 Pedro A. Villa
Pedro A. Villa Nancy M. Lainez
Nancy M. Lainez Carrie R. Jonak
Carrie R. Jonak Sarah C. Berlin
Sarah C. Berlin Djurdjica Coss
Djurdjica Coss- Division of Biomedical Sciences, School of Medicine, University of California, Riverside, CA, United States
Introduction: Mutations in the Fragile X Messenger Ribonucleoprotein 1 (FMR1) gene cause Fragile X Syndrome, the most common monogenic cause of intellectual disability. Mutations of FMR1 are also associated with reproductive disorders, such as early cessation of reproductive function in females. While progress has been made in understanding the mechanisms of mental impairment, the causes of reproductive disorders are not clear. FMR1-associated reproductive disorders were studied exclusively from the endocrine perspective, while the FMR1 role in neurons that control reproduction was not addressed.
Results: Here, we demonstrate that similar to women with FMR1 mutations, female Fmr1 null mice stop reproducing early. However, young null females display larger litters, more corpora lutea in the ovaries, increased inhibin, progesterone, testosterone, and gonadotropin hormones in the circulation. Ovariectomy reveals both hypothalamic and ovarian contribution to elevated gonadotropins. Altered mRNA and protein levels of several synaptic molecules in the hypothalamus are identified, indicating reasons for hypothalamic dysregulation. Increased vascularization of corpora lutea, higher sympathetic innervation of growing follicles in the ovaries of Fmr1 nulls, and higher numbers of synaptic GABAA receptors in GnRH neurons, which are excitatory for GnRH neurons, contribute to increased FSH and LH, respectively. Unmodified and ovariectomized Fmr1 nulls have increased LH pulse frequency, suggesting that Fmr1 nulls exhibit hyperactive GnRH neurons, regardless of the ovarian feedback.
Conclusion: These results reveal Fmr1 function in the regulation of GnRH neuron secretion, and point to the role of GnRH neurons, in addition to the ovarian innervation, in the etiology of Fmr1-mediated reproductive disorders.
Introduction
Mutations in the Fragile X Messenger Ribonucleoprotein 1 (FMR1) gene lead to the most common genetic form of intellectual disability and autism, called Fragile X Syndrome (FXS) (1, 2). In addition to intellectual impairment, FMR1 gene mutations are also associated with reproductive disorders, such as early menopause in females, and macroorchidism in males (3–6). The FMR1 gene encodes FMR protein (FMRP), an mRNA binding protein that regulates protein levels of its target genes (7, 8). Target mRNAs bound by FMRP encode a variety of proteins, including transcription factors that regulate other genes (9–11). Mutation of this gene that causes FXS entails full expansion of the unstable CGG trinucleotide repeats (>200) that leads to hypermethylation, silencing of the gene and the loss of FMRP. Premutation, in a range of 50-200 repeats, causes Fragile X-associated Tremor/Ataxia Syndrome (FXTAS) and exhibits reduced FMRP levels. While the mechanisms of intellectual impairments following FMRP loss are beginning to emerge, mechanisms of reproductive disorders are not known. Although FMRP is ubiquitous, it is highly abundant in the nervous system. In the brain, FMRP binds mRNAs that encode synaptic proteins, contributing to cognitive dysfunctions in FXS (9–11). The effect of FMR1 mutations on the cortex and hippocampus have been analyzed (12, 13), however, how mutations affect hypothalamic functions have not been examined. Herein, we investigated the effects of FMRP loss in reproduction, specifically on a population of hypothalamic neurons that regulate the hypothalamus-pituitary-gonadal axis.
Given that FMR1 gene mutations are also associated with reproductive disorders (3–6), combined with increasing infertility rates (14, 15), it is critical to examine FMR1 role in the reproductive axis. Reproduction is controlled by gonadotropin-releasing hormone (GnRH)-secreting neurons from the hypothalamus (16, 17). GnRH is secreted in a pulsatile fashion into the hypophysial-portal system, to regulate synthesis and secretion of pituitary gonadotropin hormones, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), which in turn regulate gonadal function (18). Synchronization of GnRH secretion is determined by an upstream regulatory network. One population of such afferent neurons are GABAergic neurons from the mediobasal hypothalamus (19–25). Although GABA is the primary inhibitory neurotransmitter in the brain, GABA is excitatory for GnRH neurons (20, 26–28). Changes in GnRH neuron connectivity and its innervation control neuropeptide pulsatile secretion and consequently gonadotropin hormone levels.
Contrary to the hypothalamus, gonadal roles of FMRP have been analyzed in several reports. Macroorchidism in men affected with FXS (5) is caused by increased Sertoli cell proliferation during development (29). In women, mutations in the FMR1 gene comprise the largest number of cases with the known genetic causes of early cessation of reproductive function (4, 30, 31). FMR1 exhibits X-linked dominant inheritance pattern and associated disorders have a high penetrance (32), which contributes to their high incidence (33). Premature ovarian failure is an infertility disorder affecting 1% of reproductive age women that lose ovarian function before the age of 40 (34, 35). Women with early menopause experience not only early infertility, but increased risk of cardiovascular disease, osteoporosis and depression (36). Premutations of the FMR1 gene are primarily associated with the early loss of reproductive function, which may be due to the much higher prevalence of premutations compared to full mutations. Although to a lesser degree, premutation also causes lower levels of FMRP, and thus, lack of FMRP may cause pathologies in both premutation and full mutation (37). Contrary to men, premature ovarian failure in women with FMR1 mutations is not a developmental disorder. Previous studies in mice (31) and humans (6, 38–40), did not find differences in primordial follicle numbers, indicating that early follicular development is not affected. Women affected with mutations have elevated FSH (40, 41), which stimulates cyclic recruitment of a wave of ovarian follicles into the growing pool in each menstrual or estrous cycle (42). However, it is still not clear how FRM1 mutations lead to early infertility.
With a high prevalence of FMR1 mutations in women of child-bearing age, investigating FMRP-mediated effects on reproductive function will help us understand the mechanism of early menopause. Previous studies that examined ovarian hormone levels or follicle development, failed to explain early infertility. In this study, we first examined the reproductive function in female Fmr1 KO mice and determined that the mouse model lacking the Fmr1 gene mimics the reproductive deficits observed in women with FMR1 mutations associated with reduced FMRP levels. We further observed that Fmr1 KO mice had larger litters and more corpora lutea in the ovaries, but normal primordial follicle count, indicating increased recruitment. In addition, we found an increase in gonadotropin levels, inhibin B and steroid hormone levels, which indicated that ovarian feedback is present. We further analyzed ovarian vascularization and innervation, and observed changes that may explain increased FSH. Ovariectomy however, revealed a hypothalamic contribution to increased LH. We then analyzed alteration in gene expression and protein levels of critical hypothalamic molecules, and examined the consequence of FMRP loss on hypothalamic GnRH neurons, that have been neglected for their potential role in the etiology of early menopause, and show changes in GABAergic innervation. We show higher LH pulse frequency, before and after ovariectomy, which indicates higher GnRH neuron activity, correlating with alterations in GnRH neuron connectivity. Therefore, central mechanisms, most likely via alterations in the activity of GnRH neurons, in addition to ovarian effects, can contribute to increased gonadotropins and higher recruitment of follicles leading to reproductive dysfunction in women with FMR1 mutations.
Materials and methods
Animals
All animal procedures were performed with the approval from the University of California (Riverside, CA) Animal Care and Use Committee and in accordance with the National Institutes of Health Animal care and Use Guidelines. Breeding pairs of FVB.129P2-Fmr1tm1Cgr/J (Fmr1 KO) and their congenic controls (WT) mice were obtained from Jackson Laboratories and bred in-house. Mice were maintained under a 12-h light, 12-h dark cycle and received food and water ad libitum. Since we were interested in determining the mechanisms of premature ovarian failure in women with mutations in the FMR1 gene, only female mice were used for our studies. Estrous cycle stage was determined with vaginal smears and females were collected in a specific estrous cycle stage, as indicated for each analysis. For fertility studies, Fmr1 KO females and WT controls were housed with WT males and their litters and numbers of pups per litter were recorded. Ovariectomy was performed, as described before, using 8-week-old mice (43). Animals were allowed to recover and seven days later blood was collected for hormone analyses as described below.
Hormone assay
Previous studies using FXS mouse models demonstrated that these mice have heightened response to stress and altered levels of glucocorticoids (44), which can lead to a decrease in luteinizing hormone (LH) levels (45, 46). To reduce stress, animals were acclimated by daily handling and tail massage for two weeks prior to hormone measurements. For LH measurements, blood was collected from the tail and analyzed by an in house ultra-sensitive ELISA. The capture monoclonal antibody (anti-bovine LH beta subunit, 518B7) was provided by Janet Roser, University of California. The detection polyclonal antibody (rabbit LH antiserum, AFP240580Rb) was provided by the National Hormone and Peptide Program (NHPP). HRP-conjugated polyclonal antibody (goat anti-rabbit) was purchased from DakoCytomation (Glostrup, Denmark; D048701-2). Mouse LH reference prep (AFP5306A; NHPP) was used as the assay standard. Assay sensitivity is 0.016 ng/ml, while intra-assay coefficient of variation is 2.2% and inter-assay coefficient of variation was 7.3% at the low end of the curve. Other hormone assays were performed by the University of Virginia, Ligand Core using serum that was obtained from the inferior vena cava and serum prepared per their instructions. The University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core is supported by the Eunice Kennedy Shriver NICHD/NIH Grant R24HD102061. FSH was assayed by RIA using reagents provided by Dr. A.F. Parlow and the National Hormone and Peptide Program, as previously described (47). Mouse FSH reference prep AFP5308D was used for assay standards. Inhibin and steroid hormone levels were analyzed using validated commercially available assays, information for which can be found on the core’s website: http://www.medicine.virginia.edu/research/institutes-and-programs/crr/lab-facilities/assay-methods-page and reported in (48). Limits of detection were 2.4 ng/ml for FSH, 300 pg/ml for progesterone, and 100 pg/ml for testosterone. Intra- and inter-assay coefficients of variation were 6.9%/7.5%, 6.0%/11.4% and 4.4%/6.4% for FSH, progesterone (P) and testosterone (T), respectively. Each animal used for each hormone analysis is represented as a dot in the figure.
Pulsatile LH levels
Pulsatile secretion of LH strictly corresponds to GnRH secretion (49, 50). LH is used as an indicator of GnRH secretion, since GnRH secretion into median eminence cannot be measured in mice. To ascertain if LH or GnRH neuron secretion is affected, we measured LH pulses and used an ultrasensitive ELISA assay for LH that allows for LH measurement in 5 µL of whole blood (51). Mice were acclimated for 2 weeks by daily tail massage. 10µL of blood was collected every 8 min for 3 hours from the tail vain (45, 52). LH levels were analyzed using ELISA described above. LH amplitude was determined by subtracting the LH value at the peak from the basal value prior to the onset of the pulse and averaged for each mouse. Mean LH concentration was calculated by averaging LH values, while pulse frequency was determined using freeware DynPeak algorithm (53).
Nanostring analysis of hypothalamic gene expression
8-week old female mice in diestrus were perfused with ice cold PBS, brains rapidly removed and flash frozen in isopentane on dry ice. Coronal brain sections of 500 μm were obtained using vibratome, hypothalamus dissected, RNA isolated using the RNAqueous®-Micro Kit (Ambion) and quantified using Nanodrop. Gene expression in 50 ng RNA per sample was analyzed using the Nanostring instrument as described before (54), according to manufacturer’s instruction, with the nCounter Mouse Neuroinflammation Panel (770 genes, gene list available at the manufacturer’s website). The panel was customized with an addition of 30 custom probes for: Gnrh1, Kiss1, Kiss1r, Pdyn, Tac2, Tac3r, Agrp, Npy, Pomc, Cart, Mc3r, Mc4r, Hcrt, Ghrh, Crh, Trh, Oxt, Avp, Prl, Prlr, Adcyap1, Slc6a3, Slc32a1, Slc17a6, Th, Lif, Lifr, Gabra5, Gabra1, Gabrg2. Only samples with an RNA integrity number RIN over 7 were used after passing QC, with no imaging, binding, positive control, or CodeSet content normalization flags. Data analysis was performed using nSolver Analysis Software 4.0, including nCounter Advanced Analysis (version 2.0.115). Genes with the expression lower than the limit of detection after background subtraction, and compared to negative controls included in the panel, were excluded. Seven housekeeping control genes that are included in the panel, were used for normalization. A heatmap of differentially expressed genes (DEG) was created using Heatmapper software from University of Alberta (Edmonton, Canada (55);). Results are plotted in the Volcano plot as log fold change vs. log p-value, and genes with changes higher than 20% were indicated with colors in the figures: red indicates genes higher in KO compared to WT, while green indicates genes that are higher in the WT compared to KO. Genes with significant changes in expression are indicated above the dashed line. Gene ontology (GO) enrichment analysis of the DEG genes was performed using the ShinyGo 0.76.3 platform (South Dakota State University (56)). False discovery rate (FDR) cutoff was 0.05, with the pathway minimum set to 10. Data is deposited in GEO repository (accession number GSE222723).
qPCR analysis
Total RNA was extracted from ovaries using Trizol (Invitrogen, CA), and from hypothalamus and pituitary using the RNAqueous®-Micro Kit (Ambion), quantified using Nanodrop and the same amount per sample reverse transcribed using Superscript IV (Invitrogen, CA). To dissect the hypothalamus, coronal brain sections of 300 μm were obtained using vibratome. Sections containing anterior hypothalamus and posterior hypothalamus were used to dissect the 1 mm x 1 mm mediobasal portion for RNA isolation. Conditions and primers were reported before (57–61).
Histological analyses and immunohistochemistry
WT controls and Fmr1 KO mice were anesthetized, perfused with 20 ml PBS and 20 ml 4% paraformaldehyde; and tissues were collected. Ovaries were fixed in 4% paraformaldehyde, embedded in paraffin, and cut to 20 μm sections. Slides were deparaffinized in xylene, rehydrated and H&E stain was performed to count ovarian follicles. For ovarian vasculature and innervation studies, frozen floating sections were stained with antibody to CD31 (1:2000 dilution, 553370, BD Biosciences) or with antibody to tyrosine hydroxylase (TH, 1:5000, ab112, Abcam) for 48 hours at 4°C, followed by overnight incubation with goat anti-rat IgG-Alexa 488 (1:2000, A11006, Vector Laboratories, Burlingame, CA) or goat anti-rabbit IgG-Alexa 488 (1:1000, A11034, Vector Laboratories, Burlingame, CA), respectively. Vascularization of corpora lutea and antral follicles was quantified by the mean fluorescent intensity (MFI) using Fiji ImageJ. Ovarian innervation was quantified by counting the number of neuronal projections in direct contact with follicles or corpora lutea.
Hypothalami were sectioned to 100 μm sections. Sections containing organum vasculosum laminae terminalis (OVLT) where GnRH neurons are located, were blocked and stained for GnRH using rabbit anti-GnRH antibodies (1:10,000 dilution) kindly provided by Greg Anderson (University of Otago; Dunedin, New Zealand (62)), GABAγ2 receptor subunit (1:10,000 dilution, guinea pig anti-GABAγ2, Synaptic systems 224 004), VGAT (1:5,000, mouse anti-VGAT, Synaptic systems 131 011) for 72 hours at 4°C. After PBST washes, slides were incubated overnight at 4°C with secondary antibodies goat anti-rabbit IgG-Alexa 488 (1:1000, A11034, Vector Laboratories, Burlingame, CA); anti-mouse IgG-Alexa 594 (1:1000, A11032, Vector Laboratories, Burlingame, CA); anti-guinea pig–biotin (1:1000, BA-7000) followed by streptavidin-Cy5 (1:1000, 434316, Vector Laboratories, Burlingame, CA). Secondary antibody-only controls were performed to determine antibody specificity. To determine puncta density, we followed our established protocol as previously published (60, 63–66). Puncta were counted in the individual neurons, by an investigator blinded to the group, where at least 45 μm of the axon proximal to soma can be observed using z-stack acquired by confocal Leica SP2 microscope. At least 15-20 individual neurons from 4-5 different sets of mice were counted. 3–D reconstruction was performed by Imaris software (Bitplane, Inc; Concord, MA).
Immunostaining for FMRP was performed using antigen retrieval methods, as previously described (67). Slices were stained overnight with mouse anti-FMRP (1:1000; Developmental Studies Hybridoma Bank, catalog #2F5-1-s, RRID: AB_10805421). Secondary antibody was donkey anti-mouse Alexa 594 (1:300; Molecular Probes, A-21202). Slices were mounted on slides with Vectashield mounting medium containing DAPI (Vector Laboratories, H-1200).
Western blot
Whole cell lysates were obtained from the dissected hypothalami from WT controls and Fmr1 knockout mice. The same amount of protein from each sample, determined by Bradford assay, was resolved on SDS-PAGE, transferred on nitrocellulose membrane and probed for: GABAγ2 receptor subunit (1:1000, 14104-1-AP, Proteintech), NMDAR1 (1:1000, 32-0500 Invitrogen), NMDAR2B (1:1000, 07-632 EMD Millipore), postsynaptic density protein 95 (PSD-95; 1:1000, 3409, Cell Signaling), microtubule-associated protein 2 (MAP2; 1:5000, ab5392, Abcam) or β-tubulin (1:1000, sc-9104, Santa Cruz Biotechnology). Bands were quantified using ChemiDoc imaging system (Bio-Rad, Hercules, CA).
Statistical analyses
Statistical differences between WT control and Fmr1 KO mice (p < 0.05) were determined by t-test, or ANOVA when appropriate, followed by Tukey’s post-hoc test for multiple comparisons using Prism software (GraphPad, CA).
Results
Fmr1 knockout female mice experience early cessation of reproductive function
Since women with FMR1 mutation experience increased risk of early menopause, to begin investigating the role of FMRP in reproductive function, we first determined if the lack of FMRP in female Fmr1 KO mice can mimic reproductive dysfunctions observed in women with reduced levels of FMRP due to FMR1 mutation. In affected people, full expansion of the unstable CGG trinucleotide repeats (>200) leads to the loss of FMRP and FXS. CGG expansion in a range of 50-200 repeats, or FMR1 premutation, exhibits reduced FMRP levels and FXTAS. Due to differential methylation between human and mouse genes, the Fmr1 KO is a widely used mouse model to study Fragile X Syndrome and is considered a better model than putative mimics of the CGG repeat expansion (68, 69). Our study showed that Fmr1 KO female mice experienced early vaginal opening, an external sign of puberty in mice (Figure 1A, FMR1, Fmr1 knock-out (KO); WT, wild-type control; each point represents a mouse, while bars represent group average). Fmr1 KO mice demonstrated vaginal opening at postnatal day 29 (p29), compared to p31 in WT controls. At 8 weeks of age, we paired Fmr1 KO females and control WT females, with control males and recorded birth dates and number of litters, and the number of pups in each litter, until they stopped reproducing. There was no difference in the length of time between litters (Figure 1B) or in the estrous cycle duration (Figure 1C). We determined that Fmr1 KO female mice exhibited early cessation of reproductive function, which is similar to early menopause in women with FMR1 mutation or premutation (Figure 1D, each point represents a mouse) (70, 71). Fmr1 KO mice stopped having litters at 5.5 months of age and an average age of the last litter was p163 (FMR1, black squares), compared to p263 for WT control females (WT, open circles). Similar to the penetrance in women with mutations in the FMR1 gene (32), we observed different degrees of premature cessation of reproductive function. Fmr1 KO females split into two groups based on the age at last litter: females more severely affected with the mutation that stopped reproducing at p97, and less severely affected females that had the last litter at p221. Nonetheless, the difference between controls (p263) and less affected females (p221) was also statistically significant. We counted the number of pups in the litters and determined that Fmr1 KO females had larger first three litters (Figure 1E). The average litter size of the first litter was 10.6 pups for Fmr1 KO and 7.5 pups for controls; the average size of the second litter was 11.4 pups for Fmr1 KO and 8.8 pups for controls, and the average size of the third litter was 10.8 pups for Fmr1 KO and 7.1 pups for controls. However, while control females continued to produce litters for at least 10 litters, the number of Fmr1 KO females that continued to produce litters decreased after each litter, and only 5 out of 11 Fmr1 KO females had a fourth litter, while none had eighth litter. There was no difference in weight at any age between KO and control females and thus, change in reproductive function does not stem from a difference in weight. Therefore, similar to women with a mutation in the FMR1 gene, Fmr1 KO female mice experience early cessation of reproductive function.
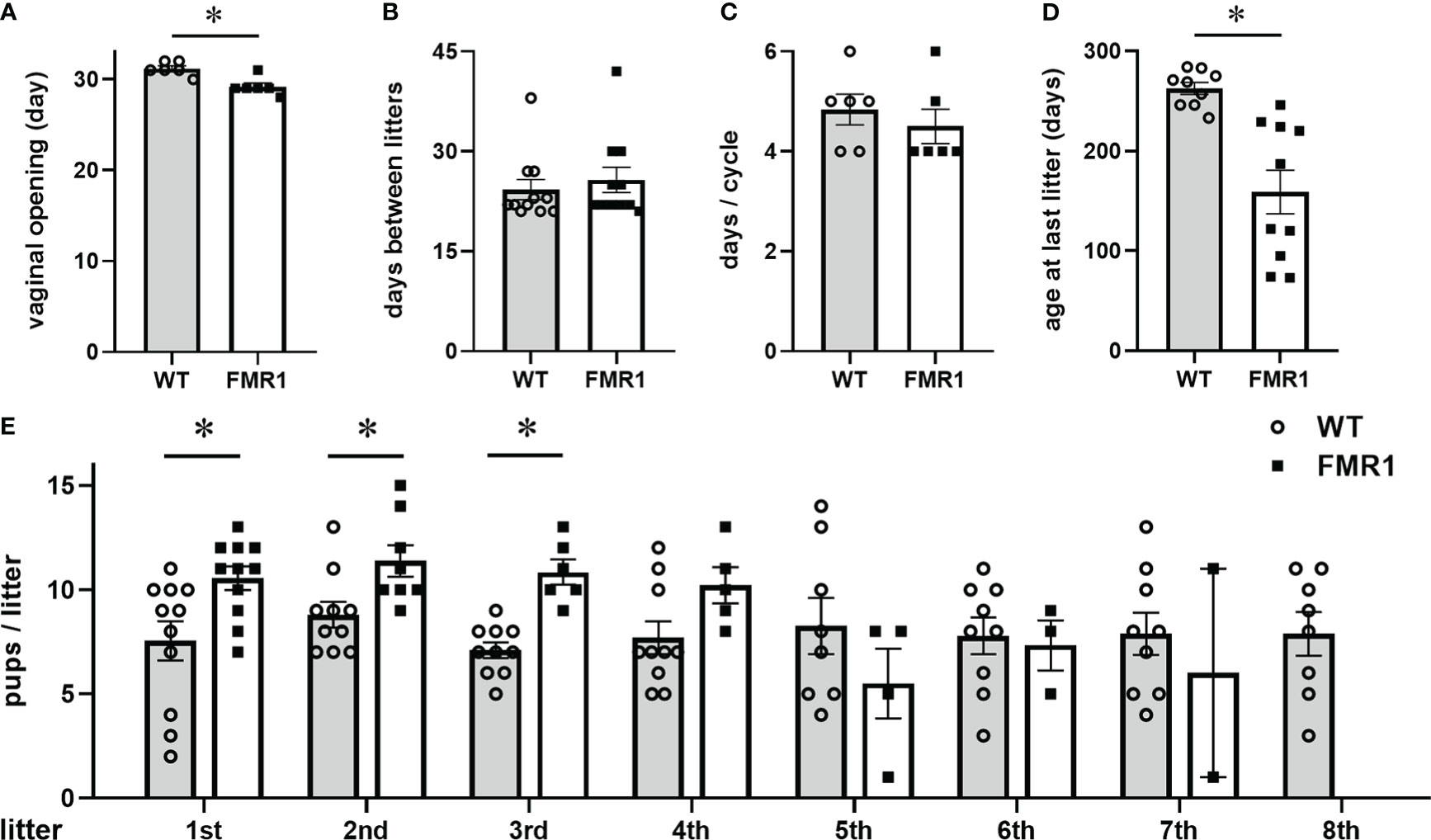
Figure 1 Fmr1 knockout (KO) females stop reproducing early. (A) Fmr1 KO females (FMR1, white bars represent group mean +/- standard error, and each black square represents one animal) experience early vaginal opening, an external sign of puberty, at postnatal day (p) 29, compared to wild type controls at p31 (WT, gray bars represent mean +/- standard error, open circles represent each animal); (B) Fmr1 KO females have litters at the same rate as WT controls; (C) No difference in the length of the estrous cycle; (D) Determined by the age at the last litter, Fmr1 KO females stop reproducing early at p163, compared to WT controls at p263; Each point represents one animal, and bars represent group means +/- standard error. Statistical significance (*, p < 0.05) was determined with t-test followed by Tukey’s post hoc test. (E) Fmr1 KO females have more pups per litter in the first three litters. Each circle represents one litter produced by WT females, while each square represents one litter produced by Fmr1 KO females, with number of pups in 1st – 8th litter indicated. The number of Fmr1 KO females that continue to produce litters, indicated by squares, decreases gradually. Bars represent group means +/- standard error, * indicates statistically significant difference between WT and KO.
To examine if early cessation of reproductive function is due to initially diminished ovarian reserve or to an accelerated loss of follicles, we counted the number of primordial follicles in pre–pubertal females at 3 weeks of age (p21, Figure 2A count, and Figure 2D representative images). Three equal size areas of the ovarian cortex were selected per mouse, primordial follicles counted, and the numbers were averaged for each mouse. There was no difference in the number of primordial follicles between Fmr1 KO and WT females, indicating that development of the initial pool is not affected by the Fmr1 loss. The analysis of the number of corpora lutea (CL) in the ovaries at 6 weeks of age (p42) showed that Fmr1 KO females had over 4 times more corpora lutea than WT controls. Fmr1 KO had 10.2 average number of corpora lutea per ovary compared to 2.2 corpora lutea per ovary in controls (Figure 2B count, Figure 2E representative images). Given that Fmr1 KO females exhibited early vaginal opening, to confirm that the increase in corpora lutea in KO females does not stem from early puberty, we counted the number of corpora lutea at 9 weeks of age (at p63; Figure 2C). At p63, an average number of corpora lutea per ovary was significantly higher in Fmr1 KO females (8.6 corpora lutea) compared to control WT mice (5.4 corpora lutea). Together, these results demonstrate that Fmr1 KO female mice stop reproducing early, have the same primordial follicle pool, but increased number of corpora lutea corresponding to the larger litter size in young animals, indicating potentially higher recruitment to the growing pool in young animals.
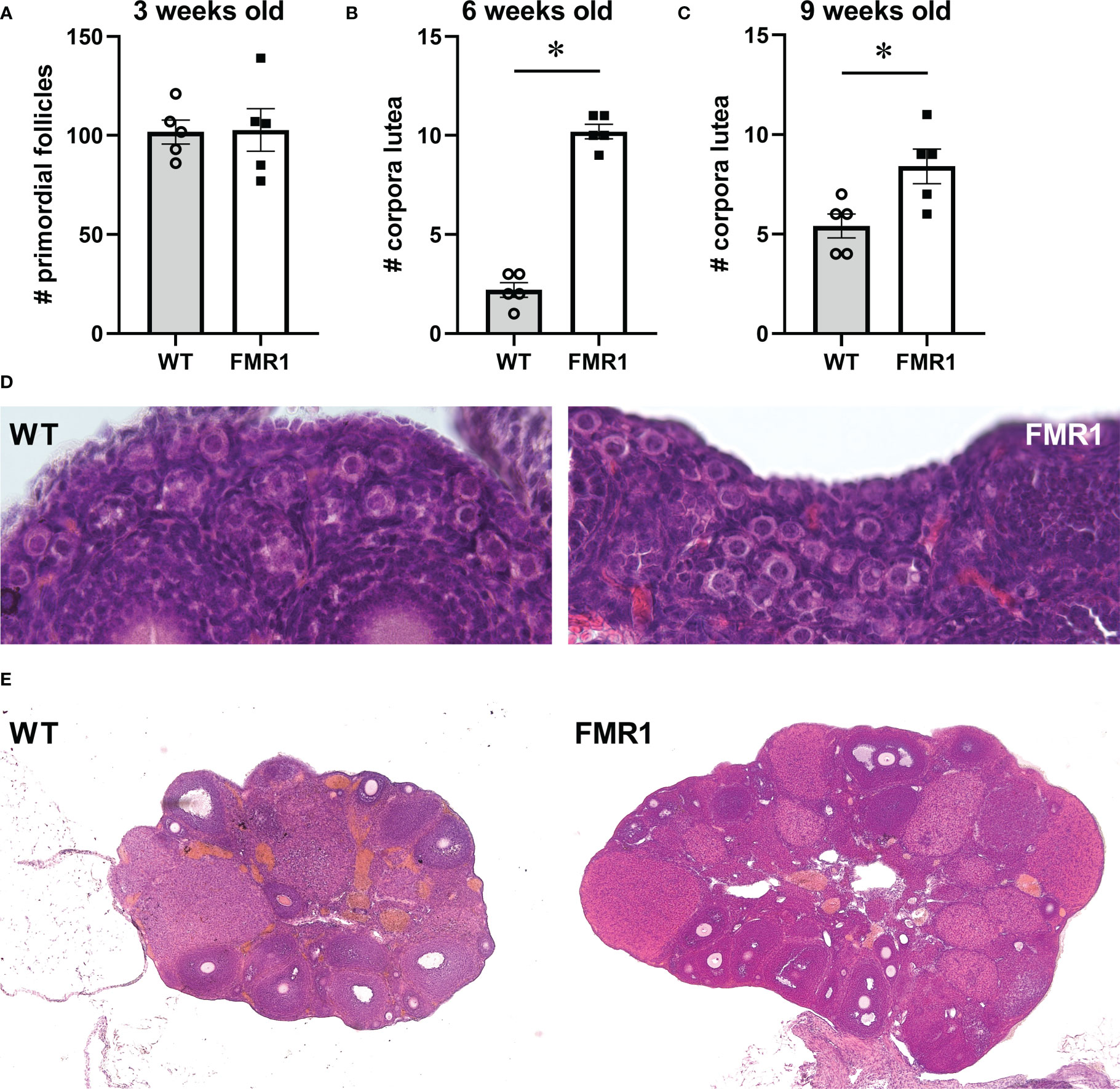
Figure 2 Ovarian histology demonstrates more corpora lutea in young Fmr1 KO mice. Fmr1 KO females (FMR1, white bars represent group mean +/- standard error while each black square represents one animal) were compared to wild type controls (WT, gray bars represent mean +/- standard error, each open circle represents one animal). (A) Primordial follicles were counted at 3 weeks of age. Each point represents one mouse, and an average of 4 separate 1x10-8 m2 areas in the ovary cortex of each mouse. (B, C) corpora lutea were counted at 6 (B) and 9 weeks of age (C) throughout each ovary. (D) representative images of ovaries from 3-week old mice to observe primordial follicles (630x). (E) representative images of ovaries at 6 weeks of age to observe numbers of corpora lutea (40x). Statistical significance, indicated with * (p < 0.05) was determined with t-test followed by Tukey’s post hoc test.
Fmr1 KO females exhibit increased gonadotropin and ovarian hormone levels
Given that ovarian function is regulated by gonadotropin hormones from the pituitary, we analyzed the hormone levels in female mice in diestrus at 9 weeks of age (Figure 3). LH and FSH levels were significantly higher in diestrus Fmr1 KO. LH doubled in KO to 0.84 ng/ml from 0.42 ng/ml in controls. Serum FSH was also higher with 4.2 ng/ml in KO, compared to 2.3 ng/ml in diestrus controls (Figure 3A). These results may demonstrate that high FSH leads to higher recruitment of follicles in the growing pool, which together with high LH results in more corpora lutea. Since LH and FSH β-subunit transcription, that is unique for each hormone, precedes changes in hormone concentration in the circulation, and fluctuations in mRNA levels in the gonadotrope correlate with concentration of the hormones (18), we analyzed pituitary mRNA levels (Supplemental Figure 1). Both Lhb (LHβ) and Fshb (FSHβ) expression was increased in Fmr1 KO mice, while expression of the common Cga (αGSU, Glycoprotein hormones common subunit alpha), Gnrhr (GnRH receptor) or other pituitary hormones was unchanged. These results indicate that concentrations of LH and FSH in the circulation correlate with β-subunit mRNA levels.
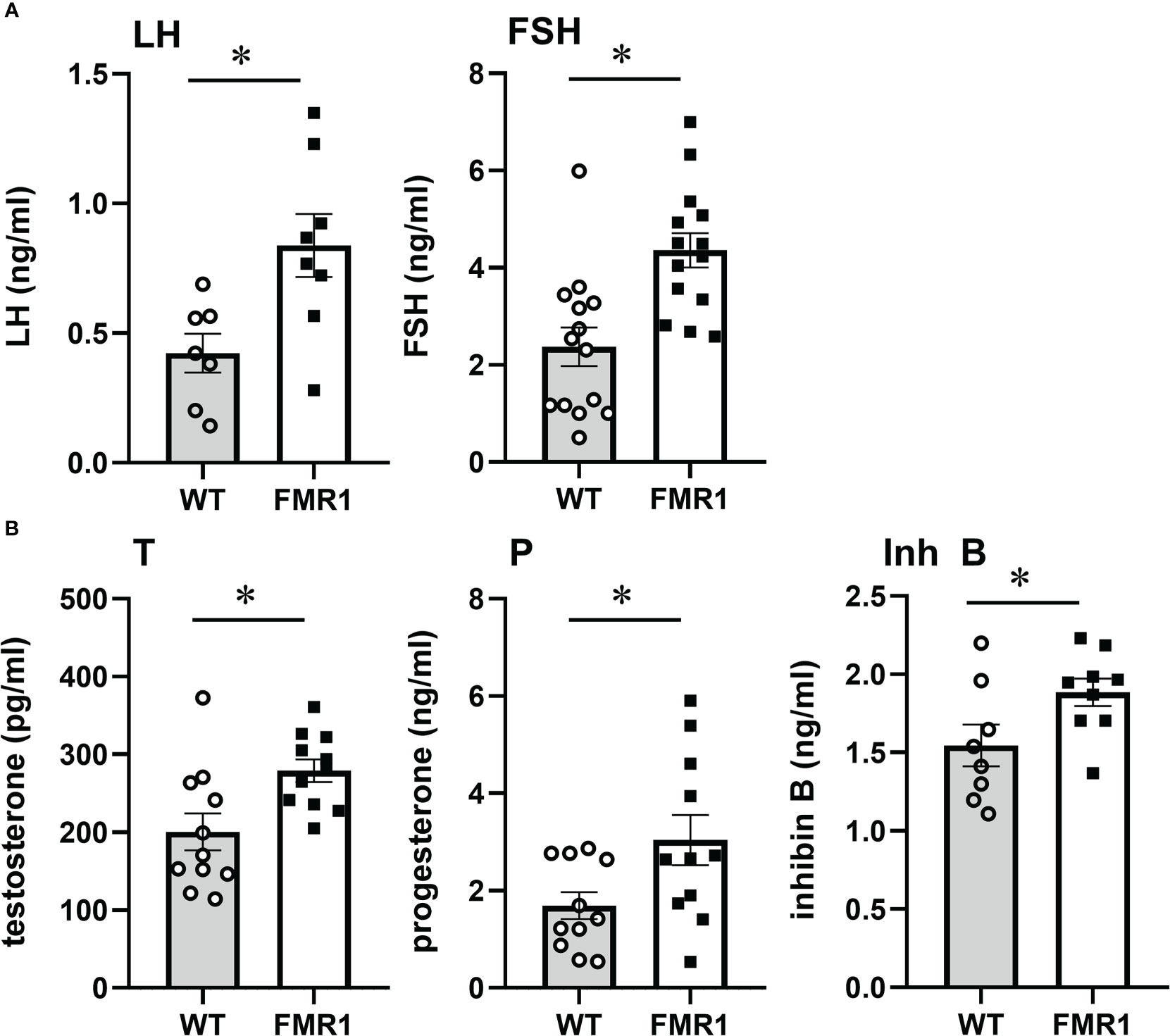
Figure 3 Fmr1 KO mice have higher LH and FSH. Serum levels of LH and FSH, (A) testosterone (T), progesterone (P) and inhibin B (Inh B) (B) in diestrus females in WT controls and Fmr1 KO. Each point represents one animal, while bars represent group means +/- standard error. LH was sampled from the tail tip after acclimatization to handling, to minimize stress and prevent exposure to isoflurane that may affect LH levels. FSH, T, P, Inh B samples are obtained from inferior vena cava. Statistical significance, indicated with a * (p < 0.05) was determined with t-test followed by Tukey’s post hoc test.
Previous studies postulated that ovarian impairment contributed to diminished negative feedback, which in turn caused increased FSH observed in affected women (40, 41). To address this possibility, we analyzed ovarian hormones that provide feedback to the hypothalamus and pituitary in 8-week-old diestrus females before cessation of reproductive function. Steroid hormones primarily provide feedback to the hypothalamus, while inhibin regulates FSH levels (72–77). Testosterone was significantly increased in Fmr1 KO female mice, 279 pg/ml in KO compared to 200 pg/ml in controls (Figure 3B, T). Progesterone was elevated as well to 3 ng/ml in Fmr1 KO from 1.7 ng/ml in controls (Figure 3B, P), demonstrating that negative feedback is present and increased LH cannot be explained by reduced negative feedback. We also analyzed inhibin B levels in diestrus females in the circulation, and determined that inhibin B was higher in KO mice, 1.9 ng/ml compared to 1.5 ng/ml in controls (Figure 3B, Inh B). Our results demonstrate that inhibin B is higher in young animals, which may be a result of larger number of follicles, or alternatively that is stimulated by higher FSH levels. This means that inhibin feedback is also present, and cannot explain elevated FSH. This implicates central mechanisms, rather than ovarian insufficiency in the reproductive phenotype of Fmr1 KO mice. Together, our results demonstrate elevated gonadotropin hormone levels, higher testosterone and progesterone, more corpora lutea, larger litters in young animals, and early cessation of reproductive function.
Ovariectomy reveals hypothalamic and ovarian contribution to endocrine changes
To discern ovarian contribution from the hypothalamic origin of the disorders, we ovariectomized (OVX) the mice and a week later analyzed LH and FSH levels. Over 10-fold higher LH and over 20-fold higher FSH confirmed the successful OVX (compare levels in Figures 3, 4). Interestingly, LH remained significantly higher in OVX KO mice (8 ng/ml) compared to OVX WT mice (6 ng/ml), while there was no difference in FSH levels between WT and Fmr1 KO females after OVX. Therefore, increased LH in unmodified animals likely stems from central dysregulation, while increased FSH involves ovaries.
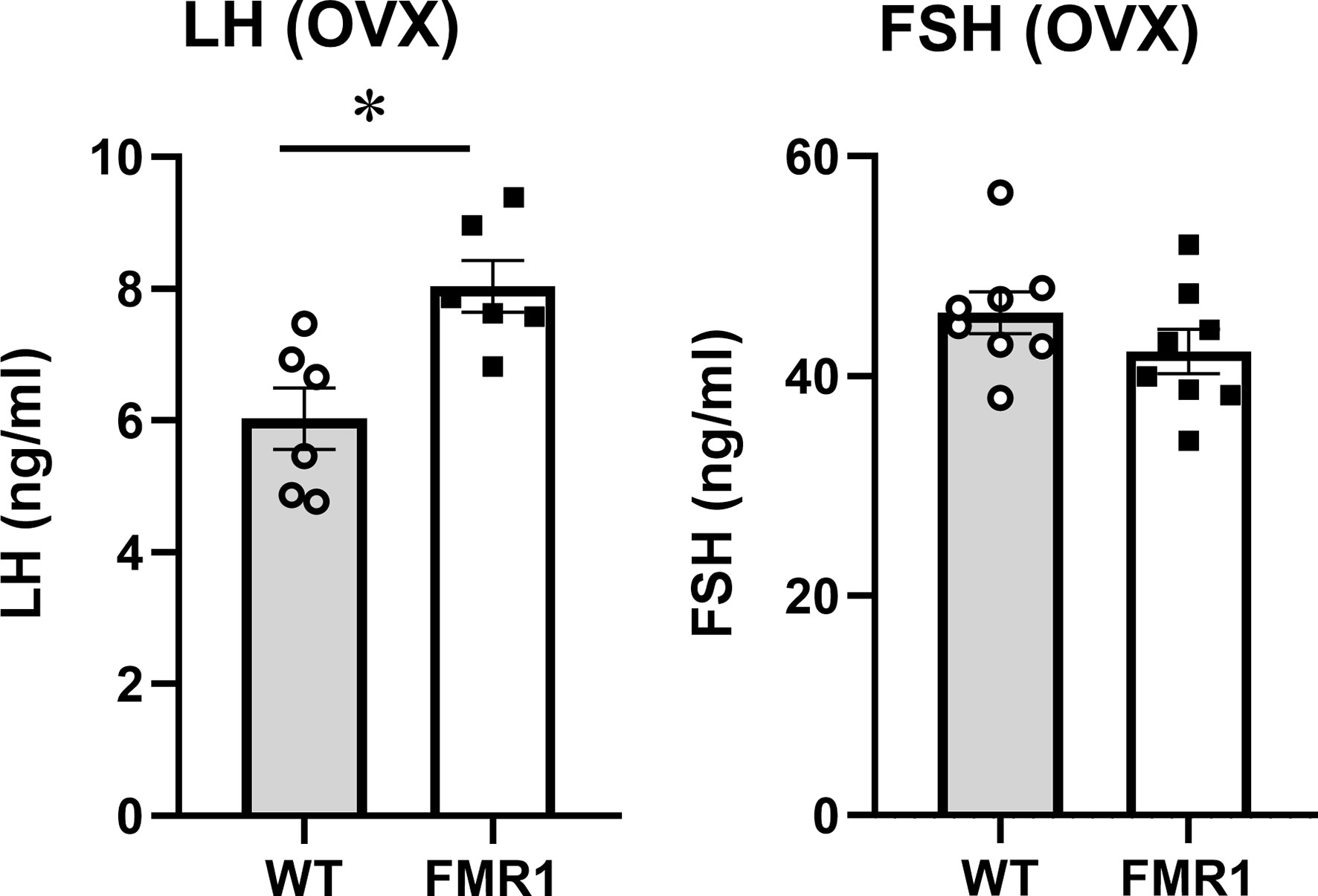
Figure 4 Ovariectomized Fmr1 KO mice have higher LH. One week after ovariectomy, LH and FSH samples were collected as in Figure 3. Each point represents one animal, while bars represent group means +/- standard error. Statistical significance, indicated with a * (p < 0.05) was determined with t-test followed by Tukey’s post hoc test.
Increased innervation and vascularization in the ovaries
To address seemingly discordant results, that Fmr1 KO females exhibit higher levels of ovarian hormones than WT controls, which can provide negative feedback, and also increased FSH, which as revealed by ovariectomy is due to ovarian dysregulation, we analyzed ovarian vascularization and innervation. Using an endothelial cell marker, CD31 we stained ovarian sections and analyzed vascularization around follicles and corpora lutea. Follicles from WT and Fmr1 KO had the same degree of vascularization (Figure 5A, representative image; Figure 5B, quantification). However, corpora lutea (CL) were more highly vascularized in Fmr1 KO than in WT mice (Figure 5C low magnification, top). Higher magnification revealed more abundant and thicker vasculature in the Fmr1 KO CL (Figure 5C, bottom, Figure 5D, quantification).
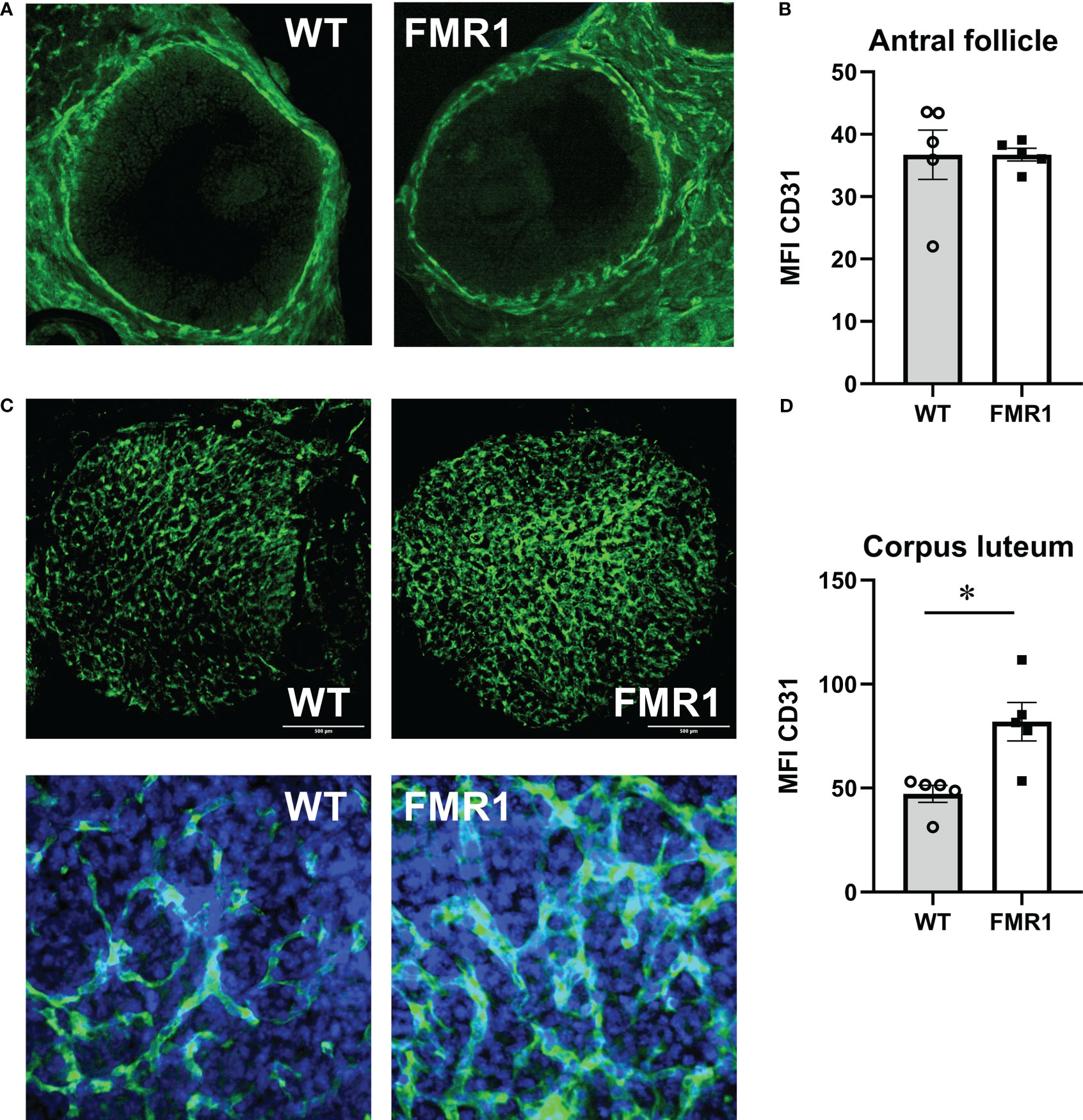
Figure 5 Fmr1 KO have increased corpus luteum vascularization. Ovaries were sectioned and stained with antibodies to CD31 (PECAM-1, Platelet endothelial cell adhesion molecule) to visualize vascularity. Mean fluorescent intensity (MFI) was determined using Fiji imageJ. Consistent areas were used to quantify fluorescence intensity. (A) Representative images of antral follicles. (B) MFI quantification. (C) Corpora lutea representative images; top, 1.6 mm x 1.6 mm area, CD31 green; bottom, 300 μm x 300 μm area, CD31, green, DAPI, blue. (D) MFI quantification. Statistical significance, indicated with a * (p < 0.05) was determined with t-test followed by Tukey’s post hoc test.
Although FMRP is expressed at high level in neurons, ovarian innervation was not previously examined to possibly explain ovarian phenotype observed in Fmr1 KO mice. Using antibodies to tyrosine hydroxylase, the rate-limiting enzyme in catecholamine synthesis, we counted numbers of neuronal fibers that reach the theca layer of growing follicles. Secondary follicles in Fmr1 KO ovaries had significantly more neuronal fibers than WT follicles; 4.8 average fibers per secondary follicle in KO compared to 2.3 average fibers in WT (Figure 6A, representative images; Figure 6B quantification). Innervation of CLs was very variable within each animal and between animals, and we did not identify significant differences between WT and KO mice (Figure 6C bottom left quarter of CL with innervation presented, Figure 6D, quantification). Together, ovarian histology demonstrated increased vascularization of corpora lutea and increased innervation of secondary follicles in Fmr1 KO mice.
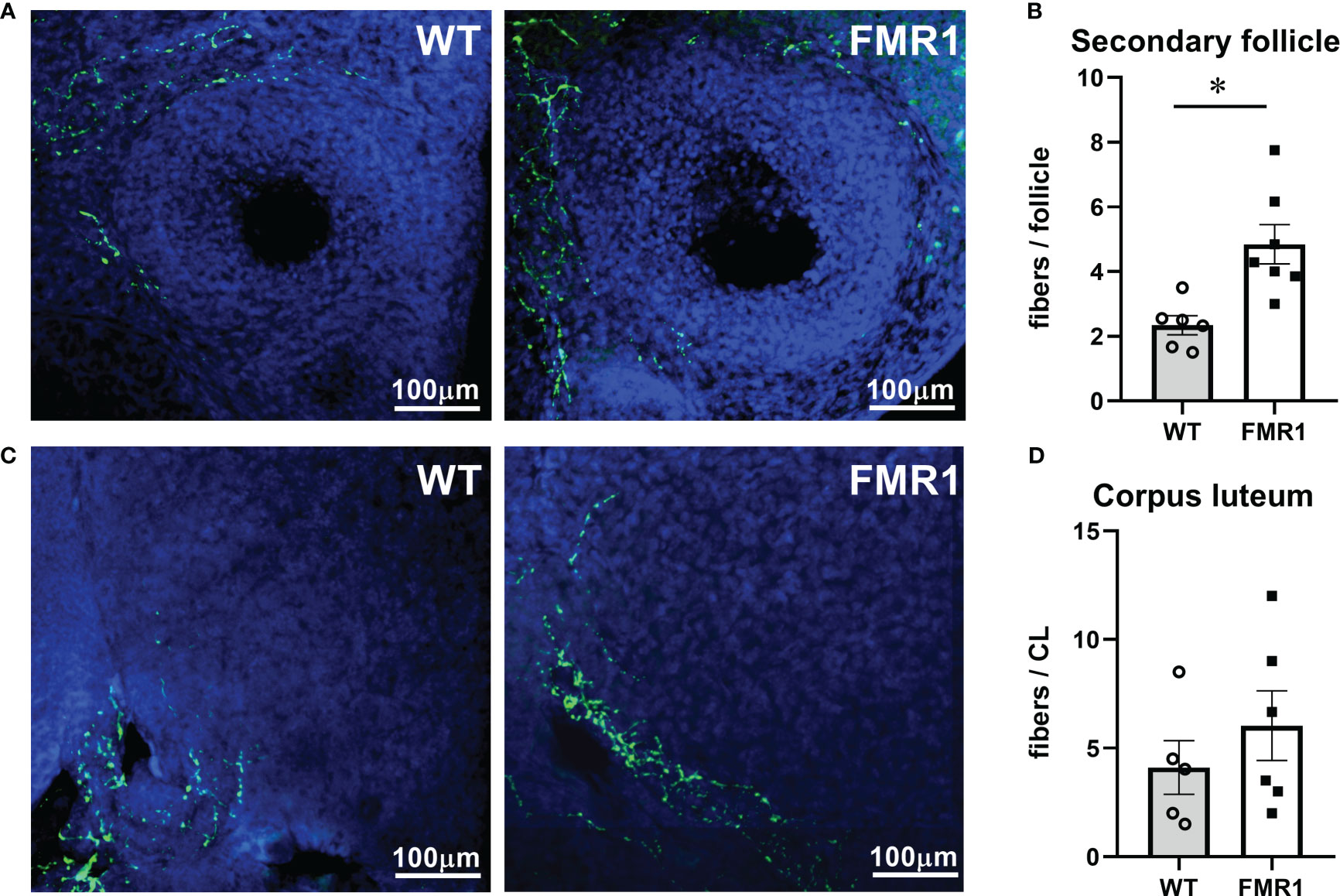
Figure 6 Fmr1 KO have higher innervation of growing follicles. Ovaries were sectioned and stained with antibodies to tyrosine hydroxylase (TH, green) to visualize innervation around secondary follicles (A) and corpora lutea (C). Fibers that surround the follicles and penetrate theca layer were counted (B, D). * indicates significant difference.
Altered levels of synaptic molecules in the hypothalamus of Fmr1 KO mice
Our results demonstrate elevated LH before and after ovariectomy, which is regulated by GnRH secretion. To address the mechanisms of changed LH, we examined changes in the hypothalamus, first focusing on gene expression changes caused by the loss of Fmr1 gene. Although FMRP is an RNA-binding protein that regulates protein levels of its targets, genome-wide changes in the RNA expression signatures can identify pathogenic pathways that may be indirectly regulated by FMRP. Especially since FMRP regulates levels of several transcription factors and other transcriptional regulators, which may exhibit extensive changes on the transcriptome. Previous analyses of Fmr1 KO transcriptomes focused on embryonic hippocampus and cortex, and identified overexpression of immune-related genes and downregulation of genes implicated in behavioral phenotype (78). For that reason, we used Nanostring neuroinflammation panel, which contains 770 genes implicated in neurological disorders, neuronal injury, neurotransmission, neuron-glia interactions, neuroplasticity, cell integrity, neuroinflammation, and metabolism; and added 30 custom probes for hypothalamic neuropeptides and their receptors. The complete list of genes that changed in KO compared to WT is presented by the heatmap (Figure 7A). There were 59 genes that were upregulated >120% from WT levels, and 39 genes that were downregulated <80% of WT levels, delineated with a dashed line. Significant changes in expression of neuropeptides and other genes of interest were highlighted in the volcano plot (Figure 7B, log fold change vs. log p-value, statistically significant change in expression indicated with a dashed line; red, upregulated genes in KO compared to WT; green, downregulated gene in KO compared to WT). Immediate early gene, transcription factors Egr1, Fos and Jun, that are used as markers of neuronal activation, were upregulated in Fmr1 KO mice. Genes encoding GABAA receptor γ2 subunit, (GABARγ2, Gabrg2) which is the obligatory subunit of the pentameric GABAA receptor (79); and PSD-95 (Dlg4), a postsynaptic scaffolding protein anchoring glutamate receptors, were upregulated in KO mice. Ppfia4, involved in neurotransmitter release, and Opalin, important for oligodendrocyte differentiation, were also upregulated. On the other hand, genes correlated with DNA repair, Ercc2; neurodegenerative disorders, Serpina3n; hypoxia, Hif1a; and apoptosis, Hcar2 and Bag4, were downregulated. Importantly, genes encoding GLAST, Slc1a3, and VGLUT2, Slc17a6, were also downregulated. Of interest, neuropeptide gene encoding GnRH, Gnrh1, was upregulated, while genes for kisspeptin, Kiss1, neurokinin B, Nkb, Tac3; and cocaine and amphetamine regulated transcript, Cart, were downregulated. We confirmed changes in Gnrh1 and Kiss1 expression by qPCR of hypothalamic lysates (Figure 7C). GO pathway analysis indicated that pathways such as AP1 complex, comprised of Fos and Jun, myelin adaxonal regulation, spine and dendrite development were upregulated, while neuropeptide binding, ubiquitin ligase binding, and receptor signaling pathways were downregulated (Figure 7D).
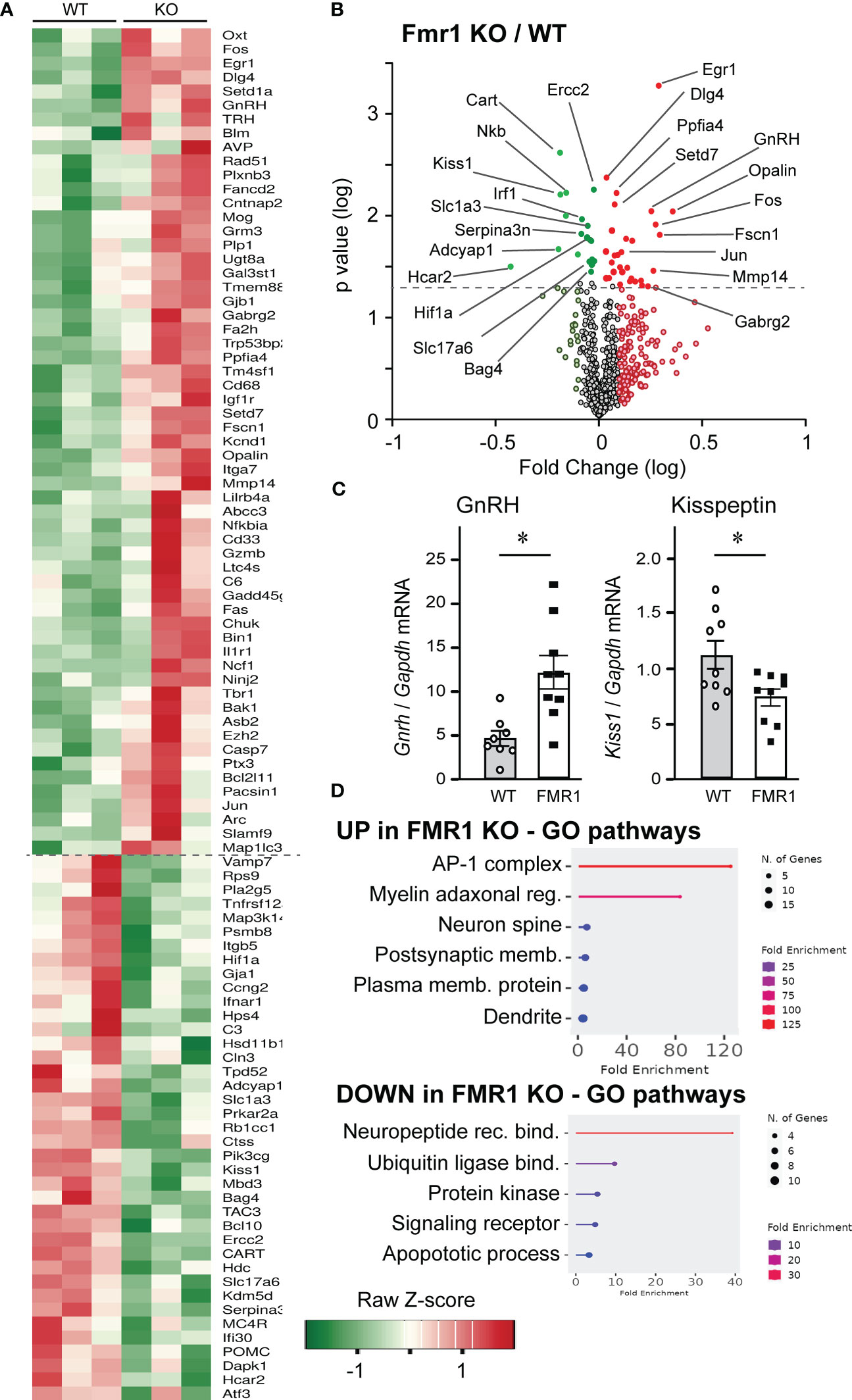
Figure 7 Nanostring analysis demonstrates significant changes in hypothalamic gene expression in Fmr1 KO mice. (A) Hypothalami from 3 mice per group were dissected and 50 ng RNA used in Nanostring analysis. Heatmap indicates gene expression changes in Fmr1 KO hypothalami that are <0.8-fold and >1.2-fold over WT. (B) Data were plotted as log fold change on x-axis vs. log p value on y-axis, and dashed line indicates significance. Red indicates genes that are increased in KO compared to WT mice, while green indicates genes that are decreased in KO compared to WT. Genes below the dotted line, light red and light green did not reach significance. (C) qPCR of the hypothalamus confirms changes in GnRH and kisspeptin expression. Each point represents one animal and bars represent group average. Statistical significance is indicated with *, determined by t-test followed by Tukey’s post hoc test. (D) Gene ontology pathway analysis indicates upregulated (top) and downregulated (bottom) pathways in the Fmr1 KO mice.
Considering changes in excitation/inhibition synaptic balance in the cortical neurons of Fmr1 KO mice (80), we next investigated the levels of several synaptic proteins in hypothalamus at the protein level. Although previous studies showed changes in neurotransmitter receptor levels, such as GABAA receptor for GABA, and NMDA receptor for glutamate, in several brain regions of male Fmr1 KO mice, no studies were done in females or in the hypothalamus. Hypothalami were dissected from diestrus female brains, and levels of synaptic molecules were measured by western blotting. We analyzed GABARγ2 (Gabrg2) levels in the hypothalami of KO and control female mice in diestrus, since GABA transmission activates GnRH neurons, via activation of GABAA receptor (26, 81), and determined that Fmr1 KO females had significantly higher levels of GABARγ2 GABAA receptor than controls (Figure 8A, representative western blots; Figure 8B quantification). Protein levels correlate with gene expression analysis, indicating that GABARγ2 may be indirectly regulated by Fmrp. We then analyzed levels of the glutamatergic NMDA receptors (NMDARs, or NRs) (82, 83), since 30-50% of GnRH neurons respond to NMDA (84, 85). NR1 is an obligatory subunit that forms a heterotetramer with either 2A or 2B subunits (other isoforms are less frequent). We determined that NR1 (Nmdar1, GluN1) levels were increased in the hypothalami of KO mice compared to controls (Figure 8C). Depending on the NR2 isoform, NMDAR is localized at synapses or extrasynaptically with different effects on long-term potentiation or negative feedback, respectively (64, 86). NR2B is localized extrasynaptically, and we determined that the levels of NR2B are lower in the KO mice compared to controls (Figure 8D). Genes encoding these proteins did not change at the transcriptional level, which indicates that they may be regulated by Fmrp at the protein level. We also analyzed levels of PSD-95, since PSD–95 anchors glutamate receptors (87), and determined that PSD-95 protein levels were the same in Fmr1 KO and controls, which is contrary to gene expression studies and again points to direct regulation by Fmrp (Figure 8E). Taken together, an increase in the synaptic GABAA and NR1 receptors, and a decrease in extrasynaptic NR2B, may contribute to altered activity of hypothalamic neurons in Fmr1 KO female mice.
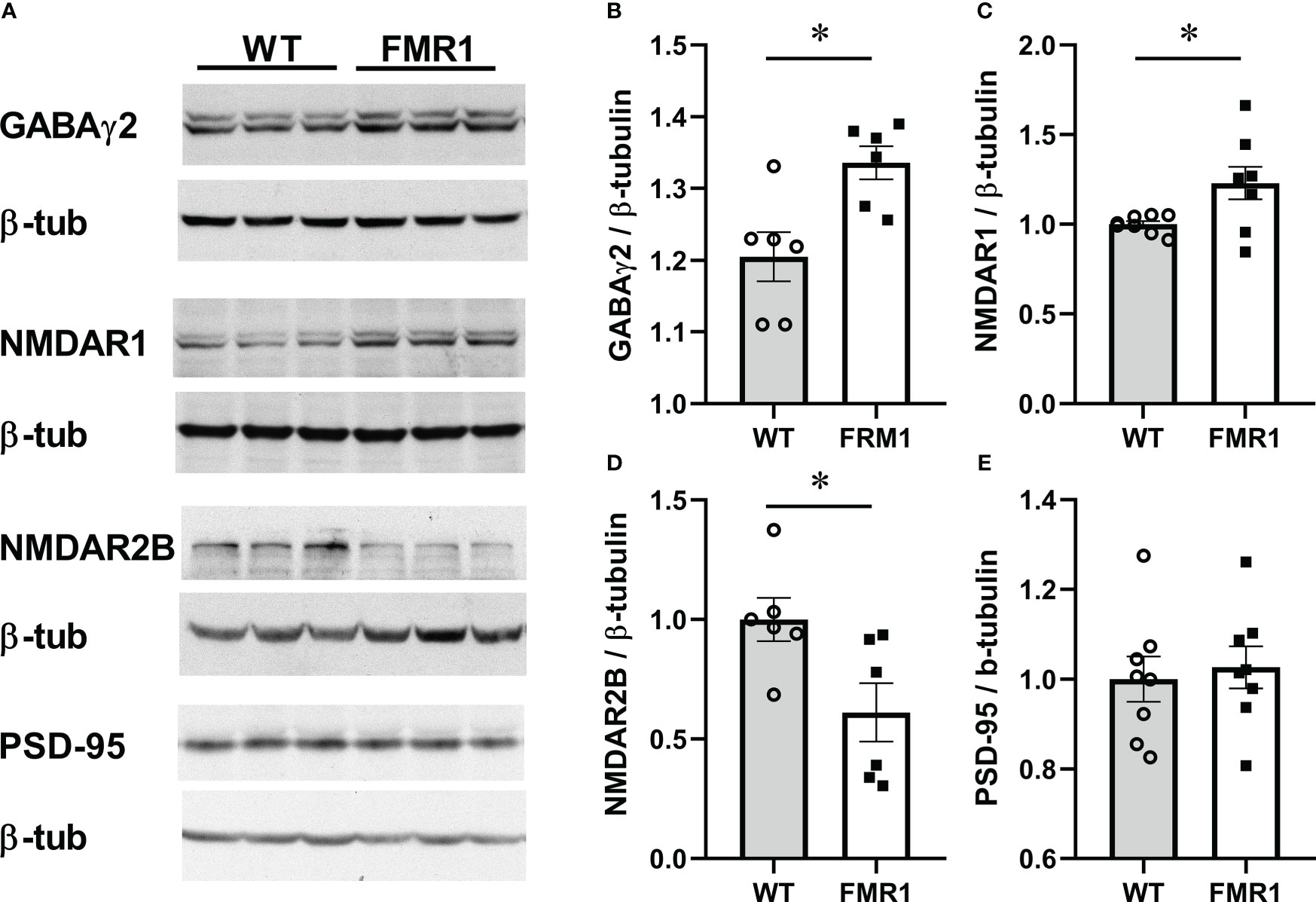
Figure 8 Fmr1 KO females have higher levels of the obligatory GABAA receptor subunit in hypothalami. Hypothalami were dissected and protein levels analyzed by western blotting. Representative blots are shown in (A). Protein levels from 6 mice per group were quantified using Chemidoc and levels of neuronal proteins normalized to β-tubulin (B–E). Statistical significance (p < 0.05), determined with t-test followed by Tukey’s post hoc test, is indicated with a *.
Lack of Fmrp changes GnRH neuron connectivity
To determine if Fmrp loss alters neurotransmitter receptor levels specifically in GnRH neurons, we first confirmed that 82% of GnRH neurons express Fmrp protein (Figure 9A, left image, confocal microscopy, right image, 3D reconstruction to demonstrate Fmrp inside GnRH neurons). Antibody specificity was determined by staining the hypothalami from Fmr1 KO mice (Supplemental Figure 2). We also analyzed GnRH neuron number in Fmr1 KO mice to compare to WT to ascertain if increased in GnRH neuron number contributes to higher LH. There was no difference in the number of GnRH neurons in WT and KO mice (Supplemental Figure 2).
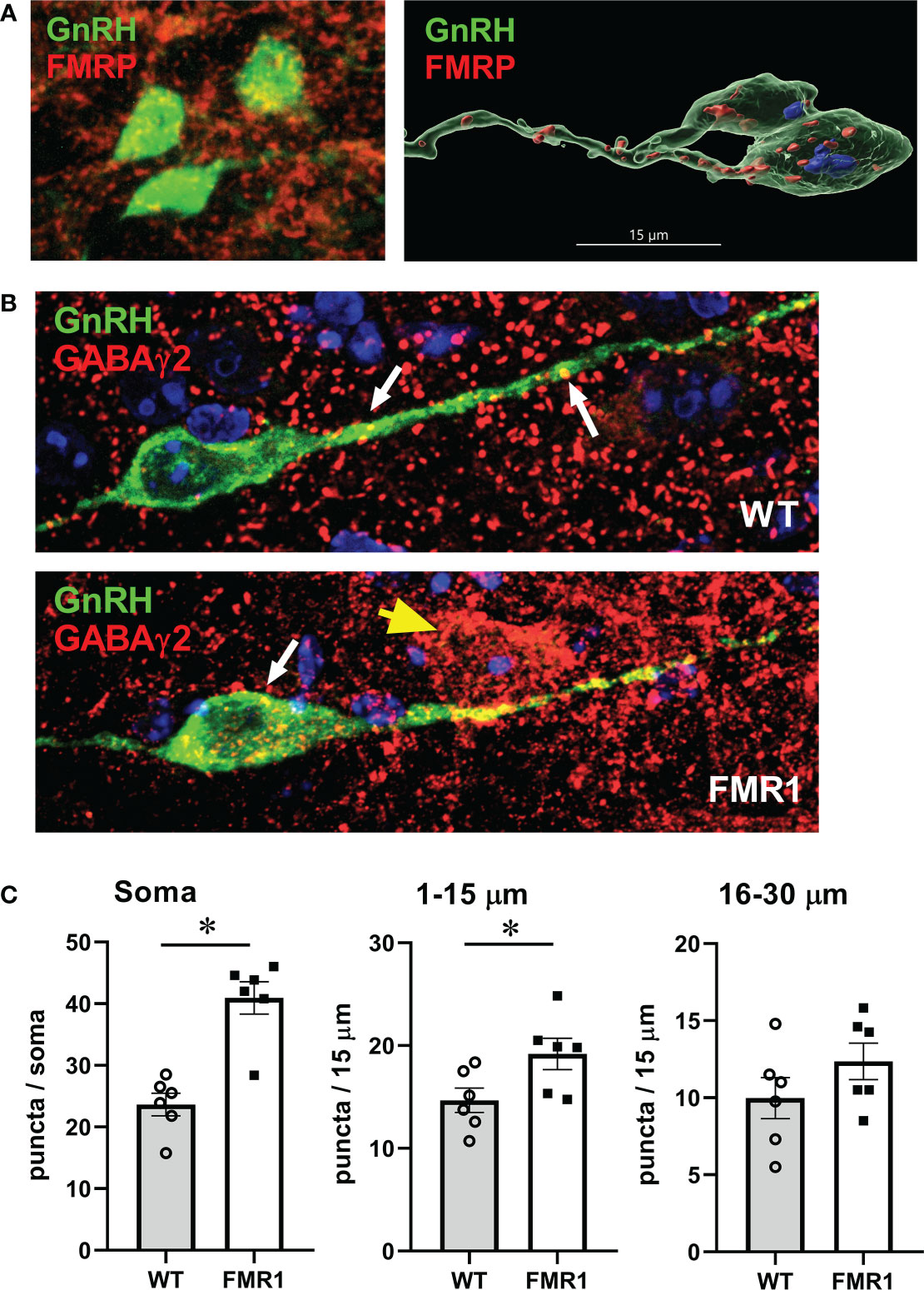
Figure 9 GnRH neurons of Fmr1 KO females have more GABAA receptors. (A) GnRH neurons (green) express Fmrp (red, top panels). (B) Representative GnRH neurons (green) from WT control (top panel) and Fmr1 KO mice (bottom panel) co-stained with GABAγ2 antibody (red). (C) Quantification of (B) Each point represents one animals and an average of 15-20 neuron per animal, and bars represent group means. Panels represent counts in the whole soma, along the first 15 μm segment of the process (1-15 μm) and second 15 μm segment (16-30 μm) from the soma, as indicated above. Statistical significance (p < 0.05), determined with t–test followed by Tukey’s post hoc test, is indicated with a *.
Since GABA can regulate GnRH neuron activity (26, 81), and we determined elevated GABAγ2 subunit of the GABAA receptor in the hypothalami of KO mice compared to controls by western blot, we analyzed if GABAA receptor immunoreactivity is increased specifically in GnRH neurons. To determine GABAA receptor distribution in GnRH neurons, we immunostained 100 μm coronal sections of the preoptic area of the hypothalamus for GABAγ2 and GnRH. After staining, sections were imaged with high-resolution confocal microscopy. GABAγ2 receptors showed puncta-like distribution in GnRH neurons. GABAγ2 receptor puncta colocalized with GnRH immunoreactivity were identified by closely apposed puncta when no black pixels were visible between two signals in optical slices, and counted blind to condition by scrolling through the series of captured z-stack images for each GnRH soma and along the process, at 15-μm intervals, for each GnRH neuron. At least 15 neurons were counted from each mouse, and the average for each mouse was calculated (represented by a dot in the Figure 9C with bars representing group average). We determined that GABAγ2 puncta numbers increased significantly in the GnRH neuron soma and in the first 15 μm of the process proximal to the soma (Figure 9C quantification, Figure 9B, representative images, top WT, bottom Fmr1 KO). The increase in GABAergic inputs in this area is significant, since this region of the neuron is the region where action potentials are initiated (88), and it exhibits synaptic plasticity during development and in different hormonal milieu (89–91). Since GABA is excitatory for GnRH neurons (20, 26–28), the alteration of the receptor levels may enhance GnRH neuron responsiveness and neuropeptide secretion, which in turn would cause changes in gonadotropin levels.
To determine GnRH innervation, we also analyzed appositions of GABAγ2 subunit of the GABAA receptor with vesicular GABA transporter (VGAT), presynaptic marker of GABAergic terminals. We performed a triple stain for GnRH, GABAγ2 and VGAT, and as above, counted number of puncta where VGAT was in a close opposition to GABAγ2 in GnRH neurons (Figure 10A). We counted at least 15-20 neurons from each mouse, 5 pairs of mice; and used Imaris software to perform 3-D modeling of VGAT-GABAA receptor appositions (Figure 10B). Fmr1 KO mice had a higher number of GABAergic appositions in GnRH neuron soma and proximal process, in the segment 1–15 μm and segment 16–30 μm from the soma, than WT controls (Figure 10C). The increase in synaptic GABAA receptors in the proximal process indicates higher innervation of GnRH neurons in the area that is plastic and receives synaptic input.
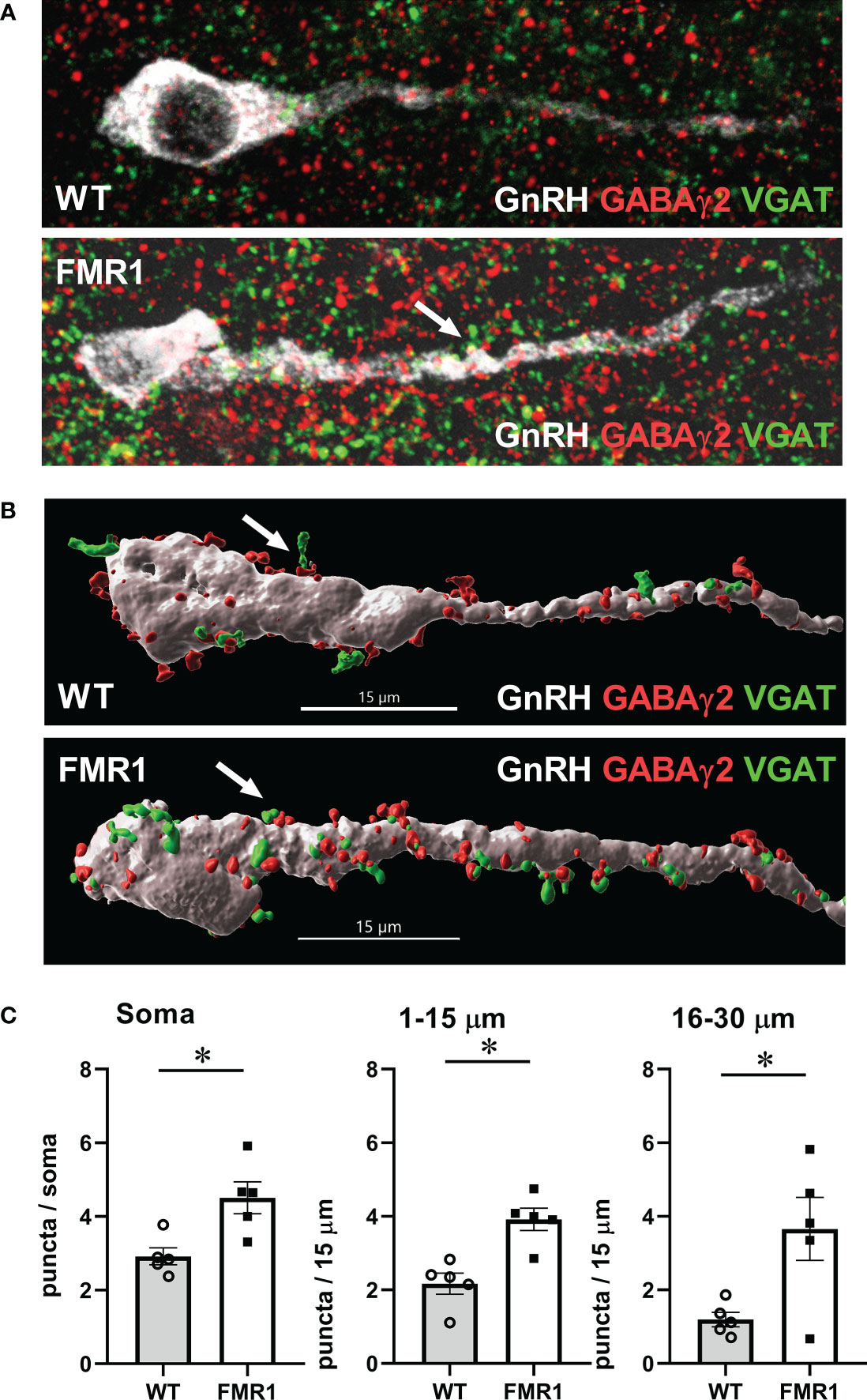
Figure 10 GnRH neurons of Fmr1 KO females have more synaptic GABAA receptors. (A) Representative images of triple stain for presynaptic VGAT (green), GABAγ2 receptor (red) and GnRH (white); (B) 3-D GnRH neuron models using Imaris software. VGAT (green) appositions to GABAγ2 receptor subunits (red) in GnRH neurons (white) were counted in 15-20 neurons per mouse, five mice from each group. (C) Quantification from different regions of the neuron, as above, is presented in panels, and significance indicated with *.
Fmrp regulates LH pulsatility
Pulsatile secretion of LH strictly corresponds to GnRH secretion (49, 50). LH is used as an indicator of GnRH secretion, since GnRH secretion into median eminence cannot be measured in mice. To ascertain whether GnRH neuron secretion was affected, we measured LH pulses and used an ultrasensitive ELISA assay for LH (51) that allows for LH measurement in 5 µl of whole blood. Mice were acclimated for 2 weeks by daily tail massage. Serial sample collection every 8 min for 3 hours from the tail vein was performed (45, 52). Representative LH pulse profile from unmodified WT and KO mice are presented in Figure 11A, right side (WT control, top; Fmr1 KO, bottom). Number of LH pulses per 2.5 hours of measurement were determined using DynPeak algorithm (53) and compared between genotypes. LH, and therefore GnRH, pulse frequency was significantly higher in Fmr1 KO mice compared to WT controls (Figure 11A, left side). Amplitude was determined by subtracting the highest LH value from the basal value prior to the onset of the pulse and averaged for each mouse (Figure 11A, middle).
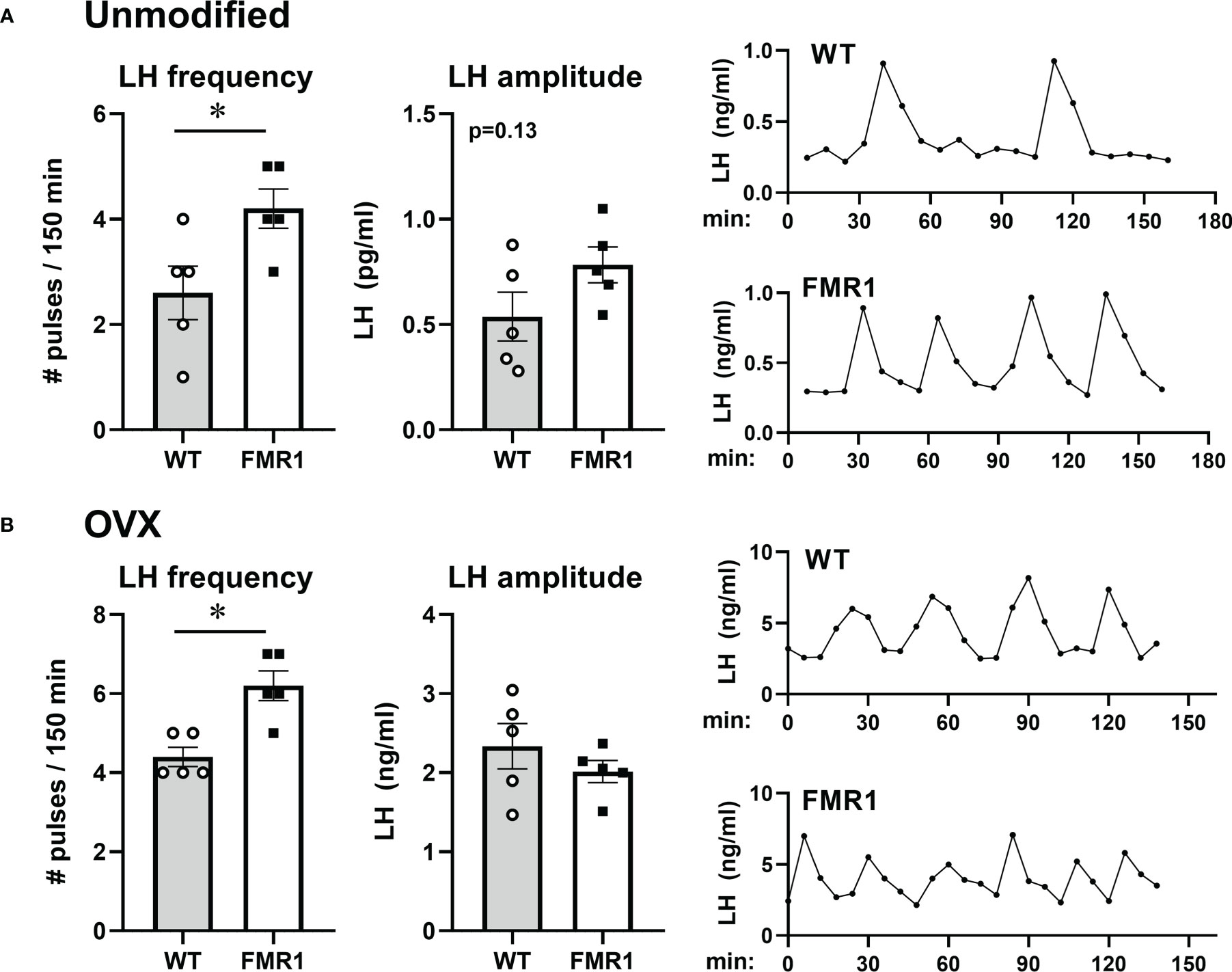
Figure 11 Higher LH/GnRH pulse frequency in Fmr1 KO females before and after ovariectomy. LH pulse frequency reflects GnRH neuron activity. (A) Frequent tail-tip whole blood sampling over 3 hours demonstrate higher pulse frequency of LH in Fmr1 KO female mice in diestrus. Representative profiles from WT control in the top and Fmr1 KO at the bottom (right side), pulse frequency calculated from pulse profiles using DynPeak (left); amplitude was determined by subtracting the LH value at the peak from the basal value prior to the onset of the pulse and averaged for each mouse (middle). (B) LH pulse in ovariectomized (OVX) animals. Statistical significance (p < 0.05), determined with t-test followed by Tukey’s post hoc test, is indicated with a *.
Pulsatile LH analyses were performed using ovariectomized animals as well. Frequency of LH secretion was faster in OVX Fmr1 KO animals compared to OVX WT mice (Figure 11B, left; representative profiles right), while pulse amplitude was the same. Representative pulse profiles are shown on the right side. These experiments determined that the lack of Fmrp increased LH pulse frequency, indicating higher GnRH neuron activity corresponding to higher GABAergic innervation of GnRH neuron. It is possible the lack of Fmrp alters GnRH neuropeptide secretion leading to the faster GnRH pulse frequency and elevated LH, which contributes to ovarian dysregulation in young animals.
Discussion
We sought out to uncover the effects of FMRP loss on hypothalamic GnRH neurons and ovarian function, which may help elucidate the mechanisms of early cessation of reproductive function in females with a mutation in the FMR1 gene. Women with FMR1 mutations comprise the majority of known genetic causes of premature ovarian failure (3, 35, 92). Premature ovarian failure or primary ovarian insufficiency is the most extreme manifestation of premature ovarian senescence and affects about 1% of women (34). Premature reproductive senescence affects approximately 10% of women and is characterized by an early depletion of ovarian follicles (34). Molecular causes of premature cessation of reproductive function in women with FMR1 mutations and mechanisms underlying reproductive dysfunctions are still unknown. The hypothalamus was especially neglected in previous studies addressing FMR1 function, or etiology of premature reproductive senescence. Our study is the first to examine the hypothalamic function of the FMR1 gene. We analyzed mechanisms of reproductive disorders associated with FMR1 mutations using the Fmr1 KO female mice, since knockout mice lack Fmrp mimicking the loss of FMRP in humans with FMR1 mutation. Fmr1 KO female mice are useful model to study reproductive disorders in women with a mutation in the FMR1 gene, as they exhibit early cessation of reproductive function similar to women with FMR1 mutations. The main findings of this study implicate central mechanisms and ovarian innervation in reproductive disorders associated with the FMRP loss. We demonstrate that Fmr1 KO female mice show higher GnRH neuron and ovarian follicle innervation, increased GnRH neuron secretion, and augmented gonadotropin levels, all of which may contribute to increased recruitment of ovarian follicles to the growing pool, corresponding to a higher number of corpora lutea and larger litters in young animals. These growing follicles exhibit increased innervation, which is associated with higher steroidogenesis. Together, our results point to hypothalamic mechanisms, specifically GnRH neuron connectivity, and ovarian innervation in the reproductive disorders associated with FMRP loss that have not been considered before.
Women with early menopause face not only infertility, but an increased risk of heart disease and osteoporosis (93–97). Most women are only diagnosed after their ovarian function has ceased, since they seek care due to infertility or amenorrhea. In that case, it is difficult to predict earlier hormonal changes, when ovarian reserves are relatively normal. As of yet, there are no screening strategies to detect women with increased risk before they are symptomatic (98). Most studies analyzing a role of FMRP have focused on males, while females are rarely included. Furthermore, cortical mechanisms attracted the attention of the investigators, because FXS is the most common monogenic cause of intellectual disability and autism. However, the mechanism underlying the dysregulation of reproductive function in FMR1 mutations was not extensively studied. Several reports demonstrate elevated FSH in women affected with FMR1 mutations showing early menopause (40, 41), which are consistent with our observations in mice. Since the primary reproductive defect in females with FMR1 mutation is premature ovarian failure, gonadal origin was proposed. We demonstrate that primordial follicle number is unaffected by Fmr1 loss, which agrees with previous studies that showed that women with FMR1 mutation and mouse models of premutation have normal pool of primordial follicles (31, 38, 39, 70). This indicates that early ovarian development is not adversely affected by the loss of FMRP. Our results using complete KO model show larger litters and more corpora lutea, while premutation mouse models have smaller litters (31, 38), but reasons for differences are not clear. Our results do not preclude other intra-ovarian defects that may contribute to the early loss of follicles and early depletion, such as increased atresia as suggested in (38). Given that there is no difference in primordial follicle development, inappropriate ovarian response to gonadotropin stimulation, compounded by changes in gonadotropin levels, likely contributes to early cessation of reproductive function in FMR1 mutations.
Removal of ovaries demonstrated that increased FSH depends on ovarian feedback. However, the increase in FSH is not a result of the lack of negative feedback as we show that in females, ovarian hormones, inhibin or steroid hormones, are higher than in controls, indicating that hormonal feedback to the pituitary and hypothalamus was present. Studies in women with FMR1 mutations are inconclusive, with one study reporting unchanged inhibin (6), while the other found decreased inhibin (40). The latter study is the only one, to our knowledge, that analyzed steroid hormone levels and found decreased progesterone (40). The discrepancy may arise due to the age of the subjects, as discussed above. Increase in inhibin B levels in our results, implies that higher FSH occurs irrespective of inhibin feedback, in fact, may lead to increased inhibin, and thus, higher FSH may be the cause rather than the consequence of reproductive disorders.
Since ovarian hormones did not provide adequate explanation, we examined ovarian innervation and vascularization, and detected increased vascularization of corpora lutea which may correlate with higher progesterone levels (99). Increased progesterone may lead to higher FSH (100, 101). Fluorescent studies with endothelial cells marker reveal that large follicle vascularization is not changed, but corpora lutea exhibit increased vascularization. Developing corpus luteum is a site of rapid angiogenesis, under the influence of the vascular endothelial growth factor (VEGF) (99, 102, 103). Studies in several species determined that angiogenesis and VEGF induction is stimulated by LH (104–106). Thus, increased LH in our study likely contributes to increased vascularization of the corpus luteum. Treatment with VEGF antagonist demonstrated decreased progesterone (107). Therefore, increased progesterone in Fmr1 KO mice we report here may be due to increased vascularization of corpora lutea. Several studies determined that progesterone could increase FSH at the transcriptional level, which is thought to be important for the specific secondary rise of FSH during luteal phase. Progesterone treatment in combination with estrogen, increased FSH, while antiprogestins blocked FSH secretion and mRNA expression during the preovulatory surge (108) and during the secondary rise (109). Therefore, our studies postulate that increased LH may cause increased angiogenesis during luteinization, which leads to higher progesterone, which in turn increases FSH, and together, may explain why increased LH is of hypothalamic origin, while increased FSH requires ovaries.
Ovaries receive sympathetic innervation via two routes (110–112). The superior ovarian nerve projections innervate the secretory component of the ovary. The fibers surround the developing follicles, but do not penetrate the granulosa layer or the corpus luteum. We determined that innervation of the follicles with fibers that originate from the superior ovarian nerve is increased. This innervation is required for steroidogenesis, since transection of the nerve reduced steroid hormone levels (113–115). Thus, higher steroid hormone levels may be a result of increased innervation. As discussed above, this may contribute to higher FSH levels in KO animals.
Increased LH likely stems from higher GnRH secretion, via increased GnRH pulse frequency, since GnRH from the hypothalamus strictly regulates LH secretion. We determined that GnRH neurons have increased GABAergic innervation, which is excitatory for GnRH neurons (20, 26–28). Therefore, it is possible that increased GABA tone leads to altered responsiveness of GnRH neurons to the pulse generator and the upstream regulatory network. Alternatively, constantly increased GABA input may lead to increased activity of GnRH neurons. The idea of enhanced activation of GnRH neurons is also supported by the observed increases in neurotransmitter receptor levels in the hypothalamus and specifically GnRH neurons. FMRP binds mRNAs that encode synaptic proteins (9–11) and previous studies reported altered levels of GABAA receptors and NMDA receptors in several brain areas. Here, we further determined that GABAA receptor abundance changes at the transcriptional level, while NMDARs change on the protein level, which may elucidate direct versus indirect regulation by FMRP. Contrary to the studies in the cortex and hippocampus, which detected decreased GABAA receptor, we determined that GABAA receptor levels are increased in the hypothalamus at the mRNA and protein levels. 30%-50% of GnRH neurons respond to NMDA (84, 85), and our studies in the hypothalamus agree with previous studies in the cortex that demonstrate increased NMDA receptors in Fmr1 KO mice. Therefore, the increase in NMDA and GABA receptors, that are both excitatory for GnRH neurons, likely changes GnRH neuron function and GnRH neuropeptide secretion.
Therefore, both the hypothalamus and ovaries contribute to endocrine disruption that may lead to larger litters in young animals. Hypothalamic contribution to the etiology of early menopause has been underappreciated. We propose that changes in GnRH neuron and ovarian innervation contribute to changes in gonadotropin hormones, LH and FSH, levels. This may cause early depletion of ovarian follicles and premature cessation of reproductive function, which will be addressed in future studies. Together, our results point to hypothalamic mechanisms and ovarian innervation in the reproductive function disorders associated with FMRP loss that have not been considered before.
Data availability statement
The data presented in the study are deposited in the Gene Expression Omnibus GEO repository, accession number GSE222723. link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE222723 these are fluorescent images and the background is meant to be black to demonstrate specific staining of the area of interest.
Ethics statement
The animal study was reviewed and approved by UCR IACUC.
Author contributions
PV and NL performed most of the experiment reported herein and data analyses. CJ determined reproductive phenotype. SB performed analyses of ovarian vasculature. IE provided valuable insight in Fragile X Syndrome pathology. DC conceived and guided the study and wrote the manuscript. PV: Conceptualization, Investigation, Formal Analysis, Visualization, Writing – Review and editing. NL: Investigation, Formal Analysis, Visualization, Writing – review and editing. CJ: Investigation, Formal Analysis, Visualization, Writing – review and editing. SB: Investigation, Formal Analysis, Visualization, Writing – review and editing. IE: Resources, Validation, Supervision, Writing – review and editing. DC: ORCid: 0000-0003-0692-1612 Conceptualization, Formal Analysis, Funding Acquisition, Project Administration, Supervision, Visualization, Writing – original draft and preparation. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by R01 HD091167 from NIH NICHD to DC. The University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core is supported by the Eunice Kennedy Shriver NICHD/NIH Grant R24HD102061.
Acknowledgments
The authors thank Dr. Greg Anderson (University of Otago, Dunedin, New Zealand) for GnRH antibody. We are grateful to Drs. Ameae Walker and Anna Kulinich for thoughtful discussion and technical support; and, Rebecca Ruggiero and Lina Morales for editorial suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1129534/full#supplementary-material
Supplementary Figure 1 | Fmr1 KO females have higher Lhb and Fshb mRNA expression. Pituitary mRNA was analyzed by RT-qPCR to determine the expression of pituitary hormones. Lhb and Fshb are increased, while expression of other hormones is unchanged. Each point represents one animal, while bars represent group means. mRNA levels were normalized to B2M mRNA. Statistical significance (p < 0.05), determined with t-test followed by Tukey’s post hoc test, is indicated with a *.
Supplementary Figure 2 | Left, specificity of antibody to FMRP is determined by staining hypothalamic sections from Fmr1 KO mice. Right, every 5th coronal section containing the preoptic area of the hypothalami from WT and Fmr1 KO female mice were stained with anti-GnRH antibodies and GnRH neurons counted. Each point represents average number of neurons from all counted sections for one animal, while bars represent group means.
References
1. Lieb-Lundell CCE. Three faces of fragile X. Phys Ther (2016) 96(11):1782–90. doi: 10.2522/ptj.20140430
2. Lozano R, Azarang A, Wilaisakditipakorn T, Hagerman RJ. Fragile X syndrome: A review of clinical management. Intractable Rare Dis Res (2016) 5(3):145–57. doi: 10.5582/irdr.2016.01048
3. Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, Holden JJ, Yang KT, Lee C, et al. Fragile X premutation is a significant risk factor for premature ovarian failure: The international collaborative POF in fragile X study–preliminary data. Am J Med Genet (1999) 83(4):322–5. doi: 10.1002/(SICI)1096-8628(19990402)83:4<322::AID-AJMG17>3.0.CO;2-B
4. Wheeler AC, Raspa M, Green A, Bishop E, Bann C, Edwards A, et al. Health and reproductive experiences of women with an FMR1 premutation with and without fragile X premature ovarian insufficiency. Front Genet (2014) 5:300. doi: 10.3389/fgene.2014.00300
5. Berkovitz GD, Wilson DP, Carpenter NJ, Brown TR, Migeon CJ. Gonadal function in men with the martin-bell (fragile-X) syndrome. Am J Med Genet (1986) 23(1-2):227–39. doi: 10.1002/ajmg.1320230118
6. Murray A, Ennis S, MacSwiney F, Webb J, Morton NE. Reproductive and menstrual history of females with fragile X expansions. Eur J Hum Genet EJHG (2000) 8(4):247–52. doi: 10.1038/sj.ejhg.5200451
7. Edbauer D, Neilson JR, Foster KA, Wang CF, Seeburg DP, Batterton MN, et al. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron. (2010) 65(3):373–84. doi: 10.1016/j.neuron.2010.01.005
8. Feng Y, Zhang F, Lokey LK, Chastain JL, Lakkis L, Eberhart D, et al. Translational suppression by trinucleotide repeat expansion at FMR1. Science. (1995) 268(5211):731–4. doi: 10.1126/science.7732383
9. Ascano M, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. (2012) 492:382. doi: 10.1038/nature11737
10. Darnell JC, Mostovetsky O, Darnell RB. FMRP RNA targets: Identification and validation. Genes brain behavior (2005) 4(6):341–9. doi: 10.1111/j.1601-183X.2005.00144.x
11. Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. (2011) 146(2):247–61. doi: 10.1016/j.cell.2011.06.013
12. Rais M, Binder DK, Razak KA, Ethell IM. Sensory processing phenotypes in fragile X syndrome. ASN neuro (2018) 10:1759091418801092. doi: 10.1177/1759091418801092
13. Booker SA, Kind PC. Mechanisms regulating input-output function and plasticity of neurons in the absence of FMRP. Brain Res bullet (2021) 175:69–80. doi: 10.1016/j.brainresbull.2021.06.025
14. Kelley AS, Qin Y, Marsh EE, Dupree JM. Disparities in accessing infertility care in the united states: Results from the national health and nutrition examination survey, 2013–16. Fertil Steril (2019) 112(3):562–8. doi: 10.1016/j.fertnstert.2019.04.044
15. Petraglia F, Serour GI, Chapron C. The changing prevalence of infertility. Int J Gynecol Obstet (2013) 123:S4–8. doi: 10.1016/j.ijgo.2013.09.005
16. Forni PE, Wray S. GnRH, anosmia and hypogonadotropic hypogonadism–where are we? Front Neuroendocrin (2015) 36:165–77. doi: 10.1016/j.yfrne.2014.09.004
17. Bliss SP, Navratil AM, Xie J, Roberson MS. GnRH signaling, the gonadotrope and endocrine control of fertility. Front Neuroendocrino (2010) 31(3):322–40. doi: 10.1016/j.yfrne.2010.04.002
18. Coss D. Regulation of reproduction via tight control of gonadotropin hormone levels. Mol Cell Endocrinol (2018) 463:116–30. doi: 10.1016/j.mce.2017.03.022
19. Wang L, Moenter SM. Differential roles of hypothalamic AVPV and arcuate kisspeptin neurons in estradiol feedback regulation of female reproduction. Neuroendocrinology. (2020) 110(3-4):172–84. doi: 10.1159/000503006
20. Herbison AE, Moenter SM. Depolarising and hyperpolarising actions of GABA(A) receptor activation on gonadotrophin-releasing hormone neurones: Towards an emerging consensus. J neuroendocrinol (2011) 23(7):557–69. doi: 10.1111/j.1365-2826.2011.02145.x
21. Pielecka-Fortuna J, Chu Z, Moenter SM. Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology. (2008) 149(4):1979–86. doi: 10.1210/en.2007-1365
22. George JT, Seminara SB. Kisspeptin and the hypothalamic control of reproduction: Lessons from the human. Endocrinology. (2012) 153(11):5130–6. doi: 10.1210/en.2012-1429
23. Seminara SB. Kisspeptin in reproduction. Semin Reprod Med (2007) 25(5):337–43. doi: 10.1055/s-2007-984739
24. Novaira HJ, Fadoju D, Diaczok D, Radovick S. Genetic mechanisms mediating kisspeptin regulation of GnRH gene expression. J Neurosci Off J Soc Neurosci (2012) 32(48):17391–400. doi: 10.1523/jneurosci.2438-12.2012
25. Novaira HJ, Sonko ML, Hoffman G, Koo Y, Ko C, Wolfe A, et al. Disrupted kisspeptin signaling in GnRH neurons leads to hypogonadotrophic hypogonadism. Mol Endocrinol (2014) 28(2):225–38. doi: 10.1210/me.2013-1319
26. Sullivan SD, Moenter SM. Gamma-aminobutyric acid neurons integrate and rapidly transmit permissive and inhibitory metabolic cues to gonadotropin-releasing hormone neurons. Endocrinology. (2004) 145(3):1194–202. doi: 10.1210/en.2003-1374
27. Sullivan SD, Moenter SM. Neurosteroids alter gamma-aminobutyric acid postsynaptic currents in gonadotropin-releasing hormone neurons: A possible mechanism for direct steroidal control. Endocrinology. (2003) 144(10):4366–75. doi: 10.1210/en.2003-0634
28. DeFazio RA, Heger S, Ojeda SR, Moenter SM. Activation of a-type gamma-aminobutyric acid receptors excites gonadotropin-releasing hormone neurons. Mol Endocrinol (2002) 16(12):2872–91. doi: 10.1210/me.2002-0163
29. Slegtenhorst-Eegdeman KE, de Rooij DG, Verhoef-Post M, van de Kant HJ, Bakker CE, Oostra BA, et al. Macroorchidism in FMR1 knockout mice is caused by increased sertoli cell proliferation during testicular development. Endocrinology. (1998) 139(1):156–62. doi: 10.1210/endo.139.1.5706
30. Sherman SL, Curnow EC, Easley CA, Jin P, Hukema RK, Tejada MI, et al. Use of model systems to understand the etiology of fragile X-associated primary ovarian insufficiency (FXPOI). J Neurodev Disord (2014) 6(1):26. doi: 10.1186/1866-1955-6-26
31. Lu C, Lin L, Tan H, Wu H, Sherman SL, Gao F, et al. Fragile X premutation RNA is sufficient to cause primary ovarian insufficiency in mice. Hum Mol Genet (2012) 21(23):5039–47. doi: 10.1093/hmg/dds348
32. Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, Xunclà M, Badenas C, Kulisevsky J, et al. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet EJHG (2009) 17(10):1359–62. doi: 10.1038/ejhg.2009.51
33. Macpherson JN, Murray A. Development of genetic testing for fragile X syndrome and associated disorders, and estimates of the prevalence of FMR1 expansion mutations. Genes (2016) 7(12):E110. doi: 10.3390/genes7120110
34. Shelling AN. Premature ovarian failure. Reproduction. (2010) 140(5):633–41. doi: 10.1530/rep-09-0567
35. Chapman C, Cree L, Shelling AN. The genetics of premature ovarian failure: Current perspectives. Int J Womens Health (2015) 7:799–810. doi: 10.2147/IJWH.S64024
36. Shuster LT, Rhodes DJ, Gostout BS, Grossardt BR, Rocca WA. Premature menopause or early menopause: Long-term health consequences. Maturitas. (2010) 65(2):161–6. doi: 10.1016/j.maturitas.2009.08.003
37. Kenneson A, Zhang F, Hagedorn CH, Warren ST. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet (2001) 10(14):1449–54. doi: 10.1093/hmg/10.14.1449
38. Hoffman GE, Le WW, Entezam A, Otsuka N, Tong Z-B, Nelson L, et al. Ovarian abnormalities in a mouse model of fragile X primary ovarian insufficiency. J Histochem Cytochem (2012) 60(6):439–56. doi: 10.1369/0022155412441002
39. Conca Dioguardi C, Uslu B, Haynes M, Kurus M, Gul M, Miao DQ, et al. Granulosa cell and oocyte mitochondrial abnormalities in a mouse model of fragile X primary ovarian insufficiency. Mol Hum reproduct (2016) 22(6):384–96. doi: 10.1093/molehr/gaw023
40. Welt CK, Smith PC, Taylor AE. Evidence of early ovarian aging in fragile X premutation carriers. J Clin Endocrinol Metab (2004) 89(9):4569–74. doi: 10.1210/jc.2004-0347
41. Pastore LM, Johnson J. The FMR1 gene, infertility, and reproductive decision-making: a review. Front Genet (2014) 5:195. doi: 10.3389/fgene.2014.00195
42. Gougeon A. Regulation of ovarian follicular development in primates: facts and hypotheses. Endocr Rev (1996) 17(2):121–55. doi: 10.1210/edrv-17-2-121
43. Kreisman MJ, McCosh RB, Tian K, Song CI, Breen KM. Estradiol enables chronic corticosterone to inhibit pulsatile luteinizing hormone secretion and suppress Kiss1 neuronal activation in female mice. Neuroendocrinology. (2020) 110(6):501–16. doi: 10.1159/000502978
44. Hardiman RL, Bratt A. Hypothalamic-pituitary-adrenal axis function in fragile X syndrome and its relationship to behaviour: A systematic review. Physiol behavior (2016) 167:341–53. doi: 10.1016/j.physbeh.2016.09.030
45. Yang JA, Song CI, Hughes JK, Kreisman MJ, Parra RA, Haisenleder DJ, et al. Acute psychosocial stress inhibits LH pulsatility and Kiss1 neuronal activation in female mice. Endocrinology. (2017) 158(11):3716–23. doi: 10.1210/en.2017-00301
46. Breen KM, Thackray VG, Hsu T, Mak-McCully RA, Coss D, Mellon PL. Stress levels of glucocorticoids inhibit LHbeta-subunit gene expression in gonadotrope cells. Mol Endocrinol (2012) 26(10):1716–31. doi: 10.1210/me.2011-1327
47. Gay VL, Midgley AR Jr., Niswender GD. Patterns of gonadotrophin secretion associated with ovulation. Fed Proc (1970) 29(6):1880–7.
48. Haisenleder DJ, Schoenfelder AH, Marcinko ES, Geddis LM, Marshall JC. Estimation of estradiol in mouse serum samples: evaluation of commercial estradiol immunoassays. Endocrinology. (2011) 152(11):4443–7. doi: 10.1210/en.2011-1501
49. Wilson RC, Kesner JS, Kaufman JM, Uemura T, Akema T, Knobil E. Central electrophysiologic correlates of pulsatile luteinizing hormone secretion in the rhesus monkey. Neuroendocrinology. (1984) 39(3):256–60. doi: 10.1159/000123988
50. Moenter SM, Brand RM, Midgley AR, Karsch FJ. Dynamics of gonadotropin-releasing hormone release during a pulse. Endocrinology. (1992) 130(1):503–10. doi: 10.1210/endo.130.1.1727719
51. Steyn FJ, Wan Y, Clarkson J, Veldhuis JD, Herbison AE, Chen C. Development of a methodology for and assessment of pulsatile luteinizing hormone secretion in juvenile and adult male mice. Endocrinology. (2013) 154(12):4939–45. doi: 10.1210/en.2013-1502
52. McCosh RB, Kreisman MJ, Breen KM. Frequent tail-tip blood sampling in mice for the assessment of pulsatile luteinizing hormone secretion. J visualized experiments JoVE (2018) 137(137):57894. doi: 10.3791/57894
53. Vidal A, Zhang Q, Médigue C, Fabre S, Clément F. DynPeak: an algorithm for pulse detection and frequency analysis in hormonal time series. PloS One (2012) 7(7):e39001–e. doi: 10.1371/journal.pone.0039001
54. Chen KE, Lainez NM, Nair MG, Coss D. Visceral adipose tissue imparts peripheral macrophage influx into the hypothalamus. J neuroinflamm (2021) 18(1):140. doi: 10.1186/s12974-021-02183-2
55. Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, et al. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res (2016) 44(W1):W147–53. doi: 10.1093/nar/gkw419
56. Ge SX, Jung D, Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics (2020) 36(8):2628–9. doi: 10.1093/bioinformatics/btz931
57. Chen KE, Lainez NM, Coss D. Sex differences in macrophage responses to obesity-mediated changes determine migratory and inflammatory traits. J Immunol (2021) 206(1):141–53. doi: 10.4049/jimmunol.2000490
58. Lainez NM, Coss D. Leukemia inhibitory factor represses GnRH gene expression via cFOS during inflammation in Male mice. Neuroendocrinology. (2019) 108(4):291–307. doi: 10.1159/000496754
59. Yeh DM, Coss D. PACAP induces FSHbeta gene expression via EPAC. Mol Cell Endocrinol (2019) 492:110438. doi: 10.1016/j.mce.2019.04.018
60. Lainez NM, Jonak CR, Nair MG, Ethell IM, Wilson EH, Carson MJ, et al. Diet-induced obesity elicits macrophage infiltration and reduction in spine density in the hypothalami of Male but not female mice. Front Immunol (2018) 9:1992(1992). doi: 10.3389/fimmu.2018.01992
61. Jonak CR, Lainez NM, Boehm U, Coss D. GnRH receptor expression and reproductive function depend on JUN in GnRH Receptor‒Expressing cells. Endocrinology. (2018) 159(3):1496–510. doi: 10.1210/en.2017-00844
62. Evans MC, Rizwan M, Mayer C, Boehm U, Anderson GM. Evidence that insulin signalling in gonadotrophin-releasing hormone and kisspeptin neurones does not play an essential role in metabolic regulation of fertility in mice. J neuroendocrinol (2014) 26(7):468–79. doi: 10.1111/jne.12166
63. Shi Y, Pontrello CG, DeFea KA, Reichardt LF, Ethell IM. Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J Neurosci Off J Soc Neurosci (2009) 29(25):8129–42. doi: 10.1523/jneurosci.4681-08.2009
64. Pontrello CG, Sun MY, Lin A, Fiacco TA, DeFea KA, Ethell IM. Cofilin under control of beta-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci United States America (2012) 109(7):E442–51. doi: 10.1073/pnas.1118803109
65. Sidhu H, Dansie LE, Hickmott PW, Ethell DW, Ethell IM. Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J Neurosci Off J Soc Neurosci (2014) 34(30):9867–79. doi: 10.1523/jneurosci.1162-14.2014
66. Koeppen J, Nguyen AQ, Nikolakopoulou AM, Garcia M, Hanna S, Woodruff S, et al. Functional consequences of synapse remodeling following astrocyte-specific regulation of ephrin-B1 in the adult hippocampus. J Neurosci Off J Soc Neurosci (2018) 38(25):5710–26. doi: 10.1523/jneurosci.3618-17.2018
67. Lovelace JW, Rais M, Palacios AR, Shuai XS, Bishay S, Popa O, et al. Deletion of Fmr1 from forebrain excitatory neurons triggers abnormal cellular, EEG, and behavioral phenotypes in the auditory cortex of a mouse model of fragile X syndrome. Cereb Cortex (2020) 30(3):969–88. doi: 10.1093/cercor/bhz141
68. Berman RF, Buijsen RA, Usdin K, Pintado E, Kooy F, Pretto D, et al. Mouse models of the fragile X premutation and fragile X-associated tremor/ataxia syndrome. J Neurodev Disord (2014) 6(1):25. doi: 10.1186/1866-1955-6-25
69. Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, Zu T, Hoogeveen-Westerveld M, Severijnen L, et al. The generation of a conditional Fmr1 knock out mouse model to study fmrp function in vivo. Neurobiol Dis (2006) 21(3):549–55. doi: 10.1016/j.nbd.2005.08.019
70. Noto V, Harrity C, Walsh D, Marron K. The impact of FMR1 gene mutations on human reproduction and development: a systematic review. J assisted Reprod Genet (2016) 33(9):1135–47. doi: 10.1007/s10815-016-0765-6
71. Sullivan AK, Marcus M, Epstein MP, Allen EG, Anido AE, Paquin JJ, et al. Association of FMR1 repeat size with ovarian dysfunction. Hum reproduct (2005) 20(2):402–12. doi: 10.1093/humrep/deh635
72. Terasawa E. Neuroestradiol in regulation of GnRH release. Hormones behavior (2018) 104:138–45. doi: 10.1016/j.yhbeh.2018.04.003
73. Radovick S, Levine JE, Wolfe A. Estrogenic regulation of the GnRH neuron. Front endocrinol (2012) 3:52. doi: 10.3389/fendo.2012.00052
74. Herbison AE. The gonadotropin-releasing hormone pulse generator. Endocrinology. (2018) 159(11):3723–36. doi: 10.1210/en.2018-00653
75. Goodman RL, Lehman MN. Kisspeptin neurons from mice to men: similarities and differences. Endocrinology. (2012) 153(11):5105–18. doi: 10.1210/en.2012-1550
76. Makanji Y, Zhu J, Mishra R, Holmquist C, Wong WPS, Schwartz NB, et al. Inhibin at 90: From discovery to clinical application, a historical review. Endocr Rev (2014) 35(5):747–94. doi: 10.1210/er.2014-1003
77. Bernard DJ, Fortin J, Wang Y, Lamba P. Mechanisms of FSH synthesis: what we know, what we don't, and why you should care. Fertil Steril (2010) 93(8):2465–85. doi: 10.1016/j.fertnstert.2010.03.034
78. Prilutsky D, Kho AT, Palmer NP, Bhakar AL, Smedemark-Margulies N, Kong SW, et al. Gene expression analysis in Fmr1KO mice identifies an immunological signature in brain tissue and mGluR5-related signaling in primary neuronal cultures. Mol Autism (2015) 6(1):66. doi: 10.1186/s13229-015-0061-9
79. Spergel DJ. Modulation of gonadotropin-releasing hormone neuron activity and secretion in mice by non-peptide neurotransmitters, gasotransmitters, and gliotransmitters. Front endocrinol (2019) 10:329. doi: 10.3389/fendo.2019.00329
80. Antoine MW, Langberg T, Schnepel P, Feldman DE. Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron. (2019) 101(4):648–61.e4. doi: 10.1016/j.neuron.2018.12.026
81. Sullivan SD, DeFazio RA, Moenter SM. Metabolic regulation of fertility through presynaptic and postsynaptic signaling to gonadotropin-releasing hormone neurons. J Neurosci Off J Soc Neurosci (2003) 23(24):8578–85. doi: 10.1523/JNEUROSCI.23-24-08578.2003
82. Yin W, Mendenhall JM, Bratton SB, Oung T, Janssen WG, Morrison JH, et al. Novel localization of NMDA receptors within neuroendocrine gonadotropin-releasing hormone terminals. Exp Biol Med (2007) 232(5):662–73.
83. Urbanski HF, Ojeda SR. A role for n-methyl-D-aspartate (NMDA) receptors in the control of LH secretion and initiation of female puberty. Endocrinology. (1990) 126:1774–6. doi: 10.1210/endo-126-3-1774
84. Iremonger KJ, Constantin S, Liu X, Herbison AE. Glutamate regulation of GnRH neuron excitability. Brain Res (2010) 1364:35–43. doi: 10.1016/j.brainres.2010.08.071
85. Christian CA, Moenter SM. The neurobiology of preovulatory and estradiol-induced gonadotropin-releasing hormone surges. Endocr Rev (2010) 31(4):544–77. doi: 10.1210/er.2009-0023
86. Seeburg PH, Burnashev N, Kohr G, Kuner T, Sprengel R, Monyer H. The NMDA receptor channel: molecular design of a coincidence detector. Recent Prog Horm Res (1995) 50:19–34. doi: 10.1016/B978-0-12-571150-0.50006-8
87. Lim WL, Idris MM, Kevin FS, Soga T, Parhar IS. Maternal dexamethasone exposure alters synaptic inputs to gonadotropin-releasing hormone neurons in the early postnatal rat. Front endocrinol (2016) 7:117. doi: 10.3389/fendo.2016.00117
88. Herde MK, Herbison AE. Morphological characterization of the action potential initiation segment in GnRH neuron dendrites and axons of Male mice. Endocrinology. (2015) 156(11):4174–86. doi: 10.1210/en.2015-1284
89. Chan H, Prescott M, Ong Z, Herde MK, Herbison AE, Campbell RE. Dendritic spine plasticity in gonadatropin-releasing hormone (GnRH) neurons activated at the time of the preovulatory surge. Endocrinology. (2011) 152(12):4906–14. doi: 10.1210/en.2011-1522
90. Cottrell EC, Campbell RE, Han SK, Herbison AE. Postnatal remodeling of dendritic structure and spine density in gonadotropin-releasing hormone neurons. Endocrinology. (2006) 147(8):3652–61. doi: 10.1210/en.2006-0296
91. Li S, Takumi K, Iijima N, Ozawa H. The increase in the number of spines on the gonadotropin-releasing hormone neuron across pubertal development in rats. Cell Tissue Res (2016) 364(2):405–14. doi: 10.1007/s00441-015-2335-0
92. Murray A. Premature ovarian failure and the FMR1 gene. Semin Reprod Med (2000) 18(1):59–66. doi: 10.1055/s-2000-13476
93. Robinson LJ, Tourkova I, Wang Y, Sharrow AC, Landau MS, Yaroslavskiy BB, et al. FSH-receptor isoforms and FSH-dependent gene transcription in human monocytes and osteoclasts. Biochem Biophys Res Commun (2010) 394(1):12–7. doi: 10.1016/j.bbrc.2010.02.112
94. Iqbal J, Sun L, Kumar TR, Blair HC, Zaidi M. Follicle-stimulating hormone stimulates TNF production from immune cells to enhance osteoblast and osteoclast formation. Proc Natl Acad Sci United States America (2006) 103(40):14925–30. doi: 10.1073/pnas.0606805103
95. Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, Papachristou DJ, et al. FSH directly regulates bone mass. Cell. (2006) 125(2):247–60. doi: 10.1016/j.cell.2006.01.051
96. Zaidi M, Blair HC, Iqbal J, Davies TF, Zhu LL, Zallone A, et al. New insights: elevated follicle-stimulating hormone and bone loss during the menopausal transition. Curr Rheumatol Rep (2009) 11(3):191–5. doi: 10.1007/s11926-009-0026-0
97. Zhu LL, Tourkova I, Yuen T, Robinson LJ, Bian Z, Zaidi M, et al. Blocking FSH action attenuates osteoclastogenesis. Biochem Biophys Res Commun (2012) 422(1):54–8. doi: 10.1016/j.bbrc.2012.04.104
98. Hale GE, Robertson DM, Burger HG. The perimenopausal woman: Endocrinology and management. J Steroid Biochem Mol Biol (2014) 142:121–31. doi: 10.1016/j.jsbmb.2013.08.015
99. Fraser HM, Wulff C. Angiogenesis in the corpus luteum. Reprod Biol Endocrinol RB&E (2003) 1:88. doi: 10.1186/1477-7827-1-88
100. Thackray VG, Mellon PL, Coss D. Hormones in synergy: Regulation of the pituitary gonadotropin genes. Mol Cell Endocrinol (2010) 314(2):192–203. doi: 10.1016/j.mce.2009.09.003
101. Thackray VG, McGillivray SM, Mellon PL. Androgens, progestins and glucocorticoids induce follicle-stimulating hormone β-subunit gene expression at the level of the gonadotrope. Mol Endocrinol (2006) 20(9):2062–79. doi: 10.1210/me.2005-0316
102. Stouffer RL, Bishop CV, Bogan RL, Xu F, Hennebold JD. Endocrine and local control of the primate corpus luteum. Reprod Biol (2013) 13(4):259–71. doi: 10.1016/j.repbio.2013.08.002
103. Stouffer RL, Xu F, Duffy DM. Molecular control of ovulation and luteinization in the primate follicle. Front biosci J virtual library (2007) 12:297–307. doi: 10.2741/2065
104. Trau HA, Davis JS, Duffy DM. Angiogenesis in the primate ovulatory follicle is stimulated by luteinizing hormone via prostaglandin E2. Biol Reprod (2015) 92(1):15. doi: 10.1095/biolreprod.114.123711
105. Robker RL, Hennebold JD, Russell DL. Coordination of ovulation and oocyte maturation: A good egg at the right time. Endocrinology. (2018) 159(9):3209–18. doi: 10.1210/en.2018-00485
106. Stocco C, Telleria C, Gibori G. The molecular control of corpus luteum formation, function, and regression. Endocr Rev (2007) 28(1):117–49. doi: 10.1210/er.2006-0022
107. Fraser HM, Dickson SE, Lunn SF, Wulff C, Morris KD, Carroll VA, et al. Suppression of luteal angiogenesis in the primate after neutralization of vascular endothelial growth factor. Endocrinology. (2000) 141(3):995–1000. doi: 10.1210/endo.141.3.7369
108. Ringstrom SJ, Szabo M, Kilen SM, Saberi S, Knox KL, Schwartz NB. The antiprogestins RU486 and ZK98299 affect follicle-stimulating hormone secretion differentially on estrus, but not on proestrus. Endocrinology. (1997) 138(6):2286–90. doi: 10.1210/endo.138.6.5161
109. Knox KL, Schwartz NB. RU486 blocks the secondary surge of follicle-stimulating hormone in the rat without blocking the drop in serum inhibin. Biol Reprod (1992) 46(2):220–5. doi: 10.1095/biolreprod46.2.220
110. Riquelme R, Ruz F, Mayerhofer A, Lara HE. Role of ovarian sympathetic nerves and cholinergic local system during cold stress. J Endocrinol (2019) 242(2):115–24. doi: 10.1530/joe-19-0125
111. Lara HE, Dorfman M, Venegas M, Luza SM, Luna SL, Mayerhofer A, et al. Changes in sympathetic nerve activity of the mammalian ovary during a normal estrous cycle and in polycystic ovary syndrome: Studies on norepinephrine release. Microsc Res technique (2002) 59(6):495–502. doi: 10.1002/jemt.10229
112. Uchida S. Sympathetic regulation of estradiol secretion from the ovary. Auton Neurosci (2015) 187:27–35. doi: 10.1016/j.autneu.2014.10.023
113. Mayerhofer A, Dissen GA, Costa ME, Ojeda SR. A role for neurotransmitters in early follicular development: Induction of functional follicle-stimulating hormone receptors in newly formed follicles of the rat ovary. Endocrinology. (1997) 138(8):3320–9. doi: 10.1210/endo.138.8.5335
114. Aguado LI, Ojeda SR. Ovarian adrenergic nerves play a role in maintaining preovulatory steroid secretion. Endocrinology. (1984) 114(5):1944–6. doi: 10.1210/endo-114-5-1944
115. Ramírez Hernández DA, Vieyra Valdez E, Rosas Gavilán G, Linares Culebro R, Espinoza Moreno JA, Chaparro Ortega A, et al. Role of the superior ovarian nerve in the regulation of follicular development and steroidogenesis in the morning of diestrus 1. J assisted Reprod Genet (2020) 37(6):1477–88. doi: 10.1007/s10815-020-01787-6
Keywords: Fragile X Syndrome, FMR1, GnRH, hypothalamus, FSH, ovary innervation
Citation: Villa PA, Lainez NM, Jonak CR, Berlin SC, Ethell IM and Coss D (2023) Altered GnRH neuron and ovarian innervation characterize reproductive dysfunction linked to the Fragile X messenger ribonucleoprotein (Fmr1) gene mutation. Front. Endocrinol. 14:1129534. doi: 10.3389/fendo.2023.1129534
Received: 22 December 2022; Accepted: 09 February 2023;
Published: 22 February 2023.
Edited by:
Neil James MacLusky, University of Guelph, CanadaReviewed by:
Pascale Crepieux, Centre National de la Recherche Scientifique (CNRS), FranceLori Raetzman, University of Illinois at Urbana-Champaign, United States
Copyright © 2023 Villa, Lainez, Jonak, Berlin, Ethell and Coss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Djurdjica Coss, ZGp1cmRqaWNhLmNvc3NAdWNyLmVkdQ==