
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 03 May 2023
Sec. Pituitary Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1123267
This article is part of the Research Topic Personalized Management of Acromegaly View all 7 articles
 Antonio Bianchi1,2
Antonio Bianchi1,2 Sabrina Chiloiro1,2*
Sabrina Chiloiro1,2* Antonella Giampietro1,2Simona Gaudino3Rosalinda Calandrelli3Ciro Mazzarella4
Antonella Giampietro1,2Simona Gaudino3Rosalinda Calandrelli3Ciro Mazzarella4 Carmelo Caldarella5Mario Rigante6Marco Gessi7,8Liverana Lauretti9,10Laura De Marinis1,2
Carmelo Caldarella5Mario Rigante6Marco Gessi7,8Liverana Lauretti9,10Laura De Marinis1,2 Alessandro Olivi9,10Alfredo Pontecorvi1,2Francesco Doglietto9,10
Alessandro Olivi9,10Alfredo Pontecorvi1,2Francesco Doglietto9,10Growth Hormone-secreting adenomas exhibits variable biological behavior and heterogeneous natural history, ranging from small adenomas and mild disease, to invasive and aggressive neoplasms with more severe clinical picture. Patients not cured or controlled after neurosurgical and first-generation somatostatin receptor ligands (SRL) therapy could require multiple surgical, medical and/or radiation treatments to achieve disease control. To date, no clinical, laboratory, histopathological, or neuroradiological markers are able to define the aggressiveness or predict the disease prognosis in patients with acromegaly. Therefore, the management of these patients requires careful evaluation of laboratory assessments, diagnostic criteria, neuroradiology examinations, and neurosurgical approaches to choose an effective and patient-tailored medical therapy. A multidisciplinary approach is particularly useful in difficult/aggressive acromegaly to schedule multimodal treatment, which includes radiation therapy, chemotherapy with temozolomide and other, recent emerging treatments. Herein, we describe the role of the different members of the multidisciplinary team according to our personal experience; a flow-chart for the therapeutic approach of difficult/aggressive acromegaly patients is proposed.
Growth hormone (GH) secreting pituitary adenomas or pituitary neuroendocrine tumors (PitNET) represent a heterogeneous group of neoplasia with complex and variable biological behavior. Although surgery and first-line medical therapy with somatostatin receptor ligands (SRL) are the cornerstones of treatment of acromegaly, a non-negligible percentage of patients (from 24 to 65%) do not reach biochemical disease control (1).
The inadequate biochemical control of acromegaly may be due to suboptimal dosing of medical therapies, poor compliance to treatments, resistance to drugs, tumor phenotype, inadequate monitoring, and uncertainty of GH and IGF-I assays (2). For these reasons, in this paper we defined “difficult” the GHomas not cured/controlled after neurosurgical (first line) and first generation SRL (second line) therapy. Concerning the term “aggressive”, at present, a clear definition of aggressive pituitary adenomas remains equivocal (3). According to clinical practice (4) and to European Society of Endocrinology (ESE) guidelines (5), GH-PitNET/adenomas are defined as aggressive if, invasive, with a high proliferative index, with refractory behavior and poor response to optimal standard treatments such as surgical, medical, and radiotherapy, and in cases of multiple local recurrences (5, 6). In our opinion, the most reliable definition is that resulting from cluster analysis (type 3 acromegalic patients) defined by Cuevas-Ramos and coworkers (4) and based on clinical, radiological, histopathological, and outcome characteristics. This definition of difficult/aggressive ranges between SRL partial responders adenomas to more aggressive and invasive one and it is a good compromise between two evidences: the definition of aggressiveness according to the presence of local invasion of surrounding structures of about 35-50% of these neoplasms based on the recent validation of the French five-tiered classification (7–11); the definition of aggressiveness based on the ESE guidelines: “the hallmark of aggressiveness is clinically relevant tumour growth despite the use of optimal standard therapies, which entails a combination of medical therapies, surgery and radiotherapy” (5).
The incidence of aggressive adenomas/PitNET ranges from 4.5 to 31% of patients and reflect the different definitions (12). A comprehensive definition of difficult/aggressive GH-secreting PitNETs probably requires multidisciplinary evaluation in a team that includes experts in all the fields of pituitary disease, including neuro-endocrinology, neurosurgery, neuropathology, neuroradiology, otolaryngology, radiation oncology and nuclear medicine (13).
In this perspective viewpoint, we critically review the different aspects that should be taken into account for multidisciplinary management of a patient with a difficult/aggressive GH-secreting adenomas/PitNET from diagnosis to the choice of treatment.
Early recognition of severe disease is crucial. At the time of diagnosis, clinical criteria for identifying a difficult/aggressive case of GH-secreting PitNETs are not univocally recognized. In clinical practice, before the treatment choice, we suspect difficult/aggressive GH-secreting adenomas/PitNET typically in cases of young patients, who might also refer signs and symptoms of hypopituitarism rather than those related to GH/IGF-I excess. This high prevalence of pituitary dysfunction in young acromegaly patients may be explained by the presence of an invasive macro- or giant adenomas/PitNET with a extrasellar extension and/or with an unusually high rate of growth (5, 14). In parallel, high GH and IGF-I levels at diagnosis are an expression of larger and invasive adenomas/PitNET (4). No data are available on comorbidities that can be identified after the diagnosis of acromegaly. However, the presence of multiple acromegaly-related complications in the same patient is considered as suggestive of more severe disease (15), due to a direct effect of high levels of circulating GH/IGF-1 (16) or to the diagnostic delay.
Magnetic resonance imaging (MRI) and computed tomography (CT) can provide significant data that should alert the clinician to a potentially aggressive behavior of the GH-secreting PitNET. Large tumor size, extra-sellar extension, and postoperative residues are generally considered to be predictors of poor outcome. A diameter that is > 15 mm has been reported in aggressive GH tumors (12) and has been detected mostly in sparsely granulated lesions (17). Aggressive GH-adenomas/PitNET show specific patterns of growth. Unlike other PitNETs, infrasellar invasion is the most common pattern of growth, with erosion of the sellar floor and clivus, that are commonly detected on preoperative CT (18, 19). The invasion of dura and sellar diaphragm is poorly identified by MRI (20). The major site of dura invasion is the medial wall of the cavernous sinus.
The tumor’s relation to the cavernous sinus is classically quantified by Knosp scoring on MRI, which measures lateral tumor extension in relation to the internal carotid arteries preoperatively (21, 22). If compared to densely granulated (DS) GH-secreting PitNETs, sparsely granulated (SG) tumors are more likely to invade the cavernous sinus (grades 3–4 of Knosp’s classification) (17).Some MRI sequences and techniques may help to predict the more SG phenotype of GH secreting PitNET: high T2 signal intensity, due to a low collagen content and low number of secreted granules, and a more avid enhancement are found in SG adenomas (17). T2WI-based texture parameters of the whole tumor appear to be able to provide more quantitative information and help predict granulation pattern better than T2 signal intensity (23). Preliminary studies showed that texture signatures based on T1WI and post-contrast T1WI of specific solid tumor areas may reflect on the biological behavior of the tumor and achieved greater diagnostic efficacy than in the entire tumor (24).
In the post-operative period and during follow-up, MRI plays a significant role in assessing surgical outcomes, as well as documenting local recurrence, progression of residual disease to surrounding tissues, and rare distant metastases both in the central nervous system and in extracranial organs.
Some pituitary carcinomas may develop from an invasive GH-adenomas/PitNET and metastasize via the subarachnoid space and lymph and blood vessels to the brain and extracranial organs, especially liver or bones, also requiring total body CT (25, 26).
Surgical treatment of acromegaly presents specific challenges, including specific anesthesiologic issues, anatomical variations of the sellar and parasellar regions, and nasal mucosa edema (27), which need to be evaluated pre-operatively to optimize treatment and decrease complications. Endoscopic transsphenoidal surgery is a relatively novel technique, in which the endoscope, together with the optimization of the transnasal corridor, provides the possibility of visualizing even the extrasellar components of pituitary adenomas. This has led to the possibility of exploring the suprasellar area and cavernous sinus with limited morbidity and higher efficacy compared to “classic” surgical approaches (28–32). Surgical experience has been demonstrated to be a significant factor for optimal outcomes, underlining the importance of centers of excellence and sub-specializations (33, 34) Other technical advancements, such as intraoperative Doppler and neuronavigation, have also led to safer and more effective surgeries. Despite these recent technical and organizational advancements, true invasiveness in GH-adenomas/PitNET remains a major limiting factor for surgical disease remission, even in case of an aggressive resection (30, 31, 35–41).
The pathological classification of pituitary adenoma/PitNETs has been recently remodeled, according to the “2017 WHO Classification of Tumors of Endocrine Organs”, and to a subsequent document by the European Pituitary Pathology Group (EPPG) on pituitary pathology (42, 43), which anticipate the new 2022 WHO Classification (44). The use of the “pituitary neuroendocrine tumors” is still matter of debate. The PANOMEN Workshop recommends that the term adenoma be retained (Ho 2021) and the 5th Edition of the WHO Classification of Endocrine and Neuroendocrine Tumors retains adenoma in duality as transition terminology (Ho 2022). According to the last WHO classification, we use the dual PitNET/adenoma term. A clinical-pathological approach was introduced based on a combination of parameters with important prognostic value, as will be seen when discussing the therapeutic approach. For this reason, in the management of adenomas/PitNET, a complete pathology report is necessary according to the criteria of the pituitary center of excellence (13). Therefore, preoperative information such as clinical and GH/IGF-I plasma levels, as well as MRI features, are mandatory. Moreover, for risk stratification, it is crucial to know the following: histology (mitotic count and histological invasion); immunohistochemistry [pituitary hormone reactivity, cytokeratin pattern (densely or sparsely granulated), proliferation markers (MIB1/Ki-67), p53 percentage, somatostatin receptos (SSTR) type and score (at least SSTR-2 and -5 if requested)] according to IRS (45) or Volante (46) Score. In selected cases, it is necessary to know O6-methylguanine-DNA methyl-transferase (MGMT) status. Unfortunately, there is currently no reliable marker of malignancy. However, the recent validation of the French five-tiered classification, which considers clinical and histological parameters, seems to be a good starting point for the introduction of a better classification of GH-secreting adenomas/PitNETs in terms of aggressiveness (7). In selected patients stratified by a risk category system, the identification of aryl hydrocarbon receptor-interacting protein (AIP) gene mutation can lead to the detection of carriers, potentially leading to a better prognosis (47). Recently, an interesting algorithm have been created to try to predict response to SRL prime line treatment from features such as age at diagnosis, sex, GH, and IGF-I levels at diagnosis and at pretreatment, SSTR-2 and -5 and cytokeratin granulation pattern (48).
The effective management of GH-secreting adenomas/PitNETs tumors firstly requires the suppression of autonomous GH secretion, normalization of the IGF-I, and removal or (at least) debulking of the pituitary tumor mass (2).
A convincing and detailed clinical definition of difficult/aggressive GH-adenomas/PitNET was proposed some years ago by the group of Shlomo Melmed (4). Based on cluster analysis, acromegaly patients were stratified into three phenotypes that range from benign (type 1) to aggressive (type 3). Patients with young age at diagnosis, short progressive disease duration, SG invasive macroadenomas, high GH and IGF-I secretion output, high Ki-67, low expression of SSTR2, and who require multiple treatment modalities belong to the latter group.
The large majority of aggressive GH-secreting adenomas/PitNET are characterized by resistance to treatment with first-generation somatostatin analogs. Current definition of resistance to SRL is based on the efficacy to control GH and IGF-I secretion and to induce tumor shrinkage (49). “Biochemical resistance” and a “tumor mass resistance” may be distinguished.
Whereas biochemical and tumoral responses are generally associated, there are some patients in which these responses are discordant (50, 51). The frequency of SRL resistance may also be influenced by the treatment setting, the duration and dosage of treatments, and by the use of optimal GH and IGF-I assay, a part of tumor phenotype (52).
Patients considered partially or completely resistant to first-generation SRL more frequently had post-surgical tumor residual, with higher secretion of GH and IGF-I (53). Tumor proliferation may predict the outcome of medical treatment in acromegaly: lower proliferative index (Ki-67) is typically identified in tumors responsive to first-generation SRL (54). SG GH-secreting tumors are more frequently resistant to SRL in contrast to DG lesions (54, 55). The hyper-intensity of the tumoral mass in T2-weighted MRIs seems to be associated with the SG cytokeratin pattern and resistance to treatment with first-generation SRL (56). Great interest is related to the role of SSTR in predicting the response to SRL (57). Several studies have demonstrated that tumors responsive to first-generation SRL showed diffuse and membranous SSTR2A expression (58, 59). In case of persistent acromegaly after neurosurgery and/or a standard dose of SRL (octreotide LAR 30 mg/28 days or lanreotide autogel 120 mg/28 days), a treatment regimen with higher dose or increased dose frequency compared to a conventional SRL may be useful, in particular in those patients who have reached a certain/partial response to the standard dose of first-generation SRL (60).
The current guidelines suggest the use of Peg-V in patients of irrelevant residual tumor and in those with alterations of glucose metabolism, while treatment with first-generation analogues with Peg-V or PAS should be chosen in the presence of important tumor concerns (60).
With regard of predictors of second-line therapies (Pegvisomant and Pasireotide Lar), a poor response to Peg-V seems related to high pre-treatment levels of GH/IGF-I, large tumor extension, and high Ki-67 values (61), while tumor extension to III ventricle, high pre-treatment GH/IGF-I levels, densely granulated PitNET, low SSTR5 score, complete resistance to first-generation SSA, and high Ki-67 values are related with a PAS poor response (62, 63).
Concerning the adenomas/PitNETs GH/IGF-I secretory output, the pharmacogenomics of the GH receptor seem to play a role: in fact, the presence of the deleted isoform of exon 3 of the receptor (d3-GHR) seems to correlate with a poor response to both pasireotide and to standard dosages of Peg-V (61, 63). Peptide receptor radionuclide therapy (PRRT) seems to be a promising treatment in selected cases, expressing SSTR and demonstrating sufficient tumor uptake of tracer by 68Ga-DOTATATE-PET/CT scan (64).
In the rare cases with full absence/low score of SSTR2 and SSTR5 expression, syndromic acromegaly should be investigated. These patients should be treated with Pegvisomant in monotherapy, even in the presence of high proliferative activity and invasive tumors, but an aggressive approach to residual tumor is mandatory, with debulking surgery if clinically appropriate or with radiotherapy/radiosurgery, also in combination with temozolomide (TMZ) (2, 63, 65). TMZ is first-line chemotherapy and recommended for treatment of aggressive adenomas/PitNETs (5, 64) that are resistant to previous therapies.
In summary, in Figure 1, we report our proposed therapeutic flowchart that is based, after unsuccessfully neurosurgery, on biochemical response to first generation SRL. We consider mandatory the knowledge of GH/IGF-I levels and tumor concerns before starting second line therapies, an molecular biomarkers such as the cytokeratin patterns, the proliferative index, expression of SSTRs We propose four clinical scenarios (Figure 1) driving the patient management from the possibility to try a high-dose SRL trial, according to ESE guidelines (in which the therapeutic schedule is monotherapy with first-generation SRL, or Pegvisomant or Pasireotide Lar and combination treatment with first- or second-generation SRL and Pegvisomant (60).
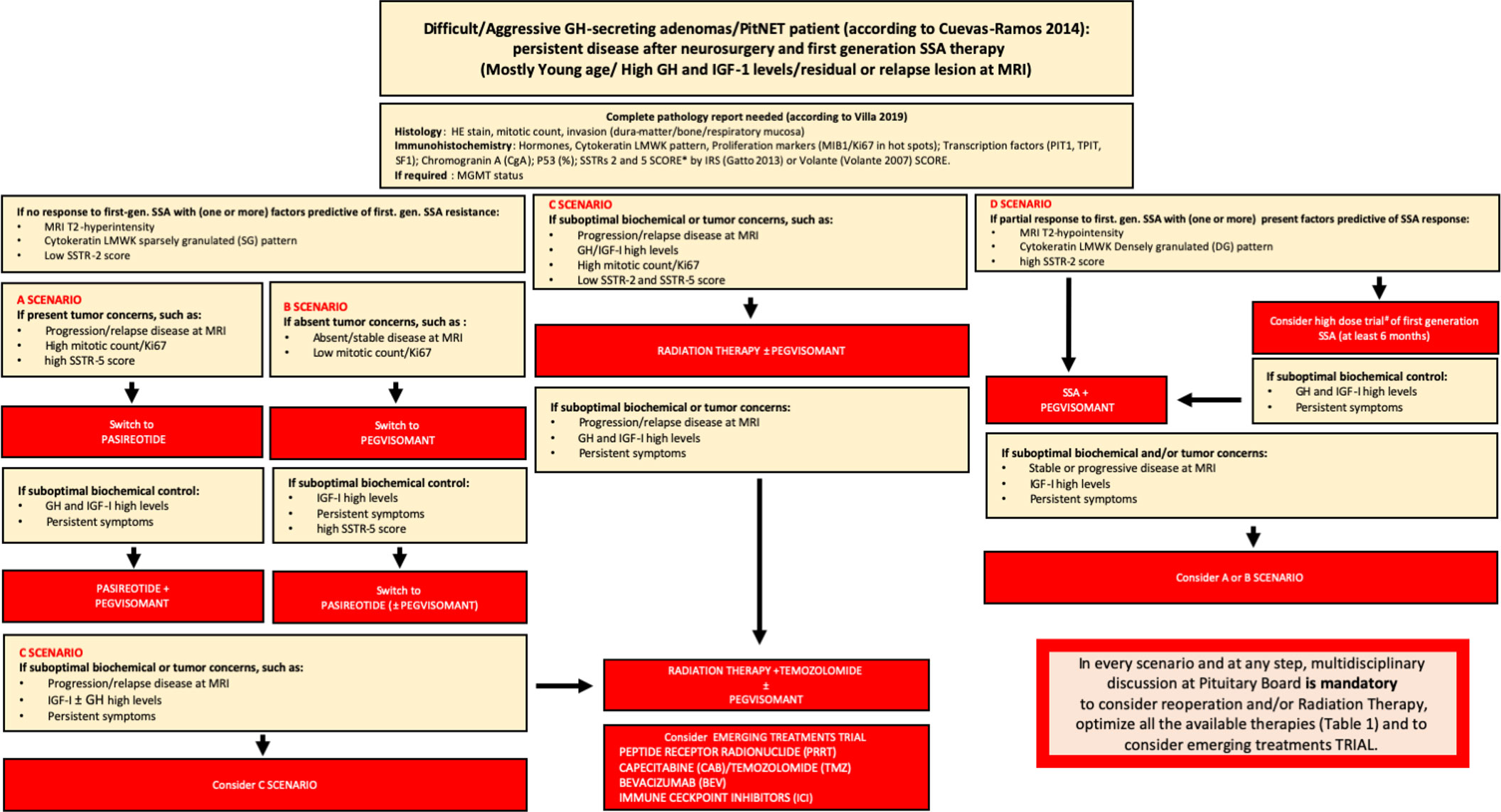
Figure 1 Flow-chart for the management of difficult/aggressive GH-secreting PitNET. After a complete pathology report according to most recent WHO classification, in the presence of no response to first-generation -SSA (and usually one or more factors predictive or first-generation SSA resistance – MRI T2-hyperintensity, cytokeratin SG pattern, low SSTR-2 score), there are 4 scenarios on the basis of tumor concerns and SSTR-5 score: A scenario (in presence of tumor concerns and high SSTR-5 score) drives the switch to PAS treatment alone or, after suboptimal biochemical control, combined to Peg-V; in B scenario (if absent tumor concerns and regardless SSTR-5 score) Peg-V alone is suggested, with the possibility to add PAS if suboptimal biochemical control is present and SSTR-5 score is favorable; in C scenario (in presence of tumor concerns and low SSTR-5 score), Peg-V + Radiation Therapy ± Temozolomide are the main treatment choices. We suggest that patients considered partially resistant to first-generation SSAs may be treated with SSA + Peg-V in cases of expected benign and favorable disease behavior, taking into account a low proliferative index, a non-detectable or not-invasive residual and in the presence of a high SSTR-2 score; alternatively, a high-dose trial of SSA is suggested (D scenario). If after SSA + Peg-V treatment, no biochemical control is achieved, on the basis of tumor concerns and SSTR-5 pattern, A or B scenario is the following step. Finally, if no disease control is obtained after Peg-V + Radiation Therapy ± Temozolomide, we suggest to consider an emerging treatment trial. In every scenario and at every step, multidisciplinary discussion at the Pituitary Board is mandatory to carefully consider reoperation and/or RT and/or every medical treatment choice.* = IRS Score: low expression of SSTRs = score > 6; high expression of SSTRs = score ³ 6; Volante Score: low expression of SSTRs = score 0 - 1; high expression of SSTRs = 2-3; IRS Score; # = lanreotide 120 mg every 14 or 21 days; lanreotide 180 mg every 28 days; or octreotide LAR 30 mg/ every 14 or 21 weeks; octreotide LAR 60 mg every 28 days.
The few patients that fail to reach disease control despite all these multimodal treatments should also be evaluated with the support of an oncologist. In this view, new target therapies are emerging, such as Capecitabine (CAP) or one of its metabolites 5-fluorouraciel (5-FU) (65); Bevacizumab (BEV), and the immune checkpoint inhibitors (ICI) (64, 66). BEV, an anti-vascular endothelial growth factor (VEGF), acts by inhibition of tumor neoangiogenesis. ICI use has been reported in a few cases of aggressive lactotroph and corticotroph adenomas/PitNETs. The effectiveness of this therapy in adenomas/PitNETs is still under investigation in pre-clinical and clinical studies (64, 66). In contrast to aggressive prolactinomas, tumors that express the Epidermal Growth Factor Receptor (EGFR) can be treated with tyrosine kinase inhibitors (TKI) (67). Everolimus (EVE), an inhibitor of the phosphatidylinositol 3-kinase/mammalian target of rapamycin (mTOR) pathway mTOR, approved to treat neuroendocrine tumors, has been demonstrated to be effective in many in vitro studies on pituitary cells. However, it showed poor efficacy in the few cases of adenomas/PitNETs reported in the literature, and only two clinical cases have been reported that described acromegaly patients being treated with VEGFR inhibitors. Clinical reports are not available for the treatment of acromegaly patients with mTOR inhibitors, TKI,or ICIs (66). Concerning new emerging drugs, such as oral somatostatin receptors ligands, no data are available on the possible use on aggressive cases. However recent preliminary data on the efficacy of paltusotine on maintain IGF-I at levels comparable to prior injected combination treatment are promising (68).
Radiation therapy (RT) (Table 1) is considered when a repeated surgery is not feasible, in case of residual active disease, or after drug therapy failure (76), or in cases with unresectable residual tumor mass. Hormonal normalization appears to be increased at 60.3% of patients if those still on pharmacological therapy after RT therapy are included. The time to reach biochemical remission varies between individual retrospective experiences and depends on pretreatment levels of GH and IGF-1 (83, 84). The choice of irradiation technique should be based on the features of the target tumor. Stereotactic radiosurgery (SRS) is a suitable treatment for patients with relatively small residual adenomas/PitNETs: the proximity to critical structures are limiting factors for its application, with 8 and 10 Gy being the maximum tolerated doses to the optic apparatus (85). Fractionated stereotactic radiation therapy (FSRT) is generally preferred for patients with larger a GH-PitNET that is not susceptible to SRS (85).
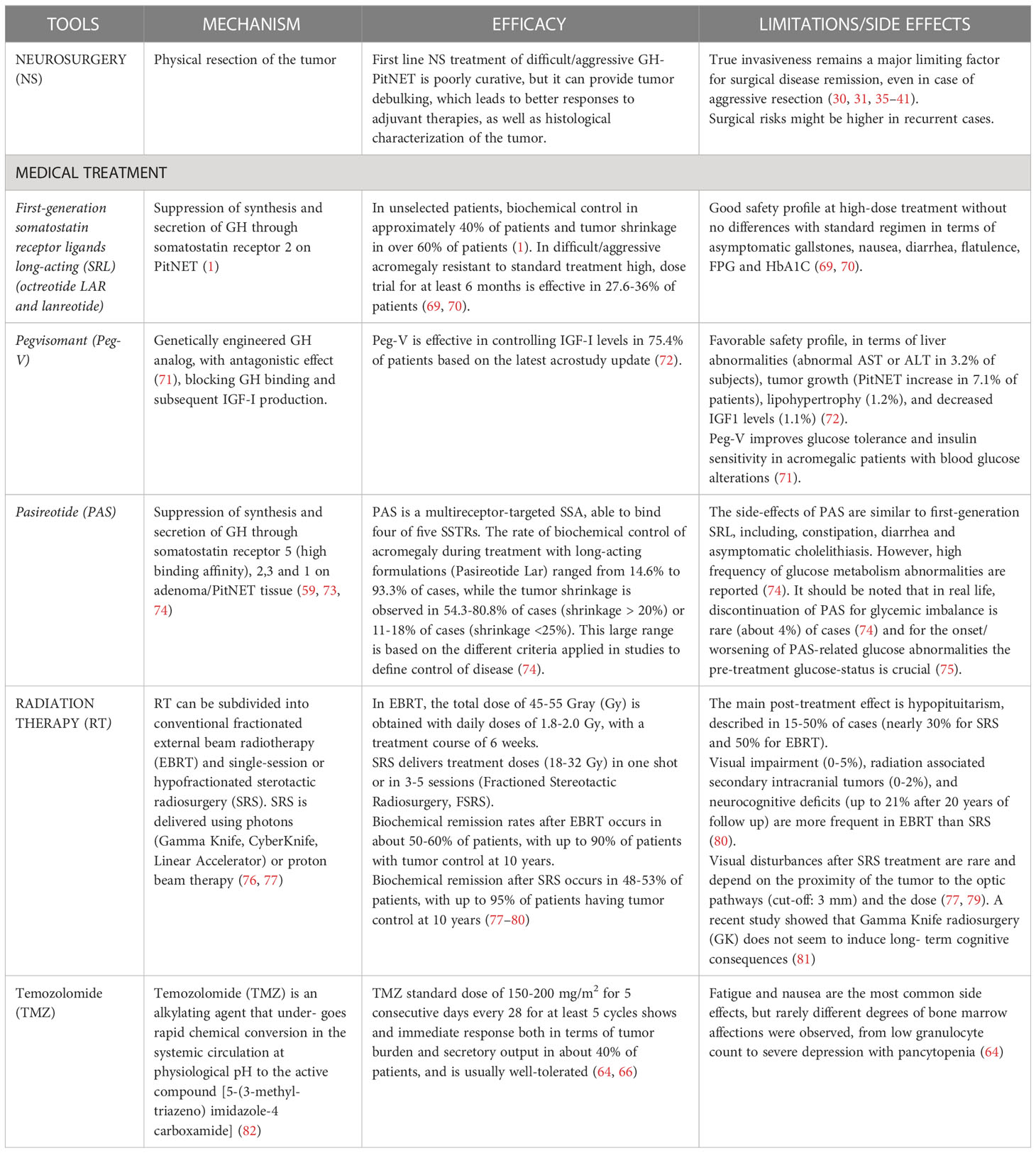
Table 1 Available treatment tools of difficult/aggressive GH-secreting PitNET.
In patients with GH-PitNET, positron emission tomography-computed tomography (PET-CT) with 18F-fluorodeoxyglucose (FDG PET-CT) has been shown to be helpful for preoperative characterization of sellar lesions, but with conflicting results (86–90). Other tracers have been developed and clinically validated in this setting, such as 68Ga-labeled somatostatin analogues (specifically, DOTATOC, DOTANOC and DOTATATE) and 11carbon-methionine (C-MET). 68Ga-labeled somatostatin analogues show high specificity in binding to SSTRs 2, 3 and 5, which are expressed by normal pituitary tissue and are hyperexpressed by GH-secreting adenoma/PitNET (91). Although in most patients, even if treatment-naïve, adenoma/PitNETs show significantly lower 68Ga-DOTATOC uptake compared to the normal pituitary gland, SSTR expression on the surface of PitNET cells (immuno-histochemically proven “a posteriori”) may lead to increased uptake in the adenoma/PitNET, which can be useful in localizing and determining response to surgical, medical, or radiation therapy. In particular, the expression of SSTRs and therefore the SUVmax is higher in patients with high 68Ga-DOTATOC uptake lesions (92); it may help to detect possible surgical failure (93); it is significantly and inversely correlated with lower circulating GH levels and complete laboratory response (94). 68Ga-labeled SRL are helpful in selecting patients who are suitable for peptide receptor radionuclide therapy (PRRT) which consists of administrating a therapeutic dosage of beta-emitting somatostatin analogue in order to carry lethal radiation to target cells: currently, 177Lu- and 90Y-labeled compounds are available for this purpose. PRRT is a novel promising treatment in patients with extensive and aggressive adenoma/PitNETs who are not suitable for surgery or refractory to medical/external radiation treatment, manifesting lower systemic adverse effects than conventional external beam radiation due to its targeted nature (67). PRRT has been used in 15 cases from 2012 to 2020 in patients with aggressive adenoma/PitNETs and carcinomas, either functioning or non-functioning, with varied radiopharmaceuticals, protocols and measured outcomes, making generalization on its effectiveness in this setting not feasible (66); only 43% patients had responded to PRRT, but most non-responders were resistant to previous temozolomide treatment and therefore had a more aggressive disease, such as a case of aggressive GH-secreting adenoma/PitNET completely resistant to other treatments and successfully treated with 90Y-DOTATATE (95). Overall, PRRT is well tolerated since it does not carry a higher risk of developing hypopituitarism (96, 97). C-MET PET-CT also plays an interesting role in diagnosing recurrent adenoma/PitNETs in patients already treated with adenomectomy or sub-total hypophysectomy, due to its ability to collect into cells with an increased amino acid intake and protein synthesis/secretion. Different from 68Ga-labeled somatostatin analogues, C-MET uptake is not affected by SSTRs expression or hormone secretion. For these reasons, in patients operated on for an aggressive GH-adenoma/PitNET with persistent or relapsed disease and with equivocal MRI, it is a promising tool to drive and facilitate neurosurgeons to perform targeted revision surgery (98).
In tertiary care pituitary centers, difficult/aggressive GH-secreting adenomas/PitNET might be more frequent than usually encountered in a primary endocrinology clinic, justifying the definition and recognition as adenomas/PitNET. A synthesis between the different positions is absolutely needed and desirable. The new WHO 2022 should be implemented with a structured pathology report, which should be the cornerstone to investigate a staging system. In our opinion, a complete pathology report is mandatory for the definition of a Pituitary Center of Excellence, together with the ability to offer a multidisciplinary approach. Recent papers have described the prognostic role on known clinical and molecular markers such as proliferation index, granulation, or SSTR expression pattern (6, 99, 100). Nonetheless, pathological analysis needs to be still standardized and validated before being included in future guidelines. In this regard, it is necessary to implement research not only on these and new prognostic factors of tumor biology and of the tumor immune microenvironment, but on the pathophysiology of GH secretion, which may represent a target for future molecular therapies (7, 101, 102). PET-CT could play a role in the diagnosis and treatment of pituitary adenoma/PitNET, and GH-secreting specifically ones, for detecting persistent or refractory disease and for selecting patients who are suitable for third-line therapies such as PRRT. In this regard, joint action of an experienced multidisciplinary team is required to progress and improve the management of acromegaly patients with difficult/aggressive GH-secreting adenomas/PitNET.
AB, SC, AG, SG, CM, CC, MR, MG, LL, FD wrote the manuscipt. LM, AO, AP reviewed the manuscript. All authors contributed to the article and approved the submitted version.
The authors would like to thank Dr. Patrick Moore for language editing, and proofreading.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gadelha MR, Wildemberg LE, Kasuki L. The future of somatostatin receptor ligands in acromegaly. J Clin Endocrinol Metab (2022) 107:297–308. doi: 10.1210/clinem/dgab726
2. Melmed S, Kleinberg DL, Bonert V, Fleseriu M. Acromegaly: assessing the disorder and navigating therapeutic options for treatment. Endocr Pract (2014) 20 Suppl 1:7–17. doi: 10.4158/EP14430.RA
3. Dekkers OM, Karavitaki N, Pereira AM. The epidemiology of aggressive pituitary tumors (and its challenges). Rev Endocr Metab Disord (2020) 21:209–12. doi: 10.1007/s11154-020-09556-7
4. Cuevas-Ramos D, Carmichael JD, Cooper O, Bonert VS, Gertych A, Mamelak AN, et al. A structural and functional acromegaly classification. J Clin Endocrinol Metab (2015) 100:122–31. doi: 10.1210/jc.2014-2468
5. Raverot G, Burman P, McCormack A, Heaney A, Petersenn S, Popovic V, et al. European Society of endocrinology clinical practice guidelines for the management of aggressive pituitary tumours and carcinomas. Eur J Endocrinol (2018) 178:G1–g24. doi: 10.1530/eje-17-0796
6. Melmed S, Kaiser UB, Lopes MB, Bertherat J, Syro LV, Raverot G, et al. Clinical biology of the pituitary adenoma. Endocr Rev (2022) 43:bnac010. doi: 10.1210/endrev/bnac010
7. Raverot G, Ilie MD, Lasolle H, Amodru V, Trouillas J, Castinetti F, et al. Aggressive pituitary tumours and pituitary carcinomas. Nat Rev Endocrinol (2021) 17:671–84. doi: 10.1038/s41574-021-00550-w
8. Asioli S, Righi A, Iommi M, Baldovini C, Ambrosi F, Guaraldi F, et al. Validation of a clinicopathological score for the prediction of post-surgical evolution of pituitary adenoma: retrospective analysis on 566 patients from a tertiary care centre. Eur J Endocrinol (2019) 180:127–34. doi: 10.1530/eje-18-0749
9. Swanson AA, Erickson D, Donegan DM, Jenkins SM, Van Gompel JJ, Atkinson JLD, et al. Clinical, biological, radiological, and pathological comparison of sparsely and densely granulated somatotroph adenomas: a single center experience from a cohort of 131 patients with acromegaly. Pituitary (2021) 24:192–206. doi: 10.1007/s11102-020-01096-2
10. Sahakian N, Appay R, Resseguier N, Graillon T, Piazzola C, Laure C, et al. Real-life clinical impact of a five-tiered classification of pituitary tumors. Eur J Endocrinol (2022) 187:893–904. doi: 10.1530/EJE-22-0812
11. Ferrés A, Reyes L, Di Somma A, Topczewski T, Mosteiro A, Guizzardi G, et al. The prognostic-based approach in growth hormone-secreting pituitary neuroendocrine tumors (PitNET): Tertiary reference center, single senior surgeon, and long-term follow-up. Cancers (Basel) (2022) 15:267. doi: 10.3390/cancers15010267
12. Donoho DA, Bose N, Zada G, Carmichael JD. Management of aggressive growth hormone secreting pituitary adenomas. Pituitary (2017) 20:169–78. doi: 10.1007/s11102-016-0781-7
13. Casanueva FF, Barkan AL, Buchfelder M, Klibanski A, Laws ER, Loeffler JS, et al. Criteria for the definition of pituitary tumor centers of excellence (PTCOE): A pituitary society statement. Pituitary (2017) 20:489–98. doi: 10.1007/s11102-017-0838-2
14. Shimon I, Jallad RS, Fleseriu M, Yedinak CG, Greenman Y, Bronstein MD. Giant GH-secreting pituitary adenomas: Management of rare and aggressive pituitary tumors. Eur J Endocrinol (2015) 172:707–13. doi: 10.1530/eje-14-1117
15. Rolla M, Jawiarczyk-Przybyłowska A, Halupczok-Żyła J, Kałużny M, Konopka BM, Błoniecka I, et al. Complications and comorbidities of acromegaly-retrospective study in polish center. Front Endocrinol (Lausanne) (2021) 12:642131. doi: 10.3389/fendo.2021.642131
16. Gadelha MR, Kasuki L, Lim DST, Fleseriu M. Systemic complications of acromegaly and the impact of the current treatment landscape: An update. Endocr Rev (2019) 40:268–332. doi: 10.1210/er.2018-00115
17. Liu C-X, Wang S-Z, Heng L-J, Han Y, Ma Y-H, Yan L-F, et al. Predicting subtype of growth hormone pituitary adenoma based on magnetic resonance imaging characteristics. J Comput Assist Tomogr (2022) 46:124–30. doi: 10.1097/RCT.0000000000001249
18. Zada G, Lin N, Laws ER. Patterns of extrasellar extension in growth hormone-secreting and nonfunctional pituitary macroadenomas. Neurosurg Focus (2010) 29:E4. doi: 10.3171/2010.7.FOCUS10155
19. Hagiwara A, Inoue Y, Wakasa K, Haba T, Tashiro T, Miyamoto T. Comparison of growth hormone-producing and non-growth hormone-producing pituitary adenomas: imaging characteristics and pathologic correlation. Radiology (2003) 228:533–8. doi: 10.1148/radiol.2282020695
20. Saeger W, Honegger J, Theodoropoulou M, Knappe UJ, Schofl C, Petersenn S, et al. Clinical impact of the current WHO classification of pituitary adenomas. Endocrine Pathol (2016) 27:104–14. doi: 10.1007/s12022-016-9418-7
21. Knosp E, Steiner E, Kitz K, Matula C. Pituitary adenomas with invasion of the cavernous sinus space: A magnetic resonance imaging classification compared with surgical findings. Neurosurgery (1993) 33:610–8. doi: 10.1227/00006123-199310000-00008
22. Micko ASG, Wöhrer A, Wolfsberger S, Knosp E. Invasion of the cavernous sinus space in pituitary adenomas: endoscopic verification and its correlation with an MRI-based classification. J Neurosurg (2015) 122:803–11. doi: 10.3171/2014.12.jns141083
23. Park YW, Kang Y, Ahn SS, Ku CR, Kim EH, Kim SH, et al. Radiomics model predicts granulation pattern in growth hormone-secreting pituitary adenomas. Pituitary (2020) 23:691–700. doi: 10.1007/s11102-020-01077-5
24. Liu C-X, Heng L-J, Han Y, Wang S-Z, Yan L-F, Yu Y, et al. Usefulness of the texture signatures based on multiparametric MRI in predicting growth hormone pituitary adenoma subtypes. Front Oncol (2021) 11:640375. doi: 10.3389/fonc.2021.640375
25. Saeger W, Koch A. Clinical implications of the new WHO classification 2017 for pituitary tumors. Exp Clin Endocrinol Diabetes (2021) 129:146–56. doi: 10.1055/a-1310-7900
26. Batisse M, Raverot G, Maqdasy S, Durando X, Sturm N, Montoriol P-F, et al. Aggressive silent GH pituitary tumor resistant to multiple treatments, including temozolomide. Cancer Invest (2013) 31:190–6. doi: 10.3109/07357907.2013.775293
27. Zada G, Cavallo LM, Esposito F, Fernandez-Jimenez JC, Tasiou A, De Angelis M, et al. Transsphenoidal surgery in patients with acromegaly: operative strategies for overcoming technically challenging anatomical variations. Neurosurg Focus (2010) 29:E8. doi: 10.3171/2010.8.FOCUS10156
28. Qiao N, Shen M, He W, He M, Zhang Z, Ye H, et al. Comparative effectiveness of endoscopic versus microscopic transsphenoidal surgery for patients with growth hormone secreting pituitary adenoma: An emulated trial. Clin Neurol Neurosurg (2021) 207:106781. doi: 10.1016/j.clineuro.2021.106781
29. Almeida JP, Ruiz-Treviño AS, Liang B, Omay SB, Shetty SR, Chen YN, et al. Reoperation for growth hormone–secreting pituitary adenomas: Report on an endonasal endoscopic series with a systematic review and meta-analysis of the literature. J Neurosurg (2018) 129:404–16. doi: 10.3171/2017.2.jns162673
30. Nishioka H, Fukuhara N, Horiguchi K, Yamada S. Aggressive transsphenoidal resection of tumors invading the cavernous sinus in patients with acromegaly: predictive factors, strategies, and outcomes. J Neurosurg (2014) 121:505–10. doi: 10.3171/2014.3.JNS132214
31. Guo X, Zhang R, Zhang D, Wang Z, Gao L, Yao Y, et al. Determinants of immediate and long-term remission after initial transsphenoidal surgery for acromegaly and outcome patterns during follow-up: a longitudinal study on 659 patients. J Neurosurg (2022) 1:1–11. doi: 10.3171/2021.11.JNS212137
32. Guo S, Wang Z, Kang X, Xin W, Li X. A meta-analysis of endoscopic vs. microscopic transsphenoidal surgery for non-functioning and functioning pituitary adenomas: Comparisons of efficacy and safety. Front Neurol (2021) 12:614382. doi: 10.3389/fneur.2021.614382
33. Leach P, Abou-Zeid AH, Kearney T, Davis J, Trainer PJ, Gnanalingham KK. Endoscopic transsphenoidal pituitary surgery: evidence of an operative learning curve. Neurosurgery (2010) 67:1205–12. doi: 10.1227/NEU.0b013e3181ef25c5
34. Goyal-Honavar A, Sarkar S, Asha HS, Kapoor N, Thomas R, Balakrishnan R, et al. Impact of experience on outcomes after endoscopic transsphenoidal surgery for acromegaly. World Neurosurg (2021) 151:e1007–15. doi: 10.1016/j.wneu.2021.05.030
35. Campbell PG, Kenning E, Andrews DW, Yadla S, Rosen M, Evans JJ. Outcomes after a purely endoscopic transsphenoidal resection of growth hormone-secreting pituitary adenomas. Neurosurg Focus (2010) 29:E5. doi: 10.3171/2010.7.FOCUS10153
36. Babu H, Ortega A, Nuno M, Dehghan A, Schweitzer A, Bonert HV, et al. Long-term endocrine outcomes following endoscopic endonasal transsphenoidal surgery for acromegaly and associated prognostic factors. Neurosurgery (2017) 81:357–66. doi: 10.1093/neuros/nyx020
37. Asha MJ, Takami H, Velasquez C, Oswari S, Almeida JP, Zadeh G, et al. Long-term outcomes of transsphenoidal surgery for management of growth hormone-secreting adenomas: single-center results. J Neurosurg (2019) 1:1–11. doi: 10.3171/2019.6.JNS191187
38. Cardinal T, Rutkowski MJ, Micko A, Shiroishi M, Jason Liu C-S, Wrobel B, et al. Impact of tumor characteristics and pre- and postoperative hormone levels on hormonal remission following endoscopic transsphenoidal surgery in patients with acromegaly. Neurosurg Focus (2020) 48:E10. doi: 10.3171/2020.3.FOCUS2080
39. Araujo-Castro M, Pascual-Corrales E, Martínez-Vaello V, Baonza Saiz G, Quiñones de Silva J, Acitores Cancela A, et al. Predictive model of surgical remission in acromegaly: age, presurgical GH levels and knosp grade as the best predictors of surgical remission. J Endocrinol Invest (2021) 44:183–93. doi: 10.1007/s40618-020-01296-4
40. Torres A, Sanmillan JL, Lau R, Gabarros A. Final outcome in growth hormone-secreting adenomas after combination of maximal tumor resection and medical treatment. World Neurosurg (2021) 154:e292–301. doi: 10.1016/j.wneu.2021.07.018
41. Serioli S, Doglietto F, Fiorindi A, Biroli A, Mattavelli D, Buffoli B, et al. Pituitary adenomas and invasiveness from anatomo-surgical, radiological, and histological perspectives: A systematic literature review. Cancers (Basel) (2019) 11:E1936. doi: 10.3390/cancers11121936
42. Villa C, Vasiljevic A, Jaffrain-Rea ML, Ansorge O, Asioli S, Barresi V, et al. A standardised diagnostic approach to pituitary neuroendocrine tumours (PitNETs): a European pituitary pathology group (EPPG) proposal. Virchows Arch (2019) 687–92 .doi: 10.1007/s00428-019-02655-0
43. Osamura R, Lopes M, Grossman A, Matsuno A, Korbonits M, Trouillas J, et al. Pituitary adenoma. In: Lloyd RV, Osamura RY, Klöppel G, Rosai J, editors. WHO classification of tumours of endocrine organs. Lyon: IARC (2017). p. 14–8.
44. Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 WHO classification of pituitary tumors. Endocr Pathol (2022) 33:6–26. doi: 10.1007/s12022-022-09703-7
45. Gatto F, Feelders RA, van der Pas R, Kros JM, Waaijers M, Sprij-Mooij D, et al. Immunoreactivity score using an anti-sst2A receptor monoclonal antibody strongly predicts the biochemical response to adjuvant treatment with somatostatin analogs in acromegaly. J Clin Endocrinol Metab (2013) 98:E66–71. doi: 10.1210/jc.2012-2609
46. Volante M, Brizzi MP, Faggiano A, La Rosa S, Rapa I, Ferrero A, et al. Somatostatin receptor type 2A immunohistochemistry in neuroendocrine tumors: a proposal of scoring system correlated with somatostatin receptor scintigraphy. Mod Pathol (2007) 20:1172–82. doi: 10.1038/modpathol.3800954
47. Bogner E-M, Daly AF, Gulde S, Karhu A, Irmler M, Beckers J, et al. miR-34a is upregulated in AIP-mutated somatotropinomas and promotes octreotide resistance. Int J Cancer (2020) 147:3523–38. doi: 10.1002/ijc.33268
48. Wildemberg LE, da Silva Camacho AH, Miranda RL, Elias PCL, de Castro Musolino NR, Nazato D, et al. Machine learning-based prediction model for treatment of acromegaly with first-generation somatostatin receptor ligands. J Clin Endocrinol Metab (2021) 106:2047–56. doi: 10.1210/clinem/dgab125
49. Gola M, Bonadonna S, Mazziotti G, Amato G, Giustina A. Resistance to somatostatin analogs in acromegaly: an evolving concept? J Endocrinol Invest (2006) 29:86–93. doi: 10.1007/BF03349183
50. Resmini E, Dadati P, Ravetti J-L, Zona G, Spaziante R, Saveanu A, et al. Rapid pituitary tumor shrinkage with dissociation between antiproliferative and antisecretory effects of a long-acting octreotide in an acromegalic patient. J Clin Endocrinol Metab (2007) 92:1592–9. doi: 10.1210/jc.2006-2084
51. Casarini AP, Pinto EM, Jallad RS, Giorgi RR, Giannella-Neto D, Bronstein MD. Dissociation between tumor shrinkage and hormonal response during somatostatin analog treatment in an acromegalic patient: preferential expression of somatostatin receptor subtype 3. J Endocrinol Invest (2006) 29:826–30. doi: 10.1007/BF03347378
52. Colao A, Auriemma RS, Lombardi G, Pivonello R. Resistance to somatostatin analogs in acromegaly. Endocr Rev (2011) 32:247–71. doi: 10.1210/er.2010-0002
53. Bianchi A, Valentini F, Iuorio R, Poggi M, Baldelli R, Passeri M, et al. Long-term treatment of somatostatin analog-refractory growth hormone-secreting pituitary tumors with pegvisomant alone or combined with long-acting somatostatin analogs: a retrospective analysis of clinical practice and outcomes. J Exp Clin Cancer Res (2013) 32:40. doi: 10.1186/1756-9966-32-40
54. Kasuki L, Wildemberg LEA, Neto LV, Marcondes J, Takiya CM, Gadelha MR. Ki-67 is a predictor of acromegaly control with octreotide LAR independent of SSTR2 status and relates to cytokeratin pattern. Eur J Endocrinol (2013) 169:217–23. doi: 10.1530/EJE-13-0349
55. Fougner SL, Casar-Borota O, Heck A, Berg JP, Bollerslev J. Adenoma granulation pattern correlates with clinical variables and effect of somatostatin analogue treatment in a large series of patients with acromegaly. Clin Endocrinol (Oxf) (2012) 76:96–102. doi: 10.1111/j.1365-2265.2011.04163.x
56. Heck A, Ringstad G, Fougner SL, Casar-Borota O, Nome T, Ramm-Pettersen J, et al. Intensity of pituitary adenoma on T2-weighted magnetic resonance imaging predicts the response to octreotide treatment in newly diagnosed acromegaly. Clin Endocrinol (Oxf) (2012) 77:72–8. doi: 10.1111/j.1365-2265.2011.04286.x
57. Ferone D, Gatto F, Arvigo M, Resmini E, Boschetti M, Teti C, et al. The clinical-molecular interface of somatostatin, dopamine and their receptors in pituitary pathophysiology. J Mol Endocrinol (2009) 42:361–70. doi: 10.1677/JME-08-0162
58. Venegas-Moreno E, Vazquez-Borrego MC, Dios E, Gros-Herguido N, Flores-Martinez A, Rivero-Cortés E, et al. Association between dopamine and somatostatin receptor expression and pharmacological response to somatostatin analogues in acromegaly. J Cell Mol Med (2018) 22:1640–9. doi: 10.1111/jcmm.13440
59. Iacovazzo D, Carlsen E, Lugli F, Chiloiro S, Piacentini S, Bianchi A, et al. Factors predicting pasireotide responsiveness in somatotroph pituitary adenomas resistant to first-generation somatostatin analogues: an immunohistochemical study. Eur J Endocrinol (2016) 174:241–50. doi: 10.1530/EJE-15-0832
60. Giustina A, Barkhoudarian G, Beckers A, Ben-Shlomo A, Biermasz N, Biller B, et al. Multidisciplinary management of acromegaly: A consensus. Rev Endocr Metab Disord (2020) 21:667–78. doi: 10.1007/s11154-020-09588-z
61. Puig Domingo M. Treatment of acromegaly in the era of personalized and predictive medicine. Clin Endocrinol (Oxf) (2015) 83:3–14. doi: 10.1111/cen.12731
62. Puig-Domingo M, Bernabéu I, Picó A, Biagetti B, Gil J, Alvarez-Escolá C, et al. Pasireotide in the personalized treatment of acromegaly. Front Endocrinol (Lausanne) (2021) 12:648411. doi: 10.3389/fendo.2021.648411
63. Chiloiro S, Bianchi A, Giampietro A, Pontecorvi A, Raverot G, Marinis LD. Second line treatment of acromegaly: Pasireotide or pegvisomant? Best Pract Res Clin Endocrinol Metab (2022), 36:101684. doi: 10.1016/j.beem.2022.101684
64. Burman P, Trouillas J, Losa M, McCormack A, Petersenn S, Popovic V, et al. Aggressive pituitary tumours and carcinomas, characteristics and management of 171 patients. Eur J Endocrinol (2022) 187:593–605. doi: 10.1530/EJE-22-0440
65. Ishida A, Shichi H, Fukuoka H, Shiramizu H, Inoshita N, Yamada S. Temozolomide and capecitabine treatment for an aggressive somatotroph pituitary tumor: A case report and literature review. Front Oncol (2022) 12:916982. doi: 10.3389/fonc.2022.916982
66. Nakano-Tateno T, Lau KJ, Wang J, McMahon C, Kawakami Y, Tateno T, et al. Multimodal non-surgical treatments of aggressive pituitary tumors. Front Endocrinol (Lausanne) (2021) 12:624686. doi: 10.3389/fendo.2021.624686
67. Cooper O, Mamelak A, Bannykh S, Carmichael J, Bonert V, Lim S, et al. Prolactinoma ErbB receptor expression and targeted therapy for aggressive tumors. Endocrine (2014) 46:318–27. doi: 10.1007/s12020-013-0093-x
68. Randeva H, Gadelha M, Gordon M, Mezosi M, Doknic M, Toth M, et al. Long-term treatment with oral paltusotine for acromegaly: Results from the ACROBAT advance study. Lyon, France: 20th Congress of the European Neuroendocrine Association (2022).
69. Giustina A, Bonadonna S, Bugari G, Colao A, Cozzi R, Cannavo S, et al. High-dose intramuscular octreotide in patients with acromegaly inadequately controlled on conventional somatostatin analogue therapy: a randomised controlled trial. Eur J Endocrinol (2009) 161:331–8. doi: 10.1530/EJE-09-0372
70. Giustina A, Mazziotti G, Cannavò S, Castello R, Arnaldi G, Bugari G, et al. High-dose and high-frequency lanreotide autogel in acromegaly: A randomized, multicenter study. J Clin Endocrinol Metab (2017) 102:2454–64. doi: 10.1210/jc.2017-00142
71. Giustina A, Arnaldi G, Bogazzi F, Cannavò S, Colao A, De Marinis L, et al. Pegvisomant in acromegaly: an update. J Endocrinol Invest (2017) 40:577–89. doi: 10.1007/s40618-017-0614-1
72. Fleseriu M, Führer-Sakel D, van der Lely AJ, De Marinis L, Brue T, van der Lans-Bussemaker J, et al. More than a decade of real-world experience of pegvisomant for acromegaly: ACROSTUDY. Eur J Endocrinol (2021) 185:525–38. doi: 10.1530/EJE-21-0239
73. Petersenn S, Unger N, Hu K, Weisshaar B, Zhang Y, Bouillaud E, et al. Pasireotide (SOM230), a novel multireceptor-targeted somatostatin analogue, is well tolerated when administered as a continuous 7-day subcutaneous infusion in healthy male volunteers. J Clin Pharmacol (2012) 52:1017–27. doi: 10.1177/0091270011408727
74. Bolanowski M, Kałużny M, Witek P, Jawiarczyk-Przybyłowska A. Pasireotide-a novel somatostatin receptor ligand after 20 years of use. Rev Endocr Metab Disord (2022) 23:601–20. doi: 10.1007/s11154-022-09710-3
75. Gadelha MR, Gu F, Bronstein MD, Brue TC, Fleseriu M, Shimon I, et al. Risk factors and management of pasireotide-associated hyperglycemia in acromegaly. Endocr Connect (2020) 9:1178–90. doi: 10.1530/EC-20-0361
76. Shih HA, Loeffler JS. Radiation therapy in acromegaly. Rev Endocr Metab Disord (2008) 9:59–65. doi: 10.1007/s11154-007-9065-x
77. Minniti G, Scaringi C, Enrici RM. Radiation techniques for acromegaly. Radiat Oncol (2011) 6:167. doi: 10.1186/1748-717X-6-167
78. Abu Dabrh AM, Asi N, Farah WH, Mohammed K, Wang Z, Farah MH, et al. Radiotherapy versus radiosurgery in treating patients with acromegaly: a systematic review and meta-analysis. Endocr Pract (2015) 21:943–56. doi: 10.4158/EP14574.OR
79. Marquez Y, Tuchman A, Zada G. Surgery and radiosurgery for acromegaly: a review of indications, operative techniques, outcomes, and complications. Int J Endocrinol (2012) 2012:386401. doi: 10.1155/2012/386401
80. Gheorghiu ML. Updates in outcomes of stereotactic radiation therapy in acromegaly. Pituitary (2017) 20:154–68. doi: 10.1007/s11102-016-0783-5
81. Castinetti F, Caron P, Raingeard I, Amodru V, Albarel F, Morange I, et al. Lack of delayed neurocognitive side effects of gamma knife radiosurgery in acromegaly: the later-Ac study. Eur J Endocrinol (2021) 186:37–44. doi: 10.1530/EJE-21-0826
82. Losa M, Bogazzi F, Cannavo S, Ceccato F, Curtò L, De Marinis L, et al. Temozolomide therapy in patients with aggressive pituitary adenomas or carcinomas. J Neurooncol (2016) 126:519–25. doi: 10.1007/s11060-015-1991-y
83. Losa M, Gioia L, Picozzi P, Franzin A, Valle M, Giovanelli M, et al. The role of stereotactic radiotherapy in patients with growth hormone-secreting pituitary adenoma. J Clin Endocrinol Metab (2008) 93:2546–52. doi: 10.1210/jc.2008-0135
84. Pollock BE, Jacob JT, Brown PD, Nippoldt TB. Radiosurgery of growth hormone-producing pituitary adenomas: factors associated with biochemical remission. J Neurosurg (2007) 106:833–8. doi: 10.3171/jns.2007.106.5.833
85. Minniti G, Osti MF, Niyazi M. Target delineation and optimal radiosurgical dose for pituitary tumors. Radiat Oncol (2016) 11:135. doi: 10.1186/s13014-016-0710-y
86. Jeong SY, Lee S-W, Lee HJ, Kang S, Seo J-H, Chun KA, et al. Incidental pituitary uptake on whole-body 18F-FDG PET/CT: a multicentre study. Eur J Nucl Med Mol Imaging (2010) 37:2334–43. doi: 10.1007/s00259-010-1571-5
87. Ju H, Zhou J, Pan Y, Lv J, Zhang Y. Evaluation of pituitary uptake incidentally identified on 18F-FDG PET/CT scan. Oncotarget (2017) 8:55544–9. doi: 10.18632/oncotarget.15417
88. Hyun SH, Choi JY, Lee K-H, Choe YS, Kim B-T. Incidental focal 18F-FDG uptake in the pituitary gland: clinical significance and differential diagnostic criteria. J Nucl Med (2011) 52:547–50. doi: 10.2967/jnumed.110.083733
89. Tosaka M, Higuchi T, Horiguchi K, Osawa T, Arisaka Y, Fujita H, et al. Preoperative evaluation of sellar and parasellar macrolesions by [18F]Fluorodeoxyglucose positron emission tomography. World Neurosurg (2017) 103:591–9. doi: 10.1016/j.wneu.2017.04.032
90. Seok H, Lee EY, Choe EY, Yang WI, Kim JY, Shin DY, et al. Analysis of 18F-fluorodeoxyglucose positron emission tomography findings in patients with pituitary lesions. Korean J Intern Med (2013) 28:81–8. doi: 10.3904/kjim.2013.28.1.81
91. Zhao X, Xiao J, Xing B, Wang R, Zhu Z, Li F. Comparison of (68)Ga DOTATATE to 18F-FDG uptake is useful in the differentiation of residual or recurrent pituitary adenoma from the remaining pituitary tissue after transsphenoidal adenomectomy. Clin Nucl Med (2014) 39:605–8. doi: 10.1097/RLU.0000000000000457
92. Tjörnstrand A, Casar-Borota O, Heurling K, Schöll M, Gjertsson P, Himmelman J, et al. Lower 68 Ga-DOTATOC uptake in nonfunctioning pituitary neuroendocrine tumours compared to normal pituitary gland-a proof-of-concept study. Clin Endocrinol (Oxf) (2020) 92:222–31. doi: 10.1111/cen.14144
93. Tjörnstrand A, Casar-Borota O, Heurling K, Schöll M, Gjertsson P, Ragnarsson O, et al. Pre- and postoperative 68 Ga-DOTATOC positron emission tomography for hormone-secreting pituitary neuroendocrine tumours. Clin Endocrinol (Oxf) (2021) 94:956–67. doi: 10.1111/cen.14425
94. Daniel KB, de Oliveira Santos A, de Andrade RA, Trentin MBF, Garmes HM. Evaluation of 68Ga-DOTATATE uptake at the pituitary region and the biochemical response to somatostatin analogs in acromegaly. J Endocrinol Invest (2021) 44:2195–202. doi: 10.1007/s40618-021-01523-6
95. Waligórska-Stachura J, Gut P, Sawicka-Gutaj N, Liebert W, Gryczyńska M, Baszko-Błaszyk D, et al. Growth hormone-secreting macroadenoma of the pituitary gland successfully treated with the radiolabeled somatostatin analog (90)Y-DOTATATE: case report. J Neurosurg (2016) 125:346–9. doi: 10.3171/2015.6.JNS15363
96. Elston MS, Love A, Kevat D, Carroll R, Siow ZR, Pattison S, et al. Pituitary function following peptide receptor radionuclide therapy for neuroendocrine tumours. Cancer Med (2021) 10:8405–11. doi: 10.1002/cam4.4345
97. Sundlöv A, Sjögreen-Gleisner K, Tennvall J, Dahl L, Svensson J, Åkesson A, et al. Pituitary function after high-dose 177Lu-DOTATATE therapy and long-term follow-up. Neuroendocrinology (2021) 111:344–53. doi: 10.1159/000507761
98. Feng Z, He D, Mao Z, Wang Z, Zhu Y, Zhang X, et al. Utility of 11C-methionine and 18F-FDG PET/CT in patients with functioning pituitary adenomas. Clin Nucl Med (2016) 41:e130–134. doi: 10.1097/RLU.0000000000001085
99. Lim DST, Fleseriu M. Personalized medical treatment of patients with acromegaly: A review. Endocr Pract (2022) 28:321–32. doi: 10.1016/j.eprac.2021.12.017
100. Fleseriu M, Langlois F, Lim DST, Varlamov EV, Melmed S. Acromegaly: pathogenesis, diagnosis, and management. Lancet Diabetes Endocrinol (2022) 10:804–26. doi: 10.1016/S2213-8587(22)00244-3
101. Zhou W, Ma C-X, Xing Y-Z, Yan Z-Y. Identification of candidate target genes of pituitary adenomas based on the DNA microarray. Mol Med Rep (2016) 13:2182–6. doi: 10.3892/mmr.2016.4785
Keywords: aggressive pituitary adenoma, acromegaly, growth hormone, multidisciplinary, pituitary adenoma, pituitary neuro-endocrine tumor
Citation: Bianchi A, Chiloiro S, Giampietro A, Gaudino S, Calandrelli R, Mazzarella C, Caldarella C, Rigante M, Gessi M, Lauretti L, De Marinis L, Olivi A, Pontecorvi A and Doglietto F (2023) Multidisciplinary management of difficult/aggressive growth-hormone pituitary neuro-endocrine tumors. Front. Endocrinol. 14:1123267. doi: 10.3389/fendo.2023.1123267
Received: 13 December 2022; Accepted: 13 March 2023;
Published: 03 May 2023.
Edited by:
Mônica Gadelha, Federal University of Rio de Janeiro, BrazilReviewed by:
Elisa Lamback, Federal University of Rio de Janeiro, BrazilCopyright © 2023 Bianchi, Chiloiro, Giampietro, Gaudino, Calandrelli, Mazzarella, Caldarella, Rigante, Gessi, Lauretti, De Marinis, Olivi, Pontecorvi and Doglietto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sabrina Chiloiro, c2FicmluYS5jaGlsb2lyb0B1bmljYXR0Lml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.