 Alessia Aureli
Alessia Aureli Sarah Bocchini1
Sarah Bocchini1 Michela Mariani
Michela Mariani Antonino Crinò
Antonino Crinò Marco Cappa
Marco Cappa Danilo Fintini
Danilo Fintini- 1Prader-Willi Reference Center, Endocrinology Unit, Bambino Gesù Children Hospital, IRCCS, Rome, Italy
- 2Center for Rare Diseases and Congenital Defects, Fondazione Policlinico Universitario A. Gemelli, IRCCS, Rome, Italy
Prader–Willi syndrome (PWS) is a rare genetic disorder resulting from lack of expression of the paternally derived chromosome 15q11–13, associated with several complications, including pubertal disorders, short stature, hyperphagia, obesity, glucose metabolism abnormalities, scoliosis, obstructive sleep apnea syndrome (OSAS) and behavioral problems. We report the case of a girl affected by PWS who presented at the age of 5.9 with premature pubarche, accelerated linear growth and advanced bone age (BA). She was subsequently diagnosed with non-classic congenital adrenal hyperplasia (CAH) confirmed by genetic analysis. Considering the clinical, biochemical, and genetic findings, hydrocortisone therapy was started to prevent rapid BA acceleration and severe compromission of final height. During infancy, short stature and low levels of insulin-like growth factor-1 (IGF-1) for age and gender led to suspicion of growth hormone deficiency (GHD), confirmed by stimulation testing (arginine and clonidine). rhGH therapy was administered and continued until final height was reached. During endocrinological follow up she developed impaired glucose tolerance with positive markers of β-cell autoimmunity (anti-glutamic acid decarboxylase antibodies, GAD Ab), which evolved over time into type 1 diabetes mellitus and insulin therapy with a basal-bolus scheme and an appropriate diet were needed.
1 Introduction
1.1 Prader-Willi Syndrome
Prader-Willi Syndrome (PWS) is a rare disease and the most frequent form of syndromic obesity, with a prevalence rate of 1 in 10,000 to 30,000 live births (1, 2). This genetic disorder is caused by the absence of the paternally expressed imprinted genes in the critical region of chromosome 15 at the locus 15q11.2-q13, which can derive from paternal deletion (del15) (65-75% of individuals), maternal uniparental disomy (UPD15) (20-30%), or an imprinting defect (ID15) (1-3%) (1, 2).
PWS is characterized by a multitude of signs and symptoms, many of which possibly linked to hypothalamic dysfunction (3), including: neonatal hypotonia accompanied by failure to thrive, followed by hyperphagia in early childhood and gradual development of morbid obesity, dysmorphic features (characteristic facial appearance with turricephaly, small hands and feet, scoliosis and kyphosis), intellectual disability, behavioral problems and multiple endocrine abnormalities, including GH deficiency (GHD) and hypogonadism (2).
1.1.1 Adrenarche and pubertal development in PWS
Early adrenarche and higher prevalence of premature pubarche (PP) have been described in PWS, although data on prevalence and age of onset are mostly based on case series (4). The estimated prevalence of PP in PWS is approximately 30% in girls and 16% in boys (4). Adrenarche is defined as the increase in production of adrenal androgens, most specifically dehydroepiandrosterone sulphate (DHEA-S) (5). It is a gradual process characterized by the appearance of adult body odor, acne and blackheads, oily hair and pubic and/or axillary hair (5) which usually occurs after the age of 8 years in girls and 9 years in boys. Early adrenarche and PP in children with PWS possibly result from earlier maturation of the zona reticularis of the adrenal glands, independently of hypothalamic-pituitary-adrenal axis activation (5, 6) which is heterogeneous in PWS. Nevertheless, PWS is characterised by a slower pubertal pace, with a higher age at Tanner stage P5 in both sexes. In girls a higher age at menarche with respect to the general population is described, whereas a minority may present with pubertal arrest and/or primary amenorrhea (7, 8). Males with PWS often present hypothalamic hypogonadism, although recently primary testicular dysfunction has been advocated as a major contributor to abnormal pubertal development (9, 10). Although puberty and sexual maturation are usually delayed or incomplete in PWS, rare occurrences of true precocious puberty are described (11, 12).
1.1.2 Diabetes mellitus in PWS
Diabetes mellitus can affects PWS patients and it typically presents with a type 2 (T2DM) phenotype (13), with a reported prevalence of 7-24% (14–16). Nonetheless, rare cases presenting as diabetes type 1 (T1DM), or a monogenic diabetes (maturity onset diabetes of the young, MODY), have also been described (17–19). Furthermore, occasional reports of islet autoantibodies in diabetic PWS subjects were associated with a T2DM phenotype (20). In a group of 23 diabetic PWS patients with age at onset <20 years (13.4 ± 3.9 years), β-cell antibodies were found in 40% of subjects (21). Besides these scarce reports, islet autoantibodies and other markers of autoimmune islet cell destruction in PWS have not been extensively studied.
In PWS it has been traditionally assumed that abnormal glucose metabolism develops as a consequence of obesity. In accordance, non-obese subjects show lower insulin and glucose values compared to obese PWS individuals, both in children and in adults (22–24). Nevertheless, the relationship between obesity and the development of diabetes is not completely elucidated and may be different than in individuals with simple obesity.
1.2 Congenital adrenal hyperplasia
Congenital adrenal hyperplasia (CAH) represents a heterogeneous group of autosomal recessive disorders caused by deficient adrenal corticosteroid biosynthesis (25, 26). It arises from enzymatic defects in the adrenal steroidogenic pathway or in the electron-providing factor, (P450 oxidoreductase - POR). Between 90% and 95% of cases of CAH are caused by 21-hydroxylase deficiency (21OHD) encoded by the CYP21A2 gene (25, 26) located on the short arm of chromosome 6 (6p21.3).
In Western societies, the incidence of classic 21OHD (present at birth with or without salt-wasting and cortisol deficiency) varies from 1 in 10.000-15.000 live births. Non-classic (or late-onset) 21OHD is more common, with an incidence of about 1 in 500 to 1.000 live births (27). The condition arises because of defective conversion of 17-hydroxyprogesterone (17OHP) to 11-deoxycortisol. Defective cortisol biosynthesis results in reduced hypothalamic-pituitary negative feedback and increased ACTH secretion; consequently, adrenal androgens are produced in excess. The genotype-phenotype correlation in CAH due to the 21OHD is well established and the clinical phenotype is dependent on residual enzymatic activity provided by the less severely mutated allele.
Several clinical variants have been identified from genital ambiguity in newborns girls to precocious pseudopuberty in both males and females. During adolescence and adulthood, females can present with a phenotype similar to polycystic ovary syndrome (PCOS) with hirsutism, primary or secondary amenorrhea, or anovulatory infertility (28). In the salt-wasting form the patients also present concomitant cortisol and aldosterone deficiency; it usually occurs after the first 2 weeks of life and neonates show hypotension, poor feeding, vomiting, lethargy, and sepsis-like symptoms.
The remaining 5-10% of CAH forms are caused by mutations in other steroidogenic enzymes involved in adrenal steroidbiosynthesis, such as: CYP11B1, CYP17A1 and HSD3B2, encoding respectively 11β-hydroxylase, 17α-hydroxylase and 3β-hydroxysteroid dehydrogenase type 2.
Association between PWS and adrenal congenital hyperplasia is very rare and only sporadic case reports are described in literature (29, 30).
2 Case report
We report the case of C.B., a 16 years-old girl affected by PWS, who presented PP in the context of CAH by 21OHD and later developed T1DM.She was born at 37 weeks by cesarean section, her birth weight was 2500 g (-0,89 standard deviation scores [SDS]). During infancy she presented with severe hypotonia, respiratory distress, weak cry, small hands and feet, which led to the suspicion of PWS. At 12 months of age the diagnosis was confirmed by methylation-specific multiplex ligation-dependent probe amplification analysis (MS-MLPA) and chromosome 15 DNA polymorphism analysis conducted on the proband and her parents, demonstrated the presence of maternal UPD. During childhood she presented neurodevelopmental disorders comprising learning and intellectual disability, language delay and impairment. Neuropsychiatric evaluation did not report behavioral disorders, although skin picking of the four limbs was found on pre-existing injuries. Food seeking behavior was controlled through parental training, providing a strict schedule of meals and avoiding the intake of sweet and high calorie foods from infancy; this is particularly relevant since the hyperphagia phase in PWS usually occurs between 2 and 4 years of age, and is then followed by an abnormal interest towards food (31). At the age of 2.4 years she presented short stature (Table 1) alongside reduced levels of insulin-like growth factor-1 (IGF-1) for age and gender (31 ng/mL, n.v. 49-289) and growth hormone (GH) stimulation tests with arginine (0.5 g/Kg iv) and clonidine (150 µg/m2 orally) were performed, leading to the diagnosis of GHD (Table 2). According to the 2006 Italian Drug Agency (AIFA) Note 39 criteria (32) rhGH therapy was started at a dose of 0.033 mg/Kg/d (0.7 mg/m²/d); thereafter the dose was titrated according to IGF-1 levels, keeping it within <2 SDS, according to guidelines for GHD treatment in PWS (31, 33). Clinical follow up was established every six months and polysomnography were performed once a year to exclude obstructive sleep apnea syndrome (OSAS) which can worsen during rhGH therapy. A controlled diet with the support of a nutrition specialist permitted to control the patient’s body mass index (BMI) keeping it in the normal weight range (Figure 1). Regular growth with a height velocity (HV) of 6-7 cm/yr was registered until the age of 5.5 (75° centile; +0.74 SDS), alongside pre-pubertal Tanner stage 1 by physical examination. At 5.5 years of age pubic hair growth was noted, without thelarche, and at the age of 5.9 an endocrinological evaluation was requested (Tables 1, 2). Six months later, elevated 17OHP and DHEA-S levels were confirmed, HV attested at 10.4 cm/yr (>97° centile; +4.7 SDS) and physical examination confirmed a Tanner stage Ph2-Pb1, with no evidence of axillary hair. Stimulation test for CAH with tetracosactide 250 µg IV was conducted showing basal and stimulated level of 17OHP >10 ng/mL, both diagnostic for 21OHD (35). The patient and her parents were referred for genetic testing (MLPA confirmed by Sanger sequencing) for non-classic CAH due to 21OHD with evidence of compound heterozygosity for CYP21A2 mutation: V281L, inherited from the mother, and A(C)656G [intron 2], inherited from the father. At the age of 7.7 years the patient confirmed accelerated HV (10 cm/aa; + 5.2 SDS), physical examination revealed Tanner stage Ph3-Pb2, initial axillary hair and clitoral enlargement; bone age (BA) was advanced corresponding to 9.5 years, according to Greulich&Pyle method, and pelvic ultrasound showed infantile uterine morphometry, with maximum ovarian volume of 1.2 mL and a thin endometrium. The evidence of pubertal progression prompted the execution of a stimulation test with 100 µg IV gonadotropin-releasing hormone (GnRH test), which showed a FSH and LH peak in accordance with pubertal activation of the hypothalamic-pituitary-gonadal axis (Table 2). According with the biochemical and clinical evidence of hyperandrogenism which led to the linear growth acceleration, and considering the risk of premature epiphyseal closure, therapy with hydrocortisone 20 mg/m2/d was started in order to promptly suppress androgen levels. Eight months after the start of therapy, Tanner stage was Ph3-Pb2, suppressed levels steroid hormones were observed, HV declined to 5 cm/yr and a left-hand X-ray documented a BA of 10.5 years (Tables 1, 2). An oral glucose tolerance test (oGTT 1.75 g/Kg) showed an impaired glucose tolerance (IGT), and therefore rhGH therapy and hydrocortisone therapy were reduced to 0.021 mg/Kg/d and 14 mg/m2/d, respectively). At the age of 9.0 the patient presented HV 3.9 cm/yr (3rd centile; -1.9 SDS) and Tanner stage did not change. Considering the patient having entered an appropriate age for pubertal start (>8 years-old), the HV and pubertal stage stability, hydrocortisone therapy was reduced to 11.5 mg/m2/d and rhGH therapy increased to 0.026 mg/Kg/d, to allow linear growth and spontaneous pubertal progression.
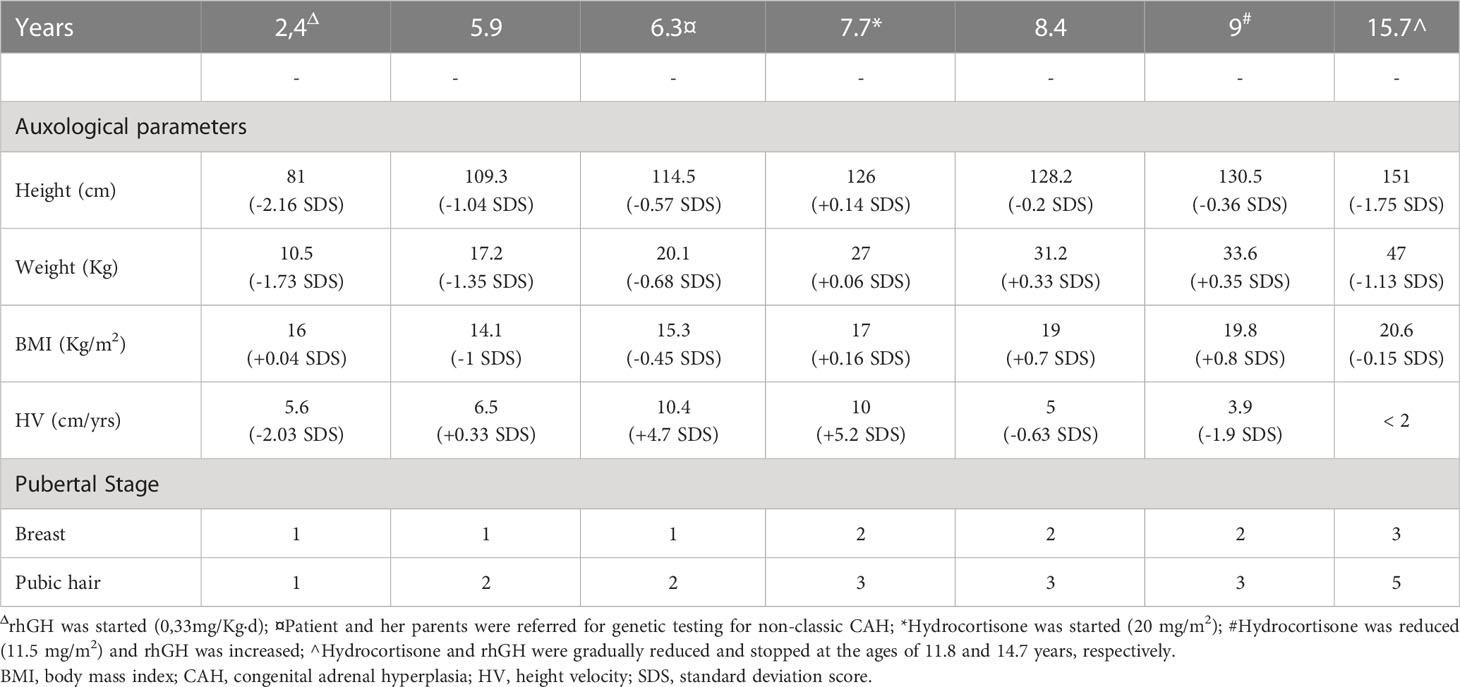
Table 1 Clinical features during endocrinological follow up.
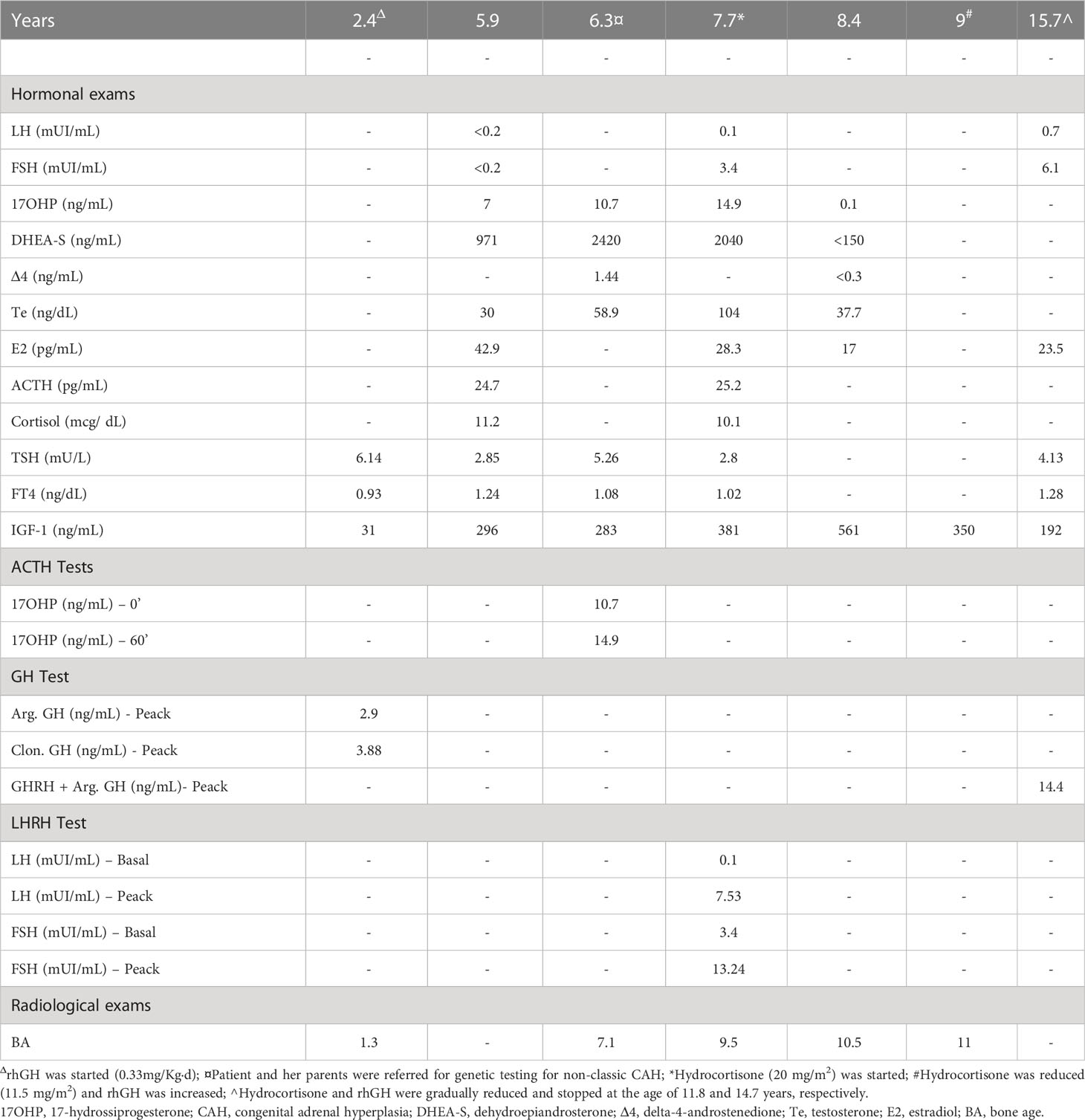
Table 2 Hormonal and radiological exams during endocrinological follow up.
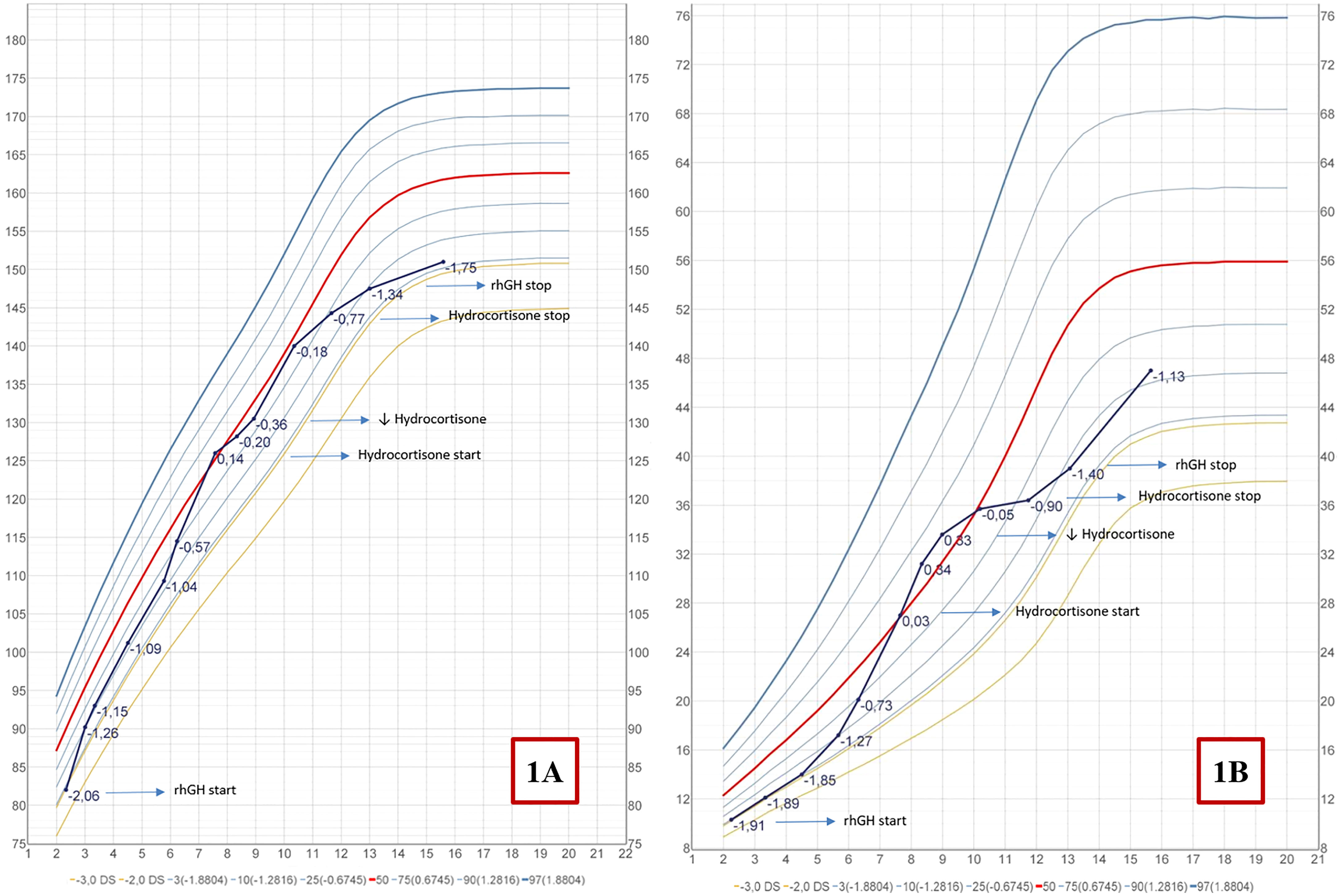
Figure 1 Italian growth chart for height (A) and weight (B) (34).
At this time the patient first presented a positive autoimmune screening for glucose metabolism disorders, with an anti-glutamic acid decarboxylase (GAD) titer of 54 U/mL (n.v. <0.9), with normal levels of fasting glucose and insulin.
During the follow-up hydrocortisone therapy was gradually reduced and then stopped at the age of 11.8. rhGH therapy was continued following IGF-1 levels and HV, gradually increased up to 0.034 mg/Kg/d and then stopped when final stature was reached at the age of 14.7 years (HV <2cm/yr). After five months from the stop of rhGH therapy, a retesting with Growth Hormone Releasing Hormone (GHRH) + arginine was performed and persistent GHD was excluded according to the current Italian Drug Agency (AIFA) Note 39 criteria (36). During the endocrinological evaluation at the age of 15.7 the patient presented: height 151 cm (-1.76 SDS), weight 47 kg (-1.13 SDS), BMI 20.6 (-0.15 SDS), Tanner stage Ph5-Pb3; biochemical and hormonal exams showed a mature hypothalamus-pituitary-gonadal axis (HPG) (Table 2), C-peptide attested at 1.14 ng/mL (in the lower part of reference range) and the IGT condition was confirmed.
At the age of 16.2 she presented with polyuria and polydipsia, weight loss and a fasting Hemoglucotest (HGT) >200 mg/dL. She referred to the emergency room, where she presented glucose levels of 382 mg/dL, HbA1c of 13.7%, C-peptide of 0.41 ng/mL, Na 141 mEq/L, K 4.8 mEq/L, glycosuria and ketonuria and an arterial blood gas analysis documented metabolic acidosis: pH 7.29, base excess -5.9, HCO3- 20.7 mmol/L, lactate 1.3 mmol/L, anion gap 19.3 mEq/L. Anti-insulin (IAA), anti-tyrosine phosphatase (IA2) and anti-ZnT8 antibodies were undetectable, whereas anti-GAD rose to 191 U/mL. Other exams showed normal thyroid function although with positive anti-thyroglobulin (Ab-Tg) and thyroid peroxidase antibodies (Ab-TPO), and a negative screening for celiac disease. Diagnosis of T1DM was made, insulin therapy with a basal-bolus scheme and an appropriate diet was started. The patient is currently followed at our Endocrine Center with a specific therapeutic program consisting in clinical and biochemical follow-up.
Written informed consent was obtained from the parents for the publication of this case report including clinical, biochemical and radiological data.
3 Discussion
Early adrenarche and higher prevalence of PP have been described in patients with PWS and in some of these cases a CAH diagnosis might be present (37). One study conducted on 120 PWS children between the ages of 2 and 17 years, described earlier adrenarche, with serum DHEA-S levels significantly higher in PWS girls and boys compared to healthy controls in the age groups from 3 to 10 years (37). Median age at onset of pubarche (Tanner stage Ph2) in children with PWS was 9.0 years in girls and 10.3 years in boys (37).
Our patient presented adrenarche and pubarche at the age of 5.5 years. In contrast with the literature, she showed fast progression in pubic hair growth; biochemical evaluation demonstrated a very high level of adrenal androgens, including 17OHP. At the first endocrinological evaluation the oestradiol (E2) concentration was also elevated, although with prepubertal levels of LH and FSH, which was interpreted as a condition of pseudopuberty due to excess of androgens and their consequent aromatization. For this reason, standard dose ACTH-stimulation test for 17OHP was performed.
PWS is a rare disease (estimated prevalence of 1:10.000-30.000) (1, 2), whereas CAH in its non-classic form is quite more common (1 in 500 to 1.000 live births in western countries) (27); as such their association may sporadically occur in the population. Nonetheless, the presence of both conditions is rare and to our knowledge only two cases are described in literature (29, 30). Patients with the non-classic form of CAH may present in early childhood with sexual precocity, pubic hair development or growth acceleration due to premature androgen excess. If left untreated, the sex steroid production stimulates premature epiphyseal closure, and final adult height is invariably reduced (38, 39). Additionally, central precocious puberty may develop in patients with CAH, possibly due to androgen activation of the HPG axis, thus exacerbating premature epiphyseal fusion (40, 41). Glucocorticoid therapy remains the standard treatment for patients with CAH. Some studies report a significant improvement in final adult height outcome in CAH children who are treated with GH, alone or in combination with GnRH analogues (42–44). rhGH therapy may counter the deceleration in growth velocity often associated with glucocorticoid therapy, whereas GnRH analogues suppress the HPG axis to prevent premature epiphyseal closure. However, since the addition of GnRH analogues has not been clearly shown to benefit GH therapy in increasing height gain, their use should be based on the age of the patient and the social impact of precocious puberty. In our patient genetic testing for 21OHD was performed with confirmation of non-classic CAH and as such hydrocortisone therapy was started. The use of corticosteroid therapy was decided based on advanced bone age, biochemical hyperandrogenism and increasing growth velocity to prevent rapid BA acceleration and severe compromission of final height, a common feature of PWS worsened by CAH (29, 44). The very high levels of adrenal androgens in our patient led us to the decision to start oral hydrocortisone at a dosage (20 mg/m2/d) above the recommended dose interval for non-classic CAH (10-15 mg/m2/d) (45) in order to suppress adrenal hyperandrogenism and prevent the effects of androgen excess on final height.
Our patient underwent clonidine and arginine GH stimulation testing, which revealed a condition of GHD (46). rhGH therapy was started at the age of 2.7 years within the recommended dose range for PWS. The prevalence of GHD in PWS is reported around 50%, nevertheless rhGH therapy produces several benefits in these subjects: increase in final height, improved body composition, muscle thickness, bone mineral density, cognitive function and behavior in PWS subjects (31, 47–49). Furthermore, rhGH therapy also influences sexual development due to the existence of a crosstalk between the HPG and the somatotropic axes (50) during development from infancy through puberty, transition age and, finally, in adult life (51–55).
Our patient did not show advanced clinical pubertal features and the pelvic ultrasound examination documented an infantile uterine feature, which prompted to avoid GnRH analogue therapy.
Autoimmune diseases are not described as a typical feature of PWS. Although thyroid dysfunction may also be observed in the syndrome, this is not the result of thyroid autoimmunity (56, 57). Nevertheless, an increased systemic low-grade inflammation was described in these patients. One paper observed that PWS subjects present overactivation of the innate immune system, independent of central adiposity and insulin-resistance (58). Among the factors that may influence this phenomenon, there are the GHD and hypogonadism described in PWS. Another frequent comorbidity in PWS patients is OSAS which may also contribute towards increased low-grade inflammation (59, 60). Nevertheless, despite their severe obesity, lower visceral fat mass, hypoinsulinemia and increased insulin sensitivity have been described in PWS patients by most (61, 62), although not all studies (22–24, 63), compared to non-syndromic obese patients (61, 62). According to early reports, lower fasting insulin levels and reduced insulin resistance (measured by HOMA-Index) have been reported at all ages in obese patients with PWS (64–66). A familial component in insulin resistance, as in the general population, could also contribute to the individual subject affected by PWS whereas the different genotypes (del or UPD15) do not appear to influence the development of altered glucose homeostasis in PWS (16, 63). Data about insulin secretion in PWS, however, are still under debate.
In our patient we observed persistent IGT although with a normal body weight, which led to a reduction of rhGH and hydrocortisone dosages. An initial improvement in glucose homeostasis was followed at the age of 9 by a positive autoimmune screening for anti-GAD with normal fasting glucose and insulin levels. At the age of 16.2 years the typical symptoms of diabetes mellitus emerged and diabetic ketoacidosis occurred as typical of type 1 phenotype (T1DM); concurrently she presented anti-thyroid antibodies, but a negative antibody screening for celiac disease. Despite the relationship between obesity and the development of diabetes being unclear in PWS, in our case it was most probable that the patient presented a genetic predisposition for autoimmune diseases, although in the absence of family history.
In conclusion, the present is the third case report of a subject with CAH associated with PWS, but the first confirmed by genetic analysis. Although CAH is a common disease, and PP a common feature in PWS, 17OHP measurement should be performed in all PWS children experiencing PP and early adrenarche to exclude a possible concurrent 21OHD. Furthermore, although PWS is most typically associated with a T2DM phenotype, anti-islet antibodies and C-peptide levels should be assayed at the occurrence of glucose metabolism impairments in children and adolescents affected by PWS to timely diagnose the sporadic T1DM cases and guide follow-up concerning disease progression and the need for insulin therapy.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the parents for the publication of this case report including clinical, biochemical and radiological data.
Author contributions
AA collected data, drafted the initial manuscript, and critically reviewed and revised the manuscript. SB and MM contributed to the original draft of the manuscript. AC and MC critically reviewed and revised the manuscript for important intellectual content. DF coordinated and supervised data collection and manuscript drafting and revised the manuscript for intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
17OHP, 17-hydroxyprogesterone; 21OHD, 21-hydroxylase deficiency; Ab-Tg, anti-thyroglobulin antibodies; Ab-TPO, thyroid peroxidase antibodies; BA, bone age; BMI, body mass index; CA, chronological age; CAH, congenital adrenal hyperplasia; del, deletion; DHEA-S, dehydroepiandrosterone sulphate; E2, oestradiol; GAD, anti-glutamic acid decarboxylase antibodies; GH, growth hormone; GHD, growth hormone deficiency; GHRH, growth hormone releasing hormone; GnRH, gonadotropin-releasing hormone; HGT, Hemoglucotest; HPG, hypothalamus-pituitary-gonadal axis; HV, height velocity; IAA, anti-insulin antibodies; IA2, anti-tyrosine phosphatase antibodies; ID, imprinting defect; IGF-1, insulin-like growth factor-1; IGT, impaired glucose tolerance; MODY, maturity onset diabetes of the young; MPH, mid parental height; MS-MLPA, methylation-specific multiplex ligation-dependent probe amplification analysis; oGTT, oral glucose tolerance test; OSAS, obstructive sleep apnea syndrome; PCOS, polycystic ovary syndrome; PP, premature pubarche; PWS, Prader-Willi Syndrome; SDS, standard deviation scores; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; UPD, uniparental disomy.
References
1. Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-willi syndrome. Genet Med (2012) 14(1):10–26. doi: 10.1038/gim.0b013e31822bead0
2. Angulo MA, Butler MG, Cataletto ME. Prader-willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. (2015) 38(12):1249–63. doi: 10.1007/s40618-015-0312-9
3. Heksch R, Kamboj M, Anglin K, Obrynba K. Review of prader-willi syndrome: The endocrine approach. Transl Pediatr (2017) 6(4):274–85. doi: 10.21037/tp.2017.09.04
4. Tauber M, Hoybye C. Endocrine disorders in prader-willi syndrome: A model to understand and treat hypothalamic dysfunction. Lancet Diabetes Endocrinol (2021) 9(4):235–46. doi: 10.1016/S2213-8587(21)00002-4
5. Palmert MR, Hayden DL, Mansfield MJ, Crigler JF Jr, Crowley WF Jr, Chandler DW, et al. The longitudinal study of adrenal maturation during gonadal suppression: Evidence that adrenarche is a gradual process. J Clin Endocrinol Metab (2001) 86(9):4536–42. doi: 10.1210/jcem.86.9.7863
6. Ibáñez L, Dimartino-Nardi J, Potau N, Saenger P. Premature adrenarche–normal variant or forerunner of adult disease? Endocr Rev (2000) 21(6):671–96. doi: 10.1210/edrv.21.6.0416
7. Pellikaan K, Ben Brahim Y, Rosenberg AGW, Davidse K, Poitou C, Coupaye M, et al. Hypogonadism in women with prader-willi syndrome-clinical recommendations based on a Dutch cohort study, review of the literature and an international expert panel discussion. J Clin Med Res (2021) 10(24). doi: 10.3390/jcm10245781
8. Crinò A, Schiaffini R, Ciampalini P, Spera S, Beccaria L, Benzi F, et al. Hypogonadism and pubertal development in prader-willi syndrome. Eur J Pediatr (2003) 162(5):327–33. doi: 10.1007/s00431-002-1132-4
9. Pellikaan K, Ben Brahim Y, Rosenberg AGW, Davidse K, Poitou C, Coupaye M, et al. Hypogonadism in adult males with prader-willi syndrome-clinical recommendations based on a Dutch cohort study, review of the literature and an international expert panel discussion. J Clin Med Res (2021) 10(19). doi: 10.3390/jcm10194361
10. Radicioni AF, Di Giorgio G, Grugni G, Cuttini M, Losacco V, Anzuini A, et al. Multiple forms of hypogonadism of central, peripheral or combined origin in males with prader-willi syndrome. Clin Endocrinol (2012) 76(1):72–7. doi: 10.1111/j.1365-2265.2011.04161.x
11. Crinò A, Di Giorgio G, Schiaffini R, Fierabracci A, Spera S, Maggioni A, et al. Central precocious puberty and growth hormone deficiency in a boy with prader-willi syndrome. Eur J Pediatr (2008) 167(12):1455–8. doi: 10.1007/s00431-008-0679-0
12. Grosso S, Anichini C, Berardi R, Balestri P, Pucci L, Morgese G. Central precocious puberty and abnormal chromosomal patterns. Endocr Pathol (2000) 11(1):69–75. doi: 10.1385/EP:11:1:69
13. Yang A, Kim J, Cho SY, Jin DK. Prevalence and risk factors for type 2 diabetes mellitus with prader-willi syndrome: A single center experience. Orphanet J Rare Dis (2017) 12(1):146. doi: 10.1186/s13023-017-0702-5
14. Vogels A, Fryns JP. Age at diagnosis, body mass index and physical morbidity in children and adults with the prader-willi syndrome. Genet Couns. (2004) 15(4):397–404.
15. Diene G, Mimoun E, Feigerlova E, Caula S, Molinas C, Grandjean H, et al. Endocrine disorders in children with prader-willi syndrome–data from 142 children of the French database. Horm Res Paediatr (2010) 74(2):121–8. doi: 10.1159/000313377
16. Fintini D, Grugni G, Bocchini S, Brufani C, Di Candia S, Corrias A, et al. Disorders of glucose metabolism in prader-willi syndrome: Results of a multicenter Italian cohort study. Nutr Metab Cardiovasc Dis (2016) 26(9):842–7. doi: 10.1016/j.numecd.2016.05.010
17. Anhalt H, Eckert KH, Hintz RL, Neely EK. Type I diabetes mellitus, ketoacidosis and thromboembolism in an adolescent with prader-willi syndrome. Acta Paediatr (1996) 85(4):516. doi: 10.1111/j.1651-2227.1996.tb14076.x
18. Bassali R, Hoffman WH, Chen H, Tuck-Muller CM. Hyperlipidemia, insulin-dependent diabetes mellitus, and rapidly progressive diabetic retinopathy and nephropathy in prader-willi syndrome with del(15)(q11.2q13). Am J Med Genet (1997) 71(3):267–70. doi: 10.1002/(SICI)1096-8628(19970822)71:3<267::AID-AJMG3>3.0.CO;2-R
19. Mariani B, Cammarata B, Grechi E, Di Candia S, Chiumello G. P2-d3-522. Poster presentations. Horm Res Paediatr (2012) 78(Suppl. 1):151–248. doi: 10.1159/000343183
20. Tomoda Y, Okauchi Y, Deguchi A, Takenoshita Y, Iwahashi H, Mineo I. Prader-willi syndrome with slowly progressive insulin-dependent diabetes mellitus. Intern Med (2021) 60(9):1423–6. doi: 10.2169/internalmedicine.5267-20
21. Schmidt F, Kapellen TM, Wiegand S, Herbst A, Wolf J, Fröhlich-Reiterer EE, et al. Diabetes mellitus in children and adolescents with genetic syndromes. Exp Clin Endocrinol Diabetes. (2012) 120(10):579–85. doi: 10.1055/s-0032-1306330
22. Brambilla P, Crinò A, Bedogni G, Bosio L, Cappa M, Corrias A, et al. Metabolic syndrome in children with prader-willi syndrome: The effect of obesity. Nutr Metab Cardiovasc Dis (2011) 21(4):269–76. doi: 10.1016/j.numecd.2009.10.004
23. Grugni G, Crinò A, Bedogni G, Cappa M, Sartorio A, Corrias A, et al. Metabolic syndrome in adult patients with prader-willi syndrome. Nutr Metab Cardiovasc Dis (2013) 23(11):1134–40. doi: 10.1016/j.numecd.2012.11.006
24. Grugni G, Licenziati MR, Valerio G, Crinò A, Maffeis C, Tanas R, et al. The rehabilitation of children and adolescents with severe or medically complicated obesity: an ISPED expert opinion document. Eating Weight Disord - Stud Anorexia Bulimia Obes (2016) 22(1):3–12. doi: 10.1007/s40519-016-0305-5
25. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev (2000) 21(3):245–91. doi: 10.1210/edrv.21.3.0398
26. Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet (2005) 365(9477):2125–36. doi: 10.1016/S0140-6736(05)66736-0
27. El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet (2017) 390(10108):2194–210. doi: 10.1016/S0140-6736(17)31431-9
28. New MI. Extensive clinical experience: Nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab (2006) 91(11):4205–14. doi: 10.1210/jc.2006-1645
29. Wasserman M, Mulvihill EM, Ganan-Soto A, Uysal S, Quintos JB. Severe short stature in an adolescent Male with prader-willi syndrome and congenital adrenal hyperplasia: A therapeutic conundrum. Case Rep Endocrinol (2017) 2017:4271978. doi: 10.1155/2017/4271978
30. Cerioni V, Grechi E, Bove M, Sogno Valin P, Di Candia S, Bosio L, et al. R-31. Read by Title. Horm Res (2008) 70(Suppl. 1):224–56. doi: 10.1159/000157534
31. Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M. Speakers contributors at the second expert meeting of the comprehensive care of patients with PWS. recommendations for the diagnosis and management of prader-willi syndrome. J Clin Endocrinol Metab (2008) 93(11):4183–97. doi: 10.1210/jc.2008-0649
32. Italian Drug Agency (AIFA). AIFA note 39 (2006). Available at: https://www.iss.it/documents/20126/6755651/Nota_39_AIFA_Suppl._G.U._10-01-2007_.pdf/35e5e5bc-8396-7ba5-5efc-08bd5292de7e?t=1646663056516.
33. Deal CL, Tony M, Höybye C, Allen DB, Tauber M, Christiansen JS, et al. GrowthHormone research society workshop summary: Consensus guidelines for recombinant human growth hormone therapy in prader-willi syndrome. J Clin Endocrinol Metab (2013) 98(6):E1072–87. doi: 10.1210/jc.2012-3888
34. Cacciari E, Milani S, Balsamo A, Spada E, Bona G, Cavallo L, et al. Italian cross-sectional growth charts for height, weight and BMI (2 to 20 yr). J Endocrinol Invest. (2006) 29(7):581–93. doi: 10.1007/BF03344156
35. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An endocrine society clinical practice guideline. J Clin Endocrinol Metab (2018) 103(11):4043–88. doi: 10.1210/jc.2018-01865
36. Nota 39 . Available at: https://www.aifa.gov.it/nota-39.
37. Siemensma EPC, de Lind van Wijngaarden RFA, Otten BJ, de Jong FH, Hokken-Koelega ACS. Pubarche and serum dehydroepiandrosterone sulphate levels in children with prader-willi syndrome. Clin Endocrinol (2011) 75(1):83–9. doi: 10.1111/j.1365-2265.2011.03989.x
38. Eugster EA, Dimeglio LA, Wright JC, Freidenberg GR, Seshadri R, Pescovitz OH. Height outcome in congenital adrenal hyperplasia caused by 21-hydroxylase deficiency: a meta-analysis. J Pediatr (2001) 138(1):26–32. doi: 10.1067/mpd.2001.110527
39. Muthusamy K, Elamin MB, Smushkin G, Murad MH, Lampropulos JF, Elamin KB, et al. Clinical review: Adult height in patients with congenital adrenal hyperplasia: a systematic review and metaanalysis. J Clin Endocrinol Metab (2010) 95(9):4161–72. doi: 10.1210/jc.2009-2616
40. Dacou-Voutetakis C, Karidis N. Congenital adrenal hyperplasia complicated by central precocious puberty: Treatment with LHRH-agonist analogue. Ann N Y Acad Sci (1993) 687:250–4. doi: 10.1111/j.1749-6632.1993.tb43873.x
41. Pescovitz OH, Comite F, Cassorla F, Dwyer AJ, Poth MA, Sperling MA, et al. True precocious puberty complicating congenital adrenal hyperplasia: Treatment with a luteinizing hormone-releasing hormone analog. J Clin Endocrinol Metab (1984) 58(5):857–61. doi: 10.1210/jcem-58-5-857
42. Quintos JB, Vogiatzi MG, Harbison MD, New MI. Growth hormone therapy alone or in combination with gonadotropin-releasing hormone analog therapy to improve the height deficit in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab (2001) 86(4):1511–7. doi: 10.1210/jcem.86.4.7412
43. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab (2005) 90(6):3318–25. doi: 10.1210/jc.2004-2128
44. Lin-Su K, Harbison MD, Lekarev O, Vogiatzi MG, New MI. Final adult height in children with congenital adrenal hyperplasia treated with growth hormone. J Clin Endocrinol Metab (2011) 96(6):1710–7. doi: 10.1210/jc.2010-2699
45. Grigorescu-Sido A, Bettendorf M, Schulze E, Duncea I, Heinrich U. Growth analysis in patients with 21-hydroxylase deficiency influence of glucocorticoid dosage, age at diagnosis, phenotype and genotype on growth and height outcome. Horm Res (2003) 60(2):84–90. doi: 10.1159/000071876
46. Di Giorgio G, Grugni G, Fintini D, Bocchini S, Spera S, Cuttini M, et al. Growth hormone response to standard provocative stimuli and combined tests in very young children with prader-willi syndrome. Horm Res Paediatr (2014) 81(3):189–95. doi: 10.1159/000356927
47. Reus L, Pillen S, Pelzer BJ, van Alfen-van der Velden JAAEM, Hokken-Koelega ACS, Zwarts M, et al. Growth hormone therapy, muscle thickness, and motor development in prader-willi syndrome: an RCT. Pediatrics (2014) 134(6):e1619–27. doi: 10.1542/peds.2013-3607
48. Siemensma EPC, Tummers-de Lind van Wijngaarden RFA, Festen DAM, Troeman ZCE, van Alfen-van der Velden AAEMJ, Otten BJ, et al. Beneficial effects of growth hormone treatment on cognition in children with prader-willi syndrome: A randomized controlled trial and longitudinal study. J Clin Endocrinol Metab (2012) 97(7):2307–14. doi: 10.1210/jc.2012-1182
49. Lo ST, Siemensma EPC, Festen DAM, Collin PJL, Hokken-Koelega ACS. Behavior in children with prader-willi syndrome before and during growth hormone treatment: A randomized controlled trial and 8-year longitudinal study. Eur Child Adolesc Psychiatry (2015) 24(9):1091–101. doi: 10.1007/s00787-014-0662-4
50. Tenuta M, Carlomagno F, Cangiano B, Kanakis G, Pozza C, Sbardella E, et al. Somatotropic-testicular axis: A crosstalk between GH/IGF-I and gonadal hormones during development, transition, and adult age. Andrology (2021) 9(1):168–84. doi: 10.1111/andr.12918
51. Lee PA, Mazur T, Houk CP, Blizzard RM. Growth hormone deficiency causing micropenis: Lessons learned from a well-adjusted adult. Pediatrics (2018) 142(1). doi: 10.1542/peds.2017-4168
52. Laron Z, Klinger B. Effect of insulin-like growth factor-I treatment on serum androgens and testicular and penile size in males with laron syndrome (primary growth hormone resistance). Eur J Endocrinol (1998) 138(2):176–80. doi: 10.1530/eje.0.1380176
53. Leschek EW, Troendle JF, Yanovski JA, Rose SR, Bernstein DB, Cutler GB Jr, et al. Effect of growth hormone treatment on testicular function, puberty, and adrenarche in boys with non-growth hormone-deficient short stature: A randomized, double-blind, placebo-controlled trial. J Pediatr (2001) 138(3):406–10. doi: 10.1067/mpd.2001.111332
54. Burns EC, Tanner JM, Preece MA, Cameron N. Final height and pubertal development in 55 children with idiopathic growth hormone deficiency, treated for between 2 and 15 years with human growth hormone. Eur J Pediatr (1981) 137(2):155–64. doi: 10.1007/BF00441309
55. Price DA. Puberty in children with idiopathic growth hormone deficiency on growth hormone treatment: Preliminary analysis of the data from the kabi pharmacia international growth study. Acta Paediatr Scand Suppl. (1991) 379:117–24; discussion 125. doi: 10.1111/j.1651-2227.1991.tb12061.x
56. Kyritsi EM, Kanaka-Gantenbein C. Autoimmune thyroid disease in specific genetic syndromes in childhood and adolescence. Front Endocrinol (2020) 11:543. doi: 10.3389/fendo.2020.00543
57. Iughetti L, Vivi G, Balsamo A, Corrias A, Crinò A, Delvecchio M, et al. Thyroid function in patients with prader-willi syndrome: an Italian multicenter study of 339 patients. J Pediatr Endocrinol Metab (2019) 32(2):159–65. doi: 10.1515/jpem-2018-0388
58. Viardot A, Sze L, Purtell L, Sainsbury A, Loughnan G, Smith E, et al. Prader-willi syndrome is associated with activation of the innate immune system independently of central adiposity and insulin resistance. J Clin Endocrinol Metab (2010) 95(7):3392–9. doi: 10.1210/jc.2009-2492
59. Chorostowska-Wynimko J, Radomska D, Pływaczewski R, Jończak L, Stepniewska A, Górecka D, et al. Disturbed angiogenic activity in sera from obstructive sleep apnea patients. J Physiol Pharmacol (2005) 56 Suppl 4:71–7.
60. Minoguchi K, Yokoe T, Tazaki T, Minoguchi H, Tanaka A, Oda N, et al. Increased carotid intima-media thickness and serum inflammatory markers in obstructive sleep apnea. Am J Respir Crit Care Med (2005) 172(5):625–30. doi: 10.1164/rccm.200412-1652OC
61. Goldstone AP, Thomas EL, Brynes AE, Bell JD, Frost G, Saeed N, et al. Visceral adipose tissue and metabolic complications of obesity are reduced in prader-willi syndrome female adults: Evidence for novel influences on body fat distribution. J Clin Endocrinol Metab (2001) 86(9):4330–8. doi: 10.1210/jcem.86.9.7814
62. Crinò A, Grugni G. Update on diabetes mellitus and glucose metabolism alterations in prader-willi syndrome. Curr Diabetes Rep (2020) 20(2):7. doi: 10.1007/s11892-020-1284-5
63. Valerio G, Maffeis C, Saggese G, Ambruzzi MA, Balsamo A, Bellone S, et al. Diagnosis, treatment and prevention of pediatric obesity: Consensus position statement of the Italian society for pediatric endocrinology and diabetology and the Italian society of pediatrics. Ital J Pediatr (2018) 44(1):1–21. doi: 10.1186/s13052-018-0525-6
64. Lacroix D, Moutel S, Coupaye M, Huvenne H, Faucher P, Pelloux V, et al. Metabolic and adipose tissue signatures in adults with prader-willi syndrome: A model of extreme adiposity. J Clin Endocrinol Metab (2015) 100(3):850–9. doi: 10.1210/jc.2014-3127
65. Haqq AM, Muehlbauer MJ, Newgard CB, Grambow S, Freemark M. The metabolic phenotype of prader-willi syndrome (PWS) in childhood: Heightened insulin sensitivity relative to body mass index. J Clin Endocrinol Metab (2011) 96(1):E225–32. doi: 10.1210/jc.2010-1733
Keywords: Prader-Willi Syndrome, adrenarche, premature pubarche, pubertal development, growth hormone deficiency, congenital adrenal hyperplasia, diabetes mellitus
Citation: Aureli A, Bocchini S, Mariani M, Crinò A, Cappa M and Fintini D (2023) A rare occurrence of non-classic congenital adrenal hyperplasia and type 1 diabetes mellitus in a girl with Prader-Willi Syndrome: Case report and review of the literature. Front. Endocrinol. 14:1148318. doi: 10.3389/fendo.2023.1148318
Received: 19 January 2023; Accepted: 23 March 2023;
Published: 12 April 2023.
Edited by:
Katja Dumic Kubat, University of Zagreb, CroatiaReviewed by:
Gerdi Tuli, Regina Margherita Hospital, ItalyMatteo Spaziani, Division of Medical Pathophysiology, Sapienza University of Rome, Italy
Copyright © 2023 Aureli, Bocchini, Mariani, Crinò, Cappa and Fintini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danilo Fintini, ZGFuaWxvLmZpbnRpbmlAb3BiZy5uZXQ=