
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 20 March 2023
Sec. Molecular and Structural Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1145392
This article is part of the Research Topic Interaction of Endocrine Disorders and Metabolic Associated Fatty Liver Disease: The Genetic and Epigenetic Basis View all 6 articles
 Chen Huang1†
Chen Huang1† Youlian Zhou1,2†
Youlian Zhou1,2† Jiemin Cheng1,2
Jiemin Cheng1,2 Xue Guo1,2
Xue Guo1,2 Diwen Shou1,2
Diwen Shou1,2 Ying Quan1,2
Ying Quan1,2 Hanqing Chen1,2*
Hanqing Chen1,2* Huiting Chen1*
Huiting Chen1* Yongjian Zhou1,2*
Yongjian Zhou1,2*Nonalcoholic fatty liver disease (NAFLD) is characterized by excessive lipid accumulation and has become the leading chronic liver disease worldwide. NAFLD is viewed as the hepatic manifestation of metabolic syndrome, ranging from simple steatosis and nonalcoholic steatohepatitis (NASH) to advanced fibrosis, eventually leading to cirrhosis and hepatocellular carcinoma (HCC). The pathogenesis of NAFLD progression is still not clear. Pattern recognition receptor (PRR)-mediated innate immune responses play a critical role in the initiation of NAFLD and the progression of NAFLD-related HCC. Toll-like receptors (TLRs) and the cyclic GMP-AMP (cGAMP) synthase (cGAS) are the two major PRRs in hepatocytes and resident innate immune cells in the liver. Increasing evidence indicates that the overactivation of TLRs and the cGAS signaling pathways may contribute to the development of liver disorders, including NAFLD progression. However, induction of PRRs is critical for the release of type I interferons (IFN-I) and the maturation of dendritic cells (DCs), which prime systemic antitumor immunity in HCC therapy. In this review, we will summarize the emerging evidence regarding the molecular mechanisms of TLRs and cGAS in the development of NAFLD and HCC. The dysfunction of PRR-mediated innate immune response is a critical determinant of NAFLD pathology; targeting and selectively inhibiting TLRs and cGAS signaling provides therapeutic potential for treating NALF-associated diseases in humans.
Nonalcoholic fatty liver disease (NAFLD), which is newly defined metabolic associated fatty liver disease (MAFLD) is characterized by abnormal hepatic lipid accumulation and inflammatory syndrome, in addition to one of the following three criteria: overweight/obesity, type 2 diabetes mellitus (T2DM), or evidence of metabolic dysregulation (1–4). Recently, NAFLD is emerging as the rising public health problem that endangers human health worldwide (5, 6). The meta-analysis reveals that, the global prevalence of NAFLD has been approximately 25.2%, with the highest prevalence in Southeast Asia (42.0%) and the lowest rates reported from Africa (13.5%) (7, 8). The rising rates of obesity, insulin resistance, type 2 diabetes, and hyperlipidaemia contribute to the development of NAFLD (9). Thus, the prevalence of NAFLD is more than 70% among obese and diabetic patients (10). The hepatic metabolic disorder leads to chronic inflammation and compensatory tissue repair, which result in steatohepatitis with or without fibrosis. Current clinical studies indicated that around 10-20% of NAFLD patients would gradually develop into nonalcoholic steatohepatitis (NASH). NASH is characterized by liver inflammation, hepatocellular injury, and different degrees of hepatic fibrosis, which is mediated by innate immune responses. There are up to one-third of NASH cases might progress to advanced fibrosis or cirrhosis, then eventually develop into hepatocellular carcinoma (HCC) (11–14). The current mechanism studies indicate that lipotoxicity, inflammation, oxidative stress, mitochondrial dysfunction, and gut-liver axis are associated with the development of NAFLD to NASH and HCC (15–19). However, the pathogenesis of NAFLD progression is still not clear, and there are still no FDA-approved effective therapies available for end-stage NAFLD except liver transplantation (20, 21).
Growing evidence suggests that hepatic innate immune response plays an important role in triggering NAFLD initiation and progression. Although the innate immune system is considered to be a metabolic sensor against metabolic-related stresses, an overactive innate immune response becomes pathological. This scenario occurs in the development from NAFLD to NASH (22, 23). Pattern recognition receptors (PRRs) constitute the first line of defense in the host immune system, and are mainly expressed in the cell membrane, endosome, or cytoplasm of innate immune cells, such as macrophages, Dendritic cells (DCs), neutrophils, natural killer (NK) cells, and even in hepatocytes (24–26). During the initial event of the innate immune response, PRRs detect the evolutionarily conserved structures on invaded bacteria or viruses from cell death and tissue damage by pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), which lead to the activation of multiple intracellular signaling pathways and trigger the production of inflammatory cytokines and chemokines (24, 27, 28). Toll-like receptors (TLRs), a well-known family of endocytic PRRs, are widely expressed on all types of cells in the liver, including Kupffer cells (29, 30), hepatocytes (29, 31), hepatic stellate cells (HSCs) (32, 33) and biliary epithelial cells (29) (Table 1). cGAS is a newly discovered DNA sensor, which is considered to involve in the development of diverse liver diseases, such as viral hepatitis, NAFLD, drug-induced liver injury (DILI), and HCC (35, 43, 44). In this Review, we will summarize the currently available information regarding the role of PRRs on the pathogenesis and progression of NAFLD.

Table 1 The expression and action of TLRs and cGAS-STING in a variety of liver cells.
TLRs are one of the earliest discovered PRRs, which form the cornerstone of in innate immune responses. To data, ten and twelve known functional TLRs have been identified and characterized in humans (TLR1-10) and twelve (TLR1-9 and 11-13) in mice, respectively. Some TLRs, such as TLR1, 2, 4, 5, 6 and 10, are found on the plasma membrane in the form of homodimers or heterodimers, which mainly recognize the membrane components of pathogenic microorganisms, including LPS, lipid A, lipoproteins, flagellin. The other TLRs including TLR3, 7, 8, and 9 are expressed on the intracellular membrane to recognize the microbial nucleic acid ligands. After binding with its ligand, the PRRs are activated and then recruit the downstream adaptors to elicit the assembly of signaling complexes by initiating the signaling cascades in MyD88-dependent or TRIF-dependent manner. The transcriptional factors, such as IRF3/IRF7, NF-κB, and AP-1, are activated and then translocated into the nucleus, resulting in the production of inflammatory cytokines, chemokines, and type I interferons. Recent accumulating evidence has demonstrated the association between NAFLD progression and TLR signaling pathway activation, particularly TLR4, TLR2 and TLR9 (Figure 1).
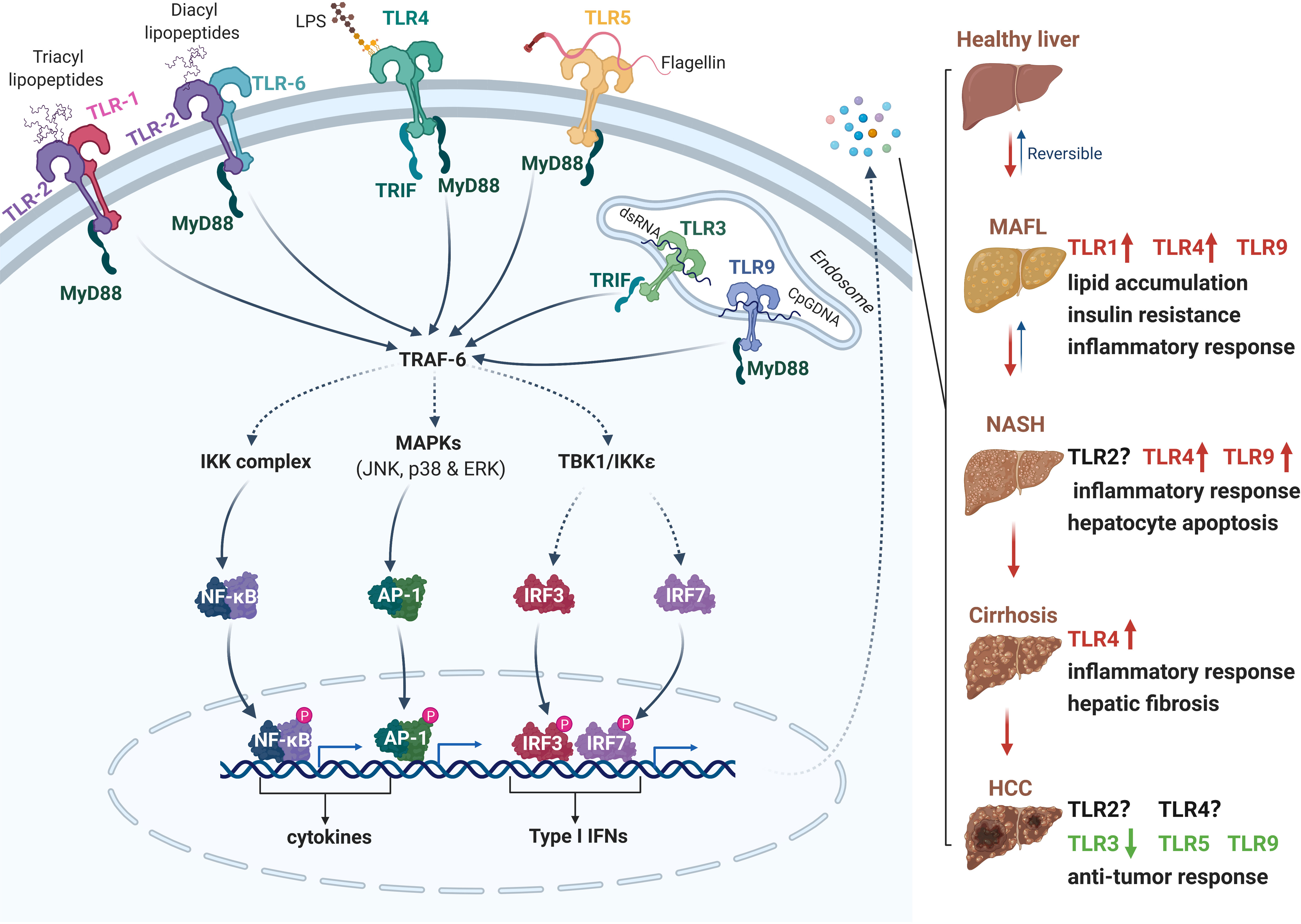
Figure 1 The role of Toll-like receptors (TLRs) in the pathogenesis of NAFLD and HCC. Following excessive high-fat diet intake and liver injury, PAMPs (e.g., LPS, flagellin, lipopeptides and DNA) from microbes and DAMPs released from damaged or dying cells are accumulated in the liver. TLRs recognize PAMPs and DAMPs and initiate downstream cascades in TRIF- or MyD88-dependent manner. The activation of IKK, MAPKs and TBK1 kinases trigger dimerization and translocation of NF-κB, AP-1 and IRF3/7 respectively, then induce the expression of several cytokines, chemokines, type I interferons, and other pro-apoptotic genes. The secretion of these active factors may contribute to insulin resistance, lipid accumulation, inflammation, hepatocyte apoptosis or anti-cancer responses. Different TLRs play diverse roles in the process of NAFLD to HCC. PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; TRIF, TIR-domain-containing adapter-inducing interferon β; MyD88, myeloid differentiation primary response 88; IKK, IκB kinase; MAPK, mitogen-activated protein kinase; TBK1, TANK-binding kinase 1; NF- κB, nuclear factor kappa B; AP-1, activator protein 1; IRF, interferon regulatory factor. Red, accelerative effect; Green, protective effect.
The mRNA levels of TLR 1-10 were determined in the liver samples of 11 patients with NAFLD and 11 health persons (45). The mRNA expression of TLR 1 to 5 was significantly higher in NAFLD patients, and the mRNA level of TLR 6 to 10 mRNAs was comparable between NAFLD patients and healthy controls. Another study found that the protein level of TLR6, but not TLR2, was gradually increased in the hepatocytes from the cohorts of NAFLD and NASH patients compared to those from normal liver biopsy, indicating that TLR6 contributed to inflammatory responses in the development of NASH (46). However, the effect of TLR2 on different models of NASH seems to be controversial. TLR2 deficient mouse exhibited severe hepatocellular damage and accelerated MCD diet-induced steatohepatitis and fibrosis (47). However, TLR2 deficiency has been shown to decrease the infiltration of inflammatory cells and the activation of inflammasome in choline-deficient amino acid-defined (CDAA) diet induced NASH model (48), indicating the critical role of TLR2 in the initiation of liver inflammation. TLR1 deficiency displayed significantly protective effect from the development of diet-induced NAFLD when compared to that in wild-type mice. Moreover, TLR1 expression was positively correlated with the level of Holdemanella genus, and was negative associated with the content of Gemmiger and Ruminococcus genera in NAFLD patients (49). Thus, TLR1 may be acted as a potential target in the treatment of NAFLD patients along with TLR1 high expression.
TLR4, a receptor of LPS, is widely expressed on the surface of Kupffer cells and hepatocytes. In the progression of NAFLD, metabolic alterations were usually accompanied by the increased intestinal permeability and gut microbiota dysbiosis (50, 51), which resulted in the elevating levels of circulating LPS in NAFLD/NASH patients and HFD or MCD-induced animal models (52, 53). The level of free fatty acids (FFAs), such as palmitic acid and stearic acid, was higher in NAFLD patients, indicating the association with the activation of TLR4 activation (54, 55). Moreover, the mRNA and protein levels of TLR4 were increased in liver samples from patients with hepatitis and cirrhosis (56). Overactivation of TLR4 increased the phosphorylation of NF-κB, MAPK and IRF3, and promoted the production pro-inflammatory cytokines in MyD88-dependent manner, which triggered severe liver injury and promoted the development of NAFLD. Genetic ablation of TLR4 resulted in a marked attenuation of liver inflammation in preclinical NASH models (57). In addition, absence of TLR4 downstream molecules IKKϵ and TBK1, or treatment with an IKKε- and TBK1-specific inhibitors showed a protective effect against hepatic steatosis (58, 59). These data demonstrate that TLR4 signaling promotes NAFLD progression, and inhibition of LPS release from the intestinal microbiota or usage of TLR4 signaling antagonism may be a feasible strategy for the prevention or treatment of NAFLD/NASH.
Similar with TLR4, the mRNA level of TLR9 was significantly upregulated in patients with NASH, but not in patients with hepatic steatosis (60). As a CpG DNA sensor, TLR9 could be activated by circulating mitochondrial DNA (mtDNA), which exhibited higher level in plasma from mice and patients with NASH (61–63). Pharmacological inhibition of TLR9 prevented the hepatic histopathological injury, proinflammatory cytokine production, and plasma ALT activity in NASH models (61). Furthermore, TLR9 deficiency significantly reduced liver weights and ameliorated hepatic steatosis in HFD-induced NASH mice, indicating the protective role of TLR9 in hepatic dysmetabolism (64).
Approximately 80% of HCCs are developed by chronic liver disease, hepatic fibrosis and cirrhosis (65). Liver inflammation is considered as a risk factor to induce the development of HCC. TLRs has been reported to be a critical point in the pathogenesis and progression of HCC. Lin et al. reported that TLR4 was functionally expressed on two human hepatoma cell lines HepG2 and H7402. The activation of TLR4 induced by LPS significantly enhances COX-2/PGE2/STAT3 positive feedback loop and then triggers the proliferation of hepatoma cells and the occurrence of multidrug resistance in HCC chemotherapy (66). Unlike the higher expression of TLR4 in hepatitis and cirrhosis, the expression of TLR4 in liver samples of patients with hepatocarcinoma does not change (56). However, the level of TLR4 is higher in liver samples from relapsed HCC patients, indicating that TLR4 expression is a potential prognostic biomarker in HCC therapy (67, 68). On the contrary, TLR3 expression is associated with longer survival in HCC patients. Activation of TLR3 signaling by poly(I:C) increased intra-tumoral chemokine expression, NK-cell activation, and proliferation of tumor-infiltrating T and NK cells, which contribute to increasing cell death and decreased tumor growth. The effect of TLR3 on HCC is further confirmed by Bonnin et al. The level of TLR3 protein is downregulated in tumoral liver samples and exhibited much lower in 6 human HCC cells, including Hep3B, HepG2, and HuH7. Furthermore, the growth of tumors is accelerated in TLR3 deficiency HCC mice. Mechanistically, the absence of TLR3 prevents cell apoptosis and enhances tumor progression (69). Therefore, TLR3 and TLR4 may be potential prognostic biomarkers for HCC therapy in humans. Two SNPs in the exon of TLR2 were identified in HCC and were demonstrated to be associated with the risk of HCC in a single center-based case-control study (70). TLR2 is expressed in tumor-associated macrophages (TAM) of HCC and contributes to autophagy in the regulation of M2 macrophage polarization by high-mobility group box 1 (HMGB1)/NADPH oxidase 2 (NOX2) axis (71). TLR9 overactivation can enhance the therapeutical efficacy of PD-1 or PD-L1 on HCC treatment by promoting PD-L1 transcription and enhancing the phosphorylation of STAT3 Tyr705 (72). Recent research reported that TLR5 was an independent prognostic marker in HCC therapy, which function is similar to p53 (73). Pretreatment with TLR5 agonist effectively protects from LPS- and TNF-induced toxicity in liver and lung, and reudces the mortality, indicating that TLR5 agonist might be used as an adjuvant to enable the safe systemic application of TNF as an anticancer therapy (74).
Although TLRs have been demonstrated to be associated with chronic inflammation in the development of NAFLD/NASH, the effects of TLR signaling pathway on the progression of NAFLD-associated HCC remains unclear. TLR2 polarizes the TAMs to a pro-tumor M2 phenotype in HCC cells, and promotes the proliferation of HCC (71). However, another study performed revealed that knockdown TLR2 or downstream molecule MyD88 in B76/Huh7 cells promotes its proliferation and inhibit the apoptosis to increase the progression of hepatocarcinogenesis (75). In addition, TLR4 and TLR9 deficiency significantly reduces liver injury and blocks the progression of liver inflammation to hepatic fibrosis and HCC in a hepatic deletion of transforming growth factor-β-activated kinase 1 (Tak1ΔHep) mouse model (38), indicating that TLR3-mediated apoptosis may be a promising therapeutic target in HCC treatment (69). Taken together, further studies need to be investigated for the molecular mechanisms and therapeutic potential of TLRs in the development of HCC.
The cGAS-STING axis contains the cytosolic DNA sensor cGAS, the second messenger cyclic GMP-AMP (cGAMP) and cGAMP receptor stimulator of interferon genes (STING) (76–79). STING is expressed in non-parenchymal cells, including innate immune cells and hepatic stellate cells, and exerts an important role to maintain liver homeostasis (80). The cGAS-STING signaling is activated by pathogenic DNA from DNA virus or intracellular bacteria, and endogenous DNA, including mitochondrial and nuclear DNA in the cytosol in response to mitochondria stress, radiation therapy or autoimmune disorders (81–85). cGAS-STING axis is involved in the activation of IFN-I and pro-inflammatory responses against microbial infections via TBK1/IRF3 and NF-κB signaling, and STING- induced autophagy and lysosome-dependent cell death (86). Recently, cGAS-STING signaling has been demonstrated to be associated with various diseases, including inflammation, autoimmune diseases, metabolic disorders, and tumors (87–89). Here, we will highlight the pathogenical and therapeutical role of cGAS-STING axis in NAFLD to HCC (Figure 2).
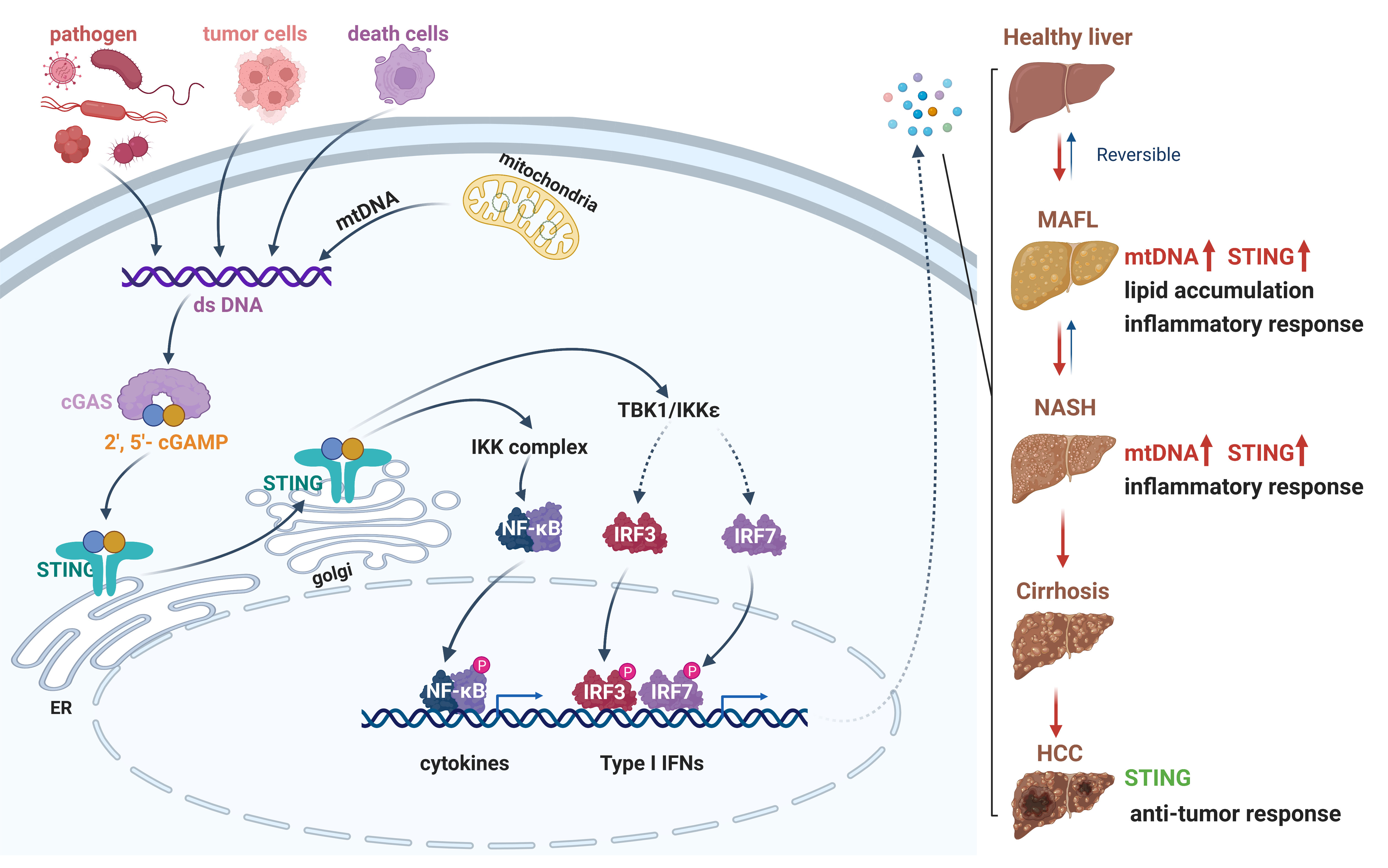
Figure 2 The cGAS-STING signaling pathway in the progression from NAFLD to HCC. A schematic detailing cytosolic dsDNA, which occurs through pathogen infection, tumor cells, death cells or cellular stress, can be recognized by cGAS. Enzymatic activation of cGAS results in the synthesis of 2′-3′ cGAMP. The binding of cGAMP enables STING translocation from ER membrane to Golgi, then recruits TBK1 and IKK complex. The phosphorylation of IRF3 and NF-κB by TBK1 and IKK enable these transcription factor dimerization and translocation to the nucleus to induce gene expression of several cytokines, type I interferons, and chemokines. Activation of cGAS-STING signaling axis could promote lipid accumulation, inflammation, or anti-cancer responses in different stage of liver disease. dsDNA, double-stranded DNA; cGAS, cyclic GMP-AMP synthase; 2′-3′ cGAMP, 2′3′ cyclic GMP-AMP; STING, stimulator of interferon genes; ER, endoplasmic reticulum; TBK1, TANK-binding kinase 1; IKK, IκB kinase; IRF3, interferon regulatory factor 3. Red, accelerative effect; Green, protective effect.
In patients with NAFLD/NASH, STING has been demonstrated to be expressed and upregulated in myeloid cells (35). The level of STING protein in hepatic non-parenchymal cells including KCs and endothelial cells from patients with NAFLD is increased compared to the healthy controls. Increased numbers of STING positive monocyte-derived macrophages (CCR2+, S100A9+), Kupffer cells (CD68+) and CD163+ macrophages are found in liver samples from NASH patients with fibrosis (90). Consistent with NAFLD patients, the expression of STING and its downstream transcriptional factor IRF3 are significantly increased in the liver of HFD-fed mice (35, 91). The phosphorylation of JNK p46 and nuclear factor κB (NF-κB) p65 are enhanced in the liver of NAFLD mice, and promotes the production of tumor necrosis factor α (TNFα), interleukin (IL)1β, and IL6. However, the phosphorylation of transcriptional factors and activation of inflammatory cytokines are significantly lower in mice with STING disruption (STINGgt). Moreover, the similar inflammatory status in myeloid cells was also found in mice with STING deficiency, indicating that myeloid-derived STING contributes to HFD-induced hepatic inflammatory response (35). Moreover, STING is deficiency in human and murine hepatocytes and the expression of STING is highly expressed in hepatic nonparenchymal cells (80, 92). However, the mechanism of HFD-triggered the activation cGAS-STING signaling is still not clear. It has been reported an abnormal liver mitochondrial function and high level of plasma mtDNA in NASH patients and HFD-induced NASH mice (61). Furthermore, STING deficiency attenuates the mtDNA-mediated TNF-α and IL-6 expression in KCs in vitro (36). Thus, cGAS-STING signaling is activated by cytoplasmic mtDNA, and then initiates the excessive inflammatory response in the liver to aggravate the liver injury in NASH patients and mice.
Indeed, inhibition of STING or reduction of its downstream signaling pathway can attenuate the NAFLD/NASH symptoms. STING deficiency reduced inflammation, steatosis and fibrosis in livers in both methionine- and choline-deficient diet (MCD)- and HFD-induced murine models of NASH (36, 93). Knockdown STING or IRF3 alleviates the hepatic lipid accumulation, liver inflammation and hepatocyte apoptosis (91, 94). Furthermore, mice with STING disruption in myeloid cells exhibited less severe NASH symptoms (35). In addition, Remdesivir (RDV, GS-5734), an antagonist of STING, considerably restrains lipid accumulation, hepatic disorder and liver inflammation in HFD-fed mice (95). Therefore, targeting cGAS-STING axis may be an effectively therapeutic strategy for NAFLD/NASH in humans.
Activation of cGAS-STING axis aggravates the NAFLD symptom and accelerates the development of NASH. Interestingly, cGAS-STING play an opposite role in HCC progression. Thomsen et al. reported that STING-deficient mice displayed higher number of large tumors than wild type mice at late stages of HCC. Furthermore, activation of STING by treatment with a cyclic dinucleotide (CDN), a traditional STING agonist, could efficiently reduce HCC tumor size (44). In addition, Lactobacillus rhamnosus GG (LGG) treatment enhanced the efficacy of immune checkpoint blockade (ICB) therapies through inducing cGAS/STING-dependent IFN-β production in colorectal cancer therapy, which suggesting that cGAS-STING axis play a vital role in anti-tumor immune responses against HCC (96).
Recently, several studies have reported that the cGAS-STING signaling is a prognostic biomarker in HCC, and can be acted as an adjuvant to enhance the anti-tumor efficacy (97–99). cGAS/STING signaling were found to be related to the stages of HCC patients. The mRNA level of STING and its downstream target molecule including IFI16, STAT6 and NLRC3 is positive associated with the overall survival of HCC patients (97), indicating the cGAS-STING signaling factors can be used as potential prognostic biomarkers and therapeutical targets for HCC treatment in humans. Increasing evidence has been demonstrated the role of cGAS-STING signaling pathway to enhance the efficacy of radiotherapy and immunotherapy (100). Radiotherapy (RT) can induce DNA damage and lead to the accumulation of cytosolic DNA, which initiate the activation of cGAS-STING signaling and increase the RT-induced antitumor immunological therapy. Besides, the radiotherapy effect was enhanced by introducing exogenous cGAMP, or combination with cGAS-STING agonists and immunotherapy (101). Wehbe et al. developed a polymer some nanoplatform encapsulated with STING (STING-NPs) to enhance cGAMP delivery in the tumor and increase the half-life of cGAMP with 40-fold. In a B16-F10 melanoma tumor model, STING-NPs increase the response rates to αPD-L1 antibodies, and lead to increase the survival time in mice (102). Furthermore, STING-NP-treated tumor-bearing mice resulted in >50% and 80% reduction in tumor burden by cGAS-STING signaling in melanoma and breast adenocarcinoma models. E7766, a macrocycle-bridged stimulator of interferon genes (STING) agonist, was demonstrated its potent antitumor activity by prolonging the immune memory response in a liver metastatic tumor mouse model (103). Conversely, STING signaling has been reported to promote the tumorigenesis and progression in some cases. Upregulation of IFNβ induced by cGAS-STING promotes the expression of immune checkpoint molecules, such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death ligand 1 (PD-L1), which result in the inhibition of T-cell activation and immune evasion (104, 105). Therefore, targeting and selectively activating cGAS-STING signaling may have therapeutic potential for treating HCC in humans.
Lastly, there are no approved therapies for NAFLD and NASH. Given that the TLR4, TLR9 and cGAS-STING signaling pathway is activated under obesity conditions in NAFLD patients and mouse models, it is tempting to speculate that TLRs and cGAS-STING contribute to increased inflammation in the progression of NAFLD to NASH. In fact, some small-molecule inhibitors have been developed to target TLRs, cGAS-STING and their downstream components over the past several years. TAK-242, a specific TLR4 inhibitor, has been used for the treatment of rheumatoid arthritis (RA) (106). RU.521 and RU.365 are found to inhibit cGAS by binding at the active site of cGAS (107). Remdesivir, an antagonist of STING, is also considered to be a candidate for the treatment of NAFLD (95). On the contrary, the PRRs play an important roles in anti-tumor immunity. The two STING agonists, DMXAA (5,6-dimethylxanthenone 4-acetic acid) and E7766, were identified as molecules that exerted potent anti-tumor activity. Thus, it is important to pay attention to the administration time of antagonists or agonists that regulate the TLRs and cGAS-STING signaling pathway in order to prevent an increased risk of HCC. In conclusion, NAFLD has emerged as one of the most prevalent form of chronic liver disease worldwide due to overnutrition, genetic predisposition, gut-liver axis and immune disorder. PRRs and their downstream signaling closely contribute to NAFLD pathogenesis and accelerate the development of NASH, hepatic fibrosis, cirrhosis, and NAFLD-related HCC. In this review, we provide the comprehensive understanding of the molecular mechanism of PRRs to control liver inflammation in physiological and pathophysiological stages of NAFLD and HCC, and highlight the therapeutic targeting PRRs for the treatment of NAFLD/NASH and HCC.
CH and YLZ conceptualized the topic of this review. JC, XG, DS, and YQ retrieved the literature and assisted in manuscript preparation. YJZ and HTC provided scholarly guidance on the topic and assisted in manuscript preparation. CH drafted the manuscript, HQC revised the manuscript. All authors have read and approved the final manuscript.
This study was supported by National Natural Science Foundation of China (82170585, 82200574, 81970507 and 32171370), The Fundamental Research Funds for the Central Universities (y2eyD2220570), the Natural Science Foundation of Guangdong Province (2022A1515010415), Funding by Science and Technology Projects in Guangzhou (SL2023A04J00596), Guangzhou High Technology Project (2019GX05), The Project of Key Medical Discipline in Guangzhou (2021-2023).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tilg H, Effenberger M. From NAFLD to MAFLD: when pathophysiology succeeds. Nat Rev Gastroenterol Hepatol (2020) 17:387–8. doi: 10.1038/s41575-020-0316-6
2. Eslam M, Sanyal AJ, George J, International Consensus P. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology (2020) 158:1999–2014.e1. doi: 10.1053/j.gastro.2019.11.312
3. Chen H. Nutrient mTORC1 signaling contributes to hepatic lipid metabolism in the pathogenesis of non-alcoholic fatty liver disease. Liver Res (2020) 4:15–22. doi: 10.1016/j.livres.2020.02.004
4. Chen H. Iron metabolism in nonalcoholic fatty liver disease: a promising therapeutic target. Liver Res (2022) 6:203–13. doi: 10.1016/j.livres.2022.09.003
5. Zhu M, Chen H, Zhou S, Zheng L, Li X, Chu R, et al. Iron oxide nanoparticles aggravate hepatic steatosis and liver injury in nonalcoholic fatty liver disease through BMP-SMAD-mediated hepatic iron overload. Nanotoxicology (2021) 15:761–78. doi: 10.1080/17435390.2021.1919329
6. Zhao R, Zhu M, Zhou S, Feng W, Chen H. Rapamycin-loaded mPEG-PLGA nanoparticles ameliorate hepatic steatosis and liver injury in non-alcoholic fatty liver disease. Front Chem (2020) 8:407. doi: 10.3389/fchem.2020.00407
7. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology (2016) 64:73–84. doi: 10.1002/hep.28431
8. Li J, Zou B, Yeo YH, Feng Y, Xie X, Lee DH, et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999-2019: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol (2019) 4:389–98. doi: 10.1016/S2468-1253(19)30039-1
9. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol (2013) 10:686–90. doi: 10.1038/nrgastro.2013.171
10. Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology (2019) 69:2672–82. doi: 10.1002/hep.30251
11. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol (2019) 16:411–28. doi: 10.1038/s41575-019-0145-7
12. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol (2013) 10:656–65. doi: 10.1038/nrgastro.2013.183
13. Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology (2010) 51:1972–8. doi: 10.1002/hep.23527
14. Marengo A, Jouness RI, Bugianesi E. Progression and natural history of nonalcoholic fatty liver disease in adults. Clin Liver Dis (2016) 20:313–24. doi: 10.1016/j.cld.2015.10.010
15. Day CP, James OF. Steatohepatitis: a tale of two "hits"? Gastroenterology (1998) 114:842–5. doi: 10.1016/S0016-5085(98)70599-2
16. Pierantonelli I, Svegliati-Baroni G. Nonalcoholic fatty liver disease: Basic pathogenetic mechanisms in the progression from NAFLD to NASH. Transplantation (2019) 103:e1–e13. doi: 10.1097/TP.0000000000002480
17. Safari Z, Gerard P. The links between the gut microbiome and non-alcoholic fatty liver disease (NAFLD). Cell Mol Life Sci (2019) 76:1541–58. doi: 10.1007/s00018-019-03011-w
18. Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci (2019) 76:99–128. doi: 10.1007/s00018-018-2947-0
19. Wen L, Li M, Lin X, Li Y, Song H, Chen H. AgNPs aggravated hepatic steatosis, inflammation, oxidative stress, and epigenetic changes in mice with NAFLD induced by HFD. Front Bioeng Biotechnol (2022) 10. doi: 10.3389/fbioe.2022.912178
20. Konerman MA, Jones JC, Harrison SA, Pharmacotherapy for NASH. Current and emerging. J Hepatol (2018) 68:362–75. doi: 10.1016/j.jhep.2017.10.015
21. Chen H, Shen F, Sherban A, Nocon A, Li Y, Wang H, et al. DEP domain–containing mTOR–interacting protein suppresses lipogenesis and ameliorates hepatic steatosis and acute-on-chronic liver injury in alcoholic liver disease. Hepatology (2018) 68:496–514. doi: 10.1002/hep.29849
22. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci (2016) 61:1294–303. doi: 10.1007/s10620-016-4049-x
23. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol (2018) 15:349–64. doi: 10.1038/s41575-018-0009-6
24. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
25. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther (2021) 6:291. doi: 10.1038/s41392-021-00687-0
26. Kesar V, Odin JA. Toll-like receptors and liver disease. Liver Int (2014) 34:184–96. doi: 10.1111/liv.12315
27. Kugelberg E. Pattern recognition receptors: curbing gut inflammation. Nat Rev Immunol (2014) 14:583. doi: 10.1038/nri3735
28. Pedra JH, Cassel SL, Sutterwala FS. Sensing pathogens and danger signals by the inflammasome. Curr Opin Immunol (2009) 21:10–6. doi: 10.1016/j.coi.2009.01.006
29. Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology (2008) 48:322–35. doi: 10.1002/hep.22306
30. Seki E, Tsutsui H, Nakano H, Tsuji N, Hoshino K, Adachi O, et al. Lipopolysaccharide-induced IL-18 secretion from murine kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1beta. J Immunol (2001) 166:2651–7. doi: 10.4049/jimmunol.166.4.2651
31. Isogawa M, Robek MD, Furuichi Y, Chisari FV. Toll-like receptor signaling inhibits hepatitis b virus replication in vivo. J Virol (2005) 79:7269–72. doi: 10.1128/JVI.79.11.7269-7272.2005
32. Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology (2003) 37:1043–55. doi: 10.1053/jhep.2003.50182
33. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med (2007) 13:1324–32. doi: 10.1038/nm1663
34. Miura K, Ishioka M, Minami S, Horie Y, Ohshima S, Goto T, et al. Toll-like receptor 4 on macrophage promotes the development of steatohepatitis-related hepatocellular carcinoma in mice. J Biol Chem (2016) 291:11504–17. doi: 10.1074/jbc.M115.709048
35. Luo X, Li H, Ma L, Zhou J, Guo X, Woo SL, et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology (2018) 155:1971–84.e4. doi: 10.1053/j.gastro.2018.09.010
36. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. STING-mediated inflammation in kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest (2019) 129:546–55. doi: 10.1172/JCI121842
37. Roh YS, Kim JW, Park S, Shon C, Kim S, Eo SK, et al. Toll-like receptor-7 signaling promotes nonalcoholic steatohepatitis by inhibiting regulatory T cells in mice. Am J Pathol (2018) 188:2574–88. doi: 10.1016/j.ajpath.2018.07.011
38. Song IJ, Yang YM, Inokuchi-Shimizu S, Roh YS, Yang L, Seki E. The contribution of toll-like receptor signaling to the development of liver fibrosis and cancer in hepatocyte-specific TAK1-deleted mice. Int J Cancer (2018) 142:81–91. doi: 10.1002/ijc.31029
39. Ma H, Kang Z, Foo TK, Shen Z, Xia B. Disrupted BRCA1-PALB2 interaction induces tumor immunosuppression and T-lymphocyte infiltration in HCC through cGAS-STING pathway. Hepatology (2023) 77:33–47. doi: 10.1002/hep.32335
40. Shen X, Zhao J, Wang Q, Chen P, Hong Y, He X, et al. The invasive potential of hepatoma cells induced by radiotherapy is related to the activation of hepatic stellate cells and could be inhibited by EGCG through the TLR4 signaling pathway. Radiat Res (2022) 197:365–75. doi: 10.1667/RADE-21-00129.1
41. Zhang Y, Li Y, Mu T, Tong N, Cheng P. Hepatic stellate cells specific liposomes with the toll-like receptor 4 shRNA attenuates liver fibrosis. J Cell Mol Med (2021) 25:1299–313. doi: 10.1111/jcmm.16209
42. Guo J, Friedman SL. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair (2010) 3:21. doi: 10.1186/1755-1536-3-21
43. Du S, Chen G, Yuan B, Hu Y, Yang P, Chen Y, et al. DNA Sensing and associated type 1 interferon signaling contributes to progression of radiation-induced liver injury. Cell Mol Immunol (2021) 18:1718–28. doi: 10.1038/s41423-020-0395-x
44. Thomsen MK, Skouboe MK, Boularan C, Vernejoul F, Lioux T, Leknes SL, et al. The cGAS-STING pathway is a therapeutic target in a preclinical model of hepatocellular carcinoma. Oncogene (2020) 39:1652–64. doi: 10.1038/s41388-019-1108-8
45. Kanuri G, Ladurner R, Skibovskaya J, Spruss A, Konigsrainer A, Bischoff SC, et al. Expression of toll-like receptors 1-5 but not TLR 6-10 is elevated in livers of patients with non-alcoholic fatty liver disease. Liver Int (2015) 35:562–8. doi: 10.1111/liv.12442
46. Arias-Loste MT, Iruzubieta P, Puente A, Ramos D, Santa Cruz C, Estebanez A, et al. Increased expression profile and functionality of TLR6 in peripheral blood mononuclear cells and hepatocytes of morbidly obese patients with non-alcoholic fatty liver disease. Int J Mol Sci (2016) 17(11):1878. doi: 10.3390/ijms17111878
47. Rivera CA, Gaskin L, Allman M, Pang J, Brady K, Adegboyega P, et al. Toll-like receptor-2 deficiency enhances non-alcoholic steatohepatitis. BMC Gastroenterol (2010) 10:52. doi: 10.1186/1471-230X-10-52
48. Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology (2013) 57:577–89. doi: 10.1002/hep.26081
49. Baumann A, Nier A, Hernandez-Arriaga A, Brandt A, Lorenzo Pisarello MJ, Jin CJ, et al. Toll-like receptor 1 as a possible target in non-alcoholic fatty liver disease. Sci Rep (2021) 11:17815. doi: 10.1038/s41598-021-97346-9
50. Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology (2009) 49:1877–87. doi: 10.1002/hep.22848
51. Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palu G, et al. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol (2007) 292:G518–25. doi: 10.1152/ajpgi.00024.2006
52. Kessoku T, Kobayashi T, Imajo K, Tanaka K, Yamamoto A, Takahashi K, et al. Endotoxins and non-alcoholic fatty liver disease. Front Endocrinol (Lausanne) (2021) 12:770986. doi: 10.3389/fendo.2021.770986
53. Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, et al. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int (2008) 28:1026–33. doi: 10.1111/j.1478-3231.2008.01723.x
54. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest (2006) 116:3015–25. doi: 10.1172/JCI28898
55. de Almeida IT, Cortez-Pinto H, Fidalgo G, Rodrigues D, Camilo ME. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin Nutr (2002) 21:219–23. doi: 10.1054/clnu.2001.0529
56. Soares JB, Pimentel-Nunes P, Afonso L, Rolanda C, Lopes P, Roncon-Albuquerque R Jr., et al. Increased hepatic expression of TLR2 and TLR4 in the hepatic inflammation-fibrosis-carcinoma sequence. Innate Immun (2012) 18:700–8. doi: 10.1177/1753425912436762
57. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol (2007) 47:571–9. doi: 10.1016/j.jhep.2007.04.019
58. Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell (2009) 138:961–75. doi: 10.1016/j.cell.2009.06.046
59. Zhao P, Wong KI, Sun X, Reilly SM, Uhm M, Liao Z, et al. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell (2018) 172:731–43.e12. doi: 10.1016/j.cell.2018.01.007
60. Mridha AR, Haczeyni F, Yeh MM, Haigh WG, Ioannou GN, Barn V, et al. TLR9 is up-regulated in human and murine NASH: pivotal role in inflammatory recruitment and cell survival. Clin Sci (Lond) (2017) 131:2145–59. doi: 10.1042/CS20160838
61. Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest (2016) 126:859–64. doi: 10.1172/JCI83885
62. An P, Wei LL, Zhao S, Sverdlov DY, Vaid KA, Miyamoto M, et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun (2020) 11:2362. doi: 10.1038/s41467-020-16092-0
63. Shepard CR. TLR9 in MAFLD and NASH: At the intersection of inflammation and metabolism. Front Endocrinol (Lausanne) (2020) 11:613639. doi: 10.3389/fendo.2020.613639
64. Revelo XS, Ghazarian M, Chng MH, Luck H, Kim JH, Zeng K, et al. Nucleic acid-targeting pathways promote inflammation in obesity-related insulin resistance. Cell Rep (2016) 16:717–30. doi: 10.1016/j.celrep.2016.06.024
65. Alderton GK. Inflammation: the gut takes a toll on liver cancer. Nat Rev Cancer (2012) 12:379. doi: 10.1038/nrc3283
66. Lin A, Wang G, Zhao H, Zhang Y, Han Q, Zhang C, et al. TLR4 signaling promotes a COX-2/PGE2/STAT3 positive feedback loop in hepatocellular carcinoma (HCC) cells. Oncoimmunology (2016) 5:e1074376. doi: 10.1080/2162402X.2015.1074376
67. Zhou S, Du R, Wang Z, Shen W, Gao R, Jiang S, et al. TLR4 increases the stemness and is highly expressed in relapsed human hepatocellular carcinoma. Cancer Med (2019) 8:2325–37. doi: 10.1002/cam4.2070
68. Kairaluoma V, Kemi N, Huhta H, Pohjanen VM, Helminen O. Prognostic role of TLR4 and TLR2 in hepatocellular carcinoma. Acta Oncol (2021) 60:554–8. doi: 10.1080/0284186X.2021.1877346
69. Bonnin M, Fares N, Testoni B, Estornes Y, Weber K, Vanbervliet B, et al. Toll-like receptor 3 downregulation is an escape mechanism from apoptosis during hepatocarcinogenesis. J Hepatol (2019) 71:763–72. doi: 10.1016/j.jhep.2019.05.031
70. Junjie X, Songyao J, Minmin S, Yanyan S, Baiyong S, Xiaxing D, et al. The association between toll-like receptor 2 single-nucleotide polymorphisms and hepatocellular carcinoma susceptibility. BMC Cancer (2012) 12:57. doi: 10.1186/1471-2407-12-57
71. Shiau DJ, Kuo WT, Davuluri GVN, Shieh CC, Tsai PJ, Chen CC, et al. Hepatocellular carcinoma-derived high mobility group box 1 triggers M2 macrophage polarization via a TLR2/NOX2/autophagy axis. Sci Rep (2020) 10:13582. doi: 10.1038/s41598-020-70137-4
72. Zhou B, Yan J, Guo L, Zhang B, Liu S, Yu M, et al. Hepatoma cell-intrinsic TLR9 activation induces immune escape through PD-L1 upregulation in hepatocellular carcinoma. Theranostics (2020) 10:6530–43. doi: 10.7150/thno.44417
73. Kairaluoma V, Kemi N, Huhta H, Pohjanen VM, Helminen O. Toll-like receptor 5 and 8 in hepatocellular carcinoma. APMIS (2021) 129:470–9. doi: 10.1111/apm.13142
74. Haderski GJ, Kandar BM, Brackett CM, Toshkov IM, Johnson CP, Paszkiewicz GM, et al. TLR5 agonist entolimod reduces the adverse toxicity of TNF while preserving its antitumor effects. PloS One (2020) 15:e0227940. doi: 10.1371/journal.pone.0227940
75. Chen Y, Huang Z, Chen X, Ye H. Activation of the tolllike receptor 2 signaling pathway inhibits the proliferation of HCC cells in vitro. Oncol Rep (2019) 42:2267–78. doi: 10.3892/or.2019.7340
76. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455:674–8. doi: 10.1038/nature07317
77. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi: 10.1126/science.1232458
78. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2',5')pA(3',5')p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell (2013) 153:1094–107. doi: 10.1016/j.cell.2013.04.046
79. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature (2013) 498:380–4. doi: 10.1038/nature12306
80. Thomsen MK, Nandakumar R, Stadler D, Malo A, Valls RM, Wang F, et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis b virus infection. Hepatology (2016) 64:746–59. doi: 10.1002/hep.28685
81. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature (2017) 550:402–6. doi: 10.1038/nature24050
82. Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol (2017) 19:1061–70. doi: 10.1038/ncb3586
83. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature (2015) 520:553–7. doi: 10.1038/nature14156
84. Riley JS, Quarato G, Cloix C, Lopez J, O'Prey J, Pearson M, et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J (2018) 37(17):e99238. doi: 10.15252/embj.201899238
85. White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell (2014) 159:1549–62. doi: 10.1016/j.cell.2014.11.036
86. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
87. Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med (2018) 215:1287–99. doi: 10.1084/jem.20180139
88. Bai J, Liu F. The cGAS-cGAMP-STING pathway: A molecular link between immunity and metabolism. Diabetes (2019) 68:1099–108. doi: 10.2337/dbi18-0052
89. Motwani M, Pesiridis S, Fitzgerald KA. DNA Sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet (2019) 20:657–74. doi: 10.1038/s41576-019-0151-1
90. Wang X, Rao H, Zhao J, Wee A, Li X, Fei R, et al. STING expression in monocyte-derived macrophages is associated with the progression of liver inflammation and fibrosis in patients with nonalcoholic fatty liver disease. Lab Invest (2020) 100:542–52. doi: 10.1038/s41374-019-0342-6
91. Qiao JT, Cui C, Qing L, Wang LS, He TY, Yan F, et al. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism (2018) 81:13–24. doi: 10.1016/j.metabol.2017.09.010
92. Lei Z, Deng M, Yi Z, Sun Q, Shapiro RA, Xu H, et al. cGAS-mediated autophagy protects the liver from ischemia-reperfusion injury independently of STING. Am J Physiol Gastrointest Liver Physiol (2018) 314:G655–67. doi: 10.1152/ajpgi.00326.2017
93. Papatheodoridi AM, Chrysavgis L, Koutsilieris M, Chatzigeorgiou A. The role of senescence in the development of nonalcoholic fatty liver disease and progression to nonalcoholic steatohepatitis. Hepatology (2020) 71:363–74. doi: 10.1002/hep.30834
94. Mao Y, Luo W, Zhang L, Wu W, Yuan L, Xu H, et al. STING-IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler Thromb Vasc Biol (2017) 37:920–9. doi: 10.1161/ATVBAHA.117.309017
95. Li YN, Su Y. Remdesivir attenuates high fat diet (HFD)-induced NAFLD by regulating hepatocyte dyslipidemia and inflammation via the suppression of STING. Biochem Biophys Res Commun (2020) 526:381–8. doi: 10.1016/j.bbrc.2020.03.034
96. Si W, Liang H, Bugno J, Xu Q, Ding X, Yang K, et al. Lactobacillus rhamnosus GG induces cGAS/STING- dependent type I interferon and improves response to immune checkpoint blockade. Gut (2022) 71(3):521–33. doi: 10.1136/gutjnl-2020-323426
97. Qi Z, Yan F, Chen D, Xing W, Li Q, Zeng W, et al. Identification of prognostic biomarkers and correlations with immune infiltrates among cGAS-STING in hepatocellular carcinoma. Biosci Rep (2020) 40(10):BSR20202603. doi: 10.1042/BSR20202603
98. Jiang M, Chen P, Wang L, Li W, Chen B, Liu Y, et al. cGAS-STING, an important pathway in cancer immunotherapy. J Hematol Oncol (2020) 13:81. doi: 10.1186/s13045-020-00916-z
99. Wang Y, Luo J, Alu A, Han X, Wei Y, Wei X. cGAS-STING pathway in cancer biotherapy. Mol Cancer (2020) 19:136. doi: 10.1186/s12943-020-01247-w
100. Waidmann O. Recent developments with immunotherapy for hepatocellular carcinoma. Expert Opin Biol Ther (2018) 18:905–10. doi: 10.1080/14712598.2018.1499722
101. Wang C, Sun Z, Zhao C, Zhang Z, Wang H, Liu Y, et al. Maintaining manganese in tumor to activate cGAS-STING pathway evokes a robust abscopal anti-tumor effect. J Control Release (2021) 331:480–90. doi: 10.1016/j.jconrel.2021.01.036
102. Wehbe M, Wang-Bishop L, Becker KW, Shae D, Baljon JJ, He X, et al. Nanoparticle delivery improves the pharmacokinetic properties of cyclic dinucleotide STING agonists to open a therapeutic window for intravenous administration. J Control Release (2021) 330:1118–29. doi: 10.1016/j.jconrel.2020.11.017
103. Kim DS, Endo A, Fang FG, Huang KC, Bao X, Choi HW, et al. E7766, a macrocycle-bridged stimulator of interferon genes (STING) agonist with potent pan-genotypic activity. ChemMedChem (2021) 16:1740–3. doi: 10.1002/cmdc.202100068
104. Morimoto Y, Kishida T, Kotani SI, Takayama K, Mazda O. Interferon-beta signal may up-regulate PD-L1 expression through IRF9-dependent and independent pathways in lung cancer cells. Biochem Biophys Res Commun (2018) 507:330–6. doi: 10.1016/j.bbrc.2018.11.035
105. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep (2019) 29:3766. doi: 10.1016/j.celrep.2019.11.113
106. Samarpita S, Kim JY, Rasool MK, Kim KS. Investigation of toll-like receptor (TLR) 4 inhibitor TAK-242 as a new potential anti-rheumatoid arthritis drug. Arthritis Res Ther (2020) 22:16. doi: 10.1186/s13075-020-2097-2
Keywords: NAFLD, HCC, PRR, innate immune signaling pathway, inflammation
Citation: Huang C, Zhou Y, Cheng J, Guo X, Shou D, Quan Y, Chen H, Chen H and Zhou Y (2023) Pattern recognition receptors in the development of nonalcoholic fatty liver disease and progression to hepatocellular carcinoma: An emerging therapeutic strategy. Front. Endocrinol. 14:1145392. doi: 10.3389/fendo.2023.1145392
Received: 16 January 2023; Accepted: 08 March 2023;
Published: 20 March 2023.
Edited by:
Alain Couvineau, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Feixia Wang, Nanjing University of Chinese Medicine, ChinaCopyright © 2023 Huang, Zhou, Cheng, Guo, Shou, Quan, Chen, Chen and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongjian Zhou, ZXl6aG91eW9uZ2ppYW5Ac2N1dC5lZHUuY24=; Huiting Chen, ZXljaGVuaHVpdGluZ0BzY3V0LmVkdS5jbg==; Hanqing Chen, Y2hlbmhxOTIxQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.