 Julian Martinez-Mayer
Julian Martinez-Mayer Maria Ines Perez-Millan
Maria Ines Perez-Millan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 08 February 2023
Sec. Neuroendocrine Science
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1132787
This article is part of the Research Topic New Advances on Hypothalamus-Pituitary Stem Cell Biology View all 6 articles
Prokineticin receptor 2 (PROKR2) encodes for a G-protein-coupled receptor that can bind PROK1 and PROK2. Mice lacking Prokr2 have been shown to present abnormal olfactory bulb formation as well as defects in GnRH neuron migration. Patients carrying mutations in PROKR2 typically present hypogonadotropic hypogonadism, anosmia/hyposmia or Kallmann Syndrome. More recently variants in PROKR2 have been linked to several other endocrine disorders. In particular, several patients with pituitary disorders have been reported, ranging from mild phenotypes, such as isolated growth hormone deficiency, to more severe ones, such as septo-optic dysplasia. Here we summarize the changing landscape of PROKR2-related disease, the variants reported to date, and discuss their origin, classification and functional assessment.
Prokineticin receptor 2 (PROKR2) encodes PROKR2, a 384 amino acids long G-protein-coupled receptor (GPCR). PROKR1 and PROKR2 share around 85% sequence identity, and share ligands, called prokineticins (PROK1 and PROK2). When the ligands bind to PROKR2, the GPCR activates several signaling pathways, including IP3/Ca2+ signaling, the MAPK pathway, and cAMP signaling. Prokineticin signaling is involved in different biological functions, including mood regulation, smooth muscle contraction, angiogenesis, neurogenesis, food intake and the circadian clock (1–6). PROKR2 shows localized distribution in the central nervous system, with particularly high expression in the suprachiasmatic nucleus and the olfactory bulbs (6, 7). Prokr2−/− knockout mice present abnormal olfactory bulb formation and severe atrophy of the reproductive system in both sexes, a phenotype similar to Kallmann Syndrome (KS) in humans (8). At the hypothalamic level, immunohistochemistry revealed a lack of neuroendocrine GnRH cells (gonadotropin-releasing hormone expressing neurons) (8). The same year Dodé et al. searched for mutations in PROKR2 in patients that presented KS, linking for the first time PROKR2 with pubertal delays and smell disorders (9).
Since then, the diminishing cost of massive parallel sequencing has allowed for more broad panels and recently, whole exome sequencing of patients not only with KS or hypogonadotropic hypogonadism (HH), but with other endocrine disorders as well. As a result of this expansion in diagnostic sequencing, PROKR2 has been recently linked to pituitary diseases, such as isolated growth hormone deficiency (IGHD) (10–14) combined pituitary hormone deficiency (CPHD), pituitary stalk interruption syndrome (15, 16) and septo-optic-dysplasia (13, 17), as well as a rare case of central precocious puberty (18).
In light of this expansion in PROKR2 phenotypes and number of patients sequenced in recent years, we aimed to collect PROKR2 reported variants in clinical cases and to establish genotype-phenotype correlations where possible. We also performed an up-to-date ACMG re-classification of each variant, in association to all functional data available.
A literature search was conducted to identify studies describing human genetic variants in PROKR2, using two databases: PubMed and SCOPUS. The complete search strategy is presented in Figure 1A. From the included literature, the following data items were collected: described patient variants, their frequency, variant type, associated pathology and country of origin of the patient. Studies were excluded if patients had been reported previously or if studies were not available in English. Studies were screened by title and abstract and potentially relevant studies were reviewed by full-text analysis. The literature search identified 294 publications after the removal of duplicates. The titles and abstracts of these publications were screened and assessed for eligibility, resulting in 121 full-text articles deemed to be included in the review. Of these, after evaluation of the full text of the publications, 41 studies were excluded. Ultimately, a total of 80 studies were included in the present review (Figure 1A).
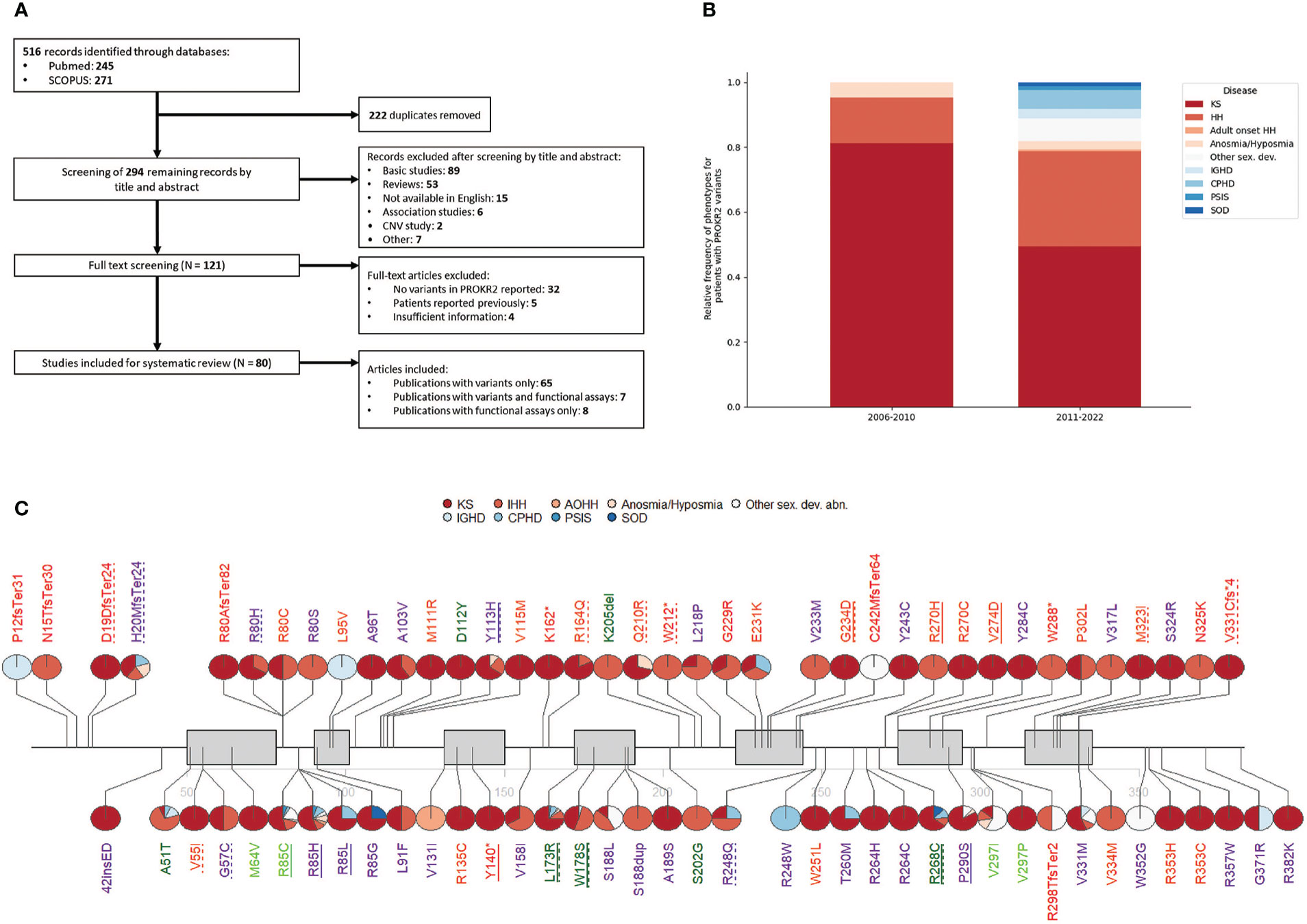
Figure 1 (A) Flow diagram showing the search and selection of studies. (B) Relative frequency of phenotypes in patients harboring PROKR2 pathogenic or likely pathogenic variants. (C) Reported PROKR2 variants. Pie charts next to each variant denote the percentage of patients carrying that variant that presented each phenotype. Each variant is coloured according to ACMG classification, where P = red, LP = orange, VUS = purple, LB = light green, B = dark green. Dotted underline means the variant was reported at least once as compound heterozygous and a full underline that it was reported at least once as homozygous. Gray boxes indicate transmembrane domains.
The absolute number of cases with PROKR2 variants in the included literature was 435, carrying a total of 453 variants, 78 of those variants being unique. KS carried the highest number of cases (236), followed by HH (108). Both diseases are highly related, as evidenced by the co-occurrence of both phenotypes in family trees (19) and their shared genetic etiology (20). Anosmia and hyposmia without pubertal delay have also been reported, accounting for a small number of cases (9, 21–24). As has been described in mice, impairment of PROKR2-mediated migration of neurons, particularly interneurons, to the olfactory bulb is sufficient to explain the anosmia or hyposmia phenotype (25). Similarly, in the hypothalamus, the neuronal migration defect can impair GnRH secretion resulting in hypogonadism (8). In recent years, reports associating PROKR2 variants to hypopituitarism and related disorders have emerged. Until now, 37 cases harboring PROKR2 mutations presented pituitary diseases: the majority were CPHD and IGHD, followed by septo-optic dysplasia and pituitary stalk interruption syndrome (Figure 1B). Given the lack of a clear molecular explanation for this novel spectrum of PROKR2-related disease, further study on the role of PROKR2 on pituitary development and disease is needed.
To date, one case of central precocious puberty has been described by Fukami et al. A female patient carried an heterozygous frameshift mutation (p.C242fsTer64), predicted to be gain of function based on Ca2+ mobilization assays. A gain of function in PROKR2 could lead to an untimely activation of the GnRH neurons and ultimately lead to CPP (18). However, another study including 31 female patients with central precocious puberty aimed to further strengthen the link between PROKR2 and CPP and no genotype-phenotype correlations were found (26). We believe the putative link deserves further study and should be taken into consideration in future precocious puberty studies.
It is important to note that the majority of patients present heterozygous PROKR2 variants. A small number of variants have been found in homozygous fashion (≃7%), and an even smaller number of cases present compound heterozygous inheritance (≃3%). Several mutations have also been found in apparently unaffected individuals, which suggests either incomplete penetrance or a digenic/oligogenic mode of inheritance in heterozygous patients (27). An example of this, is a patient presenting KS and homozygous early stop codon variant p.Y140*. Both parents are heterozygous for the same variant and are asymptomatic (28). Another two cases were reported with gain of stop codon variants: p.K162* and p.W288*, both in heterozygosity in patients with KS (29, 30). This seemingly different mode of inheritance (autosomal recessive vs autosomal dominant) for similar pathogenic variants points to either incomplete penetrance or oligogenicity as an intrinsic part of PROKR2-related disease.
With regards to oligogenicity, up to 20% of PROKR2 cases are reported together with variants in another gene: the most commonly associated genes are: FGFR1, CHD7, ANOS1, PROK2, GNRHR and SEMA3A. The first case of oligogenicity was reported by Cost-Barbosa et al., who identified a patient with KS and ligand/receptor (i.e. PROK2/PROKR2) mutations (31). Variants in these two genes affect the same pathway, a ligand that does not bind properly together with a receptor that already has impaired signaling, results in disease and explains genotype-phenotype correlation. A similar idea can explain GNRHR oligogenic cases. A weakened GnRH signaling due to PROKR2 mutations is further amplified by a defective receptor signal.
ANOS1 is involved in GnRH neuron migration, similar to PROKR2 (32). Although their mechanisms of action are different, a weakened gene regulatory network could explain how mutations in both genes at the same time could trigger a more severe disruption in neuron migration. FGFR1 plays a role in neurite extension of GnRH neurons (33). We know that despite a probable defect in GnRH neuron migration, patients carrying PROKR2 variants might still be healthy. We speculate that in patients that also carry FGFR1 mutations, the combination of migration and neurite formation defects leads to disease at an increased rate.
CHD7 has been historically linked to CHARGE syndrome (34). Although mice models show evidence for its role in olfactory bulb formation (35), its role in GnRH neurons has not been elucidated yet. Interestingly, there is also evidence that SEMA3A might play a role in CHARGE syndrome. It was shown that reduced expression CHD7 also leads to reduced expression of SEMA3A (36). Heterozygous variants in SEMA3A cause KS and HH. Recently, two cases were reported with LP variants in SEMA3A, one case with CPHD and pituitary stalk interrption syndrome, the other with CPHD and heart/skeletal abnormalities (37). Given that both genes are often reported in oligogenic cases together with PROKR2, this begs the question of whether there are also common regulatory pathways between CHD7/SEMA3A and PROKR2 that might influence GnRH neurons.
Overall oligogenicity is probably an important player in PROKR2-related disease and could help explain at least part of the incompletely penetrant phenotypes typically seen in these patients.
Most studies were performed in Asia (25 studies), Europe (22 studies) and North America (14 studies). In particular, the USA and China together amounted to more than half of the total patients studied. The low number of sequencing studies in Africa and South America is concerning, considering both continents together represent over 20% of the world population. Studies performed in these continents were also restricted geographically, to either northern Africa or just Argentina and Brazil in the case of South America. Variants circulating in these populations are currently an unexploited resource that could help reclassify known variants, as well as allow for the discovery of novel ones.
Some variants have a clear regional predominance. The p.W178S variant seems to be most prevalent in continental Asia (mainly in China, although it has also been reported in South Korea and Japan) (10, 30, 38–47). Nevertheless the variant has also been described in the USA and France, in patients of undisclosed ethnicity (9, 29). The high presence of this variant in Asian patients as well as its relatively high frequency in the east Asian population on GnomAD (1/500), point to it being a regional polymorphism (27).
Other missense variants unique to Asia are p.G57C, p.A103V and p.Y113H (23, 29–31, 38, 41–44, 46–51). The variants p.G57C, p.A103V were found in two and five patients with KS/HH respectively and both are classified as variants of uncertain significance (VUS) with a frequency of 1/1000 in this population. The variant p.Y113H is reported in twenty patients with a big range of phenotypes (KS, HH, CPHD, IGHD, SOD) and even though bioinformatic predictions mark it as pathogenic and functional studies show loss of function (LoF) (Table 1), it has high frequency in Gnomad (1/700 in East Asia), leading to its VUS classification (Figure 1C). Taking into consideration the total number of patients included in this review and their absence in other populations in GnomAD, it seems likely that these variants are indeed local. Further research is needed to determine whether these variants are pathogenic with incomplete penetrance and whether there are foundational effects.
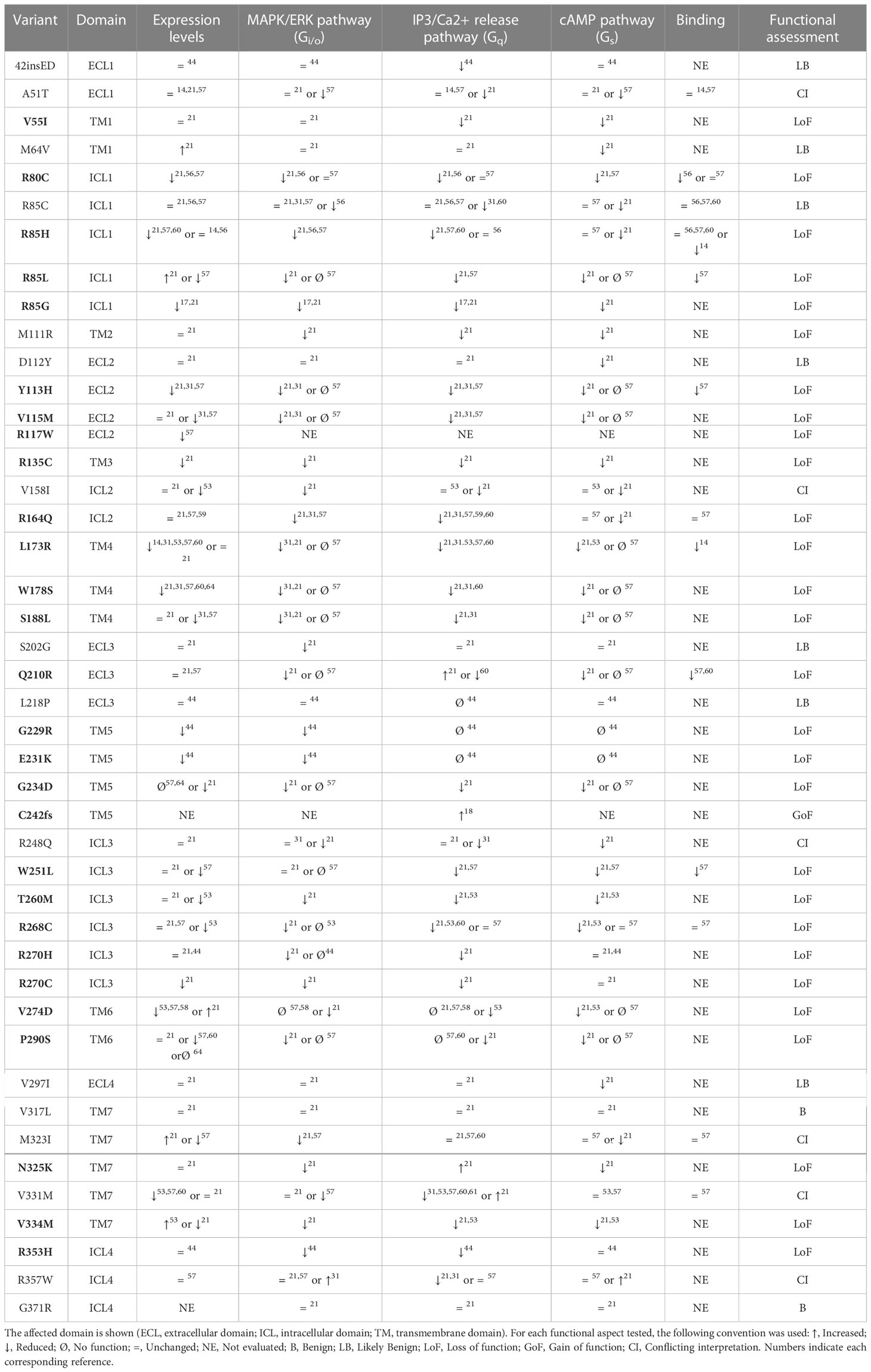
Table 1 Variants in PROKR2 and their functional assessment.
Another variant with an interesting regional pattern is p.P290S. This variant was originally reported by Dodé et al. in French patients with KS, but it has been found at an increased frequency in Maghrebian patients. France’s population also has a high population percentage of immigrants with maghrebian origins (52), meaning the variants found in french cohorts could be attributed to maghrebian immigration. Functional testing indicates the variant is a LoF (Table 1). Nevertheless the variant is currently classified as a VUS, given its high frequency in Gnomad (0.0004).
Several patients have been reported with heterozygous nonsense variants, either due to a premature stop or a frameshift that results in a truncated protein. Although these variants are deemed to be causative of the disease, the relatively high prevalence of p.H20MfsTer24 in the European and Latino/Admixed population is intriguing (27). A founder effect may be at play in the European population, given that patients with these mutations have only been reported in Italy and France (9, 53). The symptoms of these patients were quite diverse (Figure 1C), ranging from KS/HH to CPHD and anosmia. It is also interesting to note that transcripts harboring such variants would likely be subject to nonsense mediated decay (NMD). NMD can trigger genetic compensation through increased transcription of adapting genes (54). PROKR2 and PROKR1 share ligands and their distinct actions are thought to arise from specificity at the level of tissue expression more so than functional differences (55). Given their high homology degree, incomplete penetrance due to genetic compensation for this type of variants seems plausible and warrants further investigation.
PROKR2 is able to activate different G-proteins common to GPCR receptors, namely Gαi/o, Gαq and Gαs. Each of these pathways can be linked to canonical signal transduction pathways: MAPK/ERK pathway (Gαi/o), IP3/Ca2+ release pathway (Gαq) and cAMP pathway (Gαs). Several PROKR2 functional testing methodologies have been applied for each different pathway.
To study MAPK/ERK signaling two assays have been used. The most commonly used is the Egr1-luciferase assay (21, 31, 56). In this assay cells are transfected with wilde-type and mutant PROKR2 expression plasmids, together with luciferase expression vectors. In this case the plasmid carries binding sites for EGR1, a downstream effector of p-ERK. After 24 hours the cells are then stimulated with a range of PROK2 concentrations, to generate a dose-response curve. The second assay is very similar, but instead of luciferase the final outcome measured is p-ERK protein levels through Western Blot analysis (44, 57). Variants are then evaluated on the percentage of activation that they elicit compared to wilde-type.
The IP3/Ca2+ release pathway (Gαq) was measured in several ways. The most frequent assay was aequorin-based (17, 31, 44, 58, 59). This method also relies on transfection, where PROKR2 WT and mutant expression plasmids are co-transfected with an aequorin expression plasmid. Upon posterior activation of PROKR2 with PROK2, aequorin is activated by calcium release, producing light at 470 nm as an output signal. Another frequently used method was the Fluo-4 calcium reporter method (57, 60, 61). In this case Fluo-4 is added to the transfected cells, where it permeates the membrane. Once PROKR2 is activated, the subsequent intracellular calcium release triggers light emission by Fluo-4 in the 500-525 nm range.
In contrast, there are not many widely adopted cAMP pathway functional assays. Luciferase based assays have been used as described before, making use of the pGlosensor-22F cAMP sensor (21, 44). In this assay an engineered fused Luciferase is used, which carries a cAMP binding domain, and only emits light after a conformational change induced by cAMP binding (62). Other alternative functional assays have also been used, such as the commercial cAMP HTRF assay or RIA (53, 57).
Localization and expression of the receptor has been evaluated for multiple variants, mostly through Western Blot and immunofluorescence. Specific ligand binding affinity has also been tested for some variants using radioligand binding assays (14, 56, 57, 60, 63). For all reported variants where functional testing was available we took every assay into consideration and summarized the results in Table 1.
We labeled each variant as Benign (B), Likely benign (LB), conflicting interpretation (CI) or gain/loss of function (GoF/LoF), according to a qualitative consensus among studies. We considered a variant to be LoF if at least two assays showed a defect, or one assay was proven to cause loF and was tested by at least two independent publications, without conflicting results. Variants with just one defective assay were considered Likely Benign (LB), while those with none were considered Benign (B). It should be emphasized that functional assay results are not synonymous with pathogenicity. A clear example of this are variants L173R and R268C, which should be considered Benign according to ACMG criteria, despite their seemingly impaired signaling. They both present relatively high frequency in the general population, as well as homozygous individuals in GnomAD, but they could be possible risk factors given their increased frequencies in individuals with disease (64).
Despite some contradictions, generally functional assays are of great relevance for classifying most variants. Examples of this are the variants that fall in the transmembrane domain five (TM5): p.G229R, p.E231K and p.G234D. The functionally available data points to all of them being LoF, and this evidence leads to their classification as Pathogenic (P) or Likely Pathogenic (LP) (Figure 1C; Table 1). Two further variants in that same domain, p.V233M and p.Y243C, lack functional evidence and currently remain VUS. Remarkably over three quarters of the remaining VUS also lack functional testing. This exemplifies just how critical functional testing is for PROKR2 variant assessment and subsequent genetic counseling.
Since the initial report from Dodé et al. in 2006, many studies have reported PROKR2 genetic alterations in patients with neuroendocrine disorders. A vast majority of these patients have insufficiencies in the gonadal axis, commonly with hyposmia or anosmia, while a minority present diverse forms of hypopituitarism.
This review provides a comprehensive catalog of published PROKR2 variants described to date. Our catalog of predicted pathogenic variants may serve as a guideline for future genetic studies. In summary, this review represents a useful resource for clinicians overseeing PROKR2-related diseases and will help scientists to focus on the missing gaps of the literature to further cement its role in diseases concerning the hypothalamus-pituitary axis.
All authors planned and wrote the paper. JM-M designed the figures. All authors contributed to the article and approved the submitted version.
PICT 2018 4239 to MIPM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Li JD, Hu WP, Zhou QY. Disruption of the circadian output molecule prokineticin 2 results in anxiolytic and antidepressant-like effects in mice. Neuropsychopharmacology (2008) 34:367–73. doi: 10.1038/npp.2008.61
2. Li M, Bullock CM, Knauer DJ, Ehlert FJ, Zhou QY. Identification of two prokineticin cDNAs: Recombinant proteins potently contract gastrointestinal smooth muscle. Mol Pharmacol (2001) 59:692–8. doi: 10.1124/mol.59.4.692
3. Monnier J, Samson M. Prokineticins in angiogenesis and cancer. Cancer Lett (2010) 296:144–9. doi: 10.1016/j.canlet.2010.06.011
4. Ng KL, Li JD, Cheng MY, Leslie FM, Lee AG, Zhou QY. Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science (2005) 308:1923–7. doi: 10.1126/science.1112103
5. Gardiner JV, Bataveljic A, Patel NA, Bewick GA, Roy D, Campbell D, et al. Prokineticin 2 is a hypothalamic neuropeptide that potently inhibits food intake. Diabetes (2009) 59:397–406. doi: 10.2337/db09-1198
6. Cheng MY, Bullock CM, Li C, Lee AG, Bermak JC, Belluzzi J, et al. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachiasmatic nucleus. Nature (2002) 417:405–10. doi: 10.1038/417405a
7. Li JD, Hu WP, Boehmer L, Cheng MY, Lee AG, Jilek A, et al. Attenuated circadian rhythms in mice lacking the prokineticin 2 gene. J Neurosci (2006) 26:11615–23. doi: 10.1523/JNEUROSCI.3679-06.2006
8. Matsumoto SI, Yamazaki C, Masumoto KH, Nagano M, Naito M, Soga T, et al. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. PNAS (2006) 103:4140–5. doi: 10.1073/pnas.0508881103
9. Dodé C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, et al. Kallmann syndrome: Mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet (2006) 2:e175. doi: 10.1371/journal.pgen.0020175
10. Ahn J, Oh J, Suh J, Song K, Kwon A, Chae HW, et al. Next-generation sequencing-based mutational analysis of idiopathic short stature and isolated growth hormone deficiency in Korean pediatric patients. Mol Cell Endocrinol (2022) 544:111489. doi: 10.1016/j.mce.2021.111489
11. He D, Li Y, Yang W, Chen S, Sun H, Li P, et al. Molecular diagnosis for growth hormone deficiency in Chinese children and adolescents and evaluation of impact of rare genetic variants on treatment efficacy of growth hormone. Clinica Chimica Acta (2022) 524:1–10. doi: 10.1016/j.cca.2021.11.021
12. Parsons SJH, Wright NB, Burkitt-Wright E, Skae MS, Murray PG. A heterozygous microdeletion of 20p12.2-3 encompassingPROKR2andBMP2in a patient with congenital hypopituitarism and growth hormone deficiency. Am J Med Genet (2017) 173:2261–7. doi: 10.1002/ajmg.a.38306
13. McCabe MJ, Gaston-Massuet C, Gregory LC, Alatzoglou KS, Tziaferi V, Sbai O, et al. Variations inPROKR2, but NotPROK2, are associated with hypopituitarism and septo-optic dysplasia. J Clin Endocrinol Metab (2013) 98:E547–57. doi: 10.1210/jc.2012-3067
14. Reynaud R, Jayakody SA, Monnier C, Saveanu A, Bouligand J, Guedj AM, et al. PROKR2 variants in multiple hypopituitarism with pituitary stalk interruption. J Clin Endocrinol Metab (2012) 97:E1068–73. doi: 10.1210/jc.2011-3056
15. Jullien N, Saveanu A, Vergier J, Marquant E, Quentien MH, Castinetti F, et al. Clinical lessons learned in constitutional hypopituitarism from two decades of experience in a large international cohort. Clin Endocrinol (2020) 94:277–89. doi: 10.1111/cen.14355
16. McCormack SE, Li D, Kim YJ, Lee JY, Kim SH, Rapaport R, et al. Digenic inheritance of PROKR2 and WDR11 mutations in pituitary stalk interruption syndrome. J Clin Endocrinol Metab (2017) 102:2501–7. doi: 10.1210/jc.2017-00332
17. Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, et al. Genetic overlap in kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab (2012) 97:E694–9. doi: 10.1210/jc.2011-2938
18. Fukami M, Suzuki E, Izumi Y, Torii T, Narumi S, Igarashi M, et al. Paradoxical gain-of-function mutant of the G-protein-coupled receptor PROKR2 promotes early puberty. J Cell Mol Med (2017) 21:2623–6. doi: 10.1111/jcmm.13146
19. Sarfati J, Guiochon-Mantel A, Rondard P, Arnulf I, Garcia-Pinãero A, Wolczynski S, et al. A comparative phenotypic study of kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. J Clin Endocrinol Metab (2010) 95:659–69. doi: 10.1210/jc.2009-0843
20. Topaloğlu AK. Update on the genetics of idiopathic hypogonadotropic hypogonadism. J Clin Res Pediatr Endocrinol (2018) 9(Suppl 2):113–22. doi: 10.4274/jcrpe.2017.S010
21. Cox KH, Oliveira LMB, Plummer L, Corbin B, Gardella T, Balasubramanian R, et al. Modeling mutant/wild-type interactions to ascertain pathogenicity of PROKR2 missense variants in patients with isolated GnRH deficiency. Hum Mol Genet (2017) 27:338–50. doi: 10.1093/hmg/ddx404
22. Alkelai A, Olender T, Dode C, Shushan S, Tatarskyy P, Furman-Haran E, et al. Next-generation sequencing of patients with congenital anosmia. Eur J Hum Genet (2017) 25:1377–87. doi: 10.1038/s41431-017-0014-1
23. Gu WJ, Zhang Q, Wang YQ, Yang GQ, Hong TP, Zhu DL, et al. Mutation analyses in pedigrees and sporadic cases of ethnic han Chinese kallmann syndrome patients. Exp Biol Med (Maywood) (2015) 240:1480–9. doi: 10.1177/1535370215587531
24. Moya-Plana A, Villanueva C, Laccourreye O, Bonfils P, de Roux N. PROKR2 and PROK2 mutations cause isolated congenital anosmia without gonadotropic deficiency. Eur J Endocrinol (2013) 168:31–7. doi: 10.1530/EJE-12-0578
25. Wen Y, Zhang Z, Li Z, Liu G, Tao G, Song X, et al. The PROK2/PROKR2 signaling pathway is required for the migration of most olfactory bulb interneurons. J Comp Neurol (2019) 527:2931–47. doi: 10.1002/cne.24719
26. Aiello F, Cirillo G, Cassio A, Di Mase R, Tornese G, Umano GR, et al. Molecular screening of PROKR2 gene in girls with idiopathic central precocious puberty. Ital J Pediatr (2021) 47:5. doi: 10.1186/s13052-020-00951-z
27. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
28. Abreu AP, Trarbach EB, de Castro M, Frade Costa EM, Versiani B, Matias Baptista MT, et al. Loss-of-Function mutations in the genes encoding prokineticin-2 or prokineticin receptor-2 cause autosomal recessive kallmann syndrome. J Clin Endocrinol Metab (2008) 93:4113–8. doi: 10.1210/jc.2008-0958
29. Costa-Barbosa FA, Balasubramanian R, Keefe KW, Shaw ND, Al-Tassan N, Plummer L, et al. Prioritizing genetic testing in patients with kallmann syndrome using clinical phenotypes. J Clin Endocrinol Metab (2013) 98:E943–53. doi: 10.1210/jc.2012-4116
30. Li S, Zhao Y, Nie M, Yang Y, Hao M, Mao J, et al. Comparison of clinical characteristics and spermatogenesis in CHH patients caused by PROKR2 and FGFR1 mutations. Reprod Sci (2021) 28:3219–27. doi: 10.1007/s43032-021-00609-z
31. Cole LW, Sidis Y, Zhang C, Quinton R, Plummer L, Pignatelli D, et al. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: Molecular genetics and clinical spectrum. J Clin Endocrinol Metab (2008) 93:3551–9. doi: 10.1210/jc.2007-2654
32. Cariboni A, Pimpinelli F, Colamarino S, Zaninetti R, Piccolella M, Rumio C, et al. The product of X-linked kallmann’s syndrome gene (KAL1) affects the migratory activity of gonadotropin-releasing hormone (GnRH)-producing neurons. Hum Mol Genet (2004) 13:2781–91. doi: 10.1093/hmg/ddh309
33. Oleari R, Massa V, Cariboni A, Lettieri A. The differential roles for neurodevelopmental and neuroendocrine genes in shaping GnRH neuron physiology and deficiency. IJMS (2021) 22:9425. doi: 10.3390/ijms22179425
34. Vissers LELM, van Ravenswaaij CMA, Admiraal R, Hurst JA, de Vries BBA, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet (2004) 36:955–7. doi: 10.1038/ng1407
35. Layman WS, McEwen DP, Beyer LA, Lalani SR, Fernbach SD, Oh E, et al. Defects in neural stem cell proliferation and olfaction in Chd7 deficient mice indicate a mechanism for hyposmia in human CHARGE syndrome. Hum Mol Genet (2009) 18:1909–23. doi: 10.1093/hmg/ddp112
36. Ufartes R, Schwenty-Lara J, Freese L, Neuhofer C, Möller J, Wehner P, et al. Sema3a plays a role in the pathogenesis of CHARGE syndrome. Hum Mol Genet (2018) 27:1343–52. doi: 10.1093/hmg/ddy045
37. Budny B, Zemojtel T, Kaluzna M, Gut P, Niedziela M, Obara-Moszynska M, et al. SEMA3A and IGSF10 are novel contributors to combined pituitary hormone deficiency (CPHD). Front Endocrinol (2020) 11. doi: 10.3389/fendo.2020.00368
38. Wang Y, Qin M, Fan L, Gong C. Correlation analysis of genotypes and phenotypes in Chinese Male pediatric patients with congenital hypogonadotropic hypogonadism. Front Endocrinol (Lausanne) (2022) 13:846801. doi: 10.3389/fendo.2022.846801
39. Sun T, Xu W, Chen Y, Niu Y, Wang T, Wang S, et al. Reversal of idiopathic hypogonadotropic hypogonadism in a Chinese male cohort. Andrologia (2022) 54(11):e14583. doi: 10.1111/and.14583
40. Sugisawa C, Taniyama M, Sato T, Takahashi Y, Hasegawa T, Narumi S. Biallelic PROKR2 variants and congenital hypogonadotropic hypogonadism: a case report and a literature review. Endocr J (2022) 69:831–8. doi: 10.1507/endocrj.EJ21-0779
41. Liu Q, Yin X, Li P. Clinical, hormonal, and genetic characteristics of 25 Chinese patients with idiopathic hypogonadotropic hypogonadism. BMC Endocr Disord (2022) 22(1):30. doi: 10.1186/s12902-022-00940-9
42. Zhang L, Gao Y, Du Q, Liu L, Li Y, Dey SK, et al. Genetic profiles and three-year follow-up study of Chinese males with congenital hypogonadotropic hypogonadism. J Sexual Med (2021) 18:1500–10. doi: 10.1016/j.jsxm.2021.07.001
43. Tang SY, Li Z, Zhang J, Zhu XB, Li P, Lu JQ, et al. Whole exome sequencing and trio analysis to broaden the variant spectrum of genes in idiopathic hypogonadotropic hypogonadism. Asian J Androl (2021) 23:288. doi: 10.4103/aja.aja_65_20
44. Zhao Y, Wu J, Jia H, Wang X, Zheng R, Jiang F, et al. PROKR2 mutations in idiopathic hypogonadotropic hypogonadism: Selective disruption of the binding to aGα-protein leads to biased signaling. FASEB J (2018) 33:4538–46. doi: 10.1096/fj.201801575R
45. Kim JH, Seo GH, Kim GH, Huh J, Hwang IT, Jang JH, et al. Targeted gene panel sequencing for molecular diagnosis of kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Exp Clin Endocrinol Diabetes (2018) 127:538–44. doi: 10.1055/a-0681-6608
46. Zhou C, Niu Y, Xu H, Li Z, Wang T, Yang W, et al. Mutation profiles and clinical characteristics of Chinese males with isolated hypogonadotropic hypogonadism. Fertility Sterility (2018) 110:486–95.e5. doi: 10.1016/j.fertnstert.2018.04.010
47. Wang Y, Gong C, Qin M, Liu Y, Tian Y. Clinical and genetic features of 64 young male paediatric patients with congenital hypogonadotropic hypogonadism. Clin Endocrinol (2017) 87:757–66. doi: 10.1111/cen.13451
48. Ha JH, Lee S, Kim Y, Moon JI, Seo J, Jang JH, et al. Kallmann syndrome with a Tyr113His PROKR2 mutation. Medicine (2017) 96:e7974. doi: 10.1097/MD.0000000000007974
49. Shaw ND, Seminara SB, Welt CK, Au MG, Plummer L, Hughes VA, et al. Expanding the phenotype and genotype of female GnRH deficiency. J Clin Endocrinol Metab (2011) 96:E566–76. doi: 10.1210/jc.2010-2292
50. Yuan H, Deng S, Gao W, Li H, Yuan M. Identification of MMACHC and PROKR2 mutations causing coexistent cobalamin c disease and kallmann syndrome in a young woman. Metab Brain Dis (2021) 36:447–52. doi: 10.1007/s11011-020-00654-8
51. Zhang Q, He H, Janjua MU, Wang F, Yang Y, Mo Z, et al. Identification of two novel mutations in three Chinese families with kallmann syndrome using whole exome sequencing. Andrologia (2020) 52(7):e13594. doi: 10.1111/and.13594
52. Tribalat M. Mariages «mixtes» et immigration en France. Eps (2009), 203–14. doi: 10.4000/eps.3657
53. Libri DV, Kleinau G, Vezzoli V, Busnelli M, Guizzardi F, Sinisi AA, et al. Germline prokineticin receptor 2 (PROKR2) variants associated with central hypogonadism cause differential modulation of distinct intracellular pathways. J Clin Endocrinol Metab (2014) 99:E458–63. doi: 10.1210/jc.2013-2431
54. El-Brolosy MA, Kontarakis Z, Rossi A, Kuenne C, Günther S, Fukuda N, et al. Genetic compensation triggered by mutant mRNA degradation. Nature (2019) 568:193–7. doi: 10.1038/s41586-019-1064-z
55. Martin C, Balasubramanian R, Dwyer AA, Au MG, Sidis Y, Kaiser UB, et al. The role of the prokineticin 2 pathway in human reproduction: Evidence from the study of human and murine gene mutations. Endocrine Rev (2010) 32:225–46. doi: 10.1210/er.2010-0007
56. Abreu AP, Noel SD, Xu S, Carroll RS, Latronico AC, Kaiser UB. Evidence of the importance of the first intracellular loop of prokineticin receptor 2 in receptor function. Mol Endocrinol (2012) 26:1417–27. doi: 10.1210/me.2012-1102
57. Sbai O, Monnier C, Dodé C, Pin J, Hardelin J, Rondard P. Biased signaling through G-protein-coupled PROKR2 receptors harboring missense mutations. FASEB J (2014) 28:3734–44. doi: 10.1096/fj.13-243402
58. Zhou XT, Chen DN, Xie ZQ, Peng Z, Xia KD, Liu HD, et al. Functional analysis of the distal region of the third intracellular loop of PROKR2. Biochem Biophys Res Commun (2013) 439:12–7. doi: 10.1016/j.bbrc.2013.08.039
59. Peng Z, Tang Y, Luo H, Jiang F, Yang J, Sun L, et al. Disease-causing mutation in PKR2 receptor reveals a critical role of positive charges in the second intracellular loop for G-protein coupling and receptor trafficking. J Biol Chem (2011) 286:16615–22. doi: 10.1074/jbc.M111.223784
60. Monnier C, Dodé C, Fabre L, Teixeira L, Labesse G, Pin JP, et al. PROKR2 missense mutations associated with kallmann syndrome impair receptor signalling activity. Hum Mol Genet (2008) 18:75–81. doi: 10.1093/hmg/ddn318
61. Su MT, Lin SH, Chen YC, Wu LW, Kuo PL. Prokineticin receptor variants (PKR1-I379V and PKR2-V331M) are protective genotypes in human early pregnancy. Reproduction (2013) 146:63–73. doi: 10.1530/REP-13-0043
62. Buccioni M, Marucci G, Dal Ben D, Giacobbe D, Lambertucci C, Soverchia L, et al. Innovative functional cAMP assay for studying G protein-coupled receptors: application to the pharmacological characterization of GPR17. Purinergic Signalling (2011) 7:463–8. doi: 10.1007/s11302-011-9245-8
63. Chen DN, Ma YT, Liu H, Zhou QY, Li JD. Functional rescue of kallmann syndrome-associated prokineticin receptor 2 (PKR2) mutants deficient in trafficking. J Biol Chem (2014) 289:15518–26. doi: 10.1074/jbc.M114.556381
Keywords: kallmann syndrome, hypogonadotropic hypogonadism, genetic diagnosis, pituitary, functional assay
Citation: Martinez-Mayer J and Perez-Millan MI (2023) Phenotypic and genotypic landscape of PROKR2 in neuroendocrine disorders. Front. Endocrinol. 14:1132787. doi: 10.3389/fendo.2023.1132787
Received: 27 December 2022; Accepted: 25 January 2023;
Published: 08 February 2023.
Edited by:
Frédéric Gachon, The University of Queensland, AustraliaReviewed by:
Carol F Elias, University of Michigan, United StatesCopyright © 2023 Martinez-Mayer and Perez-Millan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Ines Perez-Millan, bWlwbWlsbGFuQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.