 Ji Suk Chang
Ji Suk Chang- Gene Regulation and Metabolism Laboratory, Pennington Biomedical Research Center, Baton Rouge, LA, United States
Brown adipocytes is a specialized fat cell that dissipates nutrient-derived chemical energy in the form of heat, instead of ATP synthesis. This unique feature provides a marked capacity for brown adipocyte mitochondria to oxidize substrates independent of ADP availability. Upon cold exposure, brown adipocytes preferentially oxidize free fatty acids (FFA) liberated from triacylglycerol (TAG) in lipid droplets to support thermogenesis. In addition, brown adipocytes take up large amounts of circulating glucose, concurrently increasing glycolysis and de novo FA synthesis from glucose. Given that FA oxidation and glucose-derived FA synthesis are two antagonistic mitochondrial processes in the same cell, it has long been questioned how brown adipocytes run FA oxidation and FA synthesis simultaneously. In this review, I summarize mechanisms regulating mitochondrial substrate selection and describe recent findings of two distinct populations of brown adipocyte mitochondria with different substrate preferences. I further discuss how these mechanisms may permit a concurrent increase in glycolysis, FA synthesis, and FA oxidation in brown adipocytes.
Introduction
While white adipocytes primarily store excess energy in the form of triacylglycerol (TAG), brown adipocytes located in the interscapular brown adipose tissue (BAT) have a marked capacity to oxidize nutrients and dissipate energy as heat (1). Brown-like beige adipocytes also emerge within white adipose tissue (WAT) during prolonged cold exposure or pharmacological stimulation of β3-adrenergic receptors (2–5). Notably, activation of brown and beige adipocytes in rodents (6, 7) and humans (8–14) increases energy expenditure and improves systemic glucose and lipid homeostasis. Thus, brown/beige adipocytes have emerged as an appealing target against obesity and its related metabolic disorders, such as type 2 diabetes, insulin resistance, and dyslipidemia.
Upon activation, brown adipocytes simultaneously increase glycolysis, glucose-derived de novo fatty acid synthesis (FAS), and fatty acid oxidation (FAO) (15–19), which are mutually exclusive pathways in the same cell (20). In most mammalian cells, elevated glycolysis and subsequent pyruvate oxidation in the mitochondria block mitochondrial FAO. Conversely, elevated FAO decreases the activity of glycolytic enzymes in the cytosol and pyruvate dehydrogenase (PDH) within the mitochondrial matrix, thus leading to inhibition of pyruvate production, oxidation, and de novo FAS. It is not fully understood how brown adipocytes simultaneously increase glycolysis and FAS while primarily oxidizing FA in the mitochondria. To target brown adipocytes therapeutically, it is important to understand the underlying mechanism responsible for this unique phenomenon. This mini review will focus on recent advances in our understanding of substrate utilization in brown adipocytes and discuss molecular mechanisms that may permit the concurrence of glycolysis, FAS, and FAO in brown adipocytes.
UCP1-mediated proton leak: A mechanism for high substrate oxidation in brown adipocytes
In mammalian cells, oxidation pathways of glucose, FA, and amino acids converge onto a common pathway, the tricarboxylic acid (TCA) cycle, which generates NADH and FADH2 in the mitochondrial matrix. These reduced electron carriers donate their electrons to the electron transport chain (ETC) system, which is composed of four multi-subunit complexes (I-IV) located in the inner mitochondrial membrane (IMM) and two mobile electron carriers coenzyme Q and cytochrome c (21, 22). Subsequent electron transfer through the ETC leads to pumping of protons (H+ ions) from the mitochondrial matrix to the intermembrane space, creating an electrochemical proton gradient, also known as the proton motive force (PMF). The PMF is a form of potential energy composed of an electrical charge gradient (ΔΨm) and a chemical gradient (ΔpH) across the IMM (23). The resulting PMF is used by F0F1-ATP synthase (Figure 1A; coupled respiration). The protons pass from the intermembrane space into the matrix through the F0 component, causing a conformational change in F0F1-ATP synthase so that ATP is produced from ADP and inorganic phosphate (24). For the coupling of ETC-mediated proton pumps to the ATP synthesis, the rate of substrate oxidation and electron flow is highly dependent on the availability of ADP (25). Fluctuations in coupling between ETC activity and ATP production can cause electron leak from the ETC onto oxygen, resulting in production of reactive oxygen species (ROS) (26).
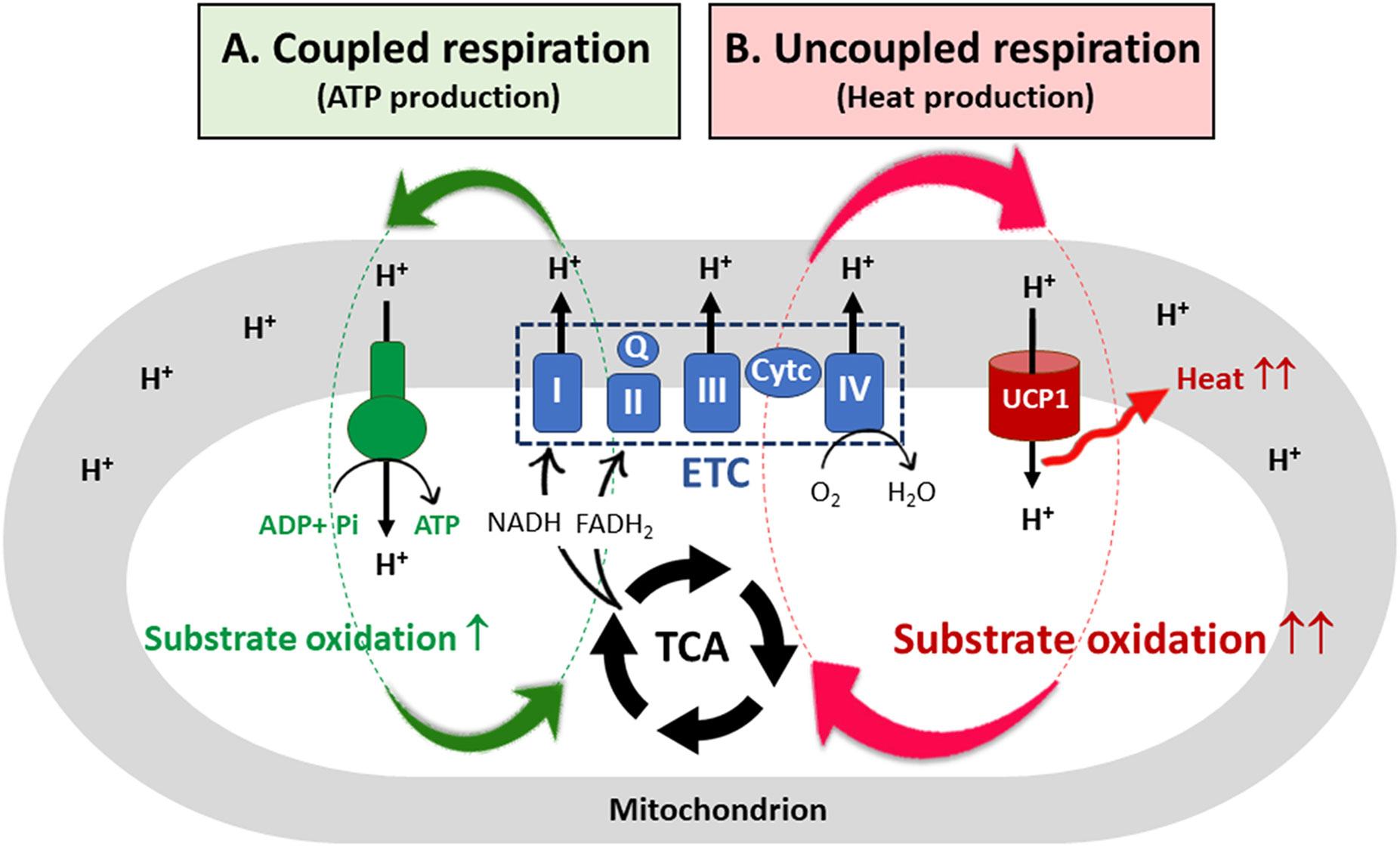
Figure 1 UCP1-mediated uncoupled respiration and its contribution to substrate oxidation. (A) In most mammalian cells, a proton motive force (PMF) created by the electron transfer chain (ETC) system is used by ATP synthase, resulting in ATP production (coupled respiration). To re-establish the electrochemical proton gradient across the inner mitochondrial membrane (IMM), the cells increase substrate oxidation, generating more NADH and FADH2 needed by the ETC. However, for coupled respiration, the rate of substrate oxidation and electron flow through the ETC is highly dependent on ADP availability. (B) In brown adipocytes, the protons pass from the intermembrane space into the mitochondrial matrix through the membrane bound UCP1. The resulting proton leak causes a drop in the PMF, releasing heat but not ATP synthesis (uncoupled respiration). The futile cycle of ETC-mediated proton pumps and UCP1-mediated proton leak provides a marked capacity for brown adipocyte mitochondria to oxidize substrates independent of ADP availability. TCA, the tricarboxylic acid cycle; I, II, III, IV, ETC multi-subunit complexes I through IV; Q, coenzyme Q; Cyt c, cytochrome c; UCP1, uncoupled protein 1.
Brown adipocytes contain a large number of mitochondria that uniquely express uncoupling protein 1 (UCP1) in the IMM (27, 28) along with abundant expression of TCA cycle enzymes and ETC complexes (I-IV) (1). Membrane-bound UCP1, an H+ transport protein, allows the re-entry of protons into the mitochondrial matrix independent of ATP synthesis (Figure 1B; uncoupled respiration) (29, 30). Thus, UCP1-mediated proton leak causes a drop in the PMF, and energy is lost as heat. As a mechanism to re-establish the electrochemical proton gradient (ΔpH) across the IMM, brown adipocytes increase the rate of substrate oxidation, generating more NADH and FADH2 needed by the ETC. Consequently, the futile cycle of ETC-mediated proton pumps and UCP1-mediated proton leak provides a marked capacity for brown adipocyte mitochondria to oxidize substrates without being affected by ADP availability. In addition, dissipation of the PMF by UCP1 has been shown to reduce mitochondrial ROS production, contributing to an increase in ETC complexes (31–33).
Mitochondrial substrate utilization in brown adipocytes: Fatty acids vs glucose?
Brown adipocytes oxidize substantial amounts of substrates due to high ETC activity (proton pumping) and UCP1 activity (proton leak) in the IMM. Upon adrenergic stimulation of the cell by cold-dependent activation of the sympathetic nervous system or β3-adrenergic receptor agonists, brown adipocytes liberate free fatty acids (FFA) by lipolysis of TAG stored in lipid droplets as well as take up extracellular nonesterified fatty acids (NEFA) from the circulation (1, 34–38). FFA released from the intracellular TAG is the main source of FA for thermogenesis. Circulating NEFA is directed toward TAG replenishment in brown adipocytes although a portion of NEFA contributes to thermogenesis (35, 39). These FAs are ligated to CoA groups before being converted to acyl-carnitine for mitochondrial import through carnitine palmitoyltransferase 1 (CPT1) located in the outer mitochondrial membrane (OMM). Subsequent FA β-oxidation in the matrix produces acetyl-CoA, which then enters the TCA cycle as citrate after condensation with oxaloacetate (OAA). FAO results in a larger increase in acetyl-CoA levels per molecule of nutrient than glucose-derived pyruvate oxidation (i.e., 1 C16-palmitic acid generates 8 acetyl-CoA; 1 glucose generates 2 acetyl-CoA). Accordingly, FAO is more efficient in generating NADH and FADH2 and FA has been viewed as the primary substrate for energy-demanding brown adipocyte mitochondria (1, 6). Moreover, it is interesting to note that FA is not only the energy substrate for thermogenesis but also activates the UCP1-mediated proton leak across the IMM (29, 40).
Surprisingly, activated brown adipocytes also take up large amounts of glucose from the circulation while primarily utilizing FA to fuel thermogenesis (19, 41–45). However, recent studies have further found that the primary function of this glucose is not to support thermogenesis (<15%) but instead fuel de novo lipogenesis (DNL) through multiple mechanisms (16, 17, 19, 38, 42, 46–49): i) Pyruvate-derived citrate serves as the precursor for de novo FA synthesis (FAS); ii) Glucose-derived glycerol-3-phosphate serves as the structural backbone for TAG synthesis; iii) Glycolytic intermediates support the pentose phosphate pathway generating NADPH needed for DNL; and iv) Cytosolic ATP production during glycolysis meets energy requirement for DNL as well as compensates for the loss of mitochondrial ATP synthesis. Therefore, it has been suggested that, while oxidizing FA to fuel thermogenesis, brown adipocytes concurrently increase glycolysis and de novo FAS to replenish intracellular TAG pool in lipid droplets (15–19). However, it is currently unclear how brown adipocytes concurrently perform FAO and FAS in the same cell because these processes are two mutually exclusive pathways in healthy cells.
More interestingly, recent studies have shown that cold-activated BAT in rodents and humans utilizes additional substrates such as branched-chain amino acids (BCAA) (50, 51), glutamate (44), and succinate (52) to support thermogenesis. Extracellular succinate contributes to thermogenic respiration in BAT by the succinate dehydrogenase (SDH)-mediated oxidation in the TCA cycle (52), although its relative contribution to thermogenesis is unclear. Similarly, BCAAs and glutamate enter the TCA cycle as acetyl-CoA/succinyl-CoA and α-ketoglutarate, respectively, to generate more reducing equivalents in BAT (44, 50, 51). It is also possible that carbons from these additional substrates replenish TCA cycle intermediates that leave the cycle for biosynthetic pathways (e.g., citrate for de novo FA synthesis).
Molecular mechanisms regulating mitochondrial substrate selection
In most mammalian cells, mitochondrial FAO suppresses glycolysis, pyruvate oxidation, and de novo FAS (20). FAO-induced increases in acetyl-CoA/CoA, NADH/NAD+, and ATP/ADP ratios inhibit the activity of pyruvate dehydrogenase (PDH) that catalyzes the conversion of glucose-derived pyruvate to acetyl-CoA in the mitochondria (53, 54). The resulting decrease in acetyl-CoA reduces the production of pyruvate-derived citrate that exits the mitochondria to serve as the precursor for FAS. Thus, FAO-dependent inhibition of PDH activity in the mitochondria is the primary mechanism preventing both pyruvate oxidation and de novo FAS from glucose. Additionally, FAO can inhibit glycolysis. A portion of excess citrate produced from FA-derived acetyl-CoA is exported to the cytosol, where it in turn inhibits glycolytic enzymes, such as phosphofructokinases (PFK1, PFK2) and pyruvate kinase (PK) (20, 55–57).
Conversely, when extracellular glucose increases, enhanced glycolysis provides more pyruvate to the mitochondria. The conversion of pyruvate to acetyl-CoA by PDH and to OAA by pyruvate carboxylase (PC) increases citrate production in the mitochondria. Under high glucose, excess citrate is exported to the cytosol and hydrolyzed back to acetyl-CoA and OAA by ATP-citrate lyase (ACLY). Acetyl-CoA is then carboxylated to malonyl-CoA by two acetyl-CoA carboxylases, ACC1 and ACC2 (58). Malonyl-CoA is the precursor of de novo synthesized FA. Remarkably, malonyl-CoA produced by ACC2 allosterically inhibits CPT1 (59, 60) that controls the entry of long-chain fatty acids from the cytosol into mitochondria. By this mechanism, glucose-derived malonyl-CoA prevents the oxidation of newly synthesized and pre-existing FAs. Thus, malonyl-CoA is a key metabolite regulating the balance between FAS and FAO.
Although ACC1 and ACC2 have same enzyme activity with over 70% protein sequence similarity, they play distinct roles in the control of FAS and FAO (58, 61, 62). ACC1 is cytosolic and directs malonyl-CoA toward de novo FA synthesis catalyzed by fatty acid synthase (FAS). In contrast, ACC2 is associated with the OMM and regulates FAO through malonyl-CoA-mediated CPT1 inhibition (59–61, 63). While lipogenic WAT predominantly expresses ACC1, BAT expresses similar amounts of ACC1 and ACC2 (64). In addition, BAT expresses CPT1β, an isoform with high sensitivity to malonyl-CoA (65–67). Despite the expression of ACC2 and CPT1β, BAT mitochondria have the highest CPT1 activity among the tissues expressing CPT1β (65–67). High FAO in BAT is surprising in light of the inhibitory effect of malonyl-CoA produced by ACC2 on CPT1β-mediated FA transport. It is unclear whether ACC2 activity or association to the mitochondria is negatively regulated by cold in brown adipocytes.
It is interesting to note that concurrent FAO and FAS have also been observed in a subset of cancer cells (68–70). Glycolytic colorectal cancer cells recruit FAO as an adaptive response to extracellular acidification associated with increased pyruvate to lactate conversion (68). A selective decrease in the transcription of ACACB gene under acidosis was in part the mechanism permitting mitochondrial FAO. However, it is unlikely that the selective decrease in ACACB gene expression provides a mechanism by which brown adipocytes concurrently perform FAO and FAS because BAT upregulates the expression of both ACACA and ACACB genes encoding ACC1 and ACC2, respectively, in response to cold (17). As another example, a subset of highly proliferating B-cell lymphomas concurrently stimulates mitochondrial FAO while increasing glycolysis and FAS (69); however, the underlying mechanism remains unknown.
Heterogeneity of brown adipocytes in BAT
Single-cell and single-nucleus RNA sequencing of BAT has uncovered the existence of multiple brown adipocyte subpopulations with large variability in their transcriptomes and with different degrees of thermogenic capacities (71–73). Compared with the high-thermogenic brown adipocytes, low-thermogenic brown adipocytes express lower levels of Ucp1 along with reduced mitochondrial respiration (71). It is considered that these subpopulations are derived from distinct precursor cells and/or represent different cell states acquired during environmental temperature changes (71–73). The co-existence of functionally different brown adipocytes within the BAT may in part explain how BAT performs FAO and FAS simultaneously. Further studies are required to delineate the location, functional specialization, and substrate utilization of these brown adipocyte subpopulations and their ratios in response to environmental stimuli.
Heterogeneity of mitochondria within the brown adipocyte: FA-oxidizing vs lipogenic mitochondria
In addition to heterogeneity of brown adipocytes, recent studies have demonstrated the presence of metabolically distinct populations of mitochondria within the same brown adipocyte: cytosolic mitochondria (CM) and peridroplet mitochondria (PDM) (74–78). PDM are found to be anchored to the lipid droplets and have reduced motility and fusion-fission dynamics that segregate PDM from the rest of the mitochondrial population (79, 80). While CM preferentially oxidize FA for theromgenesis, PDM have a higher capacity for pyruvate oxidation and ATP synthesis (74) (Figure 2). In line with increased oxidative phosphorylation, PDM is enriched with ATP synthase compared to CM (74) although UCP1 levels are comparable in PDM and CM (74, 78). More interestingly, an increase in PDM is associated with lipid droplet expansion in brown adipocytes (74). Given that coupled respiration is dependent on ADP availability, excess citrate produced from pyruvate-derived acetyl-CoA in the PDM may exit the mitochondria and be converted to malonyl-CoA by ACC1 and ACC2, thus contributing to de novo FAS for TAG synthesis and concurrently preventing FA entry into these specific subpopulations of mitochondria (Figure 2). On the contrary, in CM preferentially oxidizing FA (74), FA-derived acetyl-CoA could inhibit PDH activity, resulting in a decrease in pyruvate-derived citrate production and subsequent malonyl-CoA accumulation in close vicinity of CM (Figure 2). It is unclear whether there is a difference in ACC2 levels between PDM and CM. CM could maximize UCP1-mediated thermogenesis by producing high levels of NADH and FADH2 from FAO. The resulting rapid oxidation of FA-derived citrate through the TCA cycle may prevent citrate export to the cytosol for inhibition of glycolytic enzymes.
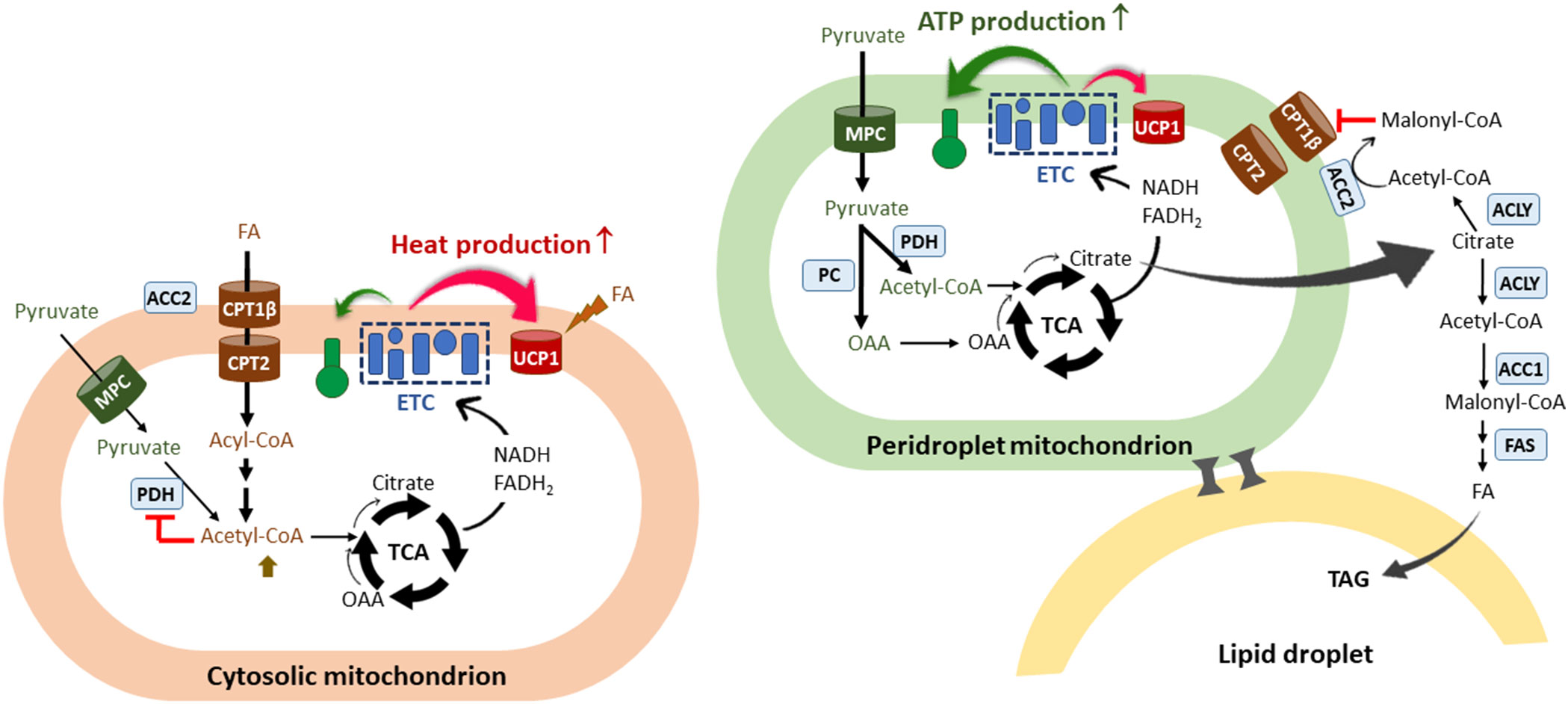
Figure 2 The co-existence of two functionally different mitochondria within the brown adipocyte. A scheme of two types of mitochondria identified in the brown adipocyte: cytosolic mitochondria (CM) and peridroplet mitochondria (PDM) (74–78). PDM are anchored to the lipid droplets and segregated from the pool of CM. CM preferentially oxidize FA and are more thermogenic compared to PDM. FAO-induced increases in acetyl-CoA/CoA and NADH/NAD+ ratios would inhibit PDH in the matrix, resulting in a decrease in pyruvate-derived citrate production and subsequent malonyl-CoA accumulation in close vicinity of CM. FA-derived citrate would be rapidly oxidized through the TCA cycle to support UCP1-mediated thermogenesis. On the contrary, PDM have a higher capacity for pyruvate oxidation and ATP synthesis compared to CM. Given that coupled respiration is dependent on ADP availability, excess citrate would escape from the mitochondria and be converted to malonyl-CoA by ACC1 and ACC2, thus contributing to de novo lipogenesis and simultaneously preventing CPT1β-mediated FA entry into PDM. The co-existence of two functionally different mitochondria within the brown adipocyte may in part explain the concurrence of glycolysis, FA synthesis, and FA oxidation in brown adipocytes. FA, fatty acids; ETC, electron transport chain; CPT, carnitine palmitoyltransferase; UCP1, uncoupled protein 1; TCA, the tricarboxylic acid cycle; OAA, oxaloacetate; MPC, mitochondrial pyruvate carrier; PDH, pyruvate dehydrogenase; PC, pyruvate carboxylase; ACLY, ATP-citrate lyase; ACC, acetyl-CoA carboxylases; FAS, fatty acid synthase; TAG, triacylglycerol.
The association between mitochondria and lipid droplets has been overserved in other tissue/cell types including skeletal muscle, heart, and adipocytes (77, 81–83). In contrast to the lipogenic role of PDM in brown adipocytes, several studies reported conflicting results that PDM promotes the oxidation of FA released from lipid droplets (77, 81). This discrepancy may imply that the role of PDM is differently regulated by the cell type, nutritional status, or cellular stress. Proteome profiling of PDM and CM in BAT has identified a subset of mitochondrial proteins differentially expressed between PDM and CM although their impact on the functional difference has not been explored (78). Additional studies are required to quantitatively characterize PDM and CM mitochondrial proteins (e.g., MPC1/2, ACC2, CPT1β) and understand the significance of relative PDM/CM ratio and the mechanism controlling this ratio in brown adipocytes.
Conclusion
Brown adipocytes have two unique features: (1) UCP1-mediated dissipation of the PMF, which provides a mechanism for maximal substrate oxidation in the mitochondria and (2) concurrence of glycolysis, de novo FAS, and FAO. Upon activation, brown adipocytes increase glycolysis and de novo FAS to replenish intracellular TAG pools that are depleted due to increased lipolysis and FAO. The co-existence of FA-oxidizing and lipogenic mitochondria within the brown adipocyte in addition to heterogeneity of brown adipocytes may in part explain the unique capacity of brown adipocytes to be involved simultaneously in FAO and FAS.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cannon B, Nedergaard J. Brown adipose tissue: Function and physiological significance. Physiol Rev (2004) 84(1):277–359. doi: 10.1152/physrev.00015.200384/1/277[pii]
2. Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell (2012) 150(2):366–76. doi: 10.1016/j.cell.2012.05.016S0092-8674(12)00595-8[pii]
3. Himms-Hagen J, Cui J, Danforth E Jr., Taatjes DJ, Lang SS, Waters BL, et al. Effect of cl-316,243, a thermogenic beta 3-agonist, on energy balance and brown and white adipose tissues in rats. Am J Physiol (1994) 266(4 Pt 2):R1371–82. doi: 10.1152/ajpregu.1994.266.4.R1371
4. Himms-Hagen J, Melnyk A, Zingaretti MC, Ceresi E, Barbatelli G, Cinti S. Multilocular fat cells in wat of cl-316243-Treated rats derive directly from white adipocytes. Am J Physiol Cell Physiol (2000) 279(3):C670–81. doi: 10.1152/ajpcell.2000.279.3.C670
5. Granneman JG, Li P, Zhu Z, Lu Y. Metabolic and cellular plasticity in white adipose tissue I: Effects of Beta3-adrenergic receptor activation. Am J Physiol Endocrinol Metab (2005) 289(4):E608–16. doi: 10.1152/ajpendo.00009.2005
6. Townsend KL, Tseng YH. Brown fat fuel utilization and thermogenesis. Trends Endocrinol Metab (2014) 25(4):168–77. doi: 10.1016/j.tem.2013.12.004
7. Stanford KI, Middelbeek RJ, Townsend KL, An D, Nygaard EB, Hitchcox KM, et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest (2013) 123(1):215–23. doi: 10.1172/JCI62308
8. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med (2009) 360(15):1500–8. doi: 10.1056/NEJMoa0808718360/15/1500[pii]
9. Chondronikola M, Volpi E, Borsheim E, Porter C, Annamalai P, Enerback S, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes (2014) 63(12):4089–99. doi: 10.2337/db14-0746
10. Ouellet V, Labbe SM, Blondin DP, Phoenix S, Guerin B, Haman F, et al. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest (2012) 122(2):545–52. doi: 10.1172/JCI6043360433[pii]
11. Hanssen MJ, Hoeks J, Brans B, van der Lans AA, Schaart G, van den Driessche JJ, et al. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat Med (2015) 21(8):863–5. doi: 10.1038/nm.3891
12. Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elia E, Kessler SH, Kahn PA, et al. Activation of human brown adipose tissue by a Beta3-adrenergic receptor agonist. Cell Metab (2015) 21(1):33–8. doi: 10.1016/j.cmet.2014.12.009
13. O'Mara AE, Johnson JW, Linderman JD, Brychta RJ, McGehee S, Fletcher LA, et al. Chronic mirabegron treatment increases human brown fat, hdl cholesterol, and insulin sensitivity. J Clin Invest (2020) 130(5):2209–19. doi: 10.1172/JCI131126
14. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med (2009) 360(15):1509–17. doi: 10.1056/NEJMoa0810780360/15/1509[pii]
15. Buckley MG, Rath EA. Regulation of fatty acid synthesis and malonyl-coa content in mouse brown adipose tissue in response to cold-exposure, starvation or re-feeding. Biochem J (1987) 243(2):437–42. doi: 10.1042/bj2430437
16. Mottillo EP, Balasubramanian P, Lee YH, Weng C, Kershaw EE, Granneman JG. Coupling of lipolysis and De novo lipogenesis in brown, beige, and white adipose tissues during chronic Beta3-adrenergic receptor activation. J Lipid Res (2014) 55(11):2276–86. doi: 10.1194/jlr.M050005
17. Yu XX, Lewin DA, Forrest W, Adams SH. Cold elicits the simultaneous induction of fatty acid synthesis and beta-oxidation in murine brown adipose tissue: Prediction from differential gene expression and confirmation in vivo. FASEB J (2002) 16(2):155–68. doi: 10.1096/fj.01-0568com16/2/155[pii]
18. Mccormack JG, Denton RM. Evidence that fatty-acid synthesis in interscapular brown adipose-tissue of cold-adapted rats is increased invivo by insulin by mechanisms involving parallel activation of pyruvate-dehydrogenase and acetyl-coenzyme-a carboxylase. Biochem J (1977) 166(3):627–30. doi: 10.1042/bj1660627
19. Jung SM, Doxsey WG, Le J, Haley JA, Mazuecos L, Luciano AK, et al. In vivo isotope tracing reveals the versatility of glucose as a brown adipose tissue substrate. Cell Rep (2021) 36(4):109459. doi: 10.1016/j.celrep.2021.109459
20. Hue L, Taegtmeyer H. The randle cycle revisited: A new head for an old hat. Am J Physiol Endocrinol Metab (2009) 297(3):E578–91. doi: 10.1152/ajpendo.00093.2009
21. Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell (2013) 155(1):160–71. doi: 10.1016/j.cell.2013.08.032
22. Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ros generation and uncoupling (Review). Int J Mol Med (2019) 44(1):3–15. doi: 10.3892/ijmm.2019.4188
23. Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature (1961) 191:144–8. doi: 10.1038/191144a0
24. Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial atp synthase: Architecture, function and pathology. J Inherit Metab Dis (2012) 35(2):211–25. doi: 10.1007/s10545-011-9382-9
25. Leverve X, Sibille B, Devin A, Piquet MA, Espie P, Rigoulet M. Oxidative phosphorylation in intact hepatocytes: Quantitative characterization of the mechanisms of change in efficiency and cellular consequences. Mol Cell Biochem (1998) 184(1-2):53–65. doi: 10.1023/A:1006810209531
26. Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem (2017) 292(41):16804–9. doi: 10.1074/jbc.R117.789271
27. Golozoubova V, Hohtola E, Matthias A, Jacobsson A, Cannon B, Nedergaard J. Only Ucp1 can mediate adaptive nonshivering thermogenesis in the cold. FASEB J (2001) 15(11):2048–50. doi: 10.1096/fj.00-0536fje00-0536fje[pii]
28. Nedergaard J, Golozoubova V, Matthias A, Asadi A, Jacobsson A, Cannon B. Ucp1: The only protein able to mediate adaptive non-shivering thermogenesis and metabolic inefficiency. Biochim Biophys Acta (2001) 1504(1):82–106. doi: 10.1016/S0005-2728(00)00247-4
29. Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-Acid-Dependent Ucp1 uncoupling in brown fat mitochondria. Cell (2012) 151(2):400–13. doi: 10.1016/j.cell.2012.09.010
30. Nicholls DG. The physiological regulation of uncoupling proteins. Biochim Biophys Acta (2006) 1757(5-6):459–66. doi: 10.1016/j.bbabio.2006.02.005
31. Dlaskova A, Clarke KJ, Porter RK. The role of ucp 1 in production of reactive oxygen species by mitochondria isolated from brown adipose tissue. Biochim Biophys Acta (2010) 1797(8):1470–6. doi: 10.1016/j.bbabio.2010.04.008
32. Oelkrug R, Kutschke M, Meyer CW, Heldmaier G, Jastroch M. Uncoupling protein 1 decreases superoxide production in brown adipose tissue mitochondria. J Biol Chem (2010) 285(29):21961–8. doi: 10.1074/jbc.M110.122861
33. Kazak L, Chouchani ET, Stavrovskaya IG, Lu GZ, Jedrychowski MP, Egan DF, et al. Ucp1 deficiency causes brown fat respiratory chain depletion and sensitizes mitochondria to calcium overload-induced dysfunction. Proc Natl Acad Sci U.S.A. (2017) 114(30):7981–6. doi: 10.1073/pnas.1705406114
34. Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med (2011) 17(2):200–5. doi: 10.1038/nm.2297nm.2297[pii]
35. Wu Q, Kazantzis M, Doege H, Ortegon AM, Tsang B, Falcon A, et al. Fatty acid transport protein 1 is required for nonshivering thermogenesis in brown adipose tissue. Diabetes (2006) 55(12):3229–37. doi: 10.2337/db06-0749
36. Chondronikola M, Volpi E, Borsheim E, Porter C, Saraf MK, Annamalai P, et al. Brown adipose tissue activation is linked to distinct systemic effects on lipid metabolism in humans. Cell Metab (2016) 23(6):1200–6. doi: 10.1016/j.cmet.2016.04.029
37. Zechner R, Kienesberger PC, Haemmerle G, Zimmermann R, Lass A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J Lipid Res (2009) 50(1):3–21. doi: 10.1194/jlr.R800031-JLR200
38. Li Y, Fromme T, Schweizer S, Schottl T, Klingenspor M. Taking control over intracellular fatty acid levels is essential for the analysis of thermogenic function in cultured primary brown and Brite/Beige adipocytes. EMBO Rep (2014) 15(10):1069–76. doi: 10.15252/embr.201438775
39. Heine M, Fischer AW, Schlein C, Jung C, Straub LG, Gottschling K, et al. Lipolysis triggers a systemic insulin response essential for efficient energy replenishment of activated brown adipose tissue in mice. Cell Metab (2018) 28(4):644–55.e4. doi: 10.1016/j.cmet.2018.06.020
40. Bertholet AM, Kirichok Y. Ucp1: A transporter for h(+) and fatty acid anions. Biochimie (2017) 134:28–34. doi: 10.1016/j.biochi.2016.10.013
41. Labbe SM, Caron A, Bakan I, Laplante M, Carpentier AC, Lecomte R, et al. In vivo measurement of energy substrate contribution to cold-induced brown adipose tissue thermogenesis. FASEB J (2015) 29(5):2046–58. doi: 10.1096/fj.14-266247
42. Ma SW, Foster DO. Uptake of glucose and release of fatty acids and glycerol by rat brown adipose tissue in vivo. Can J Physiol Pharmacol (1986) 64(5):609–14. doi: 10.1139/y86-101
43. Olsen JM, Aslund A, Bokhari MH, Hutchinson DS, Bengtsson T. Acute beta-adrenoceptor mediated glucose clearance in brown adipose tissue; a distinct pathway independent of functional insulin signaling. Mol Metab (2019) 30:240–9. doi: 10.1016/j.molmet.2019.10.004
44. Weir G, Ramage LE, Akyol M, Rhodes JK, Kyle CJ, Fletcher AM, et al. Substantial metabolic activity of human brown adipose tissue during warm conditions and cold-induced lipolysis of local triglycerides. Cell Metab (2018) 27(6):1348–55.e4. doi: 10.1016/j.cmet.2018.04.020
45. Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional brown adipose tissue in healthy adults. N Engl J Med (2009) 360(15):1518–25. doi: 10.1056/NEJMoa0808949360/15/1518[pii]]
46. Winther S, Isidor MS, Basse AL, Skjoldborg N, Cheung A, Quistorff B, et al. Restricting glycolysis impairs brown adipocyte glucose and oxygen consumption. Am J Physiol Endocrinol Metab (2018) 314(3):E214–E23. doi: 10.1152/ajpendo.00218.2017
47. Hao Q, Yadav R, Basse AL, Petersen S, Sonne SB, Rasmussen S, et al. Transcriptome profiling of brown adipose tissue during cold exposure reveals extensive regulation of glucose metabolism. Am J Physiol Endocrinol Metab (2015) 308(5):E380–92. doi: 10.1152/ajpendo.00277.2014ajpendo.00277.2014[pii]
48. Hankir MK, Klingenspor M. Brown adipocyte glucose metabolism: A heated subject. EMBO Rep (2018) 19(9). doi: 10.15252/embr.201846404
49. Isler D, Hill HP, Meier MK. Glucose metabolism in isolated brown adipocytes under beta-adrenergic stimulation. quantitative contribution of glucose to total thermogenesis. Biochem J (1987) 245(3):789–93. doi: 10.1042/bj2450789
50. Yoneshiro T, Wang Q, Tajima K, Matsushita M, Maki H, Igarashi K, et al. Bcaa catabolism in brown fat controls energy homeostasis through Slc25a44. Nature (2019) 572(7771):614–9. doi: 10.1038/s41586-019-1503-x
51. Lopez-Soriano FJ, Fernandez-Lopez JA, Mampel T, Villarroya F, Iglesias R, Alemany M. Amino acid and glucose uptake by rat brown adipose tissue. effect of cold-exposure and acclimation. Biochem J (1988) 252(3):843–9. doi: 10.1042/bj2520843
52. Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S, et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature (2018) 560(7716):102–6. doi: 10.1038/s41586-018-0353-2
53. Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans (2006) 34(Pt 2):217–22. doi: 10.1042/BST20060217
54. Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by pdks. Am J Physiol Endocrinol Metab (2003) 284(5):E855–62. doi: 10.1152/ajpendo.00526.2002
55. Garland PB, Randle PJ, Newsholme EA. Citrate as an intermediary in the inhibition of phosphofructokinase in rat heart muscle by fatty acids, ketone bodies, pyruvate, diabetes, and starvation. Nature (1963) 200:169–70. doi: 10.1038/200169a0
56. Newsholme EA, Randle PJ, Manchester KL. Inhibition of the phosphofructokinase reaction in perfused rat heart by respiration of ketone bodies, fatty acids and pyruvate. Nature (1962) 193:270–1. doi: 10.1038/193270a0
57. Cheema-Dhadli S, Robinson BH, Halperin ML. Properties of the citrate transporter in rat heart: Implications for regulation of glycolysis by cytosolic citrate. Can J Biochem (1976) 54(6):561–5. doi: 10.1139/o76-082
58. Munday MR. Regulation of mammalian acetyl-coa carboxylase. Biochem Soc Trans (2002) 30(Pt 6):1059–64. doi: 10.1042/bst0301059
59. Alam N, Saggerson ED. Malonyl-coa and the regulation of fatty acid oxidation in soleus muscle. Biochem J (1998) 334(Pt 1):233–41. doi: 10.1042/bj3340233
60. McGarry JD, Leatherman GF, Foster DW. Carnitine palmitoyltransferase i. the site of inhibition of hepatic fatty acid oxidation by malonyl-coa. J Biol Chem (1978) 253(12):4128–36. doi: 10.1016/S0021-9258(17)34693-8
61. Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-coa carboxylase 2. Science (2001) 291(5513):2613–6. doi: 10.1126/science.1056843
62. Abu-Elheiga L, Matzuk MM, Kordari P, Oh W, Shaikenov T, Gu Z, et al. Mutant mice lacking acetyl-coa carboxylase 1 are embryonically lethal. Proc Natl Acad Sci U.S.A. (2005) 102(34):12011–6. doi: 10.1073/pnas.0505714102
63. Abu-Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, Wakil SJ. The subcellular localization of acetyl-coa carboxylase 2. Proc Natl Acad Sci U.S.A. (2000) 97(4):1444–9. doi: 10.1073/pnas.97.4.1444
64. Bianchi A, Evans JL, Iverson AJ, Nordlund AC, Watts TD, Witters LA. Identification of an isozymic form of acetyl-coa carboxylase. J Biol Chem (1990) 265(3):1502–9. doi: 10.1016/S0021-9258(19)40045-8
65. Esser V, Brown NF, Cowan AT, Foster DW, McGarry JD. Expression of a cdna isolated from rat brown adipose tissue and heart identifies the product as the muscle isoform of carnitine palmitoyltransferase I (M-cpt i). m-cpt I is the predominant cpt I isoform expressed in both white (Epididymal) and brown adipocytes. J Biol Chem (1996) 271(12):6972–7. doi: 10.1074/jbc.271.12.6972
66. Yamazaki N, Shinohara Y, Shima A, Terada H. High expression of a novel carnitine palmitoyltransferase I like protein in rat brown adipose tissue and heart: Isolation and characterization of its cdna clone. FEBS Lett (1995) 363(1-2):41–5. doi: 10.1016/0014-5793(95)00277-g
67. Doh KO, Kim YW, Park SY, Lee SK, Park JS, Kim JY. Interrelation between long-chain fatty acid oxidation rate and carnitine palmitoyltransferase 1 activity with different isoforms in rat tissues. Life Sci (2005) 77(4):435–43. doi: 10.1016/j.lfs.2004.11.032
68. Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F, Feron O. Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab (2016) 24(2):311–23. doi: 10.1016/j.cmet.2016.07.003
69. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse Large b cell lymphoma. Cancer Cell (2012) 22(4):547–60. doi: 10.1016/j.ccr.2012.08.014
70. De Oliveira MP, Liesa M. The role of mitochondrial fat oxidation in cancer cell proliferation and survival. Cells (2020) 9(12). doi: 10.3390/cells9122600
71. Song A, Dai W, Jang MJ, Medrano L, Li Z, Zhao H, et al. Low- and high-thermogenic brown adipocyte subpopulations coexist in murine adipose tissue. J Clin Invest (2020) 130(1):247–57. doi: 10.1172/JCI129167
72. Sun W, Dong H, Balaz M, Slyper M, Drokhlyansky E, Colleluori G, et al. Snrna-seq reveals a subpopulation of adipocytes that regulates thermogenesis. Nature (2020) 587(7832):98–102. doi: 10.1038/s41586-020-2856-x
73. Spaethling JM, Sanchez-Alavez M, Lee J, Xia FC, Dueck H, Wang W, et al. Single-cell transcriptomics and functional target validation of brown adipocytes show their complex roles in metabolic homeostasis. FASEB J (2016) 30(1):81–92. doi: 10.1096/fj.15-273797
74. Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA, et al. Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab (2018) 27(4):869–85.e6. doi: 10.1016/j.cmet.2018.03.003
75. Yu J, Zhang S, Cui L, Wang W, Na H, Zhu X, et al. Lipid droplet remodeling and interaction with mitochondria in mouse brown adipose tissue during cold treatment. Biochim Biophys Acta (2015) 1853(5):918–28. doi: 10.1016/j.bbamcr.2015.01.020
76. Benador IY, Veliova M, Liesa M, Shirihai OS. Mitochondria bound to lipid droplets: Where mitochondrial dynamics regulate lipid storage and utilization. Cell Metab (2019) 29(4):827–35. doi: 10.1016/j.cmet.2019.02.011
77. Cui L, Mirza AH, Zhang S, Liang B, Liu P. Lipid droplets and mitochondria are anchored during brown adipocyte differentiation. Protein Cell (2019) 10(12):921–6. doi: 10.1007/s13238-019-00661-1
78. Mirza AH, Cui L, Zhang S, Liu P. Comparative proteomics reveals that lipid droplet-anchored mitochondria are more sensitive to cold in brown adipocytes. Biochim Biophys Acta Mol Cell Biol Lipids (2021) 1866(10):158992. doi: 10.1016/j.bbalip.2021.158992
79. Wang H, Sreenivasan U, Gong DW, O'Connell KA, Dabkowski ER, Hecker PA, et al. Cardiomyocyte-specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J Lipid Res (2013) 54(4):953–65. doi: 10.1194/jlr.M032466
80. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J (2008) 27(2):433–46. doi: 10.1038/sj.emboj.7601963
81. Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell (2015) 32(6):678–92. doi: 10.1016/j.devcel.2015.01.029
82. Cui L, Liu P. Two types of contact between lipid droplets and mitochondria. Front Cell Dev Biol (2020) 8:618322. doi: 10.3389/fcell.2020.618322
Keywords: brown adipocytes, fatty acid oxidation, de novo fatty acid synthesis, mitochondrial substrate utilization, uncoupled respiration
Citation: Chang JS (2023) Recent insights into the molecular mechanisms of simultaneous fatty acid oxidation and synthesis in brown adipocytes. Front. Endocrinol. 14:1106544. doi: 10.3389/fendo.2023.1106544
Received: 23 November 2022; Accepted: 10 February 2023;
Published: 21 February 2023.
Edited by:
Endre Károly Kristóf, University of Debrecen, HungaryReviewed by:
Rosemari Otton, Universidade Cruzeiro do Sul, BrazilCopyright © 2023 Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji Suk Chang, amlzdWsuY2hhbmdAcGJyYy5lZHU=