 Constanze Hoebinger
Constanze Hoebinger Dragana Rajcic
Dragana Rajcic Beatriz Silva1,2
Beatriz Silva1,2 Tim Hendrikx
Tim Hendrikx- 1Department of Laboratory Medicine, Medical University Vienna, Vienna, Austria
- 2Department of Biochemistry, Chemistry Institute, University of Sao Paulo, Sao Paulo, Brazil
Objective: Chronic ethanol consumption is known to cause alcohol-associated liver disease, which poses a global health concern as almost a quarter of heavy drinkers develop severe liver damage. Alcohol-induced liver disease ranges from a mild, reversible steatotic liver to alcoholic steatohepatitis and irreversible liver fibrosis and cirrhosis, ultimately requiring liver transplantation. While ethanol consumption is associated with dysregulated lipid metabolism and altered cholesterol homeostasis, the impact of dyslipidemia and pre-existing hypercholesterolemia on the development of alcohol-associated liver disease remains to be elucidated.
Design: To address the influence of systemic dyslipidemia on ethanol-induced liver disease, chronic-binge ethanol feeding was applied to female C57BL/6J (wild type) mice and mice deficient for the low-density lipoprotein receptor (Ldlr-/-), which display a human-like lipoprotein profile with elevated cholesterol and triglyceride levels in circulation. Respective control groups were pair-fed an isocaloric diet.
Results: Chronic-binge ethanol feeding did not alter systemic lipid levels in wild type mice. While increased systemic cholesterol levels in Ldlr-/- mice were not affected by ethanol feeding, chronic-binge ethanol diet aggravated elevated plasma triglyceride levels in Ldlr-/- mice. Despite higher circulatory triglyceride levels in Ldlr-/- mice, hepatic lipid levels and the development of hepatic steatosis were not different from wild type mice after ethanol diet, while hepatic expression of genes related to lipid metabolism (Lpl) and transport (Cd36) showed minor changes. Immunohistochemical assessment indicated a lower induction of infiltrating neutrophils in the livers of ethanol-fed Ldlr-/- mice compared to wild type mice. In line, hepatic mRNA levels of the pro-inflammatory genes Ly6g, Cd11b, Ccr2, Cxcl1 and F4/80 were reduced, indicating less inflammation in the livers of Ldlr-/- mice which was associated with reduced Tlr9 induction. While systemic ALT and hepatic MDA levels were not different, Ldlr-deficient mice showed accelerated liver fibrosis development after chronic-binge ethanol diet than wild type mice, as indicated by increased levels of Sirius Red staining and higher expression of pro-fibrotic genes Tgfb, Col1a1 and Col3a1. Ldlr-/- and wild type mice had similar plasma ethanol levels and did not show differences in the hepatic mRNA levels of Adh1 and Cyp2e1, important for ethanol metabolism.
Conclusion: Our results highlight that chronic-binge ethanol feeding enhances systemic dyslipidemia in Ldlr-/- mice which might accelerate the development of hepatic fibrosis, independent of hepatic lipid levels.
Introduction
Chronic consumption of ethanol-containing beverages has detrimental effects on tissue homeostasis and can enhance the development of various diseases in different organs, such as the liver. In fact, alcohol-associated liver disease is the most prevalent chronic liver disease worldwide and a leading cause of liver-related morbidity and mortality, and thus governs a dramatic socio-economic burden (1, 2). It encompasses a spectrum of pathologies ranging from simple steatosis (accumulation of triglycerides in hepatocytes) to alcohol-associated steatohepatitis (ASH), which is characterized by hepatic inflammation, hepatocyte injury, and ballooning (3). A persistent increase in triglyceride accumulation, inflammation, and liver injury stimulates fibrotic scarring in the liver. Subsequently, fibrosis may culminate in the development of end-stage liver diseases such as cirrhosis or hepatocellular carcinoma, ultimately requiring liver transplantation (4). Despite considerable progress in recent decades, the mechanisms involved in the pathogenesis and progression of alcohol-induced liver disease are not fully understood. The fact that up to 20% of heavy drinkers develop advanced fibrosis and cirrhosis, while for the remaining 80%, the progression of the disease is much slower, poses the need to identify the underlying factors that contribute to these variations (5).
Altered lipid metabolism and resulting dyslipidemia are key processes by which ethanol consumption contributes to the development of alcohol-related fatty liver disease (6). In the liver, it has been described that ethanol alters lipid metabolism by increasing hepatic fatty acid uptake and triglyceride synthesis through de novo lipogenesis (7). Furthermore, studies have indicated that ethanol impairs cholesterol synthesis by reducing the formation and secretion of very low density lipoprotein (VLDL). Since VLDL gets assembled in the liver from triglycerides, cholesterol, and apolipoproteins and enables lipids to enter the circulation, a decreased cholesterol synthesis promotes the storage of triglycerides as lipid droplets in the liver (8–11). Yet, some studies have reported opposite effects, namely increased hepatic cholesterol synthesis (12, 13). In parallel with altered hepatic lipid metabolism, heavy drinking is associated with systemic dyslipidemia, characterized by elevated blood low-density lipoprotein cholesterol (LDL-C), high triglyceride levels, and low levels of high-density lipoprotein cholesterol (HDL-C) (14, 15). As such, it was reported that daily moderate alcohol intake poses a risk factor for hypertriglyceridemia, whereas it was shown to be protective in terms of developing hypercholesterolemia (15). However, after binge drinking, elevated cholesterol levels were measured in humans and rats (13, 16). Despite these divergent observations, these data indicate that ethanol abuse might have important implications for diseases such as atherosclerosis, for which alterations in systemic lipid levels are established risk factors (17). While it is well-documented that alcoholic hepatitis patients have an altered lipid metabolism, the contribution of pre-existing systemic dyslipidemia to the development and progression of alcohol-associated liver disease is currently unclear. Therefore, we assessed ethanol-induced steatohepatitis in mice lacking the low-density lipoprotein receptor (Ldlr-/-), which resemble the human lipoprotein profile and are characterized by elevated cholesterol and triglyceride levels in circulation due to defective LDL clearance (18).
Materials and methods
Animal experiments
C57BL/6J mice were purchased from Charles River. Ldlr-/- mice (stock no. 002207) were originally purchased from The Jackson Laboratory and were acquired for this study from our in-house breeding facility at the Medical University of Vienna, Austria. All mice were on a C57BL/6J background and were maintained in the specific pathogen-free (SPF) facility. Mice were bred under barrier-specific pathogen-free conditions at the Department of Biomedical Research or the Department of Laboratory Animal Science and Genetics of the Medical University of Vienna, Austria, and housed in individually-ventilated cages with a 12-hour dark-/light-cycle with ad libitum access to liquid diet. All female mice used in the experiments were aged 9 to 10 weeks. All experimental studies, interventions, and sample sizes were approved by the Animal Ethics Committee of the Medical University of Vienna and the Austrian Federal Ministry of Education, Science and Research and were performed according to Good Scientific Practice guidelines (License number: 2022-0.574.136).
Dietary interventions
For chronic-binge ethanol feeding, an adapted protocol (19) of the NIAAA model was applied (20). Mice were fed Lieber–DeCarli diet for 15 days starting at day 6 with ethanol feeding. The caloric intake from EtOH was 0 on days 1–5 and 36% (6.4% (v/v)) from day 6 until the end. At day 16, mice were gavaged with one dose of ethanol (5 g/kg BW) and sacrificed 8 hours later. Pair-fed control mice received a diet with an isocaloric substitution of dextrose. The liquid diet was freshly prepared 3 times/week with an irradiated diet and was administered ad libitum in the ethanol-fed groups. The sacrifice of experimental mice occurred randomly with alternating order of genotype to prevent confounding effects of time of harvest. For all further analysis, measurements were done randomly and in a blinded fashion.
Biochemical analyses
Blood was collected in EDTA collection tubes (Greiner Bio-One, Germany), and plasma was obtained by centrifugation at 2000xg for 10 minutes. ALT plasma levels were determined using Reflotron ALT strips on a Reflotron Plus (Roche). Plasma and hepatic triglyceride and cholesterol levels were measured according to the manufacturer’s instructions using Liquid Reagents kit (GPO-PAP Triglyceride Liquicolor kit, CHOD-PAP Cholesterol Liquicolor kit, HUMAN Biochemica, and Diagnostica mbH, Wiesbaden, Germany). Protein content was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). TBARS were assessed following the manufacturer’s instructions using the Quantichrome DTBA-100 kit (BioAssay Systems, USA). Plasma ethanol levels were determined using the Ethanol assay kit (MAK076-1KT, Sigma-Aldrich, Vienna, Austria) according to the manufacturer’s protocol.
Immunohistochemistry
Mouse liver sections were embedded in OCT compound, and 7 μm frozen sections were stained and quantified for CD11b (Mac-1; Clone: M1/70; 550282, Becton Dickinson, Austria; 1:1000) for infiltrating macrophages and neutrophils, and with Oil Red O (Sigma-Aldrich, Vienna, Austria) to determine lipid content as reported previously (21). Formalin-fixed liver samples were embedded in paraffin, and 4 μm sections were stained for hematoxylin and eosin for liver morphology and steatosis and with Sirius Red for liver fibrosis detection as described previously (22). Recorded images were analyzed for the hepatic collagen content using ImageJ 1.53 software and quantified for the percentage of positive staining per liver section. At least 3 randomly selected images of the liver per mouse were analyzed. In addition, an arbitrary scoring of Sirius red staining of whole liver sections was done by an experienced experimental hepatologist. Furthermore, formalin-fixed liver samples were stained and quantified for Ly6G+ infiltrating neutrophils (Rat anti-mouse Ly6G IgG2b; NIMP-R14; MA1-40038; 1:700; ThermoFisher). In short, 4 μm thickness sections were deparaffinized and rehydrated with xylene, decreasing ethanol concentrations, and distilled water. Antigen retrieval was performed by incubating the sections in Citrate Buffer pH 6.0 at 95°C. Sections were incubated with the primary antibody overnight at 4°C. Biotinylated goat anti-rat IgG (BA-9401; VectorLABS) was used as secondary antibody. The colour was developed with diaminobenzidine, and the nuclei were counterstained with hematoxylin, followed by dehydration with increasing ethanol concentrations. The quantification of positive cells was carried out by averaging the number of positively-stained cells from 5 randomly selected high-power fields of the liver per mouse.
RNA isolation
For whole liver RNA isolation, 50mg tissue pieces of left lateral liver lobes were snap-frozen in liquid nitrogen. Frozen tissue in QIAzol lysis reagent was homogenized mechanically using 1.0 mm TriplePure M-Bio Grade High Impact Zirconium beads (Lot:44544432, Benchmark) in a Tissue Lyser II (Qiagen, Hilden, Germany). RNA was extracted by QIAzol (QIAzol lysis reagent; Cat. No./ID: 79306; Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA content and quality were assessed using Nanodrop (Peqlab).
cDNA generation and qPCR
For quantitative real-time PCR, up to 1μg of RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Thermo Fisher, Waltham, MA, USA) to generate cDNA. Real-time PCR was performed on a CFX96 Real-Time PCR System (Bio-Rad Laboratories, Hercules, CA, USA) using the KAPA SYBR FAST kit (Thermo Fisher, Waltham, MA, USA) and the primers indicated below. Gene expression was calculated using the 2–ΔΔCt method and normalized to the expression of housekeeping gene 18S.
Statistical analysis
Statistical analyses were performed using GraphPad Prism v9.1.2. Our data comprised four groups: C57BL/6J mice (ethanol feeding/isocaloric controls) and Ldlr-/- mice (ethanol feeding/isocaloric controls). Comparison of the four groups was conducted using one-way analysis of variance (ANOVAs), and significant main effects were followed up by Fisher’s least significant difference (LSD) tests to compare individual groups and conditions. In outcomes with additional pre-post comparisons, we analyzed the data using factorial ANOVAs (with the factors group and time). Significant main effects or interactions were followed up again by LSD tests. Results are expressed as mean ± standard error (SEM) unless stated otherwise. A p ≤ 0.05 was considered statistically significant.
Results
Ethanol feeding leads to increased plasma triglyceride levels in Ldlr-/- mice compared to wild type mice
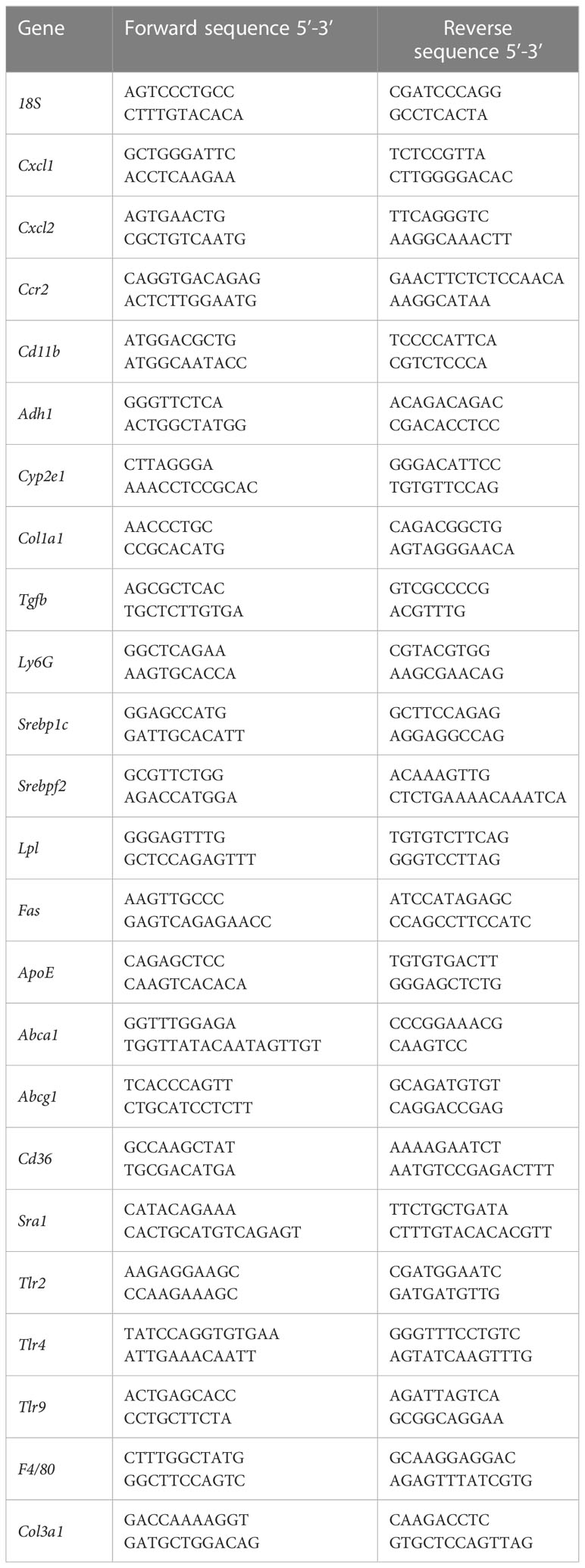
To investigate the contribution of pre-existing dyslipidemia to ethanol-induced liver disease, age-matched female Ldlr-/- mice and wild type (C57Bl6J) mice were subjected to a chronic-binge ethanol feeding model (19). Respective control mice were pair-fed an isocaloric control diet (Figure 1A). While the amount of food intake was similar between all experimental groups, ethanol-fed mice had a slightly lower body weight than control-fed mice, irrespective of the genotype. No difference in body weight between wild type and Ldlr-/- mice on ethanol diet was observed (Figures 1B, C). To assess whether ethanol consumption affects systemic lipid levels during dyslipidemia, plasma cholesterol and triglyceride levels were measured at the beginning and the end of the study. As expected, mice lacking the LDLR had significantly higher plasma cholesterol and triglyceride levels compared to wild type mice at baseline (Figures 1D, E). While Ldlr-/- mice had higher plasma cholesterol levels than wild type mice, no additional effect of ethanol feeding was observed in either genotype (Figure 1F). In contrast, Ldlr-/- mice showed increased plasma triglyceride levels after ethanol feeding compared to their respective pair-fed controls, while the amount of systemic triglycerides of wild type mice was not affected after chronic-binge ethanol feeding (Figure 1G). These data indicate that ethanol consumption exaggerates elevated plasma triglyceride levels during dyslipidemia in Ldlr-deficient mice.
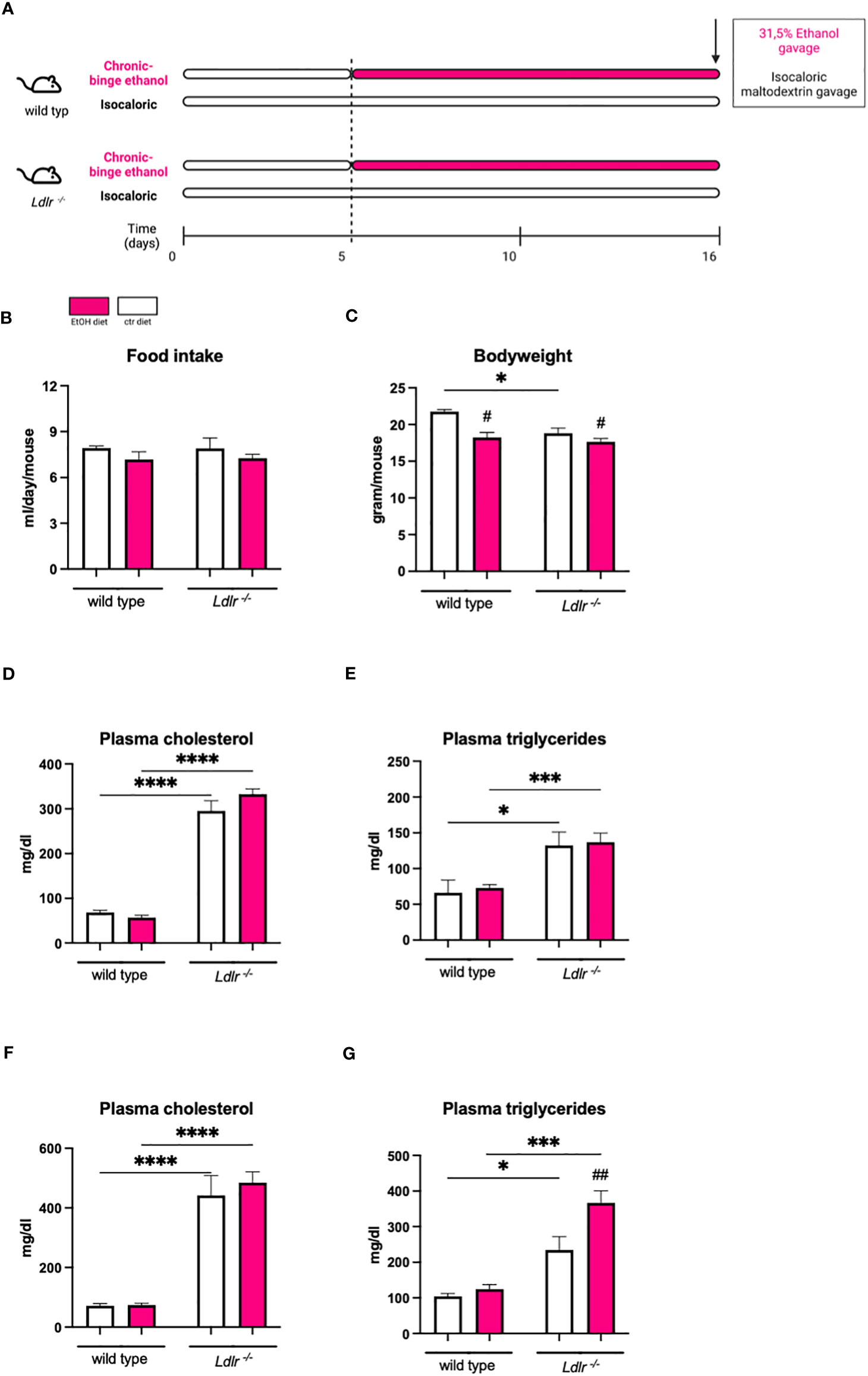
Figure 1 Chronic-binge ethanol feeding results in elevated plasma triglyceride levels in Ldlr-/- compared to wild type mice. (A) Schematic depiction of 15 days Lieber-DeCarli ethanol diet or isocaloric control diet feeding study with a single gavage of ethanol or isocaloric maltodextrin on day 16, respectively, in female C57BL/6J and female Ldlr-/- mice. Organs were harvested 8 hours after binge. (B) Amount of food intake in ml/day/mouse. (C) Body weight/mouse in grams at the end of the study. (D) Plasma cholesterol levels in mg/dl at the start of the diet intervention. (E) Plasma triglyceride levels in mg/dl at the start of the diet intervention. (F) Plasma cholesterol levels in mg/dl at the study endpoint. (G) Plasma triglyceride levels in mg/dl at the study endpoint. Data shown as mean ± SEM of n=4-17 mice/group. * indicates significant differences between wild type C57BL/6J mice and Ldlr-/- mice. # indicates significant differences between isocaloric control-fed and ethanol–fed mice. * indicates p ≤0.05, *** p≤0.001, **** p≤0.0001. # indicates p ≤0.05, ## indicates p≤0.01.
Hepatic lipid levels are similar between wild type and Ldlr-/- mice after ethanol feeding
To determine to which extent ethanol intake during Ldlr-deficiency affects liver steatosis, hepatic cholesterol and triglyceride levels were measured. While ethanol diet did not alter cholesterol levels in the liver, chronic-binge ethanol feeding resulted in increased liver weight and the accumulation of hepatic triglycerides in Ldlr-/- and wild type mice compared to their respective pair-fed controls (Figures 2A–D). Although Ldlr-/- mice display altered systemic lipid levels, no differences in the amount of hepatic cholesterol and triglyceride content were observed compared to wild types, which was confirmed by Oil Red O staining (Figures 2B–D). In addition, no major morphological differences were observed in livers of Ldlr-/- and wild type mice after ethanol feeding (Figure 2E). These data indicate that systemic dyslipidemia due to the absence of the LDLR does not alter the degree of hepatic steatosis development in mice fed the chronic-binge ethanol diet.
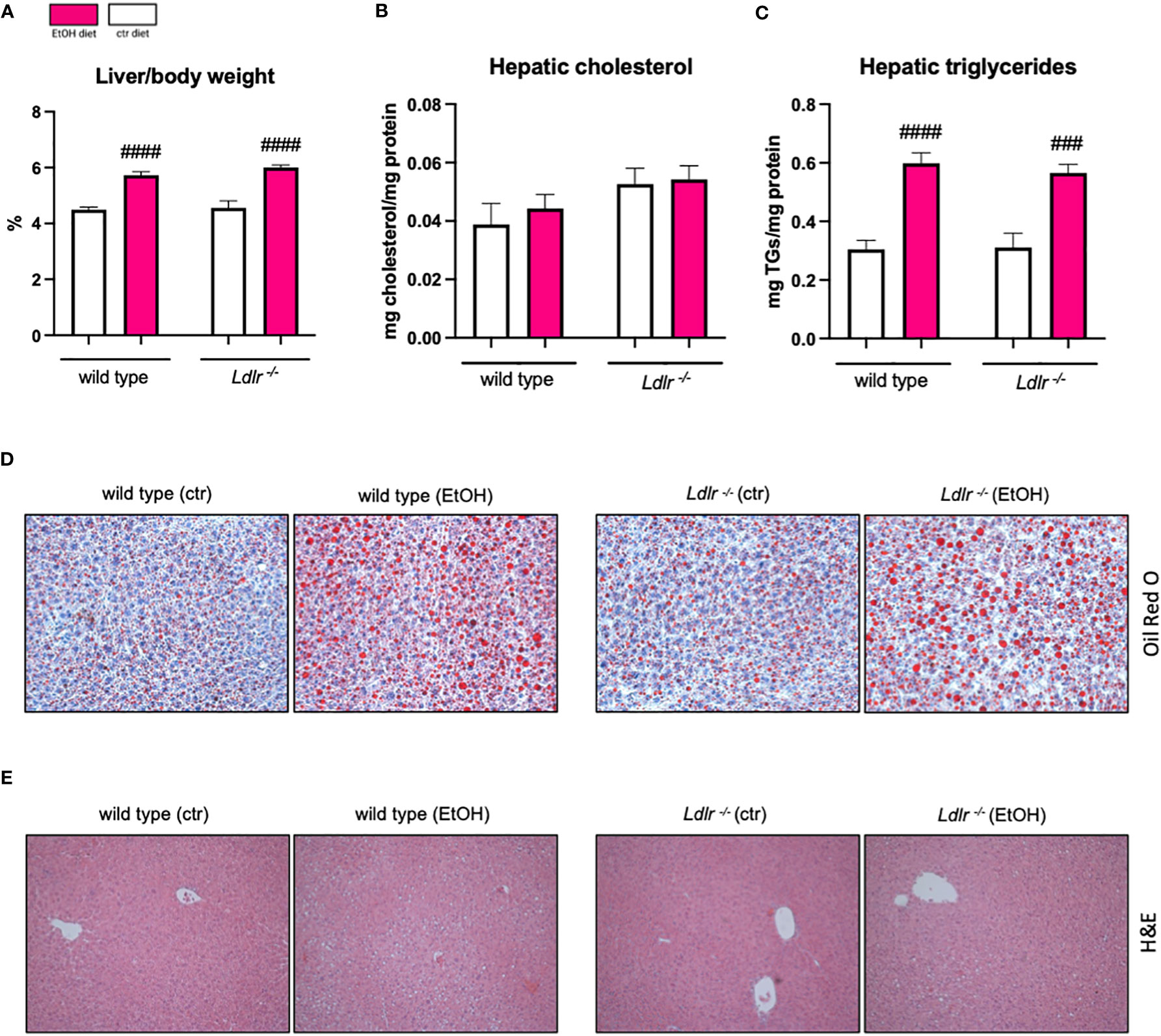
Figure 2 Wild type and Ldlr-/- mice develop hepatic steatosis to the same extent following chronic-binge ethanol feeding. (A) Liver to body weight ratio in percentage.(B) Hepatic cholesterol levels normalized to liver protein content. (C) Hepatic triglyceride levels normalized to liver protein content. (D) Representative images showing Oil Red O staining of liver sections. (magnification 20x) (E) Representative images showing H&E staining of liver sections. (magnification 10x) Data shown as mean ± SEM of n=4-17/group. # indicates significant differences between isocaloric control-fed and ethanol–fed mice. # indicates p ≤0.05, ### indicates p≤0.001, #### indicates p≤0.0001.
Next, we assessed hepatic mRNA levels of genes related to lipid metabolism and uptake in the liver, which might explain the observed accelerated hypertriglyceridemia in Ldlr-/- mice. While expression of Srebp1c, a gene involved in de novo lipogenesis, was downregulated after chronic-binge ethanol feeding in both genotypes, Srebpf2, which is crucial for cholesterol synthesis, remained unaffected by ethanol intake (Figures 3A, B). Notably, hepatic levels of Lpl mRNA were significantly higher in Ldlr-/- mice following ethanol administration than in wild type mice, while no genotype-related differences in Fas and ApoE gene expression following ethanol exposure were detectable. However, Fas and ApoE were downregulated in response to ethanol wild type but not in Ldlr-/- mice (Figures 3C–E), indicating altered lipid metabolism during Ldlr-deficiency. Interestingly, while expression of Sra1 did not differ after chronic-binge ethanol feeding, Ldlr-/- mice displayed increased hepatic expression of Cd36, important for uptake of oxidized LDL (Figures 3F, G). Further, while ethanol diet only minor affected cholesterol efflux via the expression of Abca1 and Abcg1, Ldlr-/- mice had higher Abcg1 mRNA levels in the liver after chronic-binge ethanol consumption compared to wild type mice (Figures 3H, I). Taken together, while expression levels of genes related to de novo lipogenesis seem not affected by the lack of the LDLR during ethanol consumption, increased Cd36 expression levels in mice deficient for Ldlr might enhance uptake of oxidized lipids, thereby triggering an inflammatory response in the liver.
Figure 3 Hepatic expression of genes related to lipid metabolism and transport show minor changes between wild type and Ldlr-/- mice. (A-I) mRNA levels of indicated genes in livers of ethanol- and control-fed female C57BL/6J and Ldlr-/- mice, assessed by qPCR. Data are shown relative to the wild type isocaloric control-fed mice and normalized to 18S. Data shown as mean ± SEM of n=4-17/group. # indicates significant differences between isocaloric control-fed and ethanol–fed mice. * indicates p ≤0.05, ** p≤0.01, *** p≤0.001, **** indicates p≤0.0001. # indicates p ≤0.05, ## p≤0.01, ### p≤0.001.
Ldlr-/- mice have less hepatic neutrophil recruitment compared to wild type mice after ethanol diet
Next, we determined whether elevated plasma lipid levels in mice lacking the LDLR affect ethanol-induced hepatic inflammation. Since alcohol-associated liver disease is characterized by the influx of inflammatory cells, particularly neutrophils (23), liver sections of Ldlr-/- and wild type mice were stained for infiltrating neutrophils and macrophages. In line with our expectations, chronic-binge ethanol feeding resulted in a significant induction in the recruitment of Ly6G+ neutrophils (NIMP+) to the livers of wild type mice (Figures 4A, B). A similar trend was observed upon immunohistochemical assessment of CD11b+ macrophages and neutrophils (Mac-1) in the livers of wild type mice (Figures 4C, D). Surprisingly, chronic-binge ethanol feeding resulted in less recruitment of neutrophils and macrophages to the livers in Ldlr-/- mice compared to pair-fed control mice (Figures 4A–D). In agreement with our histological observations, we found that the hepatic induction of pro-inflammatory genes Ly6g, Cd11b, Ccr2, Cxcl1, and F4/80, are significantly lower in Ldlr-/- mice than in wild type mice after ethanol diet (Figures 4E–H). Furthermore, we measured the gene expression levels of Toll-like receptors that are involved in regulating inflammatory responses. While Tlr4 was unchanged, mRNA levels of Tlr2 and Tlr9 were upregulated by ethanol intake in wild type controls (Figures 4J–L). Yet, Ldlr-/- mice failed to induce the expression of Tlr2 and Tlr9 after chronic-binge ethanol feeding. These data indicate that chronic-binge ethanol feeding in Ldlr-/- mice induces less neutrophil recruitment and inflammation in the liver compared to wild type mice, potentially as a result of diminished TLR signaling pathways.
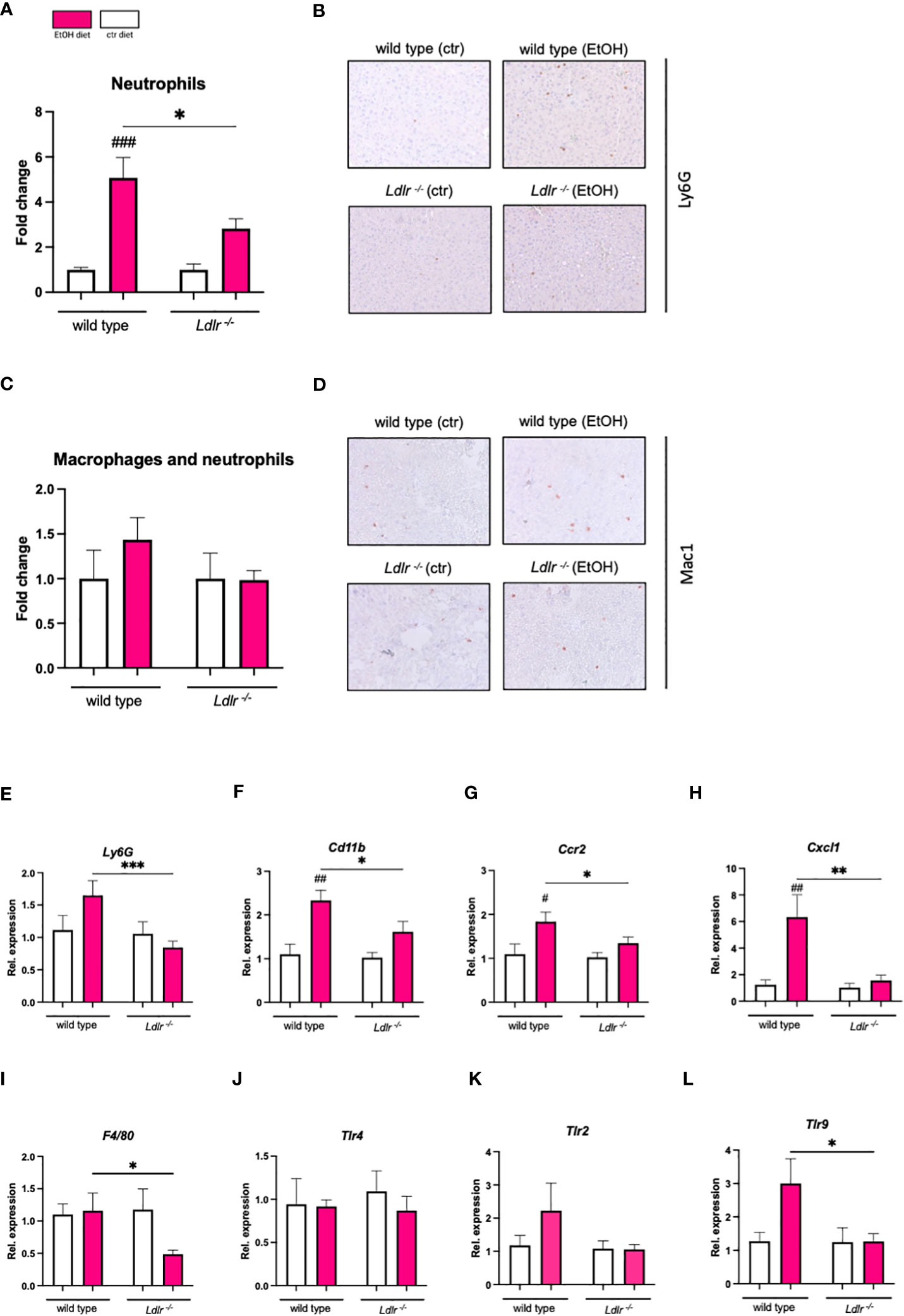
Figure 4 Ldlr-/- mice exhibit reduced hepatic inflammation compared to wild type mice following chronic-binge ethanol feeding. (A) Fold change of infiltrating neutrophils in the liver, assessed and quantified by immunohistochemical staining for NIMP. (B) Representative pictures of NIMP staining. (magnification 20x) (C) Fold change of infiltrating macrophages and neutrophils in the liver, assessed and quantified by immunohistochemical staining for Mac-1. (D) Representative pictures of Mac-1 staining. (magnification 20x) (E-L) mRNA levels of indicated genes (Ly6g, Cd11b, Ccr2, Cxcl1, F4/80, Tlr2, Tlr4, Tlr9) in livers of ethanol- and control-fed female C57BL/6J and Ldlr-/- mice, assessed by qPCR. Data are shown relative to the respective isocaloric control-fed mice and normalized to 18S. Data shown relative to the respective isocaloric control-fed mice as mean ± SEM of n=3-16/group. * indicates significant differences between wild type C57BL/6J mice and Ldlr-/- mice. # indicates significant differences between isocaloric control-fed and ethanol–fed mice.* indicates p ≤0.05, ** p≤0.01, *** p≤0.001. # indicates p ≤0.05, ## p≤0.01, ### p≤0.001.
Despite similar degrees of liver injury, Ldlr-/- mice seem to be more prone to develop hepatic fibrosis than wild type mice after chronic-binge ethanol feeding
In light of the observed attenuated hepatic inflammation in Ldlr-/- mice following chronic-binge ethanol feeding, our study sought to elucidate whether this reduction in inflammation is associated with a lower susceptibility to liver injury. Hence, we measured plasma alanine transaminase (ALT) levels, which were similar between Ldlr-/- and wild type mice (Figure 5A). Furthermore, we measured levels of thiobarbituric acid reactive substance (TBARS), a marker of oxidative stress, which is associated with hepatocellular damage, in the livers of mice subjected to chronic-binge ethanol feeding. No significant difference in TBARS levels was observed in livers of wild type and Ldlr-/- mice (Figure 5B). Next, although the chronic-binge ethanol feeding model to wild type mice is too mild and not sufficient to induce severe hepatic fibrosis, we assessed Sirius Red staining and hepatic expression of fibrosis-related genes to evaluate the effect of Ldlr-deficiency on alcohol-induced liver fibrosis. Interestingly, immunohistochemical scoring and detection of Sirius Red revealed that ethanol-fed Ldlr-/- mice developed increased collagen deposition compared to wild type mice (Figures 5C–E). In line, Ldlr-/- mice had significantly increased hepatic mRNA levels of Tgfb, Col1a1, and Col3a1 than wild type mice after chronic-binge ethanol feeding (Figures 5F–H). These data suggest that systemic dyslipidemia in Ldlr-deficient mice enhances the initiation of ethanol-induced liver fibrosis development compared with wild type mice.
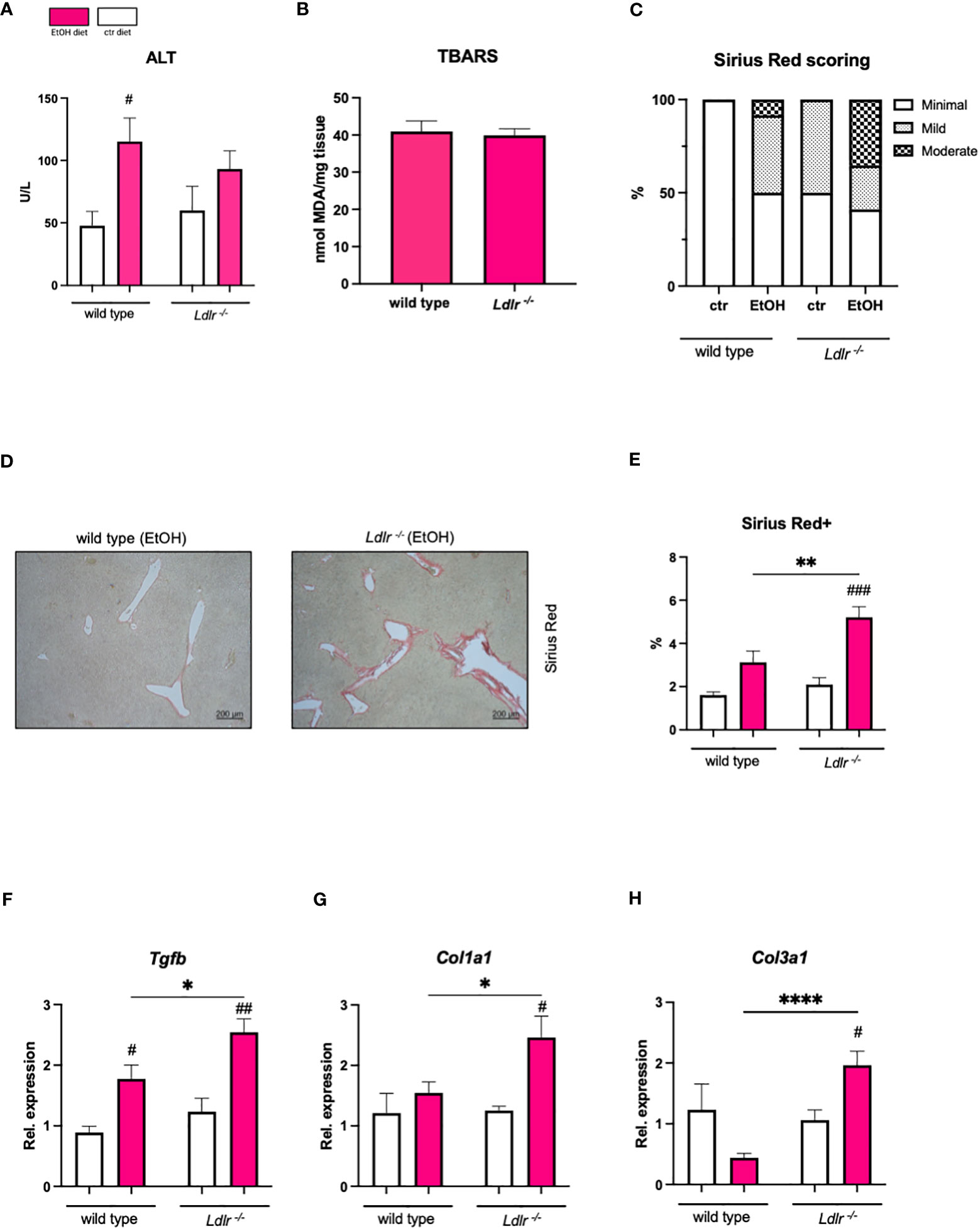
Figure 5 Despite comparable liver damage, Ldlr-/- mice exhibit increased susceptibility to ethanol-induced liver fibrosis compared with wild type mice. (A) Plasma ALT levels. (B) TBARS assay for MDA in liver tissue. (C) Percentage of mice/group with indicated arbitrary scoring for liver fibrosis based on Sirius red staining. (D) Representative pictures of Sirius Red staining of liver sections of ethanol-fed mice. (magnification 5x) (E) Quantification of immunohistochemical Sirius Red staining for liver fibrosis. (F–H) mRNA levels of indicated genes (Tgfb, Col1a1, Col3a1) in livers of ethanol- and control-fed female C57BL/6J and female Ldlr-/- mice, assessed by qPCR. Data are shown relative to the isocaloric control-fed wild type mice and normalized to 18S. Data shown as mean ± SEM of n=5-17/group. * indicates significant differences between wild type C57BL/6J mice and Ldlr-/- mice. # indicates significant differences between isocaloric control-fed and ethanol–fed mice. * indicates p ≤0.05, ** p≤0.01, **** indicates p≤0.0001. # indicates p≤0.05, ## p≤0.01, ### p≤0.001.
Ldlr-/- mice do not show altered ethanol metabolism
To assess whether our observed changes in the hepatic inflammatory and fibrotic response are related to differences in ethanol metabolism, plasma ethanol levels and expression of hepatic genes important for ethanol metabolism were measured. Plasma ethanol levels (Figure 6A), as well as hepatic mRNA levels of Adh1 and Cyp2e1, did not reveal differences between wild type and Ldlr-/- mice after ethanol feeding (Figures 6B, C). These data suggest that Ldlr-deficiency does not affect ethanol metabolism in the chronic-binge ethanol feeding model.
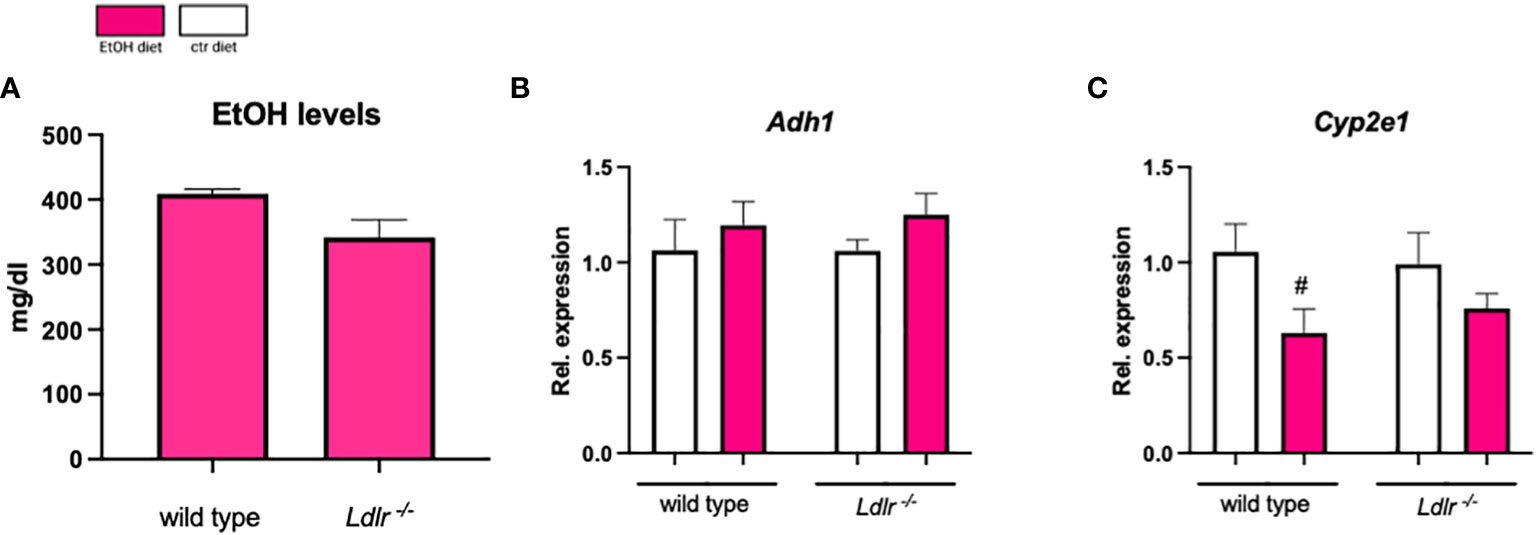
Figure 6 Ldlr-/- mice do not show alterations in ethanol absorption and metabolism. (A) Plasma ethanol levels at the study endpoint in ethanol-fed C57BL/6J and Ldlr-/- mice. (B, C) mRNA levels of indicated genes (Adh1, Cyp2e1) in livers of ethanol- and control-fed female C57BL/6J and Ldlr-/- mice, assessed by qPCR. Data are shown relative to the isocaloric control-fed mice and normalized to 18S. Data shown as mean ± SEM of n=4-16/group. # indicates significant differences between isocaloric control-fed and ethanol–fed mice. # indicates p ≤0.05.
Discussion
Chronic alcohol consumption mostly results in the development of alcohol-associated liver disease, ranging from steatosis to steatohepatitis, cirrhosis, and hepatocellular carcinoma. Since no interventions other than abstinence from alcohol are currently available and the cause for interindividual differences in the severity and progression of alcohol-related liver disease remains poorly understood, there is an urgent need to identify potential risk factors contributing to disease progression. Considering dysregulated lipid metabolism plays a significant role in all stages of the disease, we investigated the influence of pre-existing dyslipidemia on the development of alcohol-related liver disease. Our analyses revealed that chronic-binge ethanol feeding elevated systemic triglyceride levels in Ldlr-deficient mice, thereby affecting hepatic fibrosis initiation.
While we did not observe any effects of ethanol intake on systemic cholesterol levels, a study by Wang et al. reported that alcohol diet resulted in increased amounts of cholesterol in circulation due to downregulation of hepatic Ldlr expression, thereby lowering cholesterol uptake by the liver (13). Since experimental rats receiving Lieber-De Carli diet for 4 weeks were used, the difference in the animal model might explain any discrepancies observed in our study. Importantly, we found that ethanol consumption further raises high plasma triglyceride levels in the absence of LDLR. Considering that hypertriglyceridemia is a risk factor for cardiovascular disease, our results highlight that ethanol consumption among affected individuals might not only lead to alcohol-related liver disease but could further increase the risk of developing conditions affecting the heart and blood vessels (24).
Intriguingly, our findings demonstrated a decrease in hepatic neutrophil influx in Ldlr-/- mice, accompanied by indications of increased collagen deposition. In line with previously described observations that increased systemic lipid levels are associated with the severity of fibrosis (25–27), our data corroborate that systemic dyslipidemia is a key factor contributing to the progression of alcohol-related liver disease into fibrosis. Mechanistically, it is known that excessive lipid levels result in elevated oxidative stress and the formation of reactive oxygen species, thereby promoting injury and cell death. Importantly, cell death, as well as oxidative stress itself, are associated with the transdifferentiation of quiescent hepatic stellate cells (HSCs) into an activated collagen-secreting state, thereby initiating fibrosis (28–30). As such, accelerated fibrosis development in ethanol-fed dyslipidemic Ldlr-/- mice might result from elevated lipid-induced cell death and oxidative stress. Importantly, in line with our experimental data, increased LDL levels, as occurs in Ldlr-/- mice, were recently described to be an independent predictor of the severity and degree of hepatic fibrosis in patients with alcohol-associated liver disease (31). In addition, Lpl plays a pivotal function in facilitating the transfer of lipids from the bloodstream to various tissues, and due to its involvement in the regulation of lipid metabolism and energy homeostasis, increased Lpl expression has been shown to aggravate NASH. A study conducted by Teratani and colleagues (32) established a positive correlation between Lpl expression in HSCs and the worsening of fibrosis during NASH, thereby potentially explaining our observations of increased Lpl expression and hepatic deposition of collagen in Ldlr-/- mice (32). Moreover, despite the fact that our TBARS assay did not reveal any difference between wild type and Ldlr-/- mice after ethanol feeding, we observed increased hepatic expression of Cd36, which enables the uptake of oxidized lipids that enhance HSC activation, thereby potentially contributing to enhanced fibrosis. Taken together, our data confirm that metabolic profiling of patients with excessive alcohol consumption will help to identify the risk of developing alcohol-related end-stage liver disease.
Besides elevated circulatory lipid levels and altered lipid metabolism, other factors might contribute to accelerated fibrosis initiation in ethanol-fed Ldlr-/- mice. Considering alcohol consumption causes dysbiosis and increased gut leakiness (33–35), lipid-induced changes in the microbiome or altered intestinal permeability might enhance hepatic fibrosis. Indeed, Ldlr-/- mice have been shown to compose a different microbiome than wild type mice (36, 37). Hence, the translocation of specific gut-derived bacterial components in Ldlr-/- mice might promote HSCs activation and lead to more severe fibrosis after ethanol feeding (38–40). In addition, the intestinal microbiome affects bile acid metabolism and composition, which are involved in liver disease progression (41, 42). More specifically, secondary unconjugated bile acids have been shown to induce the expansion of HSCs, thereby promoting the progression of liver fibrosis (43, 44). Therefore, one might speculate that a modified bile acid composition is responsible for the increased collagen deposition in Ldlr-/- mice after ethanol diet. Further studies are needed to unravel the contribution of an altered microbiome and/or bile acid pool to aggravated collagen deposition in ethanol-fed dyslipidemic Ldlr-/- mice. Importantly, to exclude that our findings indicating enhanced systemic triglyceride levels and accelerated fibrosis after ethanol feeding during pre-existing dyslipidemia are restricted to Ldlr-deficient mice, confirmational studies using another mouse model should be undertaken, such as using ApoE-/- mice. Both Ldlr-/- mice and ApoE-/- mice exhibit impaired lipid metabolism, but the key distinction lies in the specific aspects of lipid clearance, resulting in elevated plasma cholesterol levels and altered lipid profiles. More precisely, Ldlr-deficiency is associated with increased LDL levels, while ApoE-deficiency is characterized by elevated levels of circulatory VLDL (45). Given that ApoE-/- mice also exhibit severe hypercholesterolemia and have more pronounced impaired immunoregulatory functions than Ldlr-/- mice one can speculate that ethanol feeding to ApoE-/- mice might further enhance dyslipidemia and steatohepatitis. Besides dyslipidemia in these models using genetically modified mice, it would be interesting to investigate ethanol-induced consequences during impaired lipid metabolism and systemic dyslipidemia within the context of wild type mice, i.e. combining a high-fat diet with ethanol supplementation. As such, a recent study by Chang et al. (46) introduced an experimental model that induces severe steatohepatitis through the administration of a high-fat diet for 3 days or 3 months combined with ethanol binge. While ethanol administration was shown to increase free fatty acid levels in the serum and liver, data describing triglyceride and cholesterol levels were lacking. Yet, this model seems to represent a suitable framework to examine the impact of ethanol on plasma and hepatic lipid levels in the presence of pre-existing dysregulated lipid metabolism in the setting of wild type mice.
In summary, we show that chronic-binge ethanol diet during systemic dyslipidemia in Ldlr-/- mice accelerates elevated plasma triglycerides and contributes to an early activation of a fibrotic response. Given the increased consumption of ethanol-containing beverages (47), the doubling of the prevalence of dyslipidemia between 2009 and 2019, and that familial hypercholesterolemia, which is caused by inherited mutations in the LDLR gene, affects 34 million people worldwide, our present study has important clinical implications (48, 49). Our results indicate that alcohol consumption by people with dyslipidemia or familial hypercholesterolemia may lead to more severe alcohol-related liver disease, besides enhancing their risk for cardiovascular complications. In addition, since Ldlr-/- mice display a human-like lipid profile with significant LDL-C in circulation compared to C57Bl/6J mice, our current data showing the development of ethanol-induced fibrosis in Ldlr-/- mice in the chronic-binge ethanol feeding provide evidence for a novel murine model for studying alcohol-associated liver disease (18).
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by Animal Ethics Committee of the Medical University of Vienna and the Austrian Federal Ministry of Education, Science and Research (License number: 2022-0.574.136).
Author contributions
CH performed the studies, acquired and analyzed the data, and wrote and edited the manuscript. DR, BS provided technical assistance and acquired part of the data. TH designed and performed the studies, analyzed the data, and wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
TH is funded by a ‘Right-On-Time’ grant (MLDS; W019-28) and Zukunftskollegs grant (FWF; ZK81B).
Acknowledgments
We thank all animal care takers from the Medical University of Vienna responsible for the animals used in this project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ApoE, Apolipoprotein E; Abca,: ATP-binding cassette transporter 1; Abcg1, ATP-binding cassette transporter 2; Adh1, Alcohol dehydrogenase 1; ALT, Alanine transaminase; ApoE-/,: Apolipoprotein E knock-out, ASH, Alcoholic steatohepatitis; Cd36, Cluster of differentiation 36; Ccr2, C-C Motif chemokine receptor 2; Cd11b, Integrin alpha M; Col1a1, Collagen type I alpha 1; Col3a1, Collagen type III alpha 1; Cxcl1, C-X-C Motif chemokine ligand 1; Cxcl2, C-X-C Motif chemokine ligand 2; Cyp2e1, Cytochrome P450 family 2 subfamily E member 1, Fas, Fatty acid synthase; F4/80, EGF-like module-containing mucin-like hormone receptor-like 1; HDL-C, High density lipoprotein cholesterol; HSCs, Hepatic stellate cells; H&E, Hematoxylin and eosin; LDL, Low density lipoprotein; LDL-C, Low density lipoprotein cholesterol; Ldlr-/-: LDL receptor knock-out; Lpl: Lipoproteinlipase; Mac-1, Macrophage receptor 1; NASH, Non-alcoholic steatohepatitis; Sra1, Scavenger receptor A1; Sra1, Scavenger receptor B1; Srebp1, Sterol regulatory element-binding protein 1; Srebp2, Sterol regulatory element-binding protein 2; TBARS, Thiobarbituric acid reactive substance; Tlr, Toll-like receptor; VLDL, Very low density lipoprotein.
References
1. Leggio L, Lee MR. Treatment of alcohol use disorder in patients with alcoholic liver disease. Am J Med (2017) 130(2):124–34. doi: 10.1016/j.amjmed.2016.10.004
2. Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, Lackner C, et al. Alcoholic liver disease. Nat Rev Dis Primers (2018) 4(1):16. doi: 10.1038/s41572-018-0014-7
3. Osna NA, Rasineni K, Ganesan M, Donohue TM Jr., Kharbanda KK. Pathogenesis of alcohol-associated liver disease. J Clin Exp Hepatol (2022) 12(6):1492–513. doi: 10.1016/j.jceh.2022.05.004
4. Serste T, Cornillie A, Njimi H, Pavesi M, Arroyo V, Putignano A, et al. The prognostic value of acute-on-chronic liver failure during the course of severe alcoholic hepatitis. J Hepatol (2018) 69(2):318–24. doi: 10.1016/j.jhep.2018.02.022
5. Anstee QM, Seth D, Day CP. Genetic factors that affect risk of alcoholic and nonalcoholic fatty liver disease. Gastroenterology (2016) 150(8):1728–44.e7. doi: 10.1053/j.gastro.2016.01.037
6. You M, Arteel GE. Effect of ethanol on lipid metabolism. J Hepatol (2019) 70(2):237–48. doi: 10.1016/j.jhep.2018.10.037
7. Ferdouse A, Clugston RD. Pathogenesis of alcohol-associated fatty liver: lessons from transgenic mice. Front Physiol (2022) 13:940974. doi: 10.3389/fphys.2022.940974
8. Li Q, Zhong W, Qiu Y, Kang X, Sun X, Tan X, et al. Preservation of hepatocyte nuclear factor-4α contributes to the beneficial effect of dietary medium chain triglyceride on alcohol-induced hepatic lipid dyshomeostasis in rats. Alcohol Clin Exp Res (2013) 37(4):587–98. doi: 10.1111/acer.12013
9. Tomita K, Azuma T, Kitamura N, Nishida J, Tamiya G, Oka A, et al. Pioglitazone prevents alcohol-induced fatty liver in rats through up-regulation of c-met. Gastroenterology (2004) 126(3):873–85. doi: 10.1053/j.gastro.2003.12.008
10. Cho KH, Nam HS, Kang DJ, Park MH, Kim JH. Long-term alcohol consumption caused a significant decrease in serum high-density lipoprotein (HDL)-cholesterol and apolipoprotein a-I with the atherogenic changes of HDL in middle-aged Korean women. Int J Mol Sci (2022) 23(15), 8623. doi: 10.3390/ijms23158623
11. Baraona E, Lieber CS. Effects of ethanol on lipid metabolism. J Lipid Res (1979) 20(3):289–315. doi: 10.1016/S0022-2275(20)40613-3
12. Visioli F, Monti S, Colombo C, Galli C. Ethanol enhances cholesterol synthesis and secretion in human hepatomal cells. Alcohol (1998) 15(4):299–303. doi: 10.1016/S0741-8329(97)00133-X
13. Wang Z, Yao T, Song Z. Chronic alcohol consumption disrupted cholesterol homeostasis in rats: down-regulation of low-density lipoprotein receptor and enhancement of cholesterol biosynthesis pathway in the liver. Alcohol Clin Exp Res (2010) 34(3):471–8. doi: 10.1111/j.1530-0277.2009.01111.x
14. Chait A, Mancini M, February AW, Lewis B. Clinical and metabolic study of alcoholic hyperlipidaemia. Lancet. (1972) 2(7767):62–4. doi: 10.1016/S0140-6736(72)91552-8
15. Shen Z, Munker S, Wang C, Xu L, Ye H, Chen H, et al. Association between alcohol intake, overweight, and serum lipid levels and the risk analysis associated with the development of dyslipidemia. J Clin Lipidol (2014) 8(3):273–8. doi: 10.1016/j.jacl.2014.02.003
16. Foerster M, Marques-Vidal P, Gmel G, Daeppen JB, Cornuz J, Hayoz D, et al. Alcohol drinking and cardiovascular risk in a population with high mean alcohol consumption. Am J Cardiol (2009) 103(3):361–8. doi: 10.1016/j.amjcard.2008.09.089
17. Hoebinger C, Rajcic D, Hendrikx T. Oxidized lipids: common immunogenic drivers of non-alcoholic fatty liver disease and atherosclerosis. Front Cardiovasc Med (2021) 8:824481. doi: 10.3389/fcvm.2021.824481
18. Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest (1993) 92(2):883–93. doi: 10.1172/JCI116663
19. Hendrikx T, Lang S, Rajcic D, Wang Y, McArdle S, Kim K, et al. Hepatic pIgR-mediated secretion of IgA limits bacterial translocation and prevents ethanol-induced liver disease in mice. Gut (2023). doi: 10.1136/gutjnl-2022-328265
20. Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc (2013) 8(3):627–37. doi: 10.1038/nprot.2013.032
21. Busch CJ, Hendrikx T, Weismann D, Jackel S, Walenbergh SM, Rendeiro AF, et al. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology (2017) 65(4):1181–95. doi: 10.1002/hep.28970
22. Bieghs V, Van Gorp PJ, Wouters K, Hendrikx T, Gijbels MJ, van Bilsen M, et al. LDL receptor knock-out mice are a physiological model particularly vulnerable to study the onset of inflammation in non-alcoholic fatty liver disease. PloS One (2012) 7(1):e30668. doi: 10.1371/journal.pone.0030668
23. Mookerjee RP, Stadlbauer V, Lidder S, Wright GA, Hodges SJ, Davies NA, et al. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology (2007) 46(3):831–40. doi: 10.1002/hep.21737
24. Watts GF, Catapano AL, Masana L, Zambon A, Pirillo A, Tokgözoğlu L. Hypercholesterolemia and cardiovascular disease: focus on high cardiovascular risk patients. Atheroscler Suppl (2020) 42:e30–e4. doi: 10.1016/j.atherosclerosissup.2021.01.006
25. Liu Y, Zhang P, Li J, Li H, Zhou C, Zhang Y, et al. Association between serum lipid profile and liver fibrosis in patients infected with schistosoma japonicum. Parasit Vectors (2022) 15(1):268. doi: 10.1186/s13071-022-05359-8
26. Varghese JS, Krishnaprasad K, Upadhuyay R, Revathy MS, Jayanthi V. Lipoprotein profile in cirrhosis of liver. Eur J Gastroenterol Hepatol (2007) 19(6):521–2. doi: 10.1097/MEG.0b013e3280be5ac8
27. Mena Á, Pedreira JD, Castro Á, López S, Vázquez P, Poveda E. Metabolic syndrome association with fibrosis development in chronic hepatitis b virus inactive carriers. J Gastroenterol Hepatol (2014) 29(1):173–8. doi: 10.1111/jgh.12432
28. Matsuda M, Seki E. Hepatic stellate cell-macrophage crosstalk in liver fibrosis and carcinogenesis. Semin Liver Dis (2020) 40(3):307–20. doi: 10.1055/s-0040-1708876
29. Gandhi CR. Oxidative stress and hepatic stellate cells: a PARADOXICAL RELATIONSHIP. Trends Cell Mol Biol (2012) 7:1–10.
30. De Smet V, Eysackers N, Merens V, Kazemzadeh Dastjerd M, Halder G, Verhulst S, et al. Initiation of hepatic stellate cell activation extends into chronic liver disease. Cell Death Dis (2021) 12(12):1110. doi: 10.1038/s41419-021-04377-1
31. Israelsen M, Juel HB, Detlefsen S, Madsen BS, Rasmussen DN, Larsen TR, et al. Metabolic and genetic risk factors are the strongest predictors of severity of alcohol-related liver fibrosis. Clin Gastroenterol Hepatol (2022) 20(8):1784–94.e9. doi: 10.1016/j.cgh.2020.11.038
32. Teratani T, Tomita K, Furuhashi H, Sugihara N, Higashiyama M, Nishikawa M, et al. Lipoprotein lipase up-regulation in hepatic stellate cells exacerbates liver fibrosis in nonalcoholic steatohepatitis in mice. Hepatol Commun (2019) 3(8):1098–112. doi: 10.1002/hep4.1383
33. Engen PA, Green SJ, Voigt RM, Forsyth CB, Keshavarzian A. The gastrointestinal microbiome: alcohol effects on the composition of intestinal microbiota. Alcohol Res (2015) 37(2):223–36.
34. Elamin E, Masclee A, Troost F, Pieters HJ, Keszthelyi D, Aleksa K, et al. Ethanol impairs intestinal barrier function in humans through mitogen activated protein kinase signaling: a combined in vivo and in vitro approach. PloS One (2014) 9(9):e107421. doi: 10.1371/journal.pone.0107421
35. Yan AW, Fouts DE, Brandl J, Stärkel P, Torralba M, Schott E, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology (2011) 53(1):96–105. doi: 10.1002/hep.24018
36. Gart E, Souto Lima E, Schuren F, de Ruiter CGF, Attema J, Verschuren L, et al. Diet-independent correlations between bacteria and dysfunction of gut, adipose tissue, and liver: a comprehensive microbiota analysis in feces and mucosa of the ileum and colon in obese mice with NAFLD. Int J Mol Sci (2018) 20(1). doi: 10.3390/ijms20010001
37. Kiouptsi K, Jäckel S, Pontarollo G, Grill A, Kuijpers MJE, Wilms E, et al. The microbiota promotes arterial thrombosis in low-density lipoprotein receptor-deficient mice. mBio (2019) 10(5):e02298–19. doi: 10.1128/mBio.02298-19
38. Hartmann P, Seebauer CT, Schnabl B. Alcoholic liver disease: the gut microbiome and liver cross talk. Alcohol Clin Exp Res (2015) 39(5):763–75. doi: 10.1111/acer.12704
39. Lin RS, Lee FY, Lee SD, Tsai YT, Lin HC, Lu RH, et al. Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J Hepatol (1995) 22(2):165–72. doi: 10.1016/0168-8278(95)80424-2
40. Liu B, Zhou Z, Jin Y, Lu J, Feng D, Peng R, et al. Hepatic stellate cell activation and senescence induced by intrahepatic microbiota disturbances drive progression of liver cirrhosis toward hepatocellular carcinoma. J Immunother Cancer (2022) 10(1):e003069. doi: 10.1136/jitc-2021-003069
41. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Gut microbiota, cirrhosis, and alcohol regulate bile acid metabolism in the gut. Dig Dis (2015) 33(3):338–45. doi: 10.1159/000371678
42. Zhang YL, Li ZJ, Gou HZ, Song XJ, Zhang L. The gut microbiota-bile acid axis: a potential therapeutic target for liver fibrosis. Front Cell Infect Microbiol (2022) 12:945368. doi: 10.3389/fcimb.2022.945368
43. Svegliati-Baroni G, Ridolfi F, Hannivoort R, Saccomanno S, Homan M, De Minicis S, et al. Bile acids induce hepatic stellate cell proliferation via activation of the epidermal growth factor receptor. Gastroenterology (2005) 128(4):1042–55. doi: 10.1053/j.gastro.2005.01.007
44. Saga K, Iwashita Y, Hidano S, Aso Y, Isaka K, Kido Y, et al. Secondary unconjugated bile acids induce hepatic stellate cell activation. Int J Mol Sci (2018) 19(10), 3043. doi: 10.3390/ijms19103043
45. Gisterå A, Ketelhuth DFJ, Malin SG, Hansson GK. Animal models of atherosclerosis-supportive notes and tricks of the trade. Circ Res (2022) 130(12):1869–87. doi: 10.1161/CIRCRESAHA.122.320263
46. Chang B, Xu MJ, Zhou Z, Cai Y, Li M, Wang W, et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: an important role for CXCL1. Hepatology (2015) 62(4):1070–85. doi: 10.1002/hep.27921
47. Manthey J, Shield KD, Rylett M, Hasan OSM, Probst C, Rehm J. Global alcohol exposure between 1990 and 2017 and forecasts until 2030: a modelling study. Lancet (2019) 393(10190):2493–502. doi: 10.1016/S0140-6736(18)32744-2
48. McGowan MP, Hosseini Dehkordi SH, Moriarty PM, Duell PB. Diagnosis and treatment of heterozygous familial hypercholesterolemia. J Am Heart Assoc (2019) 8(24):e013225. doi: 10.1161/JAHA.119.013225
49. Bilitou A, Were J, Farrer A, Rabe A, Ming SWY, Haq I, et al. Prevalence and patient outcomes of adult primary hypercholesterolemia and dyslipidemia in the UK: longitudinal retrospective study using a primary care dataset from 2009 to 2019. Clinicoecon Outcomes Res (2022) 14:189–203. doi: 10.2147/CEOR.S347085
Keywords: alcohol-associated liver disease (ALD), dyslipidemia, hypercholesterolemia, low-density lipoprotein receptor (LDLR), Ldlr-/- mice, alcoholic fibrosis, inflammation
Citation: Hoebinger C, Rajcic D, Silva B and Hendrikx T (2023) Chronic-binge ethanol feeding aggravates systemic dyslipidemia in Ldlr-/- mice, thereby accelerating hepatic fibrosis. Front. Endocrinol. 14:1148827. doi: 10.3389/fendo.2023.1148827
Received: 20 January 2023; Accepted: 28 June 2023;
Published: 25 July 2023.
Edited by:
Yang Yang, University of Texas Health Science Center at Houston, United StatesReviewed by:
Rebecca L. McCullough, University of Colorado, United StatesAlina Sisu, Victor Babes University of Medicine and Pharmacy, Romania
Copyright © 2023 Hoebinger, Rajcic, Silva and Hendrikx. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tim Hendrikx, dGltLmhlbmRyaWt4QG1lZHVuaXdpZW4uYWMuYXQ=