 Guzel Burganova
Guzel Burganova Anat Schonblum
Anat Schonblum Lina Sakhneny
Lina Sakhneny Alona Epshtein
Alona Epshtein Tomer Wald
Tomer Wald Mika Tzaig
Mika Tzaig Limor Landsman
Limor Landsman
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 09 March 2023
Sec. Diabetes: Molecular Mechanisms
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1142988
This article is part of the Research TopicThe Role of Pericytes in Physiology and PathophysiologyView all 6 articles
Introduction: Immune cells were recently shown to support β-cells and insulin secretion. However, little is known about how islet immune cells are regulated to maintain glucose homeostasis. Administration of various cytokines, including Interleukin-33 (IL-33), was shown to influence β-cell function. However, the role of endogenous, locally produced IL-33 in pancreatic function remains unknown. Here, we show that IL-33, produced by pancreatic pericytes, is required for glucose homeostasis.
Methods: To characterize pancreatic IL-33 production, we employed gene expression, flow cytometry, and immunofluorescence analyses. To define the role of this cytokine, we employed transgenic mouse systems to delete the Il33 gene selectively in pancreatic pericytes, in combination with the administration of recombinant IL-33. Glucose response was measured in vivo and in vitro, and morphometric and molecular analyses were used to measure β-cell mass and gene expression. Immune cells were analyzed by flow cytometry.
Resuts: Our results show that pericytes are the primary source of IL-33 in the pancreas. Mice lacking pericytic IL-33 were glucose intolerant due to impaired insulin secretion. Selective loss of pericytic IL-33 was further associated with reduced T and dendritic cell numbers in the islets and lower retinoic acid production by islet macrophages.
Discussion: Our study demonstrates the importance of local, pericytic IL-33 production for glucose regulation. Additionally, it proposes that pericytes regulate islet immune cells to support β-cell function in an IL-33-dependent manner. Our study reveals an intricate cellular network within the islet niche.
Insulin secretion is a complex and tightly regulated process that is dependent on both systemic and local signals. β-Cells respond to a variety of inputs from the islet microenvironment. The islet microenvironment comprises various cell populations, such as neuronal, immune, and vascular cells, which play a crucial role in guiding the development, replication, maturation, and function of β-cells (1–4). The activity of these cell populations needs to coordinate effectively to promote glucose regulation through insulin secretion. However, the molecular and cellular interactions between the diverse cell types within the islet microenvironment and how they orchestrate to ensure insulin secretion are still not fully understood.
While islet inflammation can contribute to diabetes, it can also play a beneficial role in glucose homeostasis by supporting β-cell mass and function (1, 5–15). Islets of healthy humans and mice contain immune cells, primarily macrophages, but also T, B, dendritic (DCs), and type 2 innate lymphoid (ILC2s) cells (7, 16). These cells form a tightly-regulated network that supports glucose regulation. Macrophages and DCs were shown to support β-cell function and mass (1, 8, 13, 17, 18), but the role of islet lymphocytes in glucose regulation is currently unclear (16, 19). Additionally, how islet inflammation is regulated to support glucose homeostasis is largely unknown.
IL-33 is a member of the interleukin-1 (IL-1) family of cytokines primarily localized in the nucleus (20, 21). Studies have shown that the localization of IL-33 in the nucleus does not affect gene transcription and that the primary function of its binding to chromatin is to regulate its own extracellular release (22–24). Thus, the biological activity of IL-33 depends on its secretion and subsequent binding to the ST2 receptor (IL-1RL1), which is mainly expressed by immune cells (21, 25). This cytokine has been dubbed “Alarmin” due to its role in activating the immune response; however, recent research has also implicated IL-33 in tissue homeostasis (26), indicating that it plays a complex role in both physiological and pathological conditions such as inflammation, tissue repair, and homeostasis.
The administration of recombinant IL-33 (rIL-33) has been shown to improve mice glucose response by affecting both adipose tissue and the pancreas (7, 27). In agreement, IL-33 null mice display glucose intolerance upon obesity (7). Additionally, rIL-33 administration has been shown to attenuate insulitis in mouse models of type 1 diabetes (28). A recent study by Dalmas et al. demonstrates that in the islets, the expression of ST2 is restricted to ILC2s, which, in response to IL-33 administration, recruit and activate macrophages and DCs (7). Subsequently, macrophages and DCs promote insulin secretion by producing retinoic acid (RA), which has various effects on β-cell function, including the induction of glucose-stimulated insulin secretion, insulin production, and glucokinase activity (7, 29). Thus, rIL-33 activates a cellular network to promote insulin secretion. Although suggested to be cells of mesenchymal origin, the pancreatic source of IL-33 is currently unknown (7). Further, the role of locally produced IL-33 in insulin secretion and glucose homeostasis remains to be elucidated.
Pericytes are contractile mural cells that wrap around capillaries and microvessels in various tissues and organs. Depending on the tissue, these cells have different origins and gene expression patterns (30, 31). In the islets, pericytes have been shown to play an essential role in regulating β-cell function and mass (2, 4, 32–38). Further, this activity can be mediated by secreted factors, including NGF, BMP4, and ECM components, independently of blood flow regulation (32, 34–37). Studies suggest that pericytes may have an immunoregulatory role in various tissues, including the brain and kidneys, by secreting cytokines to control immune cell infiltration and activation (39–44). However, whether pericytes in the islets also have this function has yet to be determined.
Here, we provide evidence for the immunoregulatory role of pancreatic pericytes. First, we identified pericytes as the predominant source of IL-33 in the pancreas. We used transgenic mouse models to selectively delete pericytic IL-33 expression and found that loss of this cytokine caused glucose intolerance due to impaired insulin secretion. Further analyses revealed that pericytic IL-33 regulates the number of immune cells in the islets and their production of RA, indicating that pericytic IL-33 plays an essential role in maintaining glucose homeostasis by regulating immune cells in the islets. Our study indicates that pancreatic pericytes are immunomodulators, revealing an additional regulatory layer of islet inflammation and insulin secretion.
Mice were maintained on a C57BL/6 background. Nkx3.2-Cre (Nkx3-2tm1(cre)Wez) mice (45) were a generous gift from Warren Zimmer (Texas A&M University, College Station, TX), and R26-EYFP (Gt(ROSA)26Sortm1(EYFP)Cos) and Il33flox/flox-eGFP (Il33tm1.1Bryc) (46) mice were obtained from the Jackson Laboratory (Table S1). Wild-type mice were purchased from Envigo Ltd. (Jerusalem, Israel). Animals were housed under specific pathogen-free conditions. All experimental protocols were approved by the Tel Aviv University Institutional Animal Care and Use Committee (IACUC). The study was carried out in compliance with the ARRIVE guidelines.
For glucose tolerance tests, mice were fasted overnight before the i.p. injection of dextrose (2 mg/gr). For insulin tolerance tests, mice were fasted 6 hours before i.p. insulin administration (0.5 U/kg). Tail vein blood glucose levels were measured at indicated time points using glucose meters (Bayer). When indicated, mice were i.p. injected every other day with either 500 ng murine rIL-33 (BioLegend; Cat #580502) or PBS to obtain a total of three doses. Mice were analyzed 24 hours after the last injection.
For islet isolation, collagenase P solution (0.8 mg/ml; Roche) was injected through the common bile duct into the pancreas of the euthanized mouse, followed by 11 minutes of incubation and a density gradient (Histopaque-1119, Sigma) separation. Islets were collected from the gradient interface and hand-picked.
To obtain single cell suspension from whole pancreas, dissected tissue was incubated in HBSS supplemented with collagenase P (0.4 mg/ml; Roche) and DNase I (0.1 ng/ml; Sigma-Aldrich). For analysis of islet cells, double-hand-picked islets were gently dispersed with Accutase (Sigma-Aldrich) solution for 5 minutes. For analysis of blood immune cells, tail vein blood was collected into EDTA-containing tubes, followed by a density gradient separation with Lymphoprep (StemCell Technologies). For analysis of splenic cells, dissected spleens were injected with collagenase D (1 mg/ml; Roche), sliced, and incubated at 37°C for 45 minutes. Then dissociated spleen was minced, strained, and washed. After supernatant aspiration, the pellet was resuspended in hypertonic ACK buffer to lyse erythrocytes for 2 minutes at room temperature.
For staining with surface markers, dispersed cells were incubated with Fc blocker (anti-CD16/CD32 antibody) for 15 minutes, followed by staining with the appropriate antibodies (Table S2) for 30 minutes on ice and incubation with DAPI (Sigma-Aldrich) to label dead cells.
Aldehyde dehydrogenase (ALDH) activity was determined using the ALDEFLUOR kit (StemCell Technologies) according to the manufacturer’s protocol. Briefly, dispersed cells from 200 islets were equally divided into two tubes, control and experimental. Control cells were incubated with ALDH inhibitor diethylaminobenzaldehyde (DEAB) for 15 minutes at 37°C to define the background fluorescence. Then, control and experimental tubes were incubated with activated fluorescent ALDH reagent for 35 minutes at 37°C. Following incubation and washes, cells were stained with appropriate antibodies.
Cells were analyzed by Cytoflex (Beckman Coulter) and analyzed with Kaluza software (Beckman Coulter).
For pancreatic pericytes isolation, pancreatic cells were collected from adult YFPPericytes (Nkx3.2-Cre;R26-EYFP) mice when pericytes were identified based on the yellow fluorescence (32, 34). Pancreatic cells were isolated according to a previously published protocol (47). Briefly, dissected pancreatic tissues were incubated in HBSS supplemented with collagenase P (0.4 mg/ml; Roche) and DNase I (0.1 ng/ml; Sigma-Aldrich) for 30 minutes at 37°C with mild agitation. Ice-cold HBSS buffer was added to stop digestion. Tubes were centrifugated at 300 g for 5 minutes at 4°C, washed, and strained through a 70 µm cell strainer (Miltenyi Biotec) to collect single cells. Cells were resuspended in PBS (without calcium chloride and magnesium chloride) supplemented with 5% fetal bovine serum and 5 mM EDTA and re-strained through a 35 µm cell strainer (Corning). When indicated, single-cell suspension from wild-type or YFPPericytes mice were incubated with Fc blocker (anti-CD16/CD32 antibody; Table S2) for 30 minutes followed by 30 minutes incubation on ice with anti-PECAM1/CD31 or anti-CD45 antibodies (Table S2) to identify endothelial and immune cells, respectively. Prior to sorting, cells were incubated with DAPI (Sigma-Aldrich) to mark and exclude dead and late apoptotic cells. Pancreatic pericytes, endothelial, and immune cells were collected using FACSAria II or FACSAria III cell sorters (BD).
For in vivo analysis, dextrose (2 mg/gr) was i.p. injected after an overnight fast. At indicated time points, tail vein blood was collected, and serum was separated. For ex vivo analysis, freshly isolated islets were pre-incubated for 30 minutes in RPMI medium with low glucose (1.67 mM). For each experiment, ten size-matched islets were hand-picked under a stereotaxic microscope and transferred in 5 µl volume to the well of 96-well U-bottom untreated plate, containing 250 µl of either low (1.67 mM) or high (16.7 mM)-glucose media, followed by 1-hour incubation at 37°C degrees and 5% CO2. Alternatively, islets were pre-incubated for an hour in Krebs-Ringer buffer containing glucose (1.67 mM), followed by a 1-hour incubation with or without KCl (30 mM). Islet insulin content was extracted by 1.5% HCl in 70% ethanol solution, followed by lysis using TissueLyser II (Qiagen). Hormone levels were measured using a mouse ultrasensitive Insulin ELISA kit (Alpco).
Dissected pancreatic tissues were fixed in 4% paraformaldehyde, followed by embedding in either O.C.T compound (Scigen) or paraffin. For paraffin-embedded tissues, heat-induced antigen retrieval in Citra buffer (BioGenex) was performed before the staining. Tissue sections were immunostained with indicated antibodies (Tables S3, S4) when control and transgenic tissues were stained in parallel. For TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) assay, the Fluorescein In Situ Cell Death Detection Kit (Roche) was used according to the manufacturer’s protocol. Images were acquired using BZ-9000 BioRevo (Keyence) and SP8 confocal (Leica microsystems) microscopes.
For analysis of functional vasculature, fluorescently labeled tomato lectin (1 mg/ml; Vector Laboratories) was intravenously injected. After 5 minutes, mice were euthanized, and their pancreas was extracted and fixed in 4% paraformaldehyde. Tissue cryosections were stained for insulin and imaged. For analysis of islet pericyte coverage, fixed pancreatic tissues of untreated mice were analyzed. Cryo-sections were immunostained for the pericytic marker Neuron-glial antigen 2 (NG2) and insulin and imaged. At least 50 islets per mouse, defined by their insulin expression, were analyzed to determine NG2- or lectin-positive areas per islet. For measurement of β-cell mass, fixed pancreatic tissues of untreated mice were analyzed. Paraffin-embedded sections were immunostained for insulin and counterstained with HCS CellMask Stain (Invitrogen) to mark the section area. Whole tissue sections, at least 100 µm apart, were automatically imaged. For each mouse, the acquired insulin-positive area was divided by the pancreatic area and multiplied by its weight. For analysis of the α-/β-cell ratio, glucagon- and insulin-positive areas were calculated for each islet when 50 islets per mouse were analyzed. For cell proliferation analysis, pancreatic tissues from postnatal day 5 pups were analyzed. Cryo-sections of at least 50 µm apart were stained for insulin and marker of proliferation Ki67 (Table S3). After imaging, at least 300 insulin-positive cells per pup were analyzed manually and the portion of the Ki67-positive cells out of the total number of insulin-positive cells was calculated. Images were acquired using Keyence BZ-9000 (Biorevo) and analyzed using ImageJ software (NIH).
Gene expression levels were detected with Taqman (Applied Biosystems) or SYBR green assays (Applied Biosystems) using indicated primers (Table S5) and normalized to GAPDH and Cyclophilin, respectively. Expression levels were determined using the StepOne cycler (Applied Biosystems). Published mouse pancreatic pericytes RNA sequencing (34) was deposited in ArrayExpress (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5325/). Published islet genomic data (48) was accessed in http://www.gaultonlab.org/pages/Islet_expression_HPAP.html.
Analysis was carried out using an unpaired two-tailed Student’s t-test (Prism software v.9; GraphPad). P values of 0.05 or less were considered to be statistically significant. Statistically significant outliers were identified according to the Grubbs’ method and excluded from the analyses.
IL-33 was shown to be produced by mesenchymal cells in the pancreas (7, 49). As pericytes are the predominant mesenchymal cell population in the islets (34, 50), we set to characterize their IL-33 expression. First, we analyzed the published transcriptome of pancreatic pericytes of naïve, healthy mice (34). As shown in Figure 1A, mouse pancreatic pericytes express IL-33 but no other members of the IL-1 family of cytokines. To define IL33 expression in human islets, we employed a published islet cell transcriptome (48). Indeed, IL33 is expressed by pericytes, identified by the pericytic markers PDGFRB, ACTA2, ENG, and CD248 (30, 48)(Figure S1).
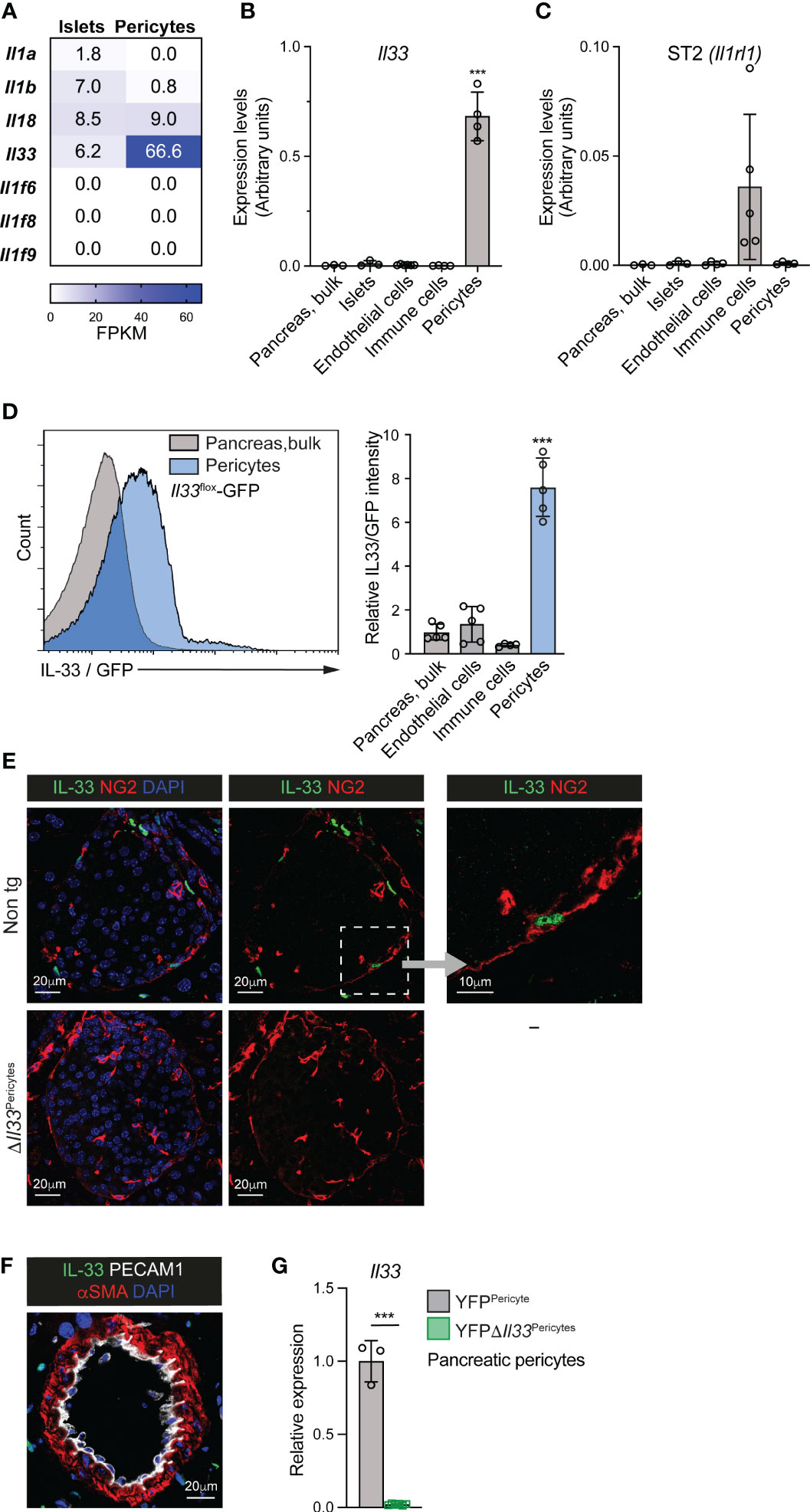
Figure 1 Pericytes are the primary source of IL-33 in the mouse pancreas and islet. (A) Analysis of IL-1 cytokine family ligands expression in isolated pancreatic pericytes and islets, employing a previously published RNAseq analysis (34). Heat maps show mean expression levels (as fragments per kilobase of exon per million aligned fragments [FPKM]) of indicated genes. N = 3. (B, C) Bar diagram (mean ± SD) shows relative levels of Il33 (B) and Il1rl1 (encodes ST2; C’) transcripts in different pancreatic cell populations, analyzed by qPCR (normalized to GAPDH). RNA was extracted from bulk pancreatic tissue, isolated islets, pancreatic endothelial cells (FACS-purified based on their PECAM1 expression) and pancreatic immune cells (FACS-purified based on their CD45 expression) of adult wild-type mice, and pancreatic pericytes (FACS-purified from Nkx3.2-Cre;R26-EYFP mice based on their yellow fluorescent labeling). N = 3-5. (D) Flow cytometry analysis of pancreatic tissue of Il33flox-eGFP mice. Green fluorescence intensity was analyzed in bulk pancreatic cells, endothelial cells (defined as PECAM1+ cells), immune cells (defined as CD45+ cells), and pericytes (defined as PDGFRβ+ cells). Left, histogram showing representative fluorescence intensities of bulk pancreatic cells (gray) and pericytes (blue). Right, bar diagram shows the relative mean fluorescence intensity (MFI) when the average of bulk pancreatic cells was set to ‘1’. N = 4-5. (E) Immunofluorescence analysis of pancreatic tissue sections of non-transgenic (top) and ΔIl33Pericytes (bottom) mice. Sections were stained for IL-33 (green) and Neuron-glial antigen 2 (NG2; red) to label pericytes and counterstained with DAPI (blue). Representative islets (defined by morphology) are shown. Right, higher magnification of the area framed by the white box in the middle panel. (F) Immunofluorescence analysis of pancreatic tissue sections of non-transgenic mice. Sections were stained for αSMA (red) to label vSMCs, PECAM1 (white) to label endothelial cells, and IL-33 (green) and counterstained with DAPI (blue). Shown is a representative field. (G) Bar diagram (mean ± SD) showing lower Il33 transcript levels in Il33-deficient pericytes. RNA was extracted from FACS-purified pancreatic pericytes from 3-month-old YFPΔIl33Pericytes (Nkx3.2-Cre;Il33flox/flox;R26-EYFP; green) and YFPPericytes (Nkx3.2-Cre;R26-EYFP; gray; the average was set to ‘1’) mice. Gene expression was analyzed by qPCR. N =3-5. ***p<0.005 (Student’s t-test). Each dot represents a single mouse.
Next, we compared Il33 transcript levels in various mouse pancreatic cell populations: bulk pancreatic tissue (contains mainly acinar cells), isolated islets (contains mainly endocrine cells), and purified pancreatic endothelial cells, immune cells, and pericytes. As shown in Figure 1B, our qPCR analysis revealed that Il33 expression in pancreatic pericytes was two orders of magnitude higher than in other analyzed cell populations. Similarly, IL33 expression in the human islets was restricted to the pericytic cell cluster (Figure S1) (48).
The expression of the IL-33 receptor ST2, encoded by Il1rl1, was previously shown to be restricted to ILC2s (7). Indeed, we detected its expression in immune cells but not in pericytes, endocrine, and endothelial cells of the mouse pancreas (Figure 1C).
To define pancreatic IL-33 expression further, we employed a transgenic mouse line in which GFP is expressed under this gene promoter, Il33flox-eGFP (46). In agreement with our qPCR analysis, pericytes, but no other analyzed pancreatic cell populations, expressed IL-33/GFP (Figure 1D). Additionally, we performed immunofluorescence analysis to determine IL-33 protein expression. As shown in Figure 1E, we detected IL-33 protein in pericytes but not in other islet cells. Notably, although vascular smooth muscle cells (vSMCs) are closely related to pericytes, these cells did not express IL-33 (Figure 1F). Thus, pericytes constitute a predominant source of IL-33 in the islets.
To elucidate the role of IL-33 in pancreatic pericytes, we specifically deleted this gene in these cells. To this end, we crossed Il33flox-eGFP mice, which allow Cre-dependent deletion of the Il33 gene (46), with Nkx3.2-Cre mice (45) to generate ΔIl33Pericytes (Nkx3.2-Cre;Il33flox/flox-eGFP) mice. As previously established, the expression of the Nkx3.2-Cre in the pancreas is restricted to the mural cell lineage (i.e., pericytes and vSMCs) and does not target epithelial or endothelial cells (32, 34, 50). Furthermore, Nkx3.2-Cre does not target immune cells in the pancreas, spleen, and blood (Figure S2). Immunofluorescence and qPCR analyses verified the loss of IL-33 expression in pancreatic pericytes of ΔIl33Pericytes mice (Figures 1E, G).
To determine if pericytic IL-33 is required for glucose regulation, we analyzed ΔIl33Pericytes and non-transgenic (Cre negative; Il33flox/flox-eGFP) age- and sex-matched mice. Of note, it was previously shown that mice carrying the Nkx3.2-Cre allele alone display normal glucose response (34). ΔIl33Pericytes and non-transgenic control mice had comparable blood glucose levels (Figure S3). However, i.p. glucose tolerance test (GTT) analysis revealed a significantly impaired response to glucose challenge of ΔIl33Pericytes male mice (Figure 2A). We did not detect glucose intolerance in transgenic female mice (Figure S3). As Il33 is expressed at similar levels in pancreatic pericytes of female and male mice (Figure S3), these differences likely reflect sex-dependent differences in glucose response (51). Correlating with their glucose intolerance, ΔIl33Pericytes male mice had significantly lower serum insulin levels after a glucose challenge than control mice (Figures 2B).
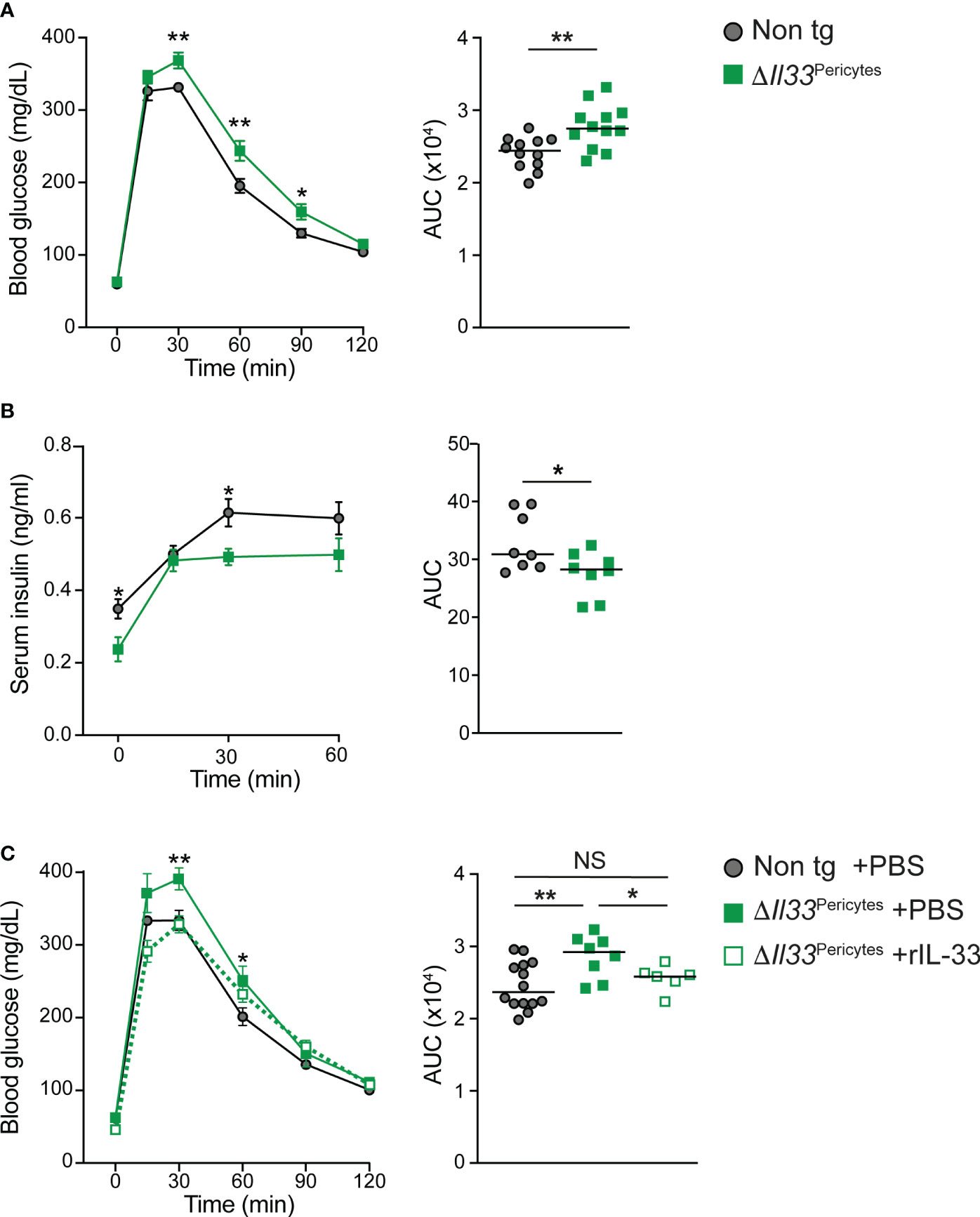
Figure 2 Impaired glucose response in mice lacking pericytic IL-33. 4-month-old ΔIl33Pericytes male mice (Nkx3.2-Cre;Il33flox/flox; green), which lack Il33 expression in their pancreatic pericytes, and non-transgenic littermates (Cre-negative; ‘Non tg’; gray) were analyzed. (A) i.p. glucose tolerance test (GTT). Left, mean (± SEM) blood glucose levels at indicated time points following glucose administration. Right, the area under the curve (AUC) of the response as shown on the left panel, when each dot represents a single mouse. N = 12. (B) In vivo GSIS. Left, mean (± SEM) serum insulin levels at indicated time points following glucose administration. Right, AUC of the response shown on the left panel, when each dot represents a single mouse. N = 8. (C) i.p. GTT after treatment of transgenic mice with exogenous IL-33. ΔIl33Pericytes mice were either i.p. injected with rIL-33 (500 ng/dose; dashed green line and empty squares) or PBS (solid green line and filled squares) every other day to obtain a total of three doses. In parallel, non-transgenic mice were i.p. injected with PBS. After an overnight fast, and 24 hours after the last treatment, mice were i.p. injected with dextrose (2 mg/g body weight), and tail vein blood glucose levels were measured at indicated times. Left, mean (± SEM) blood glucose levels at indicated time points following glucose administration. Right, AUC of the response as shown on the left panel, when each dot represents a single mouse. N = 6-14. *p<0.05; **p<0.01; NS, not significant, as compared with non-transgenic mice (Student’s t-test).
The Nkx3.2-Cre mouse line has non-pancreatic expression in the gastrointestinal mesenchyme and skeleton (45). We, therefore, analyzed for potential non-pancreatic phenotypes that may contribute to the glucose intolerance of ΔIl33Pericytes mice. Transgenic and control mice showed comparable body weight, indicating normal food uptake and digestion (Figure S3). Further, ΔIl33Pericytes male mice were insulin sensitive (Figure S3). Thus, our analysis demonstrated that pericytic IL-33 is required for proper glucose response by regulating insulin secretion.
The nuclear localization of IL-33 raised the possibility that this cytokine has a dual function and may also act cell autonomously in a receptor-independent manner (20, 21, 26). However, evidence accumulates that this cytokine acts primarily as a secreted ligand to induce signal transduction in ST2-expressing cells (22–24, 26). Notably, the loss of IL-33 affected neither islet pericytes abundance nor induced their activation (Figure S4). Further, IL-33 deficient islets had a comparable density of functional capillaries to control (Figure S4). To define if pericytic IL-33 acts as a secreted cytokine, we tested whether exogenous IL-33 rescues glucose intolerance of ΔIl33Pericytes mice. To this end, we i.p. injected rIL-33 into transgenic mice. As shown in Figure 2C, rIL-33 administration improved the glucose response of ΔIl33Pericytes mice, making it comparable to that of non-transgenic control mice. To conclude, our analysis suggests that pericytic IL-33 acts paracrine to regulate glucose response.
To define the underlying cause(s) of impaired insulin secretion of ΔIl33Pericytes mice, we analyzed their β-cells. The pancreatic mass of transgenic mice was comparable to the control when their β-cell mass was mildly, but non-significantly, lower (Figures 3A, S5). In agreement, we observed neither cell death nor expression of stress-related genes (i.e., Chop, Atf4) in ΔIl33Pericytes islets (Figure S5). Establishment of the β-cell mass relies on the proliferation of these cells in the neonatal period (52, 53). When analyzing β-cells of ΔIl33Pericytes and control pups (at postnatal day 5; p5), we observed comparable proliferation rates (Figure S5). Further, islet morphology and their β-/α-cell ratio were unaffected by the loss of pericytic IL-33 (Figures 3B, C). Thus, loss of pericytic IL-33 affected neither β-cell death nor their proliferation.
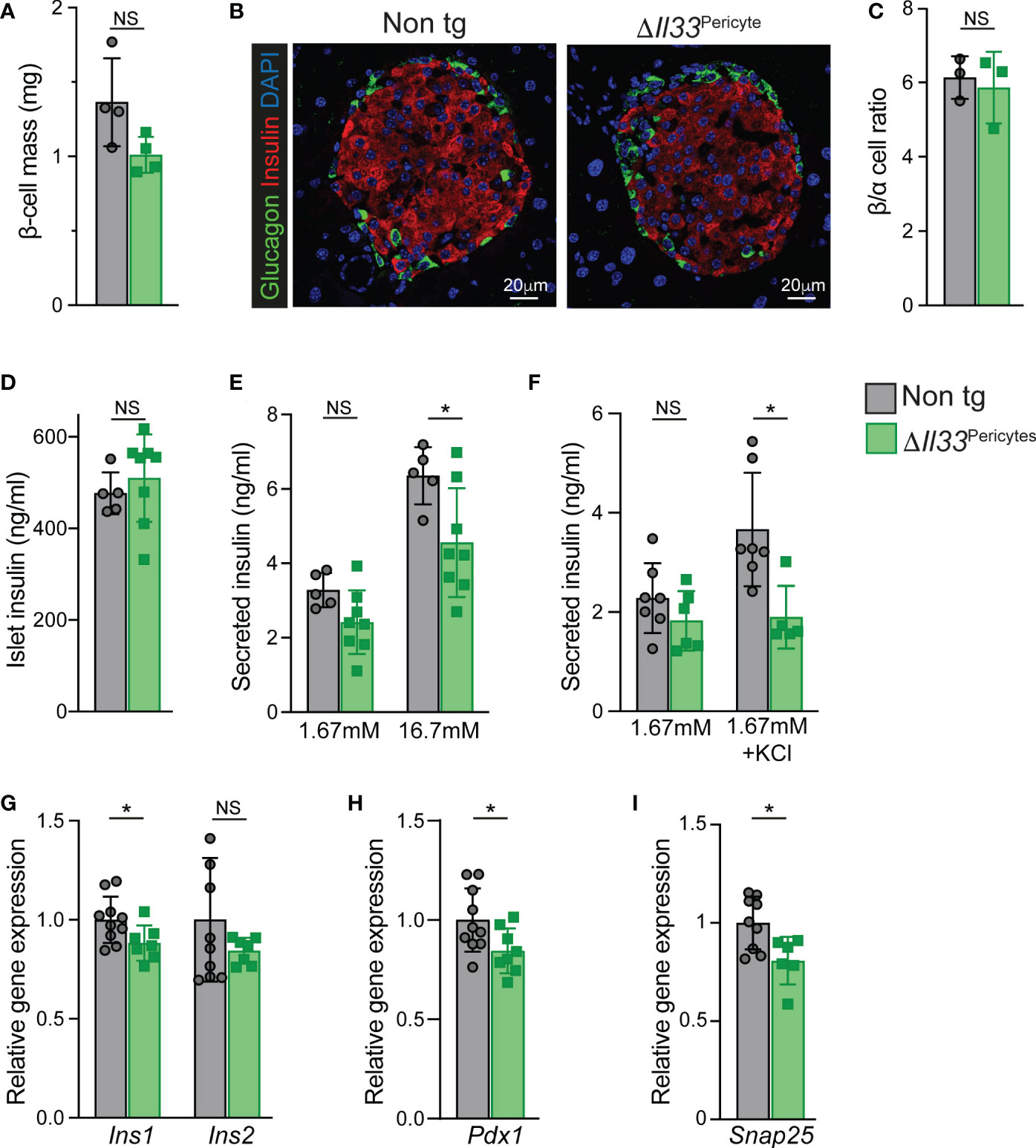
Figure 3 Impaired β-cell mass and islet insulin secretion in ΔIl33Pericytes mice. Islets and pancreatic tissues of ΔIl33Pericytes transgenic (green) and non-transgenic (‘Non tg’; gray). 4-month-old male mice were analyzed. (A) Bar diagram (mean ± SD) showing estimated β-cell mass. N = 4. (B) Pancreatic tissues of non-transgenic (left) and transgenic (right) mice were stained for insulin (red) and glucagon (green) and counterstained with DAPI (blue). Representative islets are shown. (C) Bar diagram (mean ± SD) showing the ratio between α- and β-cells (identified as glucagon and insulin-expressing cells, respectively) in adult pancreatic tissues of non-transgenic and transgenic mice. N = 3. (D) Bar diagram (mean ± SD) showing insulin content of size- and number- matched isolated islets. N = 5-8. (E) Ex vivo GSIS. Bar diagrams (mean ± SD) showing levels of insulin secreted from size-matched groups of ten isolated islets in response to 1.67 and 16.7mM glucose. N = 5-8. (F) Bar diagrams (mean ± SD) showing KCl-stimulated insulin secretion. Islets grouped into groups of ten islets were incubated with 1.67 mM glucose, either supplemented or not. N= 5-7. (G–I) Bar diagrams (mean ± SD) showing islet gene expression. Average levels in control islets were set to ‘1’. N = 6-10.*p<0.05; NS, not significant, as compared with non-transgenic control mice (Student’s t-test). Each dot represents a single mouse.
Next, we defined the islet insulin secretion ex vivo to determine blood flow-independent glucose-stimulated insulin secretion (GSIS). ΔIl33Pericytes islets displayed an impaired GSIS despite having comparable insulin content to non-transgenic islets (Figures 3D, E). Furthermore, transgenic islets secreted less insulin in response to KCl-mediated membrane depolarization (Figure 3F), pointing to abnormal islet insulin secretion in the absence of pericytic IL-33.
Gene expression analyses indicated slightly lower levels of Ins1 and Pdx1 in ΔIl33Pericytes islets but comparable levels of genes encoding other islet hormones or transcription factors (Figures 3G, H, S6). Further, we observed similar expression of genes encoding components of the GSIS machinery (Figure S6), but Snap25. This gene, which encodes a component of a SNARE complex that mediates the docking of insulin secretory granules to the plasma membrane (54), was slightly lower in ΔIl33Pericytes islets (Figure 3I). The moderately lower levels of Ins1, Pdx1, and Snap25 in IL-33-deficient islets may thus contribute to their abrogated insulin exocytosis.
Overall, our analysis indicates a requirement for IL-33 for proper insulin secretion.
Administration of rIL-33 affects islet immune composition (7). Thus, we aimed to define changes in immune cell numbers in ΔIl33Pericytes mice. First, we analyzed these mice for potential systemic effects on their immune cells by analyzing their spleen and blood. ΔIl33Pericytes and control mice had comparable portions of T cells in these tissues (Figure S7). Similarly, the levels of splenic DCs and blood monocytes (the precursors for DCs and some macrophages) were comparable in transgenic and control mice (Figure S7). Thus, we observe no systemic effect on analyzed immune cell populations in ΔIl33Pericytes mice.
Next, to define a dependency of islet immune cells on pericytic IL-33, we analyzed ΔIl33Pericytes and non-transgenic islets (Figure 4A). We found no change in the number of islet macrophages or their ratio between cells with M1 and M2-like phenotypes (Figures 4B, S7). However, the loss of pericytic IL-33 caused a significant reduction in the number of T cells and DCs in the islets (Figures 4C, D). As these cell populations do not express ST2, they are unlikely to respond to IL-33 directly but depend on ILC2s activation (7). However, the scarcity of ILC2s in islets of C57BL/6 mice hindered their reliable analysis. To conclude, our analysis indicated that pericytic IL-33 is required to establish proper islet immune cell composition locally.
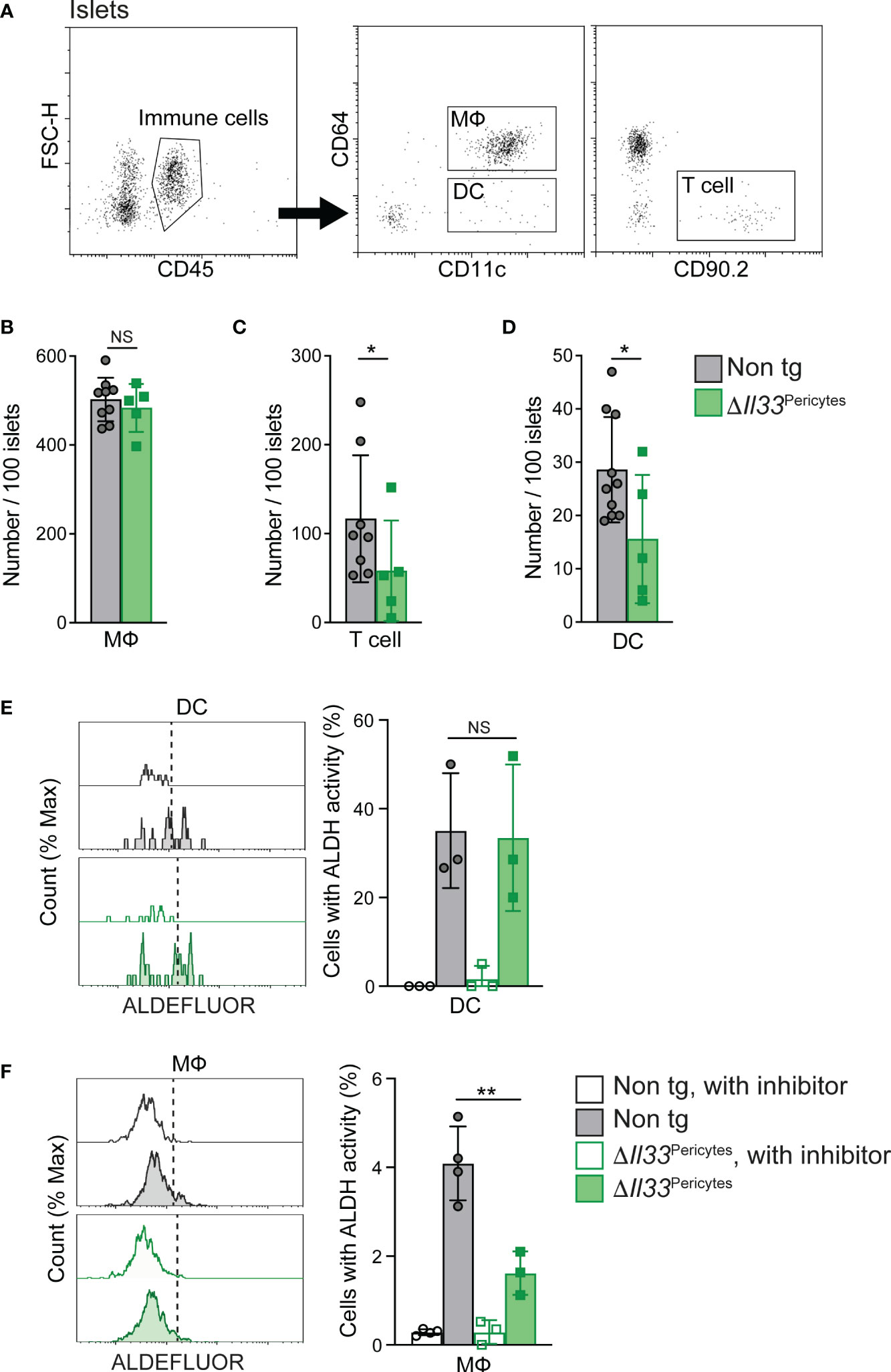
Figure 4 Loss of pericytic IL-33 affects islet immune cell number and functionIslets isolated from ΔIl33Pericytes (green) and non-transgenic (‘Non tg’; gray). 4-month-old male mice were analyzed by flow cytometry. (A) Representative dot plots indicating gate used to identify immune cells (Left panel, CD45+ cells), macrophages (MФ; Middle panel, CD45+CD11c+CD64+ cells), DCs (Middle panel, CD45+CD11c+CD64- cells), and T cells (Right panel, CD45+CD90.2+ or CD45+CD3+ cells) out of dispersed islet cells. (B–D) Bar diagrams (mean ± SD) showing the total number of macrophages (MФ; B’), T cells (C’), and DCs (D’) in 100 isolated islets. N = 5-9. (E, F) Islet cells were analyzed with ALDEFLOUR to define the frequency of DC (E’) and macrophages (F’) with ALDH activity. Islets treated with the ALDH inhibitor diethylaminobenzaldehyde (DEAB; “with inhibitor”; empty bars and histograms) were used to define the background fluorescence for each sample, as indicated in the manufacturer’s protocol. Left, representative histograms. Right, bar diagrams (mean ± SD) showing the frequencies of cells with ALDH activity. N = 3-4. *p<0.05; ** p<0.01; NS, not significant, as compared with non-transgenic control mice (Student’s t-test). Each dot represents a single sample.
rIL-33 was shown to indirectly promote islet RA synthesis to influence insulin secretion (7). RA is produced in two sequential oxidative steps: first, retinol is oxidized reversibly to retinaldehyde, and then retinaldehyde is oxidized irreversibly to RA; the latter is catalyzed by aldehyde dehydrogenases (ALDHs) (55). rIL-33 treatment enhanced ALDH activity by islet macrophages and DCs (7). To determine if pericytic IL-33 has a similar function, we measured the activity of ALDH in islet macrophages and DCs of control and ΔIl33Pericytes islets. As shown in Figure 4E, transgenic and control islets had a comparable portion of DCs with active ALDH. In contrast, the percentage of macrophages with active ALDH in ΔIl33Pericytes islets was a quarter of their portion in control islets (Figure 4F), implying that pericytic IL-33 is required for adequate RA production by these cells. Together with the overall decrease in DCs (Figure 4D), our analysis points to fewer RA-producing cells in ΔIl33Pericytes islets.
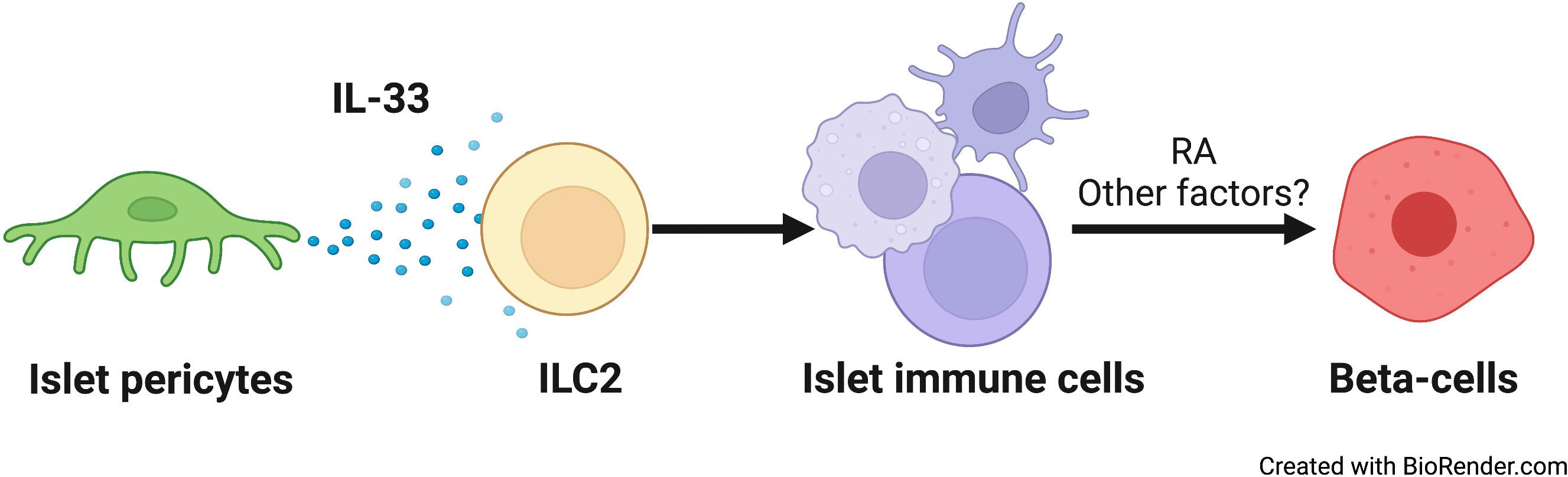
Figure 5 A schematic model of the relay effect of pericytic IL-33 on β-cells. From left to right: Islet pericytes (green) secrete IL-33. ILC2s (yellow) respond to this cytokine to affect other islet immune cells (yellow) (7). Islet T and dendritic cell numbers, as well as RA production by islet macrophages (7), are all sensitive to IL-33. IL-33-dependent RA secretion, and potentially other factors, promotes insulin secretion from β-cells.
β-Cell function relies on the islet niche. Here, we provide evidence for pericyte-regulated islet inflammation and its role in glucose homeostasis. We showed that pericytes produce IL-33 and are the predominant cell type to do so. Pericyte-selective deletion of the Il33 gene resulted in glucose intolerance due to impaired β-cell function. Pericytic IL-33 deficiency was further associated with fewer T cells and DCs in the islets. In addition, islet macrophages displayed lower ALDH activity in the absence of pericytic IL-33, implicating an impaired production of RA, which was associated with insulin secretion, by these cells. Thus, our study proposes that a cellular network comprised of pericytes and immune cells modulates β-cell function and insulin secretion (illustrated in Figure 5).
Administration of IL-33 was shown to improve glucose tolerance (7, 27). Dalmas et al. reported that systemic treatment with rIL-33 promotes insulin secretion (7). Despite some differences, our study indicated that administrated rIL-33 highly mimics the activity of the endogenous cytokine. Both exogenous and pericytic IL-33 promoted insulin secretion (7). Further, the number of islet DCs was affected by both rIL-33 and pericytic IL-33. However, while increasing IL-33 levels did not significantly affect islet T cell number (7), this cell population depends on the endogenous pericytic cytokine (Figure 5). Moreover, both endogenous and exogenous IL-33 affected RA production by islet macrophages, while RA production in DCs was only affected by increased cytokine levels (7). Thus, systemic administration of rIL-33 does not fully recapitulate its endogenous activity. Of note, systemic knockdown of Il33 did not affect the glucose response of lean mice but only of diet-induced obese animals (7). These differences highlight the importance of pancreatic-specific production of IL-33 for glucose homeostasis.
The various cell populations that make the islet microenvironment interact to ensure proper insulin secretion. Neurons were shown to regulate islet blood flow and glucose uptake by activating pericytes (38, 56). Under stress, endothelial cells regulate the number and activity of macrophages, which produce factors promoting β-cell proliferation (1, 17, 18). The various islet immune cells cooperate to induce insulin secretion when ILC2s promote recruitment and RA production by islet macrophages and DCs (7). Our study introduces an additional player in this multi-cellular network: pericytes. As pericytes express other cytokines than IL-33, these cells may modulate islet inflammation and, thus β-cell functionality by regulating the number and activity of multiple immune cell populations. Further studies into the immunoregulatory activities of pancreatic pericytes are required to decipher their role in shaping islet inflammation and subsequent glucose homeostasis.
Cells of the islet niche, including pericytes and immune cells, support postnatal β-cell development, including functional maturation and proliferation (8, 32, 33, 36, 56). We observed no significant effect on β-cell mass and neonatal proliferation in the absence of pericytic IL-33. In contrast, this deletion impaired insulin secretion from β-cells, with little effect on their mature phenotype. While the expression levels of Ins1 was slightly lower in islet lacking pericytic IL-33, this did not translate to reduced hormone levels. The expression of the transcription factor Pdx1, required for β-cell maturity, was reduced by 15% in ΔIl33Pericytes islets. While the lower Pdx1 transcript levels may point to β-cell abnormalities, it is unlikely to significantly affect β-cell gene expression or functionality. Indeed, we did not observe changes in the expression levels of genes encoding proteins required for glucose sensing and insulin production. Thus, we suggest that pericytic IL-33 affects β-cell insulin secretion with little influence on these cells’ maturation and proliferation.
Throughout the body, pericytes were shown to regulate the immune system (39–44, 57, 58). In response to tissue damage, pericytes modulate the local immune response to promote repair. Brain pericytes modulate neuroinflammation by regulating immune cell recruitment and activation (39, 41, 42, 44, 57, 58). Kidney pericytes secrete cytokines in response to tissue damage (40, 43). Pericytes may also contribute to pathologies through immune cell activation. In the tumor stroma, pericytes produce IL-33, which recruits and activates tumor-associated macrophages, ultimately promoting metastasis (59, 60). Our study ascribes an immunoregulatory ability to pancreatic pericytes mediated by IL-33 production.
The clinical onset of type 2 diabetes (T2D) occurs when pancreatic β-cells fail to secrete sufficient insulin to maintain normoglycemia in the face of insulin resistance. A growing body of evidence has established T2D as an inflammatory disease associated with deleterious islet inflammation (1, 3, 17, 61–66). In particular, T2D is associated with an increased number of immune cells, predominantly macrophages, in the islets (11, 67). The shift from beneficial to harmful islet inflammation has been suggested to play a role in the progression of diabetes (61, 68). While obesity, a leading risk of T2D, increases IL-33 production in both human and mouse islets, it also interferes with the IL-33-ILC2 axis and its ability to promote insulin secretion (7). Islet pericytes are affected by genetic and metabolic risks of T2D (34, 38, 69, 70). Thus, by highlighting their immunoregulatory role, our study proposes that pericytes may contribute to the transformation of islet inflammation associated with diabetes. However, further research is needed to understand the interplay between pericyte function and T2D fully.
We take part in the Resource Identification Initiative and use the corresponding catalog number and RRID in our manuscript.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: E-MTAB-5325 (EBI).
The animal study was reviewed and approved by Tel Aviv University Institutional Animal Care and Use Committee.
GB, AS, LS, AE, TW, and MT conducted experiments and acquired and analyzed data. LL designed and supervised research and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Israel Science Foundation (ISF; Grant agreement no. 1605/18, to LL).
This work was carried out in partial fulfillment of the requirements for a Ph.D. degree for GB from the Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1142988/full#supplementary-material
1. Brissova M, Aamodt K, Brahmachary P, Prasad N, Hong J-Y, Dai C, et al. Islet microenvironment, modulated by vascular endothelial growth factor-a signaling, promotes β cell regeneration. Cell Metab (2014) 19:498–511. doi: 10.1016/j.cmet.2014.02.001
2. Burganova G, Bridges C, Thorn P, Landsman L. The role of vascular cells in pancreatic beta-cell function. Front Endocrinol (2021) 12:667170. doi: 10.3389/fendo.2021.667170
3. Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity (2022) 55:31–55. doi: 10.1016/j.immuni.2021.12.013
4. Almaça J, Caicedo A, Landsman L. Beta cell dysfunction in diabetes: The islet microenvironment as an unusual suspect. Diabetologia (2020) 63:2076–85. doi: 10.1007/s00125-020-05186-5
5. Qian B, Yang Y, Tang N, Wang J, Sun P, Yang N, et al. M1 macrophage-derived exosomes impair beta cell insulin secretion via miR-212-5p by targeting SIRT2 and inhibiting Akt/GSK-3β/β-catenin pathway in mice. Diabetologia (2021) 64(9):2037–51. doi: 10.1007/s00125-021-05489-1
6. Xiao X, Gaffar I, Guo P, Wiersch J, Fischbach S, Peirish L, et al. M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc Natl Acad Sci (2014) 111:E1211–20. doi: 10.1073/pnas.1321347111
7. Dalmas E, Lehmann FM, Dror E, Wueest S, Thienel C, Borsigova M, et al. Interleukin-33-Activated islet-resident innate lymphoid cells promote insulin secretion through myeloid cell retinoic acid production. Immunity (2017) 47:928–942.e7. doi: 10.1016/j.immuni.2017.10.015
8. Mussar K, Pardike S, Hohl TM, Hardiman G, Cirulli V, Crisa L. A CCR2+ myeloid cell niche required for pancreatic β cell growth. JCI Insight (2017) 2:e93834. doi: 10.1172/jci.insight.93834
9. Dalmas E. Innate immune priming of insulin secretion. Curr Opin Immunol (2019) 56:44–9. doi: 10.1016/j.coi.2018.10.005
10. Ying W, Lee YS, Dong Y, Seidman JS, Yang M, Isaac R, et al. Expansion of islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab (2019) 29:457–474.e5. doi: 10.1016/j.cmet.2018.12.003
11. Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes (2007) 56:2356–70. doi: 10.2337/db06-1650
12. Chan JY, Lee K, Maxwell EL, Liang C, Laybutt DR. Macrophage alterations in islets of obese mice linked to beta cell disruption in diabetes. Diabetologia (2019) 62:993–9. doi: 10.1007/s00125-019-4844-y
13. Riley KG, Pasek RC, Maulis MF, Dunn JC, Bolus WR, Kendall PL, et al. Macrophages are essential for CTGF-mediated adult β-cell proliferation after injury. Mol Metab (2015) 4:584–91. doi: 10.1016/j.molmet.2015.05.002
14. Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab (2012) 15:518–33. doi: 10.1016/j.cmet.2012.01.023
15. Janjuha S, Singh SP, Tsakmaki A, Gharavy SNM, Murawala P, Konantz J, et al. Age-related islet inflammation marks the proliferative decline of pancreatic beta-cells in zebrafish. eLife (2018) 7:898. doi: 10.7554/elife.32965
16. Radenkovic M, Uvebrant K, Skog O, Sarmiento L, Avartsson J, Storm P, et al. Characterization of resident lymphocytes in human pancreatic islets. Clin Exp Immunol (2017) 187:418–27. doi: 10.1111/cei.12892
17. Saunders DC, Aamodt KI, Richardson TM, Hopkirk AJ, Aramandla R, Poffenberger G, et al. Coordinated interactions between endothelial cells and macrophages in the islet microenvironment promote β cell regeneration. NPJ Regener Med (2021) 6:22. doi: 10.1038/s41536-021-00129-z
18. Nackiewicz D, Dan M, Speck M, Chow SZ, Chen Y-C, Pospisilik JA, et al. Islet macrophages shift to a reparative state following pancreatic beta-cell death and are a major source of islet insulin-like growth factor-1. Iscience (2019) 23:100775. doi: 10.1016/j.isci.2019.100775
19. Whitesell JC, Lindsay RS, Olivas-Corral JG, Yannacone SF, Schoenbach MH, Lucas ED, et al. Islet lymphocytes maintain a stable regulatory phenotype under homeostatic conditions and metabolic stress. Front Immunol (2022) 13:814203. doi: 10.3389/fimmu.2022.814203
20. Cayrol C, Girard J. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol Rev (2017) 281:154–68. doi: 10.1111/imr.12619
21. Cayrol C. IL-33, an alarmin of the IL-1 family involved in allergic and non allergic inflammation: Focus on the mechanisms of regulation of its activity. Cells (2021) 11:107. doi: 10.3390/cells11010107
22. Gautier V, Cayrol C, Farache D, Roga S, Monsarrat B, Burlet-Schiltz O, et al. Extracellular IL-33 cytokine, but not endogenous nuclear IL-33, regulates protein expression in endothelial cells. Sci Rep-uk (2016) 6:34255. doi: 10.1038/srep34255
23. Travers J, Rochman M, Miracle CE, Habel JE, Brusilovsky M, Caldwell JM, et al. Chromatin regulates IL-33 release and extracellular cytokine activity. Nat Commun (2018) 9:3244. doi: 10.1038/s41467-018-05485-x
24. He Z, Chen L, Furtado GC, Lira SA. Interleukin 33 regulates gene expression in intestinal epithelial cells independently of its nuclear localization. Cytokine (2018) 111:146–53. doi: 10.1016/j.cyto.2018.08.009
25. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
26. Dwyer GK, D’Cruz LM, Turnquist HR. Emerging functions of IL-33 in homeostasis and immunity. Annu Rev Immunol (2022) 40:1–29. doi: 10.1146/annurev-immunol-101320-124243
27. Miller AM, Asquith DL, Hueber AJ, Anderson LA, Holmes WM, McKenzie AN, et al. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ Res (2010) 107:650–8. doi: 10.1161/circresaha.110.218867
28. Pavlovic S, Petrovic I, Jovicic N, Ljujic B, Kovacevic MM, Arsenijevic N, et al. IL-33 prevents MLD-STZ induction of diabetes and attenuate insulitis in prediabetic NOD mice. Front Immunol (2018) 9:2646. doi: 10.3389/fimmu.2018.02646
29. Rhee E-J, Plutzky J. Retinoid metabolism and diabetes mellitus. Diabetes Metab J (2012) 36:167–80. doi: 10.4093/dmj.2012.36.3.167
30. Armulik A, Genové G, Betsholtz C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell (2011) 21:193–215. doi: 10.1016/j.devcel.2011.07.001
31. Baek S-H, Maiorino E, Kim H, Glass K, Raby BA, Yuan K. Single cell transcriptomic analysis reveals organ specific pericyte markers and identities. Front Cardiovasc Med (2022) 9:876591. doi: 10.3389/fcvm.2022.876591
32. Sasson A, Rachi E, Sakhneny L, Baer D, Lisnyansky M, Epshtein A, et al. Islet pericytes are required for β-cell maturity. Diabetes (2016) 65:3008–14. doi: 10.2337/db16-0365
33. Epshtein A, Rachi E, Sakhneny L, Mizrachi S, Baer D, Landsman L. Neonatal pancreatic pericytes support β-cell proliferation. Mol Metab (2017) 6:1330–8. doi: 10.1016/j.molmet.2017.07.010
34. Sakhneny L, Rachi E, Epshtein A, Guez HC, Wald-Altman S, Lisnyansky M, et al. Pancreatic pericytes support β-cell function in a Tcf7l2-dependent manner. Diabetes (2018) 67:437–47. doi: 10.2337/db17-0697
35. Sakhneny L, Epshtein A, Landsman L. Pericytes contribute to the islet basement membranes to promote beta-cell gene expression. Sci Rep-uk (2021) 11:2378. doi: 10.1038/s41598-021-81774-8
36. Sakhneny L, Mueller L, Schonblum A, Azaria S, Burganova G, Epshtein A, et al. The postnatal pancreatic microenvironment guides β cell maturation through BMP4 production. Dev Cell (2021) 56:2703–2711.e5. doi: 10.1016/j.devcel.2021.08.014
37. Houtz J, Borden P, Ceasrine A, Minichiello L, Kuruvilla R. Neurotrophin signaling is required for glucose-induced insulin secretion. Dev Cell (2016) 39:329–45. doi: 10.1016/j.devcel.2016.10.003
38. Almaça J, Weitz J, Rodriguez-Diaz R, Pereira E, Caicedo A. The pericyte of the pancreatic islet regulates capillary diameter and local blood flow. Cell Metab (2018) 27:630–644.e4. doi: 10.1016/j.cmet.2018.02.016
39. Navarro R, Compte M, Álvarez-Vallina L, Sanz L. Immune regulation by pericytes: Modulating innate and adaptive immunity. Front Immunol (2016) 7:480. doi: 10.3389/fimmu.2016.00480
40. Leaf IA, Nakagawa S, Johnson BG, Cha JJ, Mittelsteadt K, Guckian KM, et al. Pericyte MyD88 and IRAK4 control inflammatory and fibrotic responses to tissue injury. J Clin Invest (2017) 127:321–34. doi: 10.1172/jci87532
41. Rustenhoven J, Jansson D, Smyth LC, Dragunow M. Brain pericytes as mediators of neuroinflammation. Trends Pharmacol Sci (2017) 38:291–304. doi: 10.1016/j.tips.2016.12.001
42. Kaushik DK, Bhattacharya A, Lozinski BM, Yong VW. Pericytes as mediators of infiltration of macrophages in multiple sclerosis. J Neuroinflamm (2021) 18:301. doi: 10.1186/s12974-021-02358-x
43. Ajay AK, Zhao L, Vig S, Fujiwara M, Thakurela S, Jadhav S, et al. Deletion of STAT3 from Foxd1 cell population protects mice from kidney fibrosis by inhibiting pericytes trans-differentiation and migration. Cell Rep (2022) 38:110473. doi: 10.1016/j.celrep.2022.110473
44. Duan L, Zhang X-D, Miao W-Y, Sun Y-J, Xiong G, Wu Q, et al. PDGFRβ cells rapidly relay inflammatory signal from the circulatory system to neurons via chemokine CCL2. Neuron (2018) 100:183–200.e8. doi: 10.1016/j.neuron.2018.08.030
45. Verzi MP, Stanfel MN, Moses KA, Kim B-M, Zhang Y, Schwartz RJ, et al. Role of the homeodomain transcription factor Bapx1 in mouse distal stomach development. Gastroenterology (2009) 136:1701–10. doi: 10.1053/j.gastro.2009.01.009
46. Han H, Roan F, Johnston LK, Smith DE, Bryce PJ, Ziegler SF. IL-33 promotes gastrointestinal allergy in a TSLP-independent manner. Mucosal Immunol (2018) 11:394–403. doi: 10.1038/mi.2017.61
47. Epshtein A, Sakhneny L, Landsman L. Isolating and analyzing cells of the pancreas mesenchyme by flow cytometry. J Vis Exp Jove (2017) (119):55344. doi: 10.3791/55344
48. Elgamal R, Kudtarkar P, Melton R, Mummey H, Benaglio P, Okino M-L, et al. An integrated map of cell type-specific gene expression in pancreatic islets. (2023). doi: 10.1101/2023.02.03.526994
49. Karlsson M, Zhang C, Méar L, Zhong W, Digre A, Katona B, et al. A single–cell type transcriptomics map of human tissues. Sci Adv (2021) 7:eabh2169. doi: 10.1126/sciadv.abh2169
50. Harari N, Sakhneny L, Khalifa-Malka L, Busch A, Hertel KJ, Hebrok M, et al. Pancreatic pericytes originate from the embryonic pancreatic mesenchyme. Dev Biol (2019) 449:14–20. doi: 10.1016/j.ydbio.2019.01.020
51. Macotela Y, Boucher J, Tran TT, Kahn CR. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes (2009) 58:803–12. doi: 10.2337/db08-1054
52. Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, et al. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab (2012) 97:3197–206. doi: 10.1210/jc.2012-1206
53. Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest (2004) 114:963–8. doi: 10.1172/jci200422098
54. Gaisano HY. Recent new insights into the role of SNARE and associated proteins in insulin granule exocytosis. Diabetes Obes Metab (2017) 19:115–23. doi: 10.1111/dom.13001
55. Rhinn M, Dollé P. Retinoic acid signalling during development. Dev (Cambridge England) (2012) 139:843–58. doi: 10.1242/dev.065938
56. Tamayo A, Gonçalves L, Rodriguez-Diaz R, Pereira E, Canales M, Caicedo A, et al. Pericyte control of blood flow in intraocular islet grafts impacts glucose homeostasis in mice. Diabetes (2022) 71(8):1679–93. doi: 10.2337/db21-1104
57. Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von BrühlM-L, et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and “instruct” them with pattern-recognition and motility programs. Nat Immunol (2013) 14:41–51. doi: 10.1038/ni.2477
58. Ogura S, Kurata K, Hattori Y, Takase H, Ishiguro-Oonuma T, Hwang Y, et al. Sustained inflammation after pericyte depletion induces irreversible blood-retina barrier breakdown. JCI Insight (2017) 2:e90905. doi: 10.1172/jci.insight.90905
59. Yang Y, Andersson P, Hosaka K, Zhang Y, Cao R, Iwamoto H, et al. The PDGF-BB-SOX7 axis-modulated IL-33 in pericytes and stromal cells promotes metastasis through tumour-associated macrophages. Nat Commun (2016) 7:11385. doi: 10.1038/ncomms11385
60. Murgai M, Ju W, Eason M, Kline J, Beury DW, Kaczanowska S, et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat Med (2017) 23:1176–90. doi: 10.1038/nm.4400
61. Donath MY, Dalmas É, Sauter NS, Böni-Schnetzler M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab (2013) 17:860–72. doi: 10.1016/j.cmet.2013.05.001
62. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol (2011) 11:98–107. doi: 10.1038/nri2925
63. Imai Y, Dobrian AD, Morris MA, Nadler JL. Islet inflammation: A unifying target for diabetes treatment? Trends Endocrinol Metab (2013) 24:351–60. doi: 10.1016/j.tem.2013.01.007
64. Marzban L. New insights into the mechanisms of islet inflammation in type 2 diabetes. Diabetes (2015) 64:1094–6. doi: 10.2337/db14-1903
65. Xiao X, Gittes GK. Concise review: New insights into the role of macrophages in β-cell proliferation. Stem Cell Transl Med (2015) 4:655–8. doi: 10.5966/sctm.2014-0248
66. Xiao Y, Shu L, Wu X, Liu Y, Cheong LY, Liao B, et al. Fatty acid binding protein 4 promotes autoimmune diabetes by recruitment and activation of pancreatic islet macrophages. JCI Insight (2021) 6:e141814. doi: 10.1172/jci.insight.141814
67. Boüni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, Prazak R, et al. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology (2009) 150:5218–29. doi: 10.1210/en.2009-0543
68. Donath MY. Multiple benefits of targeting inflammation in the treatment of type 2 diabetes. Diabetologia (2016) 59:679–82. doi: 10.1007/s00125-016-3873-z
69. Tang S-C, Chiu Y-C, Hsu C-T, Peng S-J, Fu Y-Y. Plasticity of schwann cells and pericytes in response to islet injury in mice. Diabetologia (2013) 56:2424–34. doi: 10.1007/s00125-013-2977-y
Keywords: beta-cell activity, islet vasculature, islets of Langerhans, pancreatic pericytes, Interleukin-33
Citation: Burganova G, Schonblum A, Sakhneny L, Epshtein A, Wald T, Tzaig M and Landsman L (2023) Pericytes modulate islet immune cells and insulin secretion through Interleukin-33 production in mice. Front. Endocrinol. 14:1142988. doi: 10.3389/fendo.2023.1142988
Received: 12 January 2023; Accepted: 21 February 2023;
Published: 09 March 2023.
Edited by:
Simone Baltrusch, University Hospital Rostock, GermanyReviewed by:
Florian Szymczak, Université libre de Bruxelles, BelgiumCopyright © 2023 Burganova, Schonblum, Sakhneny, Epshtein, Wald, Tzaig and Landsman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Limor Landsman, bGltb3JsQHRhdWV4LnRhdS5hYy5pbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.