 Ziwei Zhang
Ziwei Zhang Qingxia Huang
Qingxia Huang Daqing Zhao
Daqing Zhao Fengmei Lian
Fengmei Lian Xiangyan Li
Xiangyan Li Wenxiu Qi
Wenxiu Qi- 1College of Traditional Chinese Medicine, Changchun University of Chinese Medicine, Changchun, China
- 2Key Laboratory of Active Substances and Biological Mechanisms of Ginseng Efficacy, Jilin Provincial Key Laboratory of Biomacromolecules of Chinese Medicine, Ministry of Education, Northeast Asia Research Institute of Traditional Chinese Medicine, Changchun University of Chinese Medicine, Changchun, China
- 3Research Center of Traditional Chinese Medicine, The Affiliated Hospital to Changchun University of Chinese Medicine, Changchun, Jilin, China
- 4Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
Diabetes mellitus (DM) is a metabolic disease characterized by chronic hyperglycaemia, with absolute insulin deficiency or insulin resistance as the main cause, and causes damage to various target organs including the heart, kidney and neurovascular. In terms of the pathological and physiological mechanisms of DM, oxidative stress is one of the main mechanisms leading to DM and is an important link between DM and its complications. Oxidative stress is a pathological phenomenon resulting from an imbalance between the production of free radicals and the scavenging of antioxidant systems. The main site of reactive oxygen species (ROS) production is the mitochondria, which are also the main organelles damaged. In a chronic high glucose environment, impaired electron transport chain within the mitochondria leads to the production of ROS, prompts increased proton leakage and altered mitochondrial membrane potential (MMP), which in turn releases cytochrome c (cyt-c), leading to apoptosis. This subsequently leads to a vicious cycle of impaired clearance by the body’s antioxidant system, impaired transcription and protein synthesis of mitochondrial DNA (mtDNA), which is responsible for encoding mitochondrial proteins, and impaired DNA repair systems, contributing to mitochondrial dysfunction. This paper reviews the dysfunction of mitochondria in the environment of high glucose induced oxidative stress in the DM model, and looks forward to providing a new treatment plan for oxidative stress based on mitochondrial dysfunction.
1 Introduction
DM is characterized by chronic and persistent hyperglycemia and causes macrovascular and microvascular complications, which is a major cause of end-stage renal disease, blindness, and amputation today (1–4). As of 2014, there were approximately 387 million people with DM worldwide (8.3% of the world’s population) and the number of people with DM is expected to increase to 640 million by 2040 (5, 6). The cost of diabetic microvascular complications is a major component of overall treatment costs and places a heavy burden on society and families (7).
The development and progression of DM has complex pathophysiological mechanisms, with inflammation, autophagy dysregulation, oxidative stress, and hemodynamic dysregulation all involved in the progression of the disease (8–11). In this regard, oxidative stress is an important part of disease development, and chronic hyperglycaemia promotes an imbalance between the production of free radicals and the scavenging capacity of the body’s antioxidant system. Mitochondria play an important role in the process of oxidative stress in cells. Mitochondria are the main site of cellular respiration in the body and the central organelle for the production of adenosine triphosphate (ATP) (12). Continuous high glucose stimulation leads to impaired mitochondrial electron transport and promotes the production of ROS, which in turn causes damage to the mitochondria themselves and mitochondrial DNA (mtDNA), leading to impaired ATP synthesis, apoptosis, and activation of downstream inflammatory and fibrotic signaling pathways, contributing to disease progression (13–15). Scientists are now proposing that improving the antioxidant capacity of cells may be an important strategy for treating DM and complications, with some experiments in animals and humans and some results, but the evidence-based clinical support for anti-oxidative stress therapies is still insufficient (16–18). Based on this, the aim of this review is to explore together the important role of mitochondria in the process of oxidative stress in DM through abnormal mitochondrial oxidative phosphorylation (OXPHOS) and mtDNA damage, and to outline the efforts made by present-day scientists towards repairing mitochondrial function in the hope of providing new ideas for future scientific experiments.
2 Oxidative stress and ROS
The concept of oxidative stress was first introduced by the German scientist Helmut Sies and refered to the imbalance of oxidants and antioxidants that can cause damage to the organism (19). The concept of “oxidative stress” was later extended to refer to a pathological phenomenon resulting from an imbalance between the production of free radicals and the scavenging function of the antioxidant system, and is closely linked to the development of diseases such as chronic obstructive pulmonary disease, Alzheimer’s disease, cancer, DM, hypertension and age-related diseases (20–26).
Structurally, free radicals are highly reactive substances containing at least one unpaired electron and are active derivatives of ROS and reactive nitrogen species (RNS), which are closely associated with the initiation of oxidative stress (27, 28). Both endogenous and exogenous stimulation lead to pathological increases in ROS and promote oxidative stress. Endogenous factors such as metabolic factors, mitochondrial damage, immune system dysregulation, and inflammatory products induce the production of ROS (29–32). Exogenous ionizing radiation, xenobiotics, ultraviolet light, alcohol abuse and smoking contribute to the onset and progression of aging and metabolic disease by promoting ROS production in the body (33–38) (Figure 1).
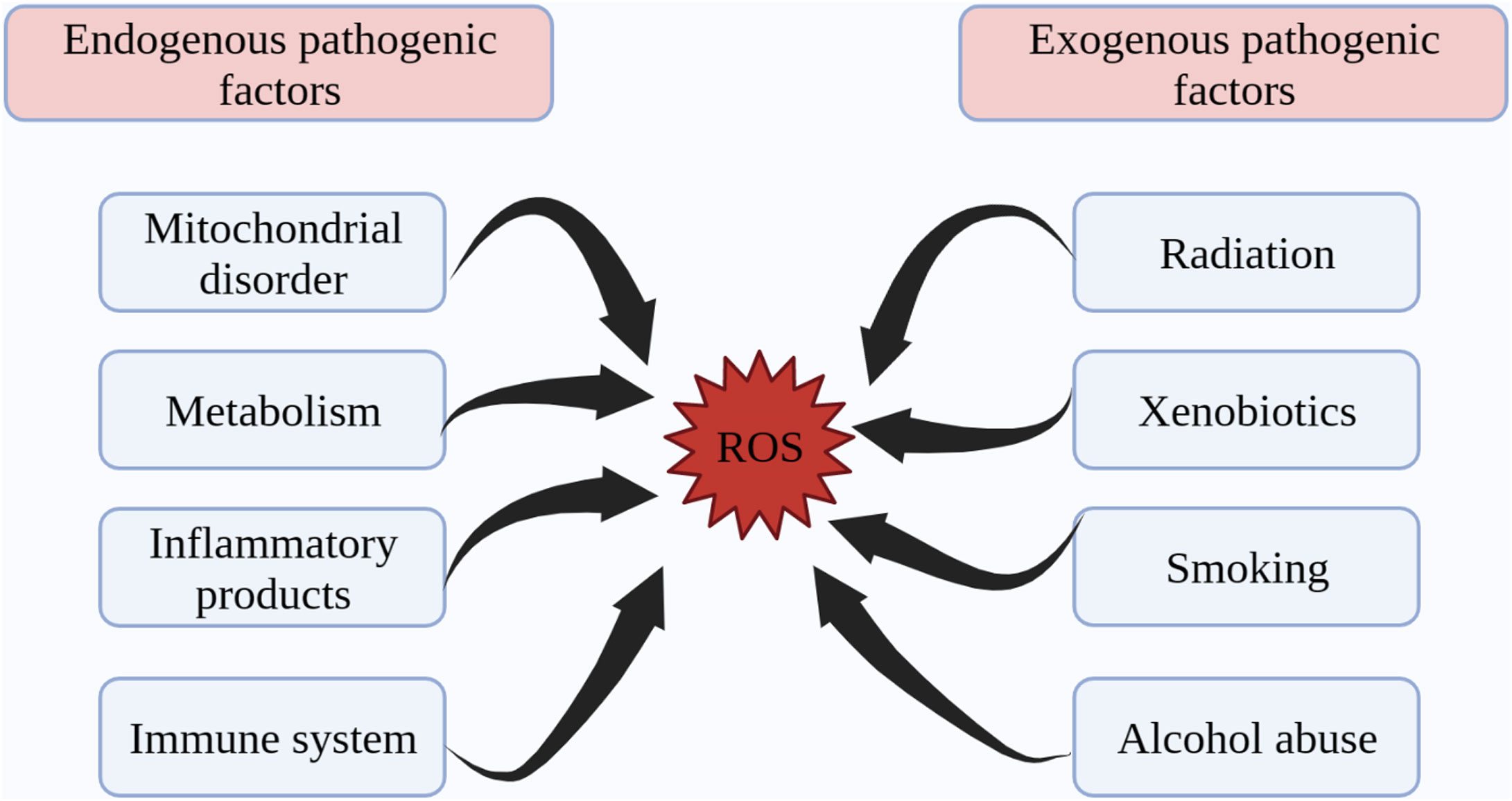
Figure 1 Endogenous and exogenous factors in ROS production. ROS generation is divided into two aspects: endogenous pathogenic factors and exogenous pathogenic factors. Endogenous pathogenic factors include mitochondrial disorder, metabolism, inflammatory products, immune system, etc. Exogenous pathogenic factors include radiation, xenobiotics, smoking, alcohol abuse, etc. ROS, reactive oxygen species.
The intracellular ROS is mainly composed of O2.- [reduced from O2 by electrons through the electron transfer chain (ETC)] and its derivatives. The three main reactive substances of ROS are superoxide anion (O2.-), hydroxyl radical (•OH) and hydrogen peroxide (H2O2), with •OH being the most reactive (39, 40). Superoxide dismutase (SOD), the first line of defense of the cellular antioxidant defense system, promotes the production of the superoxide O2.-intermediating H2O2: 2O2.-+2H+→H2O2+O2 (41). Subsequently reduced by catalase (CAT) to non-toxic H2O: 2H2O2→2H2O+O2; or catalyzed by glutathione peroxidase (GSH-Px) to produce H2O: 2GSH+H2O2→2H2O+GSSG, all of which complete the antioxidant scavenging process and constitute the antioxidant enzymatic response system of the organism, maintaining the intracellular redox balance (41, 42). Fe2+ and Cu+ are capable of redox reactions with unpaired electrons, destroying the structure and function of proteins, nucleic acids and lipids and cross-linking with these macromolecules to produce toxic substances (19). Activation of mitochondrial permeability transition pore (MPTP) promotes the release of ROS (43). Also, the vicious cycle of oxidative stress is facilitated by an increase in toxic substances such as malondialdehyde (MDA), and 4-hydroxy-2-nonenal (4-HNE), which are lipid peroxidation products (44, 45). Likewise, advanced glycosylation end products (AGEs) further promote ROS production, induce overexpression of endothelial angiopoietin (Ang)-2, promote cellular sensitivity to pro-inflammatory factors such as vascular cell-adhesion molecule (VCAM)-1, and contribute to the progression of diabetes-related vascular disease (46). AGEs are non-enzymatic glycosylated forms of free amino acids that result from the interaction of glucose with lipids or proteins, bind to receptors to promote inflammation and oxidative stress, and are important in contributing to glomerulosclerosis and mesangial hypertrophy in DKD (47–49). On the other hand, ROS acts as an agonist of NF-κB signaling pathway to initiate the activation of downstream inflammatory signaling pathways and promotes the release of inflammatory factors such as IL-1β, TNF-α, intercellular adhesion molecule-1 (ICAM-1) and monocyte chemotactic protein-1 (MCP-1) (50–54).
We know that mitochondria are the main site of ROS production and a target organelle for oxidative stress (55). The ETC is the central component of the mitochondria for functional operation, and the sequential transfer of electrons through the ETC to complex IV creates an electrochemical proton gradient that drives the F1F0 ATP synthase to produce ATP for OXPHOS (56). In addition, mitochondrial ROS production is increased and morphology is altered in high blood glucose environment of DM. Further, prolonged high glucose stimulation results in more electron donors such as NADH and FADH2 being produced in the tricarboxylic acid (TCA) cycle, and too many electron donors entering the ETC, leading to a maximum mitochondrial voltage gradient (57). Uncoupling proteins (UCPs) reduce ROS production by dissipating the proton motive force and increasing the rate of electron transfer in the ETC (58). At the same time, molecular oxygen reacts prematurely to produce superoxide O2.-, which exceeds the antioxidant scavenging capacity and leads to oxidative stress, causing impairment of cellular respiratory function, promoting apoptosis and ultimately leading to dysfunction of diseased tissues and systems (59, 60) (Figure 2). It is reported that NADH oxidoreductase (complex I) and cytochrome bc1 oxidoreductase (complex III) are the main sites for the production of ETC superoxide (61). In addition to this, much of the literature has further linked mitochondrial OXPHOS function and mtDNA damage closely to oxidative stress (55, 62). High glucose-induced oxidative stress and mitochondrial dysfunction interact to promote the progression of DM and its complications (63). One article focused on the relationship between diabetic renal tubular injury and mitochondrial dysfunction, contributing to increased ROS production and metabolic abnormalities such as abnormal mitochondrial autophagy, and the article suggested that mitochondrial dysfunction may contribute to early diabetic tubulopathy (64). In addition to mitochondria, cytoplasmic NADPH oxidase (Nox) is the main source of cytoplasmic ROS (55). Further, cytoplasmic ROS could increase mitochondrial ROS production by continuously damaging mitochondria (6). Overall, mitochondria remain the main source of endogenous ROS (43). Therefore, we will next focus on the relationship between mitochondria and oxidative stress in the next sub-section.
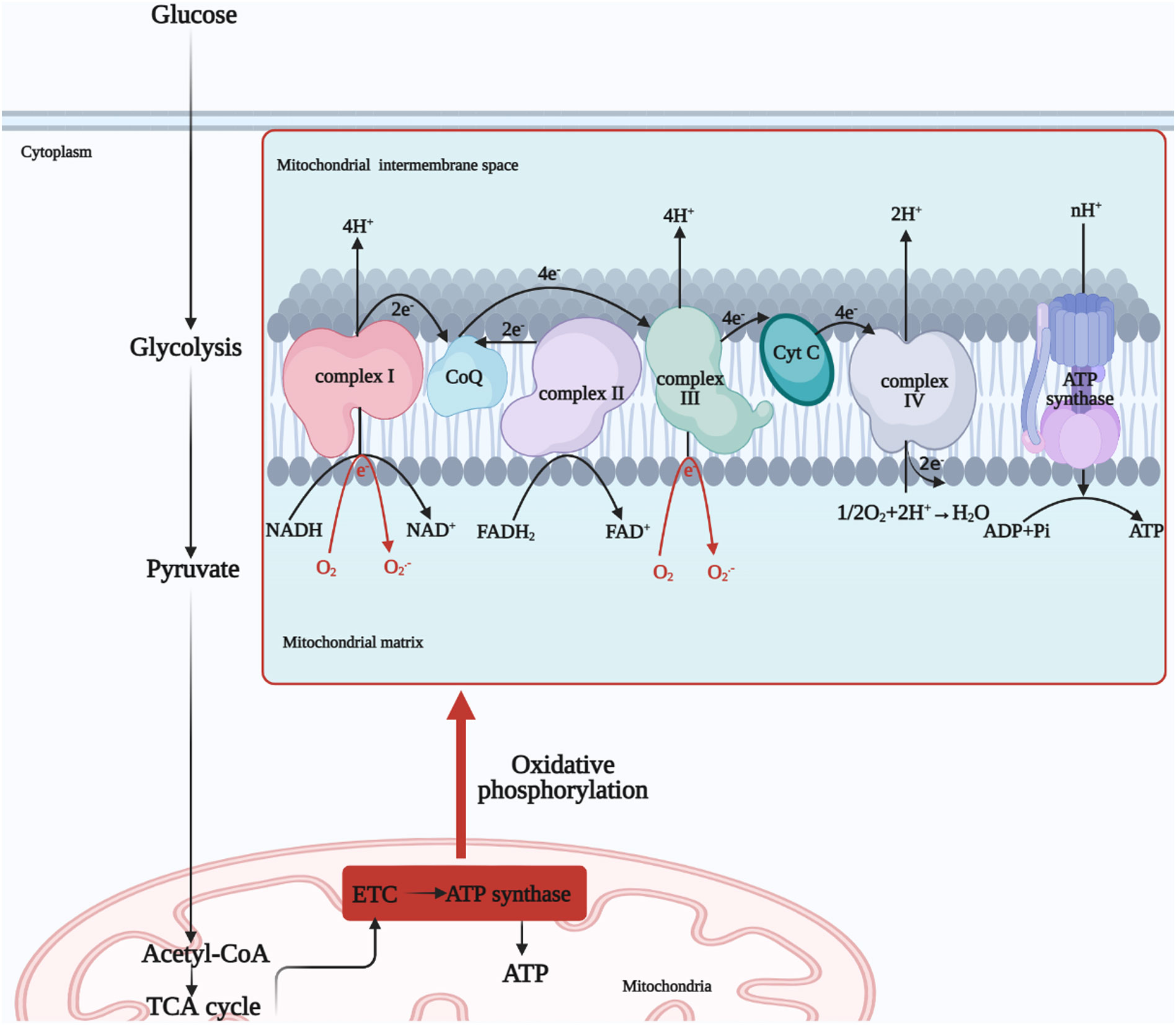
Figure 2 Mitochondrial OXPHOS process. Glucose undergoes glycolysis to produce pyruvate, which is oxidized to acetyl-coenzyme A or carboxylated to produce oxaloacetate, which enters the mitochondrial matrix for the TCA cycle. And finally NADH and FADH2, electron donors, undergo electron transfer by the ETC to produce H2O from O2 and ATP by the action of ATP synthase, completing OXPHOS process. NADH, nicotinamide adenine dinucleotide; FADH2, flavin adenine dinucleotide; ETC, electron transfer chain; TCA, tricarboxylic acid; ATP, adenosine triphosphate, ADP, adenosine diphosphate; OXPHOS, oxidative phosphorylation; Cyt C, cytochrome c; O2.-, superoxide anion.
It is important to note that the physiological level of ROS plays an important role in signaling, defense against infection and maintenance of redox homeostasis, only excessive ROS production leads to adverse effects of oxidative stress (65–67).
3 Physiological functions of mitochondria and oxidative damage
3.1 OXPHOS
In mitochondria, OXPHOS reactions use over 95% of O2 to produce ATP, and a small amount of ROS are produced daily as OXPHOS by-products (68). However, when the production of ROS exceeds the scavenging capacity of the antioxidant defense system, it can lead to oxidative stress.
Glucose is one of the main sources of energy for the body and is glycolysed in the cytoplasm to produce pyruvate, which is oxidized to acetyl-coenzyme A or carboxylated to produce oxaloacetate, which enters the mitochondrial matrix for the TCA cycle (69). Subsequent production of nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) provides the respiratory substrate for the ensuing OXPHOS process, which drives ATP production (70).
Mitochondria have a bilayer membrane structure, with the electron transport chain localized to the inner mitochondrial membrane (IMM), which is inlaid with four protein complexes, namely NADH oxidoreductase (complex I) (the largest subunits enzyme complex in the ETC), succinate dehydrogenase (complex II), cytochrome bc1 oxidoreductase (complex III) and cytochrome c oxidase (complex IV) (71). There are also two free-moving electron transport carriers on the ETC, the lipid-soluble Q and the water-soluble cyt-c, and these six components together form the ETC supercomplex (72).
Two electrons from the TCA metabolite NADH in complex I are passed to Q and reduced to QH2 (73). At the same time, the Fe-S cluster conformational change induces proton translocation and pumps four protons into the mitochondria intermembrane space (IMS) (74). Complex II is also an important carrier for the transfer of electrons, with FADH2 transferring electrons to Q via the Fe-S cluster. Protons are required for the reduction of Q, so there is no net proton increase in the IMS (75). Complex III transfers electrons of QH2 to cyt-c, cyt-c that gets electrons will be reduced, and the completion of the Q-cycle requires the pumping of four protons into the IMS (75). The reduced cytochrome carries electrons into complex IV, where they are eventually passed to the binuclear center of complex IV to complete the reduction of O2, also known as one molecule of O2 to produce two molecules of H2O (76). Eight protons in the matrix are consumed in this process, four of which are pumped into the IMS (77). At this point, the high concentration of protons in the IMS constitutes the electrochemical gradient responsible for the energy storage of the mitochondria (78). F1F0 ATP synthase, also known as complex V, transfers protons from the IMS to the matrix and controls the threshold of the MMP, at which point ATP synthase undergoes structural changes that promote ADP phosphorylation to produce ATP (79, 80). In addition, UCPs lower the membrane potential by transferring protons from the IMS to the matrix and uncouple from the ATP synthesis process, forming a switch for ATP synthesis with ATP synthase (81) (Figure 2).
3.2 Structure and function of mtDNA
mtDNA is a double-stranded circular structure (consisting of light and heavy strands), localizes in the mitochondrial matrix, closing to IMM (82). The human mtDNA is 16,569 bp in length and consists of 37 genes, 22 tRNAs, 2 rRNAs and a non-coding region displacement-loop (D-loop) (83, 84). However, the non-coding region controls the transcription and translation of mitochondrial proteins, but the high sequence mutagenicity in this region makes the mtDNA mutation rate approximately 10-20 times higher than nuclear DNA, with relevance to diseases such as aging and cancer (85–87). Moreover, due to the tight arrangement of genes in the ring structure of mtDNA, some genes overlap, and lack of histone protection, it is vulnerable to ROS generated by oxidative stress process, resulting in persistent damage to mtDNA (88, 89).
Mitochondria have a different DNA genetic system from nuclear DNA. mtDNA is responsible for encoding some of the mitochondrial proteins (such as the protein complexes that make up the ETC) and involved in mitochondrial biogenesis and signaling, with semi-autonomous genetic characteristics (82, 90, 91). Therefore, mtDNA is important for OXPHOS. Mutations in mtDNA and epigenetic changes can lead to blocked electron transport in the ETC, reducing ATP synthesis and promoting apoptosis.
Further, transcription and packaging factor (TFAM), and transcription elongation factor (TEFM), essential cofactors for mtDNA replication and transcription, play an important role in the assembly and distribution of mtDNA-protein complexes and are thought to alleviate insulin resistance-induced oxidative stress (90, 92–94).
In addition, the organism equips mtDNA with DNA repair systems, and base excision repair (BER) is considered to be the main repair mechanism (95). BER maintains the normal structure of mtDNA by eliminating base mismatches caused by methylation, oxidation and alkylation, and by cleaving, gap-filling and connecting the structure of mtDNA (96). In addition, mismatch repair (MMR), homologous recombination (HR) and non-homologous end joining (NHEJ) are also important repair pathways of mtDNA (97). It has been documented that base mismatches caused by elevated ROS can be recognized and excised by the BER pathway to maintain the functional and structural integrity of mtDNA (98–100). Similarly, in vitro and in vivo studies of the MMR pathway in high glucose environments have shown that high glucose environments induce damage to mtDNA and also impair the repair of the MMR pathway (101) (Figure 3).
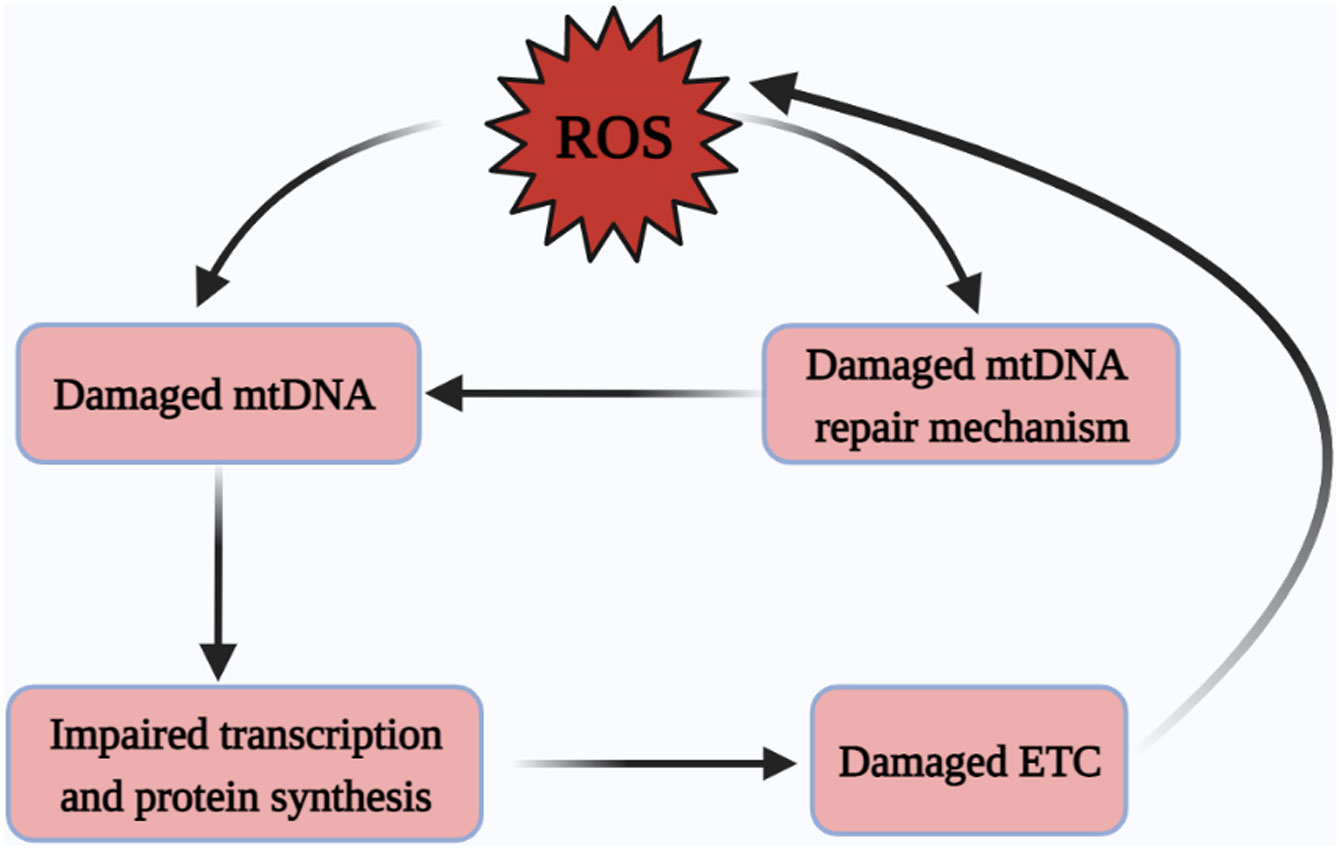
Figure 3 Excessive ROS damages mtDNA. ROS drives damage to mtDNA and the mtDNA repair systems, yet the damage of mtDNA repair systems is also an important contributor to mtDNA damage, followed by damage to mitochondrial protein-related transcription and synthesis pathways, ultimately leading to a vicious cycle of ETC abnormalities and subsequent the excessive generation of ROS. ROS, reactive oxygen species; mtDNA, mitochondrial DNA; ETC, electron transfer chain.
4 The relationship between oxidative stress-induced mitochondrial damage and diabetic microvascular complications
Oxidative stress, an important trigger for the development of DM and its complications, is characterized by excessive ROS production and intracellular oxidative damage. Oxidative stress can lead to alterations in mitochondrial morphology and function, inducing structural changes and functional abnormalities in macromolecules such as proteins, lipids and nucleic acids, ultimately leading to apoptosis and accelerating the progression of diabetic microvascular complications such as diabetic retinopathy (22, 102–105). In the following we will focus on mitochondria as an entry point to explore the impact of oxidative stress-induced mitochondrial dysfunction on diabetic microvascular complications.
4.1 Impaired OXPHOS and recovery in diabetic microvascular complications
OXPHOS occurs in mitochondria and is the main process involved in supplying energy for cellular respiration and ATP synthesis (106). ETC is the central element of the OXPHOS process, with the sequence of complexes (I-IV) working to achieve electron and proton transfer, creating MMP to store energy for the next work of ATP synthase (75). However, prolonged hyperglycaemic stimulation leads to abnormal electron transfer, resulting in increased production of ROS such as superoxide, inducing the onset of oxidative stress (107). Therefore, excessive ROS is one of the main factors leading to impaired OXPHOS function.
4.1.1 Diabetes kidney disease (DKD)
In high glucose induced podocytes, the superoxide levels were found to be increased while MMP expression and mitochondrial number were found to be decreased. However, overexpression of SIRT6 was shown to reverse this phenomenon (108). Dioscin effectively reduced blood glucose and markers of renal impairment in diabetic rats, reversed mitochondrial respiratory chain disorders, increased the activity of SOD and CAT antioxidant enzymes and reduced the level of ROS (109). Jujuboside A modulated mitochondrial respiratory chain complex protein expression in T2DM rats, improved respiratory chain function, reduced ROS levels, increased SOD, CAT and GPX expression, and downregulated apoptotic protein expression (110). In a study evaluating Abroma augusta L. (Malvaceae) leaf extract on T2DM-related nephropathy and cardiomyopathy in experimental rats, it was observed that redox homeostasis was disrupted, intracellular NAD and ATP levels were reduced and mitochondria-dependent apoptotic pathways were activated in T2DM state (111). Studies have shown that soluble klotho protein (referred to as rKL, known as an inhibitor of aging) reduced albuminuria, restored mitochondrial function and reduced ROS production in db/db mice. Moreover, in high glucose-induced mouse proximal tubular cells, rKL treatment alleviated OXPHOS impairment and induced mitochondrial repair via the PGC-1α-AMPK pathway (112). To investigate the effects of a high-fat diet on oxidative stress in wild-type and RAGE (receptors for AGEs) deficient mice, it was shown that RAGE can regulate mitochondrial respiratory chain function and oxidative stress in flounder muscle (113). Metformin, a classical hypoglycemic agent, promoted normalization of energy status and biochemical alterations, elevated ATP and lowered AMP, inhibited TNF-α and IL-6 pro-inflammatory gene expression, and exerted protective function of kidney in DKD rats (114). C3a induced mitochondrial fragmentation in podocytes, promoted mitochondrial depolarization, decreased SOD expression and increased ROS production, contributing to abnormal cellular energy metabolism, but this phenomenon could be inhibited by SS-31 (115). Knockdown of heat shock protein 60 (HSP60) in high-glucose-induced canine renal tubular cells showed that HSP60 regulated protein aggregation and ATP production in renal tubular cells (116). In high glucose and angiotensin II (ANG II)-induced HK-2 cells, increased p66Shc (promoter of apoptosis) and p-p66Shc were accompanied by increased ROS. The researchers made in-depth research and concluded that p66Shc mediated high glucose and ANG II-induced mitochondrial dysfunction via protein kinase C (PKC)-Β and peptidyl-prolyl isomerase (Pin1) pathways, decreased MMP, promoted cyt-c leakage and increased the apoptotic protein caspase-9 (117). In another study, one of the mechanisms by which p66Shc promoted DKD was that p66Shc promoted disruption of mitochondrial dynamics, enhanced Mfn1-Bak interactions leading to loss of mitochondrial voltage potential, cyt-c release, excessive ROS production and apoptosis (118). In contrast, coagulation protease activated protein C and normalized MMP through epigenetic inhibition of p66Shc (119); also Obacunone exhibited nephroprotective effects that inhibited oxidative stress and mitochondrial dysfunction (120). Purple Rice Husk improved mitochondrial function through the PGC-1α/SIRT3/SOD2 signaling pathway and reduced oxidative damage in renal tissue (121). Knockdown of the mitochondrial uncoupling protein UCP-2 increased uncoupling through adenine nucleotide transport proteins and reduced oxidative stress in the diabetic kidney in rat models (122). In STZ-induced diabetic mice, phillyrin reduced blood glucose and serum creatinine levels, increased Bcl-2/Bax ratio, reduced cyt-c leakage into the cytoplasm and inhibited apoptosis through the PI3K/Akt/GSK-3β signaling pathway (123). Genistein protected podocytes integrity, increased MMP, improved mitochondrial function and inflammatory status in rats with diabetic nephropathy by inhibiting the MAPK/NF-κB pathway (124). Telmisartan increased the MMP of glomerular endothelial cells induced by high glucose, and reduced the levels of 8-hydroxy-2-deoxyguanosine (8-OHdG) and MDA to alleviate oxidative stress (125). The complex I inhibitor rotenone (ROT) reduced ROS production and increased MMP and PGC-1α-controlled mitochondrial biogenesis in STZ and different inflammatory factor-induced mouse pancreatic β-cell line Min6 cells, suggesting that inhibition of complex I might be an effective strategy to protect β-cells in T1DM (126). Another study had shown that ROT could also correct over-activated biological processes, increasing the ratio of reduced glutathione (GSH) and nicotinamide adenine dinucleotide phosphate (NADPH) to its oxidized form, leading to redox balance (127). Resveratrol alleviated proteinuria, reduced ROS and MDA levels, restored SIRT1 and PGC-1α expression in kidney tissue. Resveratrol inhibited mitochondrial oxidative stress via SIRT1/PGC-1α, improved podocytes respiratory chain complex I and III activity, increased MMP and inhibited cyt-c release from mitochondria to the cytoplasm (128). In palmitic acid (PA) and oleic acid induced podocytes, PA was found to induce mitochondrial superoxide and H2O2 production (129). To investigate the role of SIRT3 deficiency on mitochondrial damage, researchers fed SIRT3-deficient mice a high-fat diet, resulting in mitochondrial dysfunction (involving abnormalities in OXPHOS, MMP and energy metabolism) and ultrastructural changes (130). In addition, a number of studies had also shown a close pathological link between impaired OXPHOS process and DKD (131–134).
The selective SIRT1 agonist BF175 was shown to prevent high glucose-induced mitochondrial damage and reduce superoxide production (135). Salvianolate effectively inhibited the generation of superoxide derived from NOX4 (mainly located in IMM) and reduced podocyte apoptosis (136). In the STZ-induced DKD rat model, the researchers observed a significant increase in blood creatinine and urine protein, as well as in ROS and MDA levels in the model group compared to the control group (137). In H2O2-induced HBZY-1 cells, Nepeta angustifolia inhibited H2O2 by increasing MMP, reducing ROS and MDA levels while inhibiting apoptosis (138). It had been shown that in addition to elevated biochemical parameters associated with kidney damage, STZ-induced diabetic rats also increased ROS production, reduced antioxidant defenses in vivo, and ultimately initiated mitochondria-dependent apoptosis (139). Increased ROS formation, elevated lipid peroxidation products and oxidative DNA damage, and mitochondrial apoptosis were observed in kidney tissue in STZ-induced diabetic mice, but dietary eicosapentaenoic acid inhibited this phenomenon by modulating hypoxia-inducible factor (HIF)-1α (140). Another study showed that Erythropoietin alleviated mitochondrial dysfunction, inhibited mitochondrial fragmentation and ROS production, and promoted autophagic flux in vitro (141). CAT deficiency increased ROS production and fibronectin expression in DKD mice and murine mesangial cells, demonstrating that endogenous catalase played an important role in the maintenance of mitochondrial function and protected the kidney from oxidative stress (142). Ferulic acid inhibited ROS production and apoptosis and induced autophagy in STZ-induced diabetic rats (143). In addition, other researchers found that Nox4 knockdown reduced NADPH oxidase activity, accompanied by reduction in high-glucose-induced superoxide, yet mitochondrial Nox4 expression was increased in the renal cortex of diabetic rats, demonstrating the role for Nox4 in the regulation of mitochondrial function (144). Adropin improved lipid metabolism and renal function in diabetic mice, regulated blood glucose and lipids, inhibited ROS production, improved lipid deposition and down-regulated lipoprotein expression (145). G Protein-Coupled Bile Acid Receptor TGR5 improved indicators of renal injury in db/db mice, upregulated regulators of mitochondrial biogenesis, reduced lipid accumulation and H2O2 production and increased SOD2 activity; similarly, similar results were observed in high glucose-induced podocytes (146).
4.1.2 Diabetic retinopathy (DR)
In a high glucose-induced retinal ganglion cells (RGC) model, Hesperidin (Citrus Flavonone) restored mitochondrial function, prevented loss of MMP and cyt-c release into the cytoplasm, prevented ROS production, increased intracellular levels of antioxidant enzymes and inhibited apoptosis (147). A study on metabolic memory of mitochondrial oxidative damage found that in primary rat retinal endothelial cells (rRECs) cultured in high glucose, MMP and cyt-c levels decreased and ROS levels increased in the model group compared to the control group as the duration of high glucose culture increased, suggesting that metabolic memory of mitochondrial oxidative damage can lead to DR (148). Another study on Berberine (BBR) showed that BBR alleviated oxidative stress (inhibited cyt-c leakage and ROS production and increased antioxidant enzyme levels) in diabetic rats and high glucose-induced Müller cells by inhibiting the NF-κB signaling pathway, thereby preventing DR (149). Leakage of cyt-c and increased accumulation of Bax in mitochondria in STZ-induced diabetic rats and high glucose cultured retinal endothelial cells and pericytes, which were inhibited by SOD and its mimics (150). In another study, in addition to demonstrating the protective effect of manganese superoxide dismutase (MnSOD) on the retina, it was also demonstrated that complex III might be a more significant source of superoxide compared to complex I (151). It had been suggested that MTP-131 (a novel mitochondrial targeting peptide) alleviated H2O2-induced oxidative stress in RGC-5 (blocking MMP depolarization and cyt-c release, reducing ROS production and preventing apoptosis) (152). NaHS (donor of H2S) blocked retinal abnormalities in diabetic rats and alleviated DR by inhibiting mitochondrial dysfunction and NF-κB activation (153).
In high glucose-induced and platelet-derived growth factor-induced retinal pigment epithelial cells, researchers found that SIRT3 knockdown led to epithelial-mesenchymal transition and migration of epithelial cells, which was alleviated by overexpression of SIRT3. Further studies revealed that the cause was knockdown of SIRT3 leading to overproduction of mitochondrial ROS, suggesting the role for SIRT3 in inhibiting mitochondrial oxidative stress (154). In a vitro study, 670 nm photobiomodulation reduced high glucose-induced ROS production and maintained mitochondrial integrity in rat Müller glial cells (155). In an STZ-induced mouse experiment, STZ induced an increase in cytoplasmic and mitochondrial ROS, accompanied by lipid peroxidation and apoptosis, and a decrease in GSH and GSH-Px as well as optic nerve activity and vitamin A levels, which could be reversed by selenium and resveratrol (156). Some scientists investigated the mechanism of oxidative stress induced by high glucose in RGC-5, and concluded that high glucose induced ROS production, disrupted mitochondrial mechanisms (MMP, mtDNA and mitochondrial mass damage) and antioxidant mechanisms, and triggered the production of downstream inflammatory factors and neurodegenerative markers (157). A study on green tea (Camellia sinensis) and antioxidant vitamins showed that green tea and vitamins reduced retinal superoxide production and that green tea improved inhibition of ETC and complex III activity, but promoted tissue collagen matrix glyco-oxidation (150).
4.1.3 Diabetic peripheral neuropathy (DPN)
Phosphocreatine (PCr, a high-energy phosphate compound) prevented oxidative stress and promoted normalization of mitochondrial function in vivo and vitro experiments: PCr acted on complex I and complex II of the mitochondrial respiratory chain to increase cellular respiration and reduce ROS, and might be a potential drug for the treatment of diabetes-related neurodegenerative diseases (158). Salvianolic Acid A (SalA) inhibited high glucose-induced mitochondrial damage in Schwann RSC96 cells by modulating nuclear factor erythroid 2-related factor 2 (Nrf2): SalA scavenged mitochondrial ROS, reduced MMP, increased ATP production and upregulated OXPHOS-related gene expression; and alleviated abnormal glucolipid metabolism in KK-Ay mice, exerting peripheral neuroprotective effects (159). In contrast, high glucose induction led to abnormal changes on mitochondrial superoxide, MMP and neurosynaptic growth in Neuro2a cells, STZ-induced abnormalities in motor/nerve conduction and neuroblood supply in diabetic rats, and polydatin improved mitochondrial dysfunction and biogenesis via SIRT1/Nrf2 (160). Long chain fatty acids induced mitochondrial dysfunction of Schwann cells, while overexpression of long chain acyl CoA synthetase 1 improved mitochondrial coupling efficiency, reduced proton leakage, and improved mitochondrial function (161). Human neuroblastoma SH-SY5Y cells exposed to high glucose levels reduced neuropil numbers, downregulated uncoupling protein (UCP) 3, increased MMP and ROS levels, while insulin-like growth factor type 1 normalized these changes (162).
An in vitro study of quercetin showed that quercetin reduced high glucose-induced ROS production in RSC96 cells and improved mitochondrial morphological abnormalities and DNA damage, as well as peripheral nerve hypofunction in lesioned mice (163). In high glucose-induced Schwann cells, puerarin inhibited ROS production and mitochondrial depolarization and prevented apoptosis (164). Additionally, it had also been shown that Fuzi protected Schwann cells induced by high glucose, prevented excessive ROS production and apoptosis, and had neuroprotective effects (165).
4.2 mtDNA damage and recovery in diabetic microvascular complications
4.2.1 DKD
Increased urinary 8-OHdG was detected in DKD-sensitive DBA/2J mice and human DKD specimens and showed a correlation between glomerular endothelin-1 receptor type A expression and increased mtDNA damage (166). The combination of dietary fenugreek (Trigonella foenum-graecum) seeds and onion (Allium cepa) was effective in reducing STZ-induced oxidative stress, lowering triglyceride and total cholesterol levels, reducing 8-OHdG and DNA fragmentation, and eliminating mtDNA deletions (167). In addition, salidroside alleviated renal fibrosis and kidney damage in DKD mice, and promoted the increase of mtDNA copy number and mitochondrial biogenesis (168).
4.2.2 DR
In high glucose-induced retinal endothelial cells, researchers found increased damage to mtDNA and DNA repair mechanisms and decreased expression of genes responsible for encoding the ETC protein complex, however, overexpression of MnTBAP or MnSOD suppressed this phenomenon (169). Similarly, another study also pointed out that high glucose-induced mtDNA damage led to excessive ROS production and further promoted mtDNA damage, leading to a vicious cycle of oxidative stress (170). Hydrogen sulfide is an endogenous gastransmitter signaling molecule with antioxidant properties, and its donor GYY4137 exhibited antioxidant effects in STZ-induced diabetic mice by resisting mtDNA damage, promoting Cytb transcription, limiting ROS production and inhibiting increased mitochondrial membrane permeability (171). An interesting study found that mtDNA and its repair/replication mechanism were significantly associated with the course of DM: early mtDNA repair/replication enzymes increased compensatorily, and as the disease progressed the repair/replication mechanism was disrupted and the mtDNA copy number decreased significantly (172).
4.2.3 DPN
A study comparing differences in mtDNA and transcript levels between diabetic and PGC-1α(-/-) diabetic mice found that PGC-1α(-/-) exacerbated neurological abnormalities in diseased mice, promoted mtDNA damage and protein oxidation, and led to more severe mitochondrial degeneration, demonstrating that modulation of PGC-1α may be a strategy for treating DPN (173). TFAM overexpression upregulated mtDNA and total TFAM levels, prevented the reduction of mtDNA copy number and inhibited motor and sensory nerve conduction abnormalities in diseased mice (174). A study on the neurological evaluation of 125 Italian T2DM patients noted that mtDNA was reduced in T2DM patients, this result was more significant in DPN patients and was associated with the MIR499A gene polymorphism (175).
4.3 Inactivation and recovery of antioxidant defense systems in diabetic microvascular complications
4.3.1 DKD
CD38 inhibitor apigenin upregulated NAD/NADH ratio and SIRT3-mediated mitochondrial antioxidant enzyme activity, while knockdown of CD38 inhibited SIRT3 activity, suggesting a correlation between CD38 and SIRT3 in oxidative stress mechanisms (176). In vitro experiments using high glucose-induced glomerular mesangial cells revealed that high glucose stimulated ROS production, decreased SOD and GSH levels, increased NADPH oxidase activity and promoted an increase in apoptotic factors which was also verified in diabetic rats (177). Antioxidant peptide SS31 inhibited the reduction of MnSOD and CAT activity, inhibited NADPH oxidase and NF-κB p65 activity in db/db mice and high glucose induced HK-2 cells (178). It had also been shown that exercise increased the expression of SOD and reduced oxidative damage (179). In addition, the activity of antioxidant enzymes in the body changed with the duration of diabetic hyperglycemia. The mRNA expression and activity of heme oxygenase-1 (HO-1) and MnSOD increased, and GSH-Px activity increased during short-term hyperglycemia; as the disease progressed the mRNA expression and activity of both decreased, accompanied by an increase in MDA and a decrease in GSH levels (180). The use of fluorofenidone in db/db mice showed that fluorofenidone alleviated oxidative stress-induced renal injury by blocking RAGE/AGEs/NOX and PKC/NOX signaling, down-regulating NADPH oxidase and up-regulating GSH-Px and SOD (181). In another study using STZ to create a model of DM in rats, MDA, CAT and GSH-Px were significantly different compared with the control group and tempol treatment restored GSH-Px levels (182). Intervention with carnosine in H2O2-induced HK-2 cells concluded that carnosine increased total SOD activity, decreased NOX4 expression and ROS levels, and alleviated oxidative stress (183). The use of honokiol in BTBR ob/ob mice with T2DM resulted in the conclusion that honokiol ameliorated renal damage and maintained mitochondrial function by activating SIRT3 and thereby restoring SOD2 and PGC-1α expression (184). In STZ-induced diabetic rats, Rap1 significantly ameliorated mitochondrial dysfunction and oxidative stress injury in renal tubular cells, modulated C/EBP-β binding to the endogenous PGC-1α promoter, and the interaction of PGC-1α with CAT or SOD (185).
4.3.2 DR
Exendin-4 (a glucagon-like protein) increased GSH and magnesium superoxide dismutase levels, decreased NADPH oxidase levels, inhibited ROS production and cyt-c release, and prevented apoptosis in high glucose-induced adult human retinal pigment epithelial-19 cells by inhibiting p66Shc expression and activation (186). In a study on the relationship between retinal neuronal apoptosis and MnSOD in diabetic rats, it was noted that apoptosis increased in diabetic rats at 8 and 12 weeks, and the number of RGC cells decreased at 12 weeks, while MnSOD activity and mRNA levels decreased at 4, 8 and 12 weeks, indicating a close relationship between MnSOD and RGC apoptosis (187). Similarly, two other studies had shown that MnSOD overexpression inhibited the increase in 8-OHdG and nitrotyrosine levels, prevented the decrease in GSH and total antioxidant capacity caused by DR (188, 189). An interesting study explored the response of knockdown of the Sigma 1 receptor (σ1RKO) on primary retinal Müller glial cells, showing that SOD1, CAT and GPX1 expression and protein levels were reduced in the σ1RKO group, as well as GSH and GSH/GSSG ratios, demonstrating that the neuroprotective effects of σ1R are related to the inhibition of oxidative stress (190).
4.3.3 DPN
Aldose reductase inhibitors corrected neurological and metabolic abnormalities, restored GSH and ascorbic acid levels, and inhibited lipid peroxidation in diabetic rats (191). Berberine (BBR) increased Nrf-2-mediated antioxidant defense system, ameliorated mitochondrial damage and neurotransmission abnormalities in diabetic rats, and upregulated PGC-1α-mediated mitochondrial biogenesis in high glucose-induced N2A cells, demonstrating the important role of BBR in DPN treatment (192).
5 Abnormalities in metabolic pathways of oxidative stress
High glucose-induced activation of the AGE, PKC, polyol and hexosamine pathways, as well as the formation of ROS in the mitochondria and cytoplasm, contribute to increased ROS production, and promote mitochondrial dysfunction and induce oxidative stress, mediating cellular dysfunction and accelerating the disease process (32, 193–197) (Figure 4). We have previously addressed the formation of ROS in the cytoplasm and mitochondria, so the following section focuses on the other four metabolic pathways.
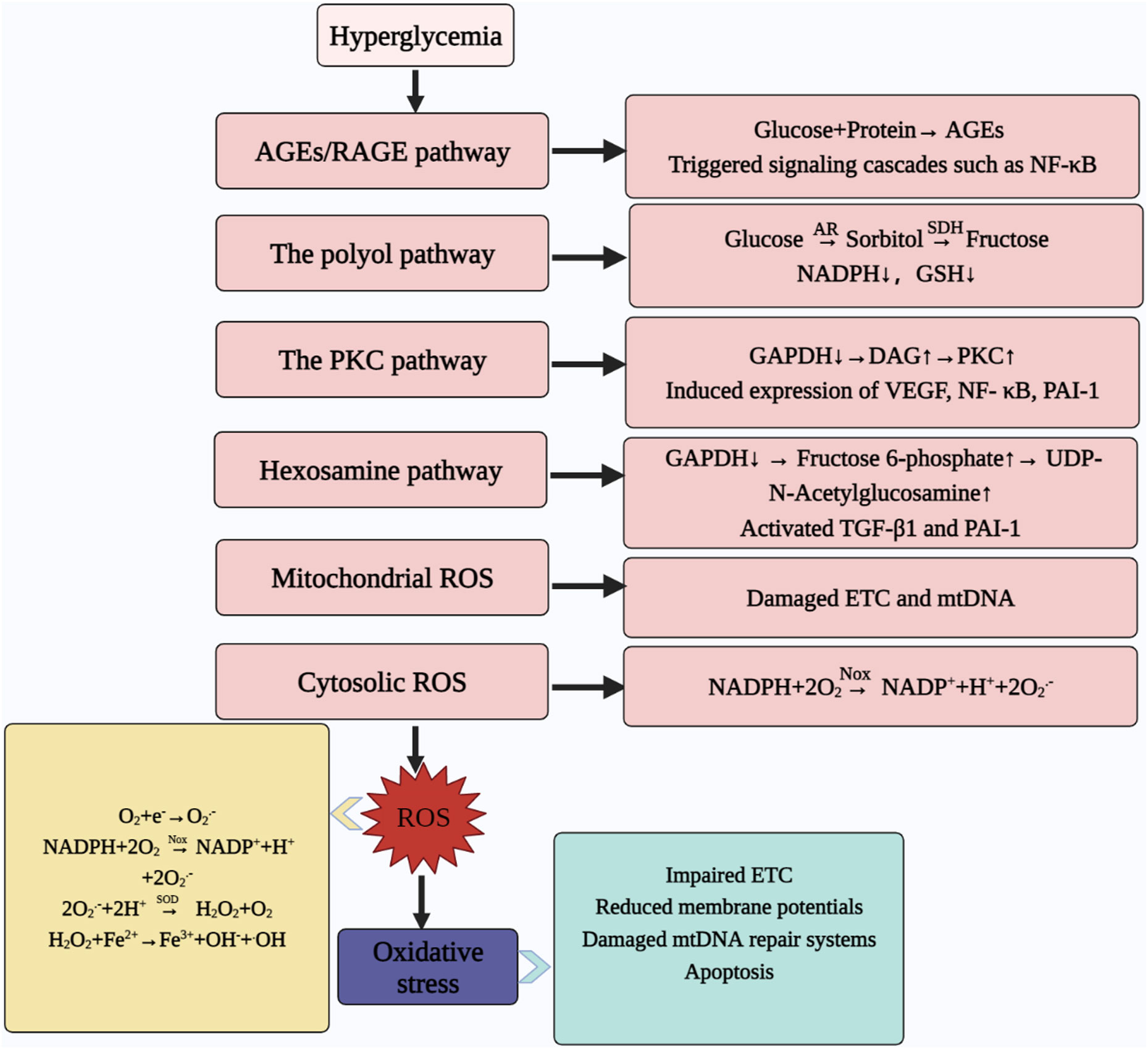
Figure 4 Abnormal metabolic pathways caused by hyperglycaemia. Hyperglycemia contributes to ROS production through the AGEs pathway, hexosamine pathway, PKC pathway, and polyol pathway; meanwhile, mitochondria and cytoplasm are also important sites for ROS production, which ultimately leads to oxidative stress. At the same time, oxidative stress can contribute to ETC abnormalities, reduce MMP, damage the mtDNA repair system and promote apoptosis. AGEs, advanced glycosylation end products; RAGE, the receptor for AGEs; NF-ΚB, nuclear factor kappa-light-chain-enhancer of activated B cells; AR, aldose reductase; SDH, sorbitol dehydrogenase; NADPH, nicotinamide adenine dinucleotide phosphate; GSH, reduced glutathione; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; DAG, diacylglycerol; PKC, protein kinase C; VEGF, vascular endothelial growth factor; PAI-1, plasminogen activator inhibitor-1; TGF-Β1, transforming growth factor; ROS, reactive oxygen species; ETC, electron transfer chain; mtDNA, mitochondrial DNA; NOX, NADPH oxidases; NADP+, nicotinamide adenine dinucleotide phosphate oxidized; O2.-, superoxide anion; SOD, superoxide dismutase; H2O2, hydrogen peroxide; •OH: hydroxyl radical.
5.1 AGEs/RAGE pathway
Non-enzymatic glycosylation of proteins and other macromolecules caused by prolonged high glucose levels, resulting in a series of dehydration and fracture reactions leading to the production of AGEs, resulting in abnormal protein structure and function, and consequently abnormal physiological function (198). AGEs promote oxidative stress by impairing the ETC to promote ROS formation (199). At the same time, the production of ROS can in turn stimulate the production of AGEs, thus creating a vicious circle (200). In addition, AGEs mediate the activation of downstream inflammatory and fibrotic signaling pathways by binding to cell surface receptors (RAGE) (201–203).
5.2 The polyol pathway
High glucose environment promoted activation of the polyol pathway (204). Glucose is converted to sorbitol by aldose reductase (AR) and subsequently oxidised to fructose by sorbitol dehydrogenase (SDH), during which NADPH is consumed as an electron donor (205). However, the reduction of the antioxidant GSH is dependent on NADPH, and the high glucose state accelerates the depletion of NADPH and reduces the antioxidant capacity of the body (205). At the same time, sorbitol can increase the osmotic pressure of cells, or act as a precursor substance for the formation of AGEs to promote the body’s sensitivity to oxidative stress, leading to DPN or DR (23, 206, 207). In addition to this, some researchers verified the relationship between AR and NLRP3 inflammasome: AR inhibitors inhibited the activation of NLRP3 inflammasome, reduced the production of inflammatory factors and mitigated the production of ROS during oxidative stress. It proved that AR participated in the innate immune response induced by NLRP3 inflammasome (208, 209).
5.3 The PKC pathway
High glucose promotes increased glycolysis, leading to greater diacylglycerol (DAG) production, while inhibition of the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) increases DAG activity and activates the PKC pathway (210). Activation of the PKC pathway is often accompanied by increased production of inflammatory factors and vascular endothelial growth factor (VEGF), and is closely associated with the development of diabetic complications (193, 211–215).
5.4 The hexosamine pathway
The hexosamine pathway is one of the pathways that promote the development of DM and its complications (195). Similarly, high glucose acts as a trigger switch for ROS production, resulting in the inhibition of GAPDH activity and the conversion of increased fructose-6-phosphate to the end product diphosphate uracil-N-acetylglucosamine (UDP-GlcNAc) (216). This is accompanied by an increase in ROS and fibrogenic factors downstream of the pathway, causing oxidative stress in mitochondria and is closely associated with thickening of the basement membrane of DKD (23, 217, 218).
6 Discussion
Oxidative stress is an imbalance in the redox state of the body, where excessive production of free radicals or damage to the antioxidant system, leads to a pathological outcome that is closely linked to the development of diseases such as cancer and metabolic disorders (219). ROS is a major component of free radicals, mainly produced in small amounts during OXPHOS in mitochondria, and plays an important role in cell signaling, cell proliferation and antibacterial immunity (220, 221).
However, the prolonged and persistent hyperglycaemic state of DM leads to an increase in cellular respiratory substrates entering the mitochondria, with excess electron donors impairing ETC, contributing to ROS production, mediating the breakdown of the proton electrochemical gradient, impaired MMP, increased cyt-c leakage and inadequate ATP synthesis (56). Due to the lack of histone protection of mtDNA, the high mutability of the non-coding region and the restriction of its loop structure, ROS can further damage mtDNA, leading to a reduction in the copy number of mtDNA, damage the genes responsible for encoding some mitochondrial proteins and impair the function of mitochondria (87, 222, 223). At the same time, the increased ROS can damage the repair system of mtDNA, further deepening the damage to mtDNA and causing functional impairment of mitochondria (224, 225). In addition, the instability of ROS encourages cross-linking with macromolecular proteins, DNA and lipids, altering the structure and function of macromolecules and having toxic effects, further affecting cell function (226). However, ROS can also mediate the activation of downstream signaling pathways such as inflammation and fibrosis, leading to the progression of diabetic microvascular complications such as DKD and DPN (227–229).
Studies have shown that some herbal active ingredients (puerarin, polydatin, quercetin, etc.), vitamin C, vitamin E, α-lipoic acid are important antioxidant strategies (160, 163, 164, 230–233). Targeting mitochondria to overexpress catalase in mice extends lifespan and alleviates oxidative stress in diseases such as metabolic syndrome (234). A clinical trial of the drug elamipretide (a mitochondrial tetrapeptide that interacts with cardiolipin) showed that elamipretide significantly improved clinical symptoms and skeletal muscle performance in Barth syndrome (235). In addition, animal models have demonstrated that enzymatic antioxidants mimics (SOD mimics, GPX mimics and CAT mimics) can scavenge superoxide and inhibit oxidative stress (236–238). Recent studies have shown that bioadhesive hydrogel can promote oral wound healing in DM rats; novel nanoparticle can accelerate wound healing in DM and is an emerging and effective treatment strategy (239, 240). Some combinations of antioxidants have also been shown to have antioxidant effects (241–243). The relevant literatures state that the combinations of ferulic acid and metformin have been shown to improve DM (244). And the combinations of superoxide dismutase, α-lipoic acid, acetyl-L-carnitine, and vitamin B12 have been shown to improve sural nerve conduction velocity, amplitude and pain in patients with DPN (233). Therefore, antioxidants play a positive therapeutic role in the treatment of DM.
However, many antioxidants suffer from poor solubility, unstable storage and gastrointestinal degradation, thus limiting the use of oxidants in clinical practice (245). In recent years antioxidant drugs have mainly focused on animal studies and have not been adequately tested in clinical trials, therefore, poorly supported by clinical data. The few drugs that have been clinically studied have not yielded satisfactory results either, and achieving effective drug concentrations in the body is an important issue for modern science. In addition to this, mtDNA, a key structure involved in oxidative stress, is under-researched for drugs targeting mtDNA, leading to a lack of development of antioxidant drugs. We hope to be able to characterize mitochondrial dysfunction in the high glucose state and look forward to providing a bit of new ideas for future experimental studies.
7 Conclusion
Hyperglycemia causes redox imbalance and massive production of ROS leading to impairment of OXPHOS, mtDNA function, mitochondrial dysfunction and oxidation of macromolecules and in turn accelerates apoptosis and disease progression. Therefore, oxidative stress is an important mechanism that promotes the development of DM and its complications. Targeted development of antioxidants and the combination of multiple acting antioxidant components may be a strategy for the treatment of DM.
Author contributions
ZZ is responsible for writing and the conception of the article, QH is responsible for drawing and document sorting, and DZ is responsible for sorting out the ideas of the article. FL, XL, and WQ are responsible for controlling the overall quality of the article and revising the article. All authors contributed to the article and approved the submitted version.
Funding
Authors’ work has been supported by Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (No: ZYYCXTD-D-202001), and 2015 Traditional Chinese Medicine Scientific Research (No: 201507001-11).
Acknowledgments
At the same time, we would like to thank the BioRender app for its help in creating figures of the article (https://app.biorender.com/gallery).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: Challenges, progress, and possibilities. Clin J Am Soc Nephrol (2017) 12(12):2032–45. doi: 10.2215/cjn.11491116
2. Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet (2010) 376(9735):124–36. doi: 10.1016/s0140-6736(09)62124-3
3. Sloan G, Selvarajah D, Tesfaye S. Pathogenesis, diagnosis and clinical management of diabetic sensorimotor peripheral neuropathy. Nat Rev Endocrinol (2021) 17(7):400–20. doi: 10.1038/s41574-021-00496-z
4. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature (2001) 414(6865):813–20. doi: 10.1038/414813a
5. Klein KR, Buse JB. The trials and tribulations of determining Hba(1c) targets for diabetes mellitus. Nat Rev Endocrinol (2020) 16(12):717–30. doi: 10.1038/s41574-020-00425-6
6. Kowluru RA, Kowluru A, Mishra M, Kumar B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res (2015) 48:40–61. doi: 10.1016/j.preteyeres.2015.05.001
7. Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, et al. Idf diabetes atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract (2017) 128:40–50. doi: 10.1016/j.diabres.2017.03.024
8. Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity (2022) 55(1):31–55. doi: 10.1016/j.immuni.2021.12.013
9. Kitada M, Koya D. Autophagy in metabolic disease and ageing. Nat Rev Endocrinol (2021) 17(11):647–61. doi: 10.1038/s41574-021-00551-9
10. Zhang P, Li T, Wu X, Nice EC, Huang C, Zhang Y. Oxidative stress and diabetes: Antioxidative strategies. Front Med (2020) 14(5):583–600. doi: 10.1007/s11684-019-0729-1
11. Agnoletti D, Lieber A, Zhang Y, Protogerou AD, Borghi C, Blacher J, et al. Central hemodynamic modifications in diabetes mellitus. Atherosclerosis (2013) 230(2):315–21. doi: 10.1016/j.atherosclerosis.2013.07.054
12. Ng MYW, Wai T, Simonsen A. Quality control of the mitochondrion. Dev Cell (2021) 56(7):881–905. doi: 10.1016/j.devcel.2021.02.009
13. Faria A, Persaud SJ. Cardiac oxidative stress in diabetes: Mechanisms and therapeutic potential. Pharmacol Ther (2017) 172:50–62. doi: 10.1016/j.pharmthera.2016.11.013
14. Fetterman JL, Holbrook M, Westbrook DG, Brown JA, Feeley KP, Bretón-Romero R, et al. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovasc Diabetol (2016) 15:53. doi: 10.1186/s12933-016-0372-y
15. Yaribeygi H, Sathyapalan T, Atkin SL, Sahebkar A. Molecular mechanisms linking oxidative stress and diabetes mellitus. Oxid Med Cell Longev (2020) 2020:8609213. doi: 10.1155/2020/8609213
16. Sheweita SA, Mashaly S, Newairy AA, Abdou HM, Eweda SM. Changes in oxidative stress and antioxidant enzyme activities in streptozotocin-induced diabetes mellitus in rats: role of alhagi maurorum extracts. Oxid Med Cell Longev (2016) 2016:5264064. doi: 10.1155/2016/5264064
17. Du L, Wang L, Wang B, Wang J, Hao M, Chen YB, et al. A novel compound Ab38b attenuates oxidative stress and ecm protein accumulation in kidneys of diabetic mice through modulation of Keap1/Nrf2 signaling. Acta Pharmacol Sin (2020) 41(3):358–72. doi: 10.1038/s41401-019-0297-6
18. Shen W, Teo KYC, Wood JPM, Vaze A, Chidlow G, Ao J, et al. Preclinical and clinical studies of photobiomodulation therapy for macular oedema. Diabetologia (2020) 63(9):1900–15. doi: 10.1007/s00125-020-05189-2
19. Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem (2017) 86:715–48. doi: 10.1146/annurev-biochem-061516-045037
20. Fratta Pasini AM, Stranieri C, Ferrari M, Garbin U, Cazzoletti L, Mozzini C, et al. Oxidative stress and nrf2 expression in peripheral blood mononuclear cells derived from copd patients: an observational longitudinal study. Respir Res (2020) 21(1):37. doi: 10.1186/s12931-020-1292-7
21. Plascencia-Villa G, Perry G. Preventive and therapeutic strategies in alzheimer's disease: focus on oxidative stress, redox metals, and ferroptosis. Antioxid Redox Signal (2021) 34(8):591–610. doi: 10.1089/ars.2020.8134
22. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discovery (2013) 12(12):931–47. doi: 10.1038/nrd4002
23. Ighodaro OM. Molecular pathways associated with oxidative stress in diabetes mellitus. BioMed Pharmacother (2018) 108:656–62. doi: 10.1016/j.biopha.2018.09.058
24. Kang Q, Yang C. Oxidative stress and diabetic retinopathy: molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol (2020) 37:101799. doi: 10.1016/j.redox.2020.101799
25. Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, et al. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res (2010) 107(1):106–16. doi: 10.1161/circresaha.109.214601
26. Skulachev VP, Anisimov VN, Antonenko YN, Bakeeva LE, Chernyak BV, Erichev VP, et al. An attempt to prevent senescence: A mitochondrial approach. Biochim Biophys Acta (2009) 1787(5):437–61. doi: 10.1016/j.bbabio.2008.12.008
27. Zarkovic N. Roles and functions of ros and rns in cellular physiology and pathology. Cells (2020) 9(3). doi: 10.3390/cells9030767
28. Liochev SI. Reactive oxygen species and the free radical theory of aging. Free Radic Biol Med (2013) 60:1–4. doi: 10.1016/j.freeradbiomed.2013.02.011
29. Liu S, Li L, Lou P, Zhao M, Wang Y, Tang M, et al. Elevated branched-chain α-keto acids exacerbate macrophage oxidative stress and chronic inflammatory damage in type 2 diabetes mellitus. Free Radic Biol Med (2021) 175:141–54. doi: 10.1016/j.freeradbiomed.2021.08.240
30. Wu T, Liang X, Liu X, Li Y, Wang Y, Kong L, et al. Induction of ferroptosis in response to graphene quantum dots through mitochondrial oxidative stress in microglia. Part Fibre Toxicol (2020) 17(1):30. doi: 10.1186/s12989-020-00363-1
31. Wu Z, Huang H, Han Q, Hu Z, Teng XL, Ding R, et al. Senp7 senses oxidative stress to sustain metabolic fitness and antitumor functions of Cd8+ t cells. J Clin Invest (2022) 132(7). doi: 10.1172/jci155224
32. Sul OJ, Ra SW. Quercetin prevents lps-induced oxidative stress and inflammation by modulating Nox2/Ros/Nf-kb in lung epithelial cells. Molecules (2021) 26(22). doi: 10.3390/molecules26226949
33. Ruzza P, Honisch C, Hussain R, Siligardi G. Free radicals and ros induce protein denaturation by uv photostability assay. Int J Mol Sci (2021) 22(12). doi: 10.3390/ijms22126512
34. Rai Y, Anita, Kumari N, Singh S, Kalra N, Soni R, et al. Mild mitochondrial uncoupling protects from ionizing radiation induced cell death by attenuating oxidative stress and mitochondrial damage. Biochim Biophys Acta Bioenerg (2021) 1862(1):148325. doi: 10.1016/j.bbabio.2020.148325
35. An R, Wang X, Yang L, Zhang J, Wang N, Xu F, et al. Polystyrene microplastics cause granulosa cells apoptosis and fibrosis in ovary through oxidative stress in rats. Toxicology (2021) 449:152665. doi: 10.1016/j.tox.2020.152665
36. Yan T, Zhao Y. Acetaldehyde induces phosphorylation of dynamin-related protein 1 and mitochondrial dysfunction Via elevating intracellular ros and Ca(2+) levels. Redox Biol (2020) 28:101381. doi: 10.1016/j.redox.2019.101381
37. Madhu NR, Sarkar B, Slama P, Jha NK, Ghorai SK, Jana SK, et al. Effect of environmental stressors, xenobiotics, and oxidative stress on male reproductive and sexual health. Adv Exp Med Biol (2022) 1391:33–58. doi: 10.1007/978-3-031-12966-7_3
38. Lee J, Kim MH, Kim H. Anti-oxidant and anti-inflammatory effects of astaxanthin on gastrointestinal diseases. Int J Mol Sci (2022) 23(24). doi: 10.3390/ijms232415471
39. Nakai K, Tsuruta D. What are reactive oxygen species, free radicals, and oxidative stress in skin diseases? Int J Mol Sci (2021) 22(19). doi: 10.3390/ijms221910799
40. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell (2012) 48(2):158–67. doi: 10.1016/j.molcel.2012.09.025
41. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ (2015) 22(3):377–88. doi: 10.1038/cdd.2014.150
42. Poprac P, Jomova K, Simunkova M, Kollar V, Rhodes CJ, Valko M. Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol Sci (2017) 38(7):592–607. doi: 10.1016/j.tips.2017.04.005
43. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (Ros) and ros-induced ros release. Physiol Rev (2014) 94(3):909–50. doi: 10.1152/physrev.00026.2013
44. Del Rio D, Stewart AJ, Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis (2005) 15(4):316–28. doi: 10.1016/j.numecd.2005.05.003
45. Dalleau S, Baradat M, Guéraud F, Huc L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (Hne) in the balance. Cell Death Differ (2013) 20(12):1615–30. doi: 10.1038/cdd.2013.138
46. Yao D, Taguchi T, Matsumura T, Pestell R, Edelstein D, Giardino I, et al. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of Msin3a. J Biol Chem (2007) 282(42):31038–45. doi: 10.1074/jbc.M704703200
47. Lee J, Yun JS, Ko SH. Advanced glycation end products and their effect on vascular complications in type 2 diabetes mellitus. Nutrients (2022) 14(15). doi: 10.3390/nu14153086
48. Anil Kumar P, Welsh GI, Saleem MA, Menon RK. Molecular and cellular events mediating glomerular podocyte dysfunction and depletion in diabetes mellitus. Front Endocrinol (Lausanne) (2014) 5:151. doi: 10.3389/fendo.2014.00151
49. Rabbani N, Thornalley PJ. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int (2018) 93(4):803–13. doi: 10.1016/j.kint.2017.11.034
50. Wu M, Yang Z, Zhang C, Shi Y, Han W, Song S, et al. Inhibition of Nlrp3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism (2021) 118:154748. doi: 10.1016/j.metabol.2021.154748
51. Li M, Deng L, Xu G. Mettl14 promotes glomerular endothelial cell injury and diabetic nephropathy Via M6a modification of α-klotho. Mol Med (2021) 27(1):106. doi: 10.1186/s10020-021-00365-5
52. Su SC, Hung YJ, Huang CL, Shieh YS, Chien CY, Chiang CF, et al. Cilostazol inhibits hyperglucose-induced vascular smooth muscle cell dysfunction by modulating the Rage/Erk/Nf-κb signaling pathways. J BioMed Sci (2019) 26(1):68. doi: 10.1186/s12929-019-0550-9
53. Namgung S, Yoon JJ, Yoon CS, Han BH, Choi ES, Oh H, et al. Prunella vulgaris attenuates diabetic renal injury by suppressing glomerular fibrosis and inflammation. Am J Chin Med (2017) 45(3):475–95. doi: 10.1142/s0192415x1750029x
54. Chang M, Nguyen TT. Strategy for treatment of infected diabetic foot ulcers. Acc Chem Res (2021) 54(5):1080–93. doi: 10.1021/acs.accounts.0c00864
55. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol (2015) 6:472–85. doi: 10.1016/j.redox.2015.09.005
56. Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol (2020) 37:101674. doi: 10.1016/j.redox.2020.101674
57. Brownlee M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes (2005) 54(6):1615–25. doi: 10.2337/diabetes.54.6.1615
58. Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys (1996) 29(2):169–202. doi: 10.1017/s0033583500005795
59. Fiorentino TV, Prioletta A, Zuo P, Folli F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr Pharm Des (2013) 19(32):5695–703. doi: 10.2174/1381612811319320005
60. Shopit A, Niu M, Wang H, Tang Z, Li X, Tesfaldet T, et al. Protection of diabetes-induced kidney injury by phosphocreatine Via the regulation of Erk/Nrf2/Ho-1 signaling pathway. Life Sci (2020) 242:117248. doi: 10.1016/j.lfs.2019.117248
61. Cadenas S. Ros and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic Biol Med (2018) 117:76–89. doi: 10.1016/j.freeradbiomed.2018.01.024
62. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature (2006) 443(7113):787–95. doi: 10.1038/nature05292
63. Alnahdi A, John A, Raza H. Augmentation of glucotoxicity, oxidative stress, apoptosis and mitochondrial dysfunction in Hepg2 cells by palmitic acid. Nutrients (2019) 11(9). doi: 10.3390/nu11091979
64. Yao L, Liang X, Qiao Y, Chen B, Wang P, Liu Z. Mitochondrial dysfunction in diabetic tubulopathy. Metabolism (2022) 131:155195. doi: 10.1016/j.metabol.2022.155195
65. Dröge W. Free radicals in the physiological control of cell function. Physiol Rev (2002) 82(1):47–95. doi: 10.1152/physrev.00018.2001
66. Schieber M, Chandel NS. Ros function in redox signaling and oxidative stress. Curr Biol (2014) 24(10):R453–62. doi: 10.1016/j.cub.2014.03.034
67. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol (2007) 39(1):44–84. doi: 10.1016/j.biocel.2006.07.001
68. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol (2003) 552(Pt 2):335–44. doi: 10.1113/jphysiol.2003.049478
69. Fernie AR, Carrari F, Sweetlove LJ. Respiratory metabolism: Glycolysis, the tca cycle and mitochondrial electron transport. Curr Opin Plant Biol (2004) 7(3):254–61. doi: 10.1016/j.pbi.2004.03.007
70. Fernandez-Caggiano M, Eaton P. Heart failure-emerging roles for the mitochondrial pyruvate carrier. Cell Death Differ (2021) 28(4):1149–58. doi: 10.1038/s41418-020-00729-0
71. Vercellino I, Sazanov LA. The assembly, regulation and function of the mitochondrial respiratory chain. Nat Rev Mol Cell Biol (2022) 23(2):141–61. doi: 10.1038/s41580-021-00415-0
72. Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ros generation and uncoupling (Review). Int J Mol Med (2019) 44(1):3–15. doi: 10.3892/ijmm.2019.4188
73. Sazanov LA, Hinchliffe P. Structure of the hydrophilic domain of respiratory complex i from thermus thermophilus. Science (2006) 311(5766):1430–6. doi: 10.1126/science.1123809
74. Berrisford JM, Sazanov LA. Structural basis for the mechanism of respiratory complex i. J Biol Chem (2009) 284(43):29773–83. doi: 10.1074/jbc.M109.032144
75. Rich PR, Maréchal A. The mitochondrial respiratory chain. Essays Biochem (2010) 47:1–23. doi: 10.1042/bse0470001
76. Shimada S, Shinzawa-Itoh K, Baba J, Aoe S, Shimada A, Yamashita E, et al. Complex structure of cytochrome c-cytochrome c oxidase reveals a novel protein-protein interaction mode. EMBO J (2017) 36(3):291–300. doi: 10.15252/embj.201695021
77. Wikstrom MK. Proton pump coupled to cytochrome c oxidase in mitochondria. Nature (1977) 266(5599):271–3. doi: 10.1038/266271a0
78. Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, et al. Mitochondrial membrane potential. Anal Biochem (2018) 552:50–9. doi: 10.1016/j.ab.2017.07.009
79. Kühlbrandt W. Structure and mechanisms of f-type atp synthases. Annu Rev Biochem (2019) 88:515–49. doi: 10.1146/annurev-biochem-013118-110903
80. Trumpower BL. The protonmotive q cycle. energy transduction by coupling of proton translocation to electron transfer by the cytochrome Bc1 complex. J Biol Chem (1990) 265(20):11409–12.
81. Demine S, Renard P, Arnould T. Mitochondrial uncoupling: A key controller of biological processes in physiology and diseases. Cells (2019) 8(8). doi: 10.3390/cells8080795
82. Garcia I, Jones E, Ramos M, Innis-Whitehouse W, Gilkerson R. The little big genome: The organization of mitochondrial DNA. Front Biosci (Landmark Ed) (2017) 22(4):710–21. doi: 10.2741/4511
83. Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol (2013) 5(11):a021220. doi: 10.1101/cshperspect.a021220
84. Farge G, Falkenberg M. Organization of DNA in mammalian mitochondria. Int J Mol Sci (2019) 20(11). doi: 10.3390/ijms20112770
85. Yu M, Zhou Y, Shi Y, Ning L, Yang Y, Wei X, et al. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in chinese breast cancer patients. IUBMB Life (2007) 59(7):450–7. doi: 10.1080/15216540701509955
86. Cassano P, Lezza AM, Leeuwenburgh C, Cantatore P, Gadaleta MN. Measurement of the 4,834-bp mitochondrial DNA deletion level in aging rat liver and brain subjected or not to caloric restriction diet. Ann N Y Acad Sci (2004) 1019:269–73. doi: 10.1196/annals.1297.045
87. Sharma P, Sampath H. Mitochondrial DNA integrity: Role in health and disease. Cells (2019) 8(2). doi: 10.3390/cells8020100
88. Chen XJ, Butow RA. The organization and inheritance of the mitochondrial genome. Nat Rev Genet (2005) 6(11):815–25. doi: 10.1038/nrg1708
89. Protasoni M, Zeviani M. Mitochondrial structure and bioenergetics in normal and disease conditions. Int J Mol Sci (2021) 22(2). doi: 10.3390/ijms22020586
90. Gustafsson CM, Falkenberg M, Larsson NG. Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem (2016) 85:133–60. doi: 10.1146/annurev-biochem-060815-014402
91. Fontana GA, Gahlon HL. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res (2020) 48(20):11244–58. doi: 10.1093/nar/gkaa804
92. Koh JH, Johnson ML, Dasari S, LeBrasseur NK, Vuckovic I, Henderson GC, et al. Tfam enhances fat oxidation and attenuates high-fat diet-induced insulin resistance in skeletal muscle. Diabetes (2019) 68(8):1552–64. doi: 10.2337/db19-0088
93. Mulder H. Transcribing β-cell mitochondria in health and disease. Mol Metab (2017) 6(9):1040–51. doi: 10.1016/j.molmet.2017.05.014
94. Alam TI, Kanki T, Muta T, Ukaji K, Abe Y, Nakayama H, et al. Human mitochondrial DNA is packaged with tfam. Nucleic Acids Res (2003) 31(6):1640–5. doi: 10.1093/nar/gkg251
95. Stein A, Sia EA. Mitochondrial DNA repair and damage tolerance. Front Biosci (Landmark Ed) (2017) 22(5):920–43. doi: 10.2741/4525
96. Robertson AB, Klungland A, Rognes T, Leiros I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell Mol Life Sci (2009) 66(6):981–93. doi: 10.1007/s00018-009-8736-z
97. Dahal S, Raghavan SC. Mitochondrial genome stability in human: Understanding the role of DNA repair pathways. Biochem J (2021) 478(6):1179–97. doi: 10.1042/bcj20200920
98. Ferrando B, Furlanetto A, Gredilla R, Havelund JF, Hebelstrup KH, Møller IM, et al. DNA repair in plant mitochondria - a complete base excision repair pathway in potato tuber mitochondria. Physiol Plant (2019) 166(2):494–512. doi: 10.1111/ppl.12801
99. Gredilla R, Sánchez-Román I, Gómez A, López-Torres M, Barja G. Mitochondrial base excision repair positively correlates with longevity in the liver and heart of mammals. Geroscience (2020) 42(2):653–65. doi: 10.1007/s11357-020-00158-4
100. Yang C, Sengupta S, Hegde PM, Mitra J, Jiang S, Holey B, et al. Regulation of oxidized base damage repair by chromatin assembly factor 1 subunit a. Nucleic Acids Res (2017) 45(2):739–48. doi: 10.1093/nar/gkw1024
101. Mishra M, Kowluru RA. Retinal mitochondrial DNA mismatch repair in the development of diabetic retinopathy, and its continued progression after termination of hyperglycemia. Invest Ophthalmol Vis Sci (2014) 55(10):6960–7. doi: 10.1167/iovs.14-15020
102. Alfarhan M, Jafari E, Narayanan SP. Acrolein: A potential mediator of oxidative damage in diabetic retinopathy. Biomolecules (2020) 10(11). doi: 10.3390/biom10111579
103. Jha JC, Banal C, Chow BS, Cooper ME, Jandeleit-Dahm K. Diabetes and kidney disease: Role of oxidative stress. Antioxid Redox Signal (2016) 25(12):657–84. doi: 10.1089/ars.2016.6664
104. Pop-Busui R, Sima A, Stevens M. Diabetic neuropathy and oxidative stress. Diabetes Metab Res Rev (2006) 22(4):257–73. doi: 10.1002/dmrr.625
105. Vitale G, Salvioli S, Franceschi C. Oxidative stress and the ageing endocrine system. Nat Rev Endocrinol (2013) 9(4):228–40. doi: 10.1038/nrendo.2013.29
106. Wilson DF. Oxidative phosphorylation: Regulation and role in cellular and tissue metabolism. J Physiol (2017) 595(23):7023–38. doi: 10.1113/jp273839
107. Kuznetsov AV, Margreiter R, Ausserlechner MJ, Hagenbuchner J. The complex interplay between mitochondria, ros and entire cellular metabolism. Antioxidants (Basel) (2022) 11(10). doi: 10.3390/antiox11101995
108. Fan Y, Yang Q, Yang Y, Gao Z, Ma Y, Zhang L, et al. Sirt6 suppresses high glucose-induced mitochondrial dysfunction and apoptosis in podocytes through ampk activation. Int J Biol Sci (2019) 15(3):701–13. doi: 10.7150/ijbs.29323
109. Zhong Y, Liu J, Sun D, Guo T, Yao Y, Xia X, et al. Dioscin relieves diabetic nephropathy Via suppressing oxidative stress and apoptosis, and improving mitochondrial quality and quantity control. Food Funct (2022) 13(6):3660–73. doi: 10.1039/d1fo02733f
110. Zhong Y, Luo R, Liu Q, Zhu J, Lei M, Liang X, et al. Jujuboside a ameliorates high fat diet and streptozotocin induced diabetic nephropathy Via suppressing oxidative stress, apoptosis, and enhancing autophagy. Food Chem Toxicol (2022) 159:112697. doi: 10.1016/j.fct.2021.112697
111. Khanra R, Dewanjee S T, Sahu R, Gangopadhyay M, De Feo V, Abroma Augusta L. (Malvaceae) leaf extract attenuates diabetes induced nephropathy and cardiomyopathy Via inhibition of oxidative stress and inflammatory response. J Transl Med (2015) 13:6. doi: 10.1186/s12967-014-0364-1
112. Lee J, Tsogbadrakh B, Yang S, Ryu H, Kang E, Kang M, et al. Klotho ameliorates diabetic nephropathy Via Lkb1-Ampk-Pgc1α-Mediated renal mitochondrial protection. Biochem Biophys Res Commun (2021) 534:1040–6. doi: 10.1016/j.bbrc.2020.10.040
113. Velayoudom-Cephise FL, Cano-Sanchez M, Bercion S, Tessier F, Yu Y, Boulanger E, et al. Receptor for advanced glycation end products modulates oxidative stress and mitochondrial function in the soleus muscle of mice fed a high-fat diet. Appl Physiol Nutr Metab (2020) 45(10):1107–17. doi: 10.1139/apnm-2019-0936
114. Alhaider AA, Korashy HM, Sayed-Ahmed MM, Mobark M, Kfoury H, Mansour MA. Metformin attenuates streptozotocin-induced diabetic nephropathy in rats through modulation of oxidative stress genes expression. Chem Biol Interact (2011) 192(3):233–42. doi: 10.1016/j.cbi.2011.03.014
115. Morigi M, Perico L, Corna D, Locatelli M, Cassis P, Carminati CE, et al. C3a receptor blockade protects podocytes from injury in diabetic nephropathy. JCI Insight (2020) 5(5). doi: 10.1172/jci.insight.131849
116. Aluksanasuwan S, Sueksakit K, Fong-Ngern K, Thongboonkerd V. Role of Hsp60 (Hspd1) in diabetes-induced renal tubular dysfunction: Regulation of intracellular protein aggregation, atp production, and oxidative stress. FASEB J (2017) 31(5):2157–67. doi: 10.1096/fj.201600910RR
117. Sun L, Xiao L, Nie J, Liu FY, Ling GH, Zhu XJ, et al. P66shc mediates high-glucose and angiotensin ii-induced oxidative stress renal tubular injury Via mitochondrial-dependent apoptotic pathway. Am J Physiol Renal Physiol (2010) 299(5):F1014–25. doi: 10.1152/ajprenal.00414.2010
118. Zhan M, Usman I, Yu J, Ruan L, Bian X, Yang J, et al. Perturbations in mitochondrial dynamics by P66shc lead to renal tubular oxidative injury in human diabetic nephropathy. Clin Sci (Lond) (2018) 132(12):1297–314. doi: 10.1042/cs20180005
119. Bock F, Shahzad K, Wang H, Stoyanov S, Wolter J, Dong W, et al. Activated protein c ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme P66shc. Proc Natl Acad Sci U.S.A. (2013) 110(2):648–53. doi: 10.1073/pnas.1218667110
120. Zhou J, Wang T, Wang H, Jiang Y, Peng S. Obacunone attenuates high glucose-induced oxidative damage in nrk-52e cells by inhibiting the activity of gsk-3β. Biochem Biophys Res Commun (2019) 513(1):226–33. doi: 10.1016/j.bbrc.2019.03.201
121. Wongmekiat O, Lailerd N, Kobroob A, Peerapanyasut W. Protective effects of purple rice husk against diabetic nephropathy by modulating pgc-1α/Sirt3/Sod2 signaling and maintaining mitochondrial redox equilibrium in rats. Biomolecules (2021) 11(8). doi: 10.3390/biom11081224
122. Friederich-Persson M, Aslam S, Nordquist L, Welch WJ, Wilcox CS, Palm F. Acute knockdown of uncoupling protein-2 increases uncoupling Via the adenine nucleotide transporter and decreases oxidative stress in diabetic kidneys. PloS One (2012) 7(7):e39635. doi: 10.1371/journal.pone.0039635
123. Wang T, Wen X, Zhang Z, Xie M, Zhou J. Phillyrin ameliorates diabetic nephropathy through the Pi3k/Akt/Gsk-3β signalling pathway in streptozotocin-induced diabetic mice. Hum Exp Toxicol (2021) 40(12_suppl):S487–s96. doi: 10.1177/09603271211051598
124. Li Y, Ou S, Liu Q, Gan L, Zhang L, Wang Y, et al. Genistein improves mitochondrial function and inflammatory in rats with diabetic nephropathy Via inhibiting Mapk/Nf-κb pathway. Acta Cir Bras (2022) 37(6):e370601. doi: 10.1590/acb370601
125. Zhan X, Chen W, Chen J, Lei C, Wei L. Telmisartan mitigates high-Glucose-Induced injury in renal glomerular endothelial cells (Rgecs) and albuminuria in diabetes mice. Chem Res Toxicol (2021) 34(9):2079–86. doi: 10.1021/acs.chemrestox.1c00159
126. Wu M, Chen W, Zhang S, Huang S, Zhang A, Zhang Y, et al. Rotenone protects against β-cell apoptosis and attenuates type 1 diabetes mellitus. Apoptosis (2019) 24(11-12):879–91. doi: 10.1007/s10495-019-01566-4
127. Wu M, Li S, Yu X, Chen W, Ma H, Shao C, et al. Mitochondrial activity contributes to impaired renal metabolic homeostasis and renal pathology in stz-induced diabetic mice. Am J Physiol Renal Physiol (2019) 317(3):F593–f605. doi: 10.1152/ajprenal.00076.2019
128. Zhang T, Chi Y, Kang Y, Lu H, Niu H, Liu W, et al. Resveratrol ameliorates podocyte damage in diabetic mice Via Sirt1/Pgc-1α mediated attenuation of mitochondrial oxidative stress. J Cell Physiol (2019) 234(4):5033–43. doi: 10.1002/jcp.27306
129. Lee E, Choi J, Lee HS. Palmitate induces mitochondrial superoxide generation and activates ampk in podocytes. J Cell Physiol (2017) 232(12):3209–17. doi: 10.1002/jcp.25867
130. Locatelli M, Macconi D, Corna D, Cerullo D, Rottoli D, Remuzzi G, et al. Sirtuin 3 deficiency aggravates kidney disease in response to high-fat diet through lipotoxicity-induced mitochondrial damage. Int J Mol Sci (2022) 23(15). doi: 10.3390/ijms23158345
131. Yuan S, Liu X, Zhu X, Qu Z, Gong Z, Li J, et al. The role of Tlr4 on pgc-1α-Mediated oxidative stress in tubular cell in diabetic kidney disease. Oxid Med Cell Longev (2018) 2018:6296802. doi: 10.1155/2018/6296802
132. Li C, Matavelli LC, Akhtar S, Siragy HM. (Pro)Renin receptor contributes to renal mitochondria dysfunction, apoptosis and fibrosis in diabetic mice. Sci Rep (2019) 9(1):11667. doi: 10.1038/s41598-019-47055-1
133. Wei J, Wu H, Zhang H, Li F, Chen S, Hou B, et al. Anthocyanins inhibit high glucose-induced renal tubular cell apoptosis caused by oxidative stress in Db/Db mice. Int J Mol Med (2018) 41(3):1608–18. doi: 10.3892/ijmm.2018.3378
134. Serralha RS, Rodrigues IF, Bertolini A, Lima DY, Nascimento M, Mouro MG, et al. Esculin reduces P2x7 and reverses mitochondrial dysfunction in the renal cortex of diabetic rats. Life Sci (2020) 254:117787. doi: 10.1016/j.lfs.2020.117787
135. Hong Q, Zhang L, Das B, Li Z, Liu B, Cai G, et al. Increased podocyte sirtuin-1 function attenuates diabetic kidney injury. Kidney Int (2018) 93(6):1330–43. doi: 10.1016/j.kint.2017.12.008
136. Liang Y, Liu H, Fang Y, Lin P, Lu Z, Zhang P, et al. Salvianolate ameliorates oxidative stress and podocyte injury through modulation of Nox4 activity in Db/Db mice. J Cell Mol Med (2021) 25(2):1012–23. doi: 10.1111/jcmm.16165
137. Chen X, Fang M. Oxidative stress mediated mitochondrial damage plays roles in pathogenesis of diabetic nephropathy rat. Eur Rev Med Pharmacol Sci (2018) 22(16):5248–54. doi: 10.26355/eurrev_201808_15723
138. Huang S, Tan M, Guo F, Dong L, Liu Z, Yuan R, et al. Nepeta angustifolia c. y. wu improves renal injury in Hfd/Stz-induced diabetic nephropathy and inhibits oxidative stress-induced apoptosis of mesangial cells. J Ethnopharmacol (2020) 255:112771. doi: 10.1016/j.jep.2020.112771
139. Pal PB, Sinha K, Sil PC. Mangiferin attenuates diabetic nephropathy by inhibiting oxidative stress mediated signaling cascade, tnfα related and mitochondrial dependent apoptotic pathways in streptozotocin-induced diabetic rats. PloS One (2014) 9(9):e107220. doi: 10.1371/journal.pone.0107220
140. Taneda S, Honda K, Tomidokoro K, Uto K, Nitta K, Oda H. Eicosapentaenoic acid restores diabetic tubular injury through regulating oxidative stress and mitochondrial apoptosis. Am J Physiol Renal Physiol (2010) 299(6):F1451–61. doi: 10.1152/ajprenal.00637.2009
141. Yi X, Yan W, Guo T, Liu N, Wang Z, Shang J, et al. Erythropoietin mitigates diabetic nephropathy by restoring Pink1/Parkin-mediated mitophagy. Front Pharmacol (2022) 13:883057. doi: 10.3389/fphar.2022.883057
142. Hwang I, Lee J, Huh JY, Park J, Lee HB, Ho YS, et al. Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes (2012) 61(3):728–38. doi: 10.2337/db11-0584
143. Chowdhury S, Ghosh S, Das AK, Sil PC. Ferulic acid protects hyperglycemia-induced kidney damage by regulating oxidative insult, inflammation and autophagy. Front Pharmacol (2019) 10:27. doi: 10.3389/fphar.2019.00027
144. Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U.S.A. (2009) 106(34):14385–90. doi: 10.1073/pnas.0906805106
145. Yu M, Wang D, Zhong D, Xie W, Luo J. Adropin carried by reactive oxygen species-responsive nanocapsules ameliorates renal lipid toxicity in diabetic mice. ACS Appl Mater Interfaces (2022) 14(33):37330–44. doi: 10.1021/acsami.2c06957
146. Wang XX, Edelstein MH, Gafter U, Qiu L, Luo Y, Dobrinskikh E, et al. G protein-coupled bile acid receptor Tgr5 activation inhibits kidney disease in obesity and diabetes. J Am Soc Nephrol (2016) 27(5):1362–78. doi: 10.1681/asn.2014121271
147. Liu WY, Liou SS, Hong TY, Liu IM. Protective effects of hesperidin (Citrus flavonone) on high glucose induced oxidative stress and apoptosis in a cellular model for diabetic retinopathy. Nutrients (2017) 9(12). doi: 10.3390/nu9121312
148. Wang Z, Zhao H, Guan W, Kang X, Tai X, Shen Y. Metabolic memory in mitochondrial oxidative damage triggers diabetic retinopathy. BMC Ophthalmol (2018) 18(1):258. doi: 10.1186/s12886-018-0921-0
149. Zhai J, Li Z, Zhang H, Ma L, Ma Z, Zhang Y, et al. Berberine protects against diabetic retinopathy by inhibiting cell apoptosis Via deactivation of the Nf-κb signaling pathway. Mol Med Rep (2020) 22(5):4227–35. doi: 10.3892/mmr.2020.11505
150. Mustata GT, Rosca M, Biemel KM, Reihl O, Smith MA, Viswanathan A, et al. Paradoxical effects of green tea (Camellia sinensis) and antioxidant vitamins in diabetic rats: Improved retinopathy and renal mitochondrial defects but deterioration of collagen matrix glycoxidation and cross-linking. Diabetes (2005) 54(2):517–26. doi: 10.2337/diabetes.54.2.517
151. Kanwar M, Chan PS, Kern TS, Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: Possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci (2007) 48(8):3805–11. doi: 10.1167/iovs.06-1280
152. Chen M, Liu B, Ma J, Ge J, Wang K. Protective effect of Mitochondria-Targeted peptide Mtp-131 against oxidative Stress-Induced apoptosis in Rgc-5 cells. Mol Med Rep (2017) 15(4):2179–85. doi: 10.3892/mmr.2017.6271
153. Si YF, Wang J, Guan J, Zhou L, Sheng Y, Zhao J. Treatment with hydrogen sulfide alleviates streptozotocin-induced diabetic retinopathy in rats. Br J Pharmacol (2013) 169(3):619–31. doi: 10.1111/bph.12163
154. Wang JX, Yang Y, Li WY. Sirt3 deficiency increases mitochondrial oxidative stress and promotes migration of retinal pigment epithelial cells. Exp Biol Med (Maywood) (2021) 246(8):877–87. doi: 10.1177/1535370220976073
155. Nonarath HJ, Hall AE, SenthilKumar G, Abroe B, Eells JT, Liedhegner ES. 670nm photobiomodulation modulates bioenergetics and oxidative stress, in rat müller cells challenged with high glucose. PloS One (2021) 16(12):e0260968. doi: 10.1371/journal.pone.0260968
156. Daldal H, Nazıroğlu M. Selenium and resveratrol attenuated diabetes mellitus-mediated oxidative retinopathy and apoptosis Via the modulation of Trpm2 activity in mice. Biol Trace Elem Res (2022) 200(5):2283–97. doi: 10.1007/s12011-022-03203-9
157. Pal S, Rao GN, Pal A. High glucose-induced ros accumulation is a critical regulator of Erk1/2-Akt-Tuberin-Mtor signalling in rgc-5 cells. Life Sci (2020) 256:117914. doi: 10.1016/j.lfs.2020.117914
158. Li H, Tang Z, Chu P, Song Y, Yang Y, Sun B, et al. Neuroprotective effect of phosphocreatine on oxidative stress and mitochondrial dysfunction induced apoptosis in vitro and in vivo: Involvement of dual Pi3k/Akt and Nrf2/Ho-1 pathways. Free Radic Biol Med (2018) 120:228–38. doi: 10.1016/j.freeradbiomed.2018.03.014
159. Xu C, Hou B, He P, Ma P, Yang X, Yang X, et al. Neuroprotective effect of salvianolic acid a against diabetic peripheral neuropathy through modulation of Nrf2. Oxid Med Cell Longev (2020) 2020:6431459. doi: 10.1155/2020/6431459
160. Bheereddy P, Yerra VG, Kalvala AK, Sherkhane B, Kumar A. Sirt1 activation by polydatin alleviates oxidative damage and elevates mitochondrial biogenesis in experimental diabetic neuropathy. Cell Mol Neurobiol (2021) 41(7):1563–77. doi: 10.1007/s10571-020-00923-1
161. Hinder LM, Figueroa-Romero C, Pacut C, Hong Y, Vivekanandan-Giri A, Pennathur S, et al. Long-chain acyl coenzyme a synthetase 1 overexpression in primary cultured schwann cells prevents long chain fatty acid-induced oxidative stress and mitochondrial dysfunction. Antioxid Redox Signal (2014) 21(4):588–600. doi: 10.1089/ars.2013.5248
162. Gustafsson H, Söderdahl T, Jönsson G, Bratteng JO, Forsby A. Insulin-like growth factor type 1 prevents hyperglycemia-induced uncoupling protein 3 down-regulation and oxidative stress. J Neurosci Res (2004) 77(2):285–91. doi: 10.1002/jnr.20142
163. Zhang Q, Song W, Zhao B, Xie J, Sun Q, Shi X, et al. Quercetin attenuates diabetic peripheral neuropathy by correcting mitochondrial abnormality Via activation of Ampk/Pgc-1α pathway in vivo and in vitro. Front Neurosci (2021) 15:636172. doi: 10.3389/fnins.2021.636172
164. Xue B, Wang L, Zhang Z, Wang R, Xia XX, Han PP, et al. Puerarin may protect against schwann cell damage induced by glucose fluctuation. J Nat Med (2017) 71(3):472–81. doi: 10.1007/s11418-016-1067-0
165. Han J, Tan P, Li Z, Wu Y, Li C, Wang Y, et al. Fuzi attenuates diabetic neuropathy in rats and protects schwann cells from apoptosis induced by high glucose. PloS One (2014) 9(1):e86539. doi: 10.1371/journal.pone.0086539
166. Qi H, Casalena G, Shi S, Yu L, Ebefors K, Sun Y, et al. Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic kidney disease susceptibility. Diabetes (2017) 66(3):763–78. doi: 10.2337/db16-0695
167. Pradeep SR, Srinivasan K. Alleviation of oxidative stress-mediated nephropathy by dietary fenugreek (Trigonella foenum-graecum) seeds and onion (Allium cepa) in streptozotocin-induced diabetic rats. Food Funct (2018) 9(1):134–48. doi: 10.1039/c7fo01044c
168. Xue H, Li P, Luo Y, Wu C, Liu Y, Qin X, et al. Salidroside stimulates the Sirt1/Pgc-1α axis and ameliorates diabetic nephropathy in mice. Phytomedicine (2019) 54:240–7. doi: 10.1016/j.phymed.2018.10.031
169. Madsen-Bouterse SA, Zhong Q, Mohammad G, Ho YS, Kowluru RA. Oxidative damage of mitochondrial DNA in diabetes and its protection by manganese superoxide dismutase. Free Radic Res (2010) 44(3):313–21. doi: 10.3109/10715760903494168
170. Xie L, Zhu X, Hu Y, Li T, Gao Y, Shi Y, et al. Mitochondrial DNA oxidative damage triggering mitochondrial dysfunction and apoptosis in high glucose-induced hrecs. Invest Ophthalmol Vis Sci (2008) 49(9):4203–9. doi: 10.1167/iovs.07-1364
171. Mohammad G, Radhakrishnan R, Kowluru RA. Hydrogen sulfide: A potential therapeutic target in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci (2020) 61(14):35. doi: 10.1167/iovs.61.14.35
172. Santos JM, Tewari S, Kowluru RA. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic Biol Med (2012) 53(9):1729–37. doi: 10.1016/j.freeradbiomed.2012.08.588
173. Choi J, Chandrasekaran K, Inoue T, Muragundla A, Russell JW. Pgc-1α regulation of mitochondrial degeneration in experimental diabetic neuropathy. Neurobiol Dis (2014) 64:118–30. doi: 10.1016/j.nbd.2014.01.001
174. Chandrasekaran K, Anjaneyulu M, Inoue T, Choi J, Sagi AR, Chen C, et al. Mitochondrial transcription factor a regulation of mitochondrial degeneration in experimental diabetic neuropathy. Am J Physiol Endocrinol Metab (2015) 309(2):E132–41. doi: 10.1152/ajpendo.00620.2014
175. Latini A, Borgiani P, De Benedittis G, D'Amato C, Greco C, Lauro D, et al. Mitochondrial DNA copy number in peripheral blood is reduced in type 2 diabetes patients with polyneuropathy and associated with a Mir499a gene polymorphism. DNA Cell Biol (2020) 39(8):1467–72. doi: 10.1089/dna.2019.5326
176. Ogura Y, Kitada M, Xu J, Monno I, Koya D. Cd38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular Nad(+)/Nadh ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging (Albany NY) (2020) 12(12):11325–36. doi: 10.18632/aging.103410
177. Lu Q, Zhou Y, Hao M, Li C, Wang J, Shu F, et al. The mtor promotes oxidative stress-induced apoptosis of mesangial cells in diabetic nephropathy. Mol Cell Endocrinol (2018) 473:31–43. doi: 10.1016/j.mce.2017.12.012
178. Hou Y, Shi Y, Han B, Liu X, Qiao X, Qi Y, et al. The antioxidant peptide Ss31 prevents oxidative stress, downregulates Cd36 and improves renal function in diabetic nephropathy. Nephrol Dial Transplant (2018) 33(11):1908–18. doi: 10.1093/ndt/gfy021
179. Ghosh S, Khazaei M, Moien-Afshari F, Ang LS, Granville DJ, Verchere CB, et al. Moderate exercise attenuates caspase-3 activity, oxidative stress, and inhibits progression of diabetic renal disease in Db/Db mice. Am J Physiol Renal Physiol (2009) 296(4):F700–8. doi: 10.1152/ajprenal.90548.2008
180. Song F, Qi X, Chen W, Jia W, Yao P, Nussler AK, et al. Effect of momordica grosvenori on oxidative stress pathways in renal mitochondria of normal and alloxan-induced diabetic mice. Involvement Heme Oxygenase-1. Eur J Nutr (2007) 46(2):61–9. doi: 10.1007/s00394-006-0632-9
181. Qin J, Peng Z, Yuan Q, Li Q, Peng Y, Wen R, et al. Akf-pd alleviates diabetic nephropathy Via blocking the Rage/Ages/Nox and Pkc/Nox pathways. Sci Rep (2019) 9(1):4407. doi: 10.1038/s41598-018-36344-w
182. Ranjbar A, Kheiripour N, Ghasemi H, Seif Rabiei MA, Dadras F, Khoshjou F. Antioxidative effects of tempol on mitochondrial dysfunction in diabetic nephropathy. Iran J Kidney Dis (2018) 12(2):84–90.
183. Cao Y, Xu J, Cui D, Liu L, Zhang S, Shen B, et al. Protective effect of carnosine on hydrogen peroxide-induced oxidative stress in human kidney tubular epithelial cells. Biochem Biophys Res Commun (2021) 534:576–82. doi: 10.1016/j.bbrc.2020.11.037
184. Locatelli M, Zoja C, Zanchi C, Corna D, Villa S, Bolognini S, et al. Manipulating sirtuin 3 pathway ameliorates renal damage in experimental diabetes. Sci Rep (2020) 10(1):8418. doi: 10.1038/s41598-020-65423-0
185. Xiao L, Zhu X, Yang S, Liu F, Zhou Z, Zhan M, et al. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes (2014) 63(4):1366–80. doi: 10.2337/db13-1412
186. Al Sabaani N. Exendin-4 inhibits high glucose-induced oxidative stress in retinal pigment epithelial cells by modulating the expression and activation of P(66)Shc. Cutan Ocul Toxicol (2021) 40(3):175–86. doi: 10.1080/15569527.2020.1844727
187. Li X, Zhang M, Zhou H. The morphological features and mitochondrial oxidative stress mechanism of the retinal neurons apoptosis in early diabetic rats. J Diabetes Res (2014) 2014:678123. doi: 10.1155/2014/678123
188. Kowluru RA, Atasi L, Ho YS. Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci (2006) 47(4):1594–9. doi: 10.1167/iovs.05-1276
189. Kowluru RA, Kowluru V, Xiong Y, Ho YS. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic Biol Med (2006) 41(8):1191–6. doi: 10.1016/j.freeradbiomed.2006.01.012
190. Wang J, Shanmugam A, Markand S, Zorrilla E, Ganapathy V, Smith SB. Sigma 1 receptor regulates the oxidative stress response in primary retinal müller glial cells Via Nrf2 signaling and system xc(-), the na(+)-independent glutamate-cystine exchanger. Free Radic Biol Med (2015) 86:25–36. doi: 10.1016/j.freeradbiomed.2015.04.009
191. Obrosova IG, Van Huysen C, Fathallah L, Cao XC, Greene DA, Stevens MJ. An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J (2002) 16(1):123–5. doi: 10.1096/fj.01-0603fje
192. Yerra VG, Kalvala AK, Sherkhane B, Areti A, Kumar A. Adenosine monophosphate-activated protein kinase modulation by berberine attenuates mitochondrial deficits and redox imbalance in experimental diabetic neuropathy. Neuropharmacology (2018) 131:256–70. doi: 10.1016/j.neuropharm.2017.12.029
193. Hegyi B, Borst JM, Bailey LRJ, Shen EY, Lucena AJ, Navedo MF, et al. Hyperglycemia regulates cardiac k(+) channels Via o-Glcnac-Camkii and Nox2-Ros-Pkc pathways. Basic Res Cardiol (2020) 115(6):71. doi: 10.1007/s00395-020-00834-8
194. Shen H, Wang W. Effect of glutathione liposomes on diabetic nephropathy based on oxidative stress and polyol pathway mechanism. J Liposome Res (2021) 31(4):317–25. doi: 10.1080/08982104.2020.1780607
195. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res (2010) 107(9):1058–70. doi: 10.1161/circresaha.110.223545
196. Han Y, Xu X, Tang C, Gao P, Chen X, Xiong X, et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-Txnip-Nlrp3 biological axis. Redox Biol (2018) 16:32–46. doi: 10.1016/j.redox.2018.02.013
197. Yuan T, Yang T, Chen H, Fu D, Hu Y, Wang J, et al. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol (2019) 20:247–60. doi: 10.1016/j.redox.2018.09.025
198. Julius A, Non-Invasive HWA. Multi-target approach to treat diabetic retinopathy. BioMed Pharmacother (2019) 109:708–15. doi: 10.1016/j.biopha.2018.10.185
199. Akhter F, Chen D, Akhter A, Yan SF, Yan SS. Age-dependent accumulation of dicarbonyls and advanced glycation endproducts (Ages) associates with mitochondrial stress. Free Radic Biol Med (2021) 164:429–38. doi: 10.1016/j.freeradbiomed.2020.12.021
200. Moldogazieva NT, Mokhosoev IM, Mel'nikova TI, Porozov YB, Terentiev AA. Oxidative stress and advanced lipoxidation and glycation end products (Ales and ages) in aging and age-related diseases. Oxid Med Cell Longev (2019) 2019:3085756. doi: 10.1155/2019/3085756
201. Wang Y, Luo W, Han J, Khan ZA, Fang Q, Jin Y, et al. Md2 activation by direct age interaction drives inflammatory diabetic cardiomyopathy. Nat Commun (2020) 11(1):2148. doi: 10.1038/s41467-020-15978-3
202. Shen CY, Lu CH, Wu CH, Li KJ, Kuo YM, Hsieh SC, et al. The development of maillard reaction, and advanced glycation end product (Age)-receptor for age (Rage) signaling inhibitors as novel therapeutic strategies for patients with age-related diseases. Molecules (2020) 25(23). doi: 10.3390/molecules25235591
203. Kay AM, Simpson CL, Stewart JA Jr. The role of Age/Rage signaling in diabetes-mediated vascular calcification. J Diabetes Res (2016) 2016:6809703. doi: 10.1155/2016/6809703
204. Garg SS, Gupta J. Polyol pathway and redox balance in diabetes. Pharmacol Res (2022) 182:106326. doi: 10.1016/j.phrs.2022.106326
205. Niimi N, Yako H, Takaku S, Chung SK, Sango K. Aldose reductase and the polyol pathway in schwann cells: Old and new problems. Int J Mol Sci (2021) 22(3). doi: 10.3390/ijms22031031
206. Oates PJ. Polyol pathway and diabetic peripheral neuropathy. Int Rev Neurobiol (2002) 50:325–92. doi: 10.1016/s0074-7742(02)50082-9
207. Chung SS, Ho EC, Lam KS, Chung SK. Contribution of polyol pathway to diabetes-induced oxidative stress. J Am Soc Nephrol (2003) 14(8 Suppl 3):S233–6. doi: 10.1097/01.asn.0000077408.15865.06
208. Pal PB, Sonowal H, Shukla K, Srivastava SK, Ramana KV. Aldose reductase mediates Nlrp3 inflammasome-initiated innate immune response in hyperglycemia-induced Thp1 monocytes and male mice. Endocrinology (2017) 158(10):3661–75. doi: 10.1210/en.2017-00294
209. Zhou X, Liu Z, Ying K, Wang H, Liu P, Ji X, et al. Wj-39, an aldose reductase inhibitor, ameliorates renal lesions in diabetic nephropathy by activating Nrf2 signaling. Oxid Med Cell Longev (2020) 2020:7950457. doi: 10.1155/2020/7950457
210. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular death, reactive oxygen species (Ros) and diabetic complications. Cell Death Dis (2018) 9(2):119. doi: 10.1038/s41419-017-0135-z
211. Min Q, Bai Y, Zhang Y, Yu W, Zhang M, Liu D, et al. Hawthorn leaf flavonoids protect against diabetes-induced cardiomyopathy in rats Via pkc-α signaling pathway. Evid Based Complement Alternat Med (2017) 2017:2071952. doi: 10.1155/2017/2071952
212. Abdelkader NF, Ibrahim SM, Moustafa PE, Elbaset MA. Inosine mitigated diabetic peripheral neuropathy Via modulating Glo1/Ages/Rage/Nf-κb/Nrf2 and tgf-β/Pkc/Trpv1 signaling pathways. BioMed Pharmacother (2022) 145:112395. doi: 10.1016/j.biopha.2021.112395
213. Capitão M, Soares R. Angiogenesis and inflammation crosstalk in diabetic retinopathy. J Cell Biochem (2016) 117(11):2443–53. doi: 10.1002/jcb.25575
214. Choi T, Lee JW, Kim SK, Yoo KH. Diabetes mellitus promotes smooth muscle cell proliferation in mouse ureteral tissue through the p-Erk/P-Jnk/Vegf/Pkc signaling pathway. Medicina (Kaunas) (2021) 57(6). doi: 10.3390/medicina57060560
215. Pontrelli P, Ranieri E, Ursi M, Ghosh-Choudhury G, Gesualdo L, Paolo Schena F, et al. Jun-N-Terminal kinase regulates thrombin-induced pai-1 gene expression in proximal tubular epithelial cells. Kidney Int (2004) 65(6):2249–61. doi: 10.1111/j.1523-1755.2004.00644.x
216. Nie H, Yi W. O-glcnacylation, a sweet link to the pathology of diseases. J Zhejiang Univ Sci B (2019) 20(5):437–48. doi: 10.1631/jzus.B1900150
217. Daniels MC, McClain DA, Crook ED. Transcriptional regulation of transforming growth factor β1 by glucose: Investigation into the role of the hexosamine biosynthesis pathway. Am J Med Sci (2020) 359(2):79–83. doi: 10.1016/j.amjms.2019.12.013
218. Lozano L, Lara-Lemus R, Zenteno E, Alvarado-Vásquez N. The mitochondrial o-linked n-acetylglucosamine transferase (Mogt) in the diabetic patient could be the initial trigger to develop alzheimer disease. Exp Gerontol (2014) 58:198–202. doi: 10.1016/j.exger.2014.08.008
219. Forman HJ, Zhang H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat Rev Drug Discovery (2021) 20(9):689–709. doi: 10.1038/s41573-021-00233-1
220. Herb M, Schramm M. Functions of ros in macrophages and antimicrobial immunity. Antioxidants (Basel) (2021) 10(2). doi: 10.3390/antiox10020313
221. Mittler R. Ros are good. Trends Plant Sci (2017) 22(1):11–9. doi: 10.1016/j.tplants.2016.08.002
222. Cai X, Bao L, Ren J, Li Y, Zhang Z. Grape seed procyanidin B2 protects podocytes from high glucose-induced mitochondrial dysfunction and apoptosis Via the ampk-Sirt1-Pgc-1α axis in vitro. Food Funct (2016) 7(2):805–15. doi: 10.1039/c5fo01062d
223. Yan C, Duanmu X, Zeng L, Liu B, Song Z. Mitochondrial DNA: Distribution, mutations, and elimination. Cells (2019) 8(4). doi: 10.3390/cells8040379
224. Kaarniranta K, Uusitalo H, Blasiak J, Felszeghy S, Kannan R, Kauppinen A, et al. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog Retin Eye Res (2020) 79:100858. doi: 10.1016/j.preteyeres.2020.100858
225. Akhmedov AT, Marín-García J. Mitochondrial DNA maintenance: An appraisal. Mol Cell Biochem (2015) 409(1-2):283–305. doi: 10.1007/s11010-015-2532-x
226. Cutler RG. Oxidative stress profiling: Part i. its potential importance in the optimization of human health. Ann N Y Acad Sci (2005) 1055:93–135. doi: 10.1196/annals.1323.027
227. Wang X, Wu T, Ma H, Huang X, Huang K, Ye C, et al. Vx-765 ameliorates inflammation and extracellular matrix accumulation by inhibiting the Nox1/Ros/Nf-κb pathway in diabetic nephropathy. J Pharm Pharmacol (2022) 74(3):377–86. doi: 10.1093/jpp/rgab112
228. Richter K, Konzack A, Pihlajaniemi T, Heljasvaara R, Kietzmann T. Redox-fibrosis: Impact of Tgfβ1 on ros generators, mediators and functional consequences. Redox Biol (2015) 6:344–52. doi: 10.1016/j.redox.2015.08.015
229. Wei M, Li Z, Xiao L, Yang Z. Effects of ros-relative nf-κb signaling on high glucose-induced Tlr4 and mcp-1 expression in podocyte injury. Mol Immunol (2015) 68(2 Pt A):261–71. doi: 10.1016/j.molimm.2015.09.002
230. Da Silva LM, da Silva R, Maria-Ferreira D, Beltrame OC, da Silva-Santos JE, Werner MFP. Vitamin c improves gastroparesis in diabetic rats: Effects on gastric contractile responses and oxidative stress. Dig Dis Sci (2017) 62(9):2338–47. doi: 10.1007/s10620-017-4632-9
231. Yarahmadi A, Saeed Modaghegh MH, Mostafavi-Pour Z, Azarpira N, Mousavian A, Bonakdaran S, et al. The effect of platelet-rich plasma-fibrin glue dressing in combination with oral vitamin e and c for treatment of non-healing diabetic foot ulcers: A randomized, double-blind, parallel-group, clinical trial. Expert Opin Biol Ther (2021) 21(5):687–96. doi: 10.1080/14712598.2021.1897100
232. Güney M. Selenium-vitamin e combination modulates endometrial lipid peroxidation and antioxidant enzymes in streptozotocin-induced diabetic rat. Biol Trace Elem Res (2012) 149(2):234–40. doi: 10.1007/s12011-012-9426-5
233. Didangelos T, Karlafti E, Kotzakioulafi E, Kontoninas Z, Margaritidis C, Giannoulaki P, et al. Efficacy and safety of the combination of superoxide dismutase, alpha lipoic acid, vitamin B12, and carnitine for 12 months in patients with diabetic neuropathy. Nutrients (2020) 12(11). doi: 10.3390/nu12113254
234. Dai DF, Chiao YA, Martin GM, Marcinek DJ, Basisty N, Quarles EK, et al. Mitochondrial-targeted catalase: Extended longevity and the roles in various disease models. Prog Mol Biol Transl Sci (2017) 146:203–41. doi: 10.1016/bs.pmbts.2016.12.015
235. Reid Thompson W, Hornby B, Manuel R, Bradley E, Laux J, Carr J, et al. A phase 2/3 randomized clinical trial followed by an open-label extension to evaluate the effectiveness of elamipretide in barth syndrome, a genetic disorder of mitochondrial cardiolipin metabolism. Genet Med (2021) 23(3):471–8. doi: 10.1038/s41436-020-01006-8
236. Aston K, Rath N, Naik A, Slomczynska U, Schall OF, Riley DP. Computer-aided design (Cad) of Mn(Ii) complexes: Superoxide dismutase mimetics with catalytic activity exceeding the native enzyme. Inorg Chem (2001) 40(8):1779–89. doi: 10.1021/ic000958v
237. Nakamura Y, Feng Q, Kumagai T, Torikai K, Ohigashi H, Osawa T, et al. Ebselen, a glutathione peroxidase mimetic seleno-organic compound, as a multifunctional antioxidant. implication for inflammation-associated carcinogenesis. J Biol Chem (2002) 277(4):2687–94. doi: 10.1074/jbc.M109641200
238. Góth L, Eaton JW. Hereditary catalase deficiencies and increased risk of diabetes. Lancet (2000) 356(9244):1820–1. doi: 10.1016/s0140-6736(00)03238-4
239. Sun J, Chen T, Zhao B, Fan W, Shen Y, Wei H, et al. Acceleration of oral wound healing under diabetes mellitus conditions using bioadhesive hydrogel. ACS Appl Mater Interfaces (2023) 15(1):416–31. doi: 10.1021/acsami.2c17424
240. Qin W, Wu Y, Liu J, Yuan X, Gao J. A comprehensive review of the application of nanoparticles in diabetic wound healing: Therapeutic potential and future perspectives. Int J Nanomedicine (2022) 17:6007–29. doi: 10.2147/ijn.S386585
241. Hamedifard Z, Farrokhian A, Reiner Ž, Bahmani F, Asemi Z, Ghotbi M, et al. The effects of combined magnesium and zinc supplementation on metabolic status in patients with type 2 diabetes mellitus and coronary heart disease. Lipids Health Dis (2020) 19(1):112. doi: 10.1186/s12944-020-01298-4
242. Gezginci-Oktayoglu S, Basaraner H, Yanardag R, Bolkent S. The effects of combined treatment of antioxidants on the liver injury in stz diabetic rats. Dig Dis Sci (2009) 54(3):538–46. doi: 10.1007/s10620-008-0381-0
243. Motta BP, Pinheiro CG, Figueiredo ID, Cardoso FN, Oliveira JO, Machado RTA, et al. Combined effects of lycopene and metformin on decreasing oxidative stress by triggering endogenous antioxidant defenses in diet-induced obese mice. Molecules (2022) 27(23). doi: 10.3390/molecules27238503
244. Nankar R, Prabhakar PK, Doble M. Hybrid drug combination: Combination of ferulic acid and metformin as anti-diabetic therapy. Phytomedicine (2017) 37:10–3. doi: 10.1016/j.phymed.2017.10.015
Keywords: diabetes mellitus, diabetic microvascular complications, mitochondria, oxidative stress, oxidative phosphorylation, mitochondrial DNA
Citation: Zhang Z, Huang Q, Zhao D, Lian F, Li X and Qi W (2023) The impact of oxidative stress-induced mitochondrial dysfunction on diabetic microvascular complications. Front. Endocrinol. 14:1112363. doi: 10.3389/fendo.2023.1112363
Received: 30 November 2022; Accepted: 24 January 2023;
Published: 07 February 2023.
Edited by:
Pranav Kumar Prabhakar, Lovely Professional University, IndiaReviewed by:
Pabitra Bikash Pal, University of Pittsburgh, United StatesPrema Velusamy, Penn State Milton S. Hershey Medical Center, United States
Copyright © 2023 Zhang, Huang, Zhao, Lian, Li and Qi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengmei Lian, bGZtNTY1QHNvaHUuY29t; Xiangyan Li, eGlhbmd5YW5fbGkxOTgxQDE2My5jb20=; Wenxiu Qi, cWl3ZW54aXUwNTE3QDE2My5jb20=
†These authors have contributed equally to this work