 Aseel Marahleh
Aseel Marahleh Hideki Kitaura
Hideki Kitaura Fumitoshi Ohori
Fumitoshi Ohori Takahiro Noguchi2
Takahiro Noguchi2- 1Frontier Research Institute for Interdisciplinary Sciences (FRIS), Tohoku University, Sendai, Japan
- 2Division of Orthodontics and Dentofacial Orthopedics, Graduate School of Dentistry, Tohoku University, Sendai, Japan
The skeleton is an organ of dual functionality; on the one hand, it provides protection and structural competence. On the other hand, it participates extensively in coordinating homeostasis globally given that it is a mineral and hormonal reservoir. Bone is the only tissue in the body that goes through strategically consistent bouts of bone resorption to ensure its integrity and organismal survival in a temporally and spatially coordinated process, known as bone remodeling. Bone remodeling is directly enacted by three skeletal cell types, osteoclasts, osteoblasts, and osteocytes; these cells represent the acting force in a basic multicellular unit and ensure bone health maintenance. The osteocyte is an excellent mechanosensory cell and has been positioned as the choreographer of bone remodeling. It is, therefore, not surprising that a holistic grasp of the osteocyte entity in the bone is warranted. This review discusses osteocytogenesis and associated molecular and morphological changes and describes the osteocytic lacunocanalicular network (LCN) and its organization. We highlight new knowledge obtained from transcriptomic analyses of osteocytes and discuss the regulatory role of osteocytes in promoting osteoclastogenesis with an emphasis on the case of osteoclastogenesis in anosteocytic bones. We arrive at the conclusion that osteocytes exhibit several redundant means through which osteoclast formation can be initiated. However, whether osteocytes are true “orchestrators of bone remodeling” cannot be verified from the animal models used to study osteocyte biology in vivo. Results from studying osteocyte biology using current animal models should come with the caveat that these models are not osteocyte-specific, and conclusions from these studies should be interpreted cautiously.
1 Introduction
Bone remodeling is an asynchronous cellular process signaling continuously and simultaneously throughout the skeleton, ensuring skeletal integrity and conferring functional adaptation. A basic multicellular unit (BMU) is the driver of bone remodeling and gives it its quantum character (1, 2). There is a distinct coupled role for each cell group in a BMU, and the balanced activity of each cell group ensures a balanced outcome at each bone remodeling site. Osteoclast resorbing activity is coupled with the recruitment and differentiation of mature bone-laying osteoblasts. In adulthood, this produces a neutral balance, i.e., the amount of bone excavated is replaced by just enough bone to fill in the space, leading to skeletal homeostasis. However, in pathology, the balance can be negative (bone is lost) or positive (bone is gained).
Cortical bone remodeling happens in osteons where blood vessels at the center of a Haversian canal supply the cells required to carry out bone remodeling. Remodeling in cortical bones occurs in the form of tunnels consisting of three zones, the cutting cone, the reversal zone, and the closing zone (3). The cutting cone consists of a majority surge of osteoclasts and their progenitors working to erode the bone surface. The reversal zone is a mingle zone of several cell types constituting what is known as reversal cells; these cells express markers of osteogenic commitment lineage, which intensifies more when the reversal zone comes closer to osteoid seams (4). Finally, the closing zone consists of a coordinated network of osteocytes which become embedded in the mineralizing bone matrix (3). Cancellous bone remodeling happens on the surface of trabeculae at the interface between the bone marrow and the bone surface in a trench-like structure rather than a tunnel (5). The BMU in cancellous bone consists of the same sequential appearance of osteoclasts, reversal cells including osteoblasts, and then embedding osteocytes.
Osteoblast differentiation proceeds from skeletal stem cells (6) to multiple early precursor stages before forming full-fledged matrix-secreting osteoblasts (7). Then, the cell encounters one of three possible fates: 60 to 80% of osteoblasts undergo apoptosis and the remaining osteoblasts become differentiated cells, either an osteocyte embedded in osteoid or a surface bone lining cell (8). The former (osteocytes embedding in the bone) represents a significant percentage of adult bone formation since 90% of bone cells are osteocytes (9, 10), and an average of 3.33 billion osteocytes are renewed every year through remodeling; that is approximately 7 to 10% of the total number of osteocytes (11).
Osteocytes have been termed the orchestrators of bone remodeling owing to the fact that they produce RANKL, which is obligatory for osteoclast formation and bone resorption, and they produce sclerostin which inhibits osteoblast formation and consequentlybone formation. Both osteoclast and osteoblast formation are necessary events for bone remodeling to take place, and they do take place in bones without osteocytes (anosteocytic bone). Owing to the property ascribed to osteocytes as “master regulators of bone remodeling”, the observation of bone remodeling in anosteocytic bones presents us with a puzzling question: are osteocytes required for bone remodeling?
Answering this question demands a comprehensive knowledge of the osteocyte entity in the bone. Therefore, in this review, we introduce osteocytogenesis from osteoblasts. We introduce anosteocytic bone and its characteristics in terms of its cellular components and remodeling ability. We also describe the role of the osteocyte in osteoclastogenesis and bone resorption, which is the initial event that takes place in the process of bone remodeling.
2 Becoming an osteocyte
The transition from an osteoblast to an osteocyte is accompanied by distinct functional and morphological changes. An osteoblast on the surface of the bone laying down the osteoid slows down its motion concomitant with a change from a polygonal/cuboidal shape to a flat almond-shaped cell body. Cell volume decreases by 30% and the cell becomes surrounded by unmineralized collagen fibers with patches of calcification. This cell is now called an osteoid osteocyte and like its predecessor (osteoblast), contains a prominent granular endoplasmic reticulum, a well-developed Golgi apparatus, numerous free ribosomes, and many mitochondria (12–14). On the mineral-facing side (the pre-existing bone side), the podia form, which later extends towards mineralizing fronts in the process of arborization (15). At this stage, the cell is polarized, which is evident by the direction of its dendrites and the arrangement of organelles in the cytoplasm. The nucleus rests on the vascular side (facing the bone surface) and the cell body is oriented with the long axis parallel to collagen fibers in the matrix (14, 16).
Once the cell is completely embedded in a mineralized matrix (termed young osteocyte and later mature osteocyte), cell volume becomes reduced by 70%. The embedded osteocyte experiences extensive arborization, and dendrites extend to the vascular surface [for a recent review of the control of dendrite formation, check Wang and Wein et al. (2022) (17)]. This gives osteocytes their distinctive star-shaped appearance (14). Osteocyte body size varies from 5 to 20 µm depending on the maturational stage, and their processes are branched and connected to osteocytes in the same plane and adjacent planes. The intracellular landscape of the mature osteocyte includes reduced Golgi apparatus and endoplasmic reticulum, and the cytoplasm/nucleus ratio decreases with prominently condensed chromatin and one or two nucleoli (14). This is paralleled by the cell gradually reducing its biosynthesis of the major extracellular matrix proteins, collagen type I, and other non-collagenous proteins that are considered osteoblast markers such as alkaline phosphatase, osteocalcin, and bone sialoprotein (18, 19).
The specification of an osteocytic transcriptional program leading to osteocyte differentiation from the IDG-SW3 cell line has been reported to be driven by glucose restriction, AMP-activated protein kinase, and proliferator-activated receptor γ co-activator-1 (PGC-1), which means that osteocytic transitioning from osteoblasts is accompanied by changes in cell bioenergetics that favor mitochondrial function. This signifies that osteocytes are programmed to accommodate an efficient energy production machinery due to their nutrient-restricted environment (20). Gene expression profiling of osteocytes and osteoblasts distinguishes proteins exclusive to osteocytes: E11/gp38, dentine matrix protein 1 (Dmp1), matrix extracellular phosphoglycoprotein (MEPE), phosphate-regulating endopeptidase homolog X-linked (PHEX), and sclerostin; of those, E11/gp38 is required for dendritic formation as its inhibition in the MLO-Y4 osteocytic cell line impedes dendritic formation (21).
The transition from an osteoblast to an osteocyte may give the impression that osteocytes are retired osteoblasts; however, osteocytes assuming this new architectural organization are dynamic cells that exhibit motility in the form of dendrite extension and retraction and their processes are rich in proteins involved in muscle contractile function (21, 22). After transitioning, osteocytes take on the form of an extensive network that is established by an active cleavage of bone proteins. During the embedding process, osteocyte processes “invade” the surrounding mineralized matrix by periosteocytic collagen cleavage that is dependent on membrane-type matrix metalloproteinase 1 (MT1-MMP) as MT1-MMP deficiency leads to osteocytes with significantly fewer and shorter processes that cannot reach out to communicate (23). This is also evident in mice that harbor a site-directed mutation in the pro α1(I) collagen gene (Col1a1r/r mice). Col1a1r/r mice are resistant to collagenase activity and their bones exhibit osteocytes with increased apoptosis and calvaria with a significantly higher number of empty lacunae (24).
Understanding the flow of events of osteocytogenesis is important because there is no one cut-off point at which a cell can be called a young or mature osteocyte. Suppose we are talking about an osteocyte that is embedded in an osteoid (osteoid osteocyte) and one that is embedded in a newly mineralized matrix (young osteocyte), and another buried further after many bouts of remodeling (old osteocyte), in such cases, we will be describing different osteocytes in morphological, transcriptional, and molecular profiles. Franz-Odendaal et al. (2006) suggested five schemes of differentiation, four of which describe how osteoblasts may become osteocytes, and the fifth describes how osteoblasts avoid getting trapped as is the case in anosteocytic bones of higher teleosts (discussed below) (25).
3 Specificity of murine models studying osteocytes in vivo
To study the function of osteocytes in vivo, researchers used the Dmp1 promoter (the 10-kb and the 8-kb) or the Sost promoter to drive recombinase activity and study the effect of gene knockdown in osteocytes or to drive the activity of reporter genes in osteocytes. However, debate arises concerning the specificity of these models.
Using the 10-kb Dmp1 promoter in R26 reporter mice showed that not only cells embedded in bone matrix express LacZ but also cells resting on newly formed bone matrix, i.e. osteoblasts or bone lining cells (26). Additionally, mice with 10-kb Dmp1-Cre-activated tdTomato expression (Ai9 reporter mice) showed positive tdTomato expression in osteocytes of cortical and cancellous regions, osteoblasts/bone lining cells, muscles, and bone marrow cells. The 8-kb Dmp1-Cre-activated tdTomato showed similar expression, but the degree of fluorescence intensity and the proportion of expression in cells resting on bone surfaces were less than that of the 10-kb Dmp1-driven Cre activity indicating a more specific but not osteocyte-discrete expression (27).
Osteocytes obtained through digestion and fluorescent activated cell sorting (FACS) of GFP fluorescent cells of (4–9) day-old pup calvaria under the control of the 8-kb Dmp1promoter revealed that GFP was also active in cells expressing dendritic cell-specific transmembrane protein (Dcstamp) and osteoclast-associated immunoglobulin-like receptor (Oscar), indicating that using the 8-kb Dmp1-Cre model drives the expression of GFP not only in osteocytes but also in cells of hematopoietic origin (0.33% of GFP positive cells were positive for markers of hematopoietic and endothelial cells). Hematopoietic cell depletion (Lin depletion) before GFP sorting eliminated the contamination and yielded a purer osteocyte population compared to sorting without hematopoietic depletion (28).
Osteoblast enriched population (double GFP/Lin negative) showed 2-fold higher RANKL and OPG expression than the highly pure osteocyte population (osteocytes with hematopoietic depletion) (28). Interestingly enough, osteocytes obtained with or without hemopoietic cell depletion could not support osteoclastogenesis and only supported the formation of TRAP+ cells with two nuclei but not three or more. Of note, a culture of (8-kb-Dmp1/GFP) osteocytes obtained from mouse calvaria (by fractionation or by fractionation and FACS) on plastic led to the loss of well-known osteocyte markers Dmp1, Sost, Mepe, and calcitonin receptor (Calcr) within 24 hours (28). Additionally, mRNA expression of Dmp1, Sost, and Calcr of osteocytes isolated by fractionation alone was lost after 24 hours of culture (29). Those observations may indicate that cells obtained using this preparation de-differentiated or died. This may provide some insight into why osteocytes obtained through this technique could not support osteoclast formation in culture (28).
To overcome the limitation of off-target recombination, another mouse model was developed under the control of regulatory elements of the Sost gene. Sost-Cre mice were crossed with tdTomato (red) Cre-reporter mice; the resulting mice did not show fluorescence in lining cells or osteoblasts. However, large numbers of red fluorescent cells were present in their bone marrow. Flow cytometric analysis of tdTomato-positive bone marrow cells revealed that Sost-Cre transgene is active in an early hematopoietic progenitor (30).
4 The lacunocanalicular network
The bone is hierarchically structured into cortical and cancellous bone, designed for optimal stress loading distribution, much like the design of construction bridge trusses. Within the cortical bone, there are three porosity levels. The first is the vascular porosity (osteon) represented by Haversian canals that contain capillaries and nerves and are mostly filled with interstitial fluid. The second and third represent the lacunae and canaliculi, respectively. Lacunae and canaliculi form the lacunocanalicular network spaced in concentric rings around one Haversian canal. The lacunae contain the body of the osteocyte and the canaliculi house osteocyte projections, which extend and form a network connecting osteocytes to other cells and the bone surface but do not cross osteonal cement lines (31).
The LCN is a vast transport and communication network, and it can be considered a specialty transport network that senses shifts in blood and interstitial molecule levels (such as phosphate, calcium, and nutrients) better than the vascular network alone, providing it with the function of quickly achieving homeostatic concentrations at the bodily level. In appreciating the vastness of the LCN, comparisons were made with other organs in the body where the surface area represents a functional rather than just a morphological character; for example, the total surface area of the LCN is larger than the skin, the gastrointestinal tract, and the adult lung (9). Within the local environment of the bone, osteocytes within the LCN cover a 100 times larger surface area than osteoblasts and osteoclasts cover the bone surface (11).
High-resolution imaging technologies reveal that the LCN density and geometry vary considerably even within the same singular bone (32). A synchrotron X-ray phase nano-tomography (SR-PNT)-based analysis of the LCN geometry revealed that lacunae are generally ellipsoidal while canaliculi are cylindrical. Lacunar shape correlated to their location within the osteon; inner lacunae are elongated, and outer border lacunae at the cement lines are flat spheroids. The canaliculi are spatially well arranged and uniformly distributed through the extracellular matrix, and their number correlates well with lacunar size, hence osteocyte size. Additionally, the canalicular volume is three times smaller than the lacunar pore volume, but the canalicular surface area is more than three times larger than the lacunae (33).
Buenzeli and Sims (2015) quantified osteocytes and their dendritic processes in the adult human skeleton using published literature obtained by modern imaging techniques (11). They estimated the average total number of lacunae to be 44 billion and live osteocytes to be 42 billion, indicating that approximately 1% of the osteocyte population is dead. Osteocytic processes were at 3.7 trillion processes with a total length of 175,000 km, 4.2 mm per single osteocyte, and 47µm per single cell process, forming 12.7 termini and up to 1128 termini per osteocyte and a total of 23.4 trillion connections, considering that these calculations are made for processes covering a cumulative length of 47 µm from the cell body. Those numbers are difficult to imagine, but the purpose of listing them here is to give an approximate mental image of the extreme connectedness and the labyrinthian hierarchy of the LCN that allows it to cover a total surface area of more than 201 m² at a small volume of approximately 17,500 mm³. This large surface area, with a relatively small volume of extracellular fluid running through the LCN, provides evidence of the highly efficient process by which osteocytes exchange information with their surroundings. The efficiency of the LCN as a transport network is essential given the fact that mineralized bone is impermeable to solutes, and the LCN is responsible for solute transport to deliver nutrients (i.e., information) and dispose of waste (34, 35).
The total number, length, and terminal connections of osteocytes are comparable to the elaborate neuronal connections, and both networks share molecular and behavioral similarities, especially with aging (36, 37). Additionally, the osteocyte transcriptome signature proposed by Youlten et al. is enriched in terms associated with axonogenesis, axon guidance, and development, which is not surprising, given that the most prominent feature of osteocytes is their dendrites (38). The LCN can transmit extensive information via direct contact and paracrine and endocrine communications at the whole organism level (10). As an organ’s complexity and size are associated with its functions and demands, Buenzeli and Sims’ work highlighted the magnitude of the effect that can be exerted by the totality of the osteocyte network in a single regulatory role (for example, in promoting osteoclast formation).
5 Osteoclastogenesis in anosteocytic bone
One paradoxical way of appreciating the role of osteocytes in the bone is studying bone devoid of osteocytes; ancestral aquatic bone is categorized into two types, cellular and acellular, and bone in some vertebrate groups (for example, modern teleosts) completely lack osteocytes (39, 40). Although flawed, the “acellular” terminology refers to bones devoid of osteocyte lacunae found in extant teleosts, a group representing half vertebrates and bony fish. Despite this terminology, the acellular bone contains cells (osteoblasts and osteoclasts) with functional similarity to those in the cellular bone, except for one cellular component that distinguishes the two types: the absence of osteocytes and their lacunae. The term anosteocytic bone was later coined by Weiss and Watabe in 1979, which referred to bone devoid of osteocytes (41). In this review, anosteocytic and acellular bone is going to be used interchangeably.
Two hypotheses were devised to explain the anosteocytic bone phenomenon. (1) Osteocytes are present in acellular bone, but they die, and their lacunae disappear by mineralization. However, this theory was revoked for lacking tangible evidence (39, 42), but was revisited again with circumferential evidence (43). (2) Osteoblasts never get incorporated in their secreted matrix and recede from the mineralization front and never become trapped as osteocytes (25, 44). In the latter scenario, osteoblasts are polarized and move away from a single osteogenic front as one unit laying down bone matrix synchronically, resulting in the formation of acellular bone (45, 46).
Studying the differences between bone types revealed that anosteocytic bone has lower stiffness but increased toughness compared to cellular bone (47–49) (Table 1). Previous light and electron microscopic images showed that both bone types contain a heterogenous cell population with similar cell zoning between cellular and anosteocytic bone, with the only difference being the absence of osteocytes. In terms of function, both types contain osteoblasts that lay the bone matrix and osteoclasts that resorb the bone matrix; however, osteoclasts in fish (both osteocytic and anosteocytic) are structurally different in that they lack ruffled borders and are mononucleated which rendered them undetected for some time (41, 51). Anosteocytic bone can remodel and undergo resorption in response to mechanical stress to a similar degree to that of cellular bone. One study compared descaling fish (a form of demineralization or resorption induction) of both bone types to signify the role of osteocytic osteolysis on resorption in descaled fish. In osteocytic bone, osteocytes had prominent perilacunar spaces indicating resorptive activity in the form of osteocytic osteolysis. Two types of osteoclasts were described, termed osteoclasts A and B; they were absent of ruffled borders with extensive endoplasmic reticula, indicating metabolic activity. Similarly, demineralized anosteocytic bone contained the same two types of osteoclasts, A and B, with similar structures to those of cellular bone but indicating that osteocytes were not needed to initiate osteoclast formation (41).
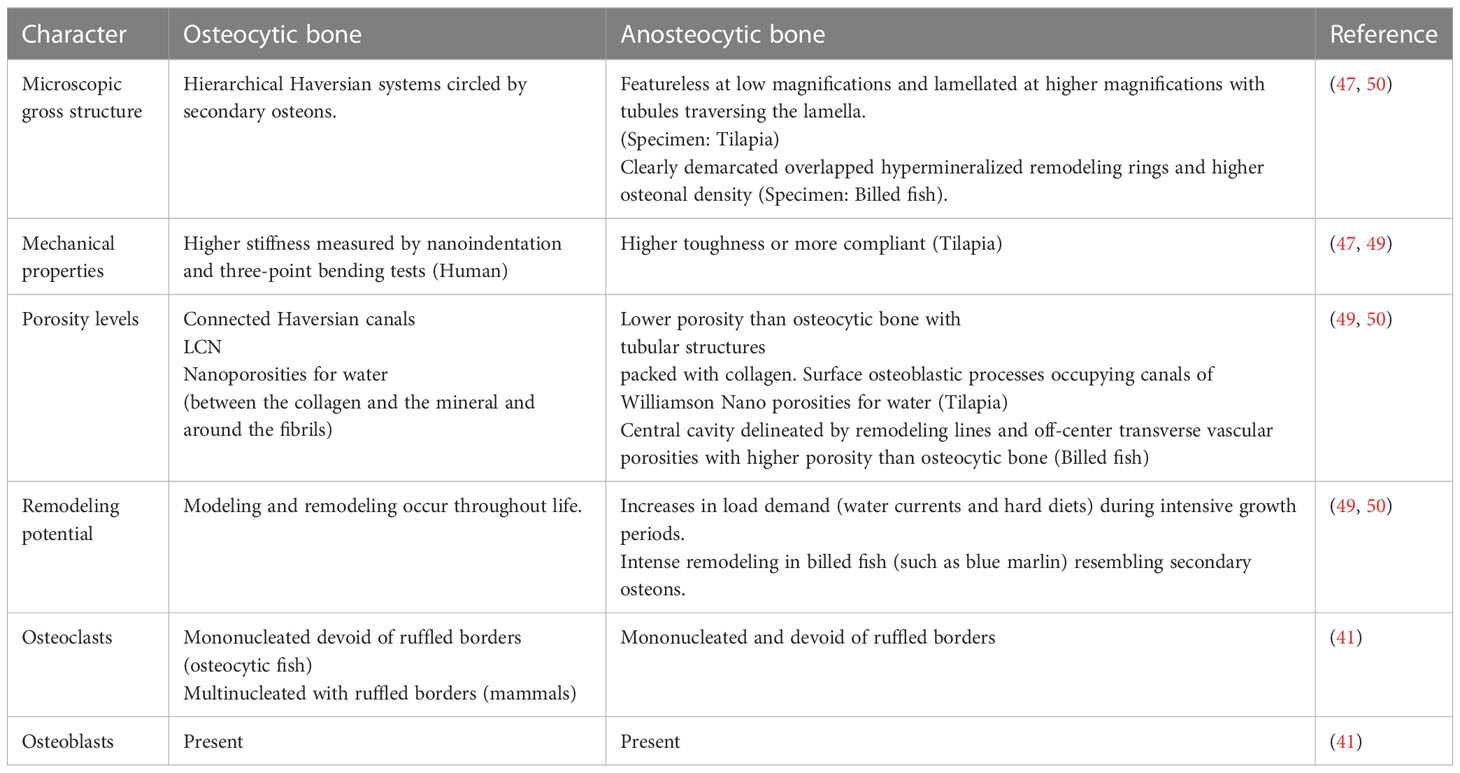
Table 1 The summary of differences between osteocytic and anosteocytic bone.
The dictum that osteocyte’s primary function is to orchestrate bone remodeling was challenged by demonstrating that anosteocytic bone of billfish rostra with heavy and cyclical cantilever loading exhibited heavily remodeled bone in the form of overlapping secondary osteons with “strike resemblance” to mammalian secondary osteons. The differentiating features were the absence of osteocytes and the higher spatial density of osteons (50). Also, anosteocytic osteons were an order of magnitude smaller than osteocytic osteons, rendering bone twice as much porous as osteocytic bone (50). Those features contribute to anosteocytic bone having an interconnected bone network that is similar to osteocytic (50) bone and can explain how anosteocytic bone of billfish can exhibit heavy remodeling since the bone surface can be readily accessed by osteoclasts for resorption. Nonetheless, the absence of osteocytes is rather perplexing since modern osteocytes are thought to respond to multiple redundant ways to initiate osteoclast formation and are major regulators of osteoclastogenesis and bone resorption. Osteocyte regulatory role in osteoclast formation (discussed below) is undeniable but may be dispensable for osteoclast formation and bone resorption.
Furthermore, acellular bone is less efficient in mineral homeostasis than osteocytic bone, partly due to the absence of osteocytic osteolysis. Osteocytic osteolysis is present in certain fish species that undergo periods of increased metabolic demand, such as in seasonal migration and during reproduction (52–54). This may indicate that mineral homeostasis is the evolutionary pressure favoring osteocytic bone, giving ancestral osteocytes the primary function of mineral homeostasis. Indeed, comparative histological data and phylogenetic distribution of both bone types highlight the role of osteocytes in mineral homeostasis through osteocytic osteolysis, which may be the primary reason for osteocyte reemergence [for a full review of phylogeny, refer to (39)].
The evolution of vertebrate endoskeleton is a milestone that conferred its species a survival advantage in the form of protection of neural tissues and vital organs, locomotion, and mineral homeostasis derived from its ground substance, calcium phosphate hydroxyapatite. Phosphorus is particularly critical for several reasons, one of which is that the most basic form of energy is a high phosphate molecule, the adenosine triphosphate (ATP). ATP utilization in aerobic glycolysis is the main pathway for energy bursts in vertebrates that would be impossible without ATP (55). In this respect, osteocytes are one of the primary regulators of systemic phosphate levels through Dmp1 and fibroblast growth factor 23 (FGF23); as in Dmp1 knockout osteocytes, FGF23 is released into the circulation, causing hypophosphatemia (56).
Mineral homeostasis is particularly important in terrestrial animals who live on land since aquatic ancestors (with cellular or acellular bone) can mobilize calcium and phosphorus from their surrounding environment through their digestive tract and gills; hence, bone is less critically active in mineral homeostasis than that of terrestrial animals in which osteocytes are majorly involved in its coordination.
In the study of fossilized animals, osteocytes can tell us a lot since osteocytes lacuna retain their shape in fossilized bone specimens as molds, and their structure (size and shape) is a proxy for the metabolic and mineral homeostatic ability of the studied animal. Using focused ion beam-scanning electron microscopy (FIB-SEM), researchers studied two taxa, osteostracans, and placoderms, of the earliest jawless and jawed vertebrates. Osteocyte lacunae in both taxa were comparable with their modern counterparts. The authors noted an area of low density contrasting the air-filled lacuna, referred to as an area of osteocytic osteolysis. This was indirect evidence of mineral metabolism of the first osteocytes (57).
6 RANKL expression by osteocytes
The role of osteocytes in altering osteoclast and osteoblast formation, thus, orchestrating bone remodeling, is known through the release of the receptor activator of nuclear factor κB ligand (RANKL) and sclerostin, respectively (58). RANKL is an obligatory cytokine that works with macrophage colony-stimulating factor (M-CSF) and pro-inflammatory cytokines to induce and maintain osteoclast formation and bone resorption (59, 60). Sclerostin is an inhibitor of Wnt coreceptors low-density lipoprotein receptor-related protein (LRP)5 and LRP6, antagonizing the canonical Wnt/β-catenin pathway that regulates the bone mass acquisition and osteoblast differentiation (61, 62).
Osteoclasts develop from hematopoietic monocytes when exposed to the myeloid and B-lymphoid specific transcription factor PU.1, which induces the expression of c-fms, the receptor for M-CSF (63). The now committed monocytes express RANK, which binds RANKL that is required to differentiate and activate osteoclasts. RANKL osteoclastogenic signaling can be neutralized by two receptors, osteoprotegerin (OPG) and leucine-rich repeat-containing G protein-coupled receptor (LRG4). OPG is a decoy-soluble RANKL receptor produced in many instances by the same cell expressing RANKL and prevents RANKL-RANK interaction. LRG4 competes with RANK and is found on the surface of osteoclast precursors and inhibits the nuclear factor of activated T cells (NFATC1) that initiates the necessary intracellular signaling cascade for osteoclast maturation and differentiation (64).
Different sources of RANKL contribute to bone remodeling and regardless of the source of RANKL, it is an obligatory osteoclastogenic factor that without osteoclast formation comes to a halt at varying degrees. Conditional deletion of RANKL in genetically distinct cell populations results in varying degrees of osteopetrosis. The deletion of RANKL from hypertrophic chondrocytes contributes to severe osteopetrosis and the presence of mineralized cartilage that is typically resorbed during the process of endochondral ossification due to the complete absence of osteoclasts (26). Conditional deletion of RANKL using osterix-Cre (active in late proliferating chondrocytes and early differentiating osteoblasts) or osteocalcin-Cre (active in mature osteoblasts) produced transgenic mice with defective tooth eruption, severe osteopetrosis, and unresorbed calcified cartilage. Conditional deletion of RANKL using Prx1-Cre mice resulted in osteoclast absence, higher bone mineral density, and severe osteopetrosis in the femurs. Additionally, the mice exhibited arrested tooth eruption, unresorbed cartilage, and widened growth plates in the distal femur, similar to RANKL or RANK germline deletion. Using Osx1-Cre mice led to a phenotype similar to that of Prx1-Cre mice. Ocn-Cre mice exhibited similar characteristics to the previous Cre models, except that some tooth eruption was observed in Ocn-Cre transgenic deletion (26).
Deletion of RANKL by targeting Cre activity under the control of the 10-kb Dmp1 promotor produced mice that were normal at 5 weeks of age and with no skeletal phenotype at birth, but which became progressively apparent with age. Dmp1-Cre mice (from 8 weeks) exhibited higher bone mineral density and cancellous bone mass in the femur and L4 vertebrae, and there was a significant (70%) decrease in the genomic RANKL content of collagenase-digested, osteocyte-enriched femoral cortical bones but only a modest (40%) decrease in whole tibias and L4 vertebrae (26, 65). This means that the contribution of RANKL produced by Dmp1-expressing cells is modest compared to the entire RANKL-expressing cell populations. Although only a modest decrease in RANKL expression was measured, this decrease led to a significant decrease in the number of osteoclasts in the cancellous bone of the distal femur of 6-month-old mice; additionally, the plasma of these mice had lower CTX and Ocn, while soluble RANKL did not change (26). As discussed above, it is important to note that since the 10-kb Dmp1 promoter is not osteocyte specific, this change may have been the result of the deletion of RANKL from subsets of osteoblasts and lining cells.
Deletion of RANKL under the regulatory elements of the Sost gene resulted in recombination in osteocytes but not in osteoblasts or lining cells. Sost-Cre mice had a higher cancellous bone mass and higher bone mineral density starting at 5 weeks of age. This phenotype increased at 22 weeks of age. Also, it resulted in a substantial decrease in RANKL genomic DNA in collagenase-digested femur shafts but not whole tibias and a significant decrease in RANKL mRNA expression in tibias, L5 vertebrae, and calvaria. Osteoclast number and surface area were significantly decreased in endocortical surfaces but increased more in cancellous regions. Those characteristics are similar to the skeletal phenotype that was generated using the 10-kb Dmp1 promoter (30).
Since the Sost-Cre transgene does not lead to recombination in lining cells or osteoblasts compared to the 10-kb Dmp1-Cre transgene (30), the Sost-Cre expressing cells (osteocytes in bone) signifies the role of osteocytes in supplying RANKL required for osteoclast formation in the context of physiological bone remodeling, but it does not mean that osteocytes are solely or exclusively responsible for supplying RANKL or supporting osteoclastogenesis. It means that they are part of the RANKL-producing cell populations and more so with increasing age. This is consolidated by the osteocyte transcriptome signature proposed by Youlten et al. (2022), which did not include (TNFSF11) the gene encoding RANKL in the signature transcriptome (38).
RANKL deletion driven by Sost-Cre showed a substantial decrease in osteoclast surface area per total bone area in the cancellous bone as opposed to endocortical regions (8 to 10% decrease vs 1 to 2%, respectively) (30), and given that 80% of remodeling happens in cancellous bone during growth, deletion of osteocytic RANKL could be recognized as an important source of RANKL in the vicinity of cancellous bone for bone remodeling during growth (66).
The sheer surface area of the osteocyte network makes it well suited to sense the need for initiating osteoclastogenesis and then act upon it. Given that osteocytes are embedded in the bone matrix, and the membrane-bound form of RANKL is the physiologically relevant form that is required for osteoclast formation, a question arises on how influential osteocyte membrane-bound RANKL is in the process of osteoclastogenesis. Research has demonstrated that osteocyte dendrites can contact cells on the bone surface and the bone marrow space (8, 11, 67). Since membrane-bound RANKL but not soluble RANKL supports osteoclastogenesis, it is of particular interest to show the contact between osteocytes and osteoclast precursors in in vivo animal models.
7 Osteoclastogenesis by osteocytes
Although it has become common wisdom that osteocytes directly initiate osteoclast formation through RANKL, osteoclastogenesis may also be initiated indirectly by other means (Table 2). Targeted ablation of osteocytes using the diphtheria toxin under the direction of the 10-kb Dmp1 promoter in mice with a constitutively active PTH/PTH-related peptide (PTHrP) type 1 receptor (PTH1R) in osteocytes increased osteoclast number and bone resorption (77, 86). Additionally, PTH1R signaling induces RANKL expression, and its deletion in osteocytes using the 10-kb Dmp1 promoter to drive recombinase activity abrogates osteocytic osteoclastogenic potential and RANKL expression (87).
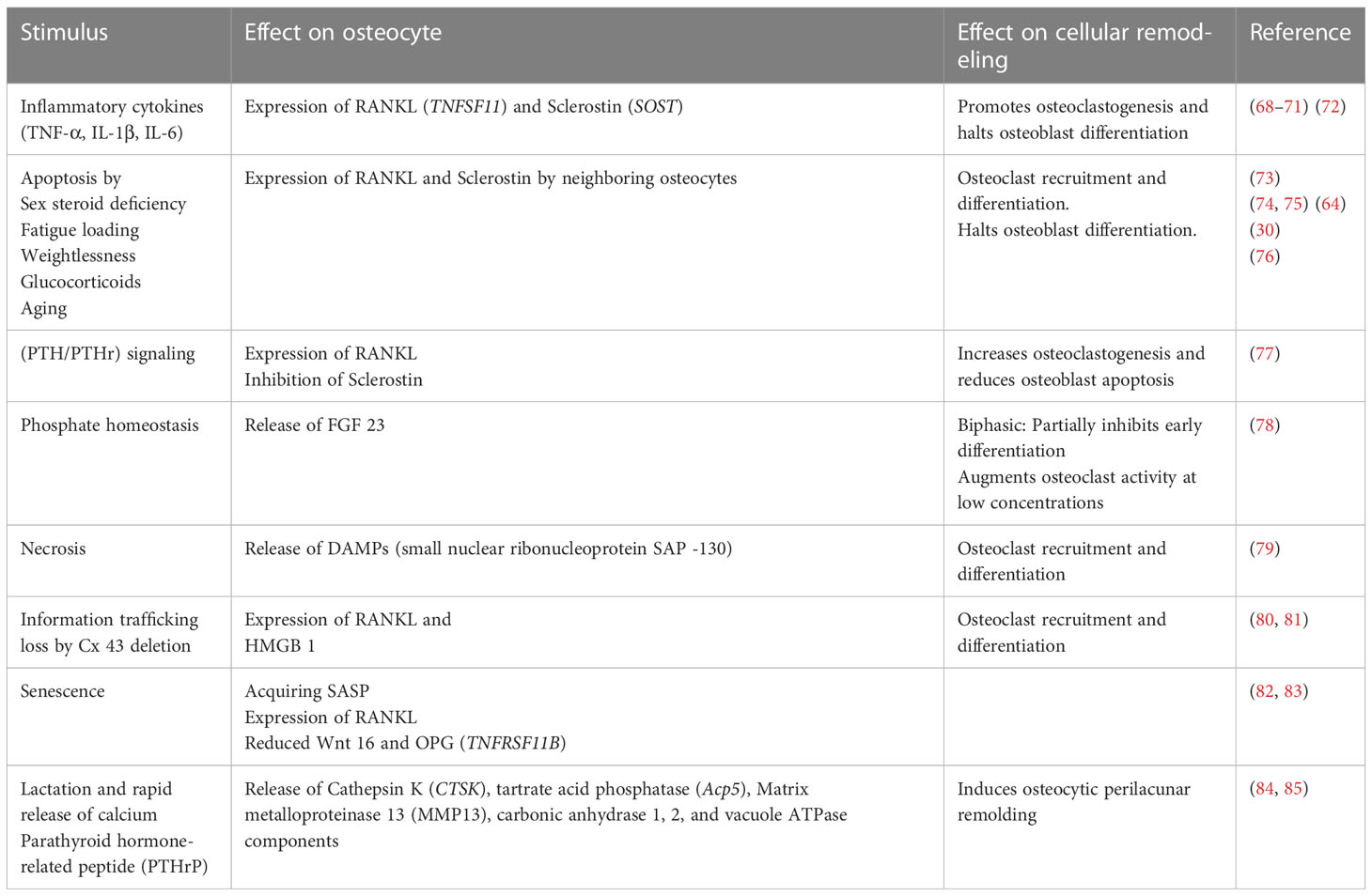
Table 2 The high degree of redundancy built in the osteocytic-driven bone resorption and stimuli impact on effector cells including osteoclasts, osteoblasts, and osteocytes themselves.
Pro-inflammatory signals such as TNF-α, IL-1β, IL-6, and IL-17, act on osteocytes to increase their RANKL expression, osteoclastogenesis, and bone remodeling or to increase sclerostin expression which contributes to reduced bone formation (Figure 1) (68–71) [for a comprehensive review of osteocyte secreted factors regulating osteoclastogenesis, check (88)]. TNF-α is a pleiotropic cytokine with a well-characterized pro-inflammatory effect; it is elevated in chronic inflammatory diseases including but not limited to obesity, diabetes, osteolytic bone diseases such as rheumatoid arthritis, and infectious inflammatory conditions such as periodontitis (89). Osteocytes express both TNF-α receptors TNFR I and II and signaling through TNFRs induced MAPKs phosphorylation and the NF-κB pathway activation to increase osteocyte RANKL and sclerostin expressions to initiate osteoclast formation and reduce osteoblast differentiation in vitro and in vivo (68, 72). Osteocytes may also control osteoclast formation independently of RANKL through FGF23. In vitro early-stage cultures of human monocytes with M-CSF and RANKL exhibited decreased osteoclastogenesis when supplemented with FGF 23, and this effect was abolished with the addition of a pan FGF23 inhibitor (78).
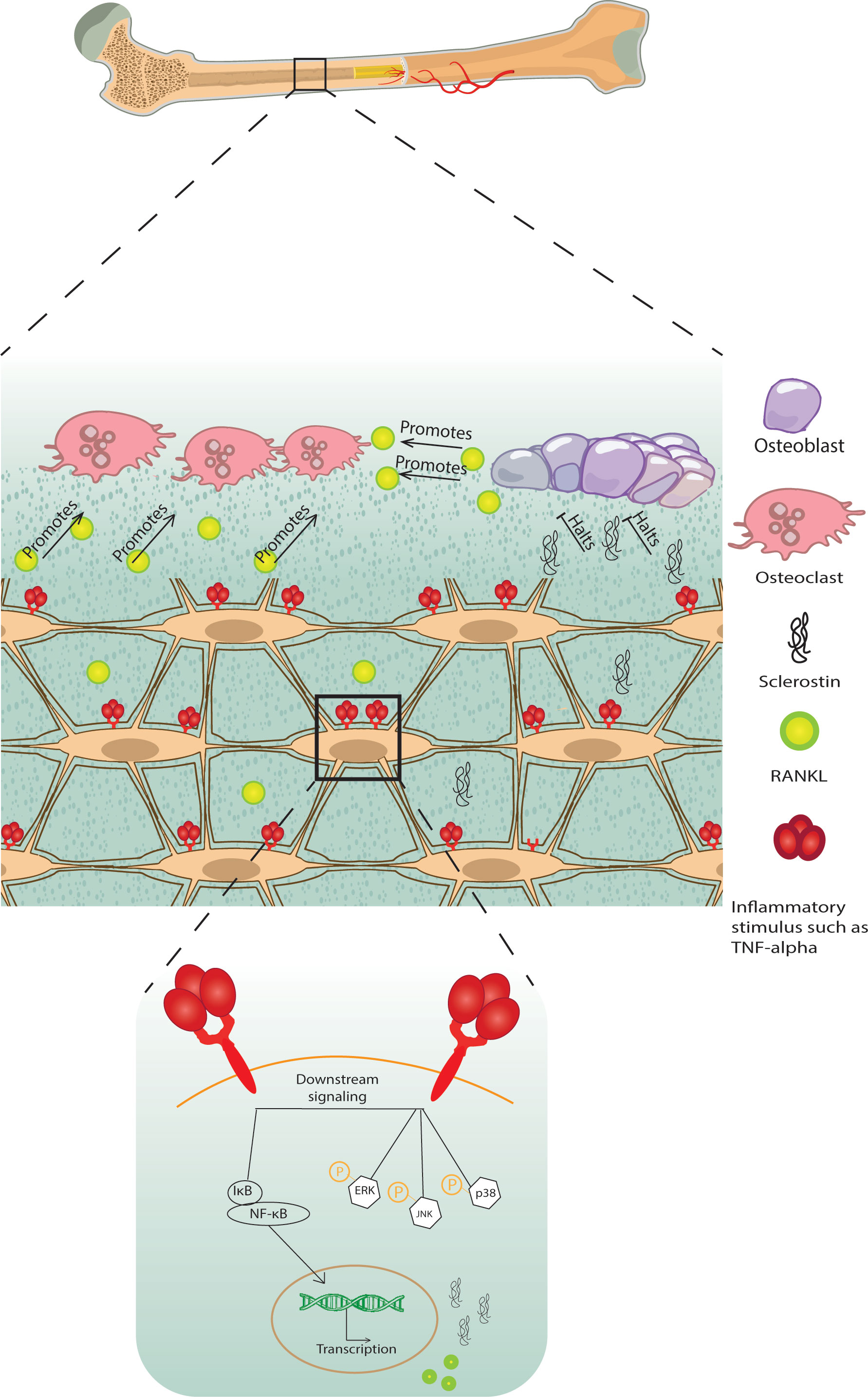
Figure 1 A schematic diagram of bone loss due to inflammatory stimuli cytokine influx. Inflammatory stimuli such as TNF-α and IL-1β lead to osteocyte and osteoblast RANKL expression and osteocyte sclerostin expression, which induces osteoclastogenesis and halts osteoblast formation, respectively, negatively modulating bone remodeling.
Osteocyte apoptosis is accompanied by bone resorption, another mode of action through which osteocytes recruit osteoclasts either by signaling neighboring cells to express RANKL or independent of RANKL by releasing apoptotic bodies, which recruit osteoclast precursors (73, 90). Additionally, apoptotic osteocytes undergo secondary necrosis due to their position in bone, which prevents their phagocytosis; this is accompanied by the release of damage-associated molecular patterns (DAMPs), which activate the monocytic release of inflammatory cytokines that amplify osteoclastogenic signals and lead to the release of RANKL in osteoblast and stromal cells (91). Of note, this mode of osteoclast recruitment and differentiation is not supported by osteoblast apoptosis or osteoblastic apoptotic bodies (90). Apoptosis of osteocytes is seen in glucocorticoid treatment and sex steroid deficiency, such as in ovariectomized rats and mice, and suppressing osteocyte apoptosis induced by sex steroid deficiency inhibits bone resorption (73, 92).
Mechanical signals conveyed to osteocytes modulate osteocyte apoptosis and osteocytes near microcracks resulting from fatigue loading the bone are apoptotic osteocytes that release ATP and signal to bystander osteocytes to express RANKL, which recruits osteoclasts to initiate bone resorption and remodeling (74, 75, 93). Contrastingly, complete weightlessness in unloading leads to osteocyte apoptosis which signals osteoclast recruitment and bone resorption (26, 94). Inducing osteocyte necrosis via the diphtheria toxin recruits osteoclasts, increases their numbers, and produces an osteoporotic phenotype. Necrotic osteocytes release DAMPs, which act as signals for pattern recognition receptors, specifically C-type lectin receptor Mincle on the surface of macrophages which triggers recruitment and differentiation to osteoclasts (79).
Aging osteocytes but not osteoblasts are noted to have a senescent character with DNA damage, mitochondrial dysfunction, telomere dysfunction-induced foci (TIFs), and senescence-associated distension of satellite DNA (SADS) (95). Senescent osteocytes acquire a distinct secretome termed senescent-associated secretory phenotype (SASP), increased RANKL expression, and decreased Wnt16 expression, which is known to inhibit osteoclastogenesis by inducing OPG expression (76, 82, 83, 96).
Administering diphtheria toxin to mice harboring a diphtheria toxin receptor guided by the 10-kb Dmp1 promoter resulted in osteocyte-less mice (OL). OL mice were not completely absent of osteocytes. At 18 weeks of age, the mice exhibited a comparable LCN fluid space, but the connectivity of the osteocyte network was markedly decreased, and the cells had a decreased nuclear size; therefore, the authors termed the mice osteocyte-less where some osteocytes were killed and ablated while others were compromised. RANKL mRNA and the number of osteoclasts were slightly elevated in OL mice; bone remodeling rate as assessed by calcein double labeling did not differ between WT and OL mice (97). The increase in RANKL mRNA and consequently osteoclast numbers in the bone of OL mice may have been due to the fact that ablated (dead) osteocytes signal to the neighboring cells that survived the ablation to release RANKL; however, this hypothesis was not tested.
In another study, the 10-kb Dmp1 promoter was used to ablate osteocytes using diphtheria toxin which caused the mice to have severe sarcopenia, osteoporosis, and degenerative kyphosis at the early stage of 13 weeks, leading to a shorter lifespan. Ablation of osteocytes in these mice caused impairment of osteogenesis and lymphopoiesis, and increased cellular senescence of total bone marrow, primary spongiosa, and cortical bone. It also increased osteoclastogenesis and expanded myeloid lineage differentiation (98).
The transgenic mice at E19.0 had no apparent skeletal phenotype indicating that embryonic development of skeletal tissue appeared to be normal. However, at 4 weeks, transgenic mice had more empty lacunae (60%) devoid of osteocytes within the cortical and cancellous bone matrix. The empty lacunae number increased at week 13 to 80%, which means that the diphtheria toxin had an accumulative effect with age. Mice at 4 weeks exhibited cancellous and cortical bone loss, which was exacerbated with age (at 13 weeks of age). At 13 weeks of age, the mice developed kyphosis due to retardation of vertebral maturation accompanied by vertebral osteoporosis, which led to the development of gait abnormalities. The mice exhibited body weight and muscle tissue loss and focal atrophy shown by examination of gastrocnemius muscle. The mice exhibited reduced osteocalcin gene expression, increased RANKL, and increased RANKL/OPG ratio at 4 weeks. Additionally, histomorphometry analysis revealed that osteoclast surface numbers (N.Oc/BS) significantly increased after osteocytes ablation, and serum collagen type I c-telopeptide (CTX), a bone resorption index, was also significantly increased, which indicated a high level of osteoclast activity (98).
However, knowing that the 10-kb Dmp1 promoter (as discussed above) is not osteocyte specific, these results should be interpreted with caution and studied in comparison to other Cre mouse models that drive recombination in cells of the bone niche since the possibility that other cell populations contributed to the developed phenotype stands still.
8 Discussion
Osteocytes have been termed master manipulators of bone remodeling. But the fact that bones that are devoid of osteocytes experience bone remodeling raises the question of how important osteocytes are for this role. In this review, we discuss osteocytogenesis and the formation of the LCN to showcase the complexity of the osteocyte entity. We discuss current Cre murine models used for the study of osteocyte osteoclastogenic potential through RANKL deletion and highlight their limitations. The importance of understanding osteocyte transitioning from osteoblasts lies in the notion that osteocyte differentiation follows a spectrum. The current murine models used to study osteocyte biology in vivo are not specific to osteocytes and drive the expression of mutations or ablations in osteoblasts, lining cells, and cells residing in the bone marrow. Studies using the Dmp1-Cre or the Sost-Cre to drive RANKL deletion have been shown to drive recombination in other cell types, as discussed above, which calls into question the size of the contribution of osteocytes in supplying RANKL required for osteoclastogenesis. The results of these studies are certainly not to be discarded, but they should be critically analyzed and compared to other Cre models targeting other cell populations within the bone niche, which has provided us with a greater understanding of osteocyte biology and their osteoclastogenic potential (99). Additionally, the observation that those models experienced said phenotypes with increasing age was consistent among the mouse models used to study osteocytes which gives credence to the likelihood of their specificity.
Osteocyte apoptosis and deterioration of the LCN are associated with increased osteoclastogenesis and contribute to bone loss; whether osteocyte apoptosis is causative was highlighted in in vitro studies using osteocyte apoptotic bodies to drive osteoclast formation, but rigorous in vivo studies using a specific osteocyte marker to drive recombination are required. Additionally, studies on bone devoid of osteocytes show that osteocytes are not necessary for osteoclast formation, as bone devoid of osteocytes undergoes resorption and remodeling. Since RANKL can be supplied from other cell types in anosteocytic bone, this means that osteocytes are not essential for this role in anosteocytic bone but contribute to it in osteocytic bone.
Paracrine and endocrine signals act on osteocytes and in turn, osteocytes relay these signals and translate them to effector molecules that control bone remodeling (99). Osteocyte molecules are a target for therapeutic discoveries not only in the physiological regulation of bone remodeling but also in the regulation of remodeling as a complication of conditions such as diabetes and cancer and as endocrine factors that affect phosphate balance, glucose metabolism, and fat mass [for a comprehensive review, check (99)]. Among the most well-characterized molecules are RANKL and sclerostin. Therapies targeting those molecules have demonstrated their success, highlighting the role of the osteocyte not only in physiological bone remodeling but also in pathological bone loss. Our understanding of osteocytes has advanced tremendously; they are regulators of bone remodeling and endocrine cells that affect distant organs; the question of how anosteocytic bone achieves the feat of bone remodeling of osteocytic bones raises the possibility of the presence of other mechanisms regulating bone remodeling independent of osteocytes. Answering this question will broaden our understanding of bone biology and the remodeling of bone beyond the osteocyte.
Author contributions
AM and HK contributed to the conceptualization, supervision, and original draft preparation. AM, HK, and IM contributed to the critical revision of the manuscript. FO and TN contributed to writing, reviewing, and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a JSPS KAKENHI grant from the Japan Society for the Promotion of Science (No. 22K10238 to HK).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Parfitt AM. The cellular basis of bone remodeling: the quantum concept reexamined in light of recent advances in the cell biology of bone. Calcif Tissue Int (1984) 36(Suppl 1):S37–45. doi: 10.1007/bf02406132
2. Parfitt AM. Quantum concept of bone remodeling and turnover: implications for the pathogenesis of osteoporosis. Calcif Tissue Int (1979) 28(1):1–5. doi: 10.1007/bf02441211
3. Bolamperti S, Villa I, Rubinacci A. Bone remodeling: an operational process ensuring survival and bone mechanical competence. Bone Res (2022) 10(1):48. doi: 10.1038/s41413-022-00219-8
4. Delaisse JM, Andersen TL, Kristensen HB, Jensen PR, Andreasen CM, Søe K. Re-thinking the bone remodeling cycle mechanism and the origin of bone loss. Bone (2020) 141:115628. doi: 10.1016/j.bone.2020.115628
5. Parfitt AM. Osteonal and hemi-osteonal remodeling: the spatial and temporal framework for signal traffic in adult human bone. J Cell Biochem (1994) 55(3):273–86. doi: 10.1002/jcb.240550303
6. Robey P. “Mesenchymal stem cells”: fact or fiction, and implications in their therapeutic use. F1000Res (2017) 6:Rev-524. doi: 10.12688/f1000research.10955.1
7. Mizoguchi T, Ono N. The diverse origin of bone-forming osteoblasts. J Bone Miner Res (2021) 36(8):1432–47. doi: 10.1002/jbmr.4410
8. Manolagas SC, Parfitt AM. What old means to bone. Trends Endocrinol Metab (2010) 21(6):369–74. doi: 10.1016/j.tem.2010.01.010
9. Marotti G. The structure of bone tissues and the cellular control of their deposition. Ital J Anat Embryol (1996) 101(4):25–79.
11. Buenzli PR, Sims NA. Quantifying the osteocyte network in the human skeleton. Bone (2015) 75:144–50. doi: 10.1016/j.bone.2015.02.016
12. Nefussi JR, Sautier JM, Nicolas V, Forest N. How osteoblasts become osteocytes: a decreasing matrix forming process. J Biol Buccale (1991) 19(1):75–82.
13. Palumbo C, Palazzini S, Zaffe D, Marotti G. Osteocyte differentiation in the tibia of newborn rabbit: an ultrastructural study of the formation of cytoplasmic processes. Acta Anat (Basel) (1990) 137(4):350–8. doi: 10.1159/000146907
14. Palumbo C. A three-dimensional ultrastructural study of osteoid-osteocytes in the tibia of chick embryos. Cell Tissue Res (1986) 246(1):125–31. doi: 10.1007/bf00219008
15. Palumbo C, Ferretti M, Marotti G. Osteocyte dendrogenesis in static and dynamic bone formation: an ultrastructural study. Anat Rec A Discovery Mol Cell Evol Biol (2004) 278A(1):474–80. doi: 10.1002/ar.a.20032
16. Marotti G. Osteocyte orientation in human lamellar bone and its relevance to the morphometry of periosteocytic lacunae. Metab Bone Dis Relat Res (1979) 1(4):325–33. doi: 10.1016/0221-8747(79)90027-4
17. Wang JS, Wein MN. Pathways controlling formation and maintenance of the osteocyte dendrite network. Curr Osteoporosis Rep (2022) 20:493–504. doi: 10.1007/s11914-022-00753-8
18. Sasano Y, Zhu JX, Kamakura S, Kusunoki S, Mizoguchi I, Kagayama M. Expression of major bone extracellular matrix proteins during embryonic osteogenesis in rat mandibles. Anat Embryol (Berl) (2000) 202(1):31–7. doi: 10.1007/pl00008242
19. Zhu JX, Sasano Y, Takahashi I, Mizoguchi I, Kagayama M. Temporal and spatial gene expression of major bone extracellular matrix molecules during embryonic mandibular osteogenesis in rats. Histochem J (2001) 33(1):25–35. doi: 10.1023/a:1017587712914
20. Sánchez-de-Diego C, Artigas N, Pimenta-Lopes C, Valer JA, Torrejon B, Gama-Pérez P, et al. Glucose restriction promotes osteocyte specification by activating a pgc-1α-Dependent transcriptional program. iScience (2019) 15:79–94. doi: 10.1016/j.isci.2019.04.015
21. Paic F, Igwe JC, Nori R, Kronenberg MS, Franceschetti T, Harrington P, et al. Identification of differentially expressed genes between osteoblasts and osteocytes. Bone (2009) 45(4):682–92. doi: 10.1016/j.bone.2009.06.010
22. Dallas SL, Bonewald LF. Dynamics of the transition from osteoblast to osteocyte. Ann N Y Acad Sci (2010) 1192:437–43. doi: 10.1111/j.1749-6632.2009.05246.x
23. Holmbeck K, Bianco P, Pidoux I, Inoue S, Billinghurst RC, Wu W, et al. The metalloproteinase Mt1-mmp is required for normal development and maintenance of osteocyte processes in bone. J Cell Sci (2005) 118(1):147–56. doi: 10.1242/jcs.01581
24. Zhao W, Byrne MH, Wang Y, Krane SM. Osteocyte and osteoblast apoptosis and excessive bone deposition accompany failure of collagenase cleavage of collagen. J Clin Invest (2000) 106(8):941–9. doi: 10.1172/jci10158
25. Franz-Odendaal TA, Hall BK, Witten PE. Buried alive: how osteoblasts become osteocytes. Dev Dyn (2006) 235(1):176–90. doi: 10.1002/dvdy.20603
26. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med (2011) 17(10):1235–41. doi: 10.1038/nm.2448
27. Lu Y, Xie Y, Zhang S, Dusevich V, Bonewald LF, Feng JQ. Dmp1-targeted cre expression in odontoblasts and osteocytes. J Dental Res (2007) 86(4):320–5. doi: 10.1177/154405910708600404
28. Chia LY, Walsh NC, Martin TJ, Sims NA. Isolation and gene expression of haematopoietic-Cell-Free preparations of highly purified murine osteocytes. Bone (2015) 72:34–42. doi: 10.1016/j.bone.2014.11.005
29. Gooi JH, Pompolo S, Karsdal MA, Kulkarni NH, Kalajzic I, McAhren SHM, et al. Calcitonin impairs the anabolic effect of pth in young rats and stimulates expression of sclerostin by osteocytes. Bone (2010) 46(6):1486–97. doi: 10.1016/j.bone.2010.02.018
30. Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, et al. Osteocytes, not osteoblasts or lining cells, are the main source of the rankl required for osteoclast formation in remodeling bone. PloS One (2015) 10(9):e0138189. doi: 10.1371/journal.pone.0138189
31. Clarke B. Normal bone anatomy and physiology. Clin J Am Soc Nephrol (2008) 3 Suppl 3(Suppl 3):S131–9. doi: 10.2215/cjn.04151206
32. Carter Y, Thomas CD, Clement JG, Peele AG, Hannah K, Cooper DM. Variation in osteocyte lacunar morphology and density in the human femur–a synchrotron radiation micro-ct study. Bone (2013) 52(1):126–32. doi: 10.1016/j.bone.2012.09.010
33. Varga P, Hesse B, Langer M, Schrof S, Männicke N, Suhonen H, et al. Synchrotron X-ray phase nano-Tomography-Based analysis of the lacunar-canalicular network morphology and its relation to the strains experienced by osteocytes in situ as predicted by case-specific finite element analysis. Biomech Model Mechanobiol (2015) 14(2):267–82. doi: 10.1007/s10237-014-0601-9
34. Ayasaka N, Kondo T, Goto T, Kido MA, Nagata E, Tanaka T. Differences in the transport systems between cementocytes and osteocytes in rats using microperoxidase as a tracer. Arch Oral Biol (1992) 37(5):363–9. doi: 10.1016/0003-9969(92)90019-5
35. Knothe Tate ML, Niederer P, Knothe U. In Vivo Tracer transport through the lacunocanalicular system of rat bone in an environment devoid of mechanical loading. Bone (1998) 22(2):107–17. doi: 10.1016/s8756-3282(97)00234-2
36. Azevedo FAC, Carvalho LRB, Grinberg LT, Farfel JM, Ferretti REL, Leite REP, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol (2009) 513(5):532–41. doi: 10.1002/cne.21974
37. Pakkenberg B, Pelvig D, Marner L, Bundgaard MJ, Gundersen HJG, Nyengaard JR, et al. Aging and the human neocortex. Exp Gerontol (2003) 38(1):95–9. doi: 10.1016/S0531-5565(02)00151-1
38. Youlten SE, Kemp JP, Logan JG, Ghirardello EJ, Sergio CM, Dack MRG, et al. Osteocyte transcriptome mapping identifies a molecular landscape controlling skeletal homeostasis and susceptibility to skeletal disease. Nat Commun (2021) 12(1):2444. doi: 10.1038/s41467-021-22517-1
39. Davesne D, Meunier FJ, Schmitt AD, Friedman M, Otero O, Benson RBJ. The phylogenetic origin and evolution of acellular bone in teleost fishes: insights into osteocyte function in bone metabolism. Biol Rev Camb Philos Soc (2019) 94(4):1338–63. doi: 10.1111/brv.12505
40. Shahar R, Dean MN. The enigmas of bone without osteocytes. Bonekey Rep (2013) 2:343. doi: 10.1038/bonekey.2013.77
41. Weiss RE, Watabe N. Studies on the biology of fish bone. iii. ultrastructure of osteogenesis and resorption in osteocytic (Cellular) and anosteocytic (Acellular) bones. Calcif Tissue Int (1979) 28(1):43–56. doi: 10.1007/bf02441217
42. Moss ML. Osteogenesis of acellular teleost fish bone. Am J Anat (1961) 108(1):99–109. doi: 10.1002/aja.1001080107
43. Ofer L, Dumont M, Rack A, Zaslansky P, Shahar R. New insights into the process of osteogenesis of anosteocytic bone. Bone (2019) 125:61–73. doi: 10.1016/j.bone.2019.05.013
44. Moss ML. Studies of the acelluar bone of teleost fish. i. morphological and systematic variations. Acta Anat (Basel) (1961) 46:343–62. doi: 10.1159/000141794
45. Ekanayake S, Hall BK. The development of acellularity of the vertebral bone of the Japanese medaka, oryzias latipes (Teleostei; cyprinidontidae). J Morphol (1987) 193(3):253–61. doi: 10.1002/jmor.1051930304
46. Ekanayake S, Hall BK. Ultrastructure of the osteogenesis of acellular vertebral bone in the Japanese medaka, oryzias latipes (Teleostei, cyprinidontidae). Am J Anat (1988) 182(3):241–9. doi: 10.1002/aja.1001820305
47. Atkins A, Reznikov N, Ofer L, Masic A, Weiner S, Shahar R. The three-dimensional structure of anosteocytic lamellated bone of fish. Acta Biomater (2015) 13:311–23. doi: 10.1016/j.actbio.2014.10.025
48. Horton JM, Summers AP. The material properties of acellular bone in a teleost fish. J Exp Biol (2009) 212(9):1413–20. doi: 10.1242/jeb.020636
49. Currey JD, Shahar R. Cavities in the compact bone in tetrapods and fish and their effect on mechanical properties. J Struct Biol (2013) 183(2):107–22. doi: 10.1016/j.jsb.2013.04.012
50. Atkins A, Dean MN, Habegger ML, Motta PJ, Ofer L, Repp F, et al. Remodeling in bone without osteocytes: billfish challenge bone structure–function paradigms. Proc Natl Acad Sci (2014) 111(45):16047–52. doi: 10.1073/pnas.1412372111
51. Witten PE, Huysseune A. A comparative view on mechanisms and functions of skeletal remodelling in teleost fish, with special emphasis on osteoclasts and their function. Biol Rev Camb Philos Soc (2009) 84(2):315–46. doi: 10.1111/j.1469-185X.2009.00077.x
52. Lopez E, Peignoux-Deville J, Lallier F, Martelly E, Milet C. Effects of calcitonin and ultimobranchialectomy (Ubx) on calcium and bone metabolism in the eel, Anguilla Anguilla l. Calcif Tissue Res (1976) 2):173–86. doi: 10.1007/bf02546406
53. Kacem A, Meunier FJ. Halastatic demineralization in the vertebrae of Atlantic salmon, during their spawning migration. J Fish Biol (2003) 63(5):1122–30. doi: 10.1046/j.1095-8649.2003.00229.x
54. Sbaihi M, Kacem A, Aroua S, Baloche S, Rousseau K, Lopez E, et al. Thyroid hormone-induced demineralisation of the vertebral skeleton of the eel, Anguilla Anguilla. Gen Comp Endocrinol (2007) 151(1):98–107. doi: 10.1016/j.ygcen.2006.12.009
55. Ruben JA, Bennett AA. The evolution of bone. Evolution (1987) 41(6):1187–97. doi: 10.1111/j.1558-5646.1987.tb02460.x
56. Feng JQ, Clinkenbeard EL, Yuan B, White KE, Drezner MK. Osteocyte regulation of phosphate homeostasis and bone mineralization underlies the pathophysiology of the heritable disorders of rickets and osteomalacia. Bone (2013) 54(2):213–21. doi: 10.1016/j.bone.2013.01.046
57. Haridy Y, Osenberg M, Hilger A, Manke I, Davesne D, Witzmann F. Bone metabolism and evolutionary origin of osteocytes: novel application of fib-sem tomography. Sci Adv (2021) 7(14):eabb9113. doi: 10.1126/sciadv.abb9113
58. Schaffler MB, Cheung WY, Majeska R, Kennedy O. Osteocytes: master orchestrators of bone. Calcif Tissue Int (2014) 94(1):5–24. doi: 10.1007/s00223-013-9790-y
59. Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev (1999) 20(3):345–57. doi: 10.1210/edrv.20.3.0367
60. Kodama H, Yamasaki A, Nose M, Niida S, Ohgame Y, Abe M, et al. Congenital osteoclast deficiency in osteopetrotic (Op/Op) mice is cured by injections of macrophage colony-stimulating factor. J Exp Med (1991) 173(1):269–72. doi: 10.1084/jem.173.1.269
61. Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, et al. Sclerostin binds to Lrp5/6 and antagonizes canonical wnt signaling*. J Biol Chem (2005) 280(20):19883–7. doi: 10.1074/jbc.M413274200
62. Tu X, Delgado-Calle J, Condon KW, Maycas M, Zhang H, Carlesso N, et al. Osteocytes mediate the anabolic actions of canonical Wnt/B-catenin signaling in bone. Proc Natl Acad Sci U.S.A. (2015) 112(5):E478–E86. doi: 10.1073/pnas.1409857112
63. Hamilton JA. Csf-1 signal transduction. J Leukoc Biol (1997) 62(2):145–55. doi: 10.1002/jlb.62.2.145
64. Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G, et al. Lgr4 is a receptor for rankl and negatively regulates osteoclast differentiation and bone resorption. Nat Med (2016) 22(5):539–46. doi: 10.1038/nm.4076
65. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through rankl expression. Nat Med (2011) 17(10):1231–4. doi: 10.1038/nm.2452
66. Langdahl B, Ferrari S, Dempster DW. Bone modeling and remodeling: potential as therapeutic targets for the treatment of osteoporosis. Ther Adv Musculoskelet Dis (2016) 8(6):225–35. doi: 10.1177/1759720x16670154
67. Guo D, Bonewald LF. Advancing our understanding of osteocyte cell biology. Ther Adv Musculoskelet Dis (2009) 1(2):87–96. doi: 10.1177/1759720x09341484
68. Marahleh A, Kitaura H, Ohori F, Kishikawa A, Ogawa S, Shen WR, et al. Tnf-A directly enhances osteocyte rankl expression and promotes osteoclast formation. Front Immunol (2019) 10:2925. doi: 10.3389/fimmu.2019.02925
69. Kulkarni RN, Bakker AD, Everts V, Klein-Nulend J. Mechanical loading prevents the stimulating effect of il-1β on osteocyte-modulated osteoclastogenesis. Biochem Biophys Res Commun (2012) 420(1):11–6. doi: 10.1016/j.bbrc.2012.02.099
70. Wu Q, Zhou X, Huang D, Ji Y, Kang F. Il-6 enhances osteocyte-mediated osteoclastogenesis by promoting Jak2 and rankl activity in vitro. Cell Physiol Biochem (2017) 41(4):1360–9. doi: 10.1159/000465455
71. Liao C, Cheng T, Wang S, Zhang C, Jin L, Yang Y. Shear stress inhibits il-17a-Mediated induction of osteoclastogenesis Via osteocyte pathways. Bone (2017) 101:10–20. doi: 10.1016/j.bone.2017.04.003
72. Ohori F, Kitaura H, Marahleh A, Kishikawa A, Ogawa S, Qi J, et al. Effect of tnf-A-Induced sclerostin on osteocytes during orthodontic tooth movement. J Immunol Res (2019) 2019:9716758. doi: 10.1155/2019/9716758
73. McCutcheon S, Majeska RJ, Spray DC, Schaffler MB, Vazquez M. Apoptotic osteocytes induce rankl production in bystanders Via purinergic signaling and activation of pannexin channels. J Bone Miner Res (2020) 35(5):966–77. doi: 10.1002/jbmr.3954
74. Verborgt O, Gibson GJ, Schaffler MB. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res (2000) 15(1):60–7. doi: 10.1359/jbmr.2000.15.1.60
75. Cheung WY, Fritton JC, Morgan SA, Seref-Ferlengez Z, Basta-Pljakic J, Thi MM, et al. Pannexin-1 and P2x7-receptor are required for apoptotic osteocytes in fatigued bone to trigger rankl production in neighboring bystander osteocytes. J Bone Miner Res (2016) 31(4):890–9. doi: 10.1002/jbmr.2740
76. Cao J, Venton L, Sakata T, Halloran BP. Expression of rankl and opg correlates with age-related bone loss in Male C57bl/6 mice. J Bone Miner Res (2003) 18(2):270–7. doi: 10.1359/jbmr.2003.18.2.270
77. O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, et al. Control of bone mass and remodeling by pth receptor signaling in osteocytes. PloS One (2008) 3(8):e2942. doi: 10.1371/journal.pone.0002942
78. Allard L, Demoncheaux N, Machuca-Gayet I, Georgess D, Coury-Lucas F, Jurdic P, et al. Biphasic effects of vitamin d and Fgf23 on human osteoclast biology. Calcif Tissue Int (2015) 97(1):69–79. doi: 10.1007/s00223-015-0013-6
79. Andreev D, Liu M, Weidner D, Kachler K, Faas M, Grüneboom A, et al. Osteocyte necrosis triggers osteoclast-mediated bone loss through macrophage-inducible c-type lectin. J Clin Invest (2020) 130(9):4811–30. doi: 10.1172/JCI134214
80. Zhang Y, Paul EM, Sathyendra V, Davison A, Sharkey N, Bronson S, et al. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PloS One (2011) 6(8):e23516. doi: 10.1371/journal.pone.0023516
81. Davis HM, Pacheco-Costa R, Atkinson EG, Brun LR, Gortazar AR, Harris J, et al. Disruption of the Cx43/Mir21 pathway leads to osteocyte apoptosis and increased osteoclastogenesis with aging. Aging Cell (2017) 16(3):551–63. doi: 10.1111/acel.12586
82. Movérare-Skrtic S, Henning P, Liu X, Nagano K, Saito H, Börjesson AE, et al. Osteoblast-derived Wnt16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat Med (2014) 20(11):1279–88. doi: 10.1038/nm.3654
83. Kim H-N, Xiong J, MacLeod RS, Iyer S, Fujiwara Y, Cawley KM, et al. Osteocyte rankl is required for cortical bone loss with age and is induced by senescence. JCI Insight (2020) 5(19):e138815. doi: 10.1172/jci.insight.138815
84. Jähn K, Kelkar S, Zhao H, Xie Y, Tiede-Lewis LM, Dusevich V, et al. Osteocytes acidify their microenvironment in response to pthrp in vitro and in lactating mice in vivo. J Bone Miner Res (2017) 32(8):1761–72. doi: 10.1002/jbmr.3167
85. Qing H, Ardeshirpour L, Pajevic PD, Dusevich V, Jähn K, Kato S, et al. Demonstration of osteocytic Perilacunar/Canalicular remodeling in mice during lactation. J Bone Miner Res (2012) 27(5):1018–29. doi: 10.1002/jbmr.1567
86. Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab (2007) 5(6):464–75. doi: 10.1016/j.cmet.2007.05.001
87. Saini V, Marengi DA, Barry KJ, Fulzele KS, Heiden E, Liu X, et al. Parathyroid hormone (Pth)/Pth-related peptide type 1 receptor (Ppr) signaling in osteocytes regulates anabolic and catabolic skeletal responses to pth. J Biol Chem (2013) 288(28):20122–34. doi: 10.1074/jbc.M112.441360
88. Kitaura H, Marahleh A, Ohori F, Noguchi T, Shen WR, Qi J, et al. Osteocyte-related cytokines regulate osteoclast formation and bone resorption. Int J Mol Sci (2020) 21(14):5169. doi: 10.3390/ijms21145169
89. Tracey KJ, Cerami A. Tumor necrosis factor, other cytokines and disease. Annu Rev Cell Biol (1993) 9:317–43. doi: 10.1146/annurev.cb.09.110193.001533
90. Kogianni G, Mann V, Noble BS. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res (2008) 23(6):915–27. doi: 10.1359/jbmr.080207
91. Messmer D, Yang H, Telusma G, Knoll F, Li J, Messmer B, et al. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J Immunol (2004) 173(1):307–13. doi: 10.4049/jimmunol.173.1.307
92. Emerton KB, Hu B, Woo AA, Sinofsky A, Hernandez C, Majeska RJ, et al. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone (2010) 46(3):577–83. doi: 10.1016/j.bone.2009.11.006
93. Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone (2012) 50(5):1115–22. doi: 10.1016/j.bone.2012.01.025
94. Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res (2006) 21(4):605–15. doi: 10.1359/jbmr.060107
95. Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, et al. Identification of senescent cells in the bone microenvironment. J Bone Miner Res (2016) 31(11):1920–9. doi: 10.1002/jbmr.2892
96. Piemontese M, Almeida M, Robling AG, Kim H-N, Xiong J, Thostenson JD, et al. Old age causes De Novo intracortical bone remodeling and porosity in mice. JCI Insight (2017) 2(17):e93771. doi: 10.1172/jci.insight.93771
97. Sato M, Asada N, Kawano Y, Wakahashi K, Minagawa K, Kawano H, et al. Osteocytes regulate primary lymphoid organs and fat metabolism. Cell Metab (2013) 18(5):749–58. doi: 10.1016/j.cmet.2013.09.014
98. Ding P, Gao C, Gao Y, Liu D, Li H, Xu J, et al. Osteocytes regulate senescence of bone and bone marrow. eLife (2022) 11:e81480. doi: 10.7554/eLife.81480
Keywords: osteocyte, osteoclast, lacunocanalicular network, anosteocytic bone, RANKL
Citation: Marahleh A, Kitaura H, Ohori F, Noguchi T and Mizoguchi I (2023) The osteocyte and its osteoclastogenic potential. Front. Endocrinol. 14:1121727. doi: 10.3389/fendo.2023.1121727
Received: 12 December 2022; Accepted: 07 April 2023;
Published: 24 May 2023.
Edited by:
Jonathan H. Tobias, University of Bristol, United KingdomReviewed by:
Natalie A. Sims, University of Melbourne, AustraliaUlf Holger Lerner, Umeå University, Sweden
Copyright © 2023 Marahleh, Kitaura, Ohori, Noguchi and Mizoguchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aseel Marahleh, YXNlZWwubWFobW91ZC5zdWxlaW1hbi5tYXJhaGxlaC5lNkB0b2hva3UuYWMuanA=; Hideki Kitaura, aGlkZWtpLmtpdGF1cmEuYjRAdG9ob2t1LmFjLmpw