 Qin Tang1
Qin Tang1 Qinhui Liu
Qinhui Liu Yanping Li
Yanping Li Li Mo
Li Mo Jinhan He
Jinhan He
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 02 March 2023
Sec. Cellular Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1117414
This article is part of the Research Topic Endoplasmic Reticulum Homeostasis and Metabolic Diseases View all 7 articles
CRELD2, a member of the cysteine-rich epidermal growth factor-like domain (CRELD) protein family, is both an endoplasmic reticulum (ER)-resident protein and a secretory factor. The expression and secretion of CRELD2 are dramatically induced by ER stress. CRELD2 is ubiquitously expressed in multiple tissues at different levels, suggesting its crucial and diverse roles in different tissues. Recent studies suggest that CRELD2 is associated with cartilage/bone metabolism homeostasis and pathological conditions involving ER stress such as chronic liver diseases, cardiovascular diseases, kidney diseases, and cancer. Herein, we first summarize ER stress and then critically review recent advances in the knowledge of the characteristics and functions of CRELD2 in various human diseases. Furthermore, we highlight challenges and present future directions to elucidate the roles of CRELD2 in human health and disease.
The endoplasmic reticulum (ER) is a multifunctional, dynamic intracellular organelle that plays an essential role in multiple physiological processes, including lipid metabolism; calcium homeostasis; and the synthesis, folding, and post-translational modifications of transmembrane and secreted proteins (1–3). The ER function is highly orchestrated by essential regulatory factors. However, various physiological and pathological stimuli can disrupt ER protein-folding capacity, thereby accumulating unfolded or misfolded proteins and disturbing ER homeostasis, which is referred to as ER stress (1–3). In response to ER stress, the unfolded protein response (UPR), which is considered as an integrated adaptive mechanism, is activated to restore protein homeostasis. However, persistent UPR activation can lead to a maladaptive cellular response, which has been implicated in various diseases including diabetes, obesity, fatty liver disease, cardiovascular diseases (CVDs), cancer, and kidney diseases (1, 3–13). Comprehensive understanding about the key factors involved in ER stress is crucial for elucidating the pathogenesis of these diseases and identifying novel therapeutic targets. Previous research has identified multiple novel proteins that play key roles during ER stress. Cysteine-rich epidermal growth factor (EGF)-like domain (CRELD) protein family member, CRELD2, was also recently identified as a putative ER stress-responsive protein (14–16).
CRELD protein family is highly conserved (17). CRELD1, the funding member of this family, is known as the AVSD2 gene as mutations in CRELD1 are associated with cardiac atrioventricular septal defects (AVSD) (17–26). CRELD2, another member of this family, is a factor that regulates the intracellular trafficking of acetylcholine receptor α4 and β2 subunits (27). Both CRELD1 and CRELD2 are multi-domain proteins, comprising varying numbers of EGF-like and calcium-binding EGF-like domains and a unique and highly conserved tryptophan-aspartic acid (WE) domain. The major structural difference between these two proteins is the presence of transmembrane domains in CRELD1 (absent in CRELD2) (15, 17, 28). This leads to distinct subcellular localizations and functions between CRELD members. Studies have revealed that CRELD1 is localized in the cytoplasm, probably mainly in the ER (29). Functional studies have demonstrated that CRELD1 is essential for heart development and immune cell function in mouse models (29–32). In contrast, CRELD2 is predominantly localized in the ER and Golgi apparatus and is spontaneously secreted (15). To date, evidence of the physiological and pathophysiological functions of CRELD2 is lacking. However, animal and cell models suggest that CRELD2 is a novel ER stress-responsive protein that might be implicated in ER homeostasis and ER stress-related diseases, including chronic liver diseases, CVDs, kidney diseases, and cancer. Herein, we summarize the characteristics and functions of CRELD2 based on current evidence and present future directions important for broadening our understanding of CRELD2 in health and disease.
The ER is an important organelle in eukaryotic cells that plays essential roles in orchestrating the homeostasis of at least one-third of the proteins within a cell and ensuring cell survival (2, 33). ER function is highly regulated by critical regulatory factors such as ER-resident enzymes and protein oxidoreductase chaperones (3). The impairment of these processes by both physiological and pathological stimuli, such as nutrient deprivation, aging, and hypoxia, may result in perturbed ER homeostasis, thereby accumulating misfolded or unfolded proteins in the ER lumen, a condition commonly known as ER stress (1–3). Following ER stress, UPR is triggered to either restore ER homeostasis or evoke cell death in cases of prolonged or unresolved ER stress. In response to mild to moderate ER stress, UPR is initiated to remove unfolded/misfolded proteins and restore ER homeostasis, resulting in “adaptive/cytoprotective” UPR. However, after severe or persistent ER stress, the UPR is hyperactivated, leading to “maladaptive/unchecked/terminal” UPR (3, 7, 12).
In mammals, the UPR comprises three signaling pathways initiated by three ER transmembrane sensors: inositol-requiring enzyme 1 (IRE1α), protein kinase R (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (34–36). Under normal conditions, glucose-regulated protein 78 (GRP78), also known as heat shock protein family A member 5 or binding-immunoglobulin protein, which is a key ER-resident chaperone, binds to the luminal domains of all three UPR sensors to keep them inactive. However, during ER stress conditions, GRP78 preferentially binds to misfolded or unfolded proteins and is dissociated from these sensors, enabling their activation (3, 37) (Figure 1).
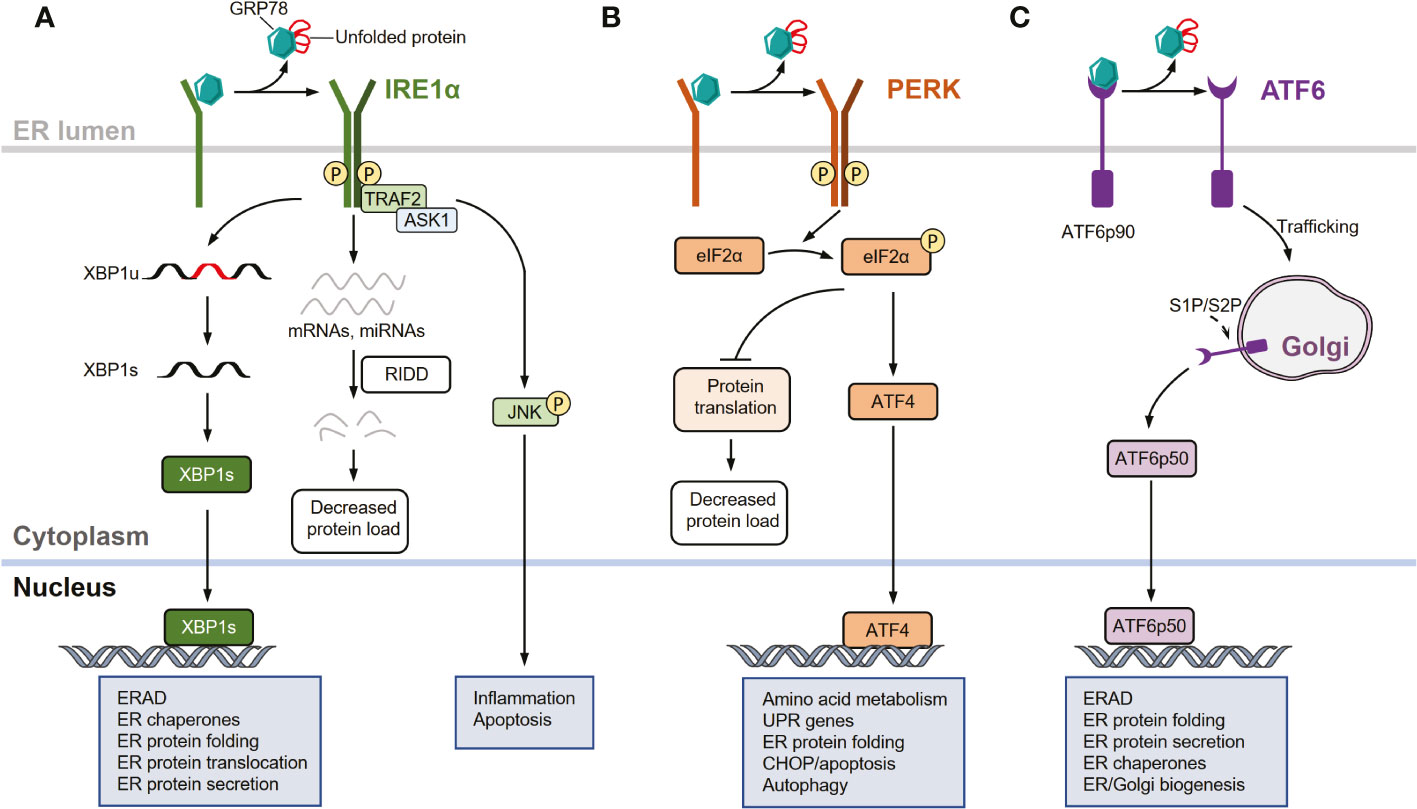
Figure 1 UPR signaling pathways. (A) Inositol-requiring enzyme 1 (IRE1α) signaling arm of the unfolded protein response (UPR). In response to endoplasmic reticulum (ER) stress, IRE1α is activated through dissociation from glucose- regulated protein 78 (GRP78), oligomerization, autophosphorylation, and subsequent allosteric activation of the cytosolic endonuclease domain. Activated IRE1α facilitates the unconventional splicing of XBP1 (XBP1u) mRNA, resulting in active transcription factor XBP1s, which drives the expression of genes involved in restoring ER homeostasis. IRE1α also degrades select mRNAs and miRNAs through regulated IRE1-dependent decay (RIDD) to reduce protein load in the ER during intensive ER stress. In response to severe or unresolved ER stress, activated IRE1α recruits and binds to tumor necrosis factor receptor-associated factor 2 (TRAF2) and apoptosis signal-regulating kinase 1 (ASK1) to promote c-Jun N-terminal kinase (JNK) signaling, leading to the activation of apoptosis. (B) Protein kinase R (PKR)-like ER kinase (PERK) arm of the UPR. In response to ER stress, PERK is activated by dissociating from GRP78 and auto-phosphorylating after dimerization. Activated PERK selectively phosphorylates eukaryotic translation initiation factor 2 subunit-α (eIF2α), thereby attenuating protein translation. The mRNA of activating transcription factor 4 (ATF4) is preferentially translated following eIF2α phosphorylation, allowing it to upregulate genes involved in restoring ER homeostasis, amino acid metabolism, apoptosis, and autophagy. (C) Activating transcription factor 6 (ATF6) arm of the UPR. In response to ER stress, GRP78 dissociates from the luminal domain of full-length of ATF6 (ATF6p90), allowing ATF6 monomers to traffic to the cis-Golgi apparatus where they are proteolytically processed and cleaved by site-1 protease (S1P) and site-2 protease (S2P). This releases an active ATF6 transcription factor fragment (ATF6p50). The transcription factor localizes to the nucleus, inducing several genes involved in restoring ER homeostasis and ER/Golgi biogenesis.
Among the UPR sensors, IRE1α is the most evolutionarily conserved arm and contains an ER stress-sensing luminal domain structure and a cytoplasmic effector domain with both RNA endonuclease (RNase) and kinase activities (37–41). Upon ER stress, IRE1α dissociates from GRP78 and is activated through several processes, including oligomerization, autophosphorylation, and subsequent allosteric activation of the cytosolic endonuclease domain (37–41). Activated IRE1α excises an inhibitory intron (an internal 26-nucleotide fragment) from cytosolic XBP1 mRNA through its endonuclease activity, generating a frameshift when translating the spliced XBP1 (XBP1s) isoform transcript (42–44). XBP1s encodes a functionally active basic leucine zipper (bZIP) transcription factor XBP1s, which regulates the transcription of multiple genes that are implicated in diverse biological pathways such as those that function in ER-associated protein degradation (ERAD) and folding proteins (37–39, 43). Additionally, the endonuclease activity of IRE1α can degrade a subset of mRNAs and miRNAs through regulated IRE1-dependent decay (RIDD), which reduces the abundance of proteins entering the ER during intensive ER stress (45–49). Moreover, activated IRE1α recruits and binds to tumor necrosis factor receptor-associated factor 2 (TRAF2) to promote c-Jun N-terminal kinase (JNK) signaling in response to severe or unresolved ER stimuli, thereby activating apoptotic programs (50, 51) (Figure 1A).
Similar to IRE1α, PERK is a type I transmembrane protein with a luminal ER stress-sensing domain and cytosolic Ser/Thr kinase domain (2, 33). Upon ER stress, PERK is activated through dissociation from GRP78 and subsequently dimerizes and auto-phosphorylates (37, 38). Activated PERK primarily acts as a kinase that phosphorylates the α subunit of eukaryotic initiation factor 2 (eIF2α), thereby attenuating protein translation and inducing activating transcription factor 4 (ATF4) (35, 37, 38, 52). Notably, PERK-mediated translational attenuation and transcriptional signaling are adaptive processes in response to acute ER stress, whereas chronic and prolonged PERK activation promotes apoptotic signaling through multiple mechanisms, including the induction of pro-apoptotic factors downstream of C/EBP homologous protein (CHOP) (33, 53) (Figure 1B).
Unlike IRE1α and PERK, ATF6 is a type II transmembrane protein with a cytosolic bZIP transcription factor dimerization domain that functions as a transcription factor rather than a kinase or endoribonuclease (1–3, 33). In response to acute ER stress, GRP78 dissociates from the luminal domain of the full-length ATF6 (ATF6p90). The increased population of ATF6 monomers traffics to the cis-Golgi apparatus, where they are proteolytically processed and cleaved by site-1 protease (S1P) and site-2 protease (S2P), releasing an active ATF6 transcription factor fragment (ATFp50) (54–56). This transcription factor localizes to the nucleus, inducing the expression of several genes involved in protein folding, lipid biogenesis, and ERAD (1, 33, 57–59) (Figure 1C).
Therefore, in response to ER stress, IRE1α, PERK, and ATF6 signaling pathways are collectively implicated in the coordination of the UPR though the following effects (1). First, these pathways attenuate translation and actively degrade RNA, reducing biosynthetic demands on the ER. Moreover, the protein refolding capacity can be enhanced to resolve misfolded/unfolded protein accumulation within the ER, and these pathways promote ubiquitination and proteasomal degradation via ERAD. Additionally, these pathways can mediate the delivery of misfolded/unfolded proteins from the ER to lysosomes for degradation via autophagy or to the cytosol for proteasomal processing. When these processes are dysregulated and not capable of restoring ER homeostasis, prolonged UPR activation and unresolved ER stress involve in cell apoptosis and death, which are associated with the pathogenesis of various diseases (1, 3–13, 60, 61).
Fluorescence in situ hybridization showed that CRELD2 maps to human chromosome 22q13 (28). Multiple splice variants are expressed by CRELD2, which encode for many transcripts that likely produce multiple protein isoforms (α, β, γ, δ, ϵ, ζ) (28). Sequence analysis revealed that each isoform has several overlapping motifs; however, they differ from each other in that they contain either tandem arrays of EGF- and/or calcium- binding EGF domains, or EGF/calcium-binding EGF domains and furin cysteine-rich domains (28). In contrast, they all have a WE-rich domain unique to the CRELD protein family, which is conserved across species (17, 28). CRELD2 is also highly conserved in orthologs and is identified in many vertebrates (28). There is also a murine Creld2 ortholog with 69% identity to human CRELD2 (17). Notably, the mouse Creld2 gene presumably corresponds to the human CRELD2α gene (14).
As the members of the CRELD family, CRELD1 and CRELD2 exhibit 51% homology in the nucleic acid sequences of the coding regions. At the amino acid level, they were 38% identical and 51% similar (17). Notably, CRELD1 and CRELD2 generally share similar domain structures, with a highly conserved WE region that has not been found in any non-homologous members, which is considered the hallmark feature of the CRELD protein family, followed by variations in EGF domains (28). In contrast to CRELD1, which has two type III transmembrane domains at the carboxyl(C)-terminus that anchors the protein to the cell surface, CRELD2 does not have any predicted transmembrane domains (15, 17, 28). The C-terminus of CRELD2 contains four (R/H)EDL amino acids, which are well conserved across species, including REDL for human, mouse, and bovine, and HEDL for rat, chicken, Xenopus, and zebrafish (15) (Figure 2A). These amino acids are reportedly similar to the C-terminal amino acids in several ER-resident proteins, such as KDEL for GRP78 (62, 63) and RTDL for mesencephalic astrocyte-derived neurotrophic factor (MANF) (64–67) (Figure 2B). In fact, many ER-resident proteins have a C-terminal KDEL (Lys-Asp-Glu-Leu) motif, which is the canonical sequence for retention of the ER chaperone including GRP78, while RTDL and (R/H)EDL are variants of KDEL sequence with similar functions (15, 66, 68). Studies have demonstrated that CRELD2 is primarily localized to the ER and Golgi apparatus and can be secreted into the extracellular space upon cellular stimuli, particularly ER stress (14, 15). The four amino acids domain in the C-terminus of CRELD2 is considered essential for the retention and secretion of CRELD2, which will be our focus in subsequent sections. Notably, studies have revealed that the amino(N)-terminus of CRELD2 is critical for its translocation into the ER-Golgi apparatus, whereas the deletion of its C-terminal region has no effect on its accumulation in the perinuclear region (15). Additionally, CRELD2 reportedly has numerous CXXC motifs (Figure 2C), and the N-terminal CXXC motif possesses protein isomerase activity (16). This pair of active protein disulfide isomerase (PDI)-like cysteine residues, CXXC, can shuttle between the disulfide and dithiol forms, thereby catalyzing thiol-disulfide oxidation, reduction, and isomerization, which are critical for correct disulfide bond formation and/or arrangement if incorrect bonds are formed (16, 69, 70). Therefore, its PDI-like activity further supports the idea that CRELD2 might participate in the quality control of proteins in the ER.
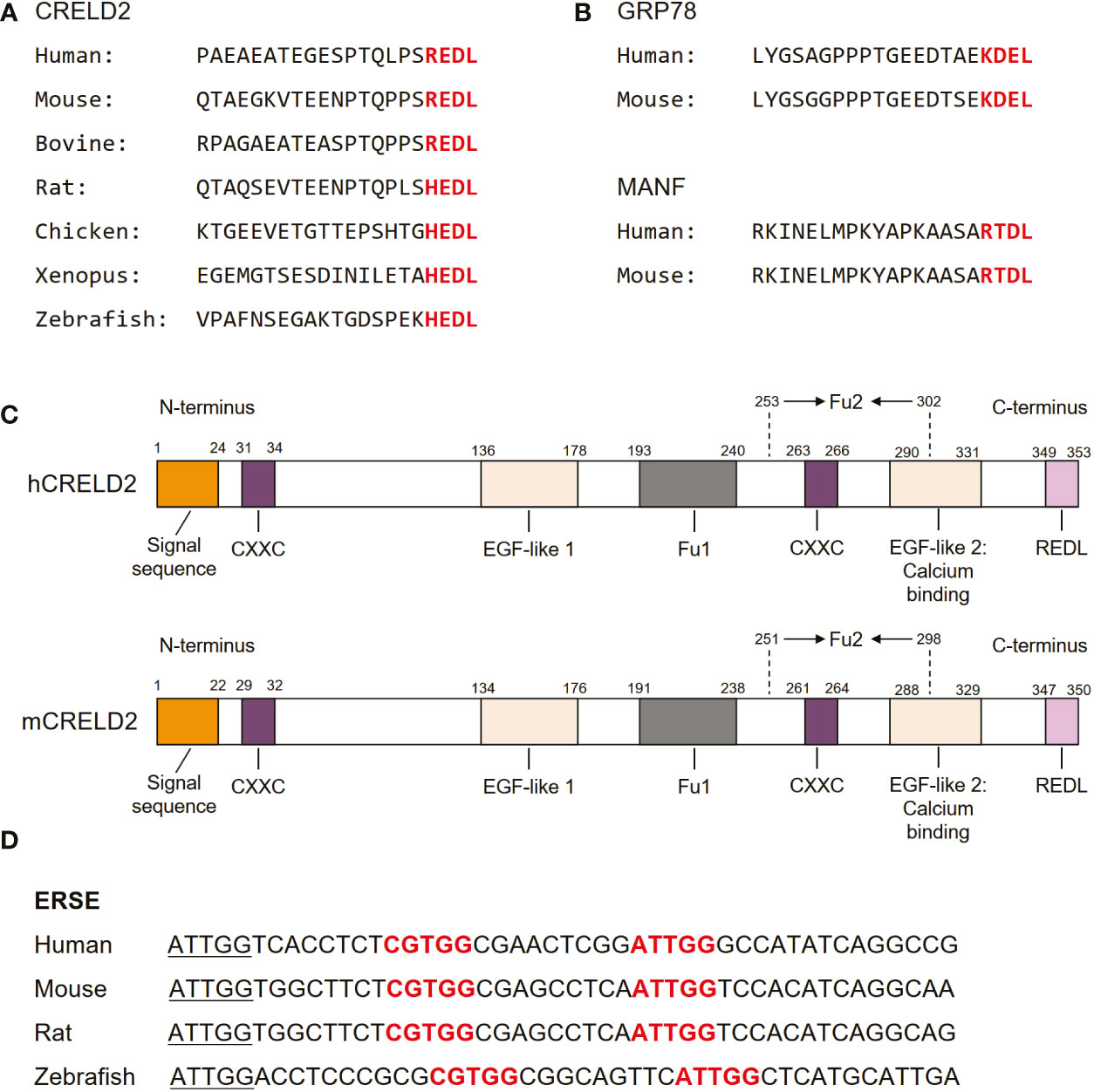
Figure 2 Molecular structure of CRELD2. (A) Comparison of amino acid sequences of CRELD2 at the C-terminus among various species. The conserved amino acids are shown in red font. (B) Four conserved C-terminal amino acids in GRP78 and mesencephalic astrocyte-derived neurotrophic factor (MANF) among species including human and mouse. The conserved amino acids are shown in red font. (C) Schematic of human and mouse CRELD2 protein. (D) Comparison of ERSE nucleotide sequences among various species. Conserved ERSE is shown in red font.
The analysis of the upstream sequences of CRELD2 revealed a functional promoter region embedded within a large CpG island (28). In 2009, Oh-hashi et al. performed a genomic sequence analysis of the promoter region of the mouse Creld2 gene and identified a putative ER stress responsible element (ERSE; CGTGG-N9-ATTGG), which is highly conserved among different species (14) (Figure 2D). This conservation further indicates that CRELD2 may play a role in regulating cellular behavior during ER stress. They first identified that CRELD2 is predominantly localized in the ER and Golgi apparatus and is a novel ER stress-inducible gene (14). Specifically, in Neuro2a cells, Creld2 mRNA was induced by treatment with the ER-inducing chemical agents, namely thapsigargin (Tg, a well-known inhibitor of Ca2+-ATPase), tunicamycin (Tm, an inhibitor of protein glycosylation), and brefeldin A (BFA, an inhibitor of ER-Golgi transport) (14). Furthermore, luciferase reporter analyses revealed that ATF6 overexpression drastically induced Creld2 mRNA expression by directly binding to the ERSE of the Creld2 gene. Mutations in the mouse Creld2 promoter ERSE considerably decreased both basal activity and responsiveness toward ER stress stimuli (14).
Additionally, further studies confirmed that CRELD2 is responsive to ER stress stimuli. For example, neonatal rat cardiomyocytes treated with Tm significantly upregulated Creld2, as well as several other genes involved in ER stress. This upregulation was significantly attenuated by salubrinal and its derivatives, which are the inhibitors of ER stress (71). Additionally, mouse podocytes treated with either Tm or Tg for 24 h remarkably induced CRELD2 expression (72), indicating that the upregulation of CRELD2 induced by ER stress stimuli is a general and not cell-type-specific response. Moreover, in vivo studies also found that CRELD2 protein expression in the mouse liver was considerably induced 24 h after intraperitoneal injection of Tm (73). These results suggest that CRELD2 may be involved in the processing of proteins and may be a novel mediator in regulating the onset and progression of various ER stress-associated diseases.
CRELD2 is both an ER-resident protein and a secretory factor (14, 15). However, the mechanisms that regulate the intracellular transportation and secretion of CRELD2 are poorly understood. In 2011, Oh-hashi et al. found that the N-terminus of CRELD2 is necessary for its translocation into the ER-Golgi apparatus (15). Additionally, the disruption of the Golgi apparatus by BFA almost completely prevents CRELD2 secretion (15, 74), suggesting that CRELD2 might be secreted via the ER-Golgi apparatus. Moreover, the overexpression of dominant negative mutant Sar1, a component of cytoplasmic vesicle coat protein complex II (COPII), drastically suppressed the spontaneous secretion of CRELD2. Sar1 plays an essential role in early step in vesicle budding and regulating vesicular transport from the ER to the Golgi (75, 76), and this further supports that COPII-mediated transport from the ER to the Golgi apparatus is the main pathway for CRELD2 trafficking (15) (Figure 3A). Despite this evidence, further studies are warranted to comprehensively understand the mechanism that regulates cellular transport of CRELD2.
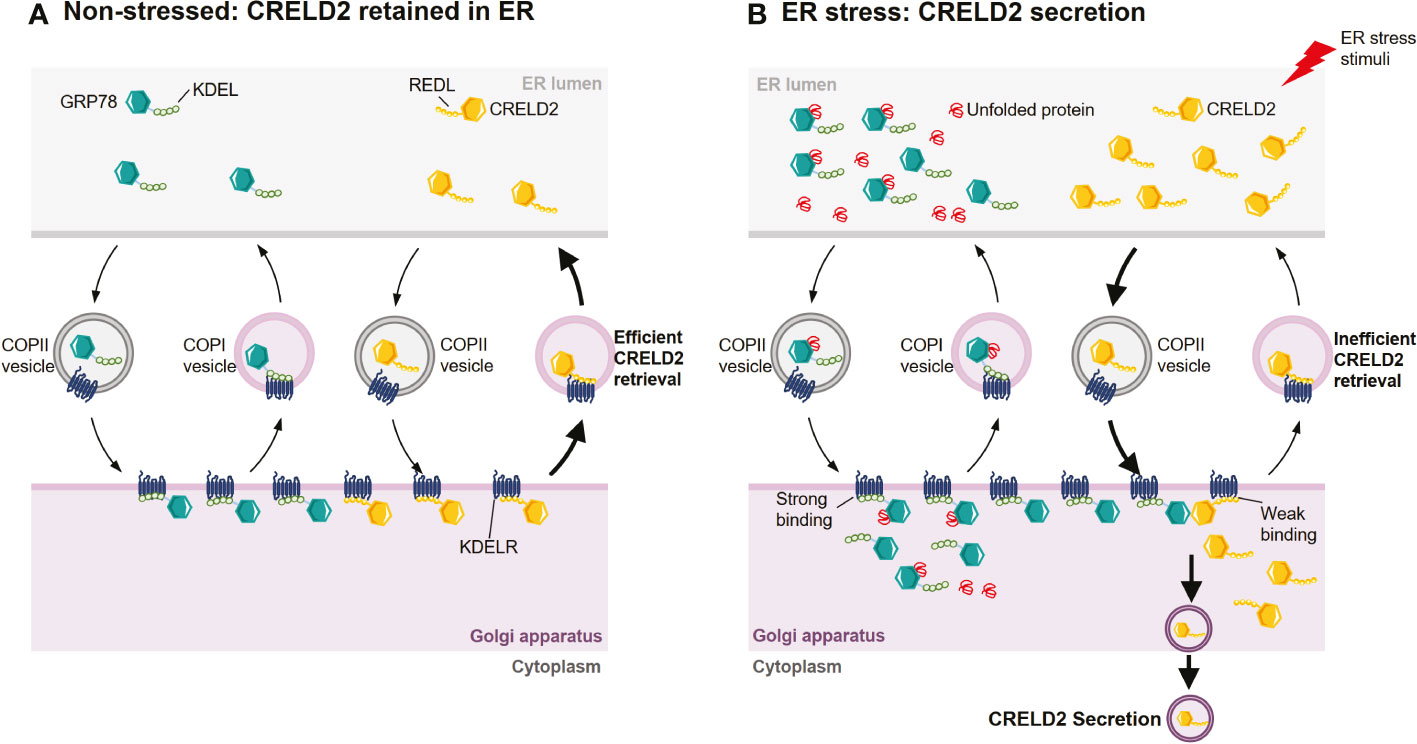
Figure 3 Hypothetical mechanism for the ER stress-induced CRELD2 secretion. Although ER-resident chaperones including GRP78 appear to be located solely in the ER lumen, they are widely distributed throughout the cell owing to a dynamic equilibrium achieved through retention and bidirectional transport between organelles. (A) Under normal conditions, GRP78 and CRELD2 can be trafficked to Golgi apparatus from ER via coat protein complexes II (COPII) vesicles. In the Golgi apparatus, the KDEL sequence of GRP78 and REDL sequence of CRELD2 can be recognized by the KDEL receptor (KDELR), which is located in the cis-Golgi. The binding of GRP78 or CRELD2 to KDELR triggers the incorporation of receptor-protein complex into vesicles such as COPI, thereby efficiently transporting them back to the ER. (B) Upon ER stress, the levels of several ER-inducible proteins, including GRP78 and CRELD2, are upregulated; however, the relative expression of KDELR is not increased. Thus, GRP78 containing perfect ER retention motifs with a high affinity for KDELR will compete for KDELR with CRELD2 containing a low affinity motif. Therefore, CRELD2 that escapes to the Golgi apparatus cannot be efficiently retrieved back to the ER, allowing CRELD2 to be secreted.
The conditions under which CRELD2 secretion is induced are still not comprehensively elucidated. However, both in vitro and in vivo studies have demonstrated that CRELD2 secretion is drastically induced upon ER stress. For example, cells subjected to ER stress by chemical agents such as Tm, Tg, and BFA, or by culturing cells in serum-free medium showed increased CRELD2 secretion (15, 73). In ER stress-related disease models, such as matrilin-3 (Matn3) and collagen type X alpha 1 chain (Col10a1) mutant growth plates, CRELD2 was also detectable at significant levels in the extracellular matrix (ECM) but not in wild-type controls (16). Additionally, CRELD2 is rapidly secreted when renal cells are subjected to ER stress in mouse models of podocyte ER stress-induced nephrotic syndrome (NS) and Tm- or I/R-induced acute kidney injury (AKI) (72). The observation that CRELD2 is an ER-resident protein and can be secreted under induced ER stress suggests that it might act as a multifunctional factor both in the ER-Golgi apparatus as well as in the extracellular space under normal and pathophysiological conditions.
Studies have revealed that the four-amino acid sequence (R/H)EDL in the C-terminal region of CRELD2 regulates its secretion. For example, in HEK293 cells, the modification of the C-terminal region, such as the deletion of four C-terminal amino acids from mouse CRELD2 or addition of tag-peptides to its C-terminus, drastically enhanced CRELD2 secretion (15). Additionally, the overexpression of wild-type GRP78, but not mutant GRP78 lacking the C-terminal KDEL sequence, remarkably enhanced CRELD2 secretion, whereas mutant CRELD2 at the C-terminus did not show any upregulation (15). Therefore, CRELD2 is a novel secretory factor regulated by GRP78, whereas the comprehensive molecular mechanism remains unresolved. However, the KDEL receptor (KDELR) competition hypothesis was also proposed (15, 16) (Figures 3A, B). The four highly conserved amino acids in CRELD2 are similar to the C-terminal amino acids in GRP78, which also possesses the well-known ER-resident signal sequence KDEL (62, 63). The secretion of many ER-resident proteins is restricted via this ER retention motif, facilitating retrieval transport from the Golgi apparatus via binding to KDELRs, which are located in the intermediate compartment or in the cis-Golgi apparatus, with high affinity (68, 77–84). Similarly, the putative ER retention motif REDL at the C-terminus enables CRELD2 to be retained in the ER (15). Under normal conditions, both REDL and KDEL trafficked to Golgi apparatus can bind with KDELRs, thereby efficiently transporting them back to the ER. However, during ER stress, the relative levels of multiple ER stress-inducible proteins such as GRP78 and CRELD2 are upregulated, whereas the relative expression of KDELRs are not increased (16, 85). Thus, GRP78, containing perfect ER retention motifs that have high affinity for the KDELRs, would compete with CRELD2, containing a motif that has a low binding affinity, thereby allowing CRELD2 to escape the ER and ultimately be secreted (15, 16) (Figures 3A, B).
Further studies have revealed that, additionally to GRP78, MANF can enhance CRELD2 secretion (86). Similar to CRELD2, MANF has recently been identified as an ER stress-responsive protein (65–67, 85, 87–92). MANF possesses ER-resident motifs at its proximal C-terminus, RTDL, which regulates MANF secretion upon ER stress (64–67). Studies have revealed that in both HEK293 and COS7 cell lines, co-transfection with CRELD2 and MANF remarkably increased the secretion of CRELD2 into the extracellular space (86). This effect highly depends on both of the four C-terminal amino acid sequences (RTDL in MANF and REDL in CRELD2) because the deletion of these four C-terminal amino acids terminated the increased secretion of CRELD2 induced by the co-expression of MANF (86). Notably, the data showed that the increase in CRELD2 secretion by MANF overexpression might not be simply due to the expression of intrinsic ER-resident chaperones or a result of competition between the two proteins for KDELRs (86). Further studies have revealed that the overexpression of mouse cerebral dopamine neurotrophic factor (CDNF), a paralogous protein of MANF that also contains the KDEL-like motif of the ER retrieval signal at its C-terminus, hardly affected the co-transfected CRELD2 secretion (93). Moreover, the inhibition of vacuolar ATPase, a proton pump that regulates the pH of several intracellular compartments, remarkably induced CRELD2 secretion without relying on its C-terminal structure (74). These results provide valuable insights into the mechanism of CRELD2 secretion regulation, which warrants further investigation.
Several studies have confirmed the ubiquitous expression of CRELD2. In 2005, through northern blotting, Ortiz et al. first revealed that the high expression levels of CRELD2 were observed in the skeletal muscle, heart, liver, kidney, and placenta of human tissues (28). In 2006, Maslen et al. conducted intensive studies in different human tissues by combining both northern blot and RNA dot blot (28). They demonstrated that CRELD2 was ubiquitously expressed during development and in mature tissues, although a broad range of signal intensities were observed. Additionally, the most prominent signals of CRELD2 in adult tissues occur in pancreas, stomach, duodenum, salivary gland, thyroid gland, appendix, and trachea, which generally overlap with the expression pattern for CRELD1 (17, 28). However, in fetal tissues, CRELD2 is highly expressed in the lung, liver, thymus, spleen, and heart (28).
Additional to its expression pattern in humans, the ubiquitous expression of CRELD2 has been confirmed in rodents (73, 94). In 2018, Oh-hashi et al. evaluated the mRNA and protein expression patterns of CRELD2 in different adult mouse tissues (73). They found that the Creld2 mRNA was present in almost all tissues tested, including the brain, heart, lung, liver, spleen, kidney, stomach, and small intestine, but not in skeletal muscles. However, CRELD2 protein expression in the heart and skeletal muscle was negligible, and that in the brain was relatively low (73). To understand the discrepancy between CRELD2 mRNA and protein expression, they further examined the stability of CRELD2 protein in Neuro2a cells through treatment with the protein synthesis inhibitor cycloheximide and/or the proteasome inhibitor MG132 together with the ER stress agent Tg. However, the protein levels of CRELD2 were hardly affected by each treatment (73). Until now, of the reason for this discrepancy remains unclear, which may be important for us to further understand the characteristics and functions of CRELD2.
As previously discussed, CRELD2 is a novel ER stress-responsive protein, and its expression and secretion can be drastically enhanced by ER stress. In this section, we provide a comprehensive overview of the recent advances in the knowledge about CRELD2 in various disease models (Table 1).
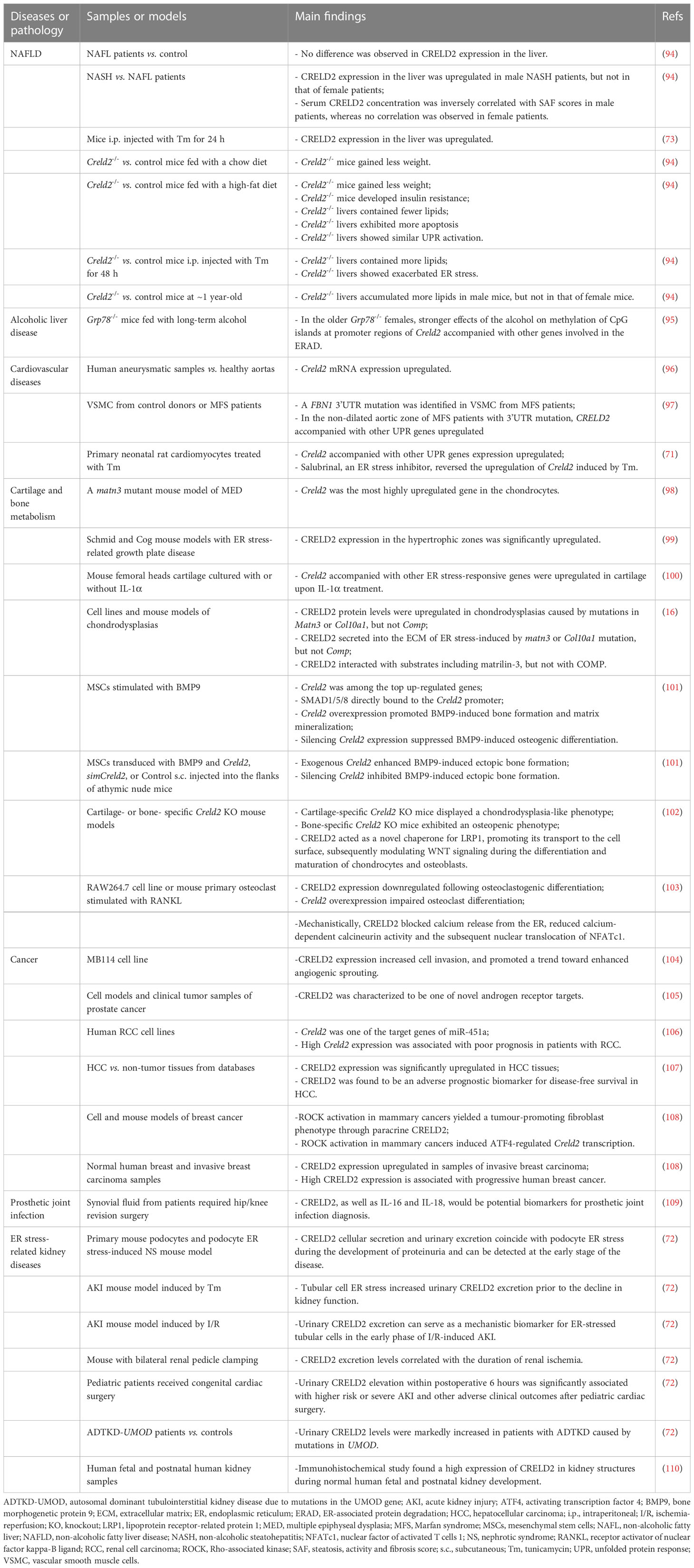
Table 1 Summary of current studies associating CRELD2 with diseases.
ER stress reportedly contributes to the onset and progression of multiple liver diseases including non-alcoholic fatty liver disease (NAFLD) and alcoholic liver diseases (3, 111–116). Although CRELD2 is involved in liver metabolism homeostasis, its exact role and molecular mechanisms remain largely undefined. The involvement of CRELD2 in liver metabolism was first reported by Nohara et al. Their results showed that gestational arsenic exposure significantly increased Creld2 expression in the liver of adult male offspring mice, accompanied by an increase in intracellular triglyceride accumulation in the liver (117). Subsequent studies further found that intraperitoneal administration of Tm for 24 h drastically induced CRELD2 protein expression in mouse livers (73). Moreover, in older Grp78-/- mice, increased effects of alcohol on the methylation of CpG islands in the promoter regions of genes involved in ERAD, including Creld2, were detected (95). However, a direct causal link between CRELD2 and liver disease has not been demonstrated in these studies.
Through intensive studies on mouse models and human samples, Kern et al. recently revealed that CRELD2 plays a critical role in liver homeostasis (94). They generated a Creld2 knockout (Creld2-/-) mouse model to investigate CRELD2 function in vivo (94). Their results showed that Creld2-/- mice exhibited a reduction in body weight under both a chow diet and high-fat diet (HFD) compared to their controls but developed insulin resistance. Additionally, Creld2-/- livers generally contained fewer lipids, which could account for lower body weight. Notably, their results clearly showed that 12 weeks of HFD did not induce ER stress in the liver, and neither transcriptome nor protein expression analysis revealed a lipid-driven ER stress response in the livers of Creld2-/- mice. Moreover, this study revealed that the adipose weights of Creld2-/- mice were significantly reduced (94). Another study also identified several genes, including Creld2, that were associated with feed efficiency using RNA sequencing in rumen papillae from steers (118). All these results suggest that CRELD2 plays a role in regulating whole-body energy balance, which remains elusive and warrants further investigation. To test whether CRELD2 is involved in the resolution of ER stress in the liver, Creld2-/- mice and littermate controls were intraperitoneally injected with Tm or vehicle. Inducing ER stress via Tm administration exacerbated ER stress in Creld2-/- mice, as indicated by upregulated GRP78 expression, and consequently increased hepatic lipid storage. Mechanistic studies have revealed that CRELD2 interacts with several chaperones and enzymes whose activity is required to overcome cellular stress, including GRP78, thioredoxin domain-containing 5, and glutathione S-transferase Mu2 (94, 119). These data further support the hypothesis that CRELD2 is involved in ER stress and the UPR, and CRELD2 function is required to maintain hepatic homeostasis under ER stress responses in mice.
Additionally, Kern et al. investigated whether human CRELD2 plays a role in the pathophysiological conditions of the liver. They found sex dimorphism in the expression levels of CRELD2 in the human liver, where both the mRNA and protein levels of CRELD2 were significantly upregulated in male patients with non-alcoholic steatohepatitis (NASH); however, expression in female patients remained unaffected (94). Notably, control patients showed expression similar to that of patients with simple steatosis. Furthermore, they found an inverse correlation between serum concentrations of CRELD2 and Steatosis, Activity, and Fibrosis (SAF) scores in male patients (94). These results indicate that CRELD2 exhibits a sex-specific function during NAFLD in humans, and upregulated CRELD2 expression in the liver is likely associated with a decrease in serum CRELD2 concentration, which is presumably associated with progression to NASH in male patients. Similarly, a sex dimorphism was also observed in aged mice, with only Creld2-/- males developing steatosis (94). Based on the results obtained from both mouse models and human samples, CRELD2 likely plays an essential role in liver metabolic homeostasis; however, further studies are warranted to validate this.
ER stress reportedly involves in the pathogenesis of CVDs, including hypertension, atherosclerosis, heart failure, and myocardial atrophy (6, 12). In primary neonatal rat cardiomyocytes, Tm treatment significantly induced the mRNA expression of Creld2, accompanied by other UPR genes, and this upregulation was remarkably reversed by treatment with salubrinal, which is an ER stress inhibitor (71). Further studies revealed high expression of CRELD2 and other ER stress-related markers in human aneurysmal samples (96). This is accompanied by exacerbated apoptosis, high reactive oxygen species production, and a reduction in mitochondrial biogenesis in the vascular wall of abdominal aortic aneurysm, a degenerative vascular disease with a complex etiology (96). Marfan syndrome (MFS) is associated with mutations in the protein fibrillin-1 (FBN1), which affects the integrity of connective tissue elastic fibers. The most severe clinical outcome of MFS is cardiovascular system abnormalities characterized by progressive aortic root enlargement and ascending aortic aneurysm (97, 120, 121). Siegert et al. found a nearly undescribed FBN1 3’UTR mutation accompanied by a well-defined gene ontological ER stress response in the non-dilated aortic zone, which was confirmed by the increased expression of MANF, GRP78, and CRELD2 (97). These results indicate that CRELD2 is associated with CVD pathogenesis. However, comprehensively elucidating the roles and mechanisms of CRELD2 in CVDs using various animal and cell models is necessary.
ER stress is also implicated in the disturbance of cartilage and bone metabolism (60, 61, 122, 123). In vitro and in vivo studies demonstrate that CRELD2 plays an important role in these processes. The association between CRELD2 and cartilage metabolism was first reported by Rajpar et al. (98). They revealed that Creld2 was the most highly upregulated gene in chondrocytes from a Matn3 mutant model of multiple epiphyseal dysplasia (MED), which is a clinically variable and genetically heterogeneous chondrodysplasia characterized by mild short stature, joint pain, and stiffness and early onset osteoarthritis (98). Further studies have also revealed that CRELD2 expression in the hypertrophic zones of ER stress-related growth plate disease mouse models (Schmid and Cog) is significantly upregulated (99). Notably, MED, Schmid, and Cog are characterized by prolonged ER stress. These data also revealed that the protein levels of CRELD2 were upregulated in cell and mouse models of chondrodysplasia caused by mutations in Matn3 or Col10a1, but not in cartilage oligomeric matrix protein (Comp) (16). Notably, upon ER stress induced by matn3 mutation in vitro and in vivo, CRELD2 was also secreted into the ECM (16). Moreover, substrate-trapping experiments confirmed that PDI-like activity enabled CRELD2 to interact with several substrates, including MATN3, laminin 5 β3, type VI collagen, and thrombospondin 1 (16). Additionally, new elements of ER stress in IL-1α-treated cartilage reportedly involve Creld2 (100), further supporting that CRELD2 might be associated with cartilage homeostasis.
Additionally, CRELD2 participates in bone development and homeostasis. For example, Creld2 is among the top upregulated genes in bone morphogenetic protein 9 (BMP9)-stimulated osteogenic differentiation of mesenchymal stem cells (101). ChIP assays revealed that SMAD1/5/8 directly binds to the Creld2 promoter in a BMP9-dependent manner. Moreover, both in vitro and in vivo studies have revealed that exogenous Creld2 promotes BMP9-induced bone formation and matrix mineralization; conversely, silencing Creld2 expression suppresses BMP9-induced osteogenic differentiation (101). These results confirmed that CRELD2 plays an important role in BMP9-induced terminal osteogenic differentiation, although the specific mechanism still needs to be thoroughly investigated (101). Dennis et al. recently found that mice with cartilage-specific deletion of Creld2 had a chondrodysplasia-like phenotype with abnormal cartilage growth plates; however, the deletion of Creld2 in bone results in osteopenia, with a low bone density and altered trabecular architecture (102). Mechanistically, CRELD2, which possesses PDI-like activity, acts as a novel chaperone for LRP1, promoting its transport to the cell surface and subsequently modulating WNT signaling during the differentiation and maturation of chondrocytes and osteoblasts (102). They recently further revealed the critical role of CRELD2 in osteoclast differentiation (103). CRELD2 expression was significantly downregulated following osteoclastogenic differentiation in both the RAW264.7 cell line and mouse primary osteoclast precursors, and the overexpression of Creld2 impaired osteoclast differentiation in vitro. Mechanistic analysis revealed that CRELD2 expression blocks calcium release from the ER. This impairs osteoclast differentiation due to reduced calcium-dependent calcineurin activity and nuclear translocation of nuclear factor of activated T cells 1 (NFATc1), a master regulator of osteoclastogenesis (103). Interestingly, another study has shown that the first member of the CRELD family of proteins, CRELD1, promotes NFATc1 dephosphorylation and its subsequent nuclear translocation by directly binding to and modulating calcineurin activity (29, 31). The opposite effect on NFATc1 translocation mediated by CRELD1 and CRELD2 indicates that these two proteins fulfill distinct cellular functions, possibly due to their structural differences. As for CRELD2, conducting in vivo studies in animal models and clinical studies in humans is necessary, as these comprehensive studies could broaden our understanding of the critical role of CRELD2 in skeletal and other human diseases.
Increasing evidence has established that ER stress and UPR pathways play essential roles in tumor progression and survival (1, 6, 124). As a novel ER stress-responsive gene, Creld2 is highly correlated with cancer development, although the detailed underlying pathophysiology remains unclear. For example, in vitro studies revealed that CRELD2 expression significantly increased MB114 cell invasion and promoted a trend toward enhanced angiogenic sprouting, thereby suggesting that CRELD2 might be a mediator of tumor angiogenesis (104). Additionally, CRELD2 was reported as a novel androgen receptor target in prostate cancer (105). Yamada et al. identified 16 putative oncogenic targets of miR-451a regulation in renal cell carcinoma. Among these, CRELD2 was one of the target genes whose expression was significantly associated with poor prognosis in patients with renal cell carcinoma by TCGA database analyses (106). Additionally, CRELD2 gene and protein expressions are significantly upregulated in hepatocellular carcinoma (HCC) than in non-tumor tissues, and CRELD2 is associated with poor overall survival and disease-free survival in HCC (107). However, the specific role of CRELD2 in carcinogenesis remains unelucidated.
Sarah et al. recently established a causal link between CRELD2 and cancer (108). Their results showed that CRELD2 is a paracrine factor that underlines PERK-mediated cancer-associated fibroblast education downstream of Rho-associated kinase (ROCK) via an analysis of tumors from patients and mice (108). They found that Creld2 is regulated by PERK-modulated ATF4 via a putative C/ebp-Atf response element (CARE) in the promoter sequences of Creld2, and CRELD2 depletion suppresses tumor progression, indicating that the paracrine ROCK-PERK-ATF4-CRELD2 axis promotes breast cancer progression. They also revealed that high CRELD2 expression was associated with progressive human breast cancer (108). Therefore, these results establish that CRELD2 is a driver of tumor progression, and targeting CRELD2 may be a suitable anticancer therapy.
The observation that CRELD2 is secreted under increased ER stress suggests that it may be exploited as a soluble extracellular biomarker of ER stress-related diseases. Emerging evidence has characterized CRELD2 as a biomarker in human body fluids for molecular phenotyping of pathogenetic processes. For example, synovial fluid CRELD2 has been identified as a potential biomarker for prosthetic joint infection (109). Moreover, male NASH patients showed an inverse correlation between serum CRELD2 concentration and NASH progression, with low CRELD2 levels at high SAF scores (94).
Moreover, CRELD2 is characterized as an early, sensitive, non-invasive, and mechanistic biomarker in the urine in various ER stress-mediated kidney diseases, including hereditary NS, ischemic AKI, and autosomal dominant tubulointerstitial kidney disease (ADTKD), due to mutations in the UMOD gene encoding uromodulin (72, 125–127). In a podocyte ER stress-induced NS mouse model, CRELD2 cellular secretion and urinary excretion coincided with podocyte ER stress during the development of proteinuria and could be detected at an early stage of the disease. Moreover, tubular cell ER stress increases urinary CRELD2 excretion prior to subsequent decline in kidney function or histologic changes due to ER stress-induced AKI. Therefore, urinary CRELD2 excretion can serve as a mechanistic biomarker for ER-stressed tubular cells in the early phase of I/R-induced AKI (72). Additionally, these data have shown that early postoperative urinary CRELD2 elevation is significantly associated with severe AKI and other adverse outcomes following pediatric cardiac surgery. Urinary CRELD2 can be used to distinguish between controls and patients with early kidney disease (72). These data suggest that CRELD2 may be a promising ER stress biomarker with potential application in early diagnosis, risk stratification, treatment response monitoring, and guiding the development of ER stress modulators in targeted patient groups. Nevertheless, further clinical studies in a multi-institutional setting with a larger patient cohort are warranted. Notably, a recent immunohistochemical study on human fetal and postnatal human kidney samples revealed a high expression of CRELD2 in kidney structures during normal human fetal and postnatal kidney development (110). This implies that CRELD2 may play an essential role in kidney homeostasis, which also warrants further investigation.
Over the past decades, increasing evidence has revealed the characteristics and functions of CRELD2. Molecular studies have demonstrated that CRELD2 acts as both an ER-resident protein and a secretory factor. Various cellular stimuli, particularly ER stress, remarkably enhance CRELD2 expression and secretion, as demonstrated by multiple in vitro and in vivo studies. Expression pattern analysis revealed that CRELD2 is ubiquitously expressed in multiple tissues to different extents, suggesting that CRELD2 may play important but diverse roles in different organs, tissues, and in different cell types. Moreover, CRELD2 is associated with various diseases and has crucial roles in various physiological and pathological processes, including liver metabolism homeostasis, cartilage and bone metabolism, and cancer progression and survival, as well as potential role as a biomarker for kidney diseases. However, despite significant progress, many outstanding concerns regarding CRELD2 remain unaddressed.
First, multiple isoforms of human CRELD2 protein cause variations in their subcellular localization, expression patterns, and functions, which have not yet been comprehensively elucidated. In addition, although there is evidence regarding the mechanism through which ER stress induces CRELD2 expression, it remains unclear whether other mechanisms are also involved in this process. Additional to the upregulation of CRELD2 expression, ER stress can drastically enhance its secretion. However, information on the changes in circulating CRELD2 levels in ER stress-related diseases, including diabetes, obesity, inflammation, NAFLD, CVDs, kidney diseases, and cancer, is still lacking. The source of circulating CRELD2 in these pathological conditions also remains unknown. Moreover, MANF overexpression significantly increased CRELD2 secretion. However, the exact mechanism underlying this process remains unclear. Whether CRELD2 plays a role in the function of MANF and whether there exists any link between these two proteins also remain unknown and require further studies. Similar to MANF, CRELD2 may possess a dual-function model (intracellular and extracellular functions). However, knowledge about its functions and mechanisms, particularly that of extracellular CRELD2, is still lacking. Current evidence has revealed an association between CRELD2 and various human diseases, whereas the direct causal link between CRELD2 and the pathogenesis of diseases remains poorly understood. Further investigations may provide clues as to whether the addition of exogenous CRELD2 or blocking endogenous CRELD2 is therapeutically beneficial for human diseases. This should also be validated in mouse models using CRELD2 peptides or specific antibodies. Addressing these gaps in knowledge would be beneficial for uncovering the pathophysiological significance of CRELD2 and its potential clinical applications.
JH contributed to the guidance of this review and gave the final approval of the version to be published. QT drafted the manuscript. QL, YL, and LM prepared the figures. All authors read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (82025007, 81930020, 82270923); Research Funding from Sichuan Province (2021YJ0458); Post-Doctoral Research Project, West China Hospital of Sichuan University (2020HXBH095); Project funded by China Postdoctoral Science Foundation (2022M710099).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AKI, acute kidney injury; ATF4, activating transcription factor 4; ATF6, activating transcription factor 6; BFA, brefeldin A; CHOP, C/EBP homologous protein; COPII, cytoplasmic vesicle coat protein complex II; CRELD, cysteine-rich epidermal growth factor-like domain; CVDs, cardiovascular diseases; ECM, extracellular matrix; EGF, epidermal growth factor; eIF2α, α subunit of eukaryotic initiation factor 2; ER, endoplasmic reticulum; ERAD, ER-associated protein degradation; ERSE, ER stress responsible element; GRP78, glucose-regulated protein 78; HCC, hepatocellular carcinoma; HFD, high-fat diet; IRE1, inositol-requiring enzyme 1; JNK, c-Jun N-terminal kinase; KDELR, KDEL receptor; MANF, mesencephalic astrocyte-derived neurotrophic factor; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; NS, nephrotic syndrome; PDI, protein disulfide isomerase; PERK, protein kinase R (PKR)-like ER kinase; RIDD, regulated IRE1-dependent decay; ROCK, rho-associated kinase; S1P, site-1 protease; S2P, site-2 protease; SAF, steatosis, activity, and fibrosis; Tg, thapsigargin; Tm, tunicamycin; TRAF2, tumor necrosis factor receptor-associated factor 2; UPR, unfolded protein response.
1. Salvagno C, Mandula JK, Rodriguez PC, Cubillos-Ruiz JR. Decoding endoplasmic reticulum stress signals in cancer cells and antitumor immunity. Trends Cancer (2022) 8(11):930–943. doi: 10.1016/j.trecan.2022.06.006
2. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature (2016) 529(7586):326–35. doi: 10.1038/nature17041
3. Ajoolabady A, Kaplowitz N, Lebeaupin C, Kroemer G, Kaufman RJ, Malhi H, et al. Endoplasmic reticulum stress in liver diseases. Hepatology (2023) 77(2):619–39. doi: 10.1002/hep.32562
4. Shih JY, Lin YW, Fisch S, Cheng JT, Kang NW, Hong CS, et al. Dapagliflozin suppresses ER stress and improves subclinical myocardial function in diabetes: From bedside to bench. Diabetes (2021) 70(1):262–7. doi: 10.2337/db20-0840
5. Yong J, Johnson JD, Arvan P, Han J, Kaufman RJ. Therapeutic opportunities for pancreatic β-cell ER stress in diabetes mellitus. Nat Rev Endocrinol (2021) 17(8):455–67. doi: 10.1038/s41574-021-00510-4
6. Marciniak SJ, Chambers JE, Ron D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat Rev Drug Discovery (2022) 21(2):115–40. doi: 10.1038/s41573-021-00320-3
7. Ajoolabady A, Liu S, Klionsky DJ, Lip GYH, Tuomilehto J, Kavalakatt S, et al. ER stress in obesity pathogenesis and management. Trends Pharmacol Sci (2022) 43(2):97–109. doi: 10.1016/j.tips.2021.11.011
8. Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, et al. The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat Immunol (2017) 18(5):519–29. doi: 10.1038/ni.3709
9. Tirosh A, Tuncman G, Calay ES, Rathaus M, Ron I, Tirosh A, et al. Intercellular transmission of hepatic ER stress in obesity disrupts systemic metabolism. Cell Metab (2021) 33(2):319–33.e316. doi: 10.1016/j.cmet.2020.11.009
10. Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, et al. ER stress drives lipogenesis and steatohepatitis via caspase-2 activation of S1P. Cell (2018) 175(1):133–45.e115. doi: 10.1016/j.cell.2018.08.020
11. Xu H, Tian Y, Tang D, Zou S, Liu G, Song J, et al. An endoplasmic reticulum stress-MicroRNA-26a feedback circuit in NAFLD. Hepatology (2021) 73(4):1327–45. doi: 10.1002/hep.31428
12. Ren J, Bi Y, Sowers JR, Hetz C, Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol (2021) 18(7):499–521. doi: 10.1038/s41569-021-00511-w
13. Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol (2017) 13(11):681–96. doi: 10.1038/nrneph.2017.129
14. Oh-hashi K, Koga H, Ikeda S, Shimada K, Hirata Y, Kiuchi K. CRELD2 is a novel endoplasmic reticulum stress-inducible gene. Biochem Biophys Res Commun (2009) 387(3):504–10. doi: 10.1016/j.bbrc.2009.07.047
15. Oh-hashi K, Kunieda R, Hirata Y, Kiuchi K. Biosynthesis and secretion of mouse cysteine-rich with EGF-like domains 2. FEBS Lett (2011) 585(15):2481–7. doi: 10.1016/j.febslet.2011.06.029
16. Hartley CL, Edwards S, Mullan L, Bell PA, Fresquet M, Boot-Handford RP, et al. Armet/Manf and Creld2 are components of a specialized ER stress response provoked by inappropriate formation of disulphide bonds: Implications for genetic skeletal diseases. Hum Mol Genet (2013) 22(25):5262–75. doi: 10.1093/hmg/ddt383
17. Rupp PA, Fouad GT, Egelston CA, Reifsteck CA, Olson SB, Knosp WM, et al. Identification, genomic organization and mRNA expression of CRELD1, the founding member of a unique family of matricellular proteins. Gene (2002) 293(1-2):47–57. doi: 10.1016/S0378-1119(02)00696-0
18. Robinson SW, Morris CD, Goldmuntz E, Reller MD, Jones MA, Steiner RD, et al. Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects. Am J Hum Genet (2003) 72(4):1047–52. doi: 10.1086/374319
19. Sarkozy A, Esposito G, Conti E, Digilio MC, Marino B, Calabrò R, et al. CRELD1 and GATA4 gene analysis in patients with nonsyndromic atrioventricular canal defects. Am J Med Genet A (2005) 139(3):236–8. doi: 10.1002/ajmg.a.31018
20. Zatyka M, Priestley M, Ladusans EJ, Fryer AE, Mason J, Latif F, et al. Analysis of CRELD1 as a candidate 3p25 atrioventicular septal defect locus (AVSD2). Clin Genet (2005) 67(6):526–8. doi: 10.1111/j.1399-0004.2005.00435.x
21. Maslen CL, Babcock D, Robinson SW, Bean LJ, Dooley KJ, Willour VL, et al. CRELD1 mutations contribute to the occurrence of cardiac atrioventricular septal defects in down syndrome. Am J Med Genet A (2006) 140(22):2501–5. doi: 10.1002/ajmg.a.31494
22. Guo Y, Shen J, Yuan L, Li F, Wang J, Sun K. Novel CRELD1 gene mutations in patients with atrioventricular septal defect. World J Pediatr (2010) 6(4):348–52. doi: 10.1007/s12519-010-0235-7
23. Ghosh P, Bhaumik P, Ghosh S, Ozbek U, Feingold E, Maslen C, et al. Polymorphic haplotypes of CRELD1 differentially predispose down syndrome and euploids individuals to atrioventricular septal defect. Am J Med Genet A (2012) 158a(11):2843–8. doi: 10.1002/ajmg.a.35626
24. Li H, Cherry S, Klinedinst D, DeLeon V, Redig J, Reshey B, et al. Genetic modifiers predisposing to congenital heart disease in the sensitized down syndrome population. Circ Cardiovasc Genet (2012) 5(3):301–8. doi: 10.1161/CIRCGENETICS.111.960872
25. Zhian S, Belmont J, Maslen CL. Specific association of missense mutations in CRELD1 with cardiac atrioventricular septal defects in heterotaxy syndrome. Am J Med Genet A (2012) 158a(8):2047–9. doi: 10.1002/ajmg.a.35457
26. Li Z, Huang J, Liang B, Zeng D, Luo S, Yan T, et al. Copy number variations in the GATA4, NKX2-5, TBX5, BMP4 CRELD1, and 22q11.2 gene regions in Chinese children with sporadic congenital heart disease. J Clin Lab Anal (2019) 33(2):e22660. doi: 10.1002/jcla.22660
27. Ortiz JA, Castillo M, del Toro ED, Mulet J, Gerber S, Valor LM, et al. The cysteine-rich with EGF-like domains 2 (CRELD2) protein interacts with the large cytoplasmic domain of human neuronal nicotinic acetylcholine receptor alpha4 and beta2 subunits. J Neurochem (2005) 95(6):1585–96. doi: 10.1111/j.1471-4159.2005.03473.x
28. Maslen CL, Babcock D, Redig JK, Kapeli K, Akkari YM, Olson SB. CRELD2: gene mapping, alternate splicing, and comparative genomic identification of the promoter region. Gene (2006) 382:111–20. doi: 10.1016/j.gene.2006.06.016
29. Mass E, Wachten D, Aschenbrenner AC, Voelzmann A, Hoch M. Murine Creld1 controls cardiac development through activation of calcineurin/NFATc1 signaling. Dev Cell (2014) 28(6):711–26. doi: 10.1016/j.devcel.2014.02.012
30. Li H, Edie S, Klinedinst D, Jeong JS, Blackshaw S, Maslen CL, et al. Penetrance of congenital heart disease in a mouse model of down syndrome depends on a trisomic potentiator of a disomic modifier. Genetics (2016) 203(2):763–70. doi: 10.1534/genetics.116.188045
31. Bonaguro L, Köhne M, Schmidleithner L, Schulte-Schrepping J, Warnat-Herresthal S, Horne A, et al. CRELD1 modulates homeostasis of the immune system in mice and humans. Nat Immunol (2020) 21(12):1517–27. doi: 10.1038/s41590-020-00811-2
32. Beckert V, Rassmann S, Kayvanjoo AH, Klausen C, Bonaguro L, Botermann DS, et al. Creld1 regulates myocardial development and function. J Mol Cell Cardiol (2021) 156:45–56. doi: 10.1016/j.yjmcc.2021.03.008
33. Wiseman RL, Mesgarzadeh JS, Hendershot LM. Reshaping endoplasmic reticulum quality control through the unfolded protein response. Mol Cell (2022) 82(8):1477–91. doi: 10.1016/j.molcel.2022.03.025
34. Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. involvement of basic leucine zipper transcription factors. J Biol Chem (1998) 273(50):33741–9. doi: 10.1074/jbc.273.50.33741
35. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature (1999) 397(6716):271–4. doi: 10.1038/16729
36. Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest (2002) 110(10):1389–98. doi: 10.1172/JCI0216886
37. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol (2020) 21(8):421–38. doi: 10.1038/s41580-020-0250-z
38. Preissler S, Ron D. Early events in the endoplasmic reticulum unfolded protein response. Cold Spring Harb Perspect Biol (2019) 11(4):a033894. doi: 10.1101/cshperspect.a033894
39. Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev (1998) 12(12):1812–24. doi: 10.1101/gad.12.12.1812
40. Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci U.S.A. (2005) 102(52):18773–84. doi: 10.1073/pnas.0509487102
41. Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, et al. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci U.S.A. (2006) 103(39):14343–8. doi: 10.1073/pnas.0606480103
42. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell (2001) 107(7):881–91. doi: 10.1016/S0092-8674(01)00611-0
43. Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature (2002) 415(6867):92–6. doi: 10.1038/415092a
44. Jurkin J, Henkel T, Nielsen AF, Minnich M, Popow J, Kaufmann T, et al. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J (2014) 33(24):2922–36. doi: 10.15252/embj.201490332
45. Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell (2009) 138(3):562–75. doi: 10.1016/j.cell.2009.07.017
46. Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science (2006) 313(5783):104–7. doi: 10.1126/science.1129631
47. Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol (2009) 186(3):323–31. doi: 10.1083/jcb.200903014
48. Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic caspase-2. Science (2012) 338(6108):818–22. doi: 10.1126/science.1226191
49. Wang JM, Qiu Y, Yang ZQ, Li L, Zhang K. Inositol-requiring enzyme 1 facilitates diabetic wound healing through modulating MicroRNAs. Diabetes (2017) 66(1):177–92. doi: 10.2337/db16-0052
50. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol (2006) 26(8):3071–84. doi: 10.1128/MCB.26.8.3071-3084.2006
51. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science (2000) 287(5453):664–6. doi: 10.1126/science.287.5453.664
52. Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U.S.A. (2004) 101(31):11269–74. doi: 10.1073/pnas.0400541101
53. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell (2018) 69(2):169–81. doi: 10.1016/j.molcel.2017.06.017
54. Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem (2000) 275(35):27013–20. doi: 10.1016/S0021-9258(19)61473-0
55. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell (1999) 10(11):3787–99. doi: 10.1091/mbc.10.11.3787
56. Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell (2000) 6(6):1355–64. doi: 10.1016/S1097-2765(00)00133-7
57. Walter F, O'Brien A, Concannon CG, Düssmann H, Prehn JHM. ER stress signaling has an activating transcription factor 6α (ATF6)-dependent "off-switch". J Biol Chem (2018) 293(47):18270–84. doi: 10.1074/jbc.RA118.002121
58. Bommiasamy H, Back SH, Fagone P, Lee K, Meshinchi S, Vink E, et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci (2009) 122(Pt 10):1626–36. doi: 10.1242/jcs.045625
59. Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol (2004) 167(1):35–41. doi: 10.1083/jcb.200406136
60. Lee SY, Wong PF, Jamal J, Roebuck MM. Naturally-derived endoplasmic reticulum stress inhibitors for osteoarthritis? Eur J Pharmacol (2022) 922:174903. doi: 10.1016/j.ejphar.2022.174903
61. Rellmann Y, Eidhof E, Dreier R. Review: ER stress-induced cell death in osteoarthritic cartilage. Cell Signal (2021) 78:109880. doi: 10.1016/j.cellsig.2020.109880
62. Munro S, Pelham HR. A c-terminal signal prevents secretion of luminal ER proteins. Cell (1987) 48(5):899–907. doi: 10.1016/0092-8674(87)90086-9
63. Takemoto H, Yoshimori T, Yamamoto A, Miyata Y, Yahara I, Inoue K, et al. Heavy chain binding protein (BiP/GRP78) and endoplasmin are exported from the endoplasmic reticulum in rat exocrine pancreatic cells, similar to protein disulfide-isomerase. Arch Biochem Biophys (1992) 296(1):129–36. doi: 10.1016/0003-9861(92)90554-A
64. Glembotski CC, Thuerauf DJ, Huang C, Vekich JA, Gottlieb RA, Doroudgar S. Mesencephalic astrocyte-derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion. J Biol Chem (2012) 287(31):25893–904. doi: 10.1074/jbc.M112.356345
65. Oh-Hashi K, Tanaka K, Koga H, Hirata Y, Kiuchi K. Intracellular trafficking and secretion of mouse mesencephalic astrocyte-derived neurotrophic factor. Mol Cell Biochem (2012) 363(1-2):35–41. doi: 10.1007/s11010-011-1155-0
66. Tang Q, Li Y, He J. MANF: an emerging therapeutic target for metabolic diseases. Trends Endocrinol Metab (2022) 33(4):236–46. doi: 10.1016/j.tem.2022.01.001
67. Tadimalla A, Belmont PJ, Thuerauf DJ, Glassy MS, Martindale JJ, Gude N, et al. Mesencephalic astrocyte-derived neurotrophic factor is an ischemia-inducible secreted endoplasmic reticulum stress response protein in the heart. Circ Res (2008) 103(11):1249–58. doi: 10.1161/CIRCRESAHA.108.180679
68. Newstead S, Barr F. Molecular basis for KDEL-mediated retrieval of escaped ER-resident proteins - SWEET talking the COPs. J Cell Sci (2020) 133(19):jcs250100. doi: 10.1242/jcs.250100
69. Ellgaard L, Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep (2005) 6(1):28–32. doi: 10.1038/sj.embor.7400311
70. Hatahet F, Ruddock LW. Substrate recognition by the protein disulfide isomerases. FEBS J (2007) 274(20):5223–34. doi: 10.1111/j.1742-4658.2007.06058.x
71. Liu CL, Zhong W, He YY, Li X, Li S, He KL. Genome-wide analysis of tunicamycin-induced endoplasmic reticulum stress response and the protective effect of endoplasmic reticulum inhibitors in neonatal rat cardiomyocytes. Mol Cell Biochem (2016) 413(1-2):57–67. doi: 10.1007/s11010-015-2639-0
72. Kim Y, Park SJ, Manson SR, Molina CA, Kidd K, Thiessen-Philbrook H, et al. Elevated urinary CRELD2 is associated with endoplasmic reticulum stress-mediated kidney disease. JCI Insight (2017) 2(23):e92896. doi: 10.1172/jci.insight.92896
73. Oh-Hashi K, Fujimura K, Norisada J, Hirata Y. Expression analysis and functional characterization of the mouse cysteine-rich with EGF-like domains 2. Sci Rep (2018) 8(1):12236. doi: 10.1038/s41598-018-30362-4
74. Oh-hashi K, Kanamori Y, Hirata Y, Kiuchi K. Characterization of V-ATPase inhibitor-induced secretion of cysteine-rich with EGF-like domains 2. Cell Biol Toxicol (2014) 30(3):127–36. doi: 10.1007/s10565-014-9274-5
75. Aridor M, Fish KN, Bannykh S, Weissman J, Roberts TH, Lippincott-Schwartz J, et al. The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J Cell Biol (2001) 152(1):213–29. doi: 10.1083/jcb.152.1.213
76. Sato K, Nakano A. Mechanisms of COPII vesicle formation and protein sorting. FEBS Lett (2007) 581(11):2076–82. doi: 10.1016/j.febslet.2007.01.091
77. Martínez-Menárguez JA, Geuze HJ, Slot JW, Klumperman J. Vesicular tubular clusters between the ER and golgi mediate concentration of soluble secretory proteins by exclusion from COPI-coated vesicles. Cell (1999) 98(1):81–90. doi: 10.1016/S0092-8674(00)80608-X
78. Bard F, Mazelin L, Péchoux-Longin C, Malhotra V, Jurdic P. Src regulates golgi structure and KDEL receptor-dependent retrograde transport to the endoplasmic reticulum. J Biol Chem (2003) 278(47):46601–6. doi: 10.1074/jbc.M302221200
79. Stauber T, Simpson JC, Pepperkok R, Vernos I. A role for kinesin-2 in COPI-dependent recycling between the ER and the golgi complex. Curr Biol (2006) 16(22):2245–51. doi: 10.1016/j.cub.2006.09.060
80. Giannotta M, Ruggiero C, Grossi M, Cancino J, Capitani M, Pulvirenti T, et al. The KDEL receptor couples to Gαq/11 to activate src kinases and regulate transport through the golgi. EMBO J (2012) 31(13):2869–81. doi: 10.1038/emboj.2012.134
81. Luini A, Mavelli G, Jung J, Cancino J. Control systems and coordination protocols of the secretory pathway. F1000Prime Rep (2014) 6:88. doi: 10.12703/P6-88
82. Wiersma VR, Michalak M, Abdullah TM, Bremer E, Eggleton P. Mechanisms of translocation of ER chaperones to the cell surface and immunomodulatory roles in cancer and autoimmunity. Front Oncol (2015) 5:7. doi: 10.3389/fonc.2015.00007
83. Jin H, Komita M, Aoe T. The role of BiP retrieval by the KDEL receptor in the early secretory pathway and its effect on protein quality control and neurodegeneration. Front Mol Neurosci (2017) 10:222. doi: 10.3389/fnmol.2017.00222
84. Kokubun H, Jin H, Aoe T. Pathogenic effects of impaired retrieval between the endoplasmic reticulum and golgi complex. Int J Mol Sci (2019) 20(22):5614. doi: 10.3390/ijms20225614
85. Glembotski CC. Functions for the cardiomyokine, MANF, in cardioprotection, hypertrophy and heart failure. J Mol Cell Cardiol (2011) 51(4):512–7. doi: 10.1016/j.yjmcc.2010.10.008
86. Oh-hashi K, Norisada J, Hirata Y, Kiuchi K. Characterization of the role of MANF in regulating the secretion of CRELD2. Biol Pharm Bull (2015) 38(5):722–31. doi: 10.1248/bpb.b14-00825
87. Mizobuchi N, Hoseki J, Kubota H, Toyokuni S, Nozaki J, Naitoh M, et al. ARMET is a soluble ER protein induced by the unfolded protein response via ERSE-II element. Cell Struct Funct (2007) 32(1):41–50. doi: 10.1247/csf.07001
88. Apostolou A, Shen Y, Liang Y, Luo J, Fang S. Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp Cell Res (2008) 314(13):2454–67. doi: 10.1016/j.yexcr.2008.05.001
89. Parkash V, Lindholm P, Peränen J, Kalkkinen N, Oksanen E, Saarma M, et al. The structure of the conserved neurotrophic factors MANF and CDNF explains why they are bifunctional. Protein Eng Des Sel (2009) 22(4):233–41. doi: 10.1093/protein/gzn080
90. Henderson MJ, Richie CT, Airavaara M, Wang Y, Harvey BK. Mesencephalic astrocyte-derived neurotrophic factor (MANF) secretion and cell surface binding are modulated by KDEL receptors. J Biol Chem (2013) 288(6):4209–25. doi: 10.1074/jbc.M112.400648
91. Neves J, Zhu J, Sousa-Victor P, Konjikusic M, Riley R, Chew S, et al. Immune modulation by MANF promotes tissue repair and regenerative success in the retina. Science (2016) 353(6294):aaf3646. doi: 10.1126/science.aaf3646
92. Yan Y, Rato C, Rohland L, Preissler S, Ron D. MANF antagonizes nucleotide exchange by the endoplasmic reticulum chaperone BiP. Nat Commun (2019) 10(1):541. doi: 10.1038/s41467-019-08450-4
93. Norisada J, Hirata Y, Amaya F, Kiuchi K, Oh-hashi K. A comparative analysis of the molecular features of MANF and CDNF. PLoS One (2016) 11(1):e0146923. doi: 10.1371/journal.pone.0146923
94. Kern P, Balzer NR, Blank N, Cygon C, Wunderling K, Bender F, et al. Creld2 function during unfolded protein response is essential for liver metabolism homeostasis. FASEB J (2021) 35(10):e21939. doi: 10.1096/fj.202002713RR
95. Han H, Hu J, Lau MY, Feng M, Petrovic LM, Ji C. Altered methylation and expression of ER-associated degradation factors in long-term alcohol and constitutive ER stress-induced murine hepatic tumors. Front Genet (2013) 4:224. doi: 10.3389/fgene.2013.00224
96. Navas-Madroñal M, Rodriguez C, Kassan M, Fité J, Escudero JR, Cañes L, et al. Enhanced endoplasmic reticulum and mitochondrial stress in abdominal aortic aneurysm. Clin Sci (Lond) (2019) 133(13):1421–38. doi: 10.1042/CS20190399
97. Siegert AM, García Díaz-Barriga G, Esteve-Codina A, Navas-Madroñal M, Gorbenko Del Blanco D, Alberch J, et al. A FBN1 3'UTR mutation variant is associated with endoplasmic reticulum stress in aortic aneurysm in marfan syndrome. Biochim Biophys Acta Mol Basis Dis (2019) 1865(1):107–14. doi: 10.1016/j.bbadis.2018.10.029
98. Nundlall S, Rajpar MH, Bell PA, Clowes C, Zeeff LA, Gardner B, et al. An unfolded protein response is the initial cellular response to the expression of mutant matrilin-3 in a mouse model of multiple epiphyseal dysplasia. Cell Stress Chaperones (2010) 15(6):835–49. doi: 10.1007/s12192-010-0193-y
99. Cameron TL, Bell KM, Tatarczuch L, Mackie EJ, Rajpar MH, McDermott BT, et al. Transcriptional profiling of chondrodysplasia growth plate cartilage reveals adaptive ER-stress networks that allow survival but disrupt hypertrophy. PLoS One (2011) 6(9):e24600. doi: 10.1371/journal.pone.0024600
100. Wilson R, Golub SB, Rowley L, Angelucci C, Karpievitch YV, Bateman JF, et al. Novel elements of the chondrocyte stress response identified using an in vitro model of mouse cartilage degradation. J Proteome Res (2016) 15(3):1033–50. doi: 10.1021/acs.jproteome.5b01115
101. Zhang J, Weng Y, Liu X, Wang J, Zhang W, Kim SH, et al. Endoplasmic reticulum (ER) stress inducible factor cysteine-rich with EGF-like domains 2 (Creld2) is an important mediator of BMP9-regulated osteogenic differentiation of mesenchymal stem cells. PLoS One (2013) 8(9):e73086. doi: 10.1371/journal.pone.0073086
102. Dennis EP, Edwards SM, Jackson RM, Hartley CL, Tsompani D, Capulli M, et al. CRELD2 is a novel LRP1 chaperone that regulates noncanonical WNT signaling in skeletal development. J Bone Miner Res (2020) 35(8):1452–69. doi: 10.1002/jbmr.4010
103. Duxfield A, Munkley J, Briggs MD, Dennis EP. CRELD2 is a novel modulator of calcium release and calcineurin-NFAT signalling during osteoclast differentiation. Sci Rep (2022) 12(1):13884. doi: 10.1038/s41598-022-17347-0
104. Albig AR, Roy TG, Becenti DJ, Schiemann WP. Transcriptome analysis of endothelial cell gene expression induced by growth on matrigel matrices: identification and characterization of MAGP-2 and lumican as novel regulators of angiogenesis. Angiogenesis (2007) 10(3):197–216. doi: 10.1007/s10456-007-9075-z
105. Jariwala U, Prescott J, Jia L, Barski A, Pregizer S, Cogan JP, et al. Identification of novel androgen receptor target genes in prostate cancer. Mol Cancer (2007) 6:39. doi: 10.1186/1476-4598-6-39
106. Yamada Y, Arai T, Sugawara S, Okato A, Kato M, Kojima S, et al. Impact of novel oncogenic pathways regulated by antitumor miR-451a in renal cell carcinoma. Cancer Sci (2018) 109(4):1239–53. doi: 10.1111/cas.13526
107. Liu GM, Zeng HD, Zhang CY, Xu JW. Key genes associated with diabetes mellitus and hepatocellular carcinoma. Pathol Res Pract (2019) 215(11):152510. doi: 10.1016/j.prp.2019.152510
108. Boyle ST, Poltavets V, Kular J, Pyne NT, Sandow JJ, Lewis AC, et al. ROCK-mediated selective activation of PERK signalling causes fibroblast reprogramming and tumour progression through a CRELD2-dependent mechanism. Nat Cell Biol (2020) 22(7):882–95. doi: 10.1038/s41556-020-0523-y
109. Chen MF, Chang CH, Yang LY, Hsieh PH, Shih HN, Ueng SWN, et al. Synovial fluid interleukin-16, interleukin-18, and CRELD2 as novel biomarkers of prosthetic joint infections. Bone Joint Res (2019) 8(4):179–88. doi: 10.1302/2046-3758.84.BJR-2018-0291.R1
110. Lasić V, Kosović I, Jurić M, Racetin A, Čurčić J, Šolić I, et al. GREB1L, CRELD2 and ITGA10 expression in the human developmental and postnatal kidneys: An immunohistochemical study. Acta Histochem (2021) 123(2):151679. doi: 10.1016/j.acthis.2021.151679
111. Xia SW, Wang ZM, Sun SM, Su Y, Li ZH, Shao JJ, et al. Endoplasmic reticulum stress and protein degradation in chronic liver disease. Pharmacol Res (2020) 161:105218. doi: 10.1016/j.phrs.2020.105218
112. Wang YL, Zhou X, Li DL, Ye JM. Role of the mTOR-autophagy-ER stress pathway in high fructose-induced metabolic-associated fatty liver disease. Acta Pharmacol Sin (2022) 43(1):10–4. doi: 10.1038/s41401-021-00629-0
113. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol (2020) 73(1):202–9. doi: 10.1016/j.jhep.2020.03.039
114. Shiha G, Korenjak M, Eskridge W, Casanovas T, Velez-Moller P, Högström S, et al. Redefining fatty liver disease: An international patient perspective. Lancet Gastroenterol Hepatol (2021) 6(1):73–9. doi: 10.1016/S2468-1253(20)30294-6
115. Sharpton SR, Schnabl B, Knight R, Loomba R. Current concepts, opportunities, and challenges of gut microbiome-based personalized medicine in nonalcoholic fatty liver disease. Cell Metab (2021) 33(1):21–32. doi: 10.1016/j.cmet.2020.11.010
116. Kucukoglu O, Sowa JP, Mazzolini GD, Syn WK, Canbay A. Hepatokines and adipokines in NASH-related hepatocellular carcinoma. J Hepatol (2021) 74(2):442–57. doi: 10.1016/j.jhep.2020.10.030
117. Nohara K, Tateishi Y, Suzuki T, Okamura K, Murai H, Takumi S, et al. Late-onset increases in oxidative stress and other tumorigenic activities and tumors with a ha-ras mutation in the liver of adult male C3H mice gestationally exposed to arsenic. Toxicol Sci (2012) 129(2):293–304. doi: 10.1093/toxsci/kfs203
118. Kern RJ, Lindholm-Perry AK, Freetly HC, Snelling WM, Kern JW, Keele JW, et al. Transcriptome differences in the rumen of beef steers with variation in feed intake and gain. Gene (2016) 586(1):12–26. doi: 10.1016/j.gene.2016.03.034
119. Horna-Terrón E, Pradilla-Dieste A, Sánchez-de-Diego C, Osada J. TXNDC5, a newly discovered disulfide isomerase with a key role in cell physiology and pathology. Int J Mol Sci (2014) 15(12):23501–18. doi: 10.3390/ijms151223501
120. Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature (1991) 352(6333):337–9. doi: 10.1038/352337a0
121. Milewicz DM, Dietz HC, Miller DC. Treatment of aortic disease in patients with marfan syndrome. Circulation (2005) 111(11):e150–157. doi: 10.1161/01.CIR.0000155243.70456.F4
122. Hughes A, Oxford AE, Tawara K, Jorcyk CL, Oxford JT. Endoplasmic reticulum stress and unfolded protein response in cartilage pathophysiology; contributing factors to apoptosis and osteoarthritis. Int J Mol Sci (2017) 18(3):665. doi: 10.3390/ijms18030665
123. Guo J, Ren R, Sun K, He J, Shao J. PERK signaling pathway in bone metabolism: Friend or foe? Cell Prolif (2021) 54(4):e13011. doi: 10.1111/cpr.13011
124. Yarmohammadi F, Hayes AW, Karimi G. The therapeutic effects of berberine against different diseases: A review on the involvement of the endoplasmic reticulum stress. Phytother Res (2022) 36(8):3215–31. doi: 10.1002/ptr.7539
125. Park SJ, Kim Y, Chen YM. Endoplasmic reticulum stress and monogenic kidney diseases in precision nephrology. Pediatr Nephrol (2019) 34(9):1493–500. doi: 10.1007/s00467-018-4031-2
126. Li C, Chen YM. Endoplasmic reticulum-associated biomarkers for molecular phenotyping of rare kidney disease. Int J Mol Sci (2021) 22(4):2161. doi: 10.3390/ijms22042161
Keywords: CRELD2, endoplasmic reticulum stress, unfolded protein response, metabolism, human diseases
Citation: Tang Q, Liu Q, Li Y, Mo L and He J (2023) CRELD2, endoplasmic reticulum stress, and human diseases. Front. Endocrinol. 14:1117414. doi: 10.3389/fendo.2023.1117414
Received: 06 December 2022; Accepted: 13 February 2023;
Published: 02 March 2023.
Edited by:
Guojun Shi, Sun Yat-sen University, ChinaReviewed by:
Lorenzo Bonaguro, University of Bonn, GermanyCopyright © 2023 Tang, Liu, Li, Mo and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhan He, amluaGFuaGVAc2N1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.