 Jun-Qing Zhang
Jun-Qing Zhang Ying-Ying Li
Ying-Ying Li Xue-Yan Zhang
Xue-Yan Zhang Zeng-Hui Tian2
Zeng-Hui Tian2 Cheng Liu
Cheng Liu Shi-Tao Wang
Shi-Tao Wang Fa-Rong Zhang
Fa-Rong Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 28 February 2023
Sec. Renal Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1085605
This article is part of the Research TopicMyriad Types of Cell Death in Nephropathy and Their Veiled PotentialView all 5 articles
Renal fibrosis (RF) is the common pathological manifestation of virtually all chronic kidney diseases (CKD) and one of the major causes of end-stage renal disease (ESRD), but the pathogenesis of which is still unclear. Renal tubulointerstitial lesions have been identified as a key pathological hallmark of RF pathology. Renal tubular epithelial cells are the resident cells of the tubulointerstitium and play an important role in kidney recovery versus renal fibrosis following injury. Studies in recent years have shown that senescence of renal tubular epithelial cells can accelerate the progression of renal fibrosis. Oxidative stress(OS), telomere attrition and DNA damage are the major causes of renal tubular epithelial cell senescence. Current interventions and therapeutic strategies for cellular senescence include calorie restriction and routine exercise, Klotho, senolytics, senostatics, and other related drugs. This paper provides an overview of the mechanisms and the key signaling pathways including Wnt/β-catenin/RAS, Nrf2/ARE and STAT-3/NF-κB pathway involved in renal tubular epithelial cell senescence in RF and therapies targeting renal tubular epithelial cell senescence future therapeutic potential for RF patients. These findings may offer promise for the further treatment of RF and CKD.
CKD is a syndrome of persistent changes in the structure, function or both of the kidneys and affects the health of the individual. There has been an increase in CKD prevalence over the past few years, with a global prevalence of nearly 13% (1) and a prevalence of 10.8% in China (2). The high prevalence, disability and mortality rates of CKD, as well as the cost of health care, have made it a global public health concern, especially as the treatment for advanced CKD by renal replacement therapy including dialysis and kidney transplantation imposes a significant burden on patients (3). RF is a hallmark feature of kinds of kidney diseases and is the ultimate pathway for progressive kidney function, and improving RF can effectively delay the progression of CKD (4, 5). The main pathological manifestations of RF are glomerulosclerosis, fibrosis of the tubular interstitium and sclerosis or blockage of blood vessels in the kidney, however, the pathogenesis is not fully understood. Therefore, an in-depth investigation into the pathogenesis of CKD and RF and the search for safe and effective drugs for early intervention and treatment can not only solve the financial and mental burden for patients, but also save more medical resources and time costs for society. Cellular senescence occurs when cells are stressed to the point of irreversible cell cycle arrest. While senescent cells do not undergo cell cycle arrest, they remain high metabolic activity and secrete large quantities of cytokines affecting the surrounding microenvironment and neighboring cells cycle, called senescence-associated secretory phenotype (SASP). The morphology of senescent cells also differs from that of living cells; Researchers have observed in vitro cell cultures that senescent cells have larger and flattened cell bodies, vacuolated cytoplasm and abnormal organelles compared to healthy cells (6). Renal tubular epithelial cells are the resident cells of the renal tubular mesenchyme and are central to kidney recovery versus renal fibrosis following injury, which play a vital part in a number of acute and chronic kidney diseases (5, 7, 8). It has been shown that cellular senescence of renal tubular epithelial cells is a driving factor in the development of RF, and that delaying it is an effective measure to inhibit RF and an important strategy to slow down the progression of CKD (9, 10). There is a close relationship between renal tubular epithelial cells senescence and RF, and the onset of cellular senescence can accelerate this process, as this paper will illustrate.
Renal cell senescence was initially described in 1992 (11). Apart from its involvement in physiological renal ageing, senescence also plays an important part in the progression of CKD and acute kidney injury (AKI). It has been reported that accumulation of senescence markers and senescent cells can be detected in several experimental animal models such as ischemia-reperfusion (IRI) (12), unilateral ureteral ligation (UUO) (13) and in renal tissues of CKD patients (14–16). Clinical studies have also shown that accelerated senescent cells have been observed in the kidneys of patients with CKD in the early stages and even in the proteinuria phenotype with normal glomerular filtration rate (16). It is notable that these cells were mainly renal tubular epithelial cells (13), suggesting that renal disease develops and progresses in the presence of renal tubular epithelial cells senescence. In addition, researchers have found that cellular senescence leads to kidney dysfunction and accelerates the progression of kidney disease (16, 17). In CKD, as cellular senescence occurs, senescent cells lose their ability to grow and repair while secreting SASP components (10, 18), including pro-inflammatory factors such as interleukin-1 (IL-1) and interleukin-6 (IL-6), and pro-fibrotic mediators such as transforming growth factor-β1 (TGF-β1) and matrix metalloproteinases (MMPs). These SASP factors, on the one hand, promote the cell’s own senescence and senesce neighboring cells in a paracrine manner. On the other hand, pro-inflammatory factors can lead to an inflammatory response and promote the phenotypic transformation of intrinsic cells, causing them to release a series of toxic factors. These toxic factors, together with the pro-fibrogenic mediators secreted by senescent cells, can contribute to Epithelial-mesenchymal transition(EMT) as well as the proliferation and differentiation of renal interstitial fibroblasts and their transformation into myofibroblasts, resulting in an increase in matrix protein synthesis and a decrease in degradation, and a large amount of extracellular matrix deposition in the renal parenchyma, destroying the normal structure of renal tissue, promoting the formation of fibrous scar, which can facilitate the progression of RF and induce irreversible structural damage to the kidney and renal hypofunction or even loss, ultimately leading to renal failure (6, 19). Moreover, the regenerative capacity of aging kidney cells is significantly reduced, contributing to maladaptive renal repair, which further expedites kidney aging and CKD progression, ultimately accounting for RF (20). Consequently, the accelerated senescence of renal tubular epithelial cells is a driving factor in the development of RF (Figure 1), and it is essential to investigate the pathogenesis of ageing in renal tubular epithelial cells and find the regulators that target the senescence of tubular epithelial cells to prevent and control the progression of RF, which can provide valuable therapeutic targets for treating CKD. This section reviews the mechanisms of renal tubular epithelial cell senescence in RF.
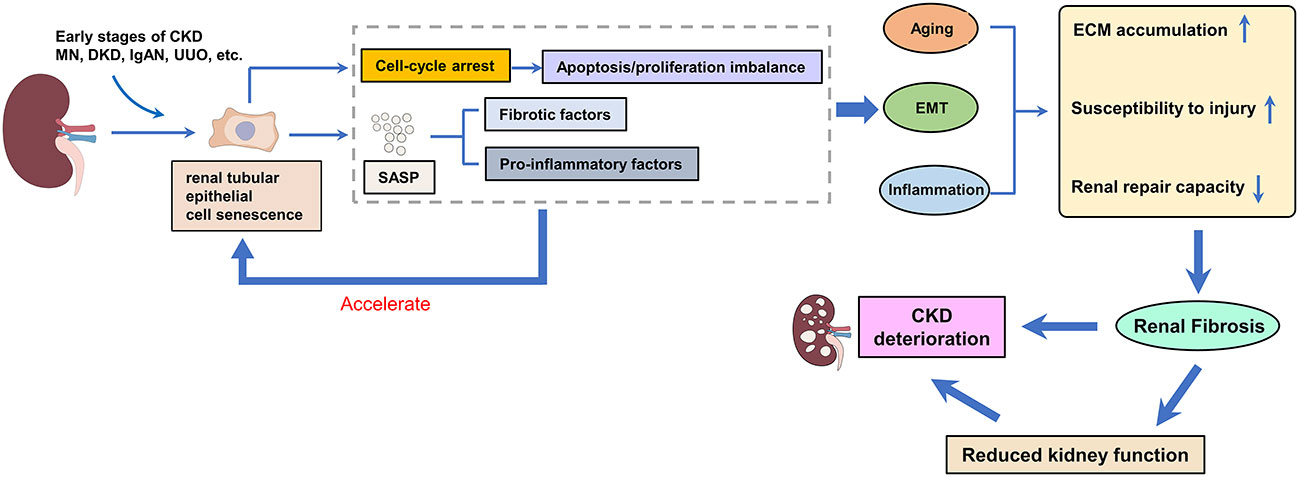
Figure 1 The effect of renal tubular epithelial cell senescence on renal fibrosis. Renal tubular epithelial cells are the main and earliest type of senescent cell to emerge. Senescent cells can secrete SASP (including pro-fibrotic and pro-inflammatory factors), which further accelerates the senescence of renal tubular epithelial cells, and the accelerated senescence of tubular epithelial cells drives the development of renal fibrosis.
OS is the driving force responsible for the loss of renal tubules, glomeruli and endothelial cells, which plays an essential part in the onset and progression of CKD and aging (21–23). OS occurs when oxidant compounds are generated in excess of antioxidant defense mechanisms as a result of external or internal irritants, creating an imbalance, favoring oxidation, resulting in the infiltration of neutrophils, the secretion of proteases, and the generation of large volumes of oxidative intermediates (24). OS is caused by free radicals and is thought to be a contributing factor to cell senescence and CKD (25). An extensive range of acute and chronic kidney diseases involve mitochondrial dysfunction first and foremost in tubular cells (26–28). Reactive oxygen species (ROS) are produced by mitochondria when their morphology changes and their function is lost which will lead to OS and inflammation, triggering DNA damage (24, 29). OS can cause accelerated telomere attrition and cellular senescence, and further exacerbates the progression of RF (30). In this review, we have focused on the crucial signaling pathways of OS, as below and in Figure 2.
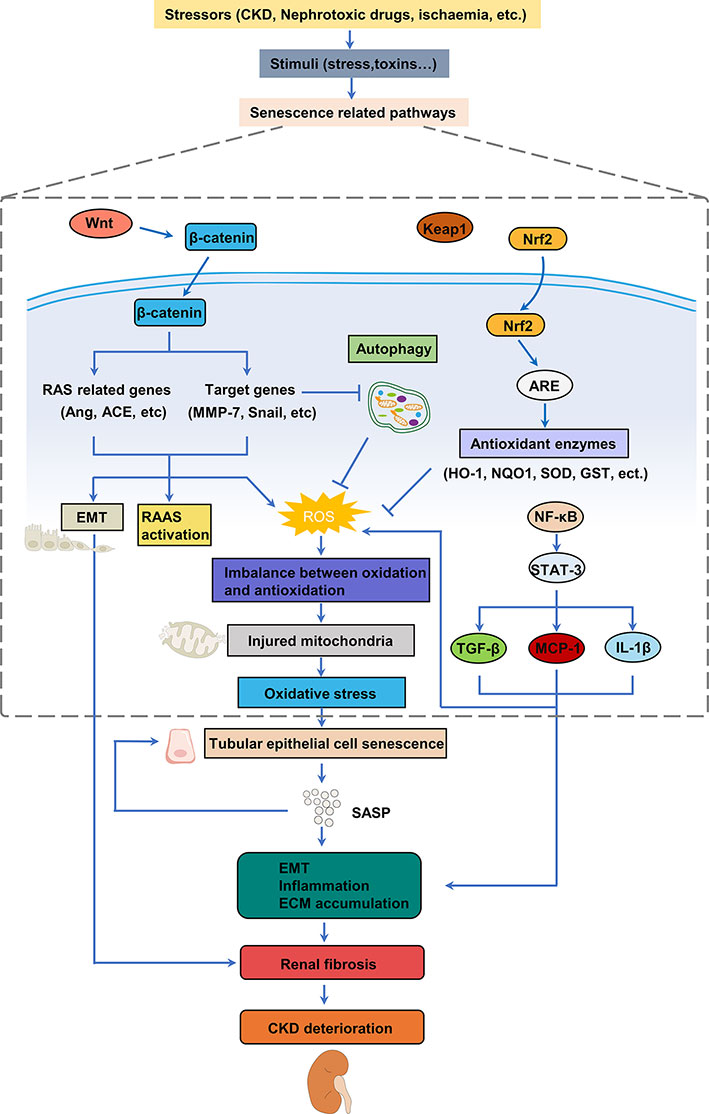
Figure 2 Mechanisms involved in oxidative stress of cellular senescence in renal fibrosis. Various stimulus can trigger oxidative stress by activating different pathways to produce ROS in the kidney, thereby leading to the senescence of renal tubular epithelial cells. Senescence of tubular epithelial cells promotes EMT and leads to the SASP, which increases inflammation and pathological accumulation of extracellular matrix, ultimately resulting in fibrosis and the development of CKD. CKD, chronic kidney disease; EMT, epithelial-mesenchymal transition; SASP, senescence-associated secretory phenotype; ECM, extracellular matrix; ROS, reactive oxygen species; TGF-β, transforming growth factor β; MCP-1, monocyte chemoattractant protein-1.
It is found that Wnt/β-catenin/RAS signaling is essential to mediate cellular senescence-driven RF and implicated in mitochondrial dysfunction (27). Wnt/β-catenin signaling is involved in organ development and tissue repair and is an evolutionarily highly conserved signaling pathway which is silent in normal adult kidneys (31). It is reactivated in different types of CKD by aberrant activation following renal injury and plays a key role in subsequent repair or disease progression following multiple injuries and its abnormal expression is highly correlated with tubulointerstitial fibrosis (12, 32, 33). Wnt/β‐catenin signaling is a notable regulatory element of RAS. The Wnt signaling pathway is activated in the pathological state, causing the disruption of the downstream β-catenin pathway of degradation and resulting in a massive upregulation of its expression. As β-catenin accumulates in large amounts, it undergoes nuclear translocation and enters the nucleus to activate downstream target genes highly relevant to renal fibrosis (fibronectin, Snail1, MMP-7, etc.) and various RAS genes (e.g. Ang, ACE, AT1R), thereby promoting cell proliferation, RAAS activation, and the occurrence of EMT, contributing to the pathological accumulation of extracellular matrix, which can lead to the initiation of RF (32, 34, 35). Consequently, repression of Wnt/β-catenin enables multiple RAS genes to be blocked simultaneously. The findings indicate that Wnt/β‐catenin/RAS signaling pathway plays a crucial role in mitochondrial dysfunction, which is likely to cause the overproduction of ROS in the body, triggering the OS and DNA damage, eventually leading to cellular aging and RF (27). Repression of Wnt/β-catenin can slow down the progression of RF by better blocking RAS and protecting mitochondrial function.
The Nuclear factor erythroid 2-related factor 2 (Nrf2)/antioxidant response element (ARE) signaling pathway is one of the most essential antioxidant regulatory pathways, playing pivotal parts in the mediation of the acclimated stress response of cells to oxidation (36). Nrf2, a redox-sensitive transcription factor encoded by the gene NFE2L2, is a primary regulator of antioxidant enzymes that protect the body from OS and inflammation. Heme oxygenase-1 (HO-1), one of the targets of Nrf2-ARE, is an essential antioxidant enzyme, which can be involved in antioxidant activities in cells and tissues and provides a protective shield against renal injury (37). NAD(P)H: quinone oxidoreductase 1 (NQO1) can suppress the reductive activity of electrons and decrease the formation of ROS through its own enzymatic effect, thus increasing the level of NQO1 is associated with a reduced susceptibility to oxidative damage. Under normal physiological conditions, Nrf2 is anchored in the cytoplasm to the proteasomal degradation by combining with Kelch-like ECH-associated protein 1 (Keap1), while Keap1 is regenerated (38). In response to oxidative damage, Nrf2 separates from Keap1 and free Nrf2 accumulates in the cytoplasm before translocating to the nucleus where it binds to the ARE and activates the transcription of downstream antioxidative enzymes, such as HO-1, NQO1, superoxide dismutase (SOD), glutathionine S-transferase (GST), glutathione reductase (GR) and glutamate-cysteine ligase catalytic subunit (GCLC), to modulate cell redox balance and reduce the extent of damage (39–41). Recent researches have revealed that activation of a variety of signaling pathways, for instance phosphatidylinositide 3-kinases (PI3K)/protein kinase B (Akt) and mitogen-activated protein kinase (MAPK), could promote the liberation of Nrf2 from Keap1 as well as its subsequent translocation to induce the downstream expression of diverse antioxidant proteases (42, 43). Downregulation of Nrf2/ARE can accelerate cellular senescence by promoting oxidative stress, which suggests that drugs targeting Nrf2 signaling can inhibit cellular senescence. In addition, studies demonstrated that the NRF2 pathway may be a critical connection between OS and ferroptosis. The reason why ferroptosis can be attenuated by suppressing mitochondrial oxidative stress through modulation of the Nrf2/ARE pathway is that it represses the capability of Nrf2 to bind ARE within GPX4 and SLC7A11 gene promoters, which in turn induces transcriptional silencing (44). Ferroptosis is characterized by intracellular iron accumulation and lipid peroxidation during cell death, which has been shown the potential association with CKD (45, 46). The connection between Nrf2/ARE and kidney diseases has been described in various reports (25, 37, 47). The lack of Nrf2 can accelerate renal injury in various models, while enhancing Nrf2 activity in renal tubules can dramatically reduce the damage associated with AKI and prevent AKI from progressing to CKD by reducing oxidative stress (48). On the other hand, activation of Nrf2 also protects against fibrosis. In mouse models of unilateral ureteral obstruction, it was observed that Keap1 was downregulated, allowing activation of Nrf2 pathway thereby preventing the production of ROS and improving fibrosis. Nevertheless, prolonged obstruction results in a gradual decrease in nuclear Nrf2, as well as a reduction in antioxidant levels, which leads to increased oxidative stress, inflammation, fibrosis and tubular damage (25). In conclusion, targeted modulation of Nrf2/ARE can ameliorate oxidative damage, in consequence delaying the aging of renal tubular epithelial cells and inhibiting the process of fibrosis and CKD.
The activation of signal transducer and activator of transcription-3 (STAT-3)/activated nuclear factor-κB (NF-κB) signaling pathway has been suggested to be regulatory mechanisms of inflammatory factors including TNFα and IL-6 and associated with the progression of CKD. It was reported that RF reduces renal antioxidant enzyme activation, while oxidative injury also plays a major role in fibrosis. After kidney injury, ROS production increases the expression of NF-κB and pro-inflammatory factors (49, 50) and activates the STAT-3 pathway (51), while NF-κB and pro-inflammatory cytokines further induce the production of excessive ROS (52). These processes lead to the disruption of cellular redox homeostasis and growth of mitochondrial damage, and stimulate the recruitment of inflammatory cells and infiltration of interstitial inflammatory macrophages, and directly augment myofibroblast activation through increasing the production of TGF-β1, IL-1β, and MCP-1 (53), causing OS and inflammation (54), leading to further damage to tubular cells and ultimately to the senescence of renal tubular epithelial cells and RF (55).
MicroRNAs (miRNAs) were first described in Caenorhabditis elegans1 (56) and are highly conserved among species. MiRNAs regulate gene expression following transcription and govern bioprocesses. MiRNAs are integral not only to growth and homeostasis, but also have a significant impact on pathophysiology in the kidney (57, 58). A number of miRNAs have been proven to either kick-start or sustain the cellular reaction that results in fibrosis (59). Studies demonstrated that miR-21 could contribute to renal fibrosis by targeting Smad7, which is a negative regulator of TGF-β1/Smad3 signaling, inducing metabolic disturbances (58). It is reported that miR-9-5P can prevent the down-regulation of genes linked to crucial pathways of metabolism, in particular mitochondrial function, oxidative phosphorylation (OXPHOS), fatty acid oxidation (FAO) and glycolysis, impede TGF-β1-induced bioenergetic disorders, reduce the expression of pro-fibrotic markers in proximal renal tubule cells, and act to delay tubular epithelial cells senescence and inhibit RF (60). In addition, overexpression of miR-214 induces apoptosis and disrupts mitochondrial oxidative phosphorylation in response to a variety of injuries. The proximal tubule-specific absence of MiR-214 impairs inflammation, apoptosis, fibrosis and mitochondrial damage (61). Such findings have highlighted the potential of miRNA as a therapeutic candidate and diagnostic biomarker for renal tubular epithelial cells senescence and RF.
Telomeres are nuclear protein structures located at the ends of eukaryotic chromosomes that maintain the integrity of the genome where telomeres prevent DNA damage and provide a key function in the manipulation of cellular senescence and physical ageing (62). The dysfunction of telomeres results from exceedingly short telomeres or altered telomere structure, and the progressive loss of telomeres contributes to the exposure of DNA ends, causing activation of DNA damage responses (DDRs) (63), ultimately culminating in replicative cellular senescence and chromosomal instability, both of which are significant markers of senescence (64). Investigations support that telomere attrition may have a causal effect on the risk of CKD and that decompensated renal function may be causative for accelerating telomere attrition (65).
Disruptor of telomeric silencing 1-like (Dot1L) is situated on the nucleosome surface (66) which is associated with a number of biological processes, for instance the regulation of transcription, DDRs, cell cycle progression and embryonic cell development (67, 68). It has been found that inhibition of Dot1L decreases ROS levels, attenuates OS and inhibits RF by reducing PI3K/AKT signaling (69). In summary, these results have demonstrated that Dot1L inhibitors could be a prospective target for treatment in RF
DNA damage is a hallmark of numerous forms of nephron damage, activating a series of cell signaling cascades called DDRs to undergo the restoration of DNA integrity, cell cycle arrest in G2/M, cell senescence and cell death, which will cause the onset and progression of RF (70, 71). DNA double-strand breaks (DSBs) are the most powerful initiators of DDRs, and p53/p21 as well as p16/p16INK4a-pRB signal pathway are the two major modulators of these responses (72). It is suggested that targeting DNA damage and repair might serve as an attractive tactic to safeguard the kidney in RF and CKD. Cellular communication network factor 2 (CCN2) was detected to exacerbate DNA damage and the consequent DDR–cellular senescence–fibrosis sequence after kidney lesions (73, 74). Multiple studies have shown that fatty acids (FA) especially saturated FA are likely to be responsible for the induction of the NLRP3 inflammasome via generating ROS, and more elevated levels of ROS can account for oxidative DNA damage (75, 76). It is beneficial to protect the renal tubular epithelial cells from DNA damage by mediating fatty acid uptake and CCN2 inhibition, thereby inhibiting cellular senescence and the progression of RF. Nicotinamide mononucleotide (NMN) has been demonstrated to display beneficial impacts on attenuating RF by repressing tubular DNA damage and cellular aging (77), and NMN administration may be a useful avenue for the prevention or treatment of RF (78).
Renal tubular epithelial cell senescence plays an essential role in the progression of renal fibrosis and CKD. Therefore, strategies to improve senescence of renal tubular epithelial cells, known as senescence therapies, are also potential therapies for these diseases (6). The available interventions for senescence include non-pharmacological therapies (such as caloric restriction and routine exercise), Klotho, drugs that selectively eliminate senescent cells (known as senolytics), drugs that inhibit SASP (known as senostatics) and other related drugs (Table 1).
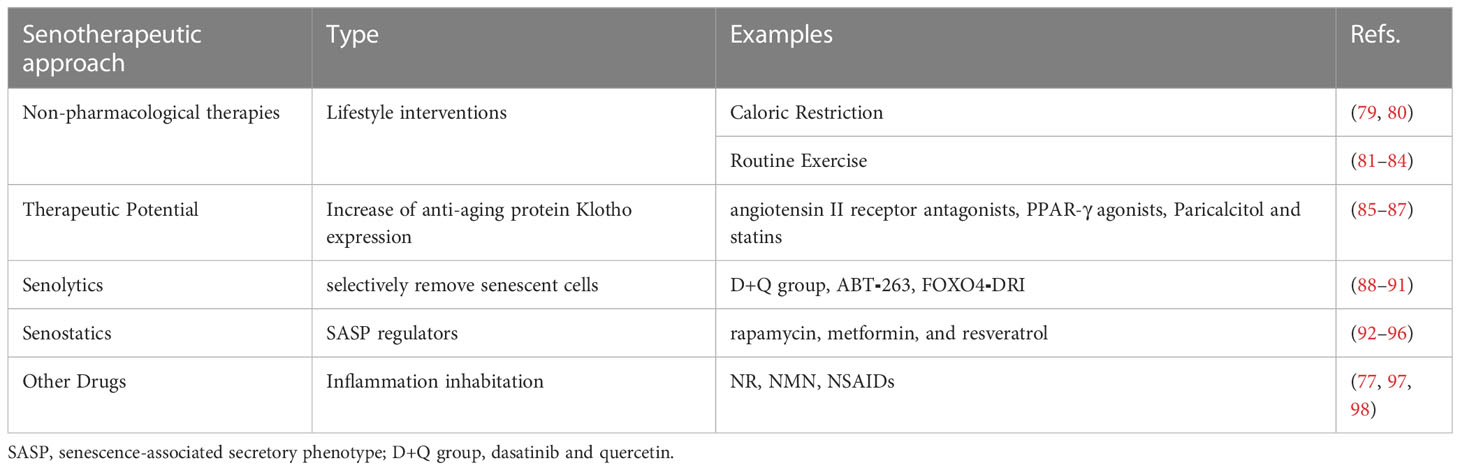
Table 1 Senotherapeutic approaches.
Caloric restriction (CR), which refers to limiting caloric intake without causing damage to the body, is recognized as an effective strategy for maintaining health and prolonging life. CR reduces the expression of aging markers in kidney, and can also delay cellular senescence and changes associated with cellular senescence within kidney, for example glomerulosclerosis, tubular atrophy and interstitial fibrosis (79). The underlying mechanisms involve inhibition of insulin-like growth factor-1 (IGF-1) activation, triggering autophagy through modulating primary metabolic signaling pathways, such as inhibition of mTOR and activation of SIRT1, AMPK, which can reduce oxidative damage (80, 99). Recently, it was found that endogenous hydrogen sulfide (H2S) mediated amino acid restriction, particularly methionine restriction, and exerted a positive effect in inhibiting the protein effect of SASP production in aged kidneys (100, 101). In addition to its antioxidant capacity, H2S also significantly reduces inflammatory cell infiltration, decreases pro-inflammatory cytokines such as TNF-α, IL-6, which inhibits progression of tubular epithelial cells senescence and RF (102–104).
Routine exercise is a promising lifestyle intervention to slow down ageing extend life span. The exercise intervention could result in participants showing measurable declines in BMI, waist circumference and fat mass, as well as a marked decrease in biomarkers of cellular ageing (81). There has been evidence that regular exercise can reduce both ROS and serum AGE levels, attenuating the oxidative stress caused by ageing (82). Interestingly, new study reveals that exercise training can enhance telomerase reverse transcriptase gene expression and telomerase activity, attenuating attrition of telomere and thereby slowing cellular ageing (83). Moreover, another study shows that the combination of melatonin and exercise attenuated metabolic syndrome and reduced anxiety and depression of type 2 diabetic rats by modulating insulin resistance, inflammatory cytokines, mitochondrial biogenesis and ATP levels (84). On the other hand, a meta-analysis reveals that exercise is beneficial in improve the blood pressure profile and significantly reduced VO2 levels of patients with renal failure (105). A latest study also demonstrates that treadmill exercise seems to be a useful intervention for inducing improved peripheral circulation. And it suggests that treadmill exercise should be performed under or near an ambient temperature of 20°C (106). Therefore, it is necessary to take long-term and regular exercise to maintain health and slow down the aging process.
The anti-aging protein Klotho, encoded by the anti-aging gene Klotho, is a single-channel transmembrane protein, mainly α-Klotho protein, divided into membrane and soluble forms. Klotho is highly expressed in normal kidneys, especially in the distal tubules, but oxidative stress, inflammation, angiotensin II, aldosterone and proteinuria, which are present during organ and organism aging and various CKD pathologies, can reduce Klotho expression (107). Klotho is a pleiotropic protein that down-regulates a variety of cytokines and growth factors, such as IGF-1, Wnt/β-catenin and TGF-β1, and plays a variety of biological functions such as regulating cellular senescence, inhibiting apoptosis, inflammation, oxidative stress and regulating calcium and phosphorus metabolism (12, 108, 109), which plays a critical role in delaying renal aging, protecting the kidney from acute and chronic injury, promoting restoration of renal function and delaying the progression of RF and CKD (85, 86, 110). Therefore, it is possible to interfere with renal senescence by upregulating Klotho expression to inhibit RF and delay CKD progression. Researches has showed that Studies have shown that many drugs such as angiotensin II receptor antagonists, PPAR-γ agonists, Paricalcitol and statins may increase endogenous Klotho expression (86, 87). However, to date, there is insufficient clinical evidence for the effects of these drugs on Klotho.
Senolytics are a class of drugs that selectively remove senescent cells, including dasatinib and quercetin (D+Q group), ABT⁃263, FOXO4⁃DRI, etc. Recent studie has found that the D+Q group can reduce the expression of senescence marker proteins such as SA⁃β⁃gal, p16, p21 and inflammatory factors such as IL⁃6 and MCP⁃1 in mouse renal tubular epithelial cells, suggesting that Senolytics could inhibit the progression of RF by delaying the senescence of renal tubular epithelial cells (9, 88). ABT ⁃263 can inhibit the activity of the anti-apoptotic proteins B lymphocytoma 2 (BCL ⁃2) and BCL ⁃XL to induce apoptosis in senescent cells and exerts an anti-aging effect (89, 90). FOXO4 ⁃DRI, a peptide that interferes with the function of FOXO4 protein, competitively inhibits the binding of FOXO4 to p53 in senescent cells and induces apoptosis to delay tubular senescence and renal hypofunction (91). These discoveries unlock an emerging and encouraging therapeutic route for the treatment of CKD and its co-morbidities through optionally targeting senescent cells.
Senostatics are drugs that inhibit the aging phenotype and reduce SASP secretion while maintaining cell viability (18),including rapamycin, metformin, and resveratrol. These compounds can activate autophagy, improve mitochondrial function and inhibit IL-6, IL-8 and other SASP expression, reduce OS (92–95), decrease SA-β-gal levels in tubular epithelial cells (96), thereby delaying the aging of renal tubular epithelial cells and inhibiting RF.
Other candidate drugs include NR and NMN that are NAD+ precursor supplements, and non-steroidal anti-inflammatory drugs (NSAIDs), which can inhibit proinflammatory signaling pathways to reduce inflammation and finally exert an anti-ageing effect (77, 97, 98).
Renal tubular epithelial cells senescence is a driver of RF, and the pathogenesis associated with it includes oxidative stress, telomere shortening, and DNA damage. These factors interact and synergistically contribute to the development of cellular senescence in RTECs. In-depth studies on the molecular mechanisms of RF associated with senescence in renal tubular epithelial cells are expected to provide potentially viable therapeutic ideas for RF, such as caloric restriction, routine exercise, endogenous Klotho, Senolytics, Senostatics, NAD+ supplementation, NSAIDs (Figure 3).
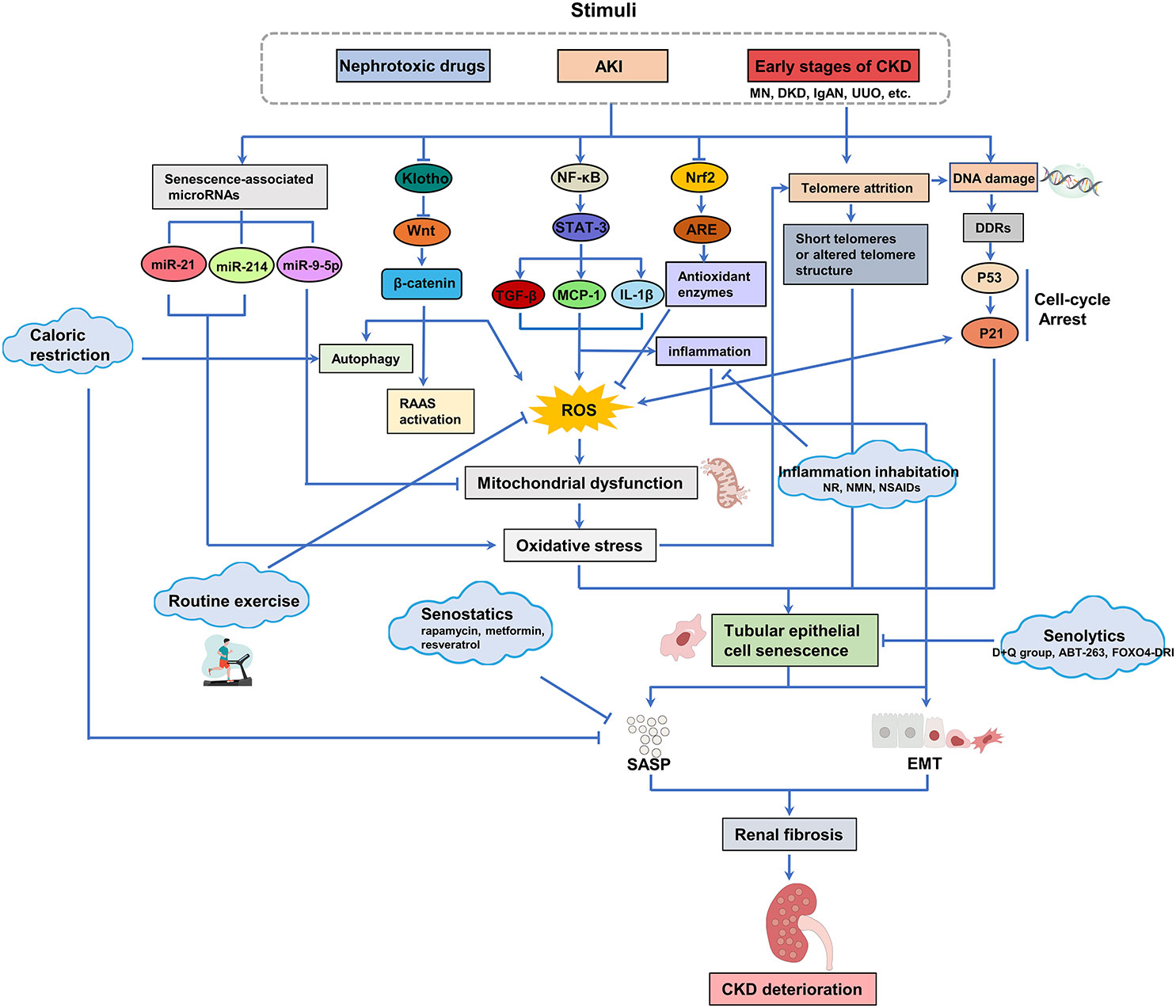
Figure 3 Oxidative stress, telomere attrition and DNA damage caused by stress, chemical or reactive oxygen species (ROS) accumulation are the main reasons for the senescence of renal tubular epithelial cells. The presence of senescent tubular epithelial cells in the kidney can be detected in the early stages of CKD. Senescent cells secrete SASP (including pro-fibrotic and pro-inflammatory factors), which further accelerates the senescence of tubular epithelial cells and drives the progression of RF. Available interventions for senescence include calorie restriction and regular exercise, Klotho, senolytics, senostatics and other related drugs, which offer promise for the treatment of renal fibrosis and CKD associated with renal tubular epithelial cell senescence.
Although the nephroprotective effects of the above therapies are hopeful, and many have been studied in animal models and in vitro, clinical trials of targeted therapeutic strategies are required to assess their safety and efficacy in patients. Moreover, many inflammatory and fibrotic factors are also SASP components and their expression is not adequate to recognize cellular senescence. Given that the available senescence indices may vary among conditions and organs, there is a need for highly sensitive and specific non-invasive methods of detecting senescence (6).In future studies, it would be beneficial to search for more therapeutic targets for RF-related cellular senescence to further facilitate the development and screening of relevant drugs, which will provide new points for the prevention and treatment of CKD and RF.
J-QZ and Y-YL decided on the topics. X-YZ and CL and S-TW reviewed the literature. J-QZ and Z-HT prepared the figures and table. J-QZ wrote the manuscript. F-RZ revised the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by the National Natural Science Foundation of China (No.81774251 and No.82274476). The funding body did not participate in the design of the study, the review of literature, or in writing the manuscript.
We thank the National Natural Science Foundation of China (No.81774251 and No.82274476) for financial support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lv J-C, Zhang L-X. Prevalence and disease burden of chronic kidney disease. Adv Exp Med Biol (2019) 1165:3–15. doi: 10.1007/978-981-13-8871-2_1
2. Zhang L, Wang F, Wang L, Wang W, Liu B, Liu J, et al. Prevalence of chronic kidney disease in China: A cross-sectional survey. Lancet (2012) 379:815–22. doi: 10.1016/S0140-6736(12)60033-6
3. Thurlow JS, Joshi M, Yan G, Norris KC, Agodoa LY, Yuan CM, et al. Global epidemiology of end-stage kidney disease and disparities in kidney replacement therapy. Am J Nephrol (2021) 52:98–107. doi: 10.1159/000514550
4. Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR. Targeting the progression of chronic kidney disease. Nat Rev Nephrol (2020) 16:269–88. doi: 10.1038/s41581-019-0248-y
5. Lin Z, Chen A, Cui H, Shang R, Su T, Li X, et al. Renal tubular epithelial cell necroptosis promotes tubulointerstitial fibrosis in patients with chronic kidney disease. FASEB J (2022) 36:e22625. doi: 10.1096/fj.202200706RR
6. Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO. Cellular senescence: The good, the bad and the unknown. Nat Rev Nephrol (2022) 18:611–27. doi: 10.1038/s41581-022-00601-z
7. Hong S, Healy H, Kassianos AJ. The emerging role of renal tubular epithelial cells in the immunological pathophysiology of lupus nephritis. Front Immunol (2020) 11:578952. doi: 10.3389/fimmu.2020.578952
8. Lu Y-A, Liao C-T, Raybould R, Talabani B, Grigorieva I, Szomolay B, et al. Single-nucleus RNA sequencing identifies new classes of proximal tubular epithelial cells in kidney fibrosis. J Am Soc Nephrol (2021) 32:2501–16. doi: 10.1681/ASN.2020081143
9. Li C, Shen Y, Huang L, Liu C, Wang J. Senolytic therapy ameliorates renal fibrosis postacute kidney injury by alleviating renal senescence. FASEB J (2021) 35:e21229. doi: 10.1096/fj.202001855RR
10. Wang Y, Wang Y, Yang M, Ma X. Implication of cellular senescence in the progression of chronic kidney disease and the treatment potencies. BioMed Pharmacother (2021) 135:111191. doi: 10.1016/j.biopha.2020.111191
11. Khandjian EW, Salomon C, Léonard N, Tremblay S, Türler H. Fibronectin gene expression in proliferating, quiescent, and SV40-infected mouse kidney cells. Exp Cell Res (1992) 202:464–70. doi: 10.1016/0014-4827(92)90100-M
12. Luo C, Zhou S, Zhou Z, Liu Y, Yang L, Liu J, et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J Am Soc Nephrol (2018) 29:1238–56. doi: 10.1681/ASN.2017050574
13. Gong W, Luo C, Peng F, Xiao J, Zeng Y, Yin B, et al. Brahma-Related gene-1 promotes tubular senescence and renal fibrosis through wnt/β-catenin/autophagy axis. Clin Sci (Lond) (2021) 135:1873–95. doi: 10.1042/CS20210447
14. Liu J, Yang J-R, He Y-N, Cai G-Y, Zhang J-G, Lin L-R, et al. Accelerated senescence of renal tubular epithelial cells is associated with disease progression of patients with immunoglobulin a (IgA) nephropathy. Transl Res (2012) 159:454–63. doi: 10.1016/j.trsl.2011.11.008
15. Sosa Peña MDP, Lopez-Soler R, Melendez JA. Senescence in chronic allograft nephropathy. Am J Physiol Renal Physiol (2018) 315:F880–9. doi: 10.1152/ajprenal.00195.2016
16. Kim SR, Puranik AS, Jiang K, Chen X, Zhu X-Y, Taylor I, et al. Progressive cellular senescence mediates renal dysfunction in ischemic nephropathy. J Am Soc Nephrol (2021) 32:1987–2004. doi: 10.1681/ASN.2020091373
17. Mylonas KJ, O'Sullivan ED, Humphries D, Baird DP, Docherty M-H, Neely SA, et al. Cellular senescence inhibits renal regeneration after injury in mice, with senolytic treatment promoting repair. Sci Transl Med (2021) 13(594):eabb0203. doi: 10.1126/scitranslmed.abb0203
18. Docherty M-H, O'Sullivan ED, Bonventre JV, Ferenbach DA. Cellular senescence in the kidney. J Am Soc Nephrol (2019) 30:726–36. doi: 10.1681/ASN.2018121251
19. Xu J, Zhou L, Liu Y. Cellular senescence in kidney fibrosis: Pathologic significance and therapeutic strategies. Front Pharmacol (2020) 11:601325. doi: 10.3389/fphar.2020.601325
20. Marquez-Exposito L, Tejedor-Santamaria L, Valentijn FA, Tejera-Muñoz A, Rayego-Mateos S, Marchant V, et al. Oxidative stress and cellular senescence are involved in the aging kidney. Antioxid (Basel) (2022) 11(2):301. doi: 10.3390/antiox11020301
21. Yuan Q, Tang B, Zhang C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct Target Ther (2022) 7:182. doi: 10.1038/s41392-022-01036-5
22. Fontecha-Barriuso M, Lopez-Diaz AM, Guerrero-Mauvecin J, Miguel V, Ramos AM, Sanchez-Niño MD, et al. Tubular mitochondrial dysfunction, oxidative stress, and progression of chronic kidney disease. Antioxid (Basel) (2022) 11(7):1356. doi: 10.3390/antiox11071356
23. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature (2000) 408:239–47. doi: 10.1038/35041687
24. Ebert T, Neytchev O, Witasp A, Kublickiene K, Stenvinkel P, Shiels PG. Inflammation and oxidative stress in chronic kidney disease and dialysis patients. Antioxid Redox Signal (2021) 35:1426–48. doi: 10.1089/ars.2020.8184
25. Stenvinkel P, Chertow GM, Devarajan P, Levin A, Andreoli SP, Bangalore S, et al. Chronic inflammation in chronic kidney disease progression: Role of Nrf2. Kidney Int Rep (2021) 6:1775–87. doi: 10.1016/j.ekir.2021.04.023
26. Chung KW, Lee EK, Lee MK, Oh GT, Yu BP, Chung HY. Impairment of PPAR and the fatty acid oxidation pathway aggravates renal fibrosis during aging. J Am Soc Nephrol (2018) 29:1223–37. doi: 10.1681/ASN.2017070802
27. Miao J, Liu J, Niu J, Zhang Y, Shen W, Luo C, et al. Wnt/β-catenin/RAS signaling mediates age-related renal fibrosis and is associated with mitochondrial dysfunction. Aging Cell (2019) 18:e13004. doi: 10.1111/acel.13004
28. Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang D, et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol (2017) 11:297–311. doi: 10.1016/j.redox.2016.12.022
29. Krata N, Zagożdżon R, Foroncewicz B, Mucha K. Oxidative stress in kidney diseases: The cause or the consequence? Arch Immunol Ther Exp (Warsz) (2018) 66:211–20. doi: 10.1007/s00005-017-0496-0
30. Maguire D, Neytchev O, Talwar D, McMillan D, Shiels PG. Telomere homeostasis: Interplay with magnesium. Int J Mol Sci (2018) 19(1):157. doi: 10.3390/ijms19010157
31. Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell (2017) 169:985–99. doi: 10.1016/j.cell.2017.05.016
32. Schunk SJ, Floege J, Fliser D, Speer T. WNT-β-catenin signalling - a versatile player in kidney injury and repair. Nat Rev Nephrol (2021) 17:172–84. doi: 10.1038/s41581-020-00343-w
33. He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol (2009) 20:765–76. doi: 10.1681/ASN.2008060566
34. Bogdanova E, Beresneva O, Galkina O, Zubina I, Ivanova G, Parastaeva M, et al. Myocardial hypertrophy and fibrosis are associated with cardiomyocyte beta-catenin and TRPC6/Calcineurin/NFAT signaling in spontaneously hypertensive rats with 5/6 nephrectomy. Int J Mol Sci (2021) 22(9):4645. doi: 10.3390/ijms22094645
35. Zhou L, Mo H, Miao J, Zhou D, Tan RJ, Hou FF, et al. Klotho ameliorates kidney injury and fibrosis and normalizes blood pressure by targeting the renin-angiotensin system. Am J Pathol (2015) 185:3211–23. doi: 10.1016/j.ajpath.2015.08.004
36. Liu S, Pi J, Zhang Q. Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway. Redox Biol (2022) 54:102389. doi: 10.1016/j.redox.2022.102389
37. Uddin MJ, Kim EH, Hannan MA, Ha H. Pharmacotherapy against oxidative stress in chronic kidney disease: Promising small molecule natural products targeting Nrf2-HO-1 signaling. Antioxid (Basel) (2021) 10(2):258. doi: 10.3390/antiox10020258
38. Ulasov AV, Rosenkranz AA, Georgiev GP, Sobolev AS. Nrf2/Keap1/ARE signaling: Towards specific regulation. Life Sci (2022) 291:120111. doi: 10.1016/j.lfs.2021.120111
39. Sun L, Zhao Q, Xiao Y, Liu X, Li Y, Zhang J, et al. Trehalose targets Nrf2 signal to alleviate d-galactose induced aging and improve behavioral ability. Biochem Biophys Res Commun (2020) 521:113–9. doi: 10.1016/j.bbrc.2019.10.088
40. Liu C, Lin H, Jiawei L, Wang Y, Wang C, Sun J, et al. Anwulignan alleviates d-galactose induced renal damage by regulating Nrf2/ARE signaling pathway in mice. Food Sci Biotechnol (2021) 30:1097–105. doi: 10.1007/s10068-021-00951-7
41. Thiruvengadam M, Venkidasamy B, Subramanian U, Samynathan R, Ali Shariati M, Rebezov M, et al. Bioactive compounds in oxidative stress-mediated diseases: Targeting the NRF2/ARE signaling pathway and epigenetic regulation. Antioxid (Basel) (2021) 10(12):1859. doi: 10.3390/antiox10121859
42. Zhuang Y, Wu H, Wang X, He J, He S, Yin Y. Resveratrol attenuates oxidative stress-induced intestinal barrier injury through PI3K/Akt-mediated Nrf2 signaling pathway. Oxid Med Cell Longev (2019) 2019:7591840. doi: 10.1155/2019/7591840
43. Wang Z, Chen Z, Jiang Z, Luo P, Liu L, Huang Y, et al. Cordycepin prevents radiation ulcer by inhibiting cell senescence via NRF2 and AMPK in rodents. Nat Commun (2019) 10:2538. doi: 10.1038/s41467-019-10386-8
44. Chen G-H, Song C-C, Pantopoulos K, Wei X-L, Zheng H, Luo Z. Mitochondrial oxidative stress mediated fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic Biol Med (2022) 180:95–107. doi: 10.1016/j.freeradbiomed.2022.01.012
45. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
46. Carney EF. Ferroptotic stress promotes the AKI to CKD transition. Nat Rev Nephrol (2021) 17:633. doi: 10.1038/s41581-021-00482-8
47. Lu Y, Sun Y, Liu Z, Lu Y, Zhu X, Lan B, et al. Activation of NRF2 ameliorates oxidative stress and cystogenesis in autosomal dominant polycystic kidney disease. Sci Transl Med (2020) 12(554):eaba3613. doi: 10.1126/scitranslmed.aba3613
48. Nezu M, Souma T, Yu L, Suzuki T, Saigusa D, Ito S, et al. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int (2017) 91:387–401. doi: 10.1016/j.kint.2016.08.023
49. Bokhari J, Khan MR. Evaluation of anti-asthmatic and antioxidant potential of boerhavia procumbens in toluene diisocyanate (TDI) treated rats. J Ethnopharmacol (2015) 172:377–85. doi: 10.1016/j.jep.2015.06.049
50. Yu L-M, Zhang W-H, Han X-X, Li Y-Y, Lu Y, Pan J, et al. Hypoxia-induced ROS contribute to myoblast pyroptosis during obstructive sleep apnea via the NF-B/HIF-1 signaling pathway. Oxid Med Cell Longev (2019) 2019:4596368. doi: 10.1155/2019/4596368
51. Xu F, Xu J, Xiong X, Deng Y. Salidroside inhibits MAPK, NF-κB, and STAT3 pathways in psoriasis-associated oxidative stress via SIRT1 activation. Redox Rep (2019) 24:70–4. doi: 10.1080/13510002.2019.1658377
52. Bang E, Kim DH, Chung HY. Protease-activated receptor 2 induces ROS-mediated inflammation through akt-mediated NF-κB and FoxO6 modulation during skin photoaging. Redox Biol (2021) 44:102022. doi: 10.1016/j.redox.2021.102022
53. Hassan NME, Shehatou GSG, Kenawy HI, Said E. Dasatinib mitigates renal fibrosis in a rat model of UUO via inhibition of Src/STAT-3/NF-κB signaling. Environ Toxicol Pharmacol (2021) 84:103625. doi: 10.1016/j.etap.2021.103625
54. Pu W-L, Bai R-Y, Zhou K, Peng Y-F, Zhang M-Y, Hottiger MO, et al. Baicalein attenuates pancreatic inflammatory injury through regulating MAPK, STAT 3 and NF-κB activation. Int Immunopharmacol (2019) 72:204–10. doi: 10.1016/j.intimp.2019.04.018
55. Hassan NME, Said E, Shehatou GSG. Nifuroxazide suppresses UUO-induced renal fibrosis in rats via inhibiting STAT-3/NF-κB signaling, oxidative stress and inflammation. Life Sci (2021) 272:119241. doi: 10.1016/j.lfs.2021.119241
56. Lee RC, Feinbaum RL, Ambros V, The C. Elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell (1993) 75:843–54. doi: 10.1016/0092-8674(93)90529-Y
57. Chandrasekaran K, Karolina DS, Sepramaniam S, Armugam A, Wintour EM, Bertram JF, et al. Role of microRNAs in kidney homeostasis and disease. Kidney Int (2012) 81:617–27. doi: 10.1038/ki.2011.448
58. Mahtal N, Lenoir O, Tinel C, Anglicheau D, Tharaux P-L. MicroRNAs in kidney injury and disease. Nat Rev Nephrol (2022) 18:643–62. doi: 10.1038/s41581-022-00608-6
59. O'Reilly S. MicroRNAs in fibrosis: Opportunities and challenges. Arthritis Res Ther (2016) 18:11. doi: 10.1186/s13075-016-0929-x
60. Fierro-Fernández M, Miguel V, Márquez-Expósito L, Nuevo-Tapioles C, Herrero JI, Blanco-Ruiz E, et al. MiR-9-5p protects from kidney fibrosis by metabolic reprogramming. FASEB J (2020) 34:410–31. doi: 10.1096/fj.201901599RR
61. Bai M, Chen H, Ding D, Song R, Lin J, Zhang Y, et al. MicroRNA-214 promotes chronic kidney disease by disrupting mitochondrial oxidative phosphorylation. Kidney Int (2019) 95:1389–404. doi: 10.1016/j.kint.2018.12.028
62. Akinnibosun OA, Maier MC, Eales J, Tomaszewski M, Charchar FJ. Telomere therapy for chronic kidney disease. Epigenomics (2022) 14:1039–54. doi: 10.2217/epi-2022-0073
63. Hockemeyer D, Palm W, Wang RC, Couto SS, de Lange T. Engineered telomere degradation models dyskeratosis congenita. Genes Dev (2008) 22:1773–85. doi: 10.1101/gad.1679208
64. Aguado J, d'Adda di Fagagna F, Wolvetang E. Telomere transcription in ageing. Ageing Res Rev (2020) 62:101115. doi: 10.1016/j.arr.2020.101115
65. Park S, Lee S, Kim Y, Cho S, Kim K, Kim YC, et al. A Mendelian randomization study found causal linkage between telomere attrition and chronic kidney disease. Kidney Int (2021) 100:1063–70. doi: 10.1016/j.kint.2021.06.041
66. Min J, Feng Q, Li Z, Zhang Y, Xu R-M. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell (2003) 112:711–23. doi: 10.1016/S0092-8674(03)00114-4
67. Wong M, Tee AEL, Milazzo G, Bell JL, Poulos RC, Atmadibrata B, et al. The histone methyltransferase DOT1L promotes neuroblastoma by regulating gene transcription. Cancer Res (2017) 77:2522–33. doi: 10.1158/0008-5472.CAN-16-1663
68. Cao K, Ugarenko M, Ozark PA, Wang J, Marshall SA, Rendleman EJ, et al. DOT1L-controlled cell-fate determination and transcription elongation are independent of H3K79 methylation. Proc Natl Acad Sci U.S.A. (2020) 117:27365–73. doi: 10.1073/pnas.2001075117
69. Yang C, Chen Z, Yu H, Liu X. Inhibition of disruptor of telomeric silencing 1-like alleviated renal ischemia and reperfusion injury-induced fibrosis by blocking PI3K/AKT-mediated oxidative stress. Drug Des Devel Ther (2019) 13:4375–87. doi: 10.2147/DDDT.S224909
70. Yang Y, Zhu X, Yu G, Ma J. Protective effect of pyxinol, one active ingredient of on cisplatin-induced nephrotoxicity via ameliorating DNA damage response. Front Pharmacol (2021) 12:735731. doi: 10.3389/fphar.2021.735731
71. Mori Y, Ajay AK, Chang J-H, Mou S, Zhao H, Kishi S, et al. KIM-1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab (2021) 33:1042–61. doi: 10.1016/j.cmet.2021.04.004
72. Yoon Y-S, You JS, Kim T-K, Ahn WJ, Kim MJ, Son KH, et al. Senescence and impaired DNA damage responses in alpha-synucleinopathy models. Exp Mol Med (2022) 54:115–28. doi: 10.1038/s12276-022-00727-x
73. Valentijn FA, Knoppert SN, Pissas G, Rodrigues-Diez RR, Marquez-Exposito L, Broekhuizen R, et al. CCN2 aggravates the immediate oxidative stress-DNA damage response following renal ischemia-reperfusion injury. Antioxid (Basel) (2021) 10(12):2020. doi: 10.3390/antiox10122020
74. Valentijn FA, Knoppert SN, Marquez-Exposito L, Rodrigues-Diez RR, Pissas G, Tang J, et al. Cellular communication network 2 (connective tissue growth factor) aggravates acute DNA damage and subsequent DNA damage response-senescence-fibrosis following kidney ischemia reperfusion injury. Kidney Int (2022) 102:1305–19. doi: 10.1016/j.kint.2022.06.030
75. Xu D, Liu L, Zhao Y, Yang L, Cheng J, Hua R, et al. Melatonin protects mouse testes from palmitic acid-induced lipotoxicity by attenuating oxidative stress and DNA damage in a SIRT1-dependent manner. J Pineal Res (2020) 69:e12690. doi: 10.1111/jpi.12690
76. Pan J, Ou Z, Cai C, Li P, Gong J, Ruan XZ, et al. Fatty acid activates NLRP3 inflammasomes in mouse kupffer cells through mitochondrial DNA release. Cell Immunol (2018) 332:111–20. doi: 10.1016/j.cellimm.2018.08.006
77. Zheng M, Cai J, Liu Z, Shu S, Wang Y, Tang C, et al. Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J Cell Mol Med (2019) 23:3995–4004. doi: 10.1111/jcmm.14285
78. Jia Y, Kang X, Tan L, Ren Y, Qu L, Tang J, et al. Nicotinamide mononucleotide attenuates renal interstitial fibrosis after AKI by suppressing tubular DNA damage and senescence. Front Physiol (2021) 12:649547. doi: 10.3389/fphys.2021.649547
79. Wang W-J, Chen X-M, Cai G-Y. Cellular senescence and the senescence-associated secretory phenotype: Potential therapeutic targets for renal fibrosis. Exp Gerontol (2021) 151:111403. doi: 10.1016/j.exger.2021.111403
80. Ning Y-C, Cai G-Y, Zhuo L, Gao J-J, Dong D, Cui S, et al. Short-term calorie restriction protects against renal senescence of aged rats by increasing autophagic activity and reducing oxidative damage. Mech Ageing Dev (2013) 134:570–9. doi: 10.1016/j.mad.2013.11.006
81. Englund DA, Sakamoto AE, Fritsche CM, Heeren AA, Zhang X, Kotajarvi BR, et al. Exercise reduces circulating biomarkers of cellular senescence in humans. Aging Cell (2021) 20:e13415. doi: 10.1111/acel.13415
82. Lee J-Y, Paik I-Y, Kim JY. Voluntary exercise reverses immune aging induced by oxidative stress in aging mice. Exp Gerontol (2019) 115:148–54. doi: 10.1016/j.exger.2018.08.009
83. Denham J, Sellami M. Exercise training increases telomerase reverse transcriptase gene expression and telomerase activity: A systematic review and meta-analysis. Ageing Res Rev (2021) 70:101411. doi: 10.1016/j.arr.2021.101411
84. Rahman M, Park S, Jeon H, Kim S. Exercise and oral melatonin attenuate anxiety and depression like behavior in type 2 diabetic rats. J Adv Biotechnol Exp Ther (2021) 4:238–47. doi: 10.5455/jabet.2021.d124
85. Miao J, Huang J, Luo C, Ye H, Ling X, Wu Q, et al. Klotho retards renal fibrosis through targeting mitochondrial dysfunction and cellular senescence in renal tubular cells. Physiol Rep (2021) 9:e14696. doi: 10.14814/phy2.14696
86. Neyra JA, Hu MC, Moe OW. Klotho in clinical nephrology: Diagnostic and therapeutic implications. Clin J Am Soc Nephrol (2020) 16:162–76. doi: 10.2215/CJN.02840320
87. Franco ML, Beyerstedt S, Rangel ÉB. Klotho and mesenchymal stem cells: A review on cell and gene therapy for chronic kidney disease and acute kidney disease. Pharmaceutics (2021) 14(1):11. doi: 10.3390/pharmaceutics14010011
88. Chen Z, Zhu Y, Lu M, Yu L, Tan S, Ren T. Effects of Rosa roxburghii tratt glycosides and quercetin on d-galactose-induced aging mice model. J Food Biochem (2022) 00:e14425. doi: 10.1111/jfbc.14425
89. Jochems F, Thijssen B, De Conti G, Jansen R, Pogacar Z, Groot K, et al. The cancer SENESCopedia: A delineation of cancer cell senescence. Cell Rep (2021) 36:109441. doi: 10.1016/j.celrep.2021.109441
90. Chang J, Wang Y, Shao L, Laberge R-M, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med (2016) 22:78–83. doi: 10.1038/nm.4010
91. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell (2017) 169:132–47. doi: 10.1016/j.cell.2017.02.031
92. Uddin MJ, Farjana M, Moni A, Hossain KS, Hannan MA, Ha H. Prospective pharmacological potential of resveratrol in delaying kidney aging. Int J Mol Sci (2021) 22(15):8258. doi: 10.3390/ijms22158258
93. Liang D, Li Z, Feng Z, Yuan Z, Dai Y, Wu X, et al. Metformin improves the senescence of renal tubular epithelial cells in a high-glucose state through E2F1. Front Pharmacol (2022) 13:926211. doi: 10.3389/fphar.2022.926211
94. Kennedy BK, Lamming DW. The mechanistic target of rapamycin: The grand ConducTOR of metabolism and aging. Cell Metab (2016) 23:990–1003. doi: 10.1016/j.cmet.2016.05.009
95. Zhang T, Chi Y, Kang Y, Lu H, Niu H, Liu W, et al. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1α mediated attenuation of mitochondrial oxidative stress. J Cell Physiol (2019) 234:5033–43. doi: 10.1002/jcp.27306
96. Jiang X, Ruan X-L, Xue Y-X, Yang S, Shi M, Wang L-N. Metformin reduces the senescence of renal tubular epithelial cells in diabetic nephropathy via the MBNL1/miR-130a-3p/STAT3 pathway. Oxid Med Cell Longev (2020) 2020:8708236. doi: 10.1155/2020/8708236
97. Cai Z, Zhang Y, Liu S, Liu X. Celecoxib, beyond anti-inflammation, alleviates tendon-derived stem cell senescence in degenerative rotator cuff tendinopathy. Am J Sports Med (2022) 50:2488–96. doi: 10.1177/03635465221098133
98. Cianciulli A, Calvello R, Ruggiero M, Panaro MA. Inflammaging and brain: Curcumin and its beneficial potential as regulator of microglia activation. Molecules (2022) 27(2):341. doi: 10.3390/molecules27020341
99. Lee G, Uddin MJ, Kim Y, Ko M, Yu I, Ha H. PGC-1α, a potential therapeutic target against kidney aging. Aging Cell (2019) 18:e12994. doi: 10.1111/acel.12994
100. Wang S-Y, Wang W-J, Liu J-Q, Song Y-H, Li P, Sun X-F, et al. Methionine restriction delays senescence and suppresses the senescence-associated secretory phenotype in the kidney through endogenous hydrogen sulfide. Cell Cycle (2019) 18:1573–87. doi: 10.1080/15384101.2019.1618124
101. Lee HJ, Feliers D, Barnes JL, Oh S, Choudhury GG, Diaz V, et al. Hydrogen sulfide ameliorates aging-associated changes in the kidney. Geroscience (2018) 40:163–76. doi: 10.1007/s11357-018-0018-y
102. Pushpakumar S, Kundu S, Weber G, Sen U. Exogenous hydrogen sulfide and miR-21 antagonism attenuates macrophage-mediated inflammation in ischemia reperfusion injury of the aged kidney. Geroscience (2021) 43:1349–67. doi: 10.1007/s11357-020-00299-6
103. Hou C-L, Wang M-J, Sun C, Huang Y, Jin S, Mu X-P, et al. Protective effects of hydrogen sulfide in the ageing kidney. Oxid Med Cell Longev (2016) 2016:7570489. doi: 10.1155/2016/7570489
104. Lee HJ, Feliers D, Barnes JL, Oh S, Choudhury GG, Diaz V, et al. Correction to: Hydrogen sulfide ameliorates aging-associated changes in the kidney. Geroscience (2021) 43:457. doi: 10.1007/s11357-021-00326-0
105. Qiu Z, Zheng K, Zhang H, Feng J, Wang L, Zhou H. Physical exercise and patients with chronic renal failure: A meta-analysis. BioMed Res Int (2017) 2017:7191826. doi: 10.1155/2017/7191826
106. Wada S, Mahbub MH, Yamaguchi N, Hase R, Nakagami Y, Takahashi H, et al. Effect of moderate-intensity treadmill exercise under different ambient temperatures on peripheral circulatory responses in young healthy adults. J Adv Biotechnol Exp Ther (2023) 6:222. doi: 10.5455/jabet.2023.d120
107. Kanbay M, Demiray A, Afsar B, Covic A, Tapoi L, Ureche C, et al. Role of klotho in the development of essential hypertension. Hypertension (2021) 77:740–50. doi: 10.1161/HYPERTENSIONAHA.120.16635
108. He P, Li Z, Yue Z, Gao H, Feng G, Wang P, et al. SIRT3 prevents angiotensin II-induced renal tubular epithelial-mesenchymal transition by ameliorating oxidative stress and mitochondrial dysfunction. Mol Cell Endocrinol (2018) 460:1–13. doi: 10.1016/j.mce.2017.04.027
109. Nagasu H, Satoh M, Kuwabara A, Yorimitsu D, Kidokoro K, Nishi Y, et al. Overexpression of klotho protein modulates uninephrectomy-induced compensatory renal hypertrophy by suppressing IGF-I signals. Biochem Biophys Res Commun (2011) 407:39–43. doi: 10.1016/j.bbrc.2011.02.089
Keywords: cellular senescence, renal fibrosis, renal tubular epithelial cells, oxidative stress, DNA damage, inflammation
Citation: Zhang J-Q, Li Y-Y, Zhang X-Y, Tian Z-H, Liu C, Wang S-T and Zhang F-R (2023) Cellular senescence of renal tubular epithelial cells in renal fibrosis. Front. Endocrinol. 14:1085605. doi: 10.3389/fendo.2023.1085605
Received: 31 October 2022; Accepted: 14 February 2023;
Published: 28 February 2023.
Edited by:
Rashmi Tupe, Symbiosis International University, IndiaReviewed by:
Jamal Uddin, Ewha Womans University, Republic of KoreaCopyright © 2023 Zhang, Li, Zhang, Tian, Liu, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fa-Rong Zhang, ZmFyb25nemhhbmdAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.