 Wilson CJ Chung
Wilson CJ Chung Pei-San Tsai
Pei-San Tsai- 1Department of Biological Sciences, Kent State University, Kent, OH, United States
- 2Department of Integrative Physiology, University of Colorado, Boulder, CO, United States
Neurons that secrete gonadotropin-releasing hormone (GnRH) drive vertebrate reproduction. Genetic lesions that disrupt these neurons in humans lead to congenital hypogonadotropic hypogonadism (CHH) and reproductive failure. Studies on CHH have largely focused on the disruption of prenatal GnRH neuronal migration and postnatal GnRH secretory activity. However, recent evidence suggests a need to also focus on how GnRH neurons initiate and maintain their identity during prenatal and postnatal periods. This review will provide a brief overview of what is known about these processes and several gaps in our knowledge, with an emphasis on how disruption of GnRH neuronal identity can lead to CHH phenotypes.
Introduction
Gonadotropin-releasing hormone (GnRH) secreted from GnRH neurons drives sexual maturation and reproduction in all vertebrates. GnRH neurons develop prenatally via a complex multi-step process that establishes their connectivity and differentiation state, ultimately allowing them to synthesize and release GnRH (1). Following their development, the continued maintenance of GnRH neurons is required for puberty and a healthy reproductive lifespan. Much of studies on GnRH neurons have focused on specific stages of their development and the control of their postnatal secretory activity, but significant gaps in our knowledge still exist regarding multiple aspects of GnRH neuronal biology. Efforts to fill these knowledge gaps are further complicated by the heterogeneity of the GnRH system that gives rise to subpopulations of GnRH neurons with seemingly different biological properties and responsiveness to signaling molecules (2).
In past decades, a large number of genes causally linked to GnRH deficiency have been uncovered in humans and rodents (3). Mutations in many of these gene in humans often lead to congenital hypogonadotropic hypogonadism (CHH), a secondary form of hypogonadism characterized by low circulating gonadal steroids, partial or absent puberty, and infertility (4, 5). Although not immediately life-threatening, CHH deleteriously and chronically impacts fertility, mental health, bone growth, and metabolism (6, 7) in ways that severely erode the patients’ quality of life. A large number of CHH-related genes are thought to control (1) prenatal GnRH neuronal migration or (2) postnatal GnRH secretion. However, a closer inspection reveals an additional need to consider genes and epigenetic factors that drive GnRH expression during prenatal and postnatal periods to ensure GnRH neuronal identity. This review aims to more specifically discuss the initiation and maintenance of GnRH neuronal identity during prenatal neurogenesis and postnatal maintenance periods with an emphasis on how the disruption of these processes can lead to reproductive disruption similar to the CHH phenotype.
The developmental stages of the mammalian GnRH system
GnRH neuronal development consists of three major phases: (1) neurogenesis to initiate GnRH neuronal identity, (2) migration to the forebrain, and (2) extension of axons to the median eminence. Studies in mice have provided much insight into these three phases. Using immunocytochemistry, in situ hybridization and birth dating techniques, two landmark mouse studies found the earliest GnRH-expressing neurons on embryonic day (E)11.5 in the medial ventral olfactory placode (OP) at the tip of the nose, from where they migrate into the forebrain (8, 9). The majority (i.e., ~80%) of mouse GnRH neurons undergo their last division between E9.5 and E10.5 (8). Taken together, the period of GnRH neuron neurogenesis has been defined to occur between E9.5 - E11.5. Following this neurogenesis phase, GnRH neurons enter a migratory phase in which they follow the olfactory, vomeronasal and terminal nerves through the cribriform plate to enter the preoptic area and hypothalamus. After the cessation of migration, GnRH neurons extend and target their axons to the external zone of the median eminence for GnRH release (10). In mice, the formation of the GnRH system is largely complete around E16.5 (9, 11–17). Postnatally, most GnRH neuronal cell bodies reside near the anterior tip of the third ventricle called organum vasculosum lamina terminalis in the preoptic area (1, 18–20). Of comparative importance, the GnRH system develops similarly in humans and non-human primates, but with additional GnRH neurons found in the tuberal hypothalamus (21–23).
CHH and Kallmann syndrome
Human and mouse studies have significantly advanced our understanding of molecular control of GnRH neuronal development and function in health and disease. Many studies have focused on understanding the pathogenesis of Kallmann syndrome (KS), a subset of CHH caused by heritable GnRH deficiency. KS has all the clinical presentations of CHH, including absent puberty and infertility, but with an additional phenotype of anosmia (5, 24). KS is believed to be caused primarily by developmental defects in GnRH neurons, whereas normosmic CHH may have additional causes involving pituitary or kisspeptin system defects. A landmark study conducted in a human fetus with KS reported GnRH neuronal migration was impaired when the gene ANOS1 was mutated. In this case, GnRH neurons were arrested anterior to the cribriform plate, just outside the brain, rather than arriving at their normal destinations in the preoptic and hypothalamic regions (23). Later studies in humans and rodents support that defects in GnRH neuronal migration is a major cause of CHH (5, 24). However, KS, as well as CHH, are highly heterogeneous in their causes and phenotypes. Increasing evidence suggests KS and CHH gene mutations may also disrupt the neurogenesis of GnRH neurons during development (1, 12, 25–36). Of note, the disruption of GnRH neuron neurogenesis has, thus far, received much less attention than GnRH neuronal migration in the context of KS or CHH.
Neurogenesis of GnRH neurons
The anatomical location of nascent GnRH neurons in various animal models suggests GnRH progenitor cells are likely derived in the embryonic medial-ventral OP. However, recent cell lineage tracing studies show a more complicated picture. The OP is an ectodermally derived structure that gives rise to both non-sensory respiratory epithelium and sensory olfactory epithelium. The olfactory epithelium eventually develops into the main olfactory and vomeronasal systems (37, 38). Ablation studies in rat and chick embryos suggest a small percentage of GnRH neurons may have come from the respiratory epithelium, whereas the majority of GnRH neurons are derived from the olfactory epithelium (39–44). Studies in mice similarly show that GnRH progenitor cells are not exclusively localized in the olfactory epithelium, but can also be detected in the respiratory epithelium by the expression of a transcription factor activator protein 2α (TFAP2α) (45). In the same study, a number of GnRH neurons are found to be positive for nestin, another marker for the respiratory epithelium. Lastly, some GnRH neurons do not express olfactory epithelium markers, such as Mash-1, Math4C/neurogenin1, and NeuroD (45). Taken together, these data suggest that both the respiratory and olfactory epithelium contribute to the GnRH progenitor pool in the OP.
In several vertebrate species, the neural crest has been implicated as a possible origin of GnRH progenitor cells (46–48). The anterior neural crest, which arises from the lateral edge of the neural plate, shares a border with the region that eventually becomes the OP. Neural crest cells have been shown to migrate into the presumptive OP, and therefore are likely to have contributed to cell populations in the developing OP (49). This notion is supported by studies in zebrafish demonstrating a fraction of neural crest cells labeled before their migration are later found to express GnRH mRNA and peptide (50–52). In mice, a small proportion (i.e., ~30%) of GnRH neurons found in the OP also have a genetic lineage similar to Wnt1- and/or Islet1/2-expressing progenitor cells that characteristically arise in the neural crest (47, 53), but the remaining 70% GnRH neurons are thought to have a placodal ectodermal origin instead of a neural crest origin (2). Taken together, GnRH neurons are currently thought to have a dual origin of both the neural crest and OP (2). This could explain, in part, the heterogeneity of GnRH neurons and why many gene knockout studies are only able to ablate a fraction of GnRH neurons (54).
Recent advances in genetic screening have identified many causal genes for CHH. There is an extensive list of up to 54 CHH causal genes according to one recent review (3). Of these, eight genes (Fgf8, Fgfr1, Fgf17, Dusp6, Flrt3, Il17rd, Klb and Spry4) are associated with fibroblast growth factor (Fgf) signaling either as a ligand, receptor, co-receptor, or synexpression gene. All genes except Spry4 and Klb are thought to be involved in the neurogenesis of GnRH neurons (3, 28, 55). The current consensus is that Fgf8, a ligand in the Fgf signaling family (56), acts as an upstream Fgf signaling factor to control the neurogenesis of GnRH neurons. Studies in humans and rodents have cemented the concept that Fgf signaling is critical for the neurogenesis of GnRH progenitor cell birth and proliferation (1, 26, 28, 32). As described above, some CHH/KS patients harbor mutations in the Fgf8 or Fgfr1 gene (28, 57). In fact, homozygous Fgf8 hypomorphic mice exhibited a complete loss of GnRH neurons as early as E11.5, at the time of their emergence, suggesting GnRH neurons did not undergo normal neurogenesis when Fgf8 was deficient (12, 28). Further, Fgfr1 hypomorphic newborn mice showed a similar reduction in the GnRH system (i.e., ~90%) (12), suggesting Fgf8 may act through one of its cognate receptors, Fgfr1, to support neurogenesis of GnRH neurons. Heterozygous Fgf8 hypomorphic mice suffered a 50% reduction in GnRH neurons but did not exhibit any migratory defect (12). Taken together, reduced Fgf8/Fgfr1 signaling caused failed neurogenesis of GnRH neuron rather than GnRH neuron migration. Later studies reported that the reduced GnRH neuronal system in these hypomorphic mice caused abnormal reproductive function (58). These results not only provided a fundamental explanation for the reproductive defects found in KS/CHH patients who harbor Fgf8/Fgfr1 mutations, they also suggested the initial neurogenesis of GnRH neurons was highly dependent on Fgf8 expression level. The four other CHH causal genes associated with Fgf signaling and thought to influence neurogenesis (Fgf17, Dusp6, Flrt3, and Il17rd) are classified as Fgf8 synexpression genes that likely modulate Fgf8 activity during development (55).
Currently, it is unknown whether Fgf8 deficiency leads to the elimination of the GnRH progenitor cells by abrogating cell proliferation or cell survival. Circumstantial evidence favors the second possibility given that increased apoptosis has been reported for the E10.5 medial-ventral OP when Fgf8 is deficient (1). Because cell lineage studies confirmed that Fgf8 mRNA expression is primarily localized in the respiratory epithelium (59), we posit that Fgf8-expressing respiratory epithelial cells secrete Fgf8 to provide trophic support for the survival of GnRH progenitors in the OP. In addition, fate specification studies in human pluripotent stem cells show that Fgf8 is also required to program these stem cells towards GnRH neuronal fate (60, 61). That said, it is also possible that newly emerged GnRH neurons may be vulnerable to cell death and require Fgf signaling for survival. An early report indicated that in mice, the number of GnRH neurons peaked to 2000 at E12.75 but were “pruned” by almost 50% around the time of birth (62). GnRH neuron-specific deletion of neuropilin 1 (Nrp1), the cognate receptor for the repulsive guidance cue semaphorin 3, resulted in excess prenatal and postnatal GnRH neurons, suggesting a defect in this pruning (63). Of interest, Fgfr1 has been shown to interact with Nrp1 (64) and may be involved in this process.
Overall, the literature suggests Fgf8/Fgfr1 signaling is critical for the survival of GnRH progenitor cells and their differentiation into GnRH neurons. However, a possible role of Fgf signaling in the survival of newly emerged GnRH neurons cannot be ruled out.
The maintenance of postnatal GnRH neurons
The prenatal development of GnRH neurons is a well-studied process with decades of insights (65). In contrast, the postnatal maintenance of GnRH neurons remains poorly understood despite its physiological significance. Understanding the maintenance of the postnatal GnRH system also imparts significant translational value because postnatal deficits in the nervous system typically occur after the critical developmental period and can theoretically be reversed with proper stimuli. This plasticity, if better understood, could be harnessed for the treatment of fertility issues in humans and other animals.
For this section, we will endeavor to address two questions. First, can a disease state such as CHH lead to the postnatal loss of fully differentiated GnRH neurons? Second, can the postnatal GnRH neuronal loss be reversed by beneficial cues? In the context of this discussion, we broadly define two mechanisms of postnatal GnRH neuronal loss as either neuronal death or the loss of GnRH neuronal identity (de-differentiation). Since the only unifying marker for differentiated GnRH neurons is GnRH itself, the long-term loss of GnRH production is viewed as neuronal de-differentiation (66).
We first ask the question if a disease state can lead to the postnatal loss of differentiated GnRH neurons without impacting their development. One interesting observation from rodent and human studies is that the number of postnatal GnRH neurons in the hypothalamic/preoptic area of healthy individuals remains largely unchanged within a broad age range examined (22, 33, 67, 68), leading to the assumption that once a mature GnRH system is established, it continues to persist during the animal’s lifespan. However, this assumption may not be true. Several lines of evidence suggest the postnatal GnRH system is indeed vulnerable to disruption and can be lost under certain disease states. The first evidence came from a mouse model of Huntington’s disease, which was born with a normal set of GnRH neurons but lost 90% of them by 12 weeks of age (69). The second evidence came from a transgenic mouse model expressing a dominant negative Fgf receptor (dnFGFR) in GnRH neurons. The expression of this transgene interferes with the function of two endogenous Fgf receptors (Fgfr1 and Fgfr3) specifically in GnRH neurons (35), resulting in GnRH neurons with reduced responsiveness to Fgf signaling. The dnFGFR mice had normal GnRH neuron number at birth but underwent a progressive GnRH neuronal loss as they aged, losing up to 70% of their GnRH neurons by 550 days of age (33). Parallel to their postnatal GnRH neuronal loss, dnFGFR mice experienced accelerated reproductive senescence (35). In a similar vein, mouse models of Down syndrome (70) and Fgfr3 deficiency (26) were both born with normal number of GnRH neurons but exhibited a significant loss of postnatal GnRH neurons in adulthood. It is currently unclear if GnRH neuronal loss from these studies resulted from cell death or de-differentiation.
Although postnatal GnRH neurons have previously been suggested to undergo apoptotic death following exposure to toxins (71, 72), death as a cause of postnatal GnRH neuronal loss has never been documented in the whole organism. More indirect evidence suggests postnatal GnRH neurons may, more frequently, undergo de-differentiation and completely lose their ability to produce GnRH. Evidence for this notion again came from studies on dnFGFR mice (33, 35). The reduced GnRH neurons and lower fertility in dnFGFR mice could, later in life, be restored back to normal by cohabitation with an opposite-sex (OS) cage mate, suggesting their GnRH neurons were not dead but instead temporarily de-differentiated and had the potential to return to normal activity. Supporting this possibility, lineage tracing studies showed that the conditional deletion of a transcription factor Vax1 (ventral anterior homeobox 1) in GnRH neurons rendered GnRH neurons unable to produce GnRH without impacting their survival (73). In addition, approximately 10% of CHH patients experienced spontaneous reversal to gain fertility after the termination of hormone replacement therapy (74–79). These patients included those harboring mutations on genes such as Fgfr1, Chd7, Prokr2 originally thought to irreversibly disrupt GnRH neuronal development and reduce postnatal GnRH neuronal population. These findings collectively suggest some GnRH neurons in these patients may be originally dormant and de-differentiated due to genetic lesions, but later became reactivated in life to drive the reproductive axis.
If de-differentiated GnRH neurons could be recovered, an important question would be: what type of signals can stimulate this recovery in the postnatal GnRH system? Robust postnatal plasticity in the GnRH system has been observed during puberty in multiple forms (80), including but not limited to the expression of receptor for a major GnRH stimulator, kisspeptin (81), firing properties (82, 83), and morphology (84–86). In this respect, the postnatal GnRH system remains plastic and may have an inherent resilience to recover from deficits generated during development and from a disease state.
Although the nature of specific cues that could restore a de-differentiated postnatal GnRH system is poorly understood, some insight could be gained from the dnFGFR mouse model (33, 35). Because the declining GnRH system and reproductive function were restored back to normal in dnFGFR mice housed with an OS, but not same sex (SS), partner (33), an intriguing hypothesis is that the postnatal reproductive brain, like the cognitive brain, is highly plastic and responsive to environmental cues, such as olfaction It is possible that OS housing in mice represents one of many forms of environmental enrichment that may alter signaling in the brain to restore the GnRH system and fertility. Indeed, the GnRH system of several vertebrates can respond acutely to environmental stimuli with a strong socio-sexual context (87–91). For example, cichlid fish showed a rapid increase in GnRH expression in response to improved social status (90), and breeding stimuli increased the number of GnRH neurons in amphibians (87) and birds (89, 91). In the musk shrew, the exposure of prepubertal females to males increased the number of GnRH neurons (88). In mice, pheromones serve as important signals that may accelerate (92, 93) or inhibit (94, 95) pubertal onset. The secretion of luteinizing hormone (LH) and testosterone (T) in male mice can also be triggered acutely by pheromones from female mice (96–101). Although humans do not exhibit pheromonal sensitivity comparable to other mammals, LH and T secretion in humans can also be acutely activated by OS interactions or sexually arousing visual cues (102–104). Perhaps OS exposure generates a combination of sensory and social cues that work in concert to upregulate protective signaling molecules in the brain capable of reversing the de-differentiated state of GnRH neurons. Identifying these cues will provide insights into the general nature of environmental signals to adopt or avoid for fertility improvement. These insights may broadly benefit fertility in individuals with a compromised GnRH system resulting from postnatal GnRH neuronal de-differentiation. A model summarizing the involvement of Fgf signaling and OS housing in the development and differentiation of GnRH neurons is presented in Figure 1.
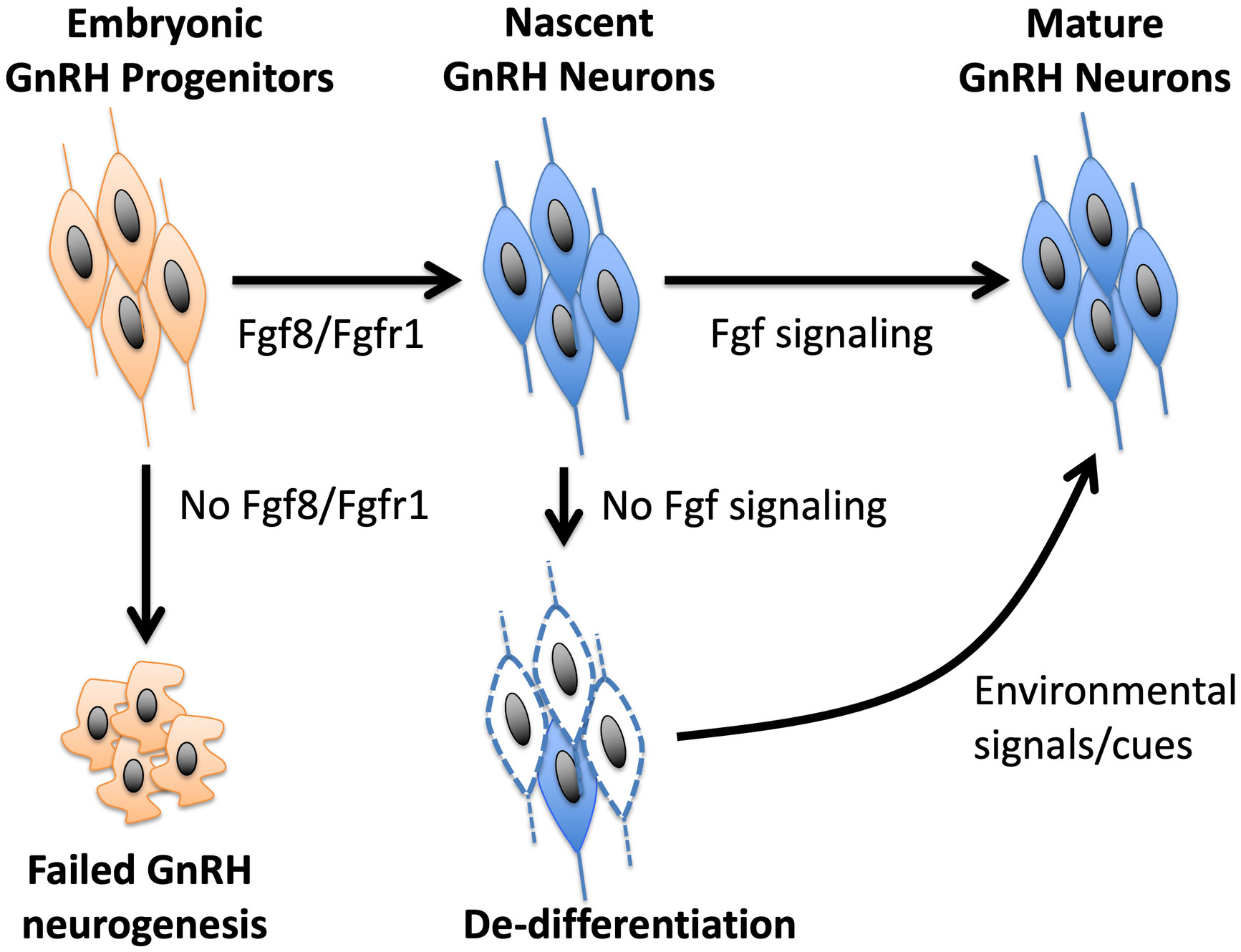
Figure 1 A model of how Fgf signaling controls the neurogenesis and differentiation of GnRH neurons. Fgf8 signaling through Fgfr1 is critical for GnRH neurogenesis from GnRH progenitors. Once nascent GnRH neurons are formed, continued exposure to Fgf signaling is required to maintain their differentiated state. In the absence of Fgf signaling, GnRH neurons fail to express GnRH and become de-differentiated. However, this de-differentiated state can be reversed by positive environmental signals, such as OS housing, that may upregulate neuroprotective molecules in the brain.
Epigenetic control of prenatal and postnatal GnRH neurons
Increasing evidence suggests GnRH gene expression, thus GnRH neuronal identity, is controlled by epigenetic mechanisms during both prenatal and postnatal periods (105–107). Importantly, environmental stimuli can epigenetically alter the expression of genes critical for cellular processes, such as cell proliferation and fate specification, to affect long-range transcriptional interactions with enhancer or silencer regulatory sequences (108). In general, definitions that describe epigenetics range from those that include heritable gene function to those stating that epigenetic changes are molecular events that remodel chromatin without altering the primary genetic code (109). To initiate and maintain the identity of GnRH neurons, the GnRH promoter must be active. The GnRH promoter has been shown to undergo major chromatin and methylation changes that resulted in the loss of their ability to produce GnRH (110). For example, GnRH promoter elements displayed higher sensitivity to DNAseI treatment, indicative of more accessible chromatin, in mature GT1-7 GnRH neuronal cell line compared to the immature GN11 GnRH neuronal cell line (110). These GnRH regulatory elements also displayed RNA polymerase II enrichment as well as the active histone marker, H3K4me3, indicating that maturation of GnRH neurons relied on chromatin changes on GnRH regulatory elements (110). Indeed, one of the earliest studies of epigenetic regulation of GnRH neuronal identity was found in Rhesus macaque monkeys (111). GnRH gene demethylation was followed by a rise in GnRH mRNA expression, indicating that the epigenetic events were critical for GnRH promoter activation. Interestingly, a similar epigenetic mechanism drove pubertal onset (111). Studies showed that GnRH gene demethylation leading to GnRH expression may have been a Ten-Eleven Translocation (TET)-dependent process. The TET family consists of three isoforms (Tet1, Tet2, Tet3) which demethylate CpG dinucleotides to 5-hydroxymethyl (5hmC), 5-formyl (5fC), and 5-carboxyl (5caC) (112). Loss of TET proteins decreased overall gene expression and demethylation at promoters of active genes (113, 114). In 2016, Kurian et al. found that Tet2 overexpression could increase GnRH mRNA by altering H3K4me3 abundance associated with the GnRH promoter (115). Moreover, GnRH-specific Tet2 knockout mice displayed lower plasma LH levels and had lower fecundity in adult males, indicating an involvement of epigenetic factors in GnRH neuronal function (115). Lastly, in polycystic ovarian syndrome (PCOS) patients with GnRH neuron hyperactivity, Tet1 was hypomethylated (over-expressed), further suggesting the importance of DNA methylation in controlling GnRH promoter activity and possibly maintaining GnRH neuronal identity (116).
Importantly, effects of environmental stimuli on the epigenetic may not only disrupt GnRH promoter activity but may also affect the transcriptional control of genes (i.e., Fgf8) required for GnRH neuron neurogenesis. For example, recent studies showed that in the olfactory region, Fgf8 transcription during the neurogenesis phase of GnRH neurons is under DNA methyltransferases (DNMT) and TET1 control (31, 117, 118). Moreover, there is evidence supporting that TET function is not limited to its enzymatic activity to catalyze CpG demethylation (112, 119), but also its ability to recruit EZH2, a polycomb repression complex 2 protein, which catalyzes H3K27 methylation. Indeed, elevated H3K27me3 association with the mouse OP Fgf8 promoter coincided with EZH2 enrichment (31). These data led to the hypothesis that the presence of TET1 on the Fgf8 promoter not only catalyzed its demethylation, but also functioned as a molecular anchor for the recruitment of epigenetic factors to facilitate H3K27me3 marks and repress gene transcription (112, 119).
Lastly, epigenetic control has been reported in postnatal peripubertal female rats exposed to DNMT inhibitor, azacitidine, which exhibited smaller ovaries and underwent delayed puberty (120, 121), indicating that abnormal demethylation disrupts reproductive health (120). Similarly, recent studies found that microRNA-mediated epigenetic changes contribute to the timing of pubertal onset (122). These results imply that epigenetic factors not only critically control prenatal GnRH neuron development, they could also mediate the maintenance of GnRH neuronal identity during postnatal GnRH neuron maturation and function.
Conclusions and perspectives
Recent advances in whole genome and exome sequencing have facilitated the identification of genes associated with CHH and KS. These genes, when validated in animal models, provide excellent mechanistic insights into the development and function of the GnRH system. That said, CHH and KS are highly heterogeneous disorders with diverse etiologies beyond the frequently described disruption in GnRH neuronal migration or secretion. Disrupted neurogenesis of GnRH neurons is one of the less described etiologies. In addition, even genetically identical probands with the same mutations in CHH causal genes could have very different clinical presentations (34), suggesting an involvement of environmental influence and epigenetic control. As such, genetic and epigenetic factors that drive GnRH neurons to initiate and maintain their identity is an underexplored area and deserve greater attention in the context of CHH and KS.
Author contributions
WC contributed to the research and writing of the developmental and epigenetic sections, and P-ST contributed to the research and writing of the postnatal, introductory, and conclusion sections. Both authors contributed equally to the integration of the ideas. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants HD058044 to WC and HD083260 to P-ST.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tsai PS, Brooks LR, Rochester JR, Kavanaugh SI, Chung WC. Fibroblast growth factor signaling in the developing neuroendocrine hypothalamus. Front Neuroendocrinol (2011) 32(1):95–107. doi: 10.1016/j.yfrne.2010.11.002
2. Shan Y, Wray S. Hidden ‘pit’falls in deciphering the gonadotropin releasing hormone neuroendocrine cell lineage. J Neuroendocrinol (2021) 33(11):e13039. doi: 10.1111/jne.13039
3. Oleari R, Massa V, Cariboni A, Lettieri A. The differential roles for neurodevelopmental and neuroendocrine genes in shaping GnRH neuron physiology and deficiency. Int J Mol Sci (2021) 22(17). doi: 10.3390/ijms22179425
4. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dode C, Dunkel L, et al. Expert consensus document: European consensus statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol (2015) 11(9):547–64. doi: 10.1038/nrendo.2015.112
5. Young J, Xu C, Papadakis GE, Acierno JS, Maione L, Hietamaki J, et al. Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev (2019) 40(2):669–710. doi: 10.1210/er.2018-00116
6. Dwyer AA, Smith N, Quinton R. Psychological aspects of congenital hypogonadotropic hypogonadism. Front Endocrinol (Lausanne) (2019) 10:353. doi: 10.3389/fendo.2019.00353
7. Garrido Oyarzun MF, Castelo-Branco C. Sexuality and quality of life in congenital hypogonadisms. Gynecol Endocrinol (2016) 32(12):947–50. doi: 10.1080/09513590.2016.1241229
8. Wray S, Grant P, Gainer H. Evidence that cells expressing luteinizing hormone-releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. Proc Natl Acad Sci U.S.A. (1989) 86(20):8132–6. doi: 10.1073/pnas.86.20.8132
9. Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature (1989) 338(6211):161–4. doi: 10.1038/338161a0
10. Rogers MC, Silverman AJ, Gibson MJ. Gonadotropin-releasing hormone axons target the median eminence: in vitro evidence for diffusible chemoattractive signals from the mediobasal hypothalamus. Endocrinology (1997) 138(9):3956–66. doi: 10.1210/endo.138.9.5416
11. Wray S, Nieburgs A, Elkabes S. Spatiotemporal cell expression of luteinizing hormone-releasing hormone in the prenatal mouse: evidence for an embryonic origin in the olfactory placode. Brain Res Dev Brain Res (1989) 46(2):309–18. doi: 10.1016/0165-3806(89)90295-2
12. Chung WC, Moyle SS, Tsai PS. Fibroblast growth factor 8 signaling through fibroblast growth factor receptor 1 is required for the emergence of gonadotropin-releasing hormone neurons. Endocrinology (2008) 149(10):4997–5003. doi: 10.1210/en.2007-1634
13. Miraoui H, Dwyer A, Pitteloud N. Role of fibroblast growth factor (FGF) signaling in the neuroendocrine control of human reproduction. Mol Cell Endocrinol (2011) 346(1-2):37–43. doi: 10.1016/j.mce.2011.05.042
14. Fueshko S, Wray S. LHRH cells migrate on peripherin fibers in embryonic olfactory explant cultures: an in vitro model for neurophilic neuronal migration. Dev Biol (1994) 166(1):331–48. doi: 10.1006/dbio.1994.1319
15. Taroc EZM, Lin JM, Tulloch AJ, Jaworski A, Forni PE. GnRH-1 neural migration from the nose to the brain is independent from Slit2, Robo3 and NELL2 signaling. Front Cell Neurosci (2019) 13:70. doi: 10.3389/fncel.2019.00070
16. Taroc EZM, Naik AS, Lin JM, Peterson NB, Keefe DL Jr., Genis E, et al. Gli3 regulates vomeronasal neurogenesis, olfactory ensheathing cell formation, and GnRH-1 neuronal migration. J Neurosci: Off J Soc Neurosci (2020) 40(2):311–26. doi: 10.1523/JNEUROSCI.1977-19.2019
17. Taroc EZM, Prasad A, Lin JM, Forni PE. The terminal nerve plays a prominent role in GnRH-1 neuronal migration independent from proper olfactory and vomeronasal connections to the olfactory bulbs. Biol Open (2017) 6(10):1552–68. doi: 10.1242/bio.029074
18. Stevenson EL, Corella KM, Chung WC. Ontogenesis of gonadotropin-releasing hormone neurons: a model for hypothalamic neuroendocrine cell development. Front Endocrinol (2013) 4:89. doi: 10.3389/fendo.2013.00089
19. King JC, Anthony EL. LHRH neurons and their projections in humans and other mammals: species comparisons. Peptides (1984) 5 Suppl 1:195–207. doi: 10.1016/0196-9781(84)90277-8
20. King JC, Tobet SA, Snavely FL, Arimura AA. LHRH immunopositive cells and their projections to the median eminence and organum vasculosum of the lamina terminalis. J Comp Neurol (1982) 209(3):287–300. doi: 10.1002/cne.902090307
21. Quanbeck C, Sherwood NM, Millar RP, Terasawa E. Two populations of luteinizing hormone-releasing hormone neurons in the forebrain of the rhesus macaque during embryonic development. J Comp Neurol (1997) 380(3):293–309. doi: 10.1002/(SICI)1096-9861(19970414)380:3<293::AID-CNE1>3.0.CO;2-Y
22. Casoni F, Malone SA, Belle M, Luzzati F, Collier F, Allet C, et al. Development of the neurons controlling fertility in humans: new insights from 3D imaging and transparent fetal brains. Development (2016) 143(21):3969–81. doi: 10.1242/dev.139444
23. Schwanzel-Fukuda M, Bick D, Pfaff DW. Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Mol Brain Res (1989) 6(4):311–26. doi: 10.1016/0169-328X(89)90076-4
24. Cho HJ, Shan Y, Whittington NC, Wray S. Nasal placode development, GnRH neuronal migration and kallmann syndrome. Front Cell Dev Biol (2019) 7:121. doi: 10.3389/fcell.2019.00121
25. Chung WC, Linscott ML, Rodriguez KM, Stewart CE. The regulation and function of fibroblast growth factor 8 and its function during gonadotropin-releasing hormone neuron development. Front Endocrinol (2016) 7:114. doi: 10.3389/fendo.2016.00114
26. Chung WC, Matthews TA, Tata BK, Tsai PS. Compound deficiencies in multiple fibroblast growth factor signalling components differentially impact the murine gonadotrophin-releasing hormone system. J Neuroendocrinol (2010) 22(8):944–50. doi: 10.1111/j.1365-2826.2010.02024.x
27. Raivio T, Cole L, Hughes V, Dwyer A, et al. FGF8 is a key ligand for FGFR1 in GnRH ontogeny: Evidence from the human disease model of idiopathic hypogonadotropic hypogonadism (IHH). 89th Annual Meeting of the Endocrine Society, Toronto ON. (2007).
28. Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest (2008) 118(8):2822–31. doi: 10.1172/JCI34538
29. Forni PE, Bharti K, Flannery EM, Shimogori T, Wray S. The indirect role of fibroblast growth factor-8 in defining neurogenic niches of the olfactory/GnRH systems. J Neurosci: Off J Soc Neurosci (2013) 33(50):19620–34. doi: 10.1523/JNEUROSCI.3238-13.2013
30. Kawauchi S, Young C, Calof A. (2007). Requirement for Fgf8 in generation of GnRH-expressing hypothalamic neurons during mouse development, in: Annual Meeting, Society for Neuroscience (2007) Vol. 33.
31. Linscott ML, Chung WCJ. TET1 regulates fibroblast growth factor 8 transcription in gonadotropin releasing hormone neurons. PloS One (2019) 14(7):e0220530. doi: 10.1371/journal.pone.0220530
32. Chung WC, Tsai PS. Role of fibroblast growth factor signaling in gonadotropin-releasing hormone neuronal system development. Front Horm Res (2010) 39:37–50. doi: 10.1159/000312692
33. Rochester JR, Chung WC, Hayes TB, Tsai PS. Opposite-sex housing reactivates the declining GnRH system in aged transgenic male mice with FGF signaling deficiency. Am J Physiol Endocrinol Metab (2012) 303(12):E1428–1439. doi: 10.1152/ajpendo.00289.2012
34. Tsai PS, Gill JC. Mechanisms of disease: insights into X-linked and autosomal-dominant kallmann syndrome. Nat Clin Pract Endocrinol Metab (2006) 2(3):160–71. doi: 10.1038/ncpendmet0119
35. Tsai PS, Moenter SM, Postigo HR, El Majdoubi M, Pak TR, Gill JC, et al. Targeted expression of a dominant-negative fibroblast growth factor (FGF) receptor in gonadotropin-releasing hormone (GnRH) neurons reduces FGF responsiveness and the size of GnRH neuronal population. Mol Endocrinol (2005) 19(1):225–36. doi: 10.1210/me.2004-0330
36. Sabado V, Barraud P, Baker CV, Streit A. Specification of GnRH-1 neurons by antagonistic FGF and retinoic acid signaling. Dev Biol (2012) 362(2):254–62. doi: 10.1016/j.ydbio.2011.12.016
37. Halpern M. The organization and function of the vomeronasal system. Annu Rev Neurosci (1987) 10:325–62. doi: 10.1146/annurev.ne.10.030187.001545
38. Farbman AI, Buchholz JA. Growth of olfactory epithelial tissue in vitro: lectin staining of axons. Microscopy Res Technique (1992) 23(2):173–80. doi: 10.1002/jemt.1070230207
39. Daikoku S, Koide I. Spatiotemporal appearance of developing LHRH neurons in the rat brain. J Comp Neurol (1998) 393(1):34–47. doi: 10.1002/(SICI)1096-9861(19980330)393:1<34::AID-CNE4>3.0.CO;2-R
40. Daikoku-Ishido H, Okamura Y, Yanaihara N, Daikoku S. Development of the hypothalamic luteinizing hormone-releasing hormone-containing neuron system in the rat: in vivo and in transplantation studies. Dev Biol (1990) 140(2):374–87. doi: 10.1016/0012-1606(90)90087-Y
41. el Amraoui A, Dubois PM. Experimental evidence for an early commitment of gonadotropin-releasing hormone neurons, with special regard to their origin from the ectoderm of nasal cavity presumptive territory. Neuroendocrinology (1993) 57(6):991–1002. doi: 10.1159/000126490
42. Akutsu S, Takada M, Ohki-Hamazaki H, Murakami S, Arai Y. Origin of luteinizing hormone-releasing hormone (LHRH) neurons in the chick embryo: effect of the olfactory placode ablation. Neurosci Lett (1992) 142(2):241–4. doi: 10.1016/0304-3940(92)90382-H
43. Arai Y, Murakami S, Seki T. Removal of olfactory placode prevents the development of LHRH neurons in the forebrain of the chick embryo: possible interaction between migrating LHRH neurons and highly polysialylated form of neural cell adhesion molecule (NCAM-h). Acta Biol Hung (1994) 45(2-4):155–68.
44. Hilal EM, Chen JH, Silverman AJ. Joint migration of gonadotropin-releasing hormone (GnRH) and neuropeptide y (NPY) neurons from olfactory placode to central nervous system. J Neurobiol (1996) 31(4):487–502. doi: 10.1002/(SICI)1097-4695(199612)31:4<487::AID-NEU8>3.0.CO;2-5
45. Kramer PR, Wray S. Midline nasal tissue influences nestin expression in nasal-placode-derived luteinizing hormone-releasing hormone neurons during development. Dev Biol (2000) 227(2):343–57. doi: 10.1006/dbio.2000.9896
46. Whitlock KE, Westerfield M. The olfactory placodes of the zebrafish form by convergence of cellular fields at the edge of the neural plate. Development (2000) 127(17):3645–53. doi: 10.1242/dev.127.17.3645
47. Forni PE, Taylor-Burds C, Melvin VS, Williams T, Wray S. Neural crest and ectodermal cells intermix in the nasal placode to give rise to GnRH-1 neurons, sensory neurons, and olfactory ensheathing cells. J Neurosci: Off J Soc Neurosci (2011) 31(18):6915–27. doi: 10.1523/JNEUROSCI.6087-10.2011
48. Katoh H, Shibata S, Fukuda K, Sato M, Satoh E, Nagoshi N, et al. The dual origin of the peripheral olfactory system: placode and neural crest. Mol Brain (2011) 4:34. doi: 10.1186/1756-6606-4-34
49. Schilling TF, Kimmel CB. Segment and cell type lineage restrictions during pharyngeal arch development in the zebrafish embryo. Development (1994) 120(3):483–94. doi: 10.1242/dev.120.3.483
50. Raible DW, Eisen JS. Restriction of neural crest cell fate in the trunk of the embryonic zebrafish. Development (1994) 120(3):495–503. doi: 10.1242/dev.120.3.495
51. Raible DW, Wood A, Hodsdon W, Henion PD, Weston JA, Eisen JS. Segregation and early dispersal of neural crest cells in the embryonic zebrafish. Dev Dyn (1992) 195(1):29–42. doi: 10.1002/aja.1001950104
52. Whitlock KE, Wolf CD, Boyce ML. Gonadotropin-releasing hormone (GnRH) cells arise from cranial neural crest and adenohypophyseal regions of the neural plate in the zebrafish, danio rerio. Dev Biol (2003) 257(1):140–52. doi: 10.1016/S0012-1606(03)00039-3
53. Forni PE, Wray S. Neural crest and olfactory system: new prospective. Mol Neurobiol (2012) 46(2):349–60. doi: 10.1007/s12035-012-8286-5
54. Duittoz AH, Forni PE, Giacobini P, Golan M, Mollard P, Negron AL, et al. Development of the gonadotropin-releasing hormone system. J Neuroendocrinol (2022) 34(5):e13087. doi: 10.1111/jne.13087
55. Miraoui H, Dwyer AA, Sykiotis GP, Plummer L, Chung W, Feng B, et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am J Hum Genet (2013) 92(5):725–43. doi: 10.1016/j.ajhg.2013.04.008
56. Dono R. Fibroblast growth factors as regulators of central nervous system development and function. Am J Physiol Regul Integr Comp Physiol (2003) 284(4):R867–881. doi: 10.1152/ajpregu.00533.2002
57. Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant kallmann syndrome. Nat Genet (2003) 33(4):463–5. doi: 10.1038/ng1122
58. Tata BK, Chung WC, Brooks LR, Kavanaugh SI, Tsai PS. Fibroblast growth factor signaling deficiencies impact female reproduction and kisspeptin neurons in mice. Biol Reprod (2012) 86(4):119. doi: 10.1095/biolreprod.111.095992
59. Bachler M, Neubuser A. Expression of members of the fgf family and their receptors during midfacial development. Mech Dev (2001) 100(2):313–6. doi: 10.1016/S0925-4773(00)00518-9
60. Lund C, Pulli K, Yellapragada V, Giacobini P, Lundin K, Vuoristo S, et al. Development of gonadotropin-releasing hormone-secreting neurons from human pluripotent stem cells. Stem Cell Rep (2016) 7(2):149–57. doi: 10.1016/j.stemcr.2016.06.007
61. Keen KL, Petersen AJ, Figueroa AG, Fordyce BI, Shin J, Yadav R, et al. Physiological characterization and transcriptomic properties of GnRH neurons derived from human stem cells. Endocrinology (2021) 162(9). doi: 10.1210/endocr/bqab120
62. Wu TJ, Gibson MJ, Rogers MC, Silverman AJ. New observations on the development of the gonadotropin-releasing hormone system in the mouse. J Neurobiol (1997) 33(7):983–98. doi: 10.1002/(SICI)1097-4695(199712)33:7<983::AID-NEU9>3.0.CO;2-4
63. Vanacker C, Trova S, Shruti S, Casoni F, Messina A, Croizier S, et al. Neuropilin-1 expression in GnRH neurons regulates prepubertal weight gain and sexual attraction. EMBO J (2020) 39(19):e104633. doi: 10.15252/embj.2020104633
64. Abdullah A, Akhand SS, Paez JSP, Brown W, Pan L, Libring S, et al. Epigenetic targeting of neuropilin-1 prevents bypass signaling in drug-resistant breast cancer. Oncogene (2021) 40(2):322–33. doi: 10.1038/s41388-020-01530-6
65. Wierman ME, Kiseljak-Vassiliades K, Tobet S. Gonadotropin-releasing hormone (GnRH) neuron migration: initiation, maintenance and cessation as critical steps to ensure normal reproductive function. Front Neuroendocrinol (2011) 32(1):43–52. doi: 10.1016/j.yfrne.2010.07.005
66. Phelps CJ. Postnatal regression of hypothalamic dopaminergic neurons in prolactin-deficient Snell dwarf mice. Endocrinology (2004) 145(12):5656–64. doi: 10.1210/en.2004-0931
67. Hoffman GE, Finch CE. LHRH neurons in the female C57BL/6J mouse brain during reproductive aging: no loss up to middle age. Neurobiol Aging (1986) 7(1):45–8. doi: 10.1016/0197-4580(86)90026-6
68. Rance NE, Young WS 3rd, McMullen NT. Topography of neurons expressing luteinizing hormone-releasing hormone gene transcripts in the human hypothalamus and basal forebrain. J Comp Neurol (1994) 339(4):573–86. doi: 10.1002/cne.903390408
69. Papalexi E, Persson A, Bjorkqvist M, Petersen A, Woodman B, Bates GP, et al. Reduction of GnRH and infertility in the R6/2 mouse model of huntington’s disease. Eur J Neurosci (2005) 22(6):1541–6. doi: 10.1111/j.1460-9568.2005.04324.x
70. Manfredi-Lozano M, Leysen V, Adamo M, Paiva I, Rovera R, Pignat JM, et al. GnRH replacement rescues cognition in down syndrome. Science (2022) 377(6610):eabq4515. doi: 10.1126/science.abq4515
71. Jin H, Hou J, Meng X, Ma T, Wang B, Liu Z, et al. Microcystin-leucine arginine induced the apoptosis of GnRH neurons by activating the endoplasmic reticulum stress resulting in a decrease of serum testosterone level in mice. Ecotoxicol Environ Saf (2021) 208:111748. doi: 10.1016/j.ecoenv.2020.111748
72. Yu Y, Zhang D, Xu J, Zhang D, Yang L, Xia R, et al. Adolescence is a sensitive period for acrylamide-induced sex hormone disruption: evidence from NHANES populations and experimental mice. Ecotoxicol Environ Saf (2023) 249:114413. doi: 10.1016/j.ecoenv.2022.114413
73. Hoffmann HM, Trang C, Gong P, Kimura I, Pandolfi EC, Mellon PL. Deletion of Vax1 from gonadotropin-releasing hormone (GnRH) neurons abolishes GnRH expression and leads to hypogonadism and infertility. J Neurosci (2016) 36(12):3506–18. doi: 10.1523/JNEUROSCI.2723-15.2016
74. Laitinen EM, Tommiska J, Sane T, Vaaralahti K, Toppari J, Raivio T. Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR mutations. PloS One (2012) 7(6):e39450. doi: 10.1371/journal.pone.0039450
75. Pitteloud N, Acierno JS Jr., Meysing AU, Dwyer AA, Hayes FJ, Crowley WF Jr. Reversible kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J Clin Endocrinol Metab (2005) 90(3):1317–22. doi: 10.1210/jc.2004-1361
76. Pitteloud N, Boepple PA, DeCruz S, Valkenburgh SB, Crowley WF Jr., Hayes FJ. The fertile eunuch variant of idiopathic hypogonadotropic hypogonadism: spontaneous reversal associated with a homozygous mutation in the gonadotropin-releasing hormone receptor. J Clin Endocrinol Metab (2001) 86(6):2470–5. doi: 10.1210/jcem.86.6.7542
77. Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med (2007) 357(9):863–73. doi: 10.1056/NEJMoa066494
78. Ribeiro RS, Vieira TC, Abucham J. Reversible kallmann syndrome: report of the first case with a KAL1 mutation and literature review. Eur J Endocrinol (2007) 156(3):285–90. doi: 10.1530/eje.1.02342
79. Sinisi AA, Asci R, Bellastella G, Maione L, Esposito D, Elefante A, et al. Homozygous mutation in the prokineticin-receptor2 gene (Val274Asp) presenting as reversible kallmann syndrome and persistent oligozoospermia: case report. Hum Reprod (2008) 23(10):2380–4. doi: 10.1093/humrep/den247
80. Naule L, Maione L, Kaiser UB. Puberty, a sensitive window of hypothalamic development and plasticity. Endocrinology (2021) 162(1). doi: 10.1210/endocr/bqaa209
81. Herbison AE, de Tassigny X, Doran J, Colledge WH. Distribution and postnatal development of Gpr54 gene expression in mouse brain and gonadotropin-releasing hormone neurons. Endocrinology (2010) 151(1):312–21. doi: 10.1210/en.2009-0552
82. Sim JA, Skynner MJ, Herbison AE. Heterogeneity in the basic membrane properties of postnatal gonadotropin-releasing hormone neurons in the mouse. J Neurosci (2001) 21(3):1067–75. doi: 10.1523/JNEUROSCI.21-03-01067.2001
83. Dulka EA, Moenter SM. Prepubertal development of gonadotropin-releasing hormone neuron activity is altered by sex, age, and prenatal androgen exposure. Endocrinology (2017) 158(11):3943–53. doi: 10.1210/en.2017-00768
84. Ybarra N, Hemond PJ, O’Boyle MP, Suter KJ. Spatially selective, testosterone-independent remodeling of dendrites in gonadotropin-releasing hormone (GnRH) neurons prepubertally in male rats. Endocrinology (2011) 152(5):2011–9. doi: 10.1210/en.2010-0871
85. Cottrell EC, Campbell RE, Han SK, Herbison AE. Postnatal remodeling of dendritic structure and spine density in gonadotropin-releasing hormone neurons. Endocrinology (2006) 147(8):3652–61. doi: 10.1210/en.2006-0296
86. Wray S, Hoffman G. Postnatal morphological changes in rat LHRH neurons correlated with sexual maturation. Neuroendocrinology (1986) 43(2):93–7. doi: 10.1159/000124516
87. Burmeister SS, Wilczynski W. Social signals regulate gonadotropin-releasing hormone neurons in the green treefrog. Brain Behav Evol (2005) 65(1):26–32. doi: 10.1159/000081108
88. Dellovade TL, Rissman EF. Gonadotropin-releasing hormone-immunoreactive cell numbers change in response to social interactions. Endocrinology (1994) 134(5):2189–97. doi: 10.1210/endo.134.5.8156921
89. Mantei KE, Ramakrishnan S, Sharp PJ, Buntin JD. Courtship interactions stimulate rapid changes in GnRH synthesis in male ring doves. Horm Behav (2008) 54(5):669–75. doi: 10.1016/j.yhbeh.2008.07.005
90. Maruska KP, Fernald RD. Plasticity of the reproductive axis caused by social status change in an african cichlid fish: II. Testicular Gene Expression Spermatogenesis Endocrinol (2011) 152(1):291–302. doi: 10.1210/en.2010-0876
91. Stevenson TJ, Ball GF. Anatomical localization of the effects of reproductive state, castration, and social milieu on cells immunoreactive for gonadotropin-releasing hormone-I in male European starlings (Sturnus vulgaris). J Comp Neurol (2009) 517(2):146–55. doi: 10.1002/cne.22159
92. Lombardi JR, Vandenbergh JG. Pheromonally induced sexual maturation in females: regulation by the social environment of the male. Science (1977) 196(4289):545–6. doi: 10.1126/science.557838
93. Vandenbergh JG. Effect of the presence of a male on the sexual maturation of female mice. Endocrinology (1967) 81(2):345–9. doi: 10.1210/endo-81-2-345
94. Van Der Lee S, Boot LM. Spontaneous pseudopregnancy in mice. II Acta Physiol Pharmacol Neerl (1956) 5(2):213–5.
95. Vandenbergh JG. The influence of the social environment on sexual maturation in male mice. J Reprod Fertil (1971) 24(3):383–90. doi: 10.1530/jrf.0.0240383
96. Bronson FH, Serum FSH. LH, and prolactin in adult ovariectomized mice bearing silastic implants of estradiol: responses to social cues. Biol Reprod (1976) 15(2):147–52. doi: 10.1095/biolreprod15.2.147
97. Bronson FH, Maruniak JA. Differential effects of male stimuli on follicle-stimulating hormone, luteinizing hormone, and prolactin secretion in prepubertal female mice. Endocrinology (1976) 98(5):1101–8. doi: 10.1210/endo-98-5-1101
98. Coquelin A, Bronson FH. Release of luteinizing hormone in male mice during exposure to females: habituation of the response. Science (1979) 206(4422):1099–101. doi: 10.1126/science.573924
99. Maruniak JA, Bronson FH. Gonadotropic responses of male mice to female urine. Endocrinology (1976) 99(4):963–9. doi: 10.1210/endo-99-4-963
100. Maruniak JA, Coquelin A, Bronson FH. The release of LH in male mice in response to female urinary odors: characteristics of the response in young males. Biol Reprod (1978) 18(2):251–5. doi: 10.1095/biolreprod18.2.251
101. Macrides F, Bartke A, Dalterio S. Strange females increase plasma testosterone levels in male mice. Science (1975) 189(4208):1104–6. doi: 10.1126/science.1162363
102. Roney JR, Lukaszewski AW, Simmons ZL. Rapid endocrine responses of young men to social interactions with young women. Horm Behav (2007) 52(3):326–33. doi: 10.1016/j.yhbeh.2007.05.008
103. Lopez HH, Hay AC, Conklin PH. Attractive men induce testosterone and cortisol release in women. Horm Behav (2009) 56(1):84–92. doi: 10.1016/j.yhbeh.2009.03.004
104. Stoleru SG, Ennaji A, Cournot A, Spira A. LH pulsatile secretion and testosterone blood levels are influenced by sexual arousal in human males. Psychoneuroendocrinology (1993) 18(3):205–18. doi: 10.1016/0306-4530(93)90005-6
105. Chung WC, Auger AP. Gender differences in neurodevelopment and epigenetics. Pflugers Archiv: Eur J Physiol (2013) 465(5):573–84. doi: 10.1007/s00424-013-1258-4
106. Aylwin CF, Toro CA, Shirtcliff E, Lomniczi A. Emerging genetic and epigenetic mechanisms underlying pubertal maturation in adolescence. J Res Adolesc: Off J Soc Res Adolesc (2019) 29(1):54–79. doi: 10.1111/jora.12385
107. Gray JD, Kogan JF, Marrocco J, McEwen BS. Genomic and epigenomic mechanisms of glucocorticoids in the brain. Nat Rev Endocrinol (2017) 13(11):661–73. doi: 10.1038/nrendo.2017.97
108. Huang Y, Chavez L, Chang X, Wang X, Pastor WA, Kang J, et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc Natl Acad Sci U States America (2014) 111(4):1361–6. doi: 10.1073/pnas.1322921111
110. Iyer AK, Brayman MJ, Mellon PL. Dynamic chromatin modifications control GnRH gene expression during neuronal differentiation and protein kinase c signal transduction. Mol Endocrinol (2011) 25(3):460–73. doi: 10.1210/me.2010-0403
111. Kurian JR, Terasawa E. Epigenetic control of gonadotropin releasing hormone neurons. Front Endocrinol (2013) 4:61. doi: 10.3389/fendo.2013.00061
112. Wu H, Zhang Y. Mechanisms and functions of tet protein-mediated 5-methylcytosine oxidation. Genes Dev (2011) 25(23):2436–52. doi: 10.1101/gad.179184.111
113. Dawlaty MM, Breiling A, Le T, Raddatz G, Barrasa MI, Cheng AW, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell (2013) 24(3):310–23. doi: 10.1016/j.devcel.2012.12.015
114. Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature (2010) 466(7310):1129–33. doi: 10.1038/nature09303
115. Kurian JR, Louis S, Keen KL, Wolfe A, Terasawa E, Levine JE. The methylcytosine dioxygenase ten-eleven translocase-2 (tet2) enables elevated GnRH gene expression and maintenance of Male reproductive function. Endocrinology (2016) 157(9):3588–603. doi: 10.1210/en.2016-1087
116. Mimouni NEH, Paiva I, Barbotin AL, Timzoura FE, Plassard D, Le Gras S, et al. Polycystic ovary syndrome is transmitted via a transgenerational epigenetic process. Cell Metab (2021) 33(3):513–530.e518. doi: 10.1016/j.cmet.2021.01.004
117. Linscott ML, Chung WCJ. Epigenomic control of gonadotrophin-releasing hormone neurone development and hypogonadotrophic hypogonadism. J Neuroendocrinol (2020) 32(6):e12860. doi: 10.1111/jne.12860
118. Linscott ML, Chung WC. Fibroblast growth factor 8 expression in GT1-7 GnRH-secreting neurons is androgen-independent, but can be upregulated by the inhibition of DNA methyltransferases. Front Cell Dev Biol (2016) 4. doi: 10.3389/fcell.2016.00034
119. Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature (2011) 473(7347):389–93. doi: 10.1038/nature09934
120. Lomniczi A, Loche A, Castellano JM, Ronnekleiv OK, Bosch M, Kaidar G, et al. Epigenetic control of female puberty. Nat Neurosci (2013) 16(3):281–9. doi: 10.1038/nn.3319
121. Lomniczi A, Ojeda SR. The emerging role of epigenetics in the regulation of female puberty. Endocr Dev (2016) 29:1–16. doi: 10.1159/000438840
Keywords: gonadotropin-releasing hormone, congenital hypogonadotropic hypogonadism, neurogenesis, neuronal de-differentiation, epigenetic factors, environmental factors
Citation: Chung WCJ and Tsai P-S (2023) The initiation and maintenance of gonadotropin-releasing hormone neuron identity in congenital hypogonadotropic hypogonadism. Front. Endocrinol. 14:1166132. doi: 10.3389/fendo.2023.1166132
Received: 14 February 2023; Accepted: 12 April 2023;
Published: 27 April 2023.
Edited by:
Roberto Oleari, University of Milan, ItalyReviewed by:
Valeria Vezzoli, Italian Auxological Institute (IRCCS), ItalyVincent Prevot, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2023 Chung and Tsai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pei-San Tsai, cGVpLXNhbi50c2FpQGNvbG9yYWRvLmVkdQ==