 Yanan Cheng
Yanan Cheng Yan Wang
Yan Wang Ruili Yin
Ruili Yin Yongsong Xu
Yongsong Xu Lijie Zhang
Lijie Zhang Yuanyuan Zhang
Yuanyuan Zhang Longyan Yang
Longyan Yang Dong Zhao
Dong Zhao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 31 March 2023
Sec. Cardiovascular Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1162754
This article is part of the Research Topic Insights in Cardiovascular Endocrinology: 2023 View all 8 articles
Diabetic cardiomyopathy (DCM), a main cardiovascular complication of diabetes, can eventually develop into heart failure and affect the prognosis of patients. Myocardial fibrosis is the main factor causing ventricular wall stiffness and heart failure in DCM. Early control of myocardial fibrosis in DCM is of great significance to prevent or postpone the progression of DCM to heart failure. A growing body of evidence suggests that cardiomyocytes, immunocytes, and endothelial cells involve fibrogenic actions, however, cardiac fibroblasts, the main participants in collagen production, are situated in the most central position in cardiac fibrosis. In this review, we systematically elaborate the source and physiological role of myocardial fibroblasts in the context of DCM, and we also discuss the potential action and mechanism of cardiac fibroblasts in promoting fibrosis, so as to provide guidance for formulating strategies for prevention and treatment of cardiac fibrosis in DCM.
Diabetes mellitus, a metabolic disease, is widely considered as a public health problem. At present, there are about 463 million people suffering from diabetes worldwide (1). Cardiovascular (CV) complications are the main causes of death in diabetic patients (2). Diabetic cardiomyopathy (DCM) is an important cardiovascular complication of diabetes mellitus, which refers to the changes of cardiac function and structure caused by diabetes, independent of any other recognized cardiac risk factors such as coronary atherosclerotic heart disease and hypertension (3, 4). The epidemiological relationship between diabetes and heart failure was confirmed 50 years ago (5). Clinical heart failure trials showed that the hospitalization rate of diabetic patients with heart failure was twice that of non-diabetic patients (6, 7). Myocardial fibrosis in DCM is a crucial factor of ventricular wall stiffness and heart failure. Therefore, early control of the progress of diabetic myocardial fibrosis is of great significance to prevent or delay the progression of DCM to heart failure.
Myocardial fibrosis, known as interstitial expansion of the myocardium caused by net accumulation of extracellular matrix (ECM) (8, 9), mainly includes three distinct forms according to the basic histopathological analysis (10). In myocardial infarction, myocardial cells that are necrotic due to ischemia are replaced by collagen-based scars, resulting in “replacement fibrosis”. With the net accumulation of ECM proteins, the endomysial and perimysial spaces were expanded, causing “interstitial fibrosis”. Also, “perivascular fibrosis”, which describes the expansion of the microvascular adventitia, is part of cardiac fibrosis (10). On the one hand, “replacement fibrosis” does not seem to occur as there is no acute loss of cardiomyocytes in DCM. However, abnormal metabolism could lead to unusual apoptosis of cardiomyocytes (11), and chronic myocardial cell injury provides a condition of “replacement fibrosis” for DCM exacerbation. This “replacement fibrosis” may be a form of self-protection of heart against chronic pathological injury in the early stage of DCM. Another manifestation of self-protection is that myofibroblasts have similar activity to macrophages, which have the function of phagocytizing apoptotic cells and secreting cytokines that suppress inflammatory responses (12, 13). On the other hand, DCM is in a persistent state of metabolic derangement (14) that leads to long-term existence of fibrotic irritants in the internal environment, increasement of proliferation and activation of fibroblasts (15), as well as transdifferentiation of other cell types into CFs (16), thereby causing interstitial and perivascular fibrosis that disrupts diastolic function and results in abnormal cardiac electrophysiological activity (17). In the late stage of DCM, myocardial cell injury and apoptosis are aggravated and replacement fibrosis is more obvious, which eventually leads to the cardiac contraction dysfunction.
Despite its severity and poor prognosis, DCM still lacks formal treatment guidelines or approved specific pharmacological drugs to treat it (18). Both in human patients and in animal models of diabetes, cardiac fibrosis is prominent characteristic of diabetic cardiomyopathy (19, 20). As the main ECM-producing cells in the heart, CFs are critically involved in all cardiac fibrotic conditions (21, 22). For these reasons, an in-depth understanding of the character of CFs in myocardial fibrosis in DCM may point the direction for formulating strategies to prevent and treat myocardial fibrosis.
DCM is a chronic disease characterized by metabolic disorder, often accompanied by fibrosis, myocardial cell apoptosis, local inflammation, oxidative stress, endoplasmic reticulum stress and mitochondrial dysfunction (23–25). It typically manifests initially with diastolic dysfunction, but with time may also evolve with systolic dysfunction, and eventually leads to heart failure (26). In 1972, the first clinical description of DCM was published, which showed the postmortem of four patients with diabetes who had died of heart failure in the absence of coronary artery disease, hypertension, or valvular heart disease (27). Myocardial hypertrophy and fibrosis were noted in the hearts of those patients, suggesting that the fibrosis is responsible for this result. These observations were supported by Regan in 1977 (28). Autopsies of 11 uncomplicated diabetic patients revealed that collagen accumulation occurred in the heart which was present as perivascular, intermuscular, or replacement fibrosis. Therefore, it is indicated that heart failure of DCM is related to cardiac fibrosis, and even to some extent, cardiac fibrosis is a basis of heart failure caused by DCM.
Diabetes-associated cardiac fibrogenesis, including cardiac fibrosis in DCM, is a multistep and multicell process originated in cellular reaction to oxidative stress, endoplasmic reticulum stress and inflammation (26). The heart tissue is composed of cardiomyocytes and non-cardiomyocytes. After acute myocardial injury, acute death of cardiomyocytes is usually an initial event in the activation of myocardial fibrotic signaling. However, deleterious stimuli in DCM may activate pathways of fibrosis in the absence of acute cell death. Several cell types have been implicated in cardiac fibrosis and remodeling in DCM, as summarized in Figure 1.
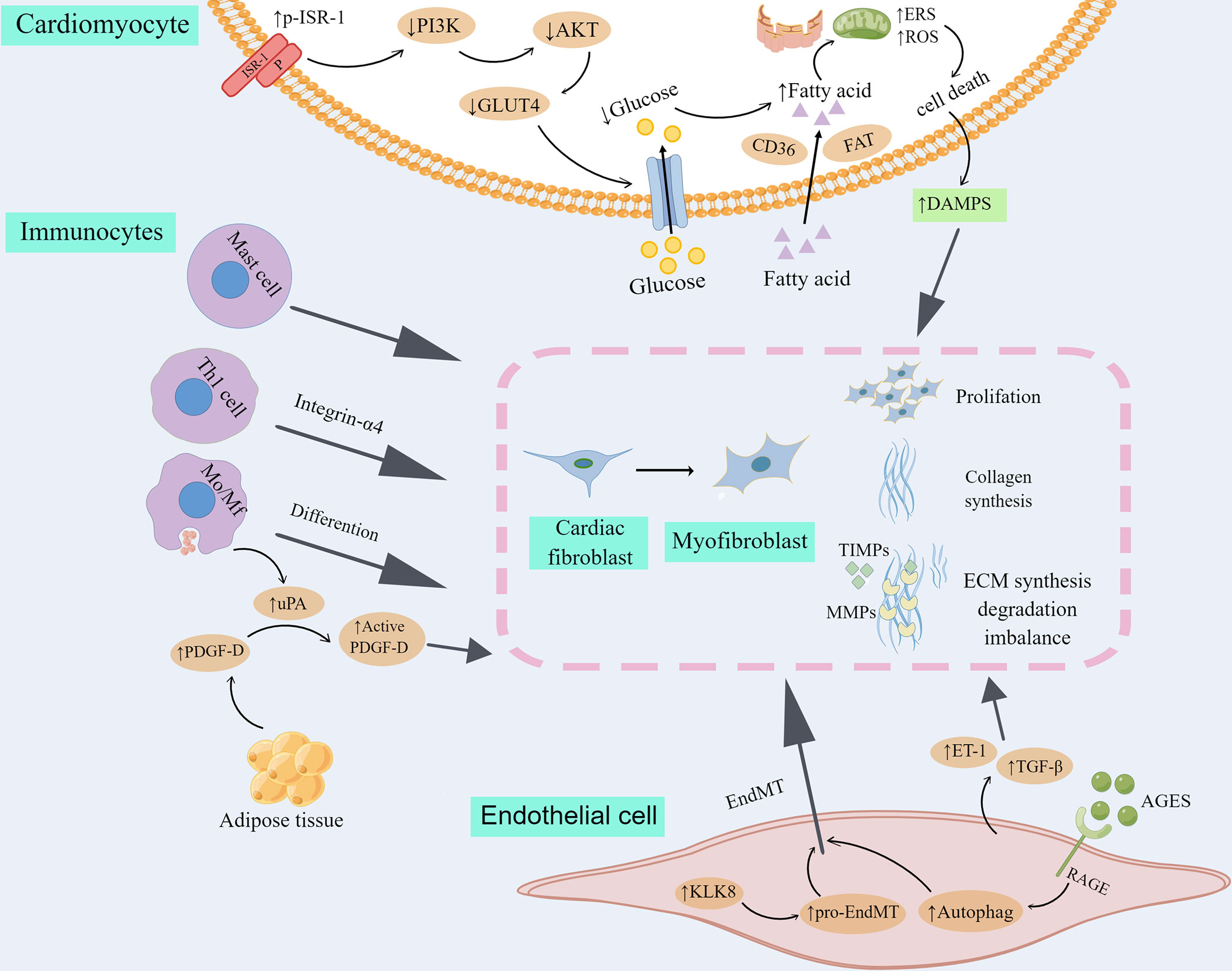
Figure 1 Cell types involved in myocardial fibrosis and remodeling in DCM. Transdifferentiation from cardiac fibroblast to myofibroblast is the core cellular event in cardiac fibrosis in DCM. 1) Myofibroblasts produce high level of collagen and other extracellular matrix proteins (ECM), and secrete matrix metalloproteinases (MMPs) as well as their inhibitors, tissue inhibitors of metalloproteinases (TIMPs), taking part in the progression to fibrotic remodeling. 2) Insulin resistance leads to the metabolic transformation of cardiomyocytes from glucose metabolism to fatty acid β oxidation, increased cell metabolic pressure, reactive oxygen species (ROS) and endoplasmic reticulum stress (ERS) and finally led to the death of cardiomyocytes. DAMPs released in inflammatory reaction by dead myocardial cells activate cardiac fibroblasts. 3) Immunocytes, including mast cell, Th1 cell and Mo/Mf could directly differentiate into myofibroblasts or indirectly promote cardiac fibroblast transdifferentiate into myofibroblast. 4) Endothelial cells were transformed into mesenchymal cells (EndMT), and further into myofibroblasts. In addition, fibrogenic mediators produced by endothelial cells participate in the proliferation and differentiation of myocardial fibroblasts. By Figdraw. ISR-1, insulin receptor substrate 1; P, phosphorylation; PI3K, phosphatidylinositol 3-kinase; GLUT4, glucose transporter 4; CD36, cluster of differentiation 36; FAT, fatty acid translocatase; DAMPS, danger-associated molecular patterns; Mo/Mf, monocytes/macrophages; PDGF-D, platelet-derived growth factor-D; uPA, urokinase plasminogen activator; KLK8, kallikrein-related peptidase 8; ET-1, endothelin 1; AGES, advanced glycation end products; RAGE, AGES receptor; TGF-β, transforming growth factor-beta.
Myofibroblasts have been demonstrated to accumulate in myocardial interstitium under different cardiac pathological conditions (29, 30). Although the mechanism of myocardial fibrosis differ in various heart diseases, the transition from CFs to myofibroblasts is a key cellular event in cardiac fibrosis (31, 32). Myofibroblast, a kind of contractile and secretory cell type, participate in cardiac fibrotic remodeling by producing proteins of ECM (21), and by secreting matrix metalloproteinases (MMPs) as well as their inhibitors, tissue inhibitors of metalloproteinases (TIMPs). The balance between synthesis and degradation of ECM is very important in cardiac fibrosis. And the main enzyme regulating ECM degradation is MMPS, whose activity is primarily regulated by TIMPs (32–34). CFs derived from type 2 diabetes(T2DM) patients exhibited high collagen synthesis (35). And CFs isolated from diabetic heart also showed increased proliferative activity and elevated collagen and protease inhibitors expression (36, 37). In vitro, high glucose (HG) treatment could promote the proliferation of CFs and the collagen formation (38–40). Moreover, hyperglycemia increased the expression of transforming growth factor β (TGF-β) in CFs (41). Smads, the main downstream medium of TGF-β signaling, play important roles in the fibrosis of liver, lung, kidney and heart (42–44). Activation of TGF-β/Smads signal transduction in DCM could induce the production of MMPs and collagens, which leads to cardiac fibrosis (45, 46).
Cardiomyocytes play a critical role in myocardial fibrosis in DCM. Long-term disturbance of glucose metabolism can lead to cardiomyocyte death. First, insulin resistance leads to the increase of serine phosphorylation of insulin receptor substrate 1 (p-IRS-1), a key insulin signal factor in myocardial cells, which in turn damages the signal transduction pathway of phosphatidylinositol 3-kinase (PI3K)- protein kinase B (also known as AKT), then blocks the shift of glucose transporter 4 (GLUT4) to the cell membrane and reduces the intake of glucose in the heart (47). Moreover, fatty acid uptake regulated by fatty acid translocase (FAT) and cluster of differentiation 36 (CD36) is increased in diabetes, which causes the metabolic transformation of cardiomyocyte from glucose metabolism to fatty acid β oxidation, increased cell metabolic pressure, and ultimately cardiomyocyte death (47). In addition, up-regulation of endoplasmic reticulum stress (ERS) and reactive oxygen species (ROS) in DCM is a major cause of cardiomyocyte apoptosis (48, 49), which may eventually impair cardiac function and induce myocardial fibrosis (50). Necrotic cardiomyocytes activate immune pathways and initiate inflammatory responses. Inflammatory signals promote the infiltration of leukocytes to clear dead cardiomyocytes, while DAMPs released by the inflammatory response activate CFs, leading to the proliferation and transdifferentiation of CFs, and the deposition of a large amount of ECM to maintain the integrity of cardiac structure, but will be accompanied by the occurrence of myocardial fibrosis (51).
Increasing evidence implicates immunocytes are involved in the regulation of cardiac fibrosis in DCM. First, the role of monocytes/macrophages (Mo/Mf) in the process of cardiac fibrosis has two sides. On the one hand, monocytes/macrophages differentiate into myofibroblasts under the action of various cytokines, producing a variety of inflammatory mediators and profibrotic factors (52). It was found that lipid metabolism disorders may lead to increased expression of full-length platelet-derived growth factor-D (PDGF-D) secreted into body fluids by adipose tissue. Urokinase plasminogen activator (uPA) produced by activated macrophages in cardiac tissue splices circulating PDGF-D into an active form that activates PI3K-AKT signaling in CFs, thereby promoting fibrosis (53, 54). The recruitment and polarization of macrophages are important processes in cardiac fibrosis (55). Traditionally, two different polarization states of macrophages are identified (29): classically activated macrophages (M1) are proinflammatory macrophages that produce proinflammatory cytokines and reactive oxygen species(ROS), and then cause the induction of inflammation and cardiac fibrosis (55, 56), whereas alternatively activated macrophages (M2) are considered as anti-inflammatory macrophages, and replacing them with M1 macrophages can inhibit cardiac fibrosis (57). On the other hand, because of their ability to devour cell debris and apoptotic cells, Mo/Mf may have anti-fibrosis effects (29, 52). Both lymphocytes and mast cells could participate in cardiac fibrosis by activating CFs. Th1 lymphocytes stimulate TGF-β to activate CFs via integrin-α4 (58, 59). Mast cells are capable of directly activating fibroblasts and stimulating fiber formation by histamine, tryptase, TGF-β and chymotrypsin (60).
Endothelial cells participation in cardiac fibrosis is suggested by the prevalence of perivascular fibrosis under the pathological conditions of infarction and pressure overload (61). Endothelial cells are the initial target of hyperglycemia damage. Sustained endothelial injury during diabetes mellitus causes endothelial-to-mesenchymal transition (EndMT), and promotes the further transformation of this endothelial cell phenotype into myofibroblasts, thus providing an additional activated fibroblasts bank and promoting cardiac fibrosis (62–64). It was found that diabetes-related EndMT and myocardial fibrosis were partly stimulated by the up-regulation of kallikrein-related peptidase 8 (KLK8). KLK8 activates platelet hemoglobin-dependent pro-EndMT signaling pathway, contributes to the evolution of EndMT and cardiac fibrosis, and accelerates the progress of cardiac dysfunction in diabetes (65). Consistently, inhibition of EndMT could reduce diabetic myocardial fibrosis (66–70), and EndMT seems to be regulated by autophagy. Endothelial autophagy deficiency induces interleukin 6 (IL-6)-dependent EndMT and cardiac fibrosis in mice with metabolic defects (71). The deletion of advanced glycation end product receptor (RAGE) has been shown to inhibit excessive autophagy and ameliorated cardiac fibrosis (72). AGES/RAGE- autophagy EndMT axis participates in the development of cardiac fibrosis, and knocking out RAGE can improve cardiac fibrosis by reducing autophagy-regulated EndMT (73). In addition, fibrogenic factors generated by endothelial cells, such as Endothelin 1 (ET-1) and TGF-β, play an important role in the pathogenesis of diabetic cardiac fibrosis (74–76). ET-1 can promote the proliferation of myocardial fibroblasts, enhance the synthesis of type I and type III collagen, resulting in the differentiation of myofibroblasts (77, 78). At the same time, ET-1 is also involved in the fibrogenic reaction of TGF-β (41, 79, 80).
Many cell types play important roles in the heart during the formation and expansion of myocardial fibrosis in DCM, and importantly, there is evidence that CFs from DCM mice and rats exhibit increased proliferation and an imbalance in ECM synthesis and degradation (Table 1), and this process is a central event in cardiac fibrosis in DCM.
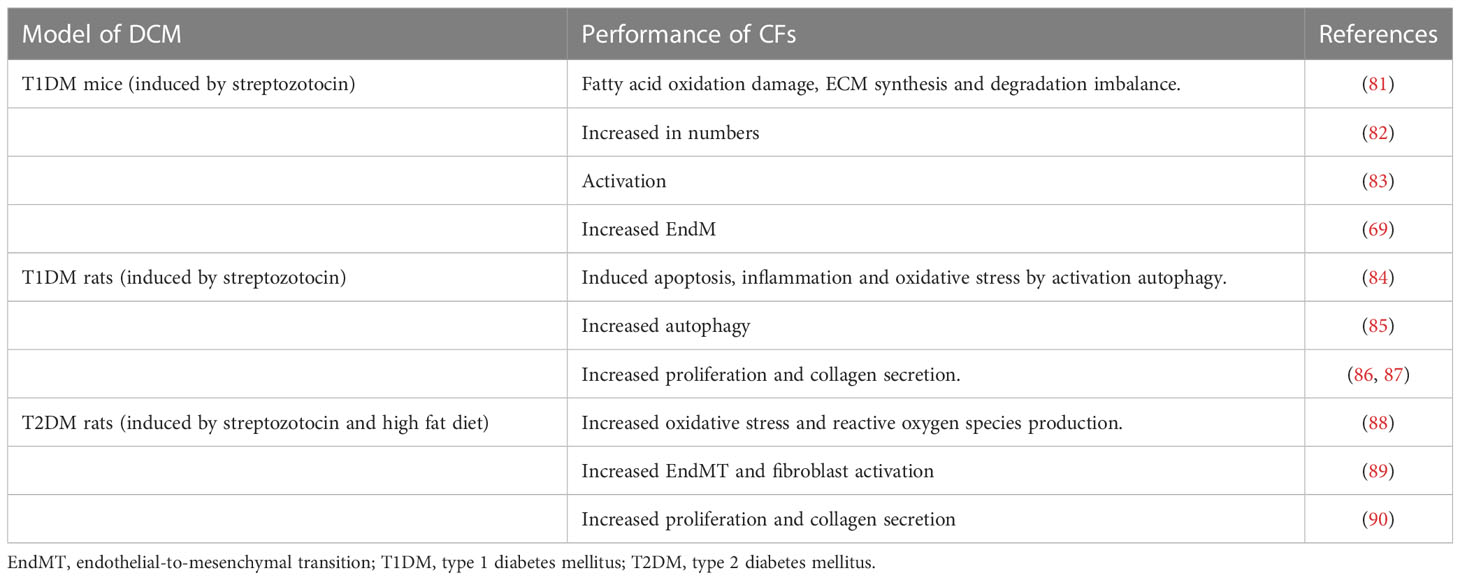
Table 1 Association between diabetic cardiomyopathy (DCM) and cardiac fibroblasts (CFs).
In the heart of adult mammals, cardiomyocytes account for about 75% of the volume of myocardium, and they are surrounded by the ECM network mainly composed of fibrous collagen (Figure 2). The main function of the cardiac ECM is to serve as the mechanical support of the heart and transmit the contraction force to ensure the normal blood pumping function of the heart (32, 91, 92). CFs are the main matrix-generating cells, forming one of the largest cell groups in the normal mammalian heart, and are usually entangled in the intima (93, 94). Methods based on histology and flow cytometry have proved that fibroblasts accounted for about 13% of mouse heart cells (95, 96). Pedigree tracing technique was used to study the origin of mouse CFs. It was found that epicardial progenitor cells differentiated into CFs and vascular smooth muscle cells during development, and endocardium differentiated into CFs through EndMT (97–104). It was estimated that approximately 85% of CFs originate from epicardial cells, while the other 15% come from endothelial cells (101). RNA analysis revealed that cells derived from epicardium and endocardium had similar expression profile (101, 104, 105), and similar multiplying activity (106). Hence, it can be inferred that there is no obvious correlation between the source and function of fibroblasts. However, it may be due to the diversity of origin and transformation that the specificity and credible marker proteins of myofibroblasts have not been reported yet (13, 31, 74, 107).
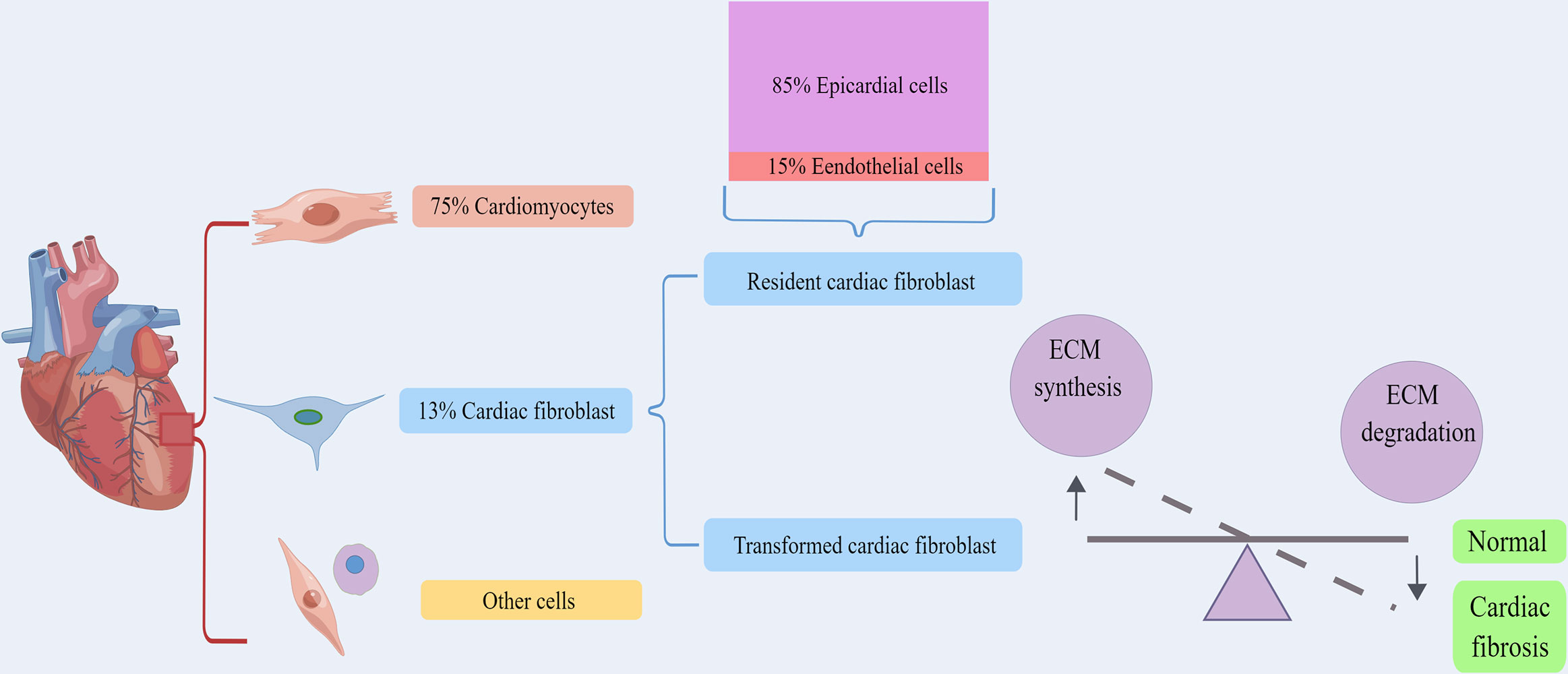
Figure 2 The origin and physiological function of cardiac fibroblasts. Cardiomyocytes account for about 75% of the volume of myocardium, and fibroblasts accounted for about 13%. Cardiac fibroblasts including resident cardiac fibroblast and transformed cardiac fibroblast. Approximately 85% of cardiac fibroblasts come from epicardial cells, while the other 15% are derived from endothelial cells. Both of them could serve to maintain the structural integrity of the ECM network and regulate collagen renewal. By Figdraw. ECM, extracellular matrix.
Resident CFs may serve to protect and support the constructional entirety of the ECM network and regulate collagen renewal, which is a continuous process of synthesis and decomposition of ECM proteins (108, 109). The Smad3 and platelet-derived growth factor receptor (PDGFR)-α signaling in fibroblasts are found to be vital for sustaining cell survival and cell function (110, 111). Regardless of ECM remodeling, fibroblasts play diverse functions in heart homeostasis and diseases, including providing support for multiple types of cardiac cells. In addition, CFs release paracrine factors, which may inhibit harmful inflammatory reactions and promote the survival of myocardial cells (112–116). In the early stage of diabetes, when DCM is not yet present, although the high glucose environment shocks cardiac cells, including CFs, which are in a stage of functional compensation and still able to balance ECM synthesis and degradation and yet provide a normal working microenvironment for cardiomyocytes, the heart does not exhibit obvious functional and pathological changes.
In diseased heart, the cellular composition has changed dramatically. One interesting question is whether the epicardium and endocardium cells of the adult heart still have the potential to transdifferentiate into CFs. Early studies detected the presence of epithelial-mesenchymal transition (EMT) in the adult heart (117–120). In response to heart injury, CFs are activated into myofibroblasts, and endothelial cells are derived into CFs (105, 121), and macrophages can also be transdifferentiated into fibroblast-like cells (122). CFs are also derived from bone marrow hematopoietic cells (123), although the contribution of these cells to the cardiac fibroblast cluster is minimal (101, 104, 105). Cells derived from different states possess diverse expression types. Active fibroblasts have high proliferation and migration activities, and their functions include recruiting inflammatory cells, stimulating the synthesis of type I and III collagen, and determining the proliferation of fibroblasts, as well as ECM degradation mediated by MMPs, which leads to a disrupted balance between ECM synthesis and degradation and results in cardiac fibrosis (Figure 2) (52, 107). Some researchers have proposed four different states of fibroblasts based on the infarcted model (13, 94, 121, 122): resting fibroblasts, active fibroblasts, myofibroblasts, and matrifibrocytes. During DCM, fibroblasts stimulated by local inflammatory cytokines in the heart get the activity of proliferation and ECM production, and enter a state of ECM expansion. Activated fibroblasts can change their phenotype into matrifibrocytes. Myofibroblasts produce ECM, while matrifibrocytes express genes related to bone and cartilage remodeling (their physiological significance in the heart is unclear). Because of their location at the scar site, matrifibrocytes may be unique to acute ischemic heart injury, and active CFs may not change to this cell type in chronic heart injury such as DCM. Based on this, during the progression of DCM, the transformation of endothelial cells, epithelial cells, macrophages into cardiac fibroblasts occurs more frequently in the heart due to the stimulation of adverse factors such as local inflammatory factors, AGES, and high glucose (23–25, 124), which, together with increased activation and proliferation of cardiac fibroblasts and impaired autophagy pathways (124–126), further creates the imbalance between ECM synthesis and degradation in cardiac tissue and causes and aggravates cardiac fibrosis in DCM.
The transport of glucose in cardiac tissue is mediated by GLUT4. Insulin binding to its receptors activates insulin receptor substrate (IRS)-1/2 and downstream AKT, which stimulates the transfer of GLUT4 to cell membrane and subsequent glucose absorption (47). In patients with diabetes mellitus, the decrease of PI3K/AKT signal transduction and the high expression and translocation of GLUT4 have been detected in ventricular tissue (127). Knockout of insulin receptor reduces cardiac glucose intake, which in turn increases the generation of cardiac reactive oxygen species (ROS), triggers mitochondrial dysfunction, and increases fibrosis and heart failure (128, 129). A large number of investigations have shown that HG can induce activation, proliferation, collagen synthesis and enhanced expression of α-SMA in neonatal CFs (130–133). Sustained hyperglycemia leads to the high activation of CFs, and persuades CFs to differentiate into myofibroblasts, leading to ECM accumulation and myocardial fibrosis in cardiac tissue (36, 37, 134). In vitro, HG treatment increased the expression of calcium-sensitive receptor (CaSR), α-SMA, collagen I/III and MMP2/9, and enhanced the generation of autophagy and the proliferation of CFs (126). CaSR is a mediator of intracellular calcium level. It is supposed that the increase of intracellular calcium concentration could attract Smad specific E3 ubiquitin protein ligase 2 (Smurf2) expression, thus degrading SKI like proto-oncogene (SnoN) and Smad family member 7 (Smad7) proteins through ubiquitin proteasome signaling pathway (135, 136). HG and CaSR agonist significantly enhance the expression of TGF-β and p-Smad2/3, and degrade Smad7, resulting in the increase of collagen secretion in CFs (126). Therefore, continuous hyperglycemia stimulation can up-regulate CaSR expression in CFs, increase intracellular Ca2+ level, stimulate Smurf2 autophagy and ubiquitin proteasome, and promote collagen secretion (126, 137). In addition, HG can increase the expression of DNA methyltransferase1 (DNMT1) in CFs, cause hypermethylation of suppression of cytokine signaling 3 (SOCS3), downregulate the expression of SOCS3, and activate signal transducer and activator of transcription 3 (Stat3), thus promoting the activation of CFs and collagen deposition (138). At the same time, HG can also regulate the expression of Methyl CpG binding protein 2 (MeCP2), resulting in the methylation of target gene promoter inhibiting the expression of ras association domain family 1 isoform A (RASSF1A) and extracellular signal-regulated kinase 1/2 (ERK1/2) pathway, thus triggering cardiac fibroblasts proliferation and DCM (86). However, it was found that hyperglycemia alone could not stimulate the activation of adult CFs, pointing out that the direct influence of hyperglycemia on fibroblasts is not the main driving factor of fibrosis and remodeling in cardiac diabetic maladjustment (139). In fact, the phenotypic changes of neonatal CFs stimulated by HG in vitro are consistent with those in vivo, which indicates that it is still important to study neonatal CFs in vitro for DCM.
In the utilization of metabolic substrates, CFs are very flexible, and relatively more dependent on fatty acid oxidation than glucose oxidation, which is similar to the integral heart (140). Obesity and insulin resistance models showed that CFs obtained enhanced myofibroblast/fibrotic gene expression and decreased reactiveness to TGF-β (141). In accordance with this, CFs isolated from db/db mice showed enhancement in collagen synthesis and decrement in TGF-β reaction (36). In response to high-fat diet (HFD), CFs have the ability to differentiate into adipocytes (142). Resistin is a hormone derived from adipocytes, which is related to obesity, insulin resistance and diabetes (143, 144). Increased resistin expression can be detected in cardiac myocytes and CFs after myocardial infarction and in neurohumoral stimulation responses (145). Studies have shown that resistin gene knockout mice fed with high fat did not exhibit relevant changes in cardiac fibrosis (146). Cardiac tissue overexpressed with resistin presented all the major markers of fibroblasts as well as the phenotypic characteristic of fibrosis, and showed augmented expression of ECM proteins including α-SMA, COL1a1, CTGF/CCN2, fibronectin, LOX, and SF2 (146). Resistin stimulates the proliferation of adult mouse CFs, activates janus kinase 2 (JAK2) by binding to toll-like receptor 4 (TLR4), and then phosphorylates Stat3, which transfers to the nucleus and activates the expression of fibroblasts target genes (146). Cell death-inducing DFFA-like effector C (CIDEC) is a vital regulator in lipid, glucose metabolism, and insulin sensitivity (147, 148). In CFs model of insulin resistance, the expression of CIDEC is increased, accompanied by nuclear translocation of CIDEC (from cytoplasm to nucleus), leading to the inhibition of AMP-activated protein kinase α (AMPK) phosphorylation and further promotes collagen synthesis (149).
Advanced glycation end products(AGES) accumulate naturally in the body, and usually exist to a lesser extent in healthy individuals (150). The accumulation of AGES occurs at a higher rate in diabetic patients (151). AGES upregulates the expression and protein level of type I collagen gene in adult rat CFs, which shows that AGES has the ability to directly promote fibrosis of CFs (152). AGES can bind to its receptor, RAGE, triggering the activation of various signal cascades, leading to downstream events, such as oxidative stress increasement, ECM reshaping and myofibroblast differentiation (153, 154). The activation of AGE/RAGE signaling pathway promotes differentiation of fibroblasts into myofibroblasts T2DM (36). The elevated AGES within diabetic collagen mediates the contraction of CFs via the increase of RAGE signal (155), and the increase of myofibroblast differentiation may lead to the increase of matrix contraction (156), in turn, the increased matrix contraction promotes the transformation of myofibroblasts, thereby increasing cardiac fibrosis (36, 153, 157). The migration level of diabetic CFs is higher than that of non-diabetic CFs, and along with the addition of AGES, the migration increases (158). Compared with non-diabetic collagen, higher levels of AGES were found in the tail collagen of diabetic mice (155). A 3D collagen matrix model made of tail collagen of diabetes mellitus was used to imitate the conditions in vivo and evaluate the changes of fibroblast function. Results showed that the increase of AGES in ECM could not only amplify the phenotypic transformation of CFs to myofibroblasts through RAGE cascade by signal transduction, but also change the cellular function and surrounding ECM, causing the increase of matrix contraction, thus accelerating the pathological remodeling of the heart (155). In addition, AGES can increase the crosslinking of matrix proteins (such as collagen) secreted by activated CFs, thus contributing to a more rigid ECM (36, 159, 160).
Thus, in the early stage of diabetes, the high glucose internal environment, the abnormal lipid metabolism of cells and the cytotoxicity generated by AGES complement each other to promote the phenotypic transformation of cardiac fibroblasts, cause the secretory dysfunction of cardiac fibroblasts, trigger cardiac fibrosis, gradually aggravate with the progression of the disease, and finally cause the occurrence of adverse outcomes of diabetic cardiomyopathy (Figure 3).
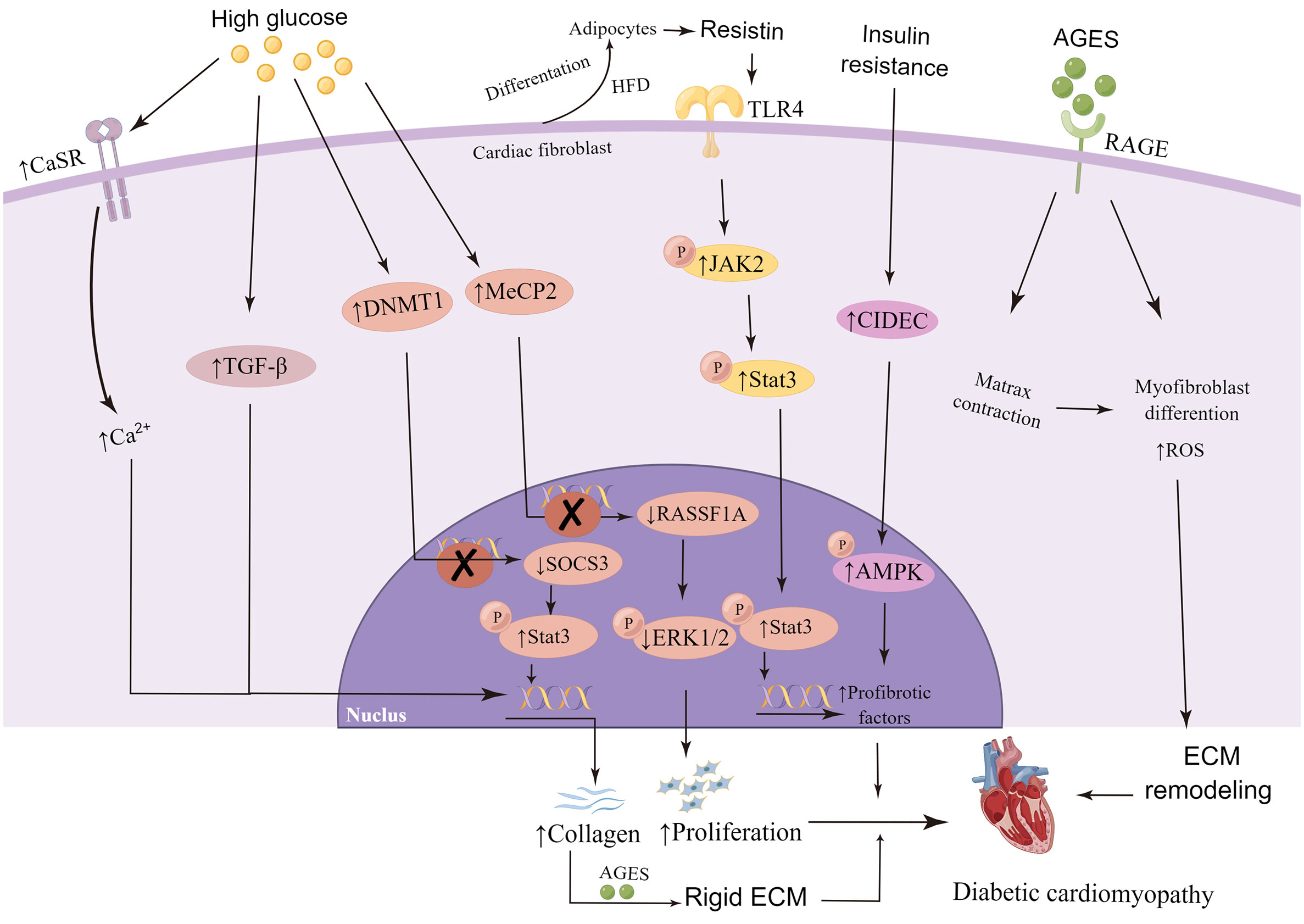
Figure 3 High glucose, abnormal lipid metabolism and advanced glycation end products (AGES) affects cardiac fibroblasts in the development of diabetic cardiomyopathy. Continuous hyperglycemia stimulation up-regulates the expression of calcium-sensitive receptor (CaSR), transforming growth factor-beta1 (TGF-β), DNA methyltransferase 1 (DNMT1) and Methyl CpG binding protein 2 (MeCP2) in cardiac fibroblast, and result in the increase of collagen secretion, deposition or proliferation, eventually promote the development of DCM. Resistin released by adipocytes increased expression of profibrotic factors. Accumulation of AGES bind to its receptor, RAGE, triggering the activation of various signal cascades, leading to downstream events, such as increased oxidative stress, extracellular matrix (ECM) remodeling and myofibroblast differentiation. By Figdraw. CaSR, calcium-sensitive receptor; TGF-β, transforming growth factor-beta; SOCS3, suppression of cytokine signaling 3; p-Stat3, phosphorylation of signal transducer and activator of transcription 3; RASSF1A, ras association domain family 1 isoform A; p-ERK1/2, phosphorylation of extracellular signal-regulated kinase 1/2; HFD, high-fat diet; TLR4, toll-like receptor 4; p-JAK2, phosphorylation of janus kinase 2; CIDEC, cell death-inducing DFFA-like effector C; p-AMPK, phosphorylation of AMP-activated protein kinase; ROS, reactive oxygen species.
TGF-β is a strong inducer involved in the differentiation of CFs into myofibroblasts, resulting in fibrosis (Figure 4) (161). Additionally, experiments in vitro also showed that HG can increase the expression and activity of TGF-β (162–165). Stimulated by TGF-β, CFs play a key role in the fibrosis process of cardiac ECM remodeling by synthesizing and secreting various components of ECM, such as type I and type III collagen (8). In the study of mice with selectively deleted TGF-β receptors Tgfbr1/2, Smad2, or Smad3 (which are important components for regulating TGF-β signal transduction) in CFs, it was found that regulating TGF-β signal transduction plays an important role in activating CFs (166). TGF-β stimulates the activation of typical Smad2/3 and AMPK signaling pathways (161). The specific deletion of Tgfbr1/2 and Smad3 notably reduce cardiac fibrosis resulting from transverse coarctation of aorta, and Smad3 is activated downstream of TGF-β receptor (166, 167). In vivo drug research found that inhibiting TGF-β/Smad2/3 pathway can inhibit the proliferation and collagen production of CFs and attenuate the degree of cardiac fibrosis (168). In recent years, there are several researches focus on how TGF-β signaling pathway regulates the phenotypic transformation and function of CFs. It was found that TGF-β activates Smad3 to regulate phosphorylation of downstream ERK1/2 and AKT, inhibits the expression of FoxO3a in CFs, and results in the increase of CFs transformation and collagen synthesis (169). This team also found that HG resulted in decreased AKT activity and AKT-mediated phosphorylation level of forkhead box O1 (FoxO1), leading to the increase of the nuclear localization and transcription activity of FoxO1, which in turn led to the phenotypic transformation of myofibroblasts and the increase of α-SMA and collagen expression (165).
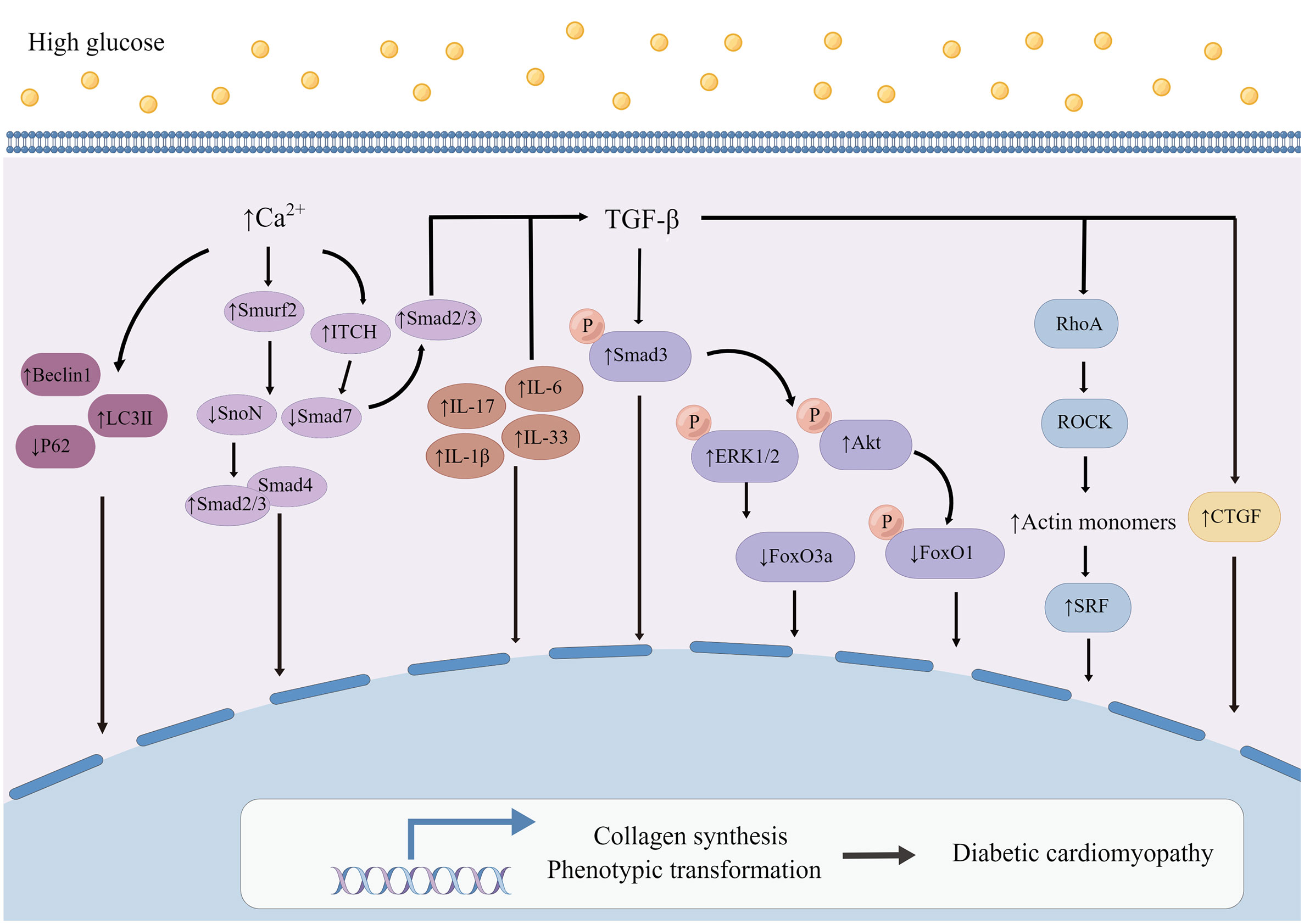
Figure 4 Mechanism of myocardial fibroblasts on myocardial fibrosis in diabetic cardiomyopathy. High glucose increases the expression and activity of TGF-β and causes a series of changes in signal pathways regulating the phenotypic transformation and synthesis function of CFs. The expressions of Interleukin (IL), including IL-6, IL-17, IL-1β and IL-33, were all up-regulated in myocardial fibroblasts attacked by HG, especially, IL-6 seems to up-regulate collagen gene by inducing transforming growth factor-beta 1 (TGF-β). At the same time, increased Ca2+ concentration regulated by CaSR could modulate the ubiquitination of Smad7 to affect DCM. By Figdraw. Smurf2, Smad specific E3 ubiquitin protein ligase 2; SnoN, SKI like proto-oncogene; Smad2/3, Smad family member 2 and Smad family member 3; Smad4, Smad family member 4; Smad7, Smad family member 7; ITCH, itchy E3 ubiquitin protein ligase; p-ERK1/2, phosphorylation of extracellular signal-regulated kinase 1/2; p-Smad3, phosphorylation of smad family member 3; p-AKT, phosphorylation of protein kinase B; p-FoxO1, phosphorylation of forkhead box O1; RhoA, ras homolog family member A; ROCK, Rho-associated coiled-coil containing kinases; SRF, serum response factor; CTGF, connective tissue growth factor.
The difference in cardiac hypertrophy responses of Tgfbr1/2 and Smad2 and Smad3 (Smad2/3) specific cardiac fibroblasts deficient mice to pressure overload stimulation indicates that atypical TGF-β signal also plays an effective role in cardiac fibroblast-mediated remodeling after chronic injury (166). It has been found that TGFβ-RhoA-ROCK pathway plays an important part in the formation of actin stress fiber, which is necessary for the differentiation of myofibroblasts (170–172). The phosphorylation of actin mediated by ROCK increases the number of actin monomers. Monomer actin loses its serum response factor (SRF) inhibitory activity. SRF and MRTF together activate the transcription of many genes, including profibrotic genes (13, 173). CFs specific deletion of ROCK2, a subtype of ROCK, significantly reduces the expression of α-SMA, decreases the expression of connective tissue growth factor (CTGF) and FGF2 in CFs, and improves cardiac function and fibrosis (174). CTGF is an important mediator in the differentiation of myofibroblasts stimulated by TGF-β (175, 176), as well as a major pathogenic factor in cardiac fibrosis (177). TGF-β has been known to increases CTGF expression in CFs (178). CTGF acts as an autocrine factor in CFs, and specific deletion of Ccn 2 (gene encoding CTGF) in CFs significantly improves cardiac function after Ang II infusion (179). Blocking the activation of TrkA, the primary receptor of CTGF, can prevent phenotype transformation and collagen production induced by HG in CFs, and CTGF knockdown shows the same result (165).
Interleukin (IL) is an important type of inflammatory cytokine. A variety of interleukin receptors are expressed in CFs, which regulate the activity of myofibroblasts (10, 22, 180, 181). It was found that the expressions of IL-6 (133), IL-17 (182), IL-1β (162) and IL-33 (183) were all up-regulated in myocardial fibroblasts attacked by HG, and loss of these inflammatory factors could reduce the collagen expression induced by HG. Under HG condition, the increased expression of IL-6 seems to up-regulate collagen gene by inducing TGF-β, which is sustained by down-regulating miR-29 by IL-6 (133). The deletion of IL-17 in diabetic mice prevents the increase of TGF-β expression (182). Therefore, interleukin mediates the ECM production of myocardial fibroblasts induced by HG (Figure 4), which seems to be related to the increase of TGF-β.
Calcium-sensitive receptor (CaSR) belongs to the superfamily of G protein-coupled receptors, whose main function is to regulate intracellular calcium level, and it is related to many diseases, including tumor, pulmonary hypertension and cardiac infarction. It was found that HG increased CaSR expression in CFs, enhancing the autophagy and proliferation of CFs (126). Activation of CaSR leads to the increase of intracellular Ca2+ concentration (126). Increased intracellular calcium promots the expression of Smurf2, which has the function of degrading SnoN and Smad7 proteins through ubiquitin proteasome signaling pathway (135, 136). Smad7 can degrade Smad2/3, prevent nuclear translocation and inhibit the activation of TGF-β/Smads signaling pathway (184). SnoN belongs to the SKI proto-oncogene family, which is able to inhibit the formation of binding complex between Smad2/3 and Smad4, thus inhibiting fibrosis (185). Up-regulation of Smurf2 degrades Smad7, which leads to the weakening of the inhibitory effect of Smad7 on TGF-β/Smad2/3 signaling pathway. Meanwhile, Smurf2-siRNA significantly decreased the expression of Beclin1 and LC3II and increases the expression of p62 in CFs, which indicated that Smurf2-siRNA can down-regulate autophagy and inhibit collagen production in CFs. Studies consistent with this have also shown that CaSR activation increases Ca2+ concentration in myocardial fibroblasts and up-regulates the expression of itchy E3 ubiquitin protein ligase (ITCH), causing an increase in the ubiquitination of Smad7 and up-regulation of p-Smad2 and p-Smad3 expressions, thus promoting fibrosis (Figure 4) (186).
The investigation of these signaling pathways of cardiac fibroblasts in diabetic cardiomyopathy could provide ideas and theoretical basis for preventing or even reversing cardiac fibrosis in DCM. Future studies will still focus on understanding the alterations of cardiac fibroblast signaling pathways in diabetic cardiomyopathy with a view to finding unique targets for cardiac fibroblasts.
To date, Clinical attention to myocardial fibrosis in DCMis far from enough, no consensus has been reached on the best management strategy for the prevention or treatment of DCM, and there is no standard drug treatment for DCM (187). But it is encouraging that the related research is increasing year by year (188). Previous studies have shown that Irbesartan inhibits CFs proliferation and ameliorates myocardial fibrosis in T2DM rats (168). Inhibition of SGLT1 expression in CFs improves DCM cardiac fibrosis by attenuating myofibroblasts activation (90), and the SGLT1/2 inhibitor sogliflozin (zynquista) has completed phase III clinical trials (189), suggesting a novel therapeutic strategy for the treatment of DCM fibrosis. It has also been found that DAPA, the new class of antidiabetic drugs, could inhibit cardiac fibroblasts activation and EndMT to prevent cardiac fibrosis via TGF-β/Smads signaling (89). A recent study showed that the diabetic basic therapeutic drug Metformin is able to attenuate profibrotic gene expression in rat aortic adventitial fibroblasts (190). It can be seen that the role of CFs in DCM fibrosis is gradually valued, and fully investigating the role of CFs in myocardial fibrosis of DCM will hopefully provide power assist on targets for the treatment and prevention of myocardial fibrosis in DCM.
Cardiac fibrosis is one of the most critical causes of morbidity and mortality in DCM, which can lead to heart failure and increase the incidence of arrhythmia events. Although DCM has been recognized for more than 40 years, the effective prevention and treatment strategies of DCM are still elusive. Enhanced CFs proliferation, increased collagen synthesis, and disordered MMPs synthesis are the results of long-term abnormalities in glucose and lipid metabolism, which disrupt the balance between ECM synthesis and degradation in DCM, and activate multiple mechanic pathways, and ultimately increase the stiffness of ventricular wall and cardiac fibrosis. Therefore, understanding sources and changes of CFs involved in the pathogenesis of cardiac fibrosis in DCM, as well as related molecular mechanism pathways, will hopefully provide the guidance for prevention and treatment of cardiac fibrosis in DCM.
YC and YW contributed equally to this work. RY, YX, LZ and YZ contributed to the manuscript revision. LY and DZ contributed to the manuscript design, discussion and revision. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (8217020350) and Natural Science Foundation of Beijing Municipality (7212055).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Johansson I, Norhammar A. Diabetes and heart failure notions from epidemiology including patterns in low-, middle- and high-income countries. Diabetes Res Clin Pract (2021) 177:108822. doi: 10.1016/j.diabres.2021.108822
2. Pearson-Stuttard J, Bennett J, Cheng Y, Vamos E, Cross A, Ezzati M, et al. Trends in predominant causes of death in individuals with and without diabetes in England from 2001 to 2018: An epidemiological analysis of linked primary care records. Lancet Diabetes Endocrinol (2021) 9(3):165–73. doi: 10.1016/s2213-8587(20)30431-9
3. Fang Z, Prins J, Marwick T. Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocr Rev (2004) 25(4):543–67. doi: 10.1210/er.2003-0012
4. Jia G, Hill M, Sowers J. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ Res (2018) 122(4):624–38. doi: 10.1161/circresaha.117.311586
5. Kannel W, Hjortland M, Castelli W. Role of diabetes in congestive heart failure: the framingham study. Am J Cardiol (1974) 34(1):29–34. doi: 10.1016/0002-9149(74)90089-7
6. Giugliano D, Bellastella G, Longo M, Scappaticcio L, Maiorino M, Chiodini P, et al. Relationship between improvement of glycaemic control and reduction of major cardiovascular events in 15 cardiovascular outcome trials: A meta-analysis with meta-regression. Diabetes Obes Metab (2020) 22(8):1397–405. doi: 10.1111/dom.14047
7. American Diabetes Association. Standards of medical care in diabetes-202110. cardiovascular disease and risk management. Diabetes Care (2021) 44:S125–50. doi: 10.2337/dc21-S010
8. Frangogiannis N. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med (2019) 65:70–99. doi: 10.1016/j.mam.2018.07.001
9. Frangogiannis N. The extracellular matrix in ischemic and nonischemic heart failure. Circ Res (2019) 125(1):117–46. doi: 10.1161/circresaha.119.311148
10. Frangogiannis N. Cardiac fibrosis. Cardiovasc Res (2021) 117(6):1450–88. doi: 10.1093/cvr/cvaa324
11. Ma T, Huang X, Zheng H, Huang G, Li W, Liu X, et al. SFRP2 improves mitochondrial dynamics and mitochondrial biogenesis, oxidative stress, and apoptosis in diabetic cardiomyopathy. Oxid Med Cell Longev (2021) 2021:9265016. doi: 10.1155/2021/9265016
12. Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature (2002) 417(6885):182–7. doi: 10.1038/417182a
14. Jia G, Whaley-Connell A, Sowers J. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia (2018) 61(1):21–8. doi: 10.1007/s00125-017-4390-4
15. Zhang Y, Cao Y, Zheng R, Xiong Z, Zhu Z, Gao F, et al. Fibroblast-specific activation of Rnd3 protects against cardiac remodeling in diabetic cardiomyopathy via suppression of notch and TGF-β signaling. Theranostics (2022) 12(17):7250–66. doi: 10.7150/thno.77043
16. Nakamura K, Miyoshi T, Yoshida M, Akagi S, Saito Y, Ejiri K, et al. Pathophysiology and treatment of diabetic cardiomyopathy and heart failure in patients with diabetes mellitus. Int J Mol Sci (2022) 23(7):3587. doi: 10.3390/ijms23073587
17. Tuleta I, Frangogiannis N. Fibrosis of the diabetic heart: Clinical significance, molecular mechanisms, and therapeutic opportunities. Adv Drug Del Rev (2021) 176:113904. doi: 10.1016/j.addr.2021.113904
18. Palomer X, Aguilar-Recarte D, García R, Nistal J, Vázquez-Carrera M. Sirtuins: To be or not to be in diabetic cardiomyopathy. Trends Mol Med (2021) 27(6):554–71. doi: 10.1016/j.molmed.2021.03.004
19. Asbun J, Villarreal F. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J Am Coll Cardiol (2006) 47(4):693–700. doi: 10.1016/j.jacc.2005.09.050
20. Russo I, Frangogiannis N. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol (2016) 90:84–93. doi: 10.1016/j.yjmcc.2015.12.011
21. Souders C, Bowers S, Baudino T. Cardiac fibroblast: the renaissance cell. Circ Res (2009) 105(12):1164–76. doi: 10.1161/circresaha.109.209809
22. Shinde A, Frangogiannis N. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol (2014) 70:74–82. doi: 10.1016/j.yjmcc.2013.11.015
23. Sanganalmath S, Dubey S, Veeranki S, Narisetty K, Krishnamurthy P. The interplay of inflammation, exosomes and Ca dynamics in diabetic cardiomyopathy. Cardiovasc Diabetol (2023) 22(1):37. doi: 10.1186/s12933-023-01755-1
24. Tian Y, Gao Z, Liu W, Li J, Jiang X, Xin Y. Unveiling the vital role of long non-coding RNAs in cardiac oxidative stress, cell death, and fibrosis in diabetic cardiomyopathy. Antioxid (Basel) (2022) 11(12):2391. doi: 10.3390/antiox11122391
25. Palomer X, Pizarro-Delgado J, Vázquez-Carrera M. Emerging actors in diabetic cardiomyopathy: Heartbreaker biomarkers or therapeutic targets? Trends Pharmacol Sci (2018) 39(5):452–67. doi: 10.1016/j.tips.2018.02.010
26. Tan Y, Zhang Z, Zheng C, Wintergerst K, Keller B, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol (2020) 17(9):585–607. doi: 10.1038/s41569-020-0339-2
27. Rubler S, Dlugash J, Yuceoglu Y, Kumral T, Branwood A, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol (1972) 30(6):595–602. doi: 10.1016/0002-9149(72)90595-4
28. Regan T, Lyons M, Ahmed S, Levinson G, Oldewurtel H, Ahmad M, et al. Evidence for cardiomyopathy in familial diabetes mellitus. J Clin Invest (1977) 60(4):884–99. doi: 10.1172/jci108843
29. Kong P, Christia P, Frangogiannis N. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci (2014) 71(4):549–74. doi: 10.1007/s00018-013-1349-6
30. Lorenzo-Almorós A, Tuñón J, Orejas M, Cortés M, Egido J, Lorenzo Ó. Diagnostic approaches for diabetic cardiomyopathy. Cardiovasc Diabetol (2017) 16(1):28. doi: 10.1186/s12933-017-0506-x
31. Jiang W, Xiong Y, Li X, Yang Y. Cardiac fibrosis: Cellular effectors, molecular pathways, and exosomal roles. Front Cardiovasc Med (2021) 8:3389/fcvm.2021.715258. doi: 10.3389/fcvm.2021.715258
32. Li L, Zhao Q, Kong W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol (2018) 68-69:490–506. doi: 10.1016/j.matbio.2018.01.013
33. Chan B, Roczkowsky A, Cho W, Poirier M, Sergi C, Keschrumrus V, et al. MMP inhibitors attenuate doxorubicin cardiotoxicity by preventing intracellular and extracellular matrix remodelling. Cardiovasc Res (2021) 117(1):188–200. doi: 10.1093/cvr/cvaa017
34. Iyer R, Patterson N, Fields G, Lindsey M. The history of matrix metalloproteinases: milestones, myths, and misperceptions. Am J Physiol Heart Circ Physiol (2012) 303(8):H919–30. doi: 10.1152/ajpheart.00577.2012. physiology, c.
35. Sedgwick B, Riches K, Bageghni S, O'Regan D, Porter K, Turner N. Investigating inherent functional differences between human cardiac fibroblasts cultured from nondiabetic and type 2 diabetic donors. Cardiovasc Pathol (2014) 23(4):204–10. doi: 10.1016/j.carpath.2014.03.004
36. Hutchinson K, Lord C, West T, Stewart J. Cardiac fibroblast-dependent extracellular matrix accumulation is associated with diastolic stiffness in type 2 diabetes. PloS One (2013) 8(8):e72080. doi: 10.1371/journal.pone.0072080
37. Fowlkes V, Clark J, Fix C, Law B, Morales M, Qiao X, et al. Type II diabetes promotes a myofibroblast phenotype in cardiac fibroblasts. Life Sci (2013) 92(11):669–76. doi: 10.1016/j.lfs.2013.01.003
38. Yang X, Zhao Z. miR-30a-5p inhibits the proliferation and collagen formation of cardiac fibroblasts in diabetic cardiomyopathy. Can J Physiol Pharmacol (2022) 100(2):167–75. doi: 10.1139/cjpp-2021-0280
39. Zhao T, Chen H, Cheng C, Zhang J, Yan Z, Kuang J, et al. Liraglutide protects high-glucose-stimulated fibroblasts by activating the CD36-JNK-AP1 pathway to downregulate P4HA1. BioMed Pharmacother (2019) 118:109224. doi: 10.1016/j.biopha.2019.109224
40. Gu X, Fang T, Kang P, Hu J, Yu Y, Li Z, et al. Effect of ALDH2 on high glucose-induced cardiac fibroblast oxidative stress, apoptosis, and fibrosis. Oxid Med Cell Longev (2017) 2017:9257967. doi: 10.1155/2017/9257967
41. Yue Y, Meng K, Pu Y, Zhang X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res Clin Pract (2017) 133:124–30. doi: 10.1016/j.diabres.2017.08.018
42. Hu H, Chen D, Wang Y, Feng Y, Cao G, Vaziri N, et al. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem Biol Interact (2018) 292:76–83. doi: 10.1016/j.cbi.2018.07.008
43. Hanna A, Humeres C, Frangogiannis N. The role of smad signaling cascades in cardiac fibrosis. Cell Signal (2021) 77:109826. doi: 10.1016/j.cellsig.2020.109826
44. Lee S, Kim D, Lee J, Lee H, Moon R, Lee Y, et al. Dapagliflozin attenuates diabetes-induced diastolic dysfunction and cardiac fibrosis by regulating SGK1 signaling. BMC Med (2022) 20(1):309. doi: 10.1186/s12916-022-02485-z
45. Ross S, Hill C. How the smads regulate transcription. Int J Biochem Cell Biol (2008) 40(3):383–408. doi: 10.1016/j.biocel.2007.09.006
46. Liu Z, Zhang Y, Wang X, Fan X, Zhang Y, Li X, et al. SIRT1 activation attenuates cardiac fibrosis by endothelial-to-mesenchymal transition. BioMed Pharmacother (2019) 118:109227. doi: 10.1016/j.biopha.2019.109227
47. Jia G, DeMarco V, Sowers J. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol (2016) 12(3):144–53. doi: 10.1038/nrendo.2015.216
48. Shirpoor A, Salami S, Khadem-Ansari M, Ilkhanizadeh B, Pakdel F, Khademvatani K, et al. Cardioprotective effect of vitamin e: rescues of diabetes-induced cardiac malfunction, oxidative stress, and apoptosis in rat. J Diabetes Complications (2009) 23(5):310–6. doi: 10.1016/j.jdiacomp.2008.02.009
49. Zhang Q, Li D, Dong X, Zhang X, Liu J, Peng L, et al. LncDACH1 promotes mitochondrial oxidative stress of cardiomyocytes by interacting with sirtuin3 and aggravates diabetic cardiomyopathy. Sci China Life Sci (2022) 65(6):1198–212. doi: 10.1007/s11427-021-1982-8
50. Ramírez E, Klett-Mingo M, Ares-Carrasco S, Picatoste B, Ferrarini A, Rupérez F, et al. Eplerenone attenuated cardiac steatosis, apoptosis and diastolic dysfunction in experimental type-II diabetes. Cardiovasc Diabetol (2013) 12:172. doi: 10.1186/1475-2840-12-172
51. Prabhu S, Frangogiannis N. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ Res (2016) 119(1):91–112. doi: 10.1161/circresaha.116.303577
52. Cojan-Minzat B, Zlibut A, Agoston-Coldea L. Non-ischemic dilated cardiomyopathy and cardiac fibrosis. Heart Fail Rev (2021) 26(5):1081–101. doi: 10.1007/s10741-020-09940-0
53. Cheng Y, Zhang Z, Lan B, Lin J, Chen X, Kong L, et al. PDGF-d activation by macrophage-derived uPA promotes AngII-induced cardiac remodeling in obese mice. J Exp Med (2021) 218(9):e20210252. doi: 10.1084/jem.20210252
54. Yepuri G, Hasan S, Schmidt A, Ramasamy R. Macrophage-adipocyte communication and cardiac remodeling. J Exp Med (2021) 218(9):e20211098. doi: 10.1084/jem.20211098
55. Yang M, Zheng J, Miao Y, Wang Y, Cui W, Guo J, et al. Serum-glucocorticoid regulated kinase 1 regulates alternatively activated macrophage polarization contributing to angiotensin II-induced inflammation and cardiac fibrosis. Arterioscler Thromb Vasc Biol (2012) 32(7):1675–86. doi: 10.1161/atvbaha.112.248732
56. Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity (2005) 23(4):344–6. doi: 10.1016/j.immuni.2005.10.001
57. Braitsch C, Kanisicak O, van Berlo J, Molkentin J, Yutzey K. Differential expression of embryonic epicardial progenitor markers and localization of cardiac fibrosis in adult ischemic injury and hypertensive heart disease. J Mol Cell Cardiol (2013) 65:108–19. doi: 10.1016/j.yjmcc.2013.10.005
58. Nevers T, Salvador A, Velazquez F, Ngwenyama N, Carrillo-Salinas F, Aronovitz M, et al. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med (2017) 214(11):3311–29. doi: 10.1084/jem.20161791
59. Bansal S, Ismahil M, Goel M, Patel B, Hamid T, Rokosh G, et al. Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ Heart Fail (2017) 10(3):e003688. doi: 10.1161/circheartfailure.116.003688
60. Legere S, Haidl I, Légaré J, Marshall J. Mast cells in cardiac fibrosis: New insights suggest opportunities for intervention. Front Immunol (2019) 10:3389/fimmu.2019.00580. doi: 10.3389/fimmu.2019.00580
61. Xia Y, Lee K, Li N, Corbett D, Mendoza L, Frangogiannis N, et al. Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol (2009) 131(4):471–81. doi: 10.1007/s00418-008-0541-5
62. Piera-Velazquez S, Jimenez S. Endothelial to mesenchymal transition: Role in physiology and in the pathogenesis of human diseases. Physiol Rev (2019) 99(2):1281–324. doi: 10.1152/physrev.00021.2018
63. Wilhelmi T, Xu X, Tan X, Hulshoff M, Maamari S, Sossalla S, et al. Serelaxin alleviates cardiac fibrosis through inhibiting endothelial-to-mesenchymal transition via RXFP1. Theranostics (2020) 10(9):3905–24. doi: 10.7150/thno.38640
64. Zeisberg E, Tarnavski O, Zeisberg M, Dorfman A, McMullen J, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med (2007) 13(8):952–61. doi: 10.1038/nm1613
65. Du J, Yu Q, Liu Y, Du S, Huang L, Xu D, et al. A novel role of kallikrein-related peptidase 8 in the pathogenesis of diabetic cardiac fibrosis. Theranostics (2021) 11(9):4207–31. doi: 10.7150/thno.48530
66. Geng H, Guan J. MiR-18a-5p inhibits endothelial-mesenchymal transition and cardiac fibrosis through the Notch2 pathway. Biochem Biophys Res Commun (2017) 491(2):329–36. doi: 10.1016/j.bbrc.2017.07.101
67. Zheng X, Peng M, Li Y, Wang X, Lu W, Wang X, et al. Cathelicidin-related antimicrobial peptide protects against cardiac fibrosis in diabetic mice heart by regulating endothelial-mesenchymal transition. Int J Biol Sci (2019) 15(11):2393–407. doi: 10.7150/ijbs.35736
68. Li Q, Yao Y, Shi S, Zhou M, Zhou Y, Wang M, et al. Inhibition of miR-21 alleviated cardiac perivascular fibrosis via repressing EndMT in T1DM. J Cell Mol Med (2020) 24(1):910–20. doi: 10.1111/jcmm.14800
69. Wang B, Wu Y, Ge Z, Zhang X, Yan Y, Xie Y, et al. NLRC5 deficiency ameliorates cardiac fibrosis in diabetic cardiomyopathy by regulating EndMT through Smad2/3 signaling pathway. Biochem Biophys Res Commun (2020) 528(3):545–53. doi: 10.1016/j.bbrc.2020.05.151
70. Wang Z, Wang Z, Gao L, Xiao L, Yao R, Du B, et al. miR-222 inhibits cardiac fibrosis in diabetic mice heart via regulating wnt/β-catenin-mediated endothelium to mesenchymal transition. J Cell Physiol (2020) 235(3):2149–60. doi: 10.1002/jcp.29119
71. Takagaki Y, Lee S, Dongqing Z, Kitada M, Kanasaki K, Koya D. Endothelial autophagy deficiency induces IL6 - dependent endothelial mesenchymal transition and organ fibrosis. Autophagy (2020) 16(10):1905–14. doi: 10.1080/15548627.2020.1713641
72. Zhang L, He J, Wang J, Liu J, Chen Z, Deng B, et al. Knockout RAGE alleviates cardiac fibrosis through repressing endothelial-to-mesenchymal transition (EndMT) mediated by autophagy. Cell Death Dis (2021) 12(5):470. doi: 10.1038/s41419-021-03750-4
73. Gao W, Zhou Z, Liang B, Huang Y, Yang Z, Chen Y, et al. Inhibiting receptor of advanced glycation end products attenuates pressure overload-induced cardiac dysfunction by preventing excessive autophagy. Front Physiol (2018) 9:3389/fphys.2018.01333. doi: 10.3389/fphys.2018.01333
74. Ma Z, Yuan Y, Wu H, Zhang X, Tang Q. Cardiac fibrosis: new insights into the pathogenesis. Int J Biol Sci (2018) 14(12):1645–57. doi: 10.7150/ijbs.28103
75. Adiarto S, Heiden S, Vignon-Zellweger N, Nakayama K, Yagi K, Yanagisawa M, et al. ET-1 from endothelial cells is required for complete angiotensin II-induced cardiac fibrosis and hypertrophy. Life Sci (2012) 91:651–7. doi: 10.1016/j.lfs.2012.02.006
76. Widyantoro B, Emoto N, Nakayama K, Anggrahini D, Adiarto S, Iwasa N, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation (2010) 121(22):2407–18. doi: 10.1161/circulationaha.110.938217
77. Piacentini L, Gray M, Honbo N, Chentoufi J, Bergman M, Karliner J, et al. Endothelin-1 stimulates cardiac fibroblast proliferation through activation of protein kinase c. J Mol Cell Cardiol (2000) 32(4):565–76. doi: 10.1006/jmcc.2000.1109
78. Shi-Wen X, Denton C, Dashwood M, Holmes A, Bou-Gharios G, Pearson J, et al. Fibroblast matrix gene expression and connective tissue remodeling: role of endothelin-1. J Invest Dermatol (2001) 116(3):417–25. doi: 10.1046/j.1523-1747.2001.01256.x
79. Shi-wen X, Kennedy L, Renzoni E, Bou-Gharios G, du Bois R, Black C, et al. Endothelin is a downstream mediator of profibrotic responses to transforming growth factor beta in human lung fibroblasts. Arthritis Rheum (2007) 56(12):4189–94. doi: 10.1002/art.23134
80. Hafizi S, Wharton J, Chester A, Yacoub M. Profibrotic effects of endothelin-1 via the ETA receptor in cultured human cardiac fibroblasts. Cell Physiol Biochem (2004) 14:285–92. doi: 10.1159/000080338
81. Qin L, Mei Y, An C, Ning R, Zhang H. Docosahexaenoic acid administration improves diabetes-induced cardiac fibrosis through enhancing fatty acid oxidation in cardiac fibroblast. J Nutr Biochem (2023) 113:109244. doi: 10.1016/j.jnutbio.2022.109244
82. Li H, Zhu X, Cao X, Lu Y, Zhou J, Zhang X. Single-cell analysis reveals lysyl oxidase (Lox) fibroblast subset involved in cardiac fibrosis of diabetic mice. J Adv Res (2023) S2090-1232(23)00031-0. doi: 10.1016/j.jare.2023.01.018
83. Cheng J, Xue F, Cheng C, Sui W, Zhang M, Qiao L, et al. viaADAM17 knockdown mitigates while ADAM17 overexpression aggravates cardiac fibrosis and dysfunction regulating ACE2 shedding and myofibroblast transformation. Front Pharmacol (2022) 13:3389/fphar.2022.997916. doi: 10.3389/fphar.2022.997916
84. Liu Q, Deng C, Xing X, Hu Y, Wang Z, Liang Y. Silencing RIPK1/mTORC1 signalling attenuated the inflammation and oxidative stress in diabetic cardiomyopathy. Exp Cell Res (2023) 422(2):113417. doi: 10.1016/j.yexcr.2022.113417
85. Tao H, Shi P, Xuan H, Ding X. DNA Methyltransferase-1 inactivation of androgen receptor axis triggers homocysteine induced cardiac fibroblast autophagy in diabetic cardiac fibrosis. Arch Biochem Biophys (2020) 692:108521. doi: 10.1016/j.abb.2020.108521
86. Tao H, Tao J, Song Z, Shi P, Wang Q, Deng Z, et al. MeCP2 triggers diabetic cardiomyopathy and cardiac fibroblast proliferation by inhibiting RASSF1A. Cell Signal (2019) 63:109387. doi: 10.1016/j.cellsig.2019.109387
87. Yuan H, Fan Y, Wang Y, Gao T, Shao Y, Zhao B, et al. Calcium−sensing receptor promotes high glucose−induced myocardial fibrosis via upregulation of the TGF−β1/Smads pathway in cardiac fibroblasts. Mol Med Rep (2019) 20(2):1093–102. doi: 10.3892/mmr.2019.10330
88. Zhang D, Li Y, Wang W, Lang X, Zhang Y, Zhao Q, et al. viaNOX1 promotes myocardial fibrosis and cardiac dysfunction activating the TLR2/NF-κB pathway in diabetic cardiomyopathy. Front Pharmacol (2022) 13:3389/fphar.2022.928762. doi: 10.3389/fphar.2022.928762
89. Tian J, Zhang M, Suo M, Liu D, Wang X, Liu M, et al. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J Cell Mol Med (2021) 25(16):7642–59. doi: 10.1111/jcmm.16601
90. Lin H, Guan L, Meng L, Uzui H, Guo H. SGLT1 knockdown attenuates cardiac fibroblast activation in diabetic cardiac fibrosis. Front Pharmacol (2021) 12:3389/fphar.2021.700366. doi: 10.3389/fphar.2021.700366
91. Bowers S, Banerjee I, Baudino T. The extracellular matrix: at the center of it all. J Mol Cell Cardiol (2010) 48(3):474–82. doi: 10.1016/j.yjmcc.2009.08.024
93. Camelliti P, Borg T, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res (2005) 65(1):40–51. doi: 10.1016/j.cardiores.2004.08.020
94. Tallquist M. Cardiac fibroblast diversity. Annu Rev Physiol (2020) 82:63–78. doi: 10.1146/annurev-physiol-021119-034527
95. Pinto A, Ilinykh A, Ivey M, Kuwabara J, D'Antoni M, Debuque R, et al. Revisiting cardiac cellular composition. Circ Res (2016) 118(3):400–9. doi: 10.1161/circresaha.115.307778
96. Zhou P, Pu W. Recounting cardiac cellular composition. Circ Res (2016) 118(3):368–70. doi: 10.1161/circresaha.116.308139
97. Cai C, Martin J, Sun Y, Cui L, Wang L, Ouyang K, et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature (2008) 454(7200):104–8. doi: 10.1038/nature06969
98. Zhou B, Ma Q, Rajagopal S, Wu S, Domian I, Rivera-Feliciano J, et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature (2008) 454(7200):109–13. doi: 10.1038/nature07060
99. Smith C, Baek S, Sung C, Tallquist M. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ Res (2011) 108(12):e15–26. doi: 10.1161/circresaha.110.235531
100. Acharya A, Baek S, Huang G, Eskiocak B, Goetsch S, Sung C, et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development (2012) 139(12):2139–49. doi: 10.1242/dev.079970
101. Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon A, Kisseleva T, Velayoudon A, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest (2014) 124(7):2921–34. doi: 10.1172/jci74783
102. Doppler S, Carvalho C, Lahm H, Deutsch M, Dreßen M, Puluca N, et al. Cardiac fibroblasts: more than mechanical support. J Thorac Dis (2017) 9:S36–51. doi: 10.21037/jtd.2017.03.122
103. Kisanuki Y, Hammer R, Miyazaki J, Williams S, Richardson J, Yanagisawa M. Tie2-cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol (2001) 230(2):230–42. doi: 10.1006/dbio.2000.0106
104. Ali S, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res (2014) 115(7):625–35. doi: 10.1161/circresaha.115.303794
105. Kanisicak O, Khalil H, Ivey M, Karch J, Maliken B, Correll R, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun (2016) 7:12260. doi: 10.1038/ncomms12260
106. Wu R, Ma F, Tosevska A, Farrell C, Pellegrini M, Deb A. Cardiac fibroblast proliferation rates and collagen expression mature early and are unaltered with advancing age. JCI Insight (2020) 5(24):e140628. doi: 10.1172/jci.insight.140628
107. Travers J, Kamal F, Robbins J, Yutzey K, Blaxall B. Cardiac fibrosis: The fibroblast awakens. Circ Res (2016) 118(6):1021–40. doi: 10.1161/circresaha.115.306565
108. Brown R, Ambler S, Mitchell M, Long C. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol (2005) 45:657–87. doi: 10.1146/annurev.pharmtox.45.120403.095802
109. Spinale F. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev (2007) 87(4):1285–342. doi: 10.1152/physrev.00012.2007
110. Huang S, Chen B, Humeres C, Alex L, Hanna A, Frangogiannis N. The role of Smad2 and Smad3 in regulating homeostatic functions of fibroblasts in vitro and in adult mice. Biochim Biophys Acta Mol Cell Res (2020) 1867(7):118703. doi: 10.1016/j.bbamcr.2020.118703
111. Ivey M, Kuwabara J, Riggsbee K, Tallquist M. Platelet-derived growth factor receptor-α is essential for cardiac fibroblast survival. Am J Physiol Heart Circ Physiol (2019) 317(2):H330–44. doi: 10.1152/ajpheart.00054.2019
112. Squiers G, McLellan M, Ilinykh A, Branca J, Rosenthal N, Pinto A. Cardiac cellularity is dependent upon biological sex and is regulated by gonadal hormones. Cardiovasc Res (2021) 117(10):2252–62. doi: 10.1093/cvr/cvaa265
113. Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair (2012) 5(1):15. doi: 10.1186/1755-1536-5-15
114. Gourdie R, Dimmeler S, Kohl P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat Rev Drug Discov (2016) 15(9):620–38. doi: 10.1038/nrd.2016.89
115. Cartledge J, Kane C, Dias P, Tesfom M, Clarke L, Mckee B, et al. Functional crosstalk between cardiac fibroblasts and adult cardiomyocytes by soluble mediators. Cardiovasc Res (2015) 105(3):260–70. doi: 10.1093/cvr/cvu264
116. Furtado M, Costa M, Pranoto E, Salimova E, Pinto A, Lam N, et al. Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ Res (2014) 114(9):1422–34. doi: 10.1161/circresaha.114.302530
117. van Tuyn J, Atsma D, Winter E, van der Velde-van Dijke I, Pijnappels D, Bax N, et al. Epicardial cells of human adults can undergo an epithelial-to-mesenchymal transition and obtain characteristics of smooth muscle cells in vitro. Stem Cells (2007) 25(2):271–8. doi: 10.1634/stemcells.2006-0366
118. Russell J, Goetsch S, Gaiano N, Hill J, Olson E, Schneider J. A dynamic notch injury response activates epicardium and contributes to fibrosis repair. Circ Res (2011) 108(1):51–9. doi: 10.1161/circresaha.110.233262
119. Zhou B, Pu W. Epicardial epithelial-to-mesenchymal transition in injured heart. J Cell Mol Med (2011) 15(12):2781–3. doi: 10.1111/j.1582-4934.2011.01450.x
120. Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, et al. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J (2012) 31(2):429–42. doi: 10.1038/emboj.2011.418
121. Fu X, Khalil H, Kanisicak O, Boyer J, Vagnozzi R, Maliken B, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest (2018) 128(5):2127–43. doi: 10.1172/jci98215
122. Haider N, Boscá L, Zandbergen H, Kovacic J, Narula N, González-Ramos S, et al. Transition of macrophages to fibroblast-like cells in healing myocardial infarction. J Am Coll Cardiol (2019) 74(25):3124–35. doi: 10.1016/j.jacc.2019.10.036
123. van Amerongen M, Bou-Gharios G, Popa E, van Ark J, Petersen A, van Dam G, et al. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J Pathol (2008) 214(3):377–86. doi: 10.1002/path.2281
124. Tuleta I, Frangogiannis N. Diabetic fibrosis. Biochim Biophys Acta Mol Basis Dis (2021) 1867(4):166044. doi: 10.1016/j.bbadis.2020.166044
125. Li Y, Lui K, Zhou B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol (2018) 15(8):445–56. doi: 10.1038/s41569-018-0023-y
126. Yuan H, Xu J, Zhu Y, Li L, Wang Q, Yu Y, et al. Activation of calcium−sensing receptor−mediated autophagy in high glucose−induced cardiac fibrosis in vitro. Mol Med Rep (2020) 22(3):2021–31. doi: 10.3892/mmr.2020.11277
127. Cook S, Varela-Carver A, Mongillo M, Kleinert C, Khan M, Leccisotti L, et al. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur Heart J (2010) 31(1):100–11. doi: 10.1093/eurheartj/ehp396
128. Bugger H, Riehle C, Jaishy B, Wende A, Tuinei J, Chen D, et al. Genetic loss of insulin receptors worsens cardiac efficiency in diabetes. J Mol Cell Cardiol (2012) 52(5):1019–26. doi: 10.1016/j.yjmcc.2012.02.001
129. Qi Y, Xu Z, Zhu Q, Thomas C, Kumar R, Feng H, et al. Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38α MAPK during insulin resistance. Diabetes (2013) 62(11):3887–900. doi: 10.2337/db13-0095
130. Fiaschi T, Magherini F, Gamberi T, Lucchese G, Faggian G, Modesti A, et al. Hyperglycemia and angiotensin II cooperate to enhance collagen I deposition by cardiac fibroblasts through a ROS-STAT3-dependent mechanism. Biochim Biophys Acta (2014) 1843(11):2603–10. doi: 10.1016/j.bbamcr.2014.07.009
131. Singh V, Baker K, Kumar R. Activation of the intracellular renin-angiotensin system in cardiac fibroblasts by high glucose: Role in extracellular matrix production. Am J Physiol Heart Circ Physiol (2008) 294(4):H1675–84. doi: 10.1152/ajpheart.91493.2007
132. Tokudome T, Horio T, Yoshihara F, Suga S, Kawano Y, Kohno M, et al. Direct effects of high glucose and insulin on protein synthesis in cultured cardiac myocytes and DNA and collagen synthesis in cardiac fibroblasts. Metabolism (2004) 53(6):710–5. doi: 10.1016/j.metabol.2004.01.006
133. Zhang Y, Wang J, Zhang Y, Wang Y, Wang J, Zhao Y, et al. Deletion of interleukin-6 alleviated interstitial fibrosis in streptozotocin-induced diabetic cardiomyopathy of mice through affecting TGFβ1 and miR-29 pathways. Sci Rep (2016) 6:23010. doi: 10.1038/srep23010
134. Cavalera M, Wang J, Frangogiannis N. Obesity, metabolic dysfunction, and cardiac fibrosis: Pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl Res (2014) 164(4):323–35. doi: 10.1016/j.trsl.2014.05.001
135. Lönn P, Vanlandewijck M, Raja E, Kowanetz M, Watanabe Y, Kowanetz K, et al. Transcriptional induction of salt-inducible kinase 1 by transforming growth factor β leads to negative regulation of type I receptor signaling in cooperation with the Smurf2 ubiquitin ligase. J Biol Chem (2012) 287(16):12867–78. doi: 10.1074/jbc.M111.307249
136. Cai Y, Shen X, Zhou C, Wang J. Abnormal expression of Smurf2 during the process of rat liver fibrosis. Chin J Dig Dis (2006) 7(4):237–45. doi: 10.1111/j.1443-9573.2006.00275.x
137. Duran J, Troncoso M, Lagos D, Ramos S, Marin G, Estrada M. GDF11 modulates Ca-dependent Smad2/3 signaling to prevent cardiomyocyte hypertrophy. Int J Mol Sci (2018) 19(5):1508. doi: 10.3390/ijms19051508
138. Tao H, Shi P, Zhao X, Xuan H, Gong W, Ding X. DNMT1 deregulation of SOCS3 axis drives cardiac fibroblast activation in diabetic cardiac fibrosis. J Cell Physiol (2021) 236(5):3481–94. doi: 10.1002/jcp.30078
139. Gorski D, Petz A, Reichert C, Twarock S, Grandoch M, Fischer J. Cardiac fibroblast activation and hyaluronan synthesis in response to hyperglycemia and diet-induced insulin resistance. Sci Rep (2019) 9(1):1827. doi: 10.1038/s41598-018-36140-6
140. Ritterhoff J, Tian R. Metabolism in cardiomyopathy: every substrate matters. Cardiovasc Res (2017) 113(4):411–21. doi: 10.1093/cvr/cvx017
141. Alex L, Russo I, Holoborodko V, Frangogiannis N. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol (2018) 315(4):H934–49. doi: 10.1152/ajpheart.00238.2018
142. Qureshi R, Kindo M, Arora H, Boulberdaa M, Steenman M, Nebigil C. Prokineticin receptor-1-dependent paracrine and autocrine pathways control cardiac tcf21 fibroblast progenitor cell transformation into adipocytes and vascular cells. Sci Rep (2017) 7(1):12804. doi: 10.1038/s41598-017-13198-2
143. Lazar M. Resistin- and obesity-associated metabolic diseases. Horm Metab Res (2007) 39(10):710–6. doi: 10.1055/s-2007-985897
144. Steppan C, Bailey S, Bhat S, Brown E, Banerjee R, Wright C, et al. The hormone resistin links obesity to diabetes. Nature (2001) 409(6818):307–12. doi: 10.1038/35053000
145. Chemaly E, Kang S, Zhang S, McCollum L, Chen J, Bénard L, et al. Differential patterns of replacement and reactive fibrosis in pressure and volume overload are related to the propensity for ischaemia and involve resistin. J Physiol (2013) 591(21):5337–55. doi: 10.1113/jphysiol.2013.258731
146. Singh R, Kaundal R, Zhao B, Bouchareb R, Lebeche D. Resistin induces cardiac fibroblast-myofibroblast differentiation through JAK/STAT3 and JNK/c-jun signaling. Pharmacol Res (2021) 167:105414. doi: 10.1016/j.phrs.2020.105414
147. Langhi C, Arias N, Rajamoorthi A, Basta J, Lee R, Baldán Á. Therapeutic silencing of fat-specific protein 27 improves glycemic control in mouse models of obesity and insulin resistance. J Lipid Res (2017) 58(1):81–91. doi: 10.1194/jlr.M069799
148. Tan X, Cao Z, Li M, Xu E, Wang J, Xiao Y. TNF-α downregulates CIDEC via MEK/ERK pathway in human adipocytes. Obes (Silver Spring) (2016) 24(5):1070–80. doi: 10.1002/oby.21436
149. Zhou H, Ti Y, Wang H, Shang Y, Liu Y, Ni X, et al. Cell death-inducing DFFA-like effector C/CIDEC gene silencing alleviates diabetic cardiomyopathy via upregulating AMPKa phosphorylation. FASEB J (2021) 35(5):e21504. doi: 10.1096/fj.202002562R
150. Simm A, Wagner J, Gursinsky T, Nass N, Friedrich I, Schinzel R, et al. Advanced glycation endproducts: a biomarker for age as an outcome predictor after cardiac surgery? Exp Gerontol (2007) 42(7):668–75. doi: 10.1016/j.exger.2007.03.006
151. Hegab Z, Gibbons S, Neyses L, Mamas M. Role of advanced glycation end products in cardiovascular disease. World J Cardiol (2012) 4(4):90–102. doi: 10.4330/wjc.v4.i4.90
152. Tang M, Zhong M, Shang Y, Lin H, Deng J, Jiang H, et al. Differential regulation of collagen types I and III expression in cardiac fibroblasts by AGEs through TRB3/MAPK signaling pathway. Cell Mol Life Sci (2008) 65(18):2924–32. doi: 10.1007/s00018-008-8255-3
153. Kato T, Yamashita T, Sekiguchi A, Tsuneda T, Sagara K, Takamura M, et al. AGEs-RAGE system mediates atrial structural remodeling in the diabetic rat. J Cardiovasc Electrophysiol (2008) 19(4):415–20. doi: 10.1111/j.1540-8167.2007.01037.x
154. Umadevi S, Gopi V, Elangovan V. Regulatory mechanism of gallic acid against advanced glycation end products induced cardiac remodeling in experimental rats. Chem Biol Interact (2014) 208:28–36. doi: 10.1016/j.cbi.2013.11.013
155. Burr S, Stewart J. Extracellular matrix components isolated from diabetic mice alter cardiac fibroblast function through the AGE/RAGE signaling cascade. Life Sci (2020) 250:117569. doi: 10.1016/j.lfs.2020.117569
156. Shinde A, Humeres C, Frangogiannis N. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim Biophys Acta Mol Basis Dis (2017) 1863(1):298–309. doi: 10.1016/j.bbadis.2016.11.006
157. Gao X, Zhang H, Schmidt A, Zhang C. AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabetic mice. Am J Physiol Heart Circ Physiol (2008) 295(2):H491–8. doi: 10.1152/ajpheart.00464.2008
158. Burr S, Harmon M. The impact of diabetic conditions and AGE/RAGE signaling on cardiac fibroblast migration. Front Cell Dev Biol (2020) 8:3389/fcell.2020.00112. doi: 10.3389/fcell.2020.00112
159. Berg T, Snorgaard O, Faber J, Torjesen P, Hildebrandt P, Mehlsen J, et al. Serum levels of advanced glycation end products are associated with left ventricular diastolic function in patients with type 1 diabetes. Diabetes Care (1999) 22(7):1186–90. doi: 10.2337/diacare.22.7.1186
160. Ramasamy R, Yan S, Schmidt A. Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci (2011) 1243:88–102. doi: 10.1111/j.1749-6632.2011.06320.x
161. Turner N. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J Mol Cell Cardiol (2016) 94:189–200. doi: 10.1016/j.yjmcc.2015.11.002
162. Che H, Wang Y, Li Y, Lv J, Li H, Liu Y, et al. Inhibition of microRNA-150-5p alleviates cardiac inflammation and fibrosis via targeting Smad7 in high glucose-treated cardiac fibroblasts. J Cell Physiol (2020) 235(11):7769–79. doi: 10.1002/jcp.29386
163. Tsai Y, Loh S, Lee C, Lee S, Chen Y, Cheng T, et al. Tanshinone IIA inhibits high glucose-induced collagen synthesis via nuclear factor erythroid 2-related factor 2 in cardiac fibroblasts. Cell Physiol Biochem (2018) 51(5):2250–61. doi: 10.1159/000495870
164. Fix C, Carver-Molina A, Chakrabarti M, Azhar M, Carver W. Effects of the isothiocyanate sulforaphane on TGF-β1-induced rat cardiac fibroblast activation and extracellular matrix interactions. J Cell Physiol (2019) 234(8):13931–41. doi: 10.1002/jcp.28075
165. Vivar R, Anfossi R, Humeres C, Catalán M, Reyes C, Cárdenas S, et al. FoxO1 is required for high glucose-dependent cardiac fibroblasts into myofibroblast phenoconversion. Cell Signal (2021) 83:109978. doi: 10.1016/j.cellsig.2021.109978
166. Khalil H, Kanisicak O, Prasad V, Correll R, Fu X, Schips T, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest (2017) 127(10):3770–83. doi: 10.1172/jci94753
167. Wang J, Gareri C, Rockman H. G-Protein-Coupled receptors in heart disease. Circ Res (2018) 123(6):716–35. doi: 10.1161/circresaha.118.311403
168. Zong M, Zhao H, Li Q, Li Y, Zhang J. Irbesartan ameliorates myocardial fibrosis in diabetic cardiomyopathy rats by inhibiting the TGFβ1/Smad2/3 pathway. Exp Ther Med (2020) 20(5):117. doi: 10.3892/etm.2020.9245
169. Vivar R, Humeres C, Anfossi R, Bolivar S, Catalán M, Hill J, et al. Role of FoxO3a as a negative regulator of the cardiac myofibroblast conversion induced by TGF-β1. Biochim Biophys Acta Mol Cell Res (2020) 1867(7):118695. doi: 10.1016/j.bbamcr.2020.118695
170. Costanza B, Umelo I, Bellier J, Castronovo V, Turtoi A. Stromal modulators of TGF-β in cancer. J Clin Med (2017) 6(1):7. doi: 10.3390/jcm6010007
171. Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell (2003) 113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x
172. Bhowmick N, Ghiassi M, Bakin A, Aakre M, Lundquist C, Engel M, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell (2001) 12(1):27–36. doi: 10.1091/mbc.12.1.27
173. Lisabeth E, Kahl D, Gopallawa I, Haynes S, Misek S, Campbell P, et al. Identification of pirin as a molecular target of the CCG-1423/CCG-203971 series of antifibrotic and antimetastatic compounds. CS Pharmacol Transl Sci (2019) 2(2):92–100. doi: 10.1021/acsptsci.8b00048
174. Shimizu T, Narang N, Chen P, Yu B, Knapp M, Janardanan J, et al. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight (2017) 2(13):e93187. doi: 10.1172/jci.insight.93187
175. Shi-wen X, Stanton L, Kennedy L, Pala D, Chen Y, Howat S, et al. CCN2 is necessary for adhesive responses to transforming growth factor-beta1 in embryonic fibroblasts. J Biol Chem (2006) 281(16):10715–26. doi: 10.1074/jbc.M511343200
176. Mori T, Kawara S, Shinozaki M, Hayashi N, Kakinuma T, Igarashi A, et al. Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: A mouse fibrosis model. J Cell Physiol (1999) 181(1):153–9. doi: 10.1002/(sici)1097-4652(199910)181:1<153::Aid-jcp16>3.0.Co;2-k
177. Daniels A, van Bilsen M, Goldschmeding R, van der Vusse G, van Nieuwenhoven F. Connective tissue growth factor and cardiac fibrosis. Acta Physiol (Oxf) (2009) 195(3):321–38. doi: 10.1111/j.1748-1716.2008.01936.x
178. Gu J, Liu X, Wang Q, Tan H, Guo M, Jiang W, et al. Angiotensin II increases CTGF expression via MAPKs/TGF-β1/TRAF6 pathway in atrial fibroblasts. Exp Cell Res (2012) 318(16):2105–15. doi: 10.1016/j.yexcr.2012.06.015
179. Dorn L, Petrosino J, Wright P, Accornero F. CTGF/CCN2 is an autocrine regulator of cardiac fibrosis. J Mol Cell Cardiol (2018) 121:205–11. doi: 10.1016/j.yjmcc.2018.07.130
180. Francis Stuart S, De Jesus N, Lindsey M, Ripplinger C. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J Mol Cell Cardiol (2016) 91:114–22. doi: 10.1016/j.yjmcc.2015.12.024
181. Turner N. Effects of interleukin-1 on cardiac fibroblast function: relevance to post-myocardial infarction remodelling. Vascul Pharmacol (2014) 60(1):1–7. doi: 10.1016/j.vph.2013.06.002
182. Zhang Y, Zhang Y, Li T, Wang J, Jiang Y, Zhao Y, et al. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J Mol Cell Cardiol (2018) 115:64–72. doi: 10.1016/j.yjmcc.2018.01.001
183. Tao A, Song J, Lan T, Xu X, Kvietys P, Kao R, et al. Cardiomyocyte-fibroblast interaction contributes to diabetic cardiomyopathy in mice: Role of HMGB1/TLR4/IL-33 axis. Biochim Biophys Acta (2015) 1852:2075–85. doi: 10.1016/j.bbadis.2015.07.015
184. Lan H. Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int J Biol Sci (2011) 7(7):1056–67. doi: 10.7150/ijbs.7.1056
185. Deheuninck J, Luo K. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res (2009) 19(1):47–57. doi: 10.1038/cr.2008.324
186. Yuan H, Xu J, Xu X, Gao T, Wang Y, Fan Y, et al. Calhex alleviates high glucose-induced myocardial fibrosis via inhibiting itch-ubiquitin proteasome pathway in vitro. Biol Pharm Bull (2019) 42(8):1337–44. doi: 10.1248/bpb.b19-00090
187. Kaludercic N, Di Lisa F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front Cardiovasc Med0 (2020) 7:3389/fcvm.2020.00012. doi: 10.3389/fcvm.2020.00012
188. Wang S, Tian C, Gao Z, Zhang B, Zhao L. Research status and trends of the diabetic cardiomyopathy in the past 10 years (2012-2021): A bibliometric analysis. Front Cardiovasc Med (2022) 9:3389/fcvm.2022.1018841. doi: 10.3389/fcvm.2022.1018841
189. Rieg T, Vallon V. Development of SGLT1 and SGLT2 inhibitors. Diabetologia (2018) 61(10):2079–86. doi: 10.1007/s00125-018-4654-7
Keywords: diabetic cardiomyopathy, cardiac fibrosis, cardiac myofibroblasts, cardiac fibroblasts, imbalance of extracellular matrix synthesis and degradation, disorder of matrix metalloproteinases synthesis
Citation: Cheng Y, Wang Y, Yin R, Xu Y, Zhang L, Zhang Y, Yang L and Zhao D (2023) Central role of cardiac fibroblasts in myocardial fibrosis of diabetic cardiomyopathy. Front. Endocrinol. 14:1162754. doi: 10.3389/fendo.2023.1162754
Received: 10 February 2023; Accepted: 20 March 2023;
Published: 31 March 2023.
Edited by:
Gaetano Santulli, Albert Einstein College of Medicine, United StatesReviewed by:
Milan M. Obradovic, VINČA Institute of Nuclear Sciences, University of Belgrade, SerbiaCopyright © 2023 Cheng, Wang, Yin, Xu, Zhang, Zhang, Yang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Longyan Yang, bHl5YW5nMTVAY2NtdS5lZHUuY24=; Dong Zhao, emhhb2RvbmdAY2NtdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.