 Delia-Maria Nicoară1*
Delia-Maria Nicoară1* Alexandra-Cristina Scutca1,2*
Alexandra-Cristina Scutca1,2* Niculina Mang1Iulius Juganaru1,2,3Andrei-Ioan Munteanu1,2Luiza Vitan4
Niculina Mang1Iulius Juganaru1,2,3Andrei-Ioan Munteanu1,2Luiza Vitan4 Otilia Mărginean1,2,3
Otilia Mărginean1,2,3- 1Department of Pediatrics, University of Medicine and Pharmacy “Victor Babes”, Timisoara, Romania
- 2Department of Pediatrics I, Children’s Emergency Hospital “Louis Turcanu”, Timisoara, Romania
- 3Research Center in Pediatrics - Disturbances of Growth and Development in Children – BELIVE, University of Medicine and Pharmacy “Victor Babes”, Timisoara, Romania
- 4Department of Endocrinology, Railway Hospital 2 Bucharest, Timisoara, Romania
Prader-Willi syndrome (PWS, OMIM176270) is a rare genetic disorder with recognizable dysmorphic features and multisystemic consequences such as endocrine, neurocognitive and metabolic ones. Although most patients with Prader-Willi syndrome exhibit hypogonadotropic hypogonadism, there is variability regarding sexual maturation, with precocious puberty occurring in rare cases. Our aim is to elaborate a thorough review of Prader-Willi patients with central precocious puberty, in order to raise awareness of such cases and to enhance our knowledge regarding the diagnosis and prompt treatment of this particular PWS patients.
Introduction
Prader-Willi syndrome (PWS, OMIM176270) is a rare genetic disorder with recognizable dysmorphic features and multisystemic consequences such as endocrine, neurocognitive and metabolic ones (1, 2).
Although described in 1956 for the first time by Swiss endocrinologists Prader, Labhart and Willi (1), based on clinical aspects, genetic confirmation was possible only since 1980, when high resolution chromosome analysis led to the discovery of the chromosomal deletion of 15q11-g13
(3–5). Most PWS patients display a paternal microdeletion of the long arm of chromosome 15 (70%), whereas 20-35% have maternal uniparental disomy 15 (mUPD), and even fewer patients have an imprinting center defect (ICD) or an unbalanced translocation (0,1%) (6).
With an estimated prevalence of 1 in 10.000-30.000 live births, Prader-Willi syndrome represents the most common genetic cause of obesity (7–9) and the first recognized disorder of human genetic imprinting (10). PWS displays great clinical variability throughout life (1), ranging from hypotonia and failure to thrive during infancy, to morbid obesity, dysmorphic features, short stature and behavioral problems such as hyperphagia and aggressive behavior (11). Patients require multidisciplinary approach, including close endocrinologic follow-up throughout their lives (12) given the hypothalamic-pituitary dysfunction that characterizes these patients. This disfunction leads in turn to multiple endocrinopathies, the most common ones being growth hormone deficiency (GHD), hypogonadism and hypothyroidism (12, 13).
Genital anomalies are common in both female and male PWS patients and are represented by pubertal development disorders (14). Hypogonadism represents a major clinical diagnostic criteria of PWS, according to Holm and Cassidy (13). In Prader-Willi syndrome puberty is most often delayed and incomplete. While PWS male patients usually present cryptorchidism and remain in a Tanner genital stage II or III (14), most female PWS patients have amenorrhea or oligomenorrhea, with only a few undergoing spontaneous menarche (15). However, exceptional cases leading up to pregnancy have been described (6), isolated premature pubarche is reported in 14% of cases (1). Despite most PWS patients having hypogonadism, a few cases of central precocious puberty (CPP) have been reported (16).
Our aim is to elaborate a thorough review of Prader-Willi patients that display central precocious puberty, in order to raise awareness of such cases and to enhance our knowledge regarding the diagnosis and prompt treatment of this particular PWS patients.
Materials and methods
Search strategy
• The literature review was based on the available papers written in English published in the electronic databases PubMed and Embase database between 1979 and January 2023.
• The search criteria used in the Medical Subject Headings (MeSH) included te following terms: “Prader-Willi” [All Fields] AND (“precocious puberty)”[MeSH Terms]
• RCTs reported as literature reviews, case reports and conference abstracts with relevant outcome data were included in the review. Articles and abstracts regarding only premature adrenarche, or cases without genetic confirmation were excluded from the study.
• Duplicates were removed.
Presentation of the results
The following data were collected from each article/abstract if available: age and gender, genetic background, Tanner Pubertal Stage, GnRH stimulation test, determination of basal and peak Luteinizing Hormone (LH) and Follicle Stimulating Hormone (FSH) serum levels, Testosterone, bone age, ultrasound of genitalia, stabilization or regression of Tanner Pubertal Stage under GnRH (gonadotropin releasing hormone) treatment according to dose and duration.
Patients were included if they fulfilled diagnostic criteria for CPP (i.e. age at pubertal onset, bone age, Tanner stage, significant value of LH, FSH and Testosterone GnRH stimulation test).
Results
We sought to summarize the following outcome measures from the reviewed articles:
▪ suppression of physical signs of puberty: breast development in females, testicular volume in males, genital development – based on modified Tanner staging
▪ incremental growth rate: cm/year
▪ suppression of GnRHa – stimulated LH: peak LH value
▪ incremental change in bone age
We identified a total of 22 children with CPP and genetically confirmed Prader-Willi syndrome mostly in case reports and abstracts, summarizing 13 children from case reports and case studies [13-22] and 9 children from Embase indexed abstracts [23-27]. There were 13 (59%) male patients and 9 (41%) female cases, with mean age at CPP onset of 7,64 +/- 1,01 years (5 – 9 years). At diagnosis they were Tanner stage II-III, mean basal LH was 1,43 +/- 1,46 UI/L (0,3 – 4,6 UI/L), and mean stimulated peak LH was 13,67 +/- 6,45 UI/L (4,6 – 29,7 UI/L). Mean basal FSH was 1,43 +/- 1,46 UI/L (0,3 – 4,6 UI/L) stimulated peak FSH was 13,67 +/- 6,45 UI/L (4,6 – 29,7 UI/L).
Auxological parameters and response to GnRH of the PWS patients with precocious puberty are displayed below in Tables 1, 2.
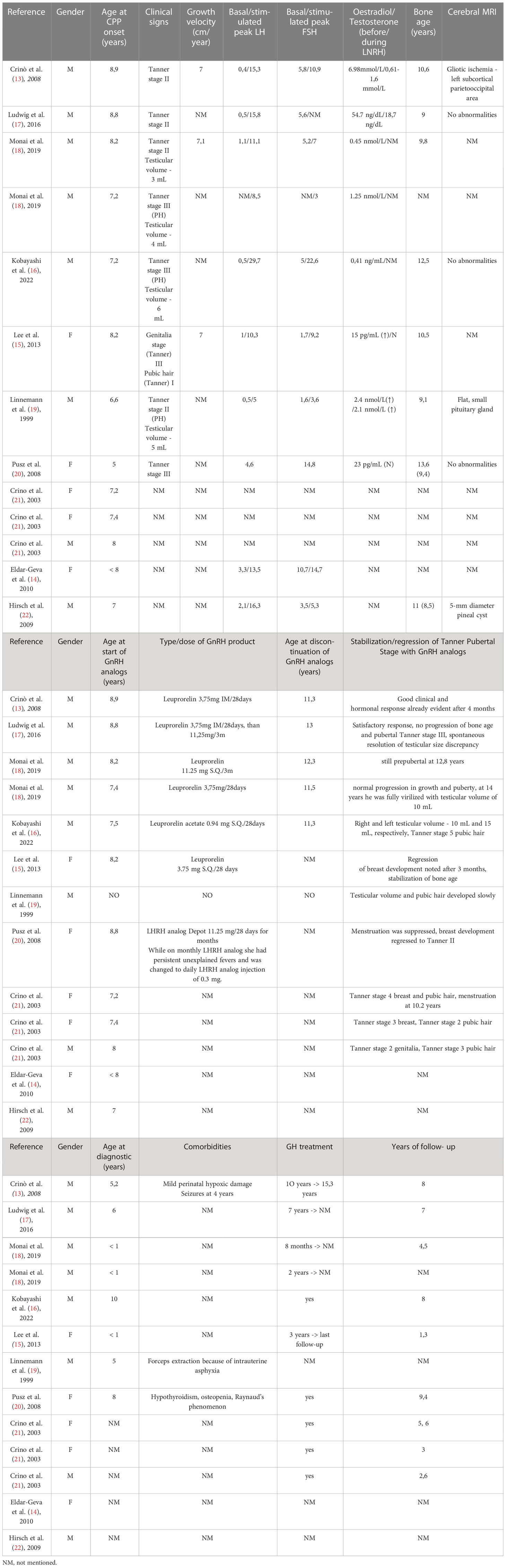
Table 1 Characteristics of PWS patients with central precocious puberty retrieved from case presentations/case studies.
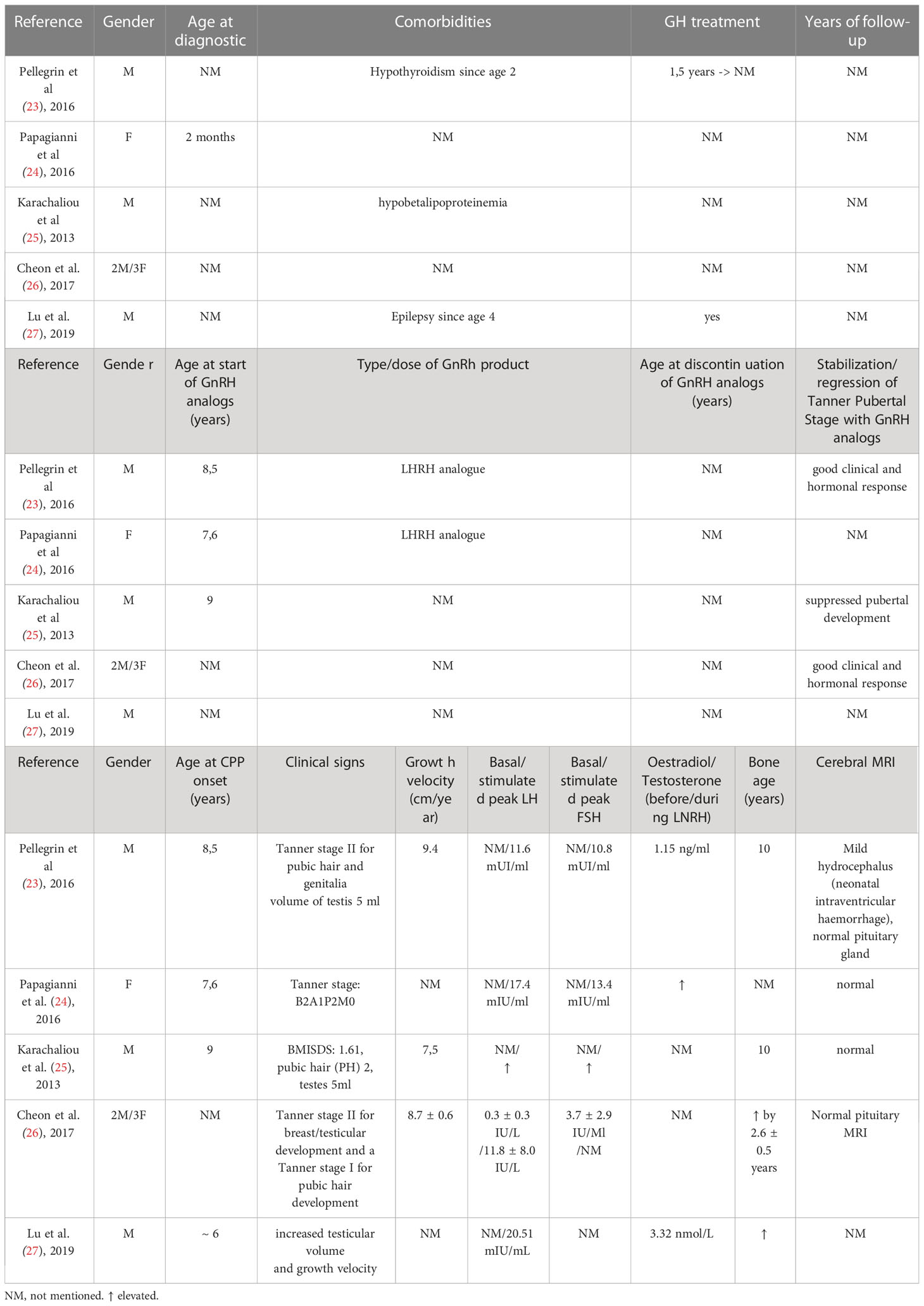
Table 2 Characteristics of PWS patients with central precocious puberty retrieved from case conference abstracts.
Discussions
Pubertal timing is a complex process, with genetic, epigenetic, endocrine, metabolic and lifestyle factors playing a role in acquiring secondary sexual characteristics, gonadal maturation and progression of linear growth (28, 29). It normally coincides with the pulsatile release of GnRH from the hypothalamic neurons (30). Normal pulsatile GnRH secretion is initially observed during fetal and neonatal periods (31, 32), suppressed during childhood, only to resume at the age of puberty, under action of Kisspeptin (33).
The first symptoms of precocious puberty are regarded in girls as onset of breast development before the age of 8 years, and in boys as testicular enlargement ≥ 4 ml before the age of 9 years (29), in accordance with the mean age at CPP onset in the reviewed cases (7,64 +/- 1,01 years). The underlying mechanism can be central (GnRH- dependent) or peripheral (GnRH-independent). In Prader-Willi cases associated with precocious puberty only central precocious puberty has been described (13–27).
Laboratory and imagistic evaluation
The most sensitive biomarker in CPP is LH, as it is untraceable before the first stages of puberty (34). Therefore determining plasma LH after exogenous GnRH or LHRH stimulation represents the reference test for diagnosing central precocious puberty (35–38). Yet peak cut-off values beyond which puberty is activated remain controversial (35, 36, 38). Values between 5 and 10 IU/L are considered acceptable cut-off points, using Chemiluminescent immunoassay (36, 38). Further assessment includes assessment of bone age (usually advanced as compared to chronological age), pelvic or testicular ultrasonography (to aid in identifying signs of peripheral precocious puberty) and brain magnetic resonance imaging in order to evaluate the hypothalamus and pituitary glands, and to exclude other brain anomalies (39). 9 cases describe imagistic evaluation through cerebral MRI, out of which 3 had abnormalities: one male patient with ischemic cerebral lesions, one with pituitary hypoplasia and one with a small pineal cyst. It can be argued that such cerebral lesions account in part for the precocious puberal onset, but additional causes related to this patients remain obscure.
Evolution and therapeutic approach
Premature activation of the hypothalamic–pituitary–gonadal (HPG) axis can have several repercussions in children and adolescents, from height deficit caused by premature fusion of epiphyses through accelerated bone maturation, to premature sexual maturation that cause body changes (34, 40). Accumulating evidence points out to the absolute or relative growth hormone deficiency in most cases of PWS patients (41–48), according to standard testing protocols (49). As such, adult height prognosis is additionally compromised in children with Prader-Willi syndrome that exhibit central precocious puberty. This reinforces the interest of detecting any central precocious puberty in Prader-Willi patients. Central precocious puberty also occurs in other diseases that associate hypopituitarism, among which lesions of the central nervous system, such as intracranial malignancies and cranial radiotherapy (50–53), severe head injury (53), arachnoid cyst and septo-optic dysplasia (54–58). Other possible situations involve genetic causes such as, combined pituitary hormone deficiency due to POU1F1 gene mutation (59, 60), Kabuki syndrome (61, 62), Williams–Beuren syndrome (63, 64), Mayer-Rokitansky-Kuster-Hauser syndrome (65) and developmental defect of the hypothalamic–pituitary area (66). The possible common etiological mechanisms in both situations should also be the subject of further studies, as the underlying mechanism of remains unclear (59).
Also, there is increased risk for metabolic comorbidities such as obesity, type 2 diabetes and for cardiovascular events (34, 67, 68). As such, there is increased need for prompt treatment. GnRH analogues are the cornerstone of treatment, and they are administered either intramuscular (1 administration every 28 days) or by subcutaneous implant (69). They have been used effectively in cases of CPP, reducing plasma gonadotrophins, gonadal steroids and peptides (34, 70). Adequate suppression of gonadotrophins can be seen after 3 – 4 months of treatment (71).
Future therapeutic options that are being evaluated as an alternative option include newer kisspeptin and neurokinin B antagonists (69, 72, 73). Clinical trials investigating the short term efficacy of GnRH analogs confirm the fact that this treatment is well tolerated and safe (71, 74–76). Also, case reports regarding PWS patients with central precocious puberty highlight the possible benefit of combined therapy (gonadotropin-releasing hormone agonist and recombinant human growth hormone) on final height, while restoring appropriate pubertal progression (16, 17).
Study limitations
Due to the exceedingly rare cases of PWS patients with precocious puberty, reviewed cases stemmed from case reports and Embase registered conference abstracts. Yet, it is our belief that such cases promote a better understanding of the variety of sexual maturation disorders in PWS patients.
Conclusions
Although most patients with Prader-Willi syndrome exhibit hypogonadotropic hypogonadism, there is variability regarding sexual maturation, with precocious puberty occurring in rare cases. Recent years have brought major improvement in scientific knowledge regarding PWS, but there is still a need for further studies to assess the pathophysiology implicated in timing and progression of pubertal onset (15), and to implement clinical guidelines (12). GnRH agonist therapy seems to be efficient and safe in such cases, although long term follow-up is of need to better address the issue (71, 74–76).
Author contributions
DMN, ACS identified potential papers, D-MN and ACS wrote original draft, D-MN, A-CS, NM, IJ, A-IM and LV analyzed selected papers, and took the lead in writing the manuscript. OM supervised the project and recommended changes. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Grechi E, Cammarata B, Mariani B, Di Candia S, Chiumello G. Prader-willi syndrome: clinical aspects. J Obes (2012) 2012:473941. doi: 10.1155/2012/473941
2. Prader A, Labhart A, Willi H. Ein syndrom von adipositas, kleinwuchs, kryptorchismus und oligophrenie nach myotonieartigem zustand im neugeborenenalter. Schweizerische Medizinische Wochenschrift (1956) 86:1260.
3. Kim Y, Wang SE, Jiang YH. Epigenetic therapy of prader-willi syndrome. Transl Res (2019) 208:105–18. doi: 10.1016/j.trsl.2019.02.012
4. Ledbetter DH, Mascarello JT, Riccardi VM, Harper VD, Airhart SD, Strobel RJ. Chromosome 15 abnormalities and the prader-willi syndrome: a follow-up report of 40 cases. Am J Hum Genet (1982) 34:278–85.
5. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. Deletions of chromosome 15 as a cause of the prader-willi syndrome. N Engl J Med (1981) 304:325–9.
6. Pellikaan K, Ben Brahim Y, Rosenberg AGW, Davidse K, Poitou C, Coupaye M, et al. Hypogonadism in women with prader-willi syndrome-clinical recommendations based on a Dutch cohort study, review of the literature and an international expert panel discussion. J Clin Med (2021) 10(24):5781. doi: 10.3390/jcm10245781
7. Angulo MA, Butler MG, Cataletto ME. Prader-willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest (2015) 38(12):1249–63. doi: 10.1007/s40618-015-0312-9
8. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion prader-willi syndrome. Nature (1989) 342(6247):281–5.
9. Napolitano L, Barone B, Morra S, Celentano G, La Rocca R, Capece M, et al. Hypogonadism in patients with prader willi syndrome: a narrative review. Int J Mol Sci (2021) 22(4):1993. doi: 10.3390/ijms22041993
10. Lecka-Ambroziak A, Wysocka-Mincewicz M, Marszałek-Dziuba K, Rudzka-Kocjan A, Szalecki M. Premature adrenarche in children with prader-willi syndrome treated with recombinant human growth hormone seems to not influence the course of central puberty and the efficacy and safety of the therapy. Life (Basel) (2020) 10(10):237. doi: 10.3390/life10100237
11. Tauber M, Barbeau C, Jouret B, Pienkowski C, Malzac P, Moncla A, et al. Auxological and endocrine evolution of 28 children with prader-willi syndrome: effect of GH therapy in 14 children. Horm Res (2000) 53(6):279–87. doi: 10.1159/000053184
12. Heksch R, Kamboj M, Anglin K, Obrynba K. Review of prader-willi syndrome: the endocrine approach. Transl Pediatr (2017) 6(4):274–85. doi: 10.21037/tp.2017.09.04
13. Crinò A, Di Giorgio G, Schiaffini R, Fierabracci A, Spera S, Maggioni A, et al. Central precocious puberty and growth hormone deficiency in a boy with prader-willi syndrome. Eur J Pediatr (2008) 167(12):1455–8. doi: 10.1007/s00431-008-0679-0
14. Eldar-Geva T, Hirsch HJ, Benarroch F, Rubinstein O, Gross-Tsur V. Hypogonadism in females with prader-willi syndrome from infancy to adulthood: variable combinations of a primary gonadal defect and hypothalamic dysfunction. Eur J Endocrinol (2010) 162(2):377–84. doi: 10.1530/EJE-09-0901
15. Lee HS, Hwang JS. Central precocious puberty in a girl with prader-willi syndrome. J Pediatr Endocrinol Metab (2013) 26(11-12):1201–4.
16. Kobayashi M, Yagasaki H, Tamaru K, Mitsui Y, Inukai T. Idiopathic central precocious puberty with prader-willi syndrome: pubertal development with discontinuation of gonadotropin-releasing hormone analog. Endocrinol Diabetes Metab Case Rep (2022) 2022:22–0244. doi: 10.1530/EDM-22-0244
17. Ludwig NG, Radaeli RF, Silva MM, Romero CM, Carrilho AJ, Bessa D, et al. A boy with prader-willi syndrome: unmasking precocious puberty during growth hormone replacement therapy. Arch Endocrinol Metab (2016) 60(6):596–600. doi: 10.1590/2359-3997000000196
18. Monai E, Johansen A, Clasen-Linde E, Rajpert-De Meyts E, Skakkebæk NE, Main KM, et al. Central precocious puberty in two boys with prader-willi syndrome on growth hormone treatment. AACE Clin Case Rep (2019) 5(6):e352–6. doi: 10.4158/ACCR-2019-0245
19. Linnemann K, Schröder C, Mix M, Krüger G, Fusch C. Prader-Labhart-Willi syndrome with central precocious puberty and empty sella syndrome. Acta Paediatr (1999) 88(11):1295–7. doi: 10.1080/080352599750030482
20. Pusz ER, Rotenstein D. Treatment of precocious puberty in a female with prader-willi syndrome. J Pediatr Endocrinol Metab (2008) 21(5):495–500. doi: 10.1515/JPEM.2008.21.5.495
21. Crinò A, Schiaffini R, Ciampalini P, Spera S, Beccaria L, Benzi F, et al. Genetic obesity study group of Italian society of pediatric endocrinology and diabetology (SIEDP). Hypogonadism and pubertal development in prader-willi syndrome. Eur J Pediatr (2003) 162(5):327–33. doi: 10.1007/s00431-002-1132-4
22. Hirsch HJ, Eldar-Geva T, Benarroch F, Rubinstein O, Gross-Tsur V. Primary testicular dysfunction is a major contributor to abnormal pubertal development in males with prader-willi syndrome. J Clin Endocrinol Metab (2009) 94(7):2262–8. doi: 10.1210/jc.2008-2760
23. Pellegrin MC, Tornese G, Faleschini E, Ventura A. A case of central precocious puberty in a patient with prader-willi syndrome. Horm Res Paediatr (2016) 86(suppl 1):1–556.
24. Papagianni M, Kosta K, Lialias I, Chatzakis C, Tsiroukidou K, Tsanakas I. Precocious puberty in a girl with prader willi syndrome. Horm Res Paediatr (2016) 86(suppl 1):1–556.
25. Karachaliou F, Kaloumenou I, Vlachopapadopoulou E, Skouma A, Fotinou A, Michalacos S. Central precocious puberty and hypobetalipoproteinaemia in a boy with prader-willi syndrome. Hormone Res Paediatrics (2013) 80(435):435.
26. Cheon CK, Kim Y-M. Idiopathic central precocious puberty in prader-willi syndrome in Korea. Hormone Res Paediatrics (2017) 88(346):346.
27. Lu W, Zheng Z, Ni J, Li X, Xi L, Luo F. Central precocious puberty in a boy with prader-willi syndrome eduring growth hormone replacement therapy. Hormone Res Paediatrics (2019) 91(587):587.
28. Canton APM, Krepischi ACV, Montenegro LR, Costa S, Rosenberg C, Steunou V, et al. Insights from the genetic characterization of central precocious puberty associated with multiple anomalies. Hum Reprod (2021) 36(2):506–18. doi: 10.1093/humrep/deaa306
29. Latronico AC, Brito VN, Carel JC. Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol (2016) 4(3):265–74. doi: 10.1016/S2213-8587(15)00380-0
30. Carli D, Riberi E, Ferrero GB, Mussa A. Syndromic disorders caused by disturbed human imprinting. J Clin Res Pediatr Endocrinol (2020) 12(1):1–16. doi: 10.4274/jcrpe.galenos.2019.2018.0249
31. Terasawa E, Fernandez DL. Neurobiological mechanisms of the onset of puberty in primates. Endocr Rev (2001) 22(1):111–51. doi: 10.1210/edrv.22.1.0418
32. Ojeda SR, Lomniczi A. Puberty in 2013: unravelling the mystery of puberty. Nat Rev Endocrinol (2014) 10(2):67–9. doi: 10.1038/nrendo.2013.233
33. Tajima T. Genetic causes of central precocious puberty. Clin Pediatr Endocrinol (2022) 31(3):101–9. doi: 10.1297/cpe.2022-0021
34. Maione L, Bouvattier C, Kaiser UB. Central precocious puberty: recent advances in understanding the aetiology and in the clinical approach. Clin Endocrinol (Oxf) (2021) 95(4):542–55. doi: 10.1111/cen.14475
35. Soriano-Guillén L, Corripio R, Labarta JI, Cañete R, Castro-Feijóo L, Espino R, et al. Central precocious puberty in children living in Spain: incidence, prevalence, and influence of adoptionand immigration. J Clin Endocrinol Metab (2010) 95:4305—13.
36. Liang JT. Value of basal serum gonadotropin levels in the diagnosis of precocious puberty in girls. Zhongguo Dang Dai Er Ke ZaZhi (2012) 14:942—5.
37. Rohani F, Salehpur S, Saffari F. Etiology of precocious puberty, 10years study in endocrine research centre (Firouzgar), Tehran. Iran J Reprod Med (2012) 10:1—6.
38. Durá-Travé T, Ortega Pérez M, Ahmed-Mohamed L, Moreno-González P, Chueca Guindulain MJ, Berrade-Zubiri S. Central precocious puberty in girls: diagnostic study and auxological response to triptorelin treatment. Endocrinol Diabetes Nutr (Engl Ed) (2019) 66(7):410–6. doi: 10.1016/j.endinu.2018.12.007
39. Howard SR. Interpretation of reproductive hormones before, during and after the pubertal transition-identifying health and disordered puberty. Clin Endocrinol (Oxf) (2021) 95(5):702–15. doi: 10.1111/cen.14578
40. Graber JA. Pubertal timing and the development of psychopathology in adolescence and beyond. Horm Behav (2013) 64(2):262–9.
41. Angulo M, Abuzzahab MJ, Pietropoli A, Ostrow V, Kelepouris N, Tauber M. Outcomes in children treated with growth hormone for prader-willi syndrome: data from the ANSWER program® and NordiNet® international outcome study. Int J Pediatr Endocrinol (2020) 2020(1):20. doi: 10.1186/s13633-020-00090-6
42. Diene G, Mimoun E, Feigerlova E, Caula S, Molinas C, Grandjean H, et al. French Reference centre for PWS. endocrine disorders in children with prader-willi syndrome–data from 142 children of the French database. Horm Res Paediatr (2010) 74(2):121–8. doi: 10.1159/000313377
43. Tauber M, Diene G. Prader-willi syndrome: hormone therapies. Handb Clin Neurol (2021) 181:351–67. doi: 10.1016/B978-0-12-820683-6.00026-9
44. Tauber M, Hoybye C. Endocrine disorders in prader-willi syndrome: a model to understand and treat hypothalamic dysfunction. Lancet Diabetes Endocrinol (2021) 9(4):235–46. doi: 10.1016/S2213-8587(21)00002-4
45. Vyas V, Menon RK. Management of short stature: use of growth hormone in GH-deficient and non-GH-Deficient conditions. Indian J Pediatr (2021) 88(12):1203–8. doi: 10.1007/s12098-021-03892-5
46. Kim SJ, Cho SY, Jin DK. Prader-willi syndrome: an update on obesity and endocrine problems. Ann Pediatr Endocrinol Metab (2021) 26(4):227–36. doi: 10.6065/apem.2142164.082
47. Lecka-Ambroziak A, Wysocka-Mincewicz M, Doleżal-Ołtarzewska K, Zygmunt-Górska A, Wędrychowicz A, Żak T, et al. On behalf of the polish coordination group for rhGH treatment. effects of recombinant human growth hormone treatment, depending on the therapy start in different nutritional phases in paediatric patients with prader-willi syndrome: a polish multicentre study. J Clin Med (2021) 10(14):3176. doi: 10.3390/jcm10143176
48. Passone CGB, Franco RR, Ito SS, Trindade E, Polak M, Damiani D, et al. Growth hormone treatment in prader-willi syndrome patients: systematic review and meta-analysis. BMJ Paediatr Open (2020) 4(1):e000630. doi: 10.1136/bmjpo-2019-000630
49. Deal CL, Tony M, Hoybye C, Allen DB, Tauber M, Christiansen JS. GrowthHormone research society workshop summary: consensus guidelines for recombinant human growth hormone therapy in prader-willi syndrome. J Clin Endocrinol Metab (2013) 98(6):E1072–87.
50. Maciel J, Dias D, Cavaco D, Donato S, Pereira MC, Simões-Pereira J. Growth hormone deficiency and other endocrinopathies after childhood brain tumors: results from a close follow-up in a cohort of 242 patients. J Endocrinol Invest (2021) 44(11):2367–74. doi: 10.1007/s40618-021-01541-4
51. Manasco PK, Pescovitz OH, Hill SC, Jones JM, Barnes KM, Hench KD, et al. Six-year results of luteinizing hormone releasing hormone (LHRH) agonist treatment in children with LHRH-dependent precocious puberty. J Pediatr (1989) 115:105–8.
52. Kreiter M, Burstein S, Rosenfield RL, Moll GW Jr, Cara JF, Yousefzadeh DK, et al. Preserving adult height potential in girls with idiopathic true precocious puberty. J Pediatr (1990) 117:364–70.
53. Rappaport R, Prevot C, Brauner R. Somatomedin-c and growth in children with precocious puberty: a study of the effect of the level of growth hormone secretion. J Clin Endocrinol Metab (1987) 65:1112–7.
54. Thamdrup E. Precocious sexual development: a clinical study of 100 children. Springfield, Illinois: Charles C Thomas (1961).
55. Sigurjonsdottir TJ, Hayles AB. Precocious puberty: a report of 96 cases. Am J Dis Child (1968) 115:309–21.
56. Comite F, Cassorla F, Barnes KM, Hench KD, Dwyer A, Skerda MC, et al. Luteinizing hormone releasing hormone analogue therapy for central precocious puberty: long-term effect on somatic growth, bone maturation, and predicted height. JAMA (1986) 255:2613–6.
57. Boepple PA, Mansfield MJ, Wierman ME, Rudlin CR, Bode HH, Crigler JF Jr, et al. Use of a potent, long-acting agonist of gonadotropin-releasing hormone in the treatment of precocious puberty. Endocr Rev (1986) 7:24–33.
58. Lee PA, Page JG, the Leuprolide Study Group. Effects of leuprolide in the treatment of central precocious puberty. J Pediatr (1989) 114:321–4.
59. Baş F, Abalı ZY, Toksoy G, Poyrazoğlu Ş, Bundak R, Güleç Ç, et al. Precocious or early puberty in patients with combined pituitary hormone deficiency due to POU1F1 gene mutation: case report and review of possible mechanisms. Hormones (Athens) (2018) 17(4):581–8. doi: 10.1007/s42000-018-0079-4
60. Jadhav S, Diwaker C, Lila AR, Gada JV, Kale S, Sarathi V, et al. POU1F1 mutations in combined pituitary hormone deficiency: differing spectrum of mutations in a Western-Indian cohort and systematic analysis of world literature. Pituitary (2021) 24(5):657–69. doi: 10.1007/s11102-021-01140-9
61. Bereket A, Turan S, Alper G, Comu S, Alpay H, Akalin F. Two patients with kabuki syndrome presenting with endocrine problems. J Pediatr Endocrinol Metab (2001) 14:215–20.
62. Devriendt K, Lemli L, Craen M, de Zegher F. Growth hormone deficiency and premature thelarche in a female infant with kabuki makeup syndrome. Horm Res (1995) 43:303–6.
63. Kim YM, Cho JH, Kang E, Kim GH, Seo EJ, Lee BH, et al. Endocrine dysfunctions in children with williams-beuren syndrome. Ann Pediatr Endocrinol Metab (2016) 21(1):15–20. doi: 10.6065/apem.2016.21.1.15
64. Levy-Shraga Y, Gothelf D, Pinchevski-Kadir S, Katz U, Modan-Moses D. Endocrine manifestations in children with williams-beuren syndrome. Acta Paediatr (2018) 107(4):678–84. doi: 10.1111/apa.14198
65. Ai Z, Zhu X, Chen H, Chen R. Precocious puberty or growth hormone deficiency as initial presentation in Mayer-Rokitansky-kuster-Hauser syndrome: a clinical report of 5 cases. BMC Pediatr (2022) 22(1):418. doi: 10.1186/s12887-022-03474-0
66. Ladjouze A, Soskin S, Garel C, Jullien M, Naud-Saudreau C, Pinto G, et al. GH deficiency with central precocious puberty: a new rare disorder associated with a developmental defect of the hypothalamic-pituitary area. Eur J Endocrinol (2007) 156(4):463–9. doi: 10.1530/EJE-06-0688
67. Jacobsen BK, Heuch I, Kvale G. Association of low age at menarche with increased all-cause mortality: a 37-year follow-up of 61,319 Norwegian women. Am J Epidemiol (2007) 166(12):1431–7.
68. Lundblad MW, Jacobsen BK. Is age at menarche associated with total mortality? the tromso study. Int J Womens Health (2018) 10:203–9.
69. Roberts SA, Kaiser UB. Genetics in endocrinology: genetic etiologies of central precocious puberty and the role of imprinted genes. Eur J Endocrinol (2020) 183(4):R107–17. doi: 10.1530/EJE-20-0103
70. Grinspon RP, Andreone L, Bedecarras P, Ropelato MG, Rey RA, Campo SM, et al. Male Central precocious puberty: serum profile of anti-mullerian hormone and inhibin b before, during, and after treatment with GnRH analogue. Int J Endocrinol (2013) 2013:823064.
71. Willemsen RH, Elleri D, Williams RM, Ong KK, Dunger DB. Pros and cons of GnRHa treatment for early puberty in girls. Nat Rev Endocrinol (2014) 10(6):352–63. doi: 10.1038/nrendo.2014.40
72. Blumenfeld Z. Investigational and experimental GnRH analogs and associated neurotransmitters. Expert Opin Investig Drugs (2017) 26:661–7.
73. Newton C, Anderson R, Millar R. Therapeutic neuroendocrine agonist and antagonist analogs of hypothalamic neuropeptides as modulators of the hypothalamic-pituitary-gonadal axis. Endocr Dev (2016) 30:106–29.
74. Carel JC, Montauban V, Teinturier C, Colle M. Treatment of central precocious puberty by subcutaneous injections of leuprorelin 3-month depot (11.25 mg). J Clin Endocrinol Metab (2002) 87:4111–6.
75. Lee PA, Klein K, Mauras N, Neely EK, Bloch CA, Larsen L, et al. Efficacy and safety of leuprolide acetate 3-month depot 11.25 milligrams or 30 milligrams for the treatment of central precocious puberty. J Clin Endocrinol Metab (2012) 97:1572–80.
Keywords: Prader-Willi syndrome, precocious puberty, endocrine, genetic, metabolic
Citation: Nicoară D-M, Scutca A-C, Mang N, Juganaru I, Munteanu A-I, Vitan L and Mărginean O (2023) Central precocious puberty in Prader-Willi syndrome: a narrative review. Front. Endocrinol. 14:1150323. doi: 10.3389/fendo.2023.1150323
Received: 24 January 2023; Accepted: 17 April 2023;
Published: 08 May 2023.
Edited by:
Katja Dumic Kubat, University of Zagreb, CroatiaReviewed by:
Marc Nicolino, Hospices Civils de Lyon, FranceNavoda Atapattu, Lady Ridgeway Hospital for Children, Sri Lanka
Copyright © 2023 Nicoară, Scutca, Mang, Juganaru, Munteanu, Vitan and Mărginean. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Delia-Maria Nicoară, bmljb2FyYS5kZWxpYUB1bWZ0LnJv; Alexandra-Cristina Scutca, c2N1dGNhLmFsZXhhbmRyYUB1bWZ0LnJv