018) 19:571–82. doi: 10.1038/s41590-018-0107-1
264. Brial F, Alzaid F, Sonomura K, Kamatani Y, Meneyrol K, Le Lay A, et al. The natural metabolite 4-Cresol improves glucose homeostasis and enhances β-cell function. Cell Rep (2020) 30:2306–2320.e5. doi: 10.1016/j.celrep.2020.01.066
265. Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U.S.A. (2009) 106:3698–703. doi: 10.1073/pnas.0812874106
266. Andersen A. Final report on the safety assessment of sodium p-chloro-m-cresol, p-chloro-m-cresol, chlorothymol, mixed cresols, m-cresol, o-cresol, p-cresol, isopropyl cresols, thymol, o-cymen-5-ol, and carvacrol. Int J Toxicol (2006) 25(Suppl 1):29–127. doi: 10.1080/10915810600716653
267. Koh A, Molinaro A, Ståhlman M, Khan MT, Schmidt C, Mannerås-Holm L, et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell (2018) 175:947–961.e17. doi: 10.1016/j.cell.2018.09.055
268. Molinaro A, Bel Lassen P, Henricsson M, Wu H, Adriouch S, Belda E, et al. Imidazole propionate is increased in diabetes and associated with dietary patterns and altered microbial ecology. Nat Commun (2020) 11:5881. doi: 10.1038/s41467-020-19589-w
Keywords: enteroendocrine cells (EEC), gut microbiota, short-chain fatty acids (SCFAs), bile acids (BAs), indoles, glucagon-like peptide-1, signal transduction
Citation: Masse KE and Lu VB (2023) Short-chain fatty acids, secondary bile acids and indoles: gut microbial metabolites with effects on enteroendocrine cell function and their potential as therapies for metabolic disease. Front. Endocrinol. 14:1169624. doi: 10.3389/fendo.2023.1169624
Received: 19 February 2023; Accepted: 05 July 2023;
Published: 25 July 2023.
Edited by:
Hannelouise Kissow, University of Copenhagen, DenmarkReviewed by:
Samuel Trammell, University of Copenhagen, DenmarkCopyright © 2023 Masse and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Van B. Lu, dmx1MjRAdXdvLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.