 Nirmal Verma
Nirmal Verma Florin Despa
Florin Despa- Department of Pharmacology and Nutritional Sciences, University of Kentucky, Lexington, KY, United States
Chronic kidney disease (CKD) is increasing worldwide and is associated with diabetic states (obesity, prediabetes and type-2 diabetes mellitus). The kidney is intrinsically susceptible to low oxygen (hypoxia) and renal hypoxia plays a vital role in the progression of CKD. Recent studies suggest an association between CKD and renal deposition of amyloid-forming amylin secreted from the pancreas. Renal accumulation of amyloid-forming amylin is associated with hypertension, mitochondrial dysfunction, increased production of reactive oxygen species (ROS) and activation of hypoxia signaling in the kidney. In this review we will discuss potential associations between renal amylin amyloid accumulation, hypertension, and mechanism of hypoxia-induced kidney dysfunction, including activation of hypoxia-inducible factors (HIFs) and mitochondrial dysfunction.
1. Introduction
The prevalence of kidney disease is increasing worldwide (1). According to the Centers for Disease Control and Prevention’s (CDC) Chronic Kidney Disease in the United States, 2021 report nearly 786,000 people in the United States are living with end-stage renal disease, with 71% on dialysis and 29% with a kidney transplant (2).
Renal hypoxia plays an important role in the progression of kidney disease through mechanisms that involve activation of hypoxia-inducible factor (HIF), a master regulator of cellular adaptation to hypoxia (1, 3, 4). Renal hypoxia is closely associated with the development of renal inflammation and fibrosis, and is common in diabetic nephropathy, anemia, cardiovascular diseases, and sarcopenia (5, 6).
The kidney is intrinsically susceptible to hypoxia. It uses only 10% of oxygen delivered by the renal artery (7). Kidney diseases are characterized by renal fibrosis and gradual decline in the glomerular filtration rate (GFR) or both. Hypoxia is a condition in which organs or cells lack a sufficient amount of oxygen supply (8). The formation of hypoxic status is determined by various factors, low oxygen supply, high energy demand, and cellular resistance to hypoxia. In the kidney, proximal tubular cells are the most sensitive to hypoxic injury and the extent of tubular injury determines the prognosis of kidney disease (9). In response to hypoxia, pericytes detach from the vessel wall and differentiate into activated myofibroblasts in interstitial space, leading to development of renal fibrosis or renal injury (10). In addition, hypoxia also induces endothelial cells activation, followed by leukocyte stasis and blocking blood supply to peritubular capillaries leading to loss of capillaries and exacerbating hypoxia and loss of nephrons (11).
Mitochondria are essential organelles and play an important role in the physiology of all organs including kidneys. Mitochondria produce cellular energy in the form of ATP which is supplied to all cells to perform their normal function. During mitochondrial metabolism, reactive oxygen species (ROS) are produced. In normal conditions ROS function as secondary messengers, inducing redox-sensitive post-translational modifications (PTM) in proteins and activating or deactivating different cell signaling pathways. However, in pathological conditions such as kidney diseases, ROS overproduction causes oxidative stress (OS) and hypoxia, inducing mitochondrial dysfunction and altering its metabolism and dynamic. The latter processes are closely related to changes in the cell redox-sensitive signaling pathways, causing inflammation and apoptosis cell death (12). For its normal function kidney is required a huge amount of energy, which is supplied by mitochondria (13, 14). Therefore, any dysfunction affecting mitochondria will also have a crucial impact on renal cellular function.
Amylin is a 37 amino acid long pancreatic hormone and co-secreted with insulin from beta cells (15–18). Studies from our lab and from other labs showed amylin have novel function in renal function and path-physiology and also in other organs (19–24). A recent study from our lab showed that red blood cell (RBC)-capillary interaction is altered by prediabetic hypersecretion of amylin that could be a potential contributing factor to renal hypoxia in diabetic kidney injury (19). Following findings from other labs also showed a link between amylin and renal physiology: 1) presence of high affinity amylin binding sites in renal cortex (20), 2) in vivo injection of radiolabeled amylin showed presence of amylin binding site on proximal tubules of kidney (22), 3) administration of amylin peptide in human and rats, stimulated plasma renin many folds (20, 25–27), 4) Amylin is a potent stimulator of sodium and water reabsorption in kidney (22), 5) Amylin acts as mitogen, stimulating hyperplasia of epithelial cells of proximal tubules (22).
In this review we will discuss potential associations between renal amylin amyloid accumulation and mechanism of hypoxia-induced kidney dysfunction, including HIF activation and mitochondrial dysfunction.
2. Hypoxia inducible factors (HIFs) and hypoxia signaling
Kidney cells as well as other cells in body are adopted for low oxygen condition or hypoxia through stabilization of HIFs. HIFs are transcription factors responsible for the induction of genes (Erythropoiesis, angiogenesis, glucose metabolism, apoptosis and immune responses) essential for survival under hypoxia conditions (Figure 1).
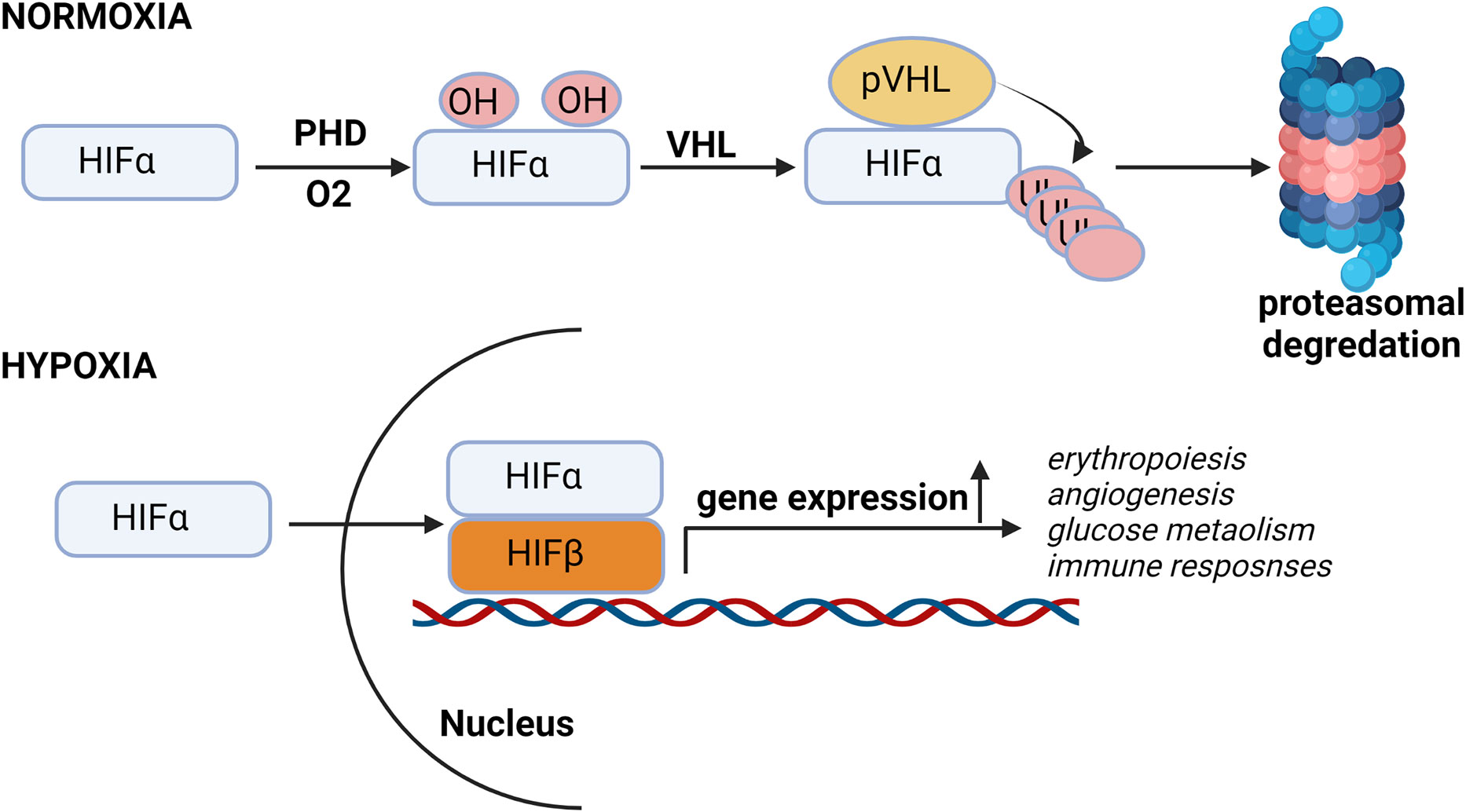
Figure 1 Hypoxia pathway under normal oxygen and hypoxic conditions. In presence of oxygen, HIF-α subunits are hydroxylated by oxygen-dependent prolyl-4-hydroxylases (PHDs) and then Von Hippel–Lindau protein (pVHL), an E3 ubiquitin ligase, binds to the hydroxylated HIF-α and, which leads to the proteasomal degradation of HIF protein. Under low oxygen conditions, HIF is stabilized and translocated into the nucleus, where it binds to its dimerization partner HIF1β and enhances the transcription of HIF target genes.
HIF is heterodimer of constitutively expressing β subunit and an oxygen regulated α subunit. The α subunit are synthesized continuously irrespective of the oxygen status of the cells. Under normal oxygen concentration or normoxia, enzyme prolyl Hydroxylase domain (PHD) hydroxylates proline residues of HIFα. Proline hydroxylated HIFα is recognized by the Von Hippel Lindau (VHL) an E3 ubiquitin ligase complex, resulting in HIFα ubiquitination and subsequent proteasomal degradation (Figure 1) (28–30).
3. Mechanism of hypoxia-induced hypoxic-ischemic injury in kidney
The renal proximal tubules are packed with mitochondria and dependent on oxidative phosphorylation, and are vulnerable to various oxidative injury like hypoxia. In response to hypoxia, tubular epithelial cells (TECs) undergoes changes and start functioning like inflammatory or fibrogenic cells (31). Transformed TECs can facilitate the inflammatory response through production of various bioactive molecules such as pro-inflammatory cytokines (Interleukins, tumor necrosis factors, colony stimulating factors and growth factors), chemokines (monocyte chemoattractant protein-1/CCL2, CXC chemokine ligand 8/IL-8 and CXC chemokine ligand 12/SDF-1), adhesion molecules (intracellular adhesion molecue-1 and selectins), reactive oxygen species and C-reactive proteins which can lead to interstitial inflammation in kidney (Figure 2A).

Figure 2 Mechanism of Hypoxia induced kidney pathologies. Figure shows hypoxia induce kidney damage involves multiple pathways, including RAGE, p38 MAPK, EMT, dysregulation of angiogenesis and inflammation. Under hypoxia condition, renal fibroblasts changed into myofibroblasts and causes increased ECM synthesis to induce renal fibrosis (A). In low oxygen endothelial also trans-differentiates in to myofibroblasts and causes kidney fibrosis. PTE cells are sensitive to hypoxic environments, and NF-κB, Wnt and Notch-1 signaling can be activated to trigger inflammatory cytokines, chemokines, adhesion molecules and peritubular inflammation to promote fibrosis (B). HIFs promote angiogenesis dysregulation by regulating the gene transcription, mRNA, and protein expression of VEGF and VEGF receptors resulted in renal damage (C). Recruitment of proinflammatory cells and cytokines, phenotypic transition of T cells induced by HIF-1α, differentiation and proliferation of regulatory T cells and dendritic cells, etc. are promoters of myofibroblast activation that affect angiogenesis, resulting in collapsing glomerulopathy, decreased capillary flow, intraluminal capillary pressure, and endothelial dysfunction, which in turn aggravates hypoxia (C). Beside Hypoxia, vascular amylin deposition also causes inflammation, endothelial dysfunction and microvascular injury resulted in amylin vasculopathy (D).
Under hypoxia condition TECs could also undergo changes in structure and phenotypes that are accompanied by altered expression and production of profibrotic factors causing tubulointerstitial fibrosis (TIF). As the fibrosis increase tubular capillary network becomes spares and leading to a decreased blood supply and declined renal function (31, 32) (Figure 2A).
Renal endothelial cells (ECs) are another main target of hypoxia. Hypoxia in kidney contribute to renal disease progression by activating the receptor for advance glycation end products (RAGE) and stimulating p38 MAPK and NFkB downstream signaling in ECs. (33) (Figure 2B). Under hypoxia injury, endothelial cells could also differentiate in to myofibroblasts (EndoMT), which further increase the production of extracellular matrix (ECM) and conversely aggravate hypoxia and hypoxia induced injury in kidney (Figure 2B).
Hypoxia also plays a critical role in epithelial-mesenchymal transition (EMT) in proximal tubular cells and activates NFkB signaling to trigger peritubular inflammation through stimulation of Wnt and Notch-1 signaling to promote kidney fibrosis (34–36) (Figure 2B).
Under hypoxia condition renal interstitial fibroblasts could also proliferate and differentiate in to myofibroblasts and promote renal scarring by accelerating extracellular matrix synthesis (Figure 2B) (34–36).
In chronic kidney disease hypoxia contributes to the development of peritubular capillary rarefaction and dysregulation of angiogenesis (37). Hypoxia induced dysregulation of angiogenesis in ECs is regulated by nitric oxide synthases, vascular endothelial growth factor (VEGF), and angiopoietins (38) and affecting the proliferation and migration of endothelial cells (Figure 2C). In addition, hypoxia induced HIF regulates angiogenesis related genes by increase activation of VEGF and internal ribosomal entry. Excessive activation of VEGF in podocytes under hypoxia condition causes collapsing glomerulopathy which resulting in decreased capillary flow and intraluminal capillary pressure. VEGF receptors (VEGFR) are predominantly expressed in endothelial cells in glomerular and peritubular capillaries and over expression of VEGFR in hypoxia also promotes endothelial dysfunction (39–42) (Figure 2C).
Under hypoxia condition, HIF-1α level is increased in T cells and induces phenotypic transition from type 1 helper T cells (Th1) to type 2 T cells (Th2) to amplify the immune response of macrophages and cytotoxic T cells. (43). Beside this increased HIF-1α in hypoxia condition also negatively regulate the adaptive immune system to protect tissues by activating the differentiation and proliferation of regulatory T cells and inhibit effector T cells (44) (Figure 2D). Hypoxia also inhibit the differentiation of dendritic cells to enhance the link between hypoxia and immune response in kidney.
4. Diabetes-associated hyperamylinemia and hypoxic-ischemic injury in kidney
Amylin from humans and a few other species, including cats, dogs, and monkeys, but not rodents, have an increased propensity to aggregate, forming amyloid (i.e., amylin dyshomeostasis) (18, 45–47). Hypersecretion of human amylin is known to activate HIF-1α, a marker of hypoxia signaling (48–50). Accumulating evidence demonstrates the presence of amylin amyloid deposition in heart, brain, and kidneys of patients with type 2 diabetes (51–57). Hypersecretion of human amylin is associated with amylin oligomers deposition in the microvasculature and red blood cells (RBCs) leading to impaired RBC capillaries interaction, increased plasma erythropoietin level and increased hypoxia markers (HIF-1α, HIF-2α, arginase-1, arginase-2 and arginase activity) causing the hypoxic-ischemic injury of renal tissues (19). Amylin deposition in kidney microvasculature also colocalized with macrophages activation which indicates that amylin dyshomeostasis injuries capillaries and associated with inflammatory responses exacerbating ischemic vascular injury in the kidney (Figure 3) (19). This indicates diabetes associated hypersecretion of amylin promotes deposition of amylin oligomers in kidney tissues which can increase hypoxia signaling pathway and inflammation leading to kidney injury and disease (Figure 3).
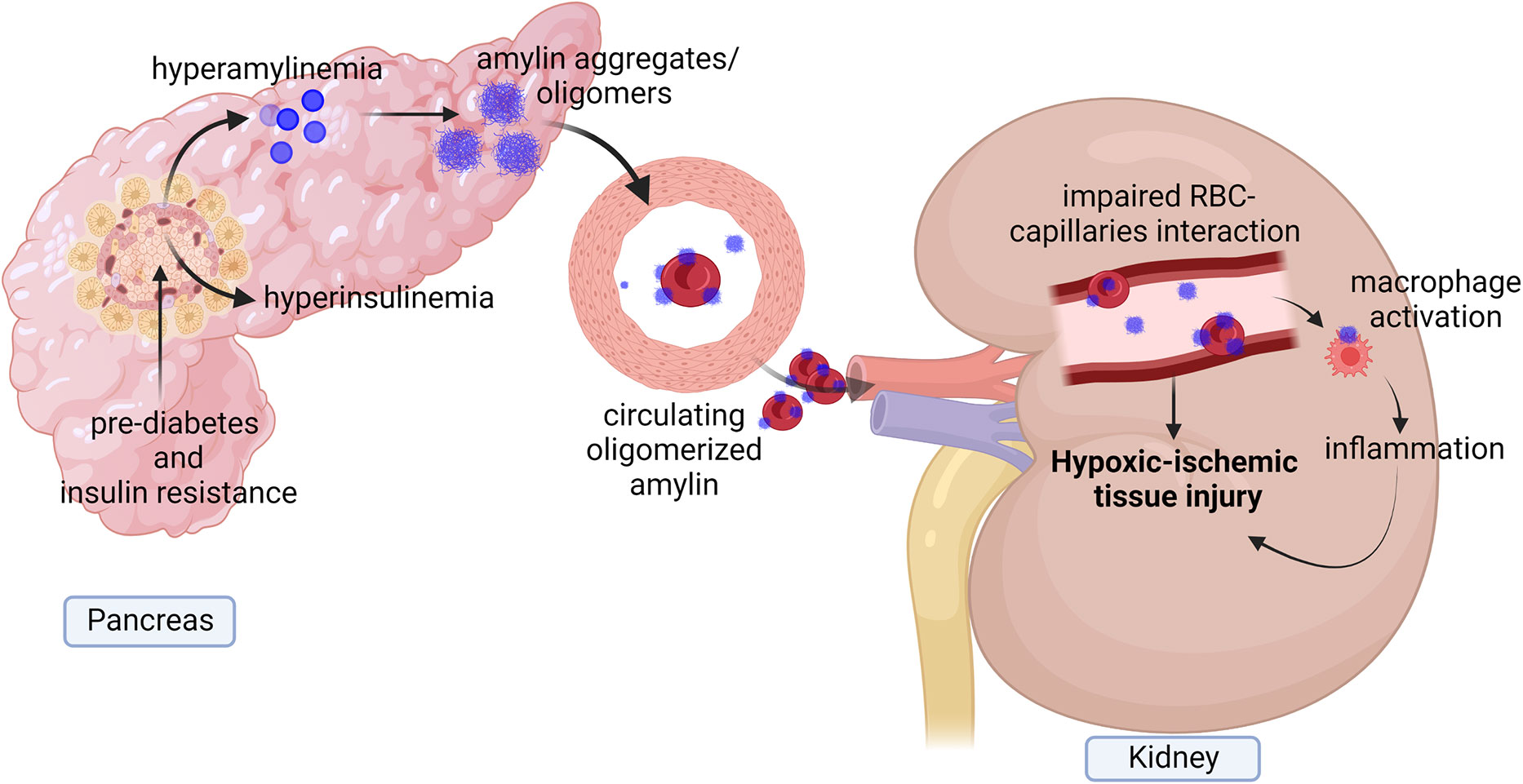
Figure 3 Diagram showing diabetes associate hyperamylinemia increases hypoxia signaling and inflammation in kidney. Prediabetes associated hyperamylinemia promotes aggregation of human amylin in pancreas. Human amylin aggregates also reach to circulation where they deposited in microvasculature and on Red blood cells (RBCs) leading to reduced RBCs capillaries interaction. Accumulation of human amylin aggregates in renal tissues and microvasculature promote elevation of EPO level, hypoxia markers and inflammation causing hypoxic injury of renal tissues.
5. Renal hypoxia is associated with amylin-induced hypertension
Systemic hypertension is caused by the chronic induction of multiple vasoconstrictions including the renin-angiotensin-aldosterone system (58, 59). As the blood vessels constrict, blood flow to kidney tissues is reduced, consequently reducing the oxygen supply to the kidneys (60–63). Besides this hypertension causes the kidney to consume more oxygen compared to normal for the transport of the same amount of sodium (61, 63). Thus, hypertension induces lower renal oxygenation or renal hypoxia through a combination of a reduced supply of oxygen caused by vasoconstriction and increased oxygen demand. It was also reported that hypertension predisposes the kidney to kidney failure by inducing renal hypoxia (64, 65), and detrimental effects of hypoxia are exacerbated by hypertension, rendering renal tissue to produce elevated levels of reactive oxygen species (66). Increased ROS in kidney tissues elevates angiotensin receptors that transduce signals to activate the pro-oxidant enzyme NADPH-oxidase (NOX) (66, 67). ROS produced by hypertension acts in the same way that generated during diabetic kidney diseases drives renal hypoxia injury. Studies with drugs against hypertension in CKD patients also showed renal hypoxia is associated with hypertension. (63, 67–71).
Previous studies have found a link between increased levels of renal amylin and amylin binding sites with increased renal hypertension and thus diabetes-associated hypersecretion of amylin could be involved in hypertension-induced renal hypoxia. High-affinity binding sites for amylin have been reported in kidneys and are involved in the genesis of hypertension (72). Rat models of hypertension, injected with labeled amylin peptide showed an increase in the density of amylin binding sites in the kidney even before the actual increase in systolic blood pressure compared to normal rats (72). These rats showed a further increase in amylin binding sites with the development of systolic blood pressure. Histological examination of kidneys from these rats showed the presence of elevated amylin binding sites in proximal tubules. Further studies in the rat models of renal ablation and hypertension showed systolic blood pressure is correlated with the density of amylin binding in the cortex. Thus, changes in amylin levels and amylin binding sites with renal hypertension showed a possible role of amylin in the development of renal hypertension. The same group of researchers also showed inhibition of angiotensin-converting enzyme reduces the density of amylin binding sites in kidney tissues besides reducing systolic blood pressure which also show a link between amylin and renal hypertension (73). Overall, we can postulate that increased levels of renal hypoxia in diabetes could be caused by amylin-induced hypertension.
6. Mitochondrial reactive oxygen species (mtROS)
Mitochondria are the major contributor to ROS production (~90% of cellular ROS) (74, 75). mtROS produce at the electron transport chain (ETC) during the oxidative phosphorylation of molecular oxygen (O2) to reduced H2O (76–78). During their transport, electron leak and interact with molecular oxygen to form superoxide (O2-.) at complex I and the Q cycle of complex III, which are major sources of superoxide and H2O2 in mitochondria (Figure 4) (78–81). In the first step of ETC, complex I transfer two electrons from nicotine adenine dinucleotide (NADH) to ubiquinone (Q) from low to high potential and reduces ubiquinone to ubiquinol (QH2) (82, 83). During this process, mtROS can be generated in the matrix by complex I (83, 84). Complex III is the major site of ROS generation at the ETC. Complex III has an inner (Qi) and outer (Qo) pools of ubiquinone oriented towards the matrix and at the intermembrane space respectively (85, 86). Ubisemiquinone at complex III is the primary direct electron donor capable of reducing O2 to superoxide. Ubisemiquinone carries a single electron which can move freely in complex III and directly leak the single electron to O2 resulting in ROS generation. Although complex I and complex III are the primary production sites in mitochondria, complex II may produce ROS to a lesser extent. The FAD site of complex II can produce O2- toward the matrix but the production rate of ROS at complex II is very low compared to complex I and complex III (Figure 4) (83, 86).
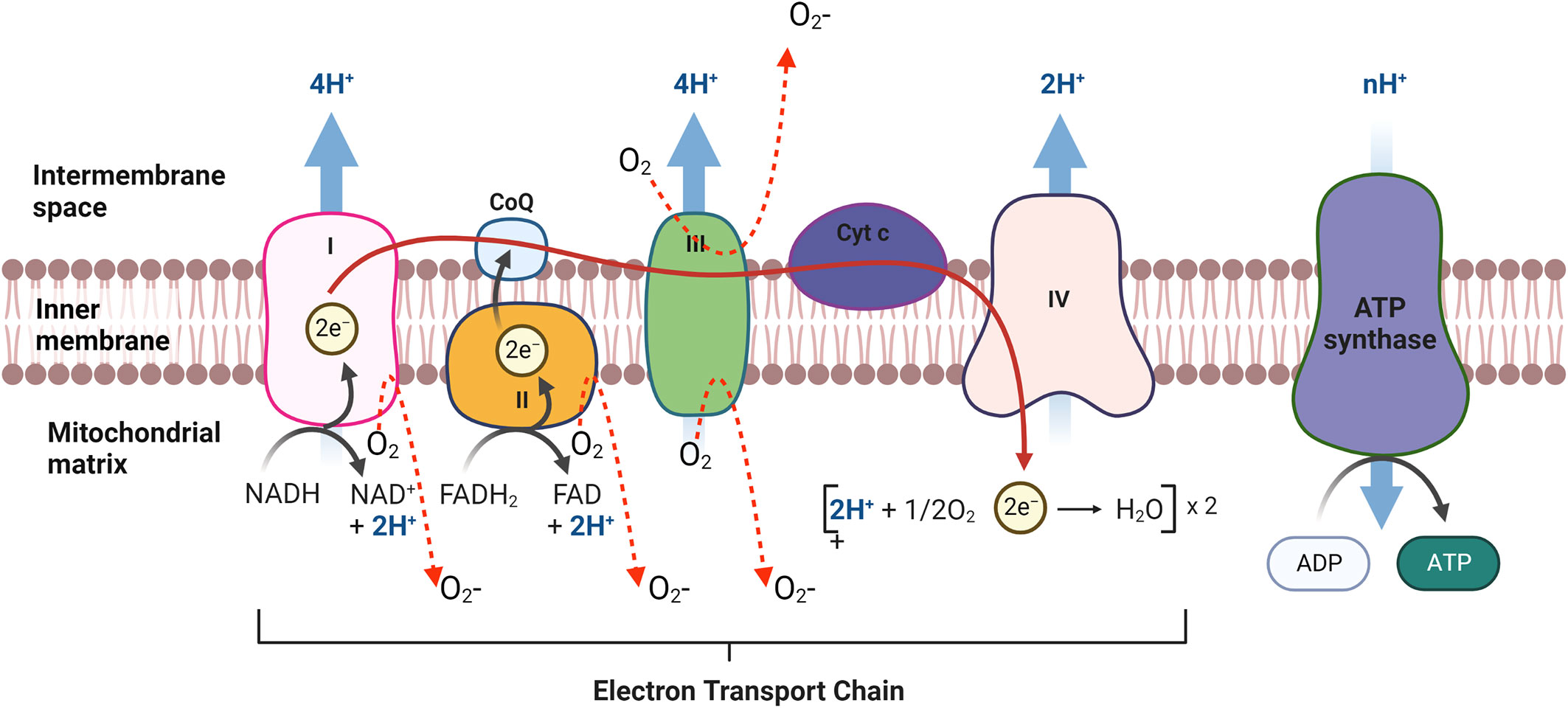
Figure 4 mtROS production in Electron transport chain (ETC) in mitochondria. The ETC is located in the mitochondrial inner membrane (IM). Complex I and II supply electrons to coenzyme Q (CoQ; ubiquinone). Sequentially, electrons are transferred from CoQ to Complex III, cytochrome c (Cyt c) and Complex IV. Oxidative stress is generated during electron transfer.
Other sites in mitochondria except for the ETC, may produce mtROS. The mitochondrial glycerol 3-phosphate dehydrogenase (mGPDH), which oxidized the glycerol 3-phosphate and reduces Q to QH2 resulting in feeding electrons into ETC is capable of generating ROS in mitochondria (87–89). Another site where electrons could escape and form ROS in mitochondria is the electron transferring flavoprotein-ubiquinone oxidoreductase (ETF-QOR) (90). Other sources of mtROS include pyruvate dehydrogenase 2-oxoglutarate dehydrogenase (Odh), dihydroorotate dehydrogenase, and p66shc/cytochrome c (87, 88).
Cellular components other than mitochondria are also capable of producing ROS in kidney. NADPH oxidases (NOX) are accepted as a major source of ROS generating in kidney (91, 92). This family is composed of seven members from NOX1 (colon), NOX2 (phagocytes), NOX3 (inner ear), NOX4, NOX5 (lymphoid tissues), DUXO1 and DUXO2 (thyroid and bronchus). NOX4 is expressed predominantly in kidney and is associated with various renal complications (91, 93, 94).
7. Mitochondrial ROS, HIF stabilization and hypoxic-ischemic injury in kidney
Kidney disease or kidney injury is a condition when the glomerular filtration rate (GFR) is decreased to less than 60ml/min per 1.73m2 or shows the presence of markers for kidney damage or both (95). The most common causes of kidney disease are diabetes and hypertension (95). The kidney needs a large amount of energy to maintain the body’s fluid composition by filtering and reabsorbing materials. Reabsorption requires a huge amount of energy in the form of ATP supplied by mitochondria (13) and thus mitochondria dysfunction will have a crucial impact on kidney function. Overproduction of mtROS in mitochondria (mtROS) is linked to mitochondrial dysfunction and oxidative stress and hypoxia, which is an early event of hypoxic-kidney injury. Mitochondrial dysfunction during kidney disease preceded podocyte fusion and proteinuria and result in epithelial cells to mesenchymal transition of renal tubular cells. (96, 97). Mitochondrial dysfunction not only precedes kidney injury but also contributes to a large increase in oxidative stress and hypoxia and to the development and progression of hypoxic-kidney injury due to loss of mitochondrial membrane potential and a drop in ATP production (98). Mitochondrial dysfunction has been linked to increasing in mtROS. Hydroxyl radicals can damage macromolecules in mitochondria such as mtDNA. Unrepaired damage of mtDNA can lead to defects in complex III, which results in an increased production of ROS and oxidative or hypoxic-kidney damage (99). Increased in oxidative damage can result in releasing intermembrane proteins to the cytosol such as cytochrome c and amplifying oxidative stress in the kidney, which gives rise to a viscous cycle of excessive mtROS production and mitochondrial dysfunction (99–101).
The first indication suggesting mitochondria act like oxygen sensors came when ρ0 Hep3B cells which are deficient in mitochondrial DNA, and thus no electron transport, are incapable of HIF-1α DNA binding activity and thus do not produce Erythropoietin (EPO) in response to low oxygen (102). Another finding where antioxidant treatment abolished the stabilization of HIF-1α under hypoxia also suggested that mitochondrial ROS is responsible for hypoxia signaling or HIFs stabilization under low oxygen conditions (102). Further, treatment of cells with H2O2 or inducing H2O2 production in cells or mutations that lead to H2O2 accumulation in cells is sufficient to increase HIF-1α even in normoxia (103). Embryonic cells lacking cytochrome c fails to stabilize HIF-1α under low oxygen condition also showed mitochondrial ROS is responsible for hypoxia-induced HIF-1α stabilization (104). The use of mitochondrial inhibitors showed ROS generation at complex III but not at complex I or II is critical for hypoxia-induced HIF-1α stabilization (105). Studies, where normal cells were fused with mitochondrial deficient cells, showed that it is not the ability of cells to convert oxygen or conduct oxidative phosphorylation but the ability of cells to produce ROS at mitochondrial complex II that is critical for the HIF-1α stabilization to downward hypoxia signaling.
Thus, an increase in mtROS production resulting from mitochondrial dysfunction in kidney disease can cause stabilization of HIF-1α and hypoxia signaling in the kidney (Figure 5).
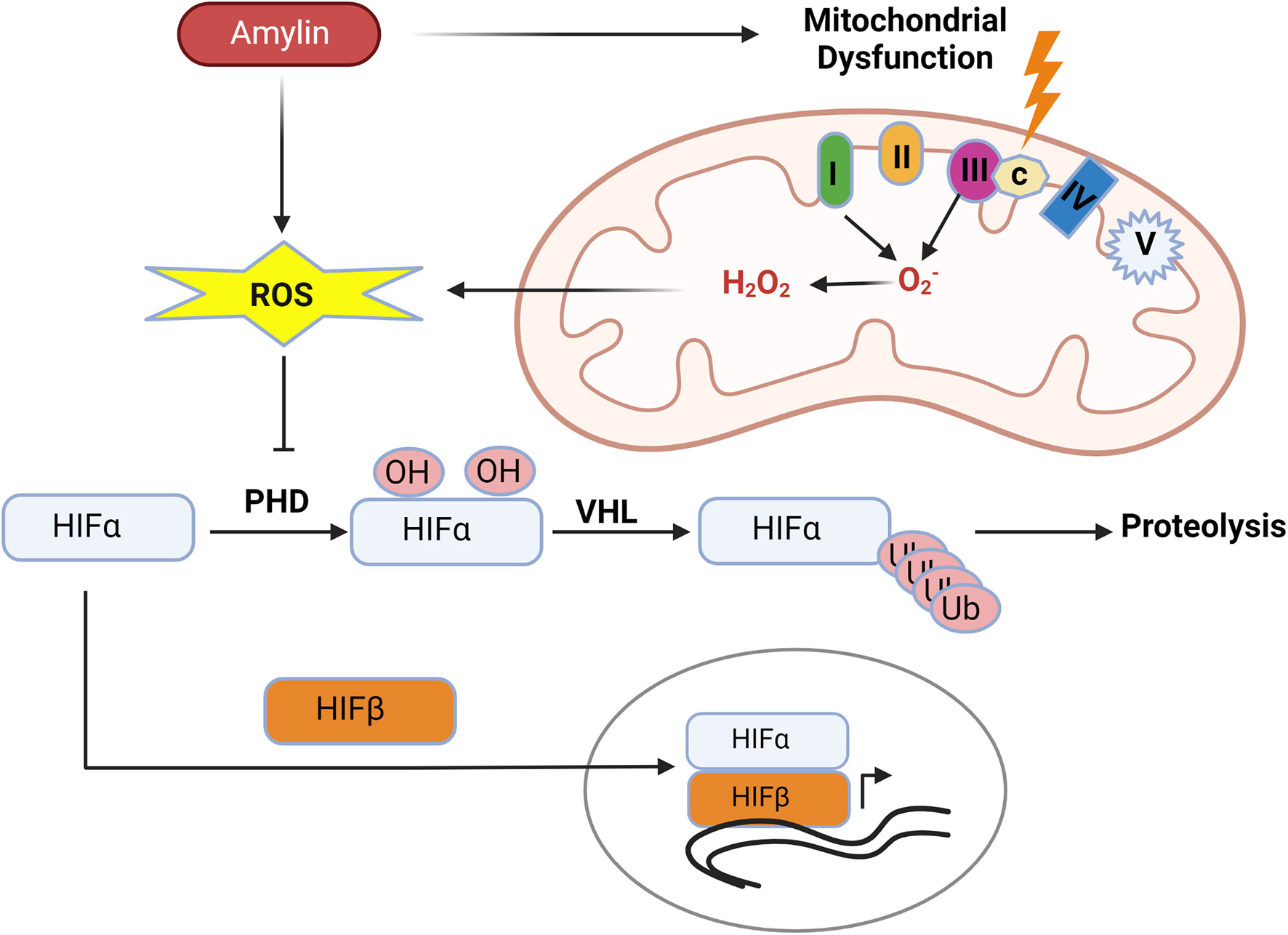
Figure 5 Human amylin induced mitochondrial dysfunction promotes hypoxia. Human amylin induced mitochondrial dysfunction increases production of mtROS which inhibits the activity of PHDs allowing for stabilization of HIF-1α subunits and HIF mediated transcription.
8. Diabetes associated hyperamylinemia, mitochondria dysfunction and mitochondrial ROS
Research has shown that human IAPP or amylin oligomers are cytotoxic and associated with endoplasmic reticulum stress, mitochondrial dysfunction, and mitochondrial ROS (106, 107). In vitro study with INS1F cells showed exogenous human amylin induces mitochondrial dysfunction and cell apoptosis (107). Mitochondrial peptidase pitrilysin regulates human amylin in beta cells’ mitochondria, and the intra-mitochondrial pool of amylin causes beta-cell apoptosis and mitochondrial dysfunction (108). Thus, diabetes-associated hyperamylinemia could promote hypoxic-renal injury by creating mitochondrial dysfunction and ROS. (Figure 5).
9. Perspectives: Pancreatic amyloid-forming amylin as a therapeutic target in CKD
Hypoxia is a critical mediator of the progression of kidney pathologies. Therefore, elucidating the response of kidney to hypoxia and factors that promote hypoxia is of a great significance to understand pathophysiology of kidney disease. Hypersecretion of amyloid-forming amylin is common in persons with prediabetes leading to deposition of aggregated amylin oligomers in the microvasculature of kidney and on RBCs. Amyloid-forming amylin impairs oxygen sensing at RBCs-capillary interface promoting activation of hypoxia signaling pathway in kidney (19), which may induce mitochondrial dysfunction through increasing mtROS generation. Future research is needed to identify inhibitors of amylin-induced hypoxia signaling in renal tissues as a potential therapeutic strategy to counteract the impact of diabetes on kidney function.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tanaka S, Tanaka T, Nangaku M. Hypoxia and hypoxia-inducible factors in chronic kidney disease. Ren Replace Ther (2016) 2:25. doi: 10.1186/s41100-016-0038-y
2. National Institute of Diabetes and Digestive and Kidney Diseases. Kidney disease statistics for the united states (2021). Available at: https://www.niddk.nih.gov/health-information/health-statistics/kidney-disease.
3. Mimura I, Nangaku M. The suffocating kidney: tubulointerstitial hypoxia in end-stage renal disease. Nat Rev Nephrol (2010) 6:667–78. doi: 10.1038/nrneph.2010.124
4. Nangaku M, Rosenberger C, Heyman SN, Eckardt KU. Regulation of hypoxia-inducible factor in kidney disease. Clin Exp Pharmacol Physiol (2013) 40:148–57. doi: 10.1111/1440-1681.12005
5. Sugahara M, Tanaka T, Nangaku M. Hypoxia-inducible factor and oxygen biology in the kidney. Kidney360 (2020) 1:(9) 1021–1031. doi: 10.34067/KID.0001302020
6. Wang B, Li ZL, Zhang YL, Wen Y, Gao YM, Liu BC. Hypoxia and chronic kidney disease. EBioMedicine (2022) 77:103942. doi: 10.1016/j.ebiom.2022.103942
7. Evans RG, Gardiner BS, Smith DW, O'Connor PM. Intrarenal oxygenation: unique challenges and the biophysical basis of homeostasis. Am J Physiol Ren Physiol (2008) 295(5):F1259–70. doi: 10.1152/ajprenal.90230.2008
8. Jager KJ, Kovesdy C, Langham R, Rosenberg M, Jha V, Zoccali C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Kidney Int (2019) 96:1048–50. doi: 10.1016/j.kint.2019.07.012
9. Liu B, Tang T, Lv L, Lan H. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int (2018) 93:568–79. doi: 10.1016/j.kint.2017.09.033
10. Shaw I, Rider S, Mullins J, Hughes J, Péault B. Pericytes in the renal vasculature: roles in health and disease. Nat Rev Nephrol. (2018) 14:521–34. doi: 10.1038/s41581-018-0032-4
11. Rabelink TJ, Wijewickrama DC, de Koning EJ. Peritubular endothelium: the Achilles heel of the kidney? Kidney Int (2007) 72:926–30. doi: 10.1038/sj.ki.5002414
12. Aranda-Rivera AK, Cruz-Gregorio A, Aparicio-Trejo OE, Pedraza-Chaverri J. Mitochondrial redox signaling and oxidative stress in kidney diseases. Biomolecules (2021) 11(8):1144. doi: 10.3390/biom11081144
13. McFarland R, Taylor RW, Turnbull DM. Mitochondrial disease–its impact, etiology, and pathology. Curr Top Dev Biol (2007) 77:113–55. doi: 10.1016/S0070-2153(06)77005-3
14. Irazabal MV, Torres VE. Reactive oxygen species and redox signaling in chronic kidney disease. Cells (2020) 9:1342. doi: 10.3390/cells9061342
15. Cooper GJS, Willis AC, Clark A, Turner RC, Sim RB, Reid KBM. Purification and characterization of a peptide from amyloid rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci USA (1987) 84:8628–32. doi: 10.1073/pnas.84.23.8628
16. Westermark P, Wernstedt C, Wilander E, Hayden DW, O’Brien TD, Johnson KD. Amyloid fibrils in human insulinoma and islets of langerhans of the diabetic cat are derived from neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci U S A (1987) 84:3811–5. doi: 10.1073/pnas.84.11.3881
17. Johnson KH, O’Brien TD, Hayden DW, Jordan K, Ghobrial HKG, Mahoney WC, et al. Immunolocalization of islet amyloid polypeptide (IAPP) in pancreatic beta cells by means of peroxidase-antiperoxidase (PAP) and protein-a gold techniques. Am J Pathol (1988) 130:1–8.
18. Kahn SE, D’Alessio DA, Schwartz MW, Fujimoto WY, Ensinck JW, Taborsky JGJ, et al. Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes (1990) 39:634–8. doi: 10.2337/diab.39.5.634
19. Verma N, Liu M, Ly H, Loria A, Campbell KS, Bush H, et al. Diabetic microcirculatory disturbances and pathologic erythropoiesis are provoked by deposition of amyloid-forming amylin in red blood cells and capillaries. Kidney Int (2020) 97(1):143–55. doi: 10.1016/j.kint.2019.07.028
20. Wookey PJ, Tikellis C, Du H-C, Qin H-F, Sexton PM, Cooper ME. Amylin binding in rat renal cortex, stimulation of adenylyl cyclase and activation of plasma renin. Am J Physiol (1996) 270:F289–94. doi: 10.1152/ajprenal.1996.270.2.F289
21. Cooper ME, Wookey PJ. Amylin: its role in the kidney. Nephrol Dial Transplant (1997) 12:8–10. doi: 10.1093/ndt/12.1.8
22. Harris PJ, Cooper ME, Hiranyachattada S, Berka JL, Kelly DJ, Nobes M, et al. Amylin stimulates proximal tubular sodium transport and cell proliferation in the rat kidney. Am J Physiol (1997) 272:F13–21. doi: 10.1152/ajprenal.1997.272.1.F13
23. Verma N, Despa F. Contributing factors to diabetic brain injury and cognitive decline. Diabetes Metab J (2019) 43(5):560–7. doi: 10.4093/dmj.2019.0153
24. Verma N, Velmurugan GV, Winford E, Coburn H, Kotiya D, Leibold N, et al. Aβ efflux impairment and inflammation linked to cerebrovascular accumulation of amyloid-forming amylin secreted from pancreas. Commun Biol (2023) 6(1):2. doi: 10.1038/s42003-022-04398-2
25. Williams B. Insulin resistance: the shape of things to come. Lancet (1994) 344:521–4. doi: 10.1016/s0140-6736(94)91904-6
26. Cooper ME, McNally PG, Phillips PA, Johnston CI. Amylin stimulates plasma renin concentration in humans. Hypertension (1995) 26:460–4. doi: 10.1161/01.hyp.26.3.460
27. Young AA, Nuttall A, Moyses C, Percy A, Vine W, Rink T. Amylin stimulates the reninangiotensin-aldosterone axis in rats and man. Diabetologia (1995) 38(suppl 1):A225.
28. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science (2001) 292:464–8. doi: 10.1126/science.1059817
29. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von hippel-lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292:468–72. doi: 10.1126/science.1059796
30. Rabinowitz MH. Inhibition of hypoxia-inducible factor prolyl hydroxylase domain oxygen sensors: Tricking the body into mounting orchestrated survival and repair responses. J Med Chem (2013) 56:9369–402. doi: 10.1021/jm400386j
31. Liu WT, Liu XQ, Jiang TT, Wang MY, Huang Y, Huang YL, et al. Using a machine learning model to predict the development of acute kidney injury in patients with heart failure. Front Cardiovasc Med (2022) 9:911987. doi: 10.3389/fcvm.2022.911987
32. Mackensen-Haen S, Bader R, Grund KE, Bohle A. Correlations between renal cortical interstitial fibrosis, atrophy of the proximal tubules and impairment of the glomerular filtration rate. Clin Nephrol. (1981) 15(4):167–71.
33. Matsui T, Oda E, Higashimoto Y, Yamagishi S. Glyceraldehyde derived pyridinium (GLAP) evokes oxidative stress and inflammatory and thrombogenic reactions in endothelial cells via the interaction with RAGE. Cardiovasc Diabetol (2015) 14:1. doi: 10.1186/s12933-014-0162-3
34. Xavier S, Vasko R, Matsumoto K, Zullo JA, Chen R, Maizel J, et al. Curtailing endothelial TGFbeta signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J Am Soc Nephrol. (2015) 26(4):817–29. doi: 10.1681/ASN.2013101137
35. Li ZL, Lv LL, Wang B, Tang TT, Feng Y, Cao JY, et al. The profibrotic effects of MK-8617 on tubulointerstitial fibrosis mediated by the KLF5 regulating pathway. FASEB J (2019) 33(11):12630–43. doi: 10.1096/fj.201901087RR
36. Kuo MC, Chang WA, Wu LY, Tsai YC, Hsu YL. Hypoxia-induced epithelial-to-mesenchymal transition in proximal tubular epithelial cells through miR-545-3p-TNFSF10. Biomolecules (2021) 11(7):1032. doi: 10.3390/biom11071032
37. Chen Q, Yu J, Rush BM, Stocker SD, Tan RJ, Kim K. Ultrasound super-resolution imaging provides a noninvasive assessment of renal microvasculature changes during mouse acute kidney injury. Kidney Int (2020) 98(2):355–65. doi: 10.1016/j.kint.2020.02.011
38. Dou YQ, Kong P, Li CL, Sun HX, Li WW, Yu Y, et al. Smooth muscle SIRT1 reprograms endothelial cells to suppress angiogenesis after ischemia. Theranostics (2020) 10(3):1197–212. doi: 10.7150/thno.39320
39. Stein I, Itin A, Einat P, Skaliter R, Grossman Z, Keshet E. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. Mol Cell Biol (1998) 18(6):3112–9. doi: 10.1128/MCB.18.6.3112
40. Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, et al. Glomerular-specific alterations of VEGF-a expression lead to distinct congenital and acquired renal diseases. J Clin Investig (2003) 111(5):707–16. doi: 10.1172/JCI17423
41. Di Marco GS, Reuter S, Hillebrand U, Amler S, König M, Larger E, et al. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J Am Soc Nephrol. (2009) 20(10):2235–45. doi: 10.1681/ASN.2009010061
42. Rudnicki M, Perco P, Enrich J, Eder S, Heininger D, Bernthaler A, et al. Hypoxia response and VEGFA expression in human proximal tubular epithelial cells in stable and progressive renal disease. Lab Investig (2009) 89(3):337–46. doi: 10.1038/labinvest.2008.158
43. Ben-Shoshan J, Afek A, Maysel-Auslender S, Barzelay A, Rubinstein A, Keren G, et al. HIF-1alpha overexpression and experimental murine atherosclerosis. Arterioscler Thromb Vasc Biol (2009) 29(5):665–70. doi: 10.1161/ATVBAHA.108.183319
44. Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur J Immunol (2008) 38(9):2412–8. doi: 10.1002/eji.200838318
45. Butler PC, Chou J, Carter WB, Wang YN, Bu BH, Chang D, et al. Effects of meal ingestion on plasma amylin concentration in NIDDM and nondiabetic humans. Diabetes (1990) 39:752–6. doi: 10.2337/diab.39.6.752
46. Westermark P, Engström U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci USA (1990) 87:5036–40. doi: 10.1073/pnas.87.13.5036
47. Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev (2011) 91:795–826. doi: 10.1152/physrev.00042.2009
48. Zraika S, Hull R.L., Udayasankar J, Aston-Mourney K, Subramanian S.L., Kisilevsky R., et al. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia (2009) 52:626–35. doi: 10.1007/s00125-008-1255-x
49. Montemurro C, Nomoto H, Pei L, Parekh VS, Vongbunyong KE, Vadrevu S, et al. IAPP toxicity activates HIF1α/PFKFB3 signaling delaying β-cell loss at the expense of β-cell function. Nat Commun (2019) 10:2679. doi: 10.1038/s41467-019-10444-1
50. Liu M, Li N, Qu C, Gao Y, Wu L, Hu LG. Amylin deposition activates HIF1α and 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase 3 (PFKFB3) signaling in failing hearts of non-human primates. Commun Biol (2021) 4(1):188. doi: 10.1038/s42003-021-01676-3
51. Gong W, Liu ZH, Zeng CH, Peng A, Chen HP, Zhou H, et al. Amylin deposition in the kidney of patients with diabetic nephropathy. Kidney Int (2007) 72:213–8. doi: 10.1038/sj.ki.5002305
52. Despa S, Margulies KB, Chen L, Knowlton AA, Havel PJ, Taegtmeyer H, et al. Hyperamylinemia contributes to cardiac dysfunction in obesity and diabetes: A study in humans and rats. Circ Res (2012) 110:598–608. doi: 10.1161/CIRCRESAHA
53. Jackson K, Barisone GA, Diaz E, Jin LW, DeCarli C, Despa F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann Neurol (2013) 74:517–26. doi: 10.1002/ana.23956
54. Despa S, Sharma S, Harris TR, Dong H, Li N, Chiamvimonvat N, et al. Cardioprotection by controlling hyperamylinemia in a “humanized” diabetic rat model. J Am Heart. Assoc (2014) 3:e001015. doi: 10.1161/JAHA.114.001015
55. Liu M, Verma N, Peng X, Srodulski S, Morris A, Chow M, et al. Hyperamylinemia increases IL-1β synthesis in the heart via peroxidative sarcolemmal injury. Diabetes (2016) 65:2772–83. doi: 10.2337/db16-0044
56. Verma N, Ly H, Liu M, Chen J, Zhu H, Chow M, et al. Intraneuronal amylin deposition, peroxidative membrane injury and increased IL-1β synthesis in brains of alzheimer’s disease patients with type-2 diabetes and in diabetic HIP rats. J Alzheimers. Dis (2016) 53:259–72. doi: 10.3233/JAD-160047
57. Ly H, Verma N, Wu F, Liu M, Saatman KE, Nelson PT, et al. Brain microvascular injury and white matter disease provoked by diabetes-associated hyperamylinemia. Ann Neurol (2017) 82:208–22. doi: 10.1002/ana.24992
58. Johnson RJ, Feig DI, Nakagawa T, Sanchez-Lozada LG, Rodriguez-Iturbe B. Pathogenesis of essential hypertension: historical paradigms and modern insights. J hypertension (2008) 26(3):381–91. doi: 10.1097/HJH.0b013e3282f29876
59. Saleh MA, Pollock DM. Endothelin in renal inflammation and hypertension. Contributions to Nephrol (2011) 172:160–70. doi: 10.1159/000328696
60. Welch WJ. Intrarenal oxygen and hypertension. Clin Exp Pharmacol Physiol (2006) 33(10):1002–5. doi: 10.1111/j.1440-1681.2006.04478
61. Welch WJ, Baumgärtl H, Lübbers D, Wilcox CS. Renal oxygenation defects in the spontaneously hypertensive rat: role of AT1 receptors. Kidney Int (2003) 63(1):202–8. doi: 10.1046/j.1523-1755.2003.00729.x
62. Welch WJ, Blau J, Xie H, Chabrashvili T, Wilcox CS. Angiotensin-induced defects in renal oxygenation: role of oxidative stress. Am J Physiol Heart Circulatory Physiol (2005) 288(1):H22–8. doi: 10.1152/ajpheart.00626.2004
63. Tanaka T, Miyata T, Inagi R, Fujita T, Nangaku M. Hypoxia in renal disease with proteinuria and/or glomerular hypertension. Am J Pathol (2004) 165(6):1979–92. doi: 10.1016/S0002-9440(10)63249-X
64. Welch WJ, Mendonca M, Aslam S, Wilcox CS. Roles of oxidative stress and AT1 receptors in renal hemodynamics and oxygenation in the postclipped 2K,1C kidney. Hypertension (2003) 41(3 Pt 2):692–6. doi: 10.1161/01.HYP.0000052945.84627.8F
65. Hansell P, Welch WJ, Blantz RC, Palm F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin Exp Pharmacol Physiol (2013) 40(2):123–37. doi: 10.1111/1440-1681.12034
66. Simão S, Gomes P, Pinto V, Silva E, Amaral JS, Igreja B, et al. Age-related changes in renal expression of oxidant and antioxidant enzymes and oxidative stress markers in male SHR and WKY rats. Exp gerontology (2011) 46(6):468–74. doi: 10.1016/j.exger.2011.02.003
67. Zhou XJ, Vaziri ND, Zhang J, Wang HW, Wang XQ. Association of renal injury with nitric oxide deficiency in aged SHR: prevention by hypertension control with AT1 blockade. Kidney Int (2002) 62(3):914–21. doi: 10.1046/j.1523-1755.2002.00516.x
68. Brezis M, Greenfel Z, Shina A, Rosen S. Angiotensin II augments medullary hypoxia and predisposes to acute renal failure. Eur J Clin Invest (1990) 20(2):199–207. doi: 10.1111/j.1365-2362.1990.tb02269.x
69. Zatz R, Bayli C. Chronic nitric oxide inhibition model six years on. Hypertension (1998) 32(6):958–64. doi: 10.1161/01.hyp.32.6.958
70. Mihailović-Stanojević N, Miloradović Z, Grujić-Milanović J, Ivanov M, Jovović D. Effects of angiotensin II type-1 receptor blocker losartan on age-related cardiovascular risk in spontaneously hypertensive rats. Gen Physiol biophysics (2009) 28:112–8.
71. Panico C, Luo Z, Damiano S, Artigiano F, Gill P, Welch WJ. Renal proximal tubular reabsorption is reduced in adult spontaneously hypertensive rats: roles of superoxide and Na+/H+ exchanger 3. Hypertension (2009) 54(6):1291–7. doi: 10.1161/HYPERTENSIONAHA.109.134783
72. Wookey PJ, Cao Z, van Geenen RC, Voskuil M, Darby IA, Komers R, et al. Increased density of renal amylin binding sites in experimental hypertension. Hypertension (1997) 30(3 Pt 1):455–60. doi: 10.1161/01.hyp.30.3.455
73. Cao Z, Wookey PJ, Wu LL, Voskuil M, van Geenen RC, Cooper ME. Renal amylin binding in normotensive and hypertensive rats: effects of angiotensin converting enzyme inhibition with perindopril. J Hypertension (1997) 15(11):1245–52. doi: 10.1097/00004872-199715110-00008
74. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell (2005) 120:483–95. doi: 10.1016/j.cell.2005.02.001
75. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res (2018) 122:877–902. doi: 10.1161/CIRCRESAHA.117.311401
76. Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res (2007) 100:460–73. doi: 10.1161/01.res.0000258450.44413.96
77. Thomas SR, Witting PK, Drummond GR. Redox control of endothelial function and dysfunction: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal (2008) 10:1713–65. doi: 10.1089/ars.2008.2027
78. Li X, Fang P, Mai J, Choi ET, Wang H, Yang XF. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol (2013) 6:19. doi: 10.1186/1756-8722-6-19
79. Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochem Soc Trans (2008) 36:976–80. doi: 10.1042/BST0360976
80. Tahara EB, Navarete FD, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med (2009) 46:1283–97. doi: 10.1016/j.freeradbiomed.2009.02.008
81. Nickel A, Kohlhaas M, Maack C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol (2014) 73:26–33. doi: 10.1016/j.yjmcc.2014.03.011
82. Ripple MO, Kim N, Springett R. Mammalian complex I pumps 4 protons per 2 electrons at high and physiological proton motive force in living cells. J Biol Chem (2013) 288:5374–80. doi: 10.1074/jbc.M112.438945
83. Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int J Mol Med (2019) 44:3–15. doi: 10.3892/ijmm.2019.4188
84. Hirst J. Towards the molecular mechanism of respiratory complex I. Biochem J (2009) 425:327–39. doi: 10.1042/BJ20091382
85. Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J (2001) 353:411–6. doi: 10.1042/0264-6021:3530411
86. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem (2003) 278:36027–31. doi: 10.1074/jbc.M304854200
87. Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol (2009) 554:165–81. doi: 10.1007/978-1-59745-521-3_11
88. Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol (2010) 45:466–72. doi: 10.1016/j.exger.2010.01.003
89. Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol (2013) 1:304–12. doi: 10.1016/j.redox.2013.04.005
90. Kao MP, Ang DS, Pall A, Struthers AD. Oxidative stress in renal dysfunction: mechanisms, clinical sequelae and therapeutic options. J Hum Hypertens (2010) 24:1–8. doi: 10.1038/jhh.2009.70
91. Sedeek M, Nasrallah R, Touyz RM, Hébert RL. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol JASN (2013) 24(10):1512–8. doi: 10.1681/ASN.2012111112
92. Muñoz M, López-Oliva ME, Rodríguez C, Martínez MP, Sáenz-Medina J, Sánchez A, et al. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol (2020) 28:101330. doi: 10.1016/j.redox.2019.101330
93. Das R, Xu S, Quan X, Nguyen TT, Kong ID, Chung CH, et al. Upregulation of mitochondrial Nox4 mediates TGF-β-induced apoptosis in cultured mouse podocytes. Am J Physiol Renal Physiol (2014) 306(2):F155–67. doi: 10.1152/ajprenal.00438.2013
94. Das R, Xu S, Nguyen TT, Quan X, Choi SK, Kim SJ, et al. Transforming growth factor β1-induced apoptosis in podocytes via the extracellular signal-regulated kinase-mammalian target of rapamycin complex 1-NADPH oxidase 4 axis. J Biol Chem (2015) 290(52):30830–42. doi: 10.1074/jbc.M115.703116
95. Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet (2017) 389:1238–52. doi: 10.1016/S0140-6736(16)32064-5
96. Zhu C, Huang S, Yuan Y, Ding G, Chen R, Liu B, et al. Mitochondrial dysfunction mediates aldosterone-induced podocyte damage: a therapeutic target of PPARγ. Am J Pathol (2011) 178:2020–31. doi: 10.1016/j.ajpath.2011.01.029
97. Su M, Dhoopun AR, Yuan Y, Huang S, Zhu C, Ding G, et al. Mitochondrial dysfunction is an early event in aldosterone-induced podocyte injury. Am J Physiol Renal Physiol (2013) 305:F520–31. doi: 10.1152/ajprenal.00570.2012
98. Bai M, Chen H, Ding D, Song R, Lin J, Zhang Y, et al. MicroRNA-214 promotes chronic kidney disease by disrupting mitochondrial oxidative phosphorylation. Kidney Int (2019) 95:1389–404. doi: 10.1016/j.kint.2018.12.028
99. Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural. Regen. Res (2013) 8:2003–14. doi: 10.3969/j.issn.1673-5374.2013.21.009
100. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J (2009) 417:1–13. doi: 10.1042/BJ20081386
101. Granata S, Dalla Gassa A, Tomei P, Lupo A, Zaza G. Mitochondria: a new therapeutic target in chronic kidney disease. Nutr Metab (2015) 12:49. doi: 10.1186/s12986-015-0044-z
102. Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. (1998) 95:11715–20. doi: 10.1073/pnas.95.20.11715
103. Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, et al. Reduces tumor angiogenesis by protecting cells from oxidative stress. Cell (2004) 118:781–94. doi: 10.1016/j.cell.2004.08.025
104. Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab (2005) 1:393–9. doi: 10.1016/j.cmet.2005.05.003
105. Agani FH, Pichiule P, Chavez JC, LaManna JC. The role of mitochondria in the regulation of hypoxia-inducible factor 1 expression during hypoxia. J Biol Chem (2000) 275:35863–7. doi: 10.1074/jbc.M005643200
106. Hoppener JWM, Lips CJM. Role of islet amyloid in type 2 diabetes mellitus. Int J Biochem Cell Biol.; (2006) 38:726–36. doi: 10.1016/j.biocel.2005.12.009
107. Li X, Chen T, Wong YS, Xu G, Fan RR, Zhao HL, et al. Involvement of mitochondrial dysfunction in human islet amyloid polypeptide-induced apoptosis in INS-1E pancreatic beta cells: an effect attenuated by phycocyanin. Int J Biochem Cell Biol.; (2011) 3:525–34. doi: 10.1016/j.biocel.2010.12.008
Keywords: kidney disease, mitochondria, reactive oxygen species, hypoxia, amylin
Citation: Verma N and Despa F (2023) The association between renal accumulation of pancreatic amyloid-forming amylin and renal hypoxia. Front. Endocrinol. 14:1104662. doi: 10.3389/fendo.2023.1104662
Received: 21 November 2022; Accepted: 06 February 2023;
Published: 16 February 2023.
Edited by:
Kyu-Sang Park, Yonsei University, Republic of KoreaReviewed by:
Akira Sugawara, Tohoku University, JapanRanjan Das, Oregon Health and Science University, United States
Copyright © 2023 Verma and Despa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nirmal Verma, bmlybWFsLnZlcm1hQHVreS5lZHU=