 Junying Hua
Junying Hua Jianping Huang2
Jianping Huang2 Gang Li
Gang Li Sien Lin
Sien Lin Liao Cui
Liao Cui
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 04 April 2023
Sec. Bone Research
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1119427
This article is part of the Research Topic New Insights into Cell Metabolism in Bone-cartilage Homeostasis View all 7 articles
Long-term or supra-physiological dose of glucocorticoid (GC) application in clinic can lead to impaired bone growth and osteoporosis. The side effects of GC on the skeletal system are particularly serious in growing children, potentially causing growth retardation or even osteoporotic fractures. Children’s bone growth is dependent on endochondral ossification of growth plate chondrocytes, and excessive GC can hinder the development of growth plate and longitudinal bone growth. Despite the availability of drugs for treating osteoporosis, they have failed to effectively prevent or treat longitudinal bone growth and development disorders caused by GCs. As of now, there is no specific drug to mitigate these severe side effects. Traditional Chinese Medicine shows potential as an alternative to the current treatments by eliminating the side effects of GC. In summary, this article comprehensively reviews the research frontiers concerning growth and development disorders resulting from supra-physiological levels of GC and discusses the future research and treatment directions for optimizing steroid therapy. This article may also provide theoretical and experimental insight into the research and development of novel drugs to prevent GC-related side effects.
Glucocorticoids (GCs) are a class of steroid hormones that are produced naturally by the adrenal gland and regulate various physiological processes in the body, including metabolism, immune response, and stress response. The endogenous physiological dose of glucocorticoid (GC) plays a key role in maintaining normal bone metabolism and osteogenic differentiation (1). Synthetic glucocorticoids are widely used in medicine as anti-inflammatory and immunosuppressive agents (2). They can be divided into long-acting, medium-acting and short-acting based on their duration of action and half-life. Short-acting glucocorticoids, such as hydrocortisone and cortisone, are primarily used for replacement therapy of adrenocortical insufficiency (3), while medium-acting glucocorticoids, such as prednisone and methylprednisolone, are mainly used for anti-rheumatic diseases, autoimmune diseases, immune transplantation therapy, etc. (4, 5). Long-acting glucocorticoids, such as dexamethasone and betamethasone, have strong anti-inflammatory potency, long duration of action, and are preferred for anti-allergy(6). Exogenous synthetic GCs are also commonly used in children with progressive muscle dystrophy (7, 8) and respiratory diseases (6). The therapeutic effect of GC has been reported very intensively, however, the side effects particularly in skeletal system caused by long-term or high-dose medication cannot be ignored. Since the skeletal system of adolescents has not yet been developed, GC can induce bone growth suppression, resulting in a significant decrease in height (9). Currently, drugs for the treatment of GC-induced osteoporosis cannot effectively reduce growth suppression(10). Therefore, there is still a lack of specific drugs to tackle GC-induced growth suppression. Recent studies have actively explored the mechanism of GC-induced bone growth retardation, which is a complex process involving multiple pathways (Figure 1). GCs affects bone growth mainly through two ways. First, GCs can affect physiological process by regulating hormone (11, 12), growth factors (13), calcium and phosphorus metabolism (14) and angiogenesis (15). Second, GC can affect bone growth in cellular behaviors by inhibiting the chondrocytes in long bone growth plate. GCs can directly inhibit chondrocyte proliferation and differentiation (16, 17), matrix proteoglycan synthesis(18), and cell apoptosis (Figure 2) (19). This review summarizes the recent studies on the pathological mechanism of glucocorticoid-induced bone growth retardation, providing valuable insights for the development of targeted therapies to address this issue.
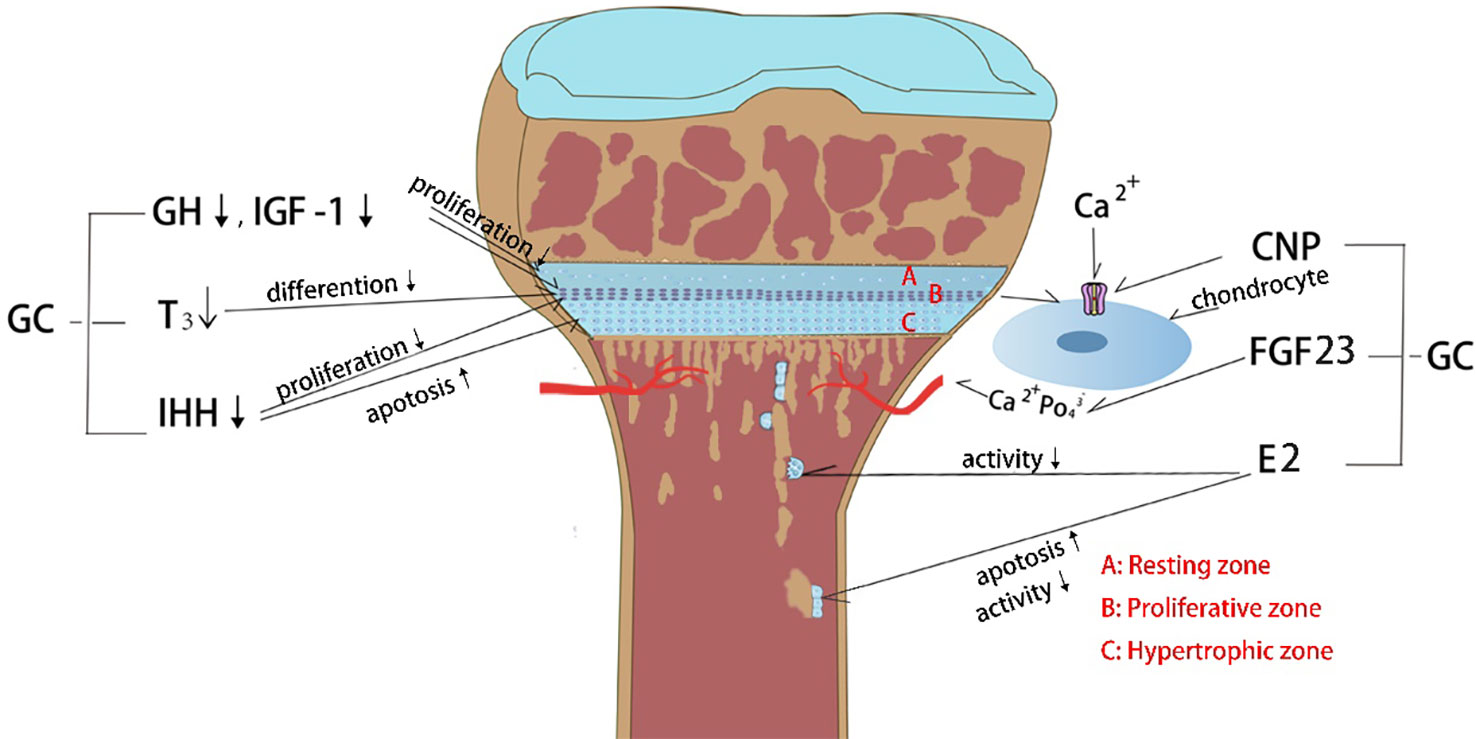
Figure 1 Effects of glucocorticoid (GC) on endocrine hormones and cytokines. (1) GC decreases growth hormone (GH) and insulin growth factor-1 (IGF-1) levels which in turn inhibits the chondrocyte proliferation. (2) GC affects thyroid hormone secretion and further inhibits the chondrocyte hypertrophy. (3) GC decreases Indian hedgehog factor (IHH) secretion and then inhibits chondrocyte proliferation and increases chondrocyte apoptosis. (4) GC constrains C-type natriuretic peptide (CNP) production, which controlling the entry of calcium ion into chondrocytes to stimulate growth. (5) GC causes hypocalcemia through upregulation of fibroblast growth factor 23 (FGF23) expression in bone and plasma, which in turn inhibits chondrocyte proliferation. (6) GC inhibits estrogen (E2) secretion, causing decreased osteoclast activity and increased osteoblast apoptosis.
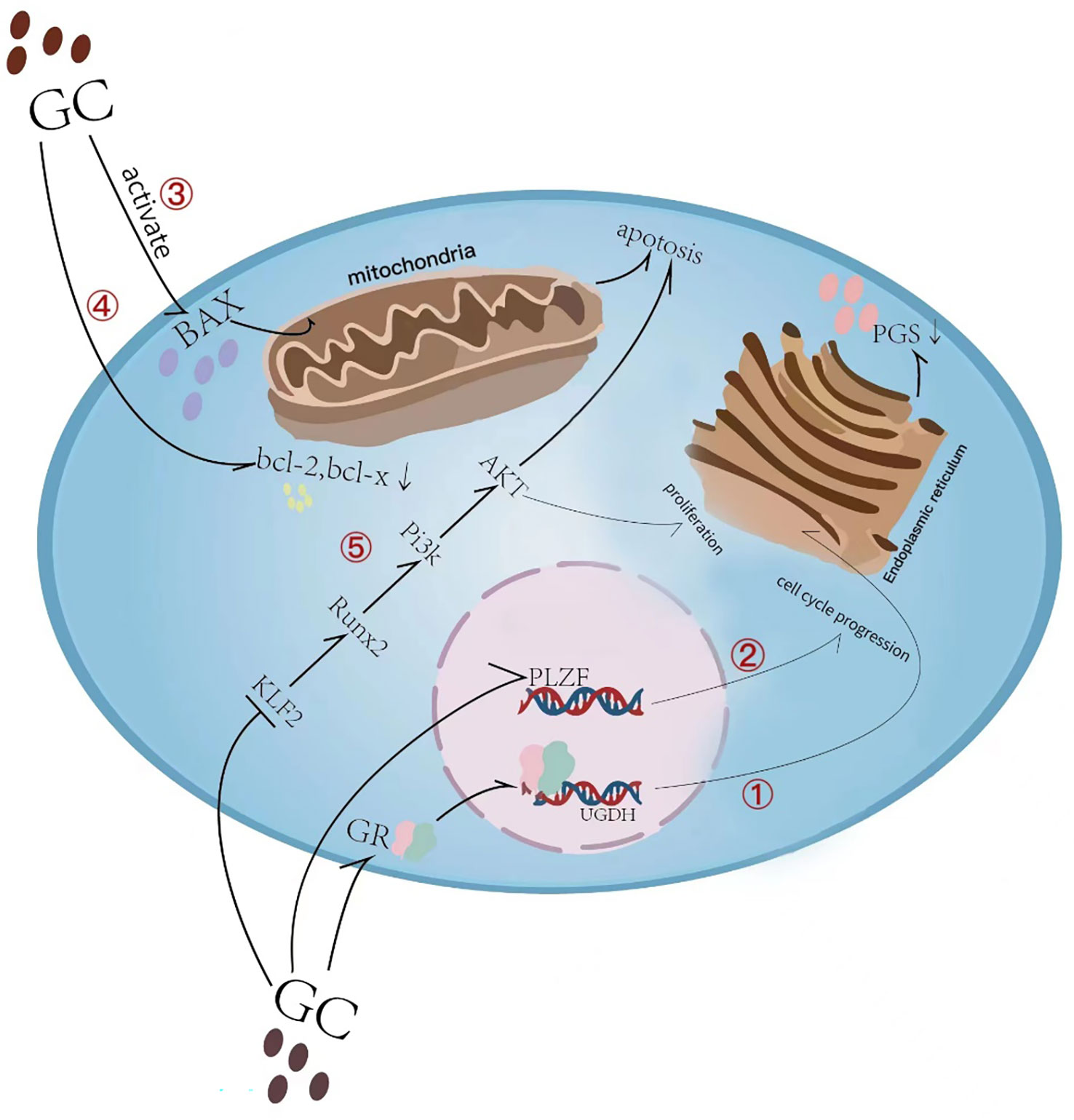
Figure 2 Overview of glucocorticoid-induced chondrocyte apoptosis and matrix synthesis reduction. (1) Glucocorticoid (GC) induces direct binding of glucocorticoid receptor (GR) to the uridine diphosphate glucose dehydrogenase (UGDH) promoter in chondrocytes and suppresses UGDH gene expression, a change that further resulted in a reduced synthesis of proteoglycans (PGs) in developing chondrocytes. (2) GC causes cell cycle inhibition by suppressing the promyelocytic leukemia zinc finger (PLZF) gene. (3) Glucocorticoid-induced activation of Bax and its translocation to the mitochondrial membrane leads to subsequent induction of apoptosis. (4) GC causes a decrease in the anti-apoptotic proteins Bcl-2 and Bcl-x in growth plate chondrocytes. (5) GC inhibits KLF2 expression which regulating the Runx2-mediated PI3K/AKT and Erk signaling pathways responsible for the chondrocyte apoptosis.
GH is secreted by pituitary cells in the human brain and is mainly regulated by growth hormone-releasing hormones secreted by the hypothalamus. GH exerts its pleiotropic effects through growth hormone receptor (GHR), which can be activated directly by tyrosine kinase activation (20) or indirectly by the induction of IGF-1 (21). Systemic corticosteroids have been shown to inhibit GH secretion (22) and IGF-1 activity (23), and this inhibitory effect is mediated by altering the regulation of somatostatin in the hypothalamus (23). Recent study have demonstrated that dexamethasone (DEX) down-regulates the mRNA expression and binding ability of GHR in growth plate chondrocytes in a dose- and time-dependent manner, and reduces the homologous increase of the IGF-1 receptor (IGFR) and GHR expression (24). In addition, after DEX treatment of pregnant SD rats, several components of the IGF-1 signaling pathway, including IGF-1 receptors, insulin receptor substrates, and serine-threonine protein kinases, were down-regulated in fetal rat growth plate chondrocytes (24). These findings provide insights into the mechanism by which GC interferes with the physiological stimulation of GH and IGF-I on the proliferation of epiphyseal chondrocytes.
Thyroid hormone (triiodothyronine, T3) is essential for bone growth after birth by binding to nuclear receptors TRα1 and TRβ1 in chondrocytes and osteoblasts (25). Impaired thyroid function can result in abnormally thin growth plates, impaired chondrocyte hypertrophy, short stature, delayed bone age, skeletal dysplasia, and delayed tooth development in childhood (26, 27). Hypothyroid rats exhibit disordered growth plates and reduced areas of hypertrophy (28), while hyperthyroidism can lead to accelerated ossification of long bones and cartilage (29). Clinical data indicate that in the serum of patients with Cushing’s syndrome, both male and female, thyroid hormone levels and free T3 are lower than those in the normal control group (30). GC can inhibit the expression of parathyroid hormone-related protein (PTHrP) in the growth plate, resulting in growth retardation (19). Studies in adult mice have shown that parathyroid hormone (PTH) can prevent GC-induced osteoporosis, possibly by blocking osteoblast and osteocyte apoptosis (31). Thyroxine (T4) combined with GC treatment can increase the total growth plate length and restore GC-induced growth inhibition (32).
Estrogen is primarily secreted by the ovaries or produced by the conversion of male hormones by aromatase. The regulation of estrogen on long bone is mainly manifested in two aspects, either through a synergistic effect with other hormones such as GH (33), or by directly binding to estrogen receptors (ERs) to control the physiological process of long bone growth plate (34, 35). Studies have shown that after GC intervention in rats, estrogen levels tend to decrease (36). Estrogen can regulate osteoblast activity and apoptosis (37, 38), effectively inhibit osteoclast-mediated bone resorption (39), and maintain the balance between osteogenesis and osteoclastogenesis. Insufficient estrogen secretion can disrupt this balance, resulting in growth plate ossification disorder. Therefore, the occurrence of growth and development disorders caused by GC may be related to its synergistic effect with estrogen.
Fibroblast growth factor 23 (FGF23) is a hormone synthesized by bone cells that regulates the ‘bone-kidney’ axis and calcium phosphate metabolism (40–42). FGF23 reduces serum phosphate levels by inhibiting proximal tubular phosphate reabsorption and intestinal phosphate absorption (43), while also reducing plasma calcitriol levels by down-regulating the expression of the renal 1-α hydroxylase and up-regulates 24-hydroxylase (44). These mechanisms explain the hypophosphatemia effect of FGF23. FGF receptors are expressed in most tissues including chondrocytes, and regulate their proliferation, differentiation, and mineralization (45–47). GC can up-regulate the expression of FGF23 in bone and plasma, activating FGFR3 receptors and contributing to GC-induced growth disorders via the FGF23/Klotho/FGFR3 pathway (48). However, some studies suggest that dexamethasone and prednisolone can down-regulate FGF23 transcription and FGF23 protein synthesis in the osteoblast-like cells and strongly reduce plasma FGF23 concentration in C57BL/6 mice (49).
IHH belongs to the hedgehog protein family and is a morphogenetic protein that plays a crucial role in embryonic formation and development. IHH is a regulator of chondrocyte differentiation rate (50) and is necessary for the osteoblast lineage in developing long bones, working in conjunction with other factors (such as BMPs) to induce osteoblast differentiation (51). However, studies have shown that GCs can inhibits the proliferation of growth plate chondrocytes and promote their apoptosis by blocking the Ihh/PTHrP signaling pathway in vitro. These findings suggest that GC-mediated IHH disorders may lead to growth plate dysplasia (52).
CNP plays an important role in cartilage growth and endochondral bone growth. CNP and its receptor guanylyl cyclase B (GC-B) are effective stimulators of endochondral bone growth. Cartilage-specific CNP or GC-B knockout mice have significantly shorter bones (53). Loss-of-function mutations in natriuretic peptide receptor (NPR)-B, which encodes for CNP receptor GC-B, have been discovered in extreme dwarfing (54), highlighting the importance of CNP in human cartilage growth. CNP promotes bone growth by facilitating the entry of calcium ions into the growth plate chondrocytes through the NPR2-PKG-BK channel and TRPM7 channel-CaMKII axis (55). Studies have shown that high doses of DEX in young male rats significantly reduced the concentration of NT-proCNP, a marker of CNP production (13, 56), and reduced the thickness of the growth plate and bone length.
The epiphysis adjacent to the hypertrophic zone is the site of vascular and osteocyte invasion and longitudinal bone growth. Vascular endothelial growth factor (VEGF) is a crucial angiogenic factor expressed in many tissues and cell types (57), including growth plate cartilage (57, 58). Studies have shown that dexamethasone affects VEGF expression in epiphyseal chondrocytes, and prednisolone treatment in piglets severely disrupts vascular invasion of the growth plates (59). Additionally, GC treatment induces senescence and increases the cell apoptosis of vascular endothelial cells in the metaphysis of the long bones (60, 61). Furthermore, GC treatment severely interferes with VEGF expression in hypertrophic chondrocytes, impairing normal invasion of blood vessels from the metaphysis to the growth plate and bone formation at the cartilage-bone junction. These effects can alter the kinetics of endochondral ossification, leading to GC-induced growth retardation (59, 62).
Recent studies have highlighted the crucial role of vascularization in regulating longitudinal bone growth during endochondral ossification. Romeo et al. reported that proteases released from type H endothelial cells, rather than osteoclasts, are critical for absorbing cartilage and promoting to longitudinal bone growth (63). This suggests that vascular-associated osteoclasts can stimulate endothelial cells to help digest cartilage templates, thereby regulating vascular growth during endochondral ossification. However, Wang et al. found that treatment with prednisolone significantly reduced the number of CD31 and Emcn double-positive H-type vessels in 8-week-old C57BL/6 mice (64). Moreover, dexamethasone inhibited the formation of H-type vessels in the bones of female offspring rats before and after birth through the PDGFRβ/FAK pathway. In vitro administration of high concentrations of dexamethasone also inhibited the angiogenesis of endothelial progenitor cells. These findings suggest that GCs can inhibit the development of H-type blood vessels in the bone tissue, leading to the inhibition of long bone development (15). Therefore, GCs can have negative effects on vascularization and angiogenesis, which are essential for normal bone growth and development.
As one of the main components of cartilage matrix, proteoglycans (PGs) play an important role in cartilage formation, matrix stability and cell proliferation during early embryonic development (65, 66). Uridine diphosphate glucose dehydrogenase (UGDH) is a key enzyme in the synthesis of PGs in various cell types and involved in maintaining the articular cartilage homeostasis and the development of osteoarthritis. Silencing UGDH gene with specific siRNA significantly reduced the PGs content in human chondrocytes (67). Studies have shown that rat maternal exposure to dexamethasone induces GR to directly bind to the UGDH promoter in fetal rat growth plate chondrocytes, recruiting HDAC1 and Sp3, inducing H3K9 deacetylation, and inhibiting UGDH gene expression. These results in reduced PGs synthesis in developing chondrocytes, leading to disrupted fetal long bone development (18). Therefore, GCs have negative effects on PG synthesis, which is essential for normal cartilage and bone development.
Chondrocytes are initially derived from limb mesenchyme during embryonic development and are located in three different areas of the growth plate: quiescent zone, proliferative zone and hypertrophic zone (68). The quiescent zone contains self-renewing, slowly proliferating chondrocytes that produce highly proliferating chondrocytes that form columns along the bone axis and form a proliferative zone (69). Growth plate senescence is caused by the qualitative and quantitative consumption of stem cell-like cells in the quiescent zone, excessive GC can preserve the proliferative capacity of the growth plate by slowing down the proliferation rate of chondrocytes in the quiescent zone and the consumption of these cells (70). GC treatment of precartilaginous cell line ATDC5 inhibited cell cycle and caused the chondrocyte growth arrest (71). Knockdown of Plzf gene by shRNA alleviated GC-induced cell cycle arrest. This explains the phenomenon of long bone catch-up growth after GC treatment cessation (72, 73). The growth of the long bone is the result of continuous downward proliferation and differentiation of growth plate chondrocytes. Different in vitro and in vivo studies have revealed the strong inhibitory effect of DEX on chondrocyte proliferation (16, 74). Dexamethasone inhibits KLF2 expression in rat tibial growth plate chondrocytes and promotes dexamethasone-induced proliferation inhibition and its apoptosis by targeting the Runx2-mediated PI3K/AKT and Erk signaling pathways (75). DEX acts in a gene-specific manner in cartilage. It promotes the expression of extracellular matrix (ECM) and metabolic transcripts necessary to maintain the phenotype of chondrocytes, and down-regulates cytokines and growth factors that stimulate cartilage to bone transformation (76). Therefore, GC-treated ATDC5 cells showed reduced nodule formation, no alkaline phosphatase (ALP) and alcian blue-positive ECM and matrix mineralization (77). The above results suggest that GC has the effect of inhibiting proliferation, differentiation, and matrix mineralization of growth plate chondrocytes.
Chondrocyte apoptosis is closely related to systemic GC treatment. The application of dexamethasone in rats can lead to apoptosis by activating caspase-3 in the three regions of the growth plate (75). In vitro DEX treatment of HCS-2/8 chondrocytes resulted in a significant increase in apoptosis, which was due to increased caspase-3 cleavage and activation of caspase-8 and -9 by cleavage of the pro-apoptotic factor Bid (78). In addition, DEX induced Bax activation and translocation to the mitochondrial membrane, which subsequently induced apoptosis (79, 80), protecting Bax-deficient mice from dexamethasone-induced apoptosis and growth retardation of growth plate chondrocytes (79). At the same time, the anti-apoptotic proteins Bcl-2 and Bcl-x in each layer of growth plate chondrocytes were significantly reduced after GC treatment (19). Therefore, anti-apoptotic proteins and pro-apoptotic proteins of the Bcl-2 family may be the key factors of DEX-induced growth plate chondrocytes apoptosis (81).
GCs have been shown to have significant effects on osteoclasts during bone growth. GCs stimulate osteoclast differentiation and activity, leading to increased bone resorption and decreased bone formation. This can result in reduced bone mineral density and increased risk of fractures, particularly in children and adolescents who are still growing (82, 83). GCs alter the expression of genes involved in osteoclast differentiation and activity, including RANKL, OPG, and NFATc1 (84). GCs activate osteoclasts via secondary hyperparathyroidism and enhance the maturation and activation of osteoclasts. GCs can both increase osteoclast lifespan by inhibiting osteoclast apoptosis and induce increased osteoclastic bone resorption (85). Recent studies highlight a crucial role of reactive oxygen species (ROS) in osteoclast formation and function by modulating receptor activator of NF-κB ligand (RANKL)-induced signaling, which eventually leading to osteoclast hyperactivity in GC-induced osteonecrosis of the femoral head (ONFH) (86), suggesting antioxidant therapy as a potential alternative to prevent GC-induced ONFH by suppressing ROS level and thereby inhibiting osteoclasts. Additionally, GC may exert diverse effects on osteoclasts when used for the treatment of inflammatory diseases. For example, temporal use of GCs may relieve rheumatoid arthritis (RA) progress, while long-term use of GCs may lead to excessive bone resorption via activating osteoclasts, suggesting a central role of immune system in regulating bone metabolism in RA (87, 88).
Since GC use is the most common iatrogenic cause of bone growth inhibition in children, but this side effect can be largely prevented. The first step is to minimize the use of oral GC in terms of dose and duration. If GC treatment is considered necessary, prevention of bone growth inhibition should also be considered. For patients who need to use GC, when the condition is stable, priority should be given to the treatment of local, short-term, low-dose and alternate-day administration, and short-acting GC with less inhibitory effect on bone growth should be selected as far as possible (89). A clinical study found that (90), compared with short-term administration once a day in the morning, taking 5 mg prednisolone once in the evening can inhibit 24-hour growth hormone secretion. Therefore, compared with taking exogenous GC at night, taking in the morning has less effect on growth rate. Follow-up of children with severe conditions requiring high-dose GC, especially in the first 1-2 months of initial treatment, the changes of plasma GH, IGF-1, C-type natriuretic peptide, amino-terminal Pro-CNP, blood glucose, insulin, blood calcium and other biochemical indicators can be monitored. Through the analysis and evaluation of endocrinologists, the growth status of children can be determined and vitamin D and calcium should be supplemented routinely (91).
At present, no drug has been approved by the FDA for clinical treatment of bone growth retardation caused by GC. However, there are medications that can be used to help mitigate the effects of glucocorticoids on bone growth. These medications may be prescribed off-label by a healthcare provider to help address the issue of bone growth retardation (Table 1).
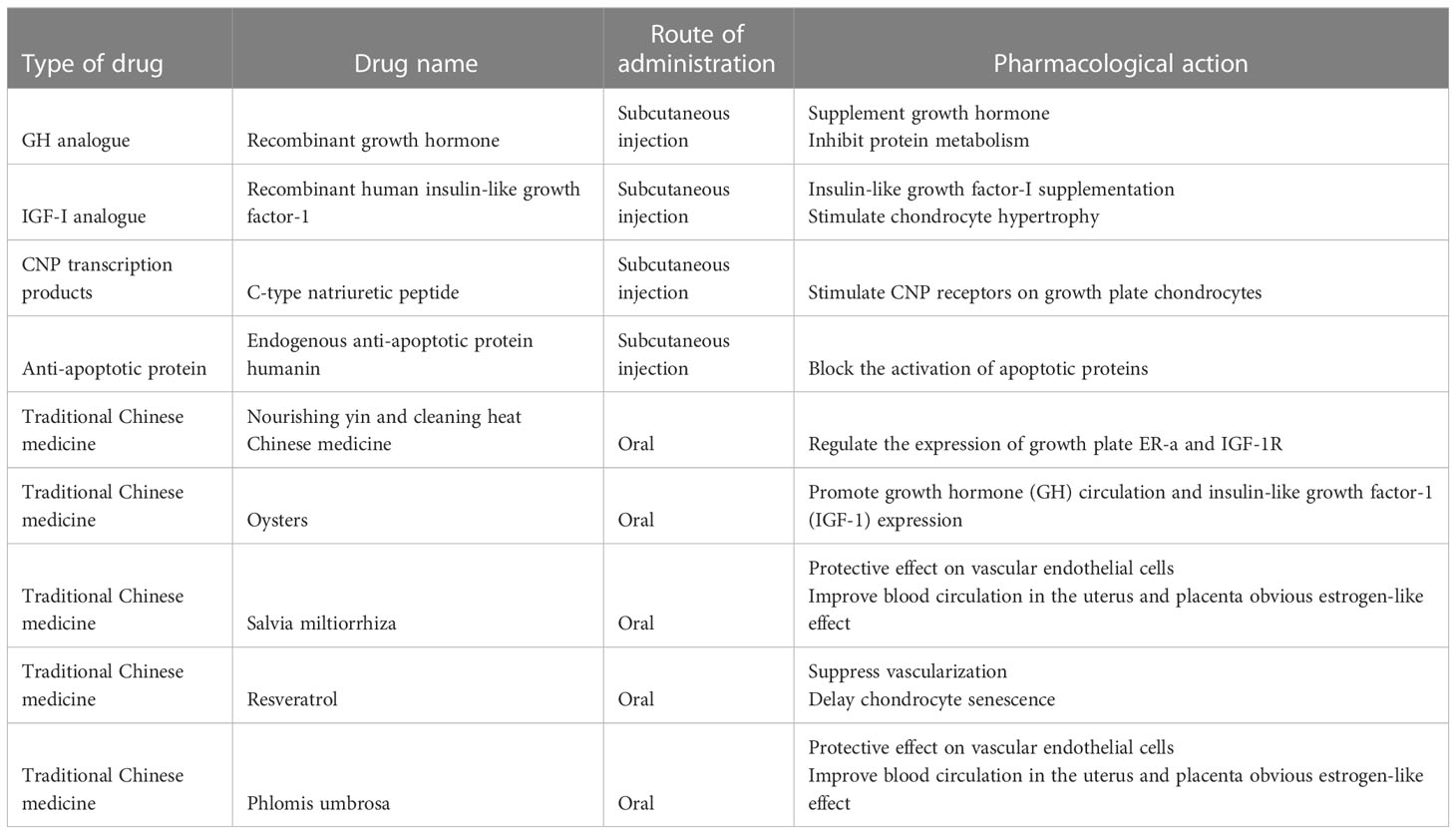
Table 1 Emerging drugs under investigation in clinical trials for bone growth inhibition.
In addition to inhibiting bone growth, long-term administration of pharmacological doses of GC can also cause muscle atrophy (92). Short-term administration of rhGH to normal volunteers has shown an inhibitory effect of prednisone on acute catabolism of protein (93, 94). Administration of rhGH significantly antagonizes the side effects of long-term GC administration, such as protein consumption, osteoporosis, and hyperlipidemia (95). rhGH can also improve growth of children receive GC treatment after liver transplantation (96). A 5-year prospective open study found that (97) children receiving GC treatment, after 36 months, rhGH group height standard deviation score change preliminary analysis significantly increased by (0.80 ± 1.03), but two patients experienced treatment-related adverse reactions: one case with poor compliance, the other case with mild hyperglycemia. Studies have shown that rhGH therapy is effective in increasing height in children with long-term GC treatment and is tolerable. However, many potential risks are still existing in rhGH treatment, including undetermined risks such as increased intracranial pressure, malignant tumors, femoral head spondylolisthesis, insulin resistance, and type 2 diabetes, which limit its large-scale use. More preclinical studies are still needed to verify the safety and efficacy of rhGH in GC-induced growth disorders (98).
Insulin-like growth factor-I (IGF-I) is naturally produced by many tissues, including liver and skeletal muscle. It is the main mediator of growth hormone (GH) for normal bone growth and is also important for muscle cell regeneration and survival (99, 100). In preclinical studies, rhIGF-1 is beneficial in animal models of muscle injury, wasting and aging (101, 102). Clinically, rhIGF-1 is approved for the treatment of growth disorders in children with severe primary IGF-I deficiency (103, 104). Studies have shown that DEX impairs longitudinal growth by inhibiting chondrocyte proliferation, while IGF-I can stimulate chondrocyte hypertrophy and reverse the inhibitory effect of DEX on growth (105). Claire L Wood et al. used growth hormone and insulin-like growth factor-1 to rescue the growth retardation of x-linked muscular dystrophy (mdx) mice treated with GC (106).
As mentioned above, CNP transcripts and their receptor NPR-B are expressed in chondrocytes of growth plates (56). Daily subcutaneous injection of CNP-53 can activate bone growth and reverse the growth inhibition caused by GC treatment in mice (107). At present, CNP-like drugs have entered phase II clinical trials in humans (108–110). Preliminary data show that recombinant CNP is safe for children and can improve the growth rate of children with chondrodysplasia. Up till now, there are no adverse reactions and allergic reactions related to bone growth. This data shed light on the clinical application of CNP in the treatment of GC-induced growth disorders.
HN is a polypeptide composed of 24 amino acids and was originally found to be a neuroprotective factor (111). HN has also been reported to exert anti-inflammatory (112) and anti-apoptotic effects by blocking the activation of pro-apoptotic proteins Bax (113) and Bak (114). HN treatment has shown promising results in preclinical models of diabetes (115), stroke(116), atherosclerosis(117) and Alzheimer’s disease(118). HN analogues (HNG) prevent bone growth retardation, chondrocyte apoptosis and proliferation inhibition by up-regulating the Hedgehog pathway without interfering with the anti-inflammatory effects of DEX(119).
According to traditional Chinese medicine theory, the kidney as the body’ s innate foundation, is also the root of the five organs of yin and yang, namely ‘yin and yang secret, spirit is the rule’. Yin and yang balance is fundamental to maintain normal physiological activities. GC is a kind of hormone secreted by adrenal cortex, which can be regarded as the nature of ‘pure yang’. GC under physiological dose has the meaning of ‘less fire generates qi’. In the case of long-term use of exogenous hormones beyond physiological dose, this kind of ‘pure yang’ medicine is easy to ‘overcome yang and consume yin’, which affects the introversion of yin essence and cannot play its nourishing role, resulting in kidney yin deficiency. When the human body are weak for a long time, it is easy to produce blood stasis, which hinders the generation and distribution of fresh blood, and reacts on deficiency, deficiency, and blood stasis, which accumulating in the body, disturbing the body’s qi, blood, yin and yang, and finally causes metabolic disorders. These in turn affect the nutrition and metabolism of the body organs(120, 121). Improving the body’s nutritional metabolism and blood stasis may have a certain preventive effect on the side effects of GC. Clinical use of traditional Chinese medicine can slow down the heat and yin caused by a large number of GC symptoms and signs, kidney strengthening medicine can promote growth and development and make children grow taller (122). Studies have shown that nourishing yin and purging fire Chinese medicine regulates bone development and maturation by regulating the expression of growth plate ER-α and IGF-1R (123). Shen Huansi et al. utilized Shengdi and tortoise shell, the traditional Chinese medicine for nourishing yin and clearing heat, to antagonize the growth retardation induced by dexamethasone in rabbits, possibly by regulating IGF-1 in the growth plate to reduce the inhibitory effect of DEX on the growth plate (124). Traditional Chinese medicine believes that oysters are beneficial to yin and yang, astringency and astringency to preserve kidney essence (125). According to the study, fermented oysters rich in γ-aminobutyric acid (GABA) promoted growth hormone (GH) circulation and insulin-like growth factor-1 (IGF-1) expression in young rats (126, 127), and increased the height and body length of the growth plate. Salvia miltiorrhiza is a commonly used traditional Chinese medicine for promoting blood circulation and removing blood stasis (128). Studies have found that Salvia miltiorrhiza has a protective effect on vascular endothelial cells in rats with blood stasis syndrome (129, 130). Clinical studies have found that Danshen can also treat pregnancy with fetal growth restriction (FGR), possibly by improving blood circulation in the uterus and placenta (131). At the same time, Danshen also has obvious estrogen-like effect, making it a potential alternative to estrogen in treating skeletal system disorders caused by estrogen reduction (132). Resveratrol is a natural antioxidant, which plays an important role in orthopedic diseases such as osteoarthritis (133), osteoporosis (134) and nerve injury repair (135). Resveratrol has been found to have the potential to improve longitudinal bone growth, which is associated with delayed growth plate fusion, resulting in increased final length (136). At the same time, resveratrol also has the effect of inhibiting the apoptosis of growth plate chondrocytes and delaying epiphyseal closure (137). Phlomis umbrosa has the functions of detumescence, muscle growth, tendon continuation and bone grafting. It was found that the mixture of Phlomis umbrosa, Astragalus membranaceus and Acanthopanax senticosus could increase the longitudinal bone growth rate of growing rats, improve the quality of bone trabeculae, and enhance the microstructure of bone trabeculae and cortical bone during growth (138). Donghun Lee et al.used the extract of Phlomis gracile to treat female adolescent rats and found that it increased the longitudinal bone growth rate and promoted the proliferation and differentiation of chondrocytes by up-regulating the expression of local IGF-1 and BMP-2 in the growth plate (139).
In summary, Chinese medicine has a comprehensive, multi-targeted and broad-spectrum effect in promoting bone growth. It can promote bone growth by affecting the GH-IGF axis, inhibiting the apoptosis of growth plate chondrocytes and delaying the closure of the growth plate. Although Chinese medicine has significant effects on promoting bone growth and development, the current studies are mostly animal experiments and lack of standardized clinical trials. In addition, compound formulas have variable effects and complex mechanisms, and the current research on single drug is mainly based on its extracts. Future studies should focus on the elaboration of multi-target Chinese medicine treatment from molecular biology and cell biology, combined with multicenter, large sample, double-blind, randomized controlled clinical studies.
This systematic review provides a comprehensive summary of recent research progress in the mechanism of GC-induced growth suppression in children. Long-term application of GCs have negative effects on the systemic endocrine system and local long bone growth plate, leading to growth suppression. Currently, there are no effective drugs to combat the negative effects of GC, and only rational application of GC and nutritional supplements are available for prevention. The complexity of its mechanism of affecting growth disorders requires further explorations and efforts to dissect its exact molecular mechanism. In conclusion, the most effective prevention and treatment method of GC-induced growth disorders remains rational GC use. Off-label use of medications, such as rhGH, may be prescribed to help address the bone growth retardation, but additional clinical trials are needed to verify their safety and efficacy.
JYH and SL carried out the literature review and drafted the manuscript. JYH, JPH, and SL revised the manuscript. SL, GL, and LC finalized and approved the manuscript for submission and provided funding support. All authors contributed to the article and approved the submitted version.
This work was funded by National Natural Science Foundation of China (82172430, 81874000, 82272505) and Discipline Construction Project of Guangdong Medical University, China (4SG21002G). This work was also partially supported by grants from Heath Medical Research Fund (HMRF) Hong Kong (16170951, 17180831, 08190416, 09203436), University Grants Committee, Research Grants Council of the Hong Kong (14121721, 14202920, 14108720, N_CUHK472/22, T13-402/17-N and AoE/M-402/20). This study also received support from the research funds from Health@InnoHK program launched by Innovation Technology Commission of the Hong Kong SAR, PR China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hardy RS, Zhou H, Seibel MJ, Cooper MS. Glucocorticoids and bone: Consequences of endogenous and exogenous excess and replacement therapy. Endocr Rev (2018) 39(5):519–48. doi: 10.1210/er.2018-00097
2. Chotiyarnwong P, McCloskey EV. Pathogenesis of glucocorticoid-induced osteoporosis and options for treatment. Nat Rev Endocrinol (2020) 16(8):437–47. doi: 10.1038/s41574-020-0341-0
3. Ceccato F, Scaroni C. Central adrenal insufficiency: open issues regarding diagnosis and glucocorticoid treatment. Clin Chem Lab Med (2019) 57(8):1125–35. doi: 10.1515/cclm-2018-0824
4. Vandewalle J, Luypaert A, De Bosscher K, Libert C. Therapeutic mechanisms of glucocorticoids. Trends Endocrinol Metab (2018) 29(1):42–54. doi: 10.1016/j.tem.2017.10.010
5. Ronchetti S, Ayroldi E, Ricci E, Gentili M, Migliorati G, Riccardi C. A glance at the use of glucocorticoids in rare inflammatory and autoimmune diseases: Still an indispensable pharmacological tool? Front Immunol (2020) 11:613435. doi: 10.3389/fimmu.2020.613435
6. Delara M, Chauhan BF, Le ML, Abou-Setta AM, Zarychanski R, tJong GW. Efficacy and safety of pulmonary application of corticosteroids in preterm infants with respiratory distress syndrome: a systematic review and meta-analysis. Arch Dis Child Fetal Neonatal Ed (2019) 104(2):F137–f144. doi: 10.1136/archdischild-2017-314046
7. Kao KT, Joseph S, Capaldi N, Brown S, Di Marco M, Dunne J, et al. Skeletal disproportion in glucocorticoid-treated boys with duchenne muscular dystrophy. Eur J Pediatr (2019) 178(5):633–40. doi: 10.1007/s00431-019-03336-5
8. Quattrocelli M, Zelikovich AS, Salamone IM, Fischer JA, McNally EM. Mechanisms and clinical applications of glucocorticoid steroids in muscular dystrophy. J Neuromuscul Dis (2021) 8(1):39–52. doi: 10.3233/jnd-200556
9. Crabtree NJ, Adams JE, Padidela R, Shaw NJ, Högler W, Roper H, et al. Growth, bone health & ambulatory status of boys with DMD treated with daily vs. intermittent oral glucocorticoid regimen. Bone (2018) 116:181–6. doi: 10.1016/j.bone.2018.07.019
10. Minnetti M, Caiulo S, Ferrigno R, Baldini-Ferroli B, Bottaro G, Gianfrilli D, et al. Abnormal linear growth in paediatric adrenal diseases: Pathogenesis, prevalence and management. Clin Endocrinol (Oxf) (2020) 92(2):98–108. doi: 10.1111/cen.14131
11. Mazziotti G, Giustina A. Glucocorticoids and the regulation of growth hormone secretion. Nat Rev Endocrinol (2013) 9(5):265–76. doi: 10.1038/nrendo.2013.5
12. Wolfgram PM, Allen DB. Factors influencing growth effects of inhaled corticosteroids in children. J Allergy Clin Immunol (2015) 136(6):1711–2.e1712. doi: 10.1016/j.jaci.2015.09.039
13. Prickett TC, Lynn AM, Barrell GK, Darlow BA, Cameron VA, Espiner EA, et al. Amino-terminal proCNP: a putative marker of cartilage activity in postnatal growth. Pediatr Res (2005) 58(2):334–40. doi: 10.1203/01.Pdr.0000169964.66260.4b
14. Kinoshita Y, Masuoka K, Miyakoshi S, Taniguchi S, Takeuchi Y. Vitamin d insufficiency underlies unexpected hypocalcemia following high dose glucocorticoid therapy. Bone (2008) 42(1):226–8. doi: 10.1016/j.bone.2007.09.042
15. Shangguan Y, Wu Z, Xie X, Zhou S, He H, Xiao H, et al. Low-activity programming of the PDGFRβ/FAK pathway mediates h-type vessel dysplasia and high susceptibility to osteoporosis in female offspring rats after prenatal dexamethasone exposure. Biochem Pharmacol (2021) 185:114414. doi: 10.1016/j.bcp.2021.114414
16. Cheng X, Chen JL, Ma ZL, Zhang ZL, Lv S, Mai DM, et al. Biphasic influence of dexamethasone exposure on embryonic vertebrate skeleton development. Toxicol Appl Pharmacol (2014) 281(1):19–29. doi: 10.1016/j.taap.2014.09.014
17. Naito M, Ohashi A, Takahashi T. Dexamethasone inhibits chondrocyte differentiation by suppression of wnt/β-catenin signaling in the chondrogenic cell line ATDC5. Histochem Cell Biol (2015) 144(3):261–72. doi: 10.1007/s00418-015-1334-2
18. Wen Y, Shi H, Wu Z, Xiao H, Wang H, Chen L. GR/Sp3/HDAC1/UGDH signaling participated in the maternal dexamethasone-induced dysplasia of the rat fetal growth plate. FASEB J (2020) 34(9):12834–46. doi: 10.1096/fj.202000106R
19. Chrysis D, Ritzen EM, Sävendahl L. Growth retardation induced by dexamethasone is associated with increased apoptosis of the growth plate chondrocytes. J Endocrinol (2003) 176(3):331–7. doi: 10.1677/joe.0.1760331
20. Campbell GS, Christian LJ, Carter-Su C. Evidence for involvement of the growth hormone receptor-associated tyrosine kinase in actions of growth hormone. J Biol Chem (1993) 268(10):7427–34.
21. Racine HL, Serrat MA. The actions of IGF-1 in the growth plate and its role in postnatal bone elongation. Curr Osteoporos Rep (2020) 18(3):210–27. doi: 10.1007/s11914-020-00570-x
22. Wehrenberg WB, Janowski BA, Piering AW, Culler F, Jones KL. Glucocorticoids: potent inhibitors and stimulators of growth hormone secretion. Endocrinology (1990) 126(6):3200–3. doi: 10.1210/endo-126-6-3200
23. Mehls O, Himmele R, Hömme M, Kiepe D, Klaus G. The interaction of glucocorticoids with the growth hormone-insulin-like growth factor axis and its effects on growth plate chondrocytes and bone cells. J Pediatr Endocrinol Metab (2001) 14 Suppl 6:1475–82.
24. Qiu J, Fan X, Ding H, Zhao M, Xu T, Lei J, et al. Antenatal dexamethasone retarded fetal long bones growth and development by down-regulating of insulin-like growth factor 1 signaling in fetal rats. Hum Exp Toxicol (2022) 41:9603271211072870. doi: 10.1177/09603271211072870
25. Desjardin C, Charles C, Benoist-Lasselin C, Riviere J, Gilles M, Chassande O, et al. Chondrocytes play a major role in the stimulation of bone growth by thyroid hormone. Endocrinology (2014) 155(8):3123–35. doi: 10.1210/en.2014-1109
26. Ghaemi N, Bagheri S, Elmi S, Mohammadzade Rezaee S, Elmi S, Erfani Sayyar R. Delayed diagnosis of hypothyroidism in children: Report of 3 cases. Iran Red Crescent Med J (2015) 17(11):e20306. doi: 10.5812/ircmj.20306
27. Williams GR, Bassett JHD. Thyroid diseases and bone health. J Endocrinol Invest (2018) 41(1):99–109. doi: 10.1007/s40618-017-0753-4
28. Stevens DA, Hasserjian RP, Robson H, Siebler T, Shalet SM, Williams GR. Thyroid hormones regulate hypertrophic chondrocyte differentiation and expression of parathyroid hormone-related peptide and its receptor during endochondral bone formation. J Bone Miner Res (2000) 15(12):2431–42. doi: 10.1359/jbmr.2000.15.12.2431
29. Wong GW, Lai J, Cheng PS. Growth in childhood thyrotoxicosis. Eur J Pediatr (1999) 158(10):776–9. doi: 10.1007/s004310051202
30. Hui H. Clinical study on the correlation between cushing's syndrome and thyroid function. master. Nanning: Guangxi Medical University (2013).
31. Weinstein RS, Jilka RL, Almeida M, Roberson PK, Manolagas SC. Intermittent parathyroid hormone administration counteracts the adverse effects of glucocorticoids on osteoblast and osteocyte viability, bone formation, and strength in mice. Endocrinology (2010) 151(6):2641–9. doi: 10.1210/en.2009-1488
32. van Buul-Offers SC, Smink JJ, Gresnigt R, Hamers N, Koedam J, Karperien M. Thyroid hormone, but not parathyroid hormone, partially restores glucocorticoid-induced growth retardation. Pediatr Nephrol (2005) 20(3):335–41. doi: 10.1007/s00467-004-1690-y
33. Birzniece V, Ho KKY. MECHANISMS IN ENDOCRINOLOGY: Paracrine and endocrine control of the growth hormone axis by estrogen. Eur J Endocrinol (2021) 184(6):R269–r278. doi: 10.1530/eje-21-0155
34. Shi S, Zheng S, Li XF, Liu ZD. The effect of estradiol on the growth plate chondrocytes of limb and spine from postnatal mice in vitro: The role of estrogen-receptor and estradiol concentration. Int J Biol Sci (2017) 13(1):100–9. doi: 10.7150/ijbs.17696
35. Chou YS, Chuang SC, Chen CH, Ho ML, Chang JK. G-Protein-Coupled estrogen receptor-1 positively regulates the growth plate chondrocyte proliferation in female pubertal mice. Front Cell Dev Biol (2021) 9:710664. doi: 10.3389/fcell.2021.710664
36. Ren H, Shen G, Jiang X, Liang D, Tang J, Cui J, et al. Changes of bone mass, bone turnover index and estrogen level in rats after glucocorticoid intervention and their correlation. Chin J Repair Reconstruction Surg (2015) 29(03):307–14.
37. Bradford PG, Gerace KV, Roland RL, Chrzan BG. Estrogen regulation of apoptosis in osteoblasts. Physiol Behav (2010) 99(2):181–5. doi: 10.1016/j.physbeh.2009.04.025
38. Gavali S, Gupta MK, Daswani B, Wani MR, Sirdeshmukh R, Khatkhatay MI. Estrogen enhances human osteoblast survival and function via promotion of autophagy. Biochim Biophys Acta Mol Cell Res (2019) 1866(9):1498–507. doi: 10.1016/j.bbamcr.2019.06.014
39. Kim HN, Ponte F, Nookaew I, Ucer Ozgurel S, Marques-Carvalho A, Iyer S, et al. Estrogens decrease osteoclast number by attenuating mitochondria oxidative phosphorylation and ATP production in early osteoclast precursors. Sci Rep (2020) 10(1):11933. doi: 10.1038/s41598-020-68890-7
40. Liu S, Quarles LD. How fibroblast growth factor 23 works. J Am Soc Nephrol (2007) 18(6):1637–47. doi: 10.1681/asn.2007010068
41. Rupp T, Butscheidt S, Vettorazzi E, Oheim R, Barvencik F, Amling M, et al. High FGF23 levels are associated with impaired trabecular bone microarchitecture in patients with osteoporosis. Osteoporos Int (2019) 30(8):1655–62. doi: 10.1007/s00198-019-04996-7
42. Bilha SC, Bilha A, Ungureanu MC, Matei A, Florescu A, Preda C, et al. FGF23 beyond the kidney: A new bone mass regulator in the general population. Horm Metab Res (2020) 52(5):298–304. doi: 10.1055/a-1151-2342
43. Takashi Y, Fukumoto S. Phosphate-sensing and regulatory mechanism of FGF23 production. J Endocrinol Invest (2020) 43(7):877–83. doi: 10.1007/s40618-020-01205-9
44. Küng CJ, Haykir B, Schnitzbauer U, Egli-Spichtig D, Hernando N, Wagner CA. Fibroblast growth factor 23 leads to endolysosomal routing of the renal phosphate cotransporters NaPi-IIa and NaPi-IIc in vivo. Am J Physiol Renal Physiol (2021) 321(6):F785–f798. doi: 10.1152/ajprenal.00250.2021
45. Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet (1996) 12(4):390–7. doi: 10.1038/ng0496-390
46. Murali SK, Roschger P, Zeitz U, Klaushofer K, Andrukhova O, Erben RG. FGF23 regulates bone mineralization in a 1,25(OH)2 D3 and klotho-independent manner. J Bone Miner Res (2016) 31(1):129–42. doi: 10.1002/jbmr.2606
47. Wen X, Li X, Tang Y, Tang J, Zhou S, Xie Y, et al. Chondrocyte FGFR3 regulates bone mass by inhibiting osteogenesis. J Biol Chem (2016) 291(48):24912–21. doi: 10.1074/jbc.M116.730093
48. Delucchi Á., Toro L, Alzamora R, Barrientos V, González M, Andaur R, et al. Glucocorticoids decrease longitudinal bone growth in pediatric kidney transplant recipients by stimulating the FGF23/FGFR3 signaling pathway. J Bone Miner Res (2019) 34(10):1851–61. doi: 10.1002/jbmr.3761
49. Feger M, Ewendt F, Strotmann J, Schäffler H, Kempe-Teufel D, Glosse P, et al. Glucocorticoids dexamethasone and prednisolone suppress fibroblast growth factor 23 (FGF23). J Mol Med (Berl) (2021) 99(5):699–711. doi: 10.1007/s00109-021-02036-8
50. Deng A, Zhang H, Hu M, Liu S, Wang Y, Gao Q, et al. The inhibitory roles of ihh downregulation on chondrocyte growth and differentiation. Exp Ther Med (2018) 15(1):789–94. doi: 10.3892/etm.2017.5458
51. Long F, Chung UI, Ohba S, McMahon J, Kronenberg HM, McMahon AP. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development (2004) 131(6):1309–18. doi: 10.1242/dev.01006
52. Peng G, Sun H, Jiang H, Wang Q, Gan L, Tian Y, et al. Exogenous growth hormone functionally alleviates glucocorticoid-induced longitudinal bone growth retardation in male rats by activating the Ihh/PTHrP signaling pathway. Mol Cell Endocrinol (2022) 545:111571. doi: 10.1016/j.mce.2022.111571
53. Nakao K, Osawa K, Yasoda A, Yamanaka S, Fujii T, Kondo E, et al. The local CNP/GC-b system in growth plate is responsible for physiological endochondral bone growth. Sci Rep (2015) 5:10554. doi: 10.1038/srep10554
54. Vasques GA, Arnhold IJ, Jorge AA. Role of the natriuretic peptide system in normal growth and growth disorders. Horm Res Paediatr (2014) 82(4):222–9. doi: 10.1159/000365049
55. Miyazaki Y, Ichimura A, Kitayama R, Okamoto N, Yasue T, Liu F, et al. C-type natriuretic peptide facilitates autonomic Ca(2+) entry in growth plate chondrocytes for stimulating bone growth. Elife (2022) 11:e71931. doi: 10.7554/eLife.71931
56. Ueda Y, Yasoda A, Yamashita Y, Kanai Y, Hirota K, Yamauchi I, et al. C-type natriuretic peptide restores impaired skeletal growth in a murine model of glucocorticoid-induced growth retardation. Bone (2016) 92:157–67. doi: 10.1016/j.bone.2016.08.026
57. Hu K, Olsen BR. The roles of vascular endothelial growth factor in bone repair and regeneration. Bone (2016) 91:30–8. doi: 10.1016/j.bone.2016.06.013
58. Stempel J, Fritsch H, Pfaller K, Blumer MJ. Development of articular cartilage and the metaphyseal growth plate: the localization of TRAP cells, VEGF, and endostatin. J Anat (2011) 218(6):608–18. doi: 10.1111/j.1469-7580.2011.01377.x
59. Smink JJ, Buchholz IM, Hamers N, van Tilburg CM, Christis C, Sakkers RJ, et al. Short-term glucocorticoid treatment of piglets causes changes in growth plate morphology and angiogenesis. Osteoarthritis Cartilage (2003) 11(12):864–71. doi: 10.1016/s1063-4584(03)00187-0
60. Liu X, Chai Y, Liu G, Su W, Guo Q, Lv X, et al. Osteoclasts protect bone blood vessels against senescence through the angiogenin/plexin-B2 axis. Nat Commun (2021) 12(1):1832. doi: 10.1038/s41467-021-22131-1
61. Yu H, Liu P, Zuo W, Sun X, Liu H, Lu F, et al. Decreased angiogenic and increased apoptotic activities of bone microvascular endothelial cells in patients with glucocorticoid-induced osteonecrosis of the femoral head. BMC Musculoskelet Disord (2020) 21(1):277. doi: 10.1186/s12891-020-03225-1
62. Chai Y, Su J, Hong W, Zhu R, Cheng C, Wang L, et al. Antenatal corticosteroid therapy attenuates angiogenesis through inhibiting osteoclastogenesis in young mice. Front Cell Dev Biol (2020) 8:601188. doi: 10.3389/fcell.2020.601188
63. Romeo SG, Alawi KM, Rodrigues J, Singh A, Kusumbe AP, Ramasamy SK. Endothelial proteolytic activity and interaction with non-resorbing osteoclasts mediate bone elongation. Nat Cell Biol (2019) 21(4):430–41. doi: 10.1038/s41556-019-0304-7
64. Wang T, Yang L, Liang Z, Bai L, Pei H, Zhang T, et al. Pulsed electromagnetic fields attenuate glucocorticoid-induced bone loss by targeting senescent LepR(+) bone marrow mesenchymal stromal cells. Biomater Adv (2022) 133:112635. doi: 10.1016/j.msec.2021.112635
65. Melrose J, Shu C, Whitelock JM, Lord MS. The cartilage extracellular matrix as a transient developmental scaffold for growth plate maturation. Matrix Biol (2016) 52-54:363–83. doi: 10.1016/j.matbio.2016.01.008
66. Chery DR, Han B, Zhou Y, Wang C, Adams SM, Chandrasekaran P, et al. Decorin regulates cartilage pericellular matrix micromechanobiology. Matrix Biol (2021) 96:1–17. doi: 10.1016/j.matbio.2020.11.002
67. Wen Y, Li J, Wang L, Tie K, Magdalou J, Chen L, et al. UDP-Glucose dehydrogenase modulates proteoglycan synthesis in articular chondrocytes: its possible involvement and regulation in osteoarthritis. Arthritis Res Ther (2014) 16(6):484. doi: 10.1186/s13075-014-0484-2
68. Ağırdil Y. The growth plate: a physiologic overview. EFORT Open Rev (2020) 5(8):498–507. doi: 10.1302/2058-5241.5.190088
69. Mizuhashi K, Ono W, Matsushita Y, Sakagami N, Takahashi A, Saunders TL, et al. Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature (2018) 563(7730):254–8. doi: 10.1038/s41586-018-0662-5
70. Schrier L, Ferns SP, Barnes KM, Emons JA, Newman EI, Nilsson O, et al. Depletion of resting zone chondrocytes during growth plate senescence. J Endocrinol (2006) 189(1):27–36. doi: 10.1677/joe.1.06489
71. Naito M, Vongsa S, Tsukune N, Ohashi A, Takahashi T. Promyelocytic leukemia zinc finger mediates glucocorticoid-induced cell cycle arrest in the chondroprogenitor cell line ATDC5. Mol Cell Endocrinol (2015) 417:114–23. doi: 10.1016/j.mce.2015.09.026
72. Lui JC, Baron J. Effects of glucocorticoids on the growth plate. Endocr Dev (2011) 20:187–93. doi: 10.1159/000321244
73. Lui JC, Nilsson O, Baron J. Growth plate senescence and catch-up growth. Endocr Dev (2011) 21:23–9. doi: 10.1159/000328117
74. Klaus G, Jux C, Fernandez P, Rodriguez J, Himmele R, Mehls O. Suppression of growth plate chondrocyte proliferation by corticosteroids. Pediatr Nephrol (2000) 14(7):612–5. doi: 10.1007/s004670000344
75. Ma Y, Peng T, Yao X, Sun C, Wang X. KLF2 reduces dexamethasone-induced injury to growth plate chondrocytes by inhibiting the Runx2-mediated PI3K/AKT and ERK signalling pathways. Autoimmunity (2023) 56(1):1–7. doi: 10.1080/08916934.2022.2141233
76. James CG, Ulici V, Tuckermann J, Underhill TM, Beier F. Expression profiling of dexamethasone-treated primary chondrocytes identifies targets of glucocorticoid signalling in endochondral bone development. BMC Genomics (2007) 8:205. doi: 10.1186/1471-2164-8-205
77. Siebler T, Robson H, Shalet SM, Williams GR. Dexamethasone inhibits and thyroid hormone promotes differentiation of mouse chondrogenic ATDC5 cells. Bone (2002) 31(4):457–64. doi: 10.1016/s8756-3282(02)00855-4
78. Chrysis D, Zaman F, Chagin AS, Takigawa M, Sävendahl L. Dexamethasone induces apoptosis in proliferative chondrocytes through activation of caspases and suppression of the akt-phosphatidylinositol 3'-kinase signaling pathway. Endocrinology (2005) 146(3):1391–7. doi: 10.1210/en.2004-1152
79. Zaman F, Chrysis D, Huntjens K, Fadeel B, Sävendahl L. Ablation of the pro-apoptotic protein bax protects mice from glucocorticoid-induced bone growth impairment. PloS One (2012) 7(3):e33168. doi: 10.1371/journal.pone.0033168
80. Simonyan L, Renault TT, Novais MJ, Sousa MJ, Côrte-Real M, Camougrand N, et al. Regulation of bax/mitochondria interaction by AKT. FEBS Lett (2016) 590(1):13–21. doi: 10.1002/1873-3468.12030
81. Zaman F, Chrysis D, Huntjens K, Chagin A, Takigawa M, Fadeel B, et al. Dexamethasone differentially regulates bcl-2 family proteins in human proliferative chondrocytes: role of pro-apoptotic bid. Toxicol Lett (2014) 224(2):196–200. doi: 10.1016/j.toxlet.2013.10.020
82. Reid IR. Bisphosphonates for prevention of bone loss in glucocorticoid-treated young people. EClinicalMedicine (2019) 12:8–9. doi: 10.1016/j.eclinm.2019.07.002
83. Ward LM, Ma J, Robinson ME, Scharke M, Ho J, Houghton K, et al. Osteoporotic fractures and vertebral body reshaping in children with glucocorticoid-treated rheumatic disorders. J Clin Endocrinol Metab (2021) 106(12):e5195–207. doi: 10.1210/clinem/dgab494
84. Chen M, Fu W, Xu H, Liu CJ. Pathogenic mechanisms of glucocorticoid-induced osteoporosis. Cytokine Growth Factor Rev (2023). doi: 10.1016/j.cytogfr.2023.03.002
85. Wang T, Liu X, He C. Glucocorticoid-induced autophagy and apoptosis in bone. Apoptosis (2020) 25(3-4):157–68. doi: 10.1007/s10495-020-01599-0
86. Chen K, Liu Y, He J, Pavlos N, Wang C, Kenny J, et al. Steroid-induced osteonecrosis of the femoral head reveals enhanced reactive oxygen species and hyperactive osteoclasts. Int J Biol Sci (2020) 16(11):1888–900. doi: 10.7150/ijbs.40917
87. Ngo D, Beaulieu E, Gu R, Leaney A, Santos L, Fan H, et al. Divergent effects of endogenous and exogenous glucocorticoid-induced leucine zipper in animal models of inflammation and arthritis. Arthritis Rheum (2013) 65(5):1203–12. doi: 10.1002/art.37858
88. Tanaka Y. Managing osteoporosis and joint damage in patients with rheumatoid arthritis: An overview. J Clin Med (2021) 10(6):1241. doi: 10.3390/jcm10061241
89. O'Connell EJ. Optimizing inhaled corticosteroid therapy in children with chronic asthma. Pediatr Pulmonol (2005) 39(1):74–83. doi: 10.1002/ppul.20126
90. Wolthers OD, Ramshanker N, Heuck C, Frystyk J. The timing of administration of exogenous glucocorticoid affects 24hour growth hormone secretion in children. Growth Horm IGF Res (2017) 35:40–4. doi: 10.1016/j.ghir.2017.06.004
91. Mingyue Y, Yuan X. New progress in glucocorticoid induced growth disorder. Chin J Endocrinol Metab (2021) 37(01):64–8.
92. Sato AY, Richardson D, Cregor M, Davis HM, Au ED, McAndrews K, et al. Glucocorticoids induce bone and muscle atrophy by tissue-specific mechanisms upstream of E3 ubiquitin ligases. Endocrinology (2017) 158(3):664–77. doi: 10.1210/en.2016-1779
93. Horber FF, Haymond MW. Human growth hormone prevents the protein catabolic side effects of prednisone in humans. J Clin Invest (1990) 86(1):265–72. doi: 10.1172/jci114694
94. Lee MK, Jeong HH, Kim MJ, Ryu H, Baek J, Lee B. Nutrients against glucocorticoid-induced muscle atrophy. Foods (2022) 11(5):687. doi: 10.3390/foods11050687
95. Giustina A, Bussi AR, Jacobello C, Wehrenberg WB. Effects of recombinant human growth hormone (GH) on bone and intermediary metabolism in patients receiving chronic glucocorticoid treatment with suppressed endogenous GH response to GH-releasing hormone. J Clin Endocrinol Metab (1995) 80(1):122–9. doi: 10.1210/jcem.80.1.7829600
96. Sarna S, Sipilä I, Rönnholm K, Koistinen R, Holmberg C. Recombinant human growth hormone improves growth in children receiving glucocorticoid treatment after liver transplantation. J Clin Endocrinol Metab (1996) 81(4):1476–82. doi: 10.1210/jcem.81.4.8636354
97. Basmaison O, Ranchin B, Zouater H, Robertson A, Gomez R, Koppiker N. Efficacy and safety of recombinant growth hormone treatment in children with growth retardation related to long-term glucocorticosteroid therapy. Ann Endocrinol (Paris) (2019) 80(4):202–10. doi: 10.1016/j.ando.2019.02.001
98. Cianfarani S. Safety of pediatric rhGH therapy: An overview and the need for long-term surveillance. Front Endocrinol (Lausanne) (2021) 12:811846. doi: 10.3389/fendo.2021.811846
99. Blum WF, Alherbish A, Alsagheir A, El Awwa A, Kaplan W, Koledova E, et al. The growth hormone-insulin-like growth factor-I axis in the diagnosis and treatment of growth disorders. Endocr Connect (2018) 7(6):R212–r222. doi: 10.1530/ec-18-0099
100. Yuan S, Larsson SC. Genetically predicted insulin-like growth factor-I in relation to muscle mass and strength. Clin Endocrinol (Oxf) (2021) 95(5):800–5. doi: 10.1111/cen.14561
101. Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, et al. Localized igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet (2001) 27(2):195–200. doi: 10.1038/84839
102. Liu HH, Wang JW, Chen X, Zhang RP, Yu HY, Jin HB, et al. In ovo administration of rhIGF-1 to duck eggs affects the expression of myogenic transcription factors and muscle mass during late embryo development. J Appl Physiol (1985) (2011) 111(6):1789–97. doi: 10.1152/japplphysiol.00551.2011
103. Backeljauw PF, Kuntze J, Frane J, Calikoglu AS, Chernausek SD. Adult and near-adult height in patients with severe insulin-like growth factor-I deficiency after long-term therapy with recombinant human insulin-like growth factor-I. Horm Res Paediatr (2013) 80(1):47–56. doi: 10.1159/000351958
104. Petriczko E, Jackowski T, Horodnicka-Józwa A, Wikiera B, Noczyńska A, Korpal-Szczyrska M, et al. Treatment of severe primary IGF-1 deficiency using rhIGF-1 preparation - first three years of polish experience. Endokrynol Pol (2019) 70(1):20–7. doi: 10.5603/EP.a2018.0074
105. Mushtaq T, Bijman P, Ahmed SF, Farquharson C. Insulin-like growth factor-I augments chondrocyte hypertrophy and reverses glucocorticoid-mediated growth retardation in fetal mice metatarsal cultures. Endocrinology (2004) 145(5):2478–86. doi: 10.1210/en.2003-1435
106. Wood CL, van 't Hof R, Dillon S, Straub V, Wong SC, Ahmed SF, et al. Combined growth hormone and insulin-like growth factor-1 rescues growth retardation in glucocorticoid-treated mdxmice but does not prevent osteopenia. J Endocrinol (2022) 253(2):63–74. doi: 10.1530/joe-21-0388
107. Ueda Y, Yasoda A, Hirota K, Yamauchi I, Yamashita T, Kanai Y, et al. Exogenous c-type natriuretic peptide therapy for impaired skeletal growth in a murine model of glucocorticoid treatment. Sci Rep (2019) 9(1):8547. doi: 10.1038/s41598-019-44975-w
108. Savarirayan R, Irving M, Bacino CA, Bostwick B, Charrow J, Cormier-Daire V, et al. C-type natriuretic peptide analogue therapy in children with achondroplasia. N Engl J Med (2019) 381(1):25–35. doi: 10.1056/NEJMoa1813446
109. Duggan S. Vosoritide: First approval. Drugs (2021) 81(17):2057–62. doi: 10.1007/s40265-021-01623-w
110. Savarirayan R, Tofts L, Irving M, Wilcox WR, Bacino CA, Hoover-Fong J, et al. Safe and persistent growth-promoting effects of vosoritide in children with achondroplasia: 2-year results from an open-label, phase 3 extension study. Genet Med (2021) 23(12):2443–7. doi: 10.1038/s41436-021-01287-7
111. Zuccato CF, Asad AS, Nicola Candia AJ, Gottardo MF, Moreno Ayala MA, Theas MS, et al. Mitochondrial-derived peptide humanin as therapeutic target in cancer and degenerative diseases. Expert Opin Ther Targets (2019) 23(2):117–26. doi: 10.1080/14728222.2019.1559300
112. Zhang P, Cui Z, Li S. The protective effects of S14G-humanin (HNG) against lipopolysaccharide (LPS)- induced inflammatory response in human dental pulp cells (hDPCs) mediated by the TLR4/MyD88/NF-κB pathway. Bioengineered (2021) 12(1):7552–62. doi: 10.1080/21655979.2021.1979914
113. Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, et al. Humanin peptide suppresses apoptosis by interfering with bax activation. Nature (2003) 423(6938):456–61. doi: 10.1038/nature01627
114. Zhai D, Luciano F, Zhu X, Guo B, Satterthwait AC, Reed JC. Humanin binds and nullifies bid activity by blocking its activation of bax and bak. J Biol Chem (2005) 280(16):15815–24. doi: 10.1074/jbc.M411902200
115. Boutari C, Pappas PD, Theodoridis TD, Vavilis D. Humanin and diabetes mellitus: A review of in vitro and in vivo studies. World J Diabetes (2022) 13(3):213–23. doi: 10.4239/wjd.v13.i3.213
116. Peng T, Wan W, Wang J, Liu Y, Fu Z, Ma X, et al. The neurovascular protective effect of S14G-humanin in a murine MCAO model and brain endothelial cells. IUBMB Life (2018) 70(7):691–9. doi: 10.1002/iub.1869
117. Gong Z, Goetzman E, Muzumdar RH. Cardio-protective role of humanin in myocardial ischemia-reperfusion. Biochim Biophys Acta Gen Subj (2022) 1866(2):130066. doi: 10.1016/j.bbagen.2021.130066
118. Zhang W, Zhang W, Li Z, Hao J, Zhang Z, Liu L, et al. S14G-humanin improves cognitive deficits and reduces amyloid pathology in the middle-aged APPswe/PS1dE9 mice. Pharmacol Biochem Behav (2012) 100(3):361–9. doi: 10.1016/j.pbb.2011.09.012
119. Zaman F, Zhao Y, Celvin B, Mehta HH, Wan J, Chrysis D, et al. Humanin is a novel regulator of hedgehog signaling and prevents glucocorticoid-induced bone growth impairment. FASEB J (2019) 33(4):4962–74. doi: 10.1096/fj.201801741R
120. Bin W. Research progress of traditional Chinese medicine on side effects of glucocorticoid. Shizhen Traditional Chin Med Traditional Chin Med (2010) 21(03):719–21.
121. Tianzhu Y, Ze H. Discussion on pathogenesis hypothesis of osteoporosis "Toxicity damaging bone collateral". Shizhen Traditional Chin Med Traditional Chin Med (2018) 29(06):1418–9.
122. Pi'an S. Key points of traditional Chinese medicine and Western medicine treatment for adverse reactions of western medicine toxicity. In: Modern Chinese medicine immunology. Beijing China: People's Medical Publishing House (2003).
123. Junping L, Depei C. Effect of Chinese herbal medicine for nourishing yin and purging fire on growth plate ER of female adolescent rats α、 regulation of IGF-1R and EGFR gene expression and protein synthesis. Chin J Integrated Traditional Western Med (2008) 08):721–4.
124. Huansi S, Chaowei C, Yumei L, Yongqiang C. Study on the mechanism of yin nourishing and heat clearing herbs antagonizing dexamethasone induced growth retardation in rabbits. Chin J Orthopedics Traumatology (2009) 17(01):11–3.
125. Miao Q. Understanding of oyster in traditional Chinese medicine. In: The elite of traditional Chinese medicine academic schools – the fourth exchange meeting of traditional Chinese medicine academic schools held by the Chinese society of traditional Chinese medicine). Harbin: Chinese Society of Traditional Chinese Medicine (2012). p. 348–50.
126. Lee H, Hwang-Bo H, Ji SY, Kim MY, Kim SY, Woo M, et al. Effect of fermented oyster extract on growth promotion in sprague-dawley rats. Integr Med Res (2020) 9(4):100412. doi: 10.1016/j.imr.2020.100412
127. Lee H, Hwangbo H, Ji SY, Kim MY, Kim SY, Kim DH, et al. Gamma aminobutyric acid-enriched fermented oyster (Crassostrea gigas) increases the length of the growth plate on the proximal tibia bone in sprague-dawley rats. Molecules (2020) 25(19):8547. doi: 10.3390/molecules25194375
128. Jing M, Qian C, Yaqian B, Zijun C, Yanjiang Q, Yanling Z. Study on the markers of salvia miltiorrhiza for promoting blood circulation and removing blood stasis based on systematic chinese pharmacy. Chin J Traditional Chin Med (2020) 45(14):3259–65. doi: 10.19540/j.cnki.cjcmm.20200210.404
129. Bi C, Li PL, Liao Y, Rao HY, Li PB, Yi J, et al. Pharmacodynamic effects of Dan-hong injection in rats with blood stasis syndrome. BioMed Pharmacother (2019) 118:109187. doi: 10.1016/j.biopha.2019.109187
130. Liu CD, Liu NN, Zhang S, Ma GD, Yang HG, Kong LL, et al. Salvianolic acid a prevented cerebrovascular endothelial injury caused by acute ischemic stroke through inhibiting the src signaling pathway. Acta Pharmacol Sin (2021) 42(3):370–81. doi: 10.1038/s41401-020-00568-2
131. Shen F, Lv H, Shi Y, Lau S, Guo F, Chen Q. A pilot study on investigating the role of salvia miltiorrhiza in fetal growth restriction. Biosci Rep (2020) 40(6):BSR20201222. doi: 10.1042/bsr20201222
132. Zhang JM, Li J, Liu EW, Wang H, Fan GW, Wang YF, et al. Danshen enhanced the estrogenic effects of Qing e formula in ovariectomized rats. BMC Complement Altern Med (2016) 16:181. doi: 10.1186/s12906-016-1146-5
133. El-Bidawy MH, Omar Hussain AB, Al-Ghamdi S, Aldossari KK, Haidara MA, Al-Ani B. Resveratrol ameliorates type 2 diabetes mellitus-induced alterations to the knee joint articular cartilage ultrastructure in rats. Ultrastruct Pathol (2021) 45(2):92–101. doi: 10.1080/01913123.2021.1882629
134. Elseweidy MM, El-Swefy SE, Shaheen MA, Baraka NM, Hammad SK. Effect of resveratrol and mesenchymal stem cell monotherapy and combined treatment in management of osteoporosis in ovariectomized rats: Role of SIRT1/FOXO3a and wnt/β-catenin pathways. Arch Biochem Biophys (2021) 703:108856. doi: 10.1016/j.abb.2021.108856
135. Recalde MD, Miguel CA, Noya-Riobó MV, González SL, Villar MJ, Coronel MF. Resveratrol exerts anti-oxidant and anti-inflammatory actions and prevents oxaliplatin-induced mechanical and thermal allodynia. Brain Res (2020) 1748:147079. doi: 10.1016/j.brainres.2020.147079
136. Yan X, Wuyin L, Weipeng X, Yuanyuan P, Na W. Screening of drugs for inhibiting apoptosis of growth plate chondrocytes and the effect of resveratrol on delayed epiphyseal closure in rats. J Chin Tissue Eng study (2022) 26(23):3644–9.
137. Chang G, Yan X, Dongdong L, Na Y, Li Z. Resveratrol inhibits apoptosis of rat growth plate chondrocytes and its effect on tibial growth %. J Shanghai J Traditional Chin Med (2021) 55(12):82–7. doi: 10.16305/j.1007-1334.2021.2106095
138. Song J, Lee SH, Lee D, Kim H. Astragalus extract mixture HT042 improves bone growth, mass, and microarchitecture in prepubertal female rats: A microcomputed tomographic study. Evid Based Complement Alternat Med (2017) 2017:5219418. doi: 10.1155/2017/5219418
Keywords: growth hormone, growth plate, bone growth suppression, glucocorticoid (GC), chondrocyte 3
Citation: Hua J, Huang J, Li G, Lin S and Cui L (2023) Glucocorticoid induced bone disorders in children: Research progress in treatment mechanisms. Front. Endocrinol. 14:1119427. doi: 10.3389/fendo.2023.1119427
Received: 09 January 2023; Accepted: 20 March 2023;
Published: 04 April 2023.
Edited by:
Jiake Xu, University of Western Australia, AustraliaReviewed by:
Qiang Guo, Third Xiangya Hospital, Central South University, ChinaCopyright © 2023 Hua, Huang, Li, Lin and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liao Cui, Y3VpbGlhb0AxNjMuY29t; Sien Lin, c2llbmxpbkBjdWhrLmVkdS5oaw==
†ORCID: Sien Lin, orcid.org/0000-0002-7292-7062
Liao Cui, orcid.org/0000-0002-1922-3066
Gang Li, orcid.org/0000-0002-3981-2239
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.