
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 28 February 2023
Sec. Bone Research
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1113547
This article is part of the Research Topic Potential Effects and Mechanisms of Bone Homeostasis on Tumor Bone Metastasis View all 6 articles
 Monika Martiniakova1†*
Monika Martiniakova1†* Vladimira Mondockova2†
Vladimira Mondockova2† Roman Biro1
Roman Biro1 Veronika Kovacova1
Veronika Kovacova1 Martina Babikova2
Martina Babikova2 Nina Zemanova2
Nina Zemanova2 Sona Ciernikova3
Sona Ciernikova3 Radoslav Omelka2*
Radoslav Omelka2*The skeleton is the third most common site of metastatic disease, which causes serious bone complications and short-term prognosis in cancer patients. Prostate and breast cancers are responsible for the majority of bone metastasis, resulting in osteolytic or osteoblastic lesions. The crosstalk between bone cells and their interactions with tumor cells are important in the development of lesions. Recently, both preclinical and clinical studies documented the clinical relevance of bone-derived factors, including osteocalcin (OC) and its undercarboxylated form (ucOC), fibroblast growth factor 23 (FGF23), sclerostin (SCL), and lipocalin 2 (LCN2) as prognostic tumor biomarkers and potential therapeutic targets in bone metastasis. Both OC and ucOC could be useful targets for the prevention of bone metastasis in breast cancer. Moreover, elevated OC level may be a metastatic marker of prostate cancer. FGF23 is particularly important for those forms of cancer that primarily affect bone and/or are characterized by bone metastasis. In other tumor entities, increased FGF23 level is enigmatic. SCL plays a significant role in the pathogenesis of both osteolytic and osteoblastic lesions, as its levels are high in metastatic breast and prostate cancers. Elevated expression levels of LCN2 have been found in aggressive subtypes of cancer. However, its role in anti-metastasis varies significantly between different cancer types. Anyway, all aforementioned bone-derived factors can be used as promising tumor biomarkers. As metastatic bone disease is generally not curable, targeting bone factors represents a new trend in the prevention of bone metastasis and patient care.
Tumor metastasis, the growth of primary tumor cells in a distant organ, involves cell migration, invasion, intra- and extravasation, survival in circulation, evasion in local immune responses, and eventual arrest at distant secondary sites (1, 2). The skeleton is considered the third most common site of metastatic disease after lung and liver. Prostate and breast cancers are responsible for most bone metastasis (up to 70%), although lung, kidney, thyroid and most adenocarcinoma primary tumors can metastasize to the bones as well (3, 4). The overall incidence of bone metastasis is not known. The relative incidence depends on the tumor type and in patients with advanced metastatic disease is: 65-75% in the prostate and breast, 60% in the thyroid, 30-40% in the lung and 20-25% in renal cell carcinoma (5). Bone metastasis influences survival rates ranging from 6 to 7 months for lung cancer to several years for breast cancer (19 to 25 months) or prostate cancer (12 to 53 months) and are a major cause of morbidity characterized by pathological fractures, spinal cord or nerve root compression, debilitating bone pain, bone marrow aplasia and hypercalcemia (2, 3).
Bone is a particularly suitable site for tumor cell metastasis mainly because it is a rich source of growth factors, neovascularization factors, and cytokines that facilitate the colonization, growth, and long-term survival of cancer cells (6). When cancer cells establish themselves in the bone microenvironment by disrupting bone homeostasis, this will result in increased bone resorption and/or bone formation. Overall, tumor cells interact with bone marrow cells and bone cells, thereby promoting tumor growth (7, 8). It is increasingly evident that osteoblasts in the bone microenvironment play a critical role in cancer cell attraction (9, 10), maintenance (11), and survival (6, 11, 12) during cancer progression in bone. The crosstalk between osteoblasts, osteocytes and osteoclasts is important in the development of bone metastasis (13).
In general, bone metastasis can be classified as osteolytic, osteoblastic, and mixed, according to the primary mechanism of interference with normal bone remodeling. Osteolytic metastasis is characterized by overactivation of bone resorption due to occurrence of osteoblast inactivation as well as the recruitment and activation of osteoclasts in the tumor-bone microenvironment (14, 15). Osteolytic lesions consist of soft parts of damaged bone with decreased bone mineral density (7). Parathyroid hormone-related peptide (PTHrP) plays a major role in their development (16). It is not clear whether the bone microenvironment induces cancer cells to express PTHrP, or if cells that metastasize to the bone have an intrinsic higher expression of PTHrP (17). The production of PTHrP up-regulates receptor activator of nuclear factor-kappa B ligand (RANKL) and down-regulates osteoprotegerin (OPG) by osteoblasts to activate osteoclastogenesis and bone resorption. Accelerated bone resorption, in turn, promotes the release of bone-derived growth factors such as transforming growth factor-beta (TGF-β), insulin-like growth factor (IGF)-1, and raises extracellular calcium concentration to further support the growth of cancer cells (Figure 1) (18, 19). In osteoblastic metastasis, on the other hand, the deposition of new bone occurs as a result of new bone formation (due to an osteoblastic response) that is not preceded by bone resorption (20). Osteoblastic lesions are the result of direct tumor stimulation of osteoblasts and are characterized by deposition of mineralized bone in tissue lesions (7). TGF- β, vascular endothelial growth factor (VEGF), bone morphogenetic proteins (BMPs) and endothelin (ET)-1 are associated with the generation of osteoblasts (21). ET-1 inhibits the expression of Dickkopf-1 (DKK-1), which normally blocks Wnt signaling. Inhibition of DKK-1 results in increased osteoblast activity supporting uncontrolled bone formation (22). Elevated bone formation drives tumor progression by releasing IGF-1 and interleukins (IL)-6 and IL-8 (Figure 1) (23). Finally, mixed bone metastasis involves a combination of both osteolytic and osteoblastic components (2).
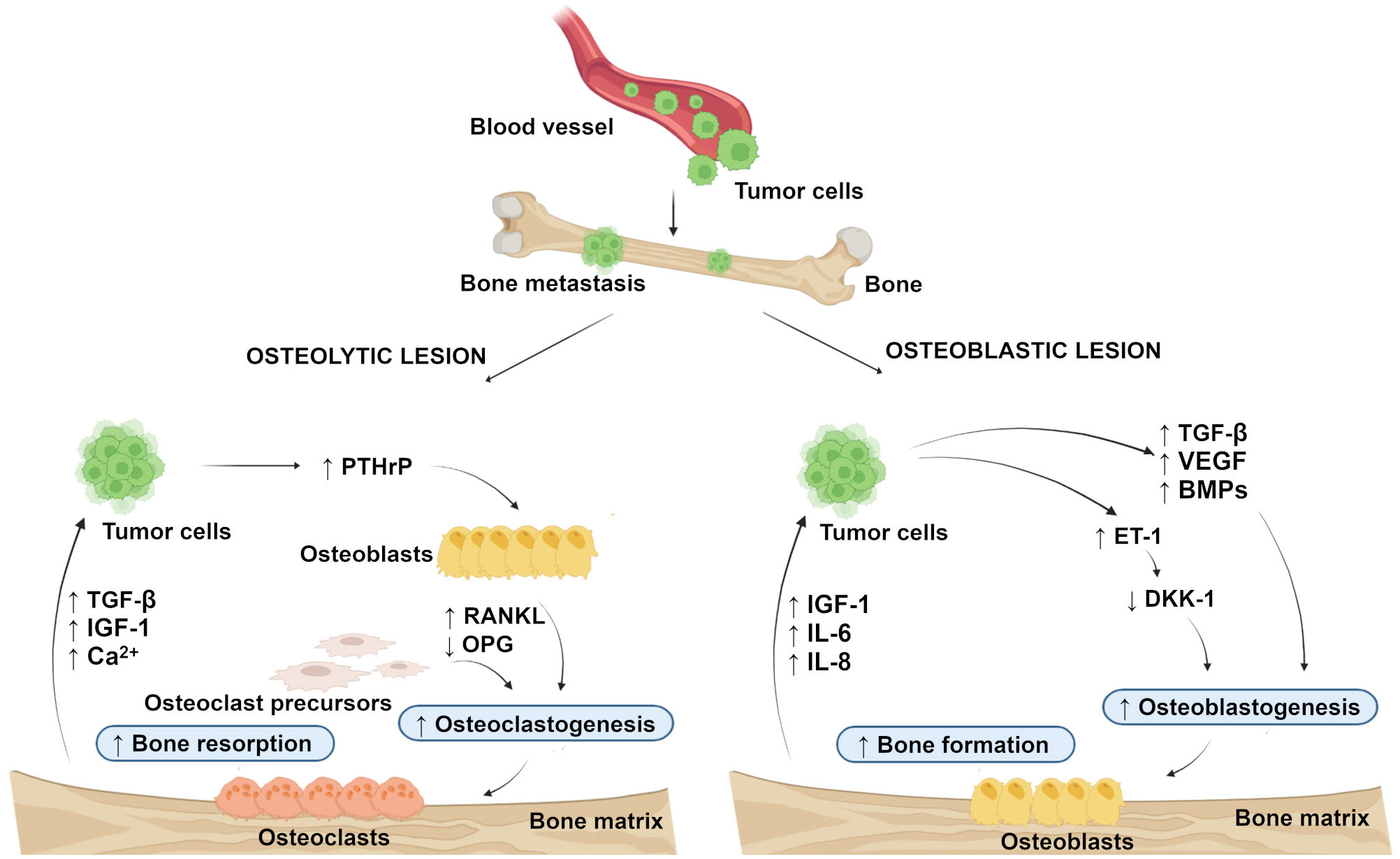
Figure 1 Schematic representation of the development of osteolytic and osteoblastic lesions. Osteolytic lesions are caused by excessive activation of bone resorption by osteoclasts in the tumor-bone microenvironment. Osteoblastic lesions are the result of direct tumor stimulation of osteoblasts, leading to uncontrolled bone formation. (Created with BioRender.com.) BMPs, bone morphogenetic proteins; DKK-1, Dickkopf-1; ET-1, endothelin-1; IGF-1, insulin-like growth factor-1; IL-6, interleukin-6; IL-8 interleukin-8; OPG, osteoprotegerin; PTHrP, parathyroid hormone-related peptide; RANKL, receptor activator of nuclear factor-kappa B ligand; TGF-β, transforming growth factor-beta; VEGF, vascular endothelial growth factor. ↑, increased; ↓, decreased.
Osteocalcin (OC), fibroblast growth factor 23 (FGF23), sclerostin (SCL), and lipocalin 2 (LCN2) are bone-derived factors secreted by bone cells that are involved in the regulation of bone homeostasis. Their implication in cancer biology has attracted research interest in recent years. This review aims to summarize the current knowledge about OC, FGF23, SCL, and LCN2 and to characterize the association between them and tumor bone metastasis.
OC is the most abundant non-collagenous protein in bone. It is secreted by osteoblasts and contains three glutamate residues that can be carboxylated, which then provide high affinity to the hydroxyapatite matrix. This modification is catalyzed by γ-glutamyl carboxylase, and uses vitamin K, O2, and CO2 as cofactors, supplied by the vitamin K cycle and circulation (24, 25). Carboxylated OC (cOC) is essential for the alignment of apatite crystals and optimal bone strength (26). Undercarboxylated OC (ucOC) with a reduced degree of carboxylation at the glutamate residues is available with less affinity for hydroxyapatite (25). When bone is resorbed by osteoclasts, the acidic pH in the resorption lacuna causes the carboxyl groups on OC to be removed and ucOC to be released into the systemic circulation. Therefore, circulating levels of ucOC are dependent on the rate of bone remodeling (27, 28). Generally, ucOC is considered an endocrinologically active form that can regulate glucose metabolism, testosterone synthesis, muscle mass, and cognitive function (29–32).
In addition, the relationship between OC and tumorigenesis has been noted (33). Pietschmann et al. (34) and Salem et al. (35) indicated significantly increased serum OC levels in breast cancer patients and subjects with bone metastasis compared to healthy controls (35). The research by Xu et al. (36) showed that ucOC facilitated the proliferation and metastasis of breast cancer cells MDA-MB-231 by accelerating the TGF-β/SMAD3 signaling pathway. Furthermore, ucOC also promoted the gene expression of IL-8 and PTHrP, which may act as osteolytic factors in breast cancer cells. Therefore, ucOC could be a useful target for the prevention of bone metastasis in breast cancer. A tumor-promoting effect of OC on human prostate cancer cell line PC-3 and pancreatic cancer cells was revealed by Ye (37) and Kayed et al. (38). OC has also been found to facilitate the growth and migration of lung tumors through neutrophil aggregation (39) and to induce the growth of prostate cancer cells via G protein-coupled receptor family C group 6 member A (GPRC6A) receptor (40). OC production correlates with prostate cancer metastasis (41). Therefore, high serum OC level may be a metastatic marker of prostate cancer (42). Relevant information related to this issue is presented in Table 1. The aforementioned studies provide an interesting basis for further research regarding the roles of OC and ucOC in tumorigenesis.
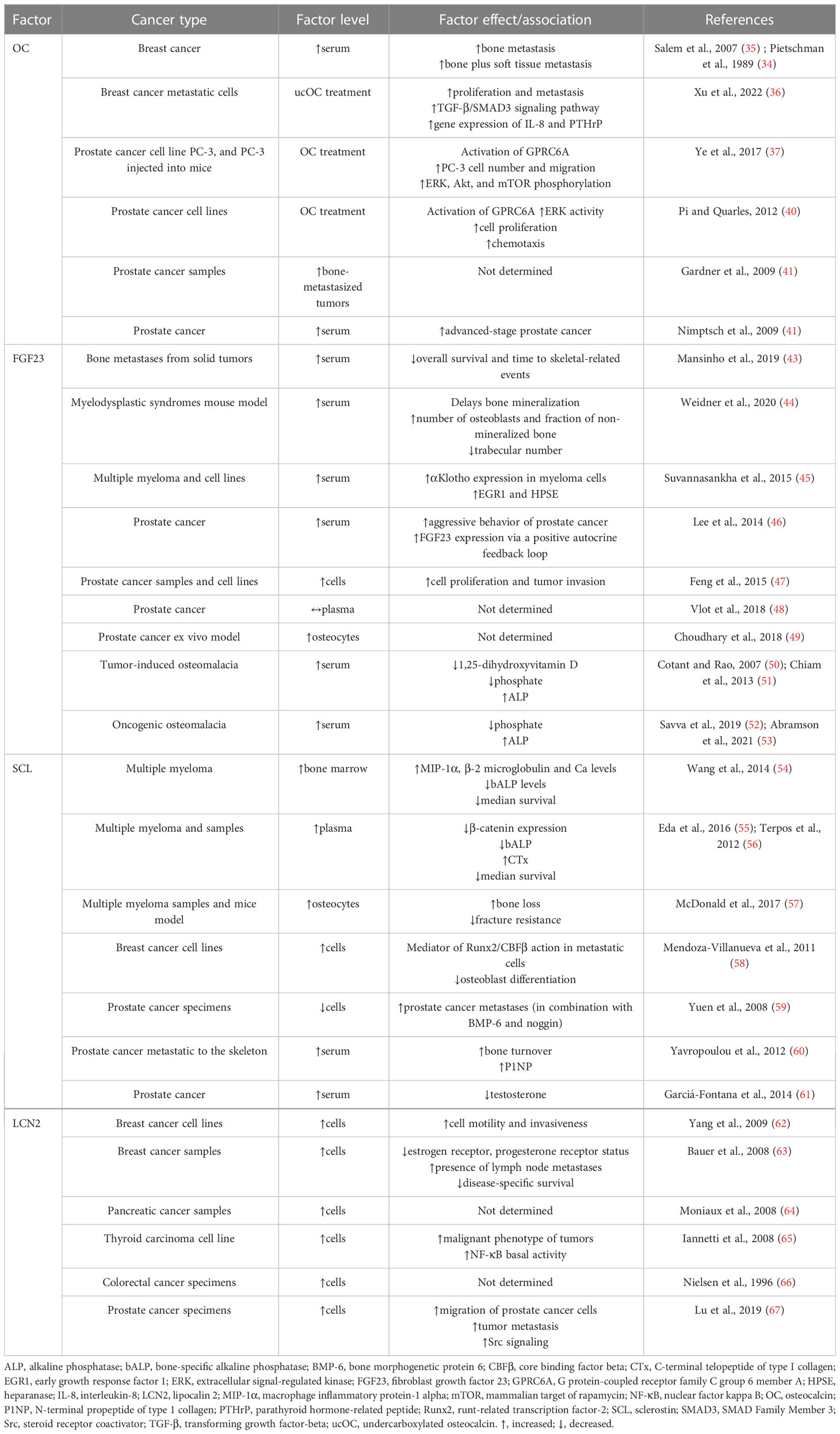
Table 1 Associations of OC, FGF23, SCL, and LCN2 with cancer.
FGF23 is a bone-derived protein predominantly secreted by osteoblasts and osteocytes (68). Its target organs include the kidney and parathyroid glands (69). In the kidney, FGF23 acts as a hormone stimulating phosphate excretion and suppressing the synthesis of 1.25(OH)2D3, an active vitamin D. In the parathyroid glands, FGF23 reduces the production and secretion of parathyroid hormone (PTH). These endocrine effects are dependent on the transmembrane protein αKlotho, which increases the binding affinity of FGF23 to FGF receptors (FGFR) (70). In this way, FGF23 is part of a hormonal circuit that additionally involves PTH and 1.25(OH)2D3 and regulates phosphate and vitamin D metabolism, as well as affecting Ca2+ (71). Locally produced FGF23 in other tissues, including the liver or heart, has additional paracrine effects without the involvement of αKlotho (70). Osteoblasts, osteocytes, and osteoclasts express the FGFR/αKlotho coreceptor. The specific knockout of αKlotho in osteocytes can lead to the osteogenic enhancement and increased bone mass (72). Several studies have also shown that FGF23 can upregulate early growth response genes in osteoblasts and the RANKL/OPG ratio in the osteoclast surface by binding to the coreceptor. Regardless, FGFR/αKlotho coreceptor activation may also account for bone remodeling (73).
In those forms of cancer that involve or originate from bone, FGF23 signaling may directly contribute to cancer biology and/or progression (70). In patients with cancer and bone metastases from solid tumors, elevated serum levels of FGF23 were associated with shorter survival, as well as time to skeletal-related events (43), providing the first evidence of the prognostic significance of FGF23 in individuals with bone metastasis. Mouse model of myelodysplastic syndrome also had increased level of FGF23 (44). Serum levels of intact FGF23 were found to be higher also in subjects with multiple myeloma (45). FGF23 expression was increased in patients with prostate cancer, as well as FGF23/FGFR1/αKlotho in various prostate cancer cell lines (46). For this aim, FGF23 can act as an autocrine factor in prostate cancer cells stimulating tumor invasion and cell proliferation (47). According to Vlot et al. (48), the serum level of FGF23 was unchanged in prostate cancer (48), although prostate cancer cells can stimulate FGF23 expression in osteocytes (49). However, bone metastasis may account for the high FGF23 levels and symptoms of tumor-induced osteomalacia identified in prostate cancer patients (50, 51). Breast cancer may also be consistent with oncogenic osteomalacia and elevated levels of FGF23 (52, 53). In all of these reported cases, including patients with bone metastases, increased concentration of FGF23 was accompanied by a decrease in the serum phosphate level. FGF23 mRNA expression was high in breast cancer cells as well (74). In addition, FGFR signaling may be highly relevant to breast cancer oncogenesis (75). Table 1 summarizes relevant studies reporting FGF23 levels and its function in several cancer types. An anti-FGF23 approach may be useful in malignancies affecting bone because they are characterized by enhanced FGF23 levels or FGF23 signaling. In other tumor entities (e.g. endometrial cancer, hepatocellular carcinoma), no change in serum FGF23 level was demonstrated (70). It is important to note that most of the above-mentioned studies report associations, not necessarily causal relationships, but FGF23 may serve as a tumor biomarker.
SCL is a glycoprotein encoded by the SOST gene and secreted mainly by osteocytes but can be widely expressed in other tissues and organs, such as the kidney, liver, and lung (76, 77). SCL expression depends on hormones, inflammatory molecules, and mechanical loading (78). SCL can suppress the activity of osteoblasts and osteoclasts, which stabilizes bone strength and toughness under normal physiological conditions (79). Lack of SCL will lead to excessive bone hardening and its overexpression will inhibit bone formation (80). SCL binds to lipoprotein receptor-related proteins (LRP) 5/6, critical coreceptors of the Wnt signaling pathway, leading to decreased bone formation (76, 81). Since the canonical Wnt signaling pathway is a key regulator of cellular functions including proliferation, differentiation and migration in various tissues, an endocrine role of SCL has been identified as well (82, 83). Moreover, the aforementioned pathway is also involved in the pathogenesis of many skeletal disorders and cancer (77).
In patients with multiple myeloma, serum and bone marrow SCL levels were elevated (54, 55) and increased serum SCL correlated with poor survival (56). Genetic deletion of the SOST gene was found to reduce osteolytic lesions in immunodeficient mice by increasing and decreasing the number of osteoblasts and osteoclasts in multiple myeloma (84). Similarly, pharmacological inhibition of SCL with an anti-SCL antibody in immunocompetent mice decreased osteolysis and elevated markers of bone formation without altering tumor growth (57). SCL was elevated in both metastatic breast cancer (MDA-MB 231) and non-metastatic (MCF-7) cell lines, due to abnormal overexpression of runt-related transcription factor-2 (Runx2), which binds to the proximal promoter of the SOST gene. Increased levels of SCL in metastatic breast cancer suppressed bone formation through inhibition of the Wnt signaling pathway, suggesting its role in the pathogenesis of osteolytic lesions (58). Yuen et al. (59) found that high expression levels of BMP-6 and low expression levels of its inhibitors SCL and noggin in primary prostate cancer significantly predicted the development of distant metastasis. SCL was found to inhibit prostate cancer migration and its deficiency led to the increased spread of prostate cancer (85). Yavropoulou et al. (60) reported a high correlation between serum SCL levels and elevated bone turnover. The authors explained this finding by a compensatory response to the increased number of osteoblasts at affected skeletal sites which may contribute to enhanced bone resorption. Elevated levels of circulating SCL have also been found in prostate cancer patients receiving androgen deprivation therapy (61). The inverse relationship between serum SCL and testosterone in these patients points to an important influence of androgens in the regulation of bone metabolism in prostate cancer. All these findings suggest that SCL could have a protective role in prostate cancer progression, and elevated serum SCL levels reflect the increased activity of osteoblasts and osteocytes in prostate cancer-induced osteoblastic lesions. Findings reporting SCL levels and its function in several types of cancer are shown in Table 1.
LCN2, also known as neutrophil gelatinase-associated lipocalin or 24p3, is a secreted glycoprotein associated with many functions such as stimulation of neutrophil migration (86), regulation of bone homeostasis (87) and skeletal muscle regeneration (88). LCN 2 was previously understood as one of the adipokines (89); however, its expression level was recently found to be at least 10-fold higher in osteoblasts compared to adipocytes. In mice with a conditional knockout of LCN2 in osteoblasts, elevated levels of plasma glucose and body fat were reported (76, 90). In bone, LCN2 is regulated by physical activity/inactivity (87) and negatively modulates bone development and turnover, as overexpression of LCN2 results in thinner cortical bone and reduced osteoblast differentiation (91). Recently, bone-derived LCN2 has been shown to have endocrine functions in appetite control and insulin secretion. According to Mosialou et al. (90), LCN2 binds to melanocortin receptor 4 (MC4R) of the paraventricular neurons in the hypothalamus and directly suppresses appetite. Moreover, the administration of LCN2 has been shown to improve glucose metabolism and energy expenditure in a mouse model (90). In patients with type 2 diabetes, higher serum LCN2 levels were associated with obesity, dyslipidemia, and insulin resistance. Therefore, LCN2 could serve as a biomarker for metabolic diseases (92). Under pathological conditions, LCN2 expression can be induced in a wide range of organs, including the kidney (93) and liver (94).
Expression levels of LCN2 have been found to be high in aggressive subtypes of cancer, including breast, pancreas, thyroid, and colon cancers (62, 64–66). In general, LCN2 may promote tumorigenesis by enhancing invasion, proliferation, and metastasis while reducing apoptosis. Some of these characteristics arise from the ability of LCN2 to facilitate iron uptake into cancer cells or its ability to form a heterodimer with matrix metalloprotease (MMP)-9, which contributes to tumor progression and metastasis (95). Studies by Yang et al. (62) and Bauer et al. (63) confirmed the presence of LCN2 in invasive breast cancer tissues and cell lines. LCN2 levels were increased in advanced stages of cancer and they were associated with decreased overall survival. Moniaux et al. (64) revealed higher LCN2 expression in neoplastic lesions of pancreatic tissue that typically develop into pancreatic cancer. In pancreatic cancer tissues, LCN2 expression was upregulated compared to tissues from healthy individuals (64, 96). According to Iannetti et al. (65), thyroid carcinoma-derived tissues also had higher LCN2 expression levels versus normal thyroid tissue. Increased expression of LCN2 in colon tumor tissues was found by Nielsen et al. (66). However, no significant differences in LCN2 expression between neoplastic and non-neoplastic tissues were noted. In metastasis, LCN2 was initially identified as a promoter to induce epithelial–mesenchymal transition (EMT) in breast cancer cells to promote tumor metastasis (62). In prostate cancer cells, LCN2 played an important role in facilitating cell migration and invasion by inducing EMT, leading to increased tumor invasion (67). However, disruption of the LCN2 gene suppressed primary mammary tumor formation in mice, while not reducing lung metastases (97). Thus, the role of LCN2 in metastasis may differ significantly between different cancer types. However, LCN2 could be a prognostic biomarker for this disease. In some types of cancer (especially breast cancer), the development of therapeutic agents targeting LCN2 may have major clinical implications for the treatment of metastasis (98). Table 1 summarizes relevant information related to this issue.
Metastatic bone disease leads to a significant decrease in quality of life and is strongly correlated with high morbidity and poor outcomes in cancer patients. Increasing evidence identified the bone marrow as a preferred metastatic site for several solid neoplasms, including breast, prostate, lung, and kidney tumors. Osteoblasts, osteocytes, and osteoclasts represent key regulators of bonehomeostasis, and their interactions with tumor cells are considered critical for metastatic colonization in the bone marrow. However, their role in remodeling the bone microenvironment, leading to a supportive niche for tumor cell proliferation and survival, needs to be evaluated. Findings from cell cultures, animal studies, and clinical trials have recently documented the importance of various bone-derived factors as prognostic biomarkers and potential therapeutic targets in bone metastasis. Large clinical studies involving metastatic/non-metastatic patients and non-cancer control subjects may bring crucial findings for more efficient therapies to combat metastasis. Further research aimed at better understanding the tumor-specific pathways associated with bone metastasis may identify potential tumor-specific therapeutic targets.
Conceptualization, MM and RO. Methodology, MM, VM and RO. Formal analysis, RB, VK, MB and NZ. Writing-original draft preparation, MM, VM, SC and RO. Writing-review and editing, MM, SC and RO. Visualization, RB, VK, MB and NZ. Supervision, MM and RO. Funding acquisition, RO. All authors contributed to the article and approved the submitted version.
This work was supported by the Ministry of Education, Science, Research and Sport of the Slovak Republic, grant number KEGA 012UKF-4/2023 and KEGA 034UKF-4/2022.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lu K-H, Lin C-W, Hsieh Y-H, Su S-C, Reiter RJ, Yang S-F. New insights into antimetastatic signaling pathways of melatonin in skeletomuscular sarcoma of childhood and adolescence. Cancer Metastasis Rev (2020) 39:303–20. doi: 10.1007/s10555-020-09845-2
2. Macedo F, Ladeira K, Pinho F, Saraiva N, Bonito N, Pinto L, et al. Bone metastases: an overview. Oncol Rev (2017) 11:321. doi: 10.4081/oncol.2017.321
3. Cecchini MG, Wetterwald A, van der PG, Thalmann GN. Molecular and biological mechanisms of bone metastasis. EAU Update Ser (2005) 3:214–26. doi: 10.1016/j.euus.2005.09.006
4. Jayarangaiah A, Kemp AK, Theetha Kariyanna P. (2022). StatPearls. Treasure Island (FL: StatPearls Publishing. Available at: http://www.ncbi.nlm.nih.gov/books/NBK507911/ (Accessed November 21, 2022).
5. Coleman RE. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat Rev (2001) 27:165–76. doi: 10.1053/ctrv.2000.0210
6. Bussard KM, Gay CV, Mastro AM. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev (2008) 27:41–55. doi: 10.1007/s10555-007-9109-4
7. Wong SK, Mohamad N-V, Giaze TR, Chin K-Y, Mohamed N, Ima-Nirwana S. Prostate cancer and bone metastases: The underlying mechanisms. Int J Mol Sci (2019) 20:2587. doi: 10.3390/ijms20102587
8. Shupp AB, Kolb AD, Mukhopadhyay D, Bussard KM. Cancer metastases to bone: Concepts, mechanisms, and interactions with bone osteoblasts. Cancers (Basel) (2018) 10:E182. doi: 10.3390/cancers10060182
9. Sun Y-X, Schneider A, Jung Y, Wang J, Dai J, Wang J, et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res (2005) 20:318–29. doi: 10.1359/JBMR.041109
10. Karlsson T, Sundar R, Widmark A, Landström M, Persson E. Osteoblast-derived factors promote metastatic potential in human prostate cancer cells, in part via non-canonical transforming growth factor β (TGFβ) signaling. Prostate (2018) 78:446–56. doi: 10.1002/pros.23489
11. Bussard KM, Venzon DJ, Mastro AM. Osteoblasts are a major source of inflammatory cytokines in the tumor microenvironment of bone metastatic breast cancer. J Cell Biochem (2010) 111:1138–48. doi: 10.1002/jcb.22799
12. Lee C, Whang YM, Campbell P, Mulcrone PL, Elefteriou F, Cho SW, et al. Dual targeting c-met and VEGFR2 in osteoblasts suppresses growth and osteolysis of prostate cancer bone metastasis. Cancer Lett (2018) 414:205–13. doi: 10.1016/j.canlet.2017.11.016
13. Maroni P, Bendinelli P. Bone, a secondary growth site of breast and prostate carcinomas: Role of osteocytes. Cancers (Basel) (2020) 12:1812. doi: 10.3390/cancers12071812
14. Selvaggi G, Scagliotti GV. Management of bone metastases in cancer: A review. Crit Rev Oncol Hematol (2005) 56:365–78. doi: 10.1016/j.critrevonc.2005.03.011
15. Taube T, Elomaa I, Blomqvist C, Beneton MN, Kanis JA. Histomorphometric evidence for osteoclast-mediated bone resorption in metastatic breast cancer. Bone (1994) 15:161–6. doi: 10.1016/8756-3282(94)90703-x
16. Southby J, Kissin MW, Danks JA, Hayman JA, Moseley JM, Henderson MA, et al. Immunohistochemical localization of parathyroid hormone-related protein in human breast cancer. Cancer Res (1990) 50:7710–6.
17. Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. RANK is essential for osteoclast and lymph node development. Genes Dev (1999) 13:2412–24. doi: 10.1101/gad.13.18.2412
18. Abou-Samra AB, Jüppner H, Force T, Freeman MW, Kong XF, Schipani E, et al. Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: A single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc Natl Acad Sci U.S.A. (1992) 89:2732–6. doi: 10.1073/pnas.89.7.2732
19. Hall CL, Kang S, MacDougald OA, Keller ET. Role of wnts in prostate cancer bone metastases. J Cell Biochem (2006) 97:661–72. doi: 10.1002/jcb.20735
20. Roodman GD. Mechanisms of bone metastasis. New Engl J Med (2004) 350:1655–64. doi: 10.1056/NEJMra030831
21. Keller ET, Zhang J, Cooper CR, Smith PC, McCauley LK, Pienta KJ, et al. Prostate carcinoma skeletal metastases: Cross-talk between tumor and bone. Cancer Metastasis Rev (2001) 20:333–49. doi: 10.1023/a:1015599831232
22. Venetis K, Piciotti R, Sajjadi E, Invernizzi M, Morganti S, Criscitiello C, et al. Breast cancer with bone metastasis: Molecular insights and clinical management. Cells (2021) 10:1377. doi: 10.3390/cells10061377
23. Furesi G, Rauner M, Hofbauer LC. Emerging players in prostate cancer–bone niche communication. Trends Cancer (2021) 7:112–21. doi: 10.1016/j.trecan.2020.09.006
24. Zoch ML, Clemens TL, Riddle RC. New insights into the biology of osteocalcin. Bone (2016) 82:42–9. doi: 10.1016/j.bone.2015.05.046
25. Rossi M, Battafarano G, Pepe J, Minisola S, Del Fattore A. The endocrine function of osteocalcin regulated by bone resorption: A lesson from reduced and increased bone mass diseases. Int J Mol Sci (2019) 20:4502. doi: 10.3390/ijms20184502
26. Komori T. Functions of osteocalcin in bone, pancreas, testis, and muscle. Int J Mol Sci (2020) 21:7513. doi: 10.3390/ijms21207513
27. Diaz−Franco MC, de Leon RFranco−Diaz, Villafan−Bernal JR. Osteocalcin−GPRC6A: An update of its clinical and biological multi−organic interactions (Review). Mol Med Rep (2019) 19:15–22. doi: 10.3892/mmr.2018.9627
28. Manolagas SC. Osteocalcin promotes bone mineralization but is not a hormone. PloS Genet (2020) 16:e1008714. doi: 10.1371/journal.pgen.1008714
29. Oury F, Sumara G, Sumara O, Ferron M, Chang H, Smith CE, et al. Endocrine regulation of Male fertility by the skeleton. Cell (2011) 144:796–809. doi: 10.1016/j.cell.2011.02.004
30. Mizokami A, Yasutake Y, Gao J, Matsuda M, Takahashi I, Takeuchi H, et al. Osteocalcin induces release of glucagon-like peptide-1 and thereby stimulates insulin secretion in mice. PloS One (2013) 8:e57375. doi: 10.1371/journal.pone.0057375
31. Mera P, Laue K, Ferron M, Confavreux C, Wei J, Galán-Díez M, et al. Osteocalcin signaling in myofibers is necessary and sufficient for optimum adaptation to exercise. Cell Metab (2016) 23:1078–92. doi: 10.1016/j.cmet.2016.05.004
32. Oury F, Khrimian L, Denny CA, Gardin A, Chamouni A, Goeden N, et al. Maternal and offspring pools of osteocalcin influence brain development and functions. Cell (2013) 155:228–41. doi: 10.1016/j.cell.2013.08.042
33. Moser SC, van der Eerden BCJ. Osteocalcin–a versatile bone-derived hormone(2019) (Accessed November 21, 2022).
34. Pietschmann P, Zielinski C, Woloszczuk W. Serum osteocalcin levels in breast cancer patients. J Cancer Res Clin Oncol (1989) 115:456–8. doi: 10.1007/BF00393337
35. Salem AM, Zohny SF, Abd El-Wahab MM, Hamdy R. Predictive value of osteocalcin and beta-CrossLaps in metastatic breast cancer. Clin Biochem (2007) 40:1201–8. doi: 10.1016/j.clinbiochem.2007.07.006
36. Xu J, Ma L, Wang D, Yang J. Uncarboxylated osteocalcin promotes proliferation and metastasis of MDA-MB-231 cells through TGF-β/SMAD3 signaling pathway. BMC Mol Cell Biol (2022) 23:18. doi: 10.1186/s12860-022-00416-7
37. Ye R, Pi M, Cox JV, Nishimoto SK, Quarles LD. CRISPR/Cas9 targeting of GPRC6A suppresses prostate cancer tumorigenesis in a human xenograft model. J Exp Clin Cancer Res (2017) 36:90. doi: 10.1186/s13046-017-0561-x
38. Kayed H, Bekasi S, Keleg S, Michalski CW, Giese T, Friess H, et al. BGLAP is expressed in pancreatic cancer cells and increases their growth and invasion. Mol Cancer (2007) 6:83. doi: 10.1186/1476-4598-6-83
39. Engblom C, Pfirschke C, Zilionis R, Da Silva Martins J, Bos SA, Courties G, et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils. Science (2017) 358:eaal5081. doi: 10.1126/science.aal5081
40. Pi M, Quarles LD. GPRC6A regulates prostate cancer progression. Prostate (2012) 72:399–409. doi: 10.1002/pros.21442
41. Gardner TA, Lee S-J, Lee S-D, Li X, Shirakawa T, Kwon DD, et al. Differential expression of osteocalcin during the metastatic progression of prostate cancer. Oncol Rep (2009) 21:903–8. doi: 10.3892/or_00000302
42. Nimptsch K, Rohrmann S, Nieters A, Linseisen J. Serum undercarboxylated osteocalcin as biomarker of vitamin K intake and risk of prostate cancer: A nested case-control study in the Heidelberg cohort of the European prospective investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev (2009) 18:49–56. doi: 10.1158/1055-9965.EPI-08-0554
43. Mansinho A, Ferreira AR, Casimiro S, Alho I, Vendrell I, Costa AL, et al. Levels of circulating fibroblast growth factor 23 (FGF23) and prognosis in cancer patients with bone metastases. Int J Mol Sci (2019) 20:E695. doi: 10.3390/ijms20030695
44. Weidner H, Baschant U, Lademann F, Ledesma Colunga MG, Balaian E, Hofbauer C, et al. Increased FGF-23 levels are linked to ineffective erythropoiesis and impaired bone mineralization in myelodysplastic syndromes. JCI Insight (2020) 5:137062. doi: 10.1172/jci.insight.137062
45. Suvannasankha A, Tompkins DR, Edwards DF, Petyaykina KV, Crean CD, Fournier PG, et al. FGF23 is elevated in multiple myeloma and increases heparanase expression by tumor cells. Oncotarget (2015) 6:19647–60. doi: 10.18632/oncotarget.3794
46. Lee EK, Martinez MCR, Blakely K, Santos KD, Hoang VC, Chow A, et al. FGF23: Mediator of poor prognosis in a sizeable subgroup of patients with castration-resistant prostate cancer presenting with severe hypophosphatemia? Med Hypotheses (2014) 83:482–7. doi: 10.1016/j.mehy.2014.08.005
47. Feng S, Wang J, Zhang Y, Creighton CJ, Ittmann M. FGF23 promotes prostate cancer progression. Oncotarget (2015) 6:17291–301. doi: 10.18632/oncotarget.4174
48. Vlot MC, Bijnsdorp I, den Heijer M, de Jonge R, van Moorselaar RJA, Heijboer AC. Plasma FGF23 is not elevated in prostate cancer. Clinica Chimica Acta (2018) 478:129–31. doi: 10.1016/j.cca.2017.12.035
49. Choudhary S, Ramasundaram P, Dziopa E, Mannion C, Kissin Y, Tricoli L, et al. Human ex vivo 3D bone model recapitulates osteocyte response to metastatic prostate cancer. Sci Rep (2018) 8:17975. doi: 10.1038/s41598-018-36424-x
50. Cotant CL, Rao PS. Elevated fibroblast growth factor 23 in a patient with metastatic prostate cancer and hypophosphatemia. Am J Kidney Dis (2007) 50:1033–6. doi: 10.1053/j.ajkd.2007.07.031
51. Chiam P, Tan HC, Bee YM, Chandran M. Oncogenic osteomalacia – hypophosphataemic spectrum from “benignancy” to “malignancy.” Bone (2013) 53:182–7. doi: 10.1016/j.bone.2012.11.040
52. Savva C, Adhikaree J, Madhusudan S, Chokkalingam K. Oncogenic osteomalacia and metastatic breast cancer: A case report and review of the literature. J Diabetes Metab Disord (2019) 18:267–72. doi: 10.1007/s40200-019-00398-y
53. Abramson M, Glezerman IG, Srinivasan M, Ross R, Flombaum C, Gutgarts V. Hypophosphatemia and FGF23 tumor-induced osteomalacia in two cases of metastatic breast cancer. Clin Nephrol (2021) 95:104–11. doi: 10.5414/CN110242
54. Wang X-T, He Y-C, Zhou S-Y, Jiang J, Huang Y-M, Liang Y-Z, et al. Bone marrow plasma macrophage inflammatory protein protein-1 alpha(MIP-1 alpha) and sclerostin in multiple myeloma: Relationship with bone disease and clinical characteristics. Leuk Res (2014) 38:525–31. doi: 10.1016/j.leukres.2014.02.010
55. Eda H, Santo L, Wein MN, Hu DZ, Cirstea DD, Nemani N, et al. Regulation of sclerostin expression in multiple myeloma by dkk-1: A potential therapeutic strategy for myeloma bone disease. J Bone Mineral Res (2016) 31:1225–34. doi: 10.1002/jbmr.2789
56. Terpos E, Christoulas D, Katodritou E, Bratengeier C, Gkotzamanidou M, Michalis E, et al. Elevated circulating sclerostin correlates with advanced disease features and abnormal bone remodeling in symptomatic myeloma: Reduction post-bortezomib monotherapy. Int J Cancer (2012) 131:1466–71. doi: 10.1002/ijc.27342
57. McDonald MM, Reagan MR, Youlten SE, Mohanty ST, Seckinger A, Terry RL, et al. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple. Blood (2017) 129:3452–64. doi: 10.1182/blood-2017-03-773341
58. Mendoza-Villanueva D, Zeef L, Shore P. Metastatic breast cancer cells inhibit osteoblast differentiation through the Runx2/CBFβ-dependent expression of the wnt antagonist, sclerostin. Breast Cancer Res (2011) 13:R106. doi: 10.1186/bcr3048
59. Yuen H-F, Chan Y-P, Cheung W-L, Wong Y-C, Wang X, Chan K-W. The prognostic significance of BMP-6 signaling in prostate cancer. Mod Pathol (2008) 21:1436–43. doi: 10.1038/modpathol.2008.94
60. Yavropoulou MP, van Lierop AH, Hamdy NAT, Rizzoli R, Papapoulos SE. Serum sclerostin levels in paget’s disease and prostate cancer with bone metastases with a wide range of bone turnover. Bone (2012) 51:153–7. doi: 10.1016/j.bone.2012.04.016
61. García-Fontana B, Morales-Santana S, Varsavsky M, García-Martín A, García-Salcedo JA, Reyes-García R, et al. Sclerostin serum levels in prostate cancer patients and their relationship with sex steroids. Osteoporos Int (2014) 25:645–51. doi: 10.1007/s00198-013-2462-y
62. Yang J, Bielenberg DR, Rodig SJ, Doiron R, Clifton MC, Kung AL, et al. Lipocalin 2 promotes breast cancer progression. Proc Natl Acad Sci (2009) 106:3913–8. doi: 10.1073/pnas.0810617106
63. Bauer M, Eickhoff JC, Gould MN, Mundhenke C, Maass N, Friedl A. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res Treat (2008) 108:389–97. doi: 10.1007/s10549-007-9619-3
64. Moniaux N, Chakraborty S, Yalniz M, Gonzalez J, Shostrom VK, Standop J, et al. Early diagnosis of pancreatic cancer: neutrophil gelatinase-associated lipocalin as a marker of pancreatic intraepithelial neoplasia. Br J Cancer (2008) 98:1540–7. doi: 10.1038/sj.bjc.6604329
65. Iannetti A, Pacifico F, Acquaviva R, Lavorgna A, Crescenzi E, Vascotto C, et al. The neutrophil gelatinase-associated lipocalin (NGAL), a NF-κB-regulated gene, is a survival factor for thyroid neoplastic cells. Proc Natl Acad Sci (2008) 105:14058–63. doi: 10.1073/pnas.0710846105
66. Nielsen BS, Borregaard N, Bundgaard JR, Timshel S, Sehested M, Kjeldsen L. Induction of NGAL synthesis in epithelial cells of human colorectal neoplasia and inflammatory bowel diseases. Gut (1996) 38:414–20. doi: 10.1136/gut.38.3.414
67. Lu Y, Dong B, Xu F, Xu Y, Pan J, Song J, et al. CXCL1-LCN2 paracrine axis promotes progression of prostate cancer via the src activation and epithelial-mesenchymal transition. Cell Communication Signaling (2019) 17:118. doi: 10.1186/s12964-019-0434-3
68. Erben RG. Pleiotropic actions of FGF23. Toxicol Pathol (2017) 45:904–10. doi: 10.1177/0192623317737469
69. Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiology-Renal Physiol (2009) 297:F282–91. doi: 10.1152/ajprenal.90742.2008
70. Ewendt F, Feger M, Föller M. Role of fibroblast growth factor 23 (FGF23) and αKlotho in cancer(2021) (Accessed November 22, 2022).
71. Blau JE, Collins MT. The PTH-vitamin d-FGF23 axis. Rev Endocr Metab Disord (2015) 16:165–74. doi: 10.1007/s11154-015-9318-z
72. Komaba H, Kaludjerovic J, Hu DZ, Nagano K, Amano K, Ide N, et al. Klotho expression in osteocytes regulates bone metabolism and controls bone formation. Kidney Int (2017) 92:599–611. doi: 10.1016/j.kint.2017.02.014
73. Kaludjerovic J, Komaba H, Sato T, Erben RG, Baron R, Olauson H, et al. Klotho expression in long bones regulates FGF23 production during renal failure. FASEB J (2017) 31:2050–64. doi: 10.1096/fj.201601036R
74. Aukes K, Forsman C, Brady NJ, Astleford K, Blixt N, Sachdev D, et al. Breast cancer cell-derived fibroblast growth factors enhance osteoclast activity and contribute to the formation of metastatic lesions. PloS One (2017) 12:e0185736. doi: 10.1371/journal.pone.0185736
75. Navid S, Fan C O, Flores-Villanueva P, Generali D, Li Y. The fibroblast growth factor receptors in breast cancer: from oncogenesis to better treatments. Int J Mol Sci (2020) 21:E2011. doi: 10.3390/ijms21062011
76. Zhou R, Guo Q, Xiao Y, Guo Q, Huang Y, Li C, et al. Endocrine role of bone in the regulation of energy metabolism. Bone Res (2021) 9:25. doi: 10.1038/s41413-021-00142-4
77. Vasiliadis ES, Evangelopoulos D-S, Kaspiris A, Benetos IS, Vlachos C, Pneumaticos SG. The role of sclerostin in bone diseases. J Clin Med (2022) 11:806. doi: 10.3390/jcm11030806
78. Compton JT, Lee FY. A review of osteocyte function and the emerging importance of sclerostin. J Bone Joint Surg Am (2014) 96:1659–68. doi: 10.2106/JBJS.M.01096
79. Li C, Wang W, Xie L, Luo X, Cao X, Wan M. Lipoprotein receptor–related protein 6 is required for parathyroid hormone–induced sost suppression. Ann N Y Acad Sci (2016) 1364:62–73. doi: 10.1111/nyas.12750
80. Liu W, Wang Z, Yang J, Wang Y, Li K, Huang B, et al. Osteocyte TSC1 promotes sclerostin secretion to restrain osteogenesis in mice. Open Biol (2019) 9:180262. doi: 10.1098/rsob.180262
81. Koide M, Kobayashi Y, Yamashita T, Uehara S, Nakamura M, Hiraoka BY, et al. Bone formation is coupled to resorption Via suppression of sclerostin expression by osteoclasts. J Bone Mineral Res (2017) 32:2074–86. doi: 10.1002/jbmr.3175
82. Kim SP, Da H, Li Z, Kushwaha P, Beil C, Mei L, et al. Lrp4 expression by adipocytes and osteoblasts differentially impacts sclerostin’s endocrine effects on body composition and glucose metabolism. J Biol Chem (2019) 294:6899–911. doi: 10.1074/jbc.RA118.006769
83. Lin X, Onda D-A, Yang C-H, Lewis JR, Levinger I, Loh K. Roles of bone-derived hormones in type 2 diabetes and cardiovascular pathophysiology. Mol Metab (2020) 40:101040. doi: 10.1016/j.molmet.2020.101040
84. Delgado-Calle J, Anderson J, Cregor MD, Condon KW, Kuhstoss SA, Plotkin LI, et al. Genetic deletion of sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia (2017) 31:2686–94. doi: 10.1038/leu.2017.152
85. Hudson BD, Hum NR, Thomas CB, Kohlgruber A, Sebastian A, Collette NM, et al. SOST inhibits prostate cancer invasion. PloS One (2015) 10:e0142058. doi: 10.1371/journal.pone.0142058
86. Schroll A, Eller K, Feistritzer C, Nairz M, Sonnweber T, Moser PA, et al. Lipocalin-2 ameliorates granulocyte functionality. Eur J Immunol (2012) 42:3346–57. doi: 10.1002/eji.201142351
87. Rucci N, Capulli M, Piperni SG, Cappariello A, Lau P, Frings-Meuthen P, et al. Lipocalin 2: A new mechanoresponding gene regulating bone homeostasis. J Bone Mineral Res (2015) 30:357–68. doi: 10.1002/jbmr.2341
88. Rebalka IA, Monaco CMF, Varah NE, Berger T, D’souza DM, Zhou S, et al. Loss of the adipokine lipocalin-2 impairs satellite cell activation and skeletal muscle regeneration. Am J Physiology-Cell Physiol (2018) 315:C714–21. doi: 10.1152/ajpcell.00195.2017
89. Yan Q-W, Yang Q, Mody N, Graham TE, Hsu C-H, Xu Z, et al. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes (2007) 56:2533–40. doi: 10.2337/db07-0007
90. Mosialou I, Shikhel S, Liu J-M, Maurizi A, Luo N, He Z, et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature (2017) 543:385–90. doi: 10.1038/nature21697
91. Costa D, Lazzarini E, Canciani B, Giuliani A, SpanÒ R, Marozzi K, et al. Altered bone development and turnover in transgenic mice over-expressing lipocalin-2 in bone. J Cell Physiol (2013) 228:2210–21. doi: 10.1002/jcp.24391
92. Abella V, Scotece M, Conde J, Gómez R, Lois A, Pino J, et al. The potential of lipocalin-2/NGAL as biomarker for inflammatory and metabolic diseases. Biomarkers (2015) 20:565–71. doi: 10.3109/1354750X.2015.1123354
93. Mishra J, Mori K, Ma Q, Kelly C, Yang J, Mitsnefes M, et al. Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin. JASN (2004) 15:3073–82. doi: 10.1097/01.ASN.0000145013.44578.45
94. Ye D, Yang K, Zang S, Lin Z, Chau H-T, Wang Y, et al. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J Hepatol (2016) 65:988–97. doi: 10.1016/j.jhep.2016.05.041
95. Santiago-Sánchez GS, Pita-Grisanti V, Quiñones-Díaz B, Gumpper K, Cruz-Monserrate Z, Vivas-Mejía PE. Biological functions and therapeutic potential of lipocalin 2 in cancer. Int J Mol Sci (2020) 21:4365. doi: 10.3390/ijms21124365
96. Gomez-Chou SB, Swidnicka-Siergiejko AK, Badi N, Chavez-Tomar M, Lesinski GB, Bekaii-Saab T, et al. Lipocalin-2 promotes pancreatic ductal adenocarcinoma by regulating inflammation in the tumor microenvironment. Cancer Res (2017) 77:2647–60. doi: 10.1158/0008-5472.CAN-16-1986
97. Berger T, Cheung CC, Elia AJ, Mak TW. Disruption of the Lcn2 gene in mice suppresses primary mammary tumor formation but does not decrease lung metastasis. Proc Natl Acad Sci (2010) 107:2995–3000. doi: 10.1073/pnas.1000101107
Keywords: osteocalcin, fibroblast growth factor 23, sclerostin, lipocalin 2, tumor bone metastasis
Citation: Martiniakova M, Mondockova V, Biro R, Kovacova V, Babikova M, Zemanova N, Ciernikova S and Omelka R (2023) The link between bone-derived factors osteocalcin, fibroblast growth factor 23, sclerostin, lipocalin 2 and tumor bone metastasis. Front. Endocrinol. 14:1113547. doi: 10.3389/fendo.2023.1113547
Received: 01 December 2022; Accepted: 13 February 2023;
Published: 28 February 2023.
Edited by:
Xiaomeng You, Brigham and Women’s Hospital and Harvard Medical School, United StatesReviewed by:
Zhanna Belaya, Endocrinology Research Center, RussiaCopyright © 2023 Martiniakova, Mondockova, Biro, Kovacova, Babikova, Zemanova, Ciernikova and Omelka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monika Martiniakova, bW1hcnRpbmlha292YUB1a2Yuc2s=; Radoslav Omelka, cm9tZWxrYUB1a2Yuc2s=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.