 Lanlan Zhang
Lanlan Zhang Faming Jiang
Faming Jiang Yingying Xie
Yingying Xie Yan Mo3
Yan Mo3 Xin Zhang
Xin Zhang Chuntao Liu
Chuntao Liu- 1Department of Respiratory and Critical Care Medicine, Division of Pulmonary Diseases, State Key Laboratory of Biotherapy, West China Hospital of Sichuan University, Chengdu, China
- 2Department of Nephrology, Seventh Affiliated Hospital, Sun Yat-sen University, Shenzhen, China
- 3Department of Neurology Medicine, The Aviation Industry Corporation of China (AVIC) 363 Hospital, Chengdu, China
- 4Department of Gastroenterology, West China Hospital of Sichuan University, Chengdu, China
Type 2 diabetes mellitus (T2DM) is a widespread metabolic condition with a high global morbidity and mortality rate that affects the whole body. Their primary consequences are mostly caused by the macrovascular and microvascular bed degradation brought on by metabolic, hemodynamic, and inflammatory variables. However, research in recent years has expanded the target organ in T2DM to include the lung. Inflammatory lung diseases also impose a severe financial burden on global healthcare. T2DM has long been recognized as a significant comorbidity that influences the course of various respiratory disorders and their disease progress. The pathogenesis of the glycemic metabolic problem and endothelial microangiopathy of the respiratory disorders have garnered more attention lately, indicating that the two ailments have a shared history. This review aims to outline the connection between T2DM related endothelial cell dysfunction and concomitant respiratory diseases, including Coronavirus disease 2019 (COVID-19), asthma, chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF).
Introduction
Monitoring other diseases associated with diabetes is necessary for patients with Type 2 diabetes mellitus (T2DM). Vascular diseases such as coronary artery disease, cerebrovascular accidents, retinopathy, nephropathy, and neuropathy are causes of morbidity and mortality in diabetes (1). Also, diabetic patients are more prone to asthma, chronic obstructive pulmonary disease (COPD), obstructive sleep apnea syndrome (OSA), acute lung injury, and respiratory infections (2). In addition, many studies have shown that various metabolic pathologies accompany respiratory diseases, and the pathophysiological mechanisms that determine the major degenerative complications of diabetes may also contribute to the leading cause of lung function deficits (3, 4).
Studies have shown that endothelial dysfunction largely influences diabetic vasculopathy’s pathophysiology. Even in the early stages of diabetic microangiopathy, endothelial dysfunction is already present (5–7). Although it is unclear whether endothelial dysfunction is a feature of diabetes itself or whether other factors are necessary to cause endothelial dysfunction, given the presence of diabetes, it is believed that endothelial dysfunction is the hallmark stage of diabetes (8). Many factors contribute to the emergence of endothelial dysfunction in diabetes. Activation of protein kinase C (PKC), increased expression of transforming growth factor-beta (TGF-β) and vascular endothelial growth factor (VEGF), non-enzymatic glycation, oxidative stress, activation of the coagulation cascade, increased expression of tumor necrosis factor-alpha (TNF-α), high levels of insulin and insulin precursor molecules, and hyperglycemic pseudohypoxia are a few common hypotheses. These factors may all contribute to endothelial dysfunction However, in the situations of diabetes and lung disease, the importance of these proposed pathways has not been evaluated (5).
Given that the lungs are anatomically covered in a significant number of vascular endothelial cells and that this increases the likelihood that endothelial cell damage brought on by diabetic lesions will result in lung disease, endothelial cells play a special role in “connecting” diabetes and lung disease. The discovery of abnormal pulmonary function in some diabetic patients raise the possibility that the lung should be viewed as a “target organ” for diabetes (8, 9). Endothelial dysfunction in chronic lung diseases has been gradually increasing in recent years, especially in Coronavirus disease 2019 (COVID-19) and idiopathic pulmonary fibrosis (IPF), COPD, and asthma, while endothelial dysfunction in diabetes leading to acute or chronic airway diseases of the respiratory is almost rarely studied (10–13). The primary purpose of this review is to examine the mechanisms of action of the vascular endothelial cells as a shared target in the co-morbidity of T2DM and respiratory disease (COVID-19,COPD, asthma, and IPF) as well as the effects of T2DM treatment on respiratory disease via the vascular endothelial cells.
Potential mechanisms of diabetes related lung disease
The traditional role of blood vessels is to carry oxygen and vital nutrients to other tissues. The primary core lesion cells in most of these vascular disorders are endothelial cells. Long believed to be only controlled by angiogenic growth factors like VEGF and other signals like Notch, research indicates that the metabolic switch in endothelial cells also has an impact on the angiogenic switch (14). The vessel wall is impacted by the metabolic environment of T2DM, which includes endothelial dysfunction, platelet overactivity, oxidative stress, and inflammation, as well as insulin resistance, hyperglycemia, and the formation of excess free fatty acids and other metabolic abnormalities. Vasoconstriction is further intensified, and thrombosis is expedited when these mechanisms are active. The endothelial dysfunction driven on by diabetes is a critical early stage that cannot be ignored in the development of vascular issues. Reduced nitric oxide (NO) release, increased oxidative stress, increased generation of inflammatory agents, aberrant angiogenesis, and impaired endothelium repair are the typical symptoms of endothelial dysfunction in microvascular issues (15).
In clinical practice, abnormal pulmonary function is observed in some diabetic patients; the most consistent abnormalities are reduced lung volume, pulmonary elastic recoil, and pulmonary diffusion impairment in both young and adult diabetic subjects, which are caused by decreased capillary blood volume in the adult group (16, 17). The existence of pulmonary microangiopathy is suggested by the histological data, which also shows thickening of the basal lamina of the pulmonary capillaries (18). The most frequent pathogenetic explanation for mechanical pulmonary dysfunction in diabetic subjects is non-enzymatic glycosylation-induced pulmonary connective tissue changes, while the most plausible explanation for impaired pulmonary microangiopathy in these patients is the presence of underlying pulmonary microangiopathy; abnormal endothelial cell function is another frequent mechanism (18, 19). It has been reported in the literature that common pathogenetic mechanisms exist between diabetes and respiratory disorders and that these mechanisms may play a significant role in the diabetes-induced decline in lung function (Table 1). However, further research is presently needed to determine the importance of endothelial cell dysfunction for respiratory disease.
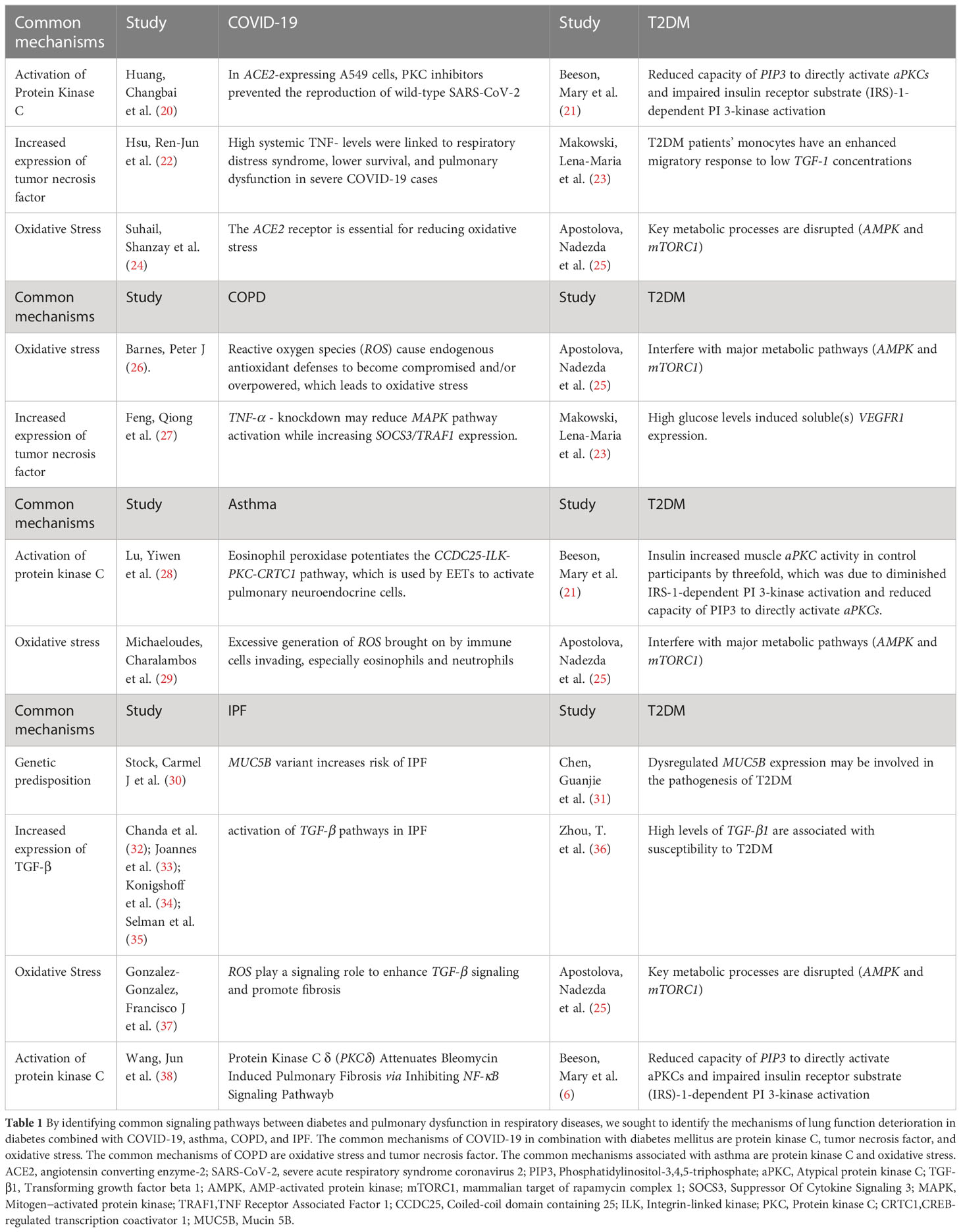
Table 1 T2DM pathophysiology shared by COVID-19, asthma, COPD and IPF.
Common etiology of T2DM related endothelial microangiopathy
Fewer research has evaluated the processes of endothelial cells as a shared target for diabetes and respiratory disorders. Different organ systems, even vascular beds in the same area of a vascular bed exhibit considerable variability in endothelial cell responses. In addition to the variances, there are shared signaling pathways. The research of Nitric oxide (NO) has focused particularly on prostacyclin and NO, two key byproducts of endothelial cells that are crucial for managing vascular homeostasis. Endothelial NO synthase (eNOS), which is crucial for the regulation of endothelial function, produces NO via an enzymatic process. Studies in models of T2DM have shown that aberrant NADPH oxidases (NOX) activation results in endothelial dysfunction and eNOS (39). Recent studies have shown that there is a common pathogenesis in targeting vascular endothelial cells for the treatment of diabetes and pulmonary diseases. Glucagon-like peptide-1 (GLP-1), for instance, are extensively used in the treatment of diabetes, and the research shows that it relates to the function of the vascular endothelial cells. GLP-1 decreased reactive oxygen species-induced senescence in human umbilical vein endothelial cells (HUVECs) in a receptor-dependent manner that included downstream Protein kinase cAMP-dependent (PKA) signaling and the activation of antioxidant genes (40). Studies conducted in vitro on HUVECs revealed that GLP-1 receptor agonists (GLP-1RAs) decreased reactive oxygen species-induced senescence in a receptor-dependent manner that included downstream PKA signaling and activation of antioxidant genes (41). Advanced glycation end products (AGEs) increased endothelial cells, however GLP-1-RAs suppressed these endothelial cells by reducing AGE receptor (RAGE) expression and upregulating VCAM-1 mRNA levels (42). Inhibiting PKC-α, NADPH oxidase, NF-κB signaling, and upregulating anti-inflammatory and antioxidant enzymes, GLP-1RAs significantly reduced inflammation and had antioxidant effects on endothelial cells (43). GLP-1RAs significantly reduced inflammation in human aortic endothelial cells. They also showed that it could raise intracellular Ca2+ and activate Calcium/calmodulin-dependent protein kinase kinase (CAMKK), which in turn could activate AMPK (44). Endothelin-1, which is also present in endothelial cells, is inhibited by GLP-1RAs via preventing the activation of nuclear factor kappa B (45). Common diseases of pulmonary dysfunction, such as COVID-19, asthma, COPD, IPF, and T2DM, have abnormal vascular endothelial cell function, and diabetes drug targets are commonly expressed in the vascular endothelial cells (Figure 1); However, in addition to common signaling pathways, lung diseases share their specific targets with T2DM when pulmonary vascular endothelial cells are used as common targets.
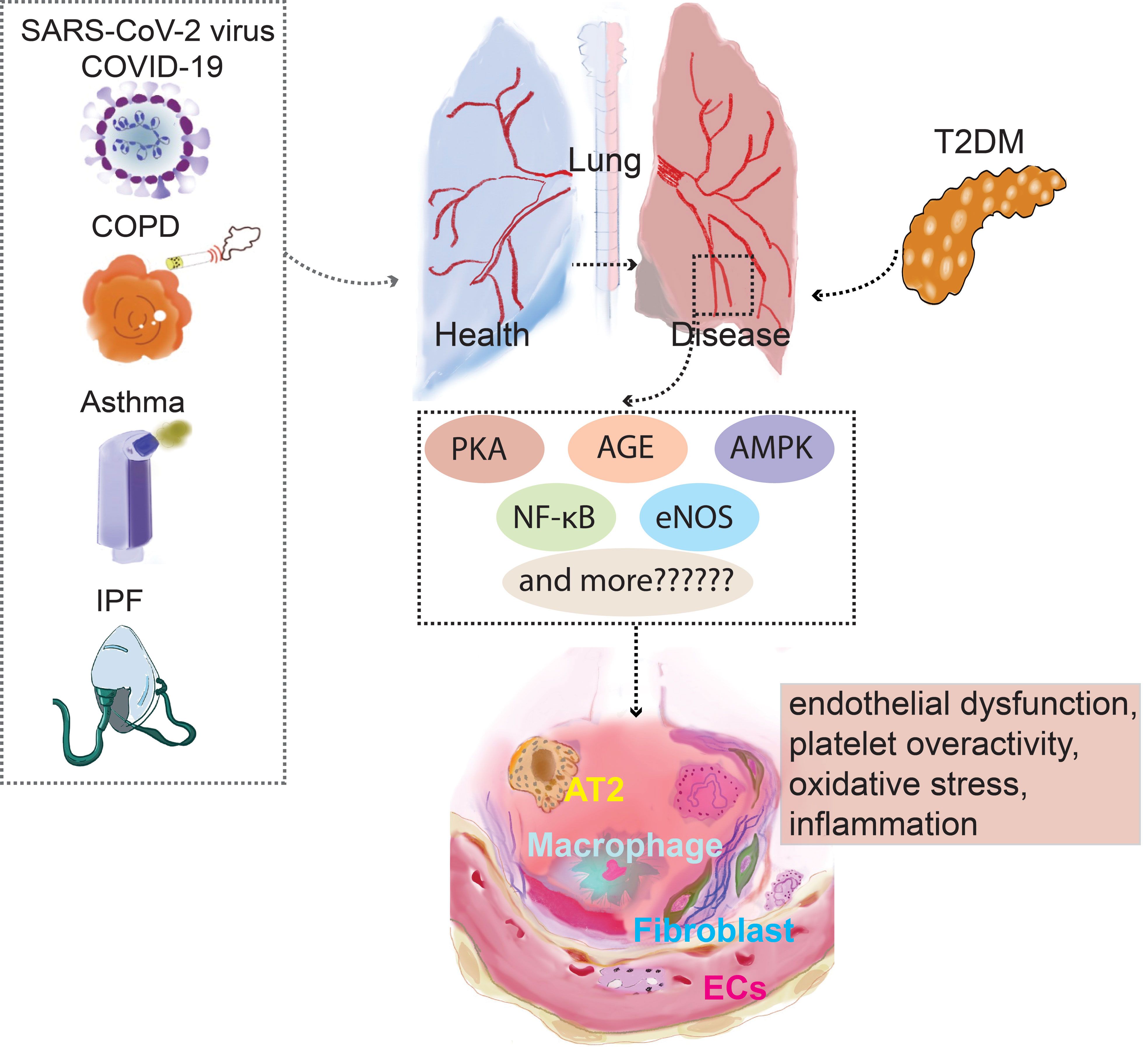
Figure 1 PKA,AGE,AMPK, NF-κB,eNOS are presented as an example for a list of prospective therapeutic targets that diabetes and pulmonary diseases potentially explore through vascular endothelial cells. NF-κB, Nuclear factor kappa B; AT2, Alveolar Type II Cells; COPD, Chronic obstructive pulmonary disease; PKC, Protein kinase C; COVID-19, Coronavirus disease 2019; eNOS, Endothelial NO synthase; AMPK, AMP-activated protein kinase.
The unique pathophysiology of pulmonary dysfunction and endothelial microangiopathy driven by T2DM
COVID-19 and T2DM
A few of the several potential causes of COVID-19 in combination with diabetes are increased inflammatory storms, immunocompromised states, disturbance of glucose homeostasis, hypercoagulable state, alveolar hyperpermeability, and vascular endothelial injury. The characteristics of patients with severe COVID-19 may be explained by the endothelial glycocalyx (Figure 2A). According to study, acquired Hpa-2 deficiency may be a possible causative factor in patients with severe COVID-19 with endothelial damage involving the integrity of the glycocalyx (46). A key element of vascular integrity and cardiovascular homeostasis is the endothelial glycocalyx (EG), which covers the apical surface of endothelial cells and floats into the lumen of the channel. Most EG-related functions include separating blood from the endothelium, regulating vascular permeability, restricting leukocyte and platelet adhesion, and improving the endothelial responsiveness to flow fluctuations through mechanosensing. Recent research findings indicate a more inventive strategy for maintaining EG levels in T2DM therapy (47). A substantial part of the glycocalyx that constitutes the vascular wall and endothelium glomerular permeability barrier is hyaluronic acid (HA). It is known that endocytosed hyaluronidase1 (HYAL1) breaks down HA into tiny pieces in a variety of cell types, including endothelial cells. Diabetes-related endothelium and glycocalyx dysfunction is facilitated by HYAL1. A novel treatment strategy to stop the vascular consequences of diabetes may use HYAL1 inhibitors (48, 49).
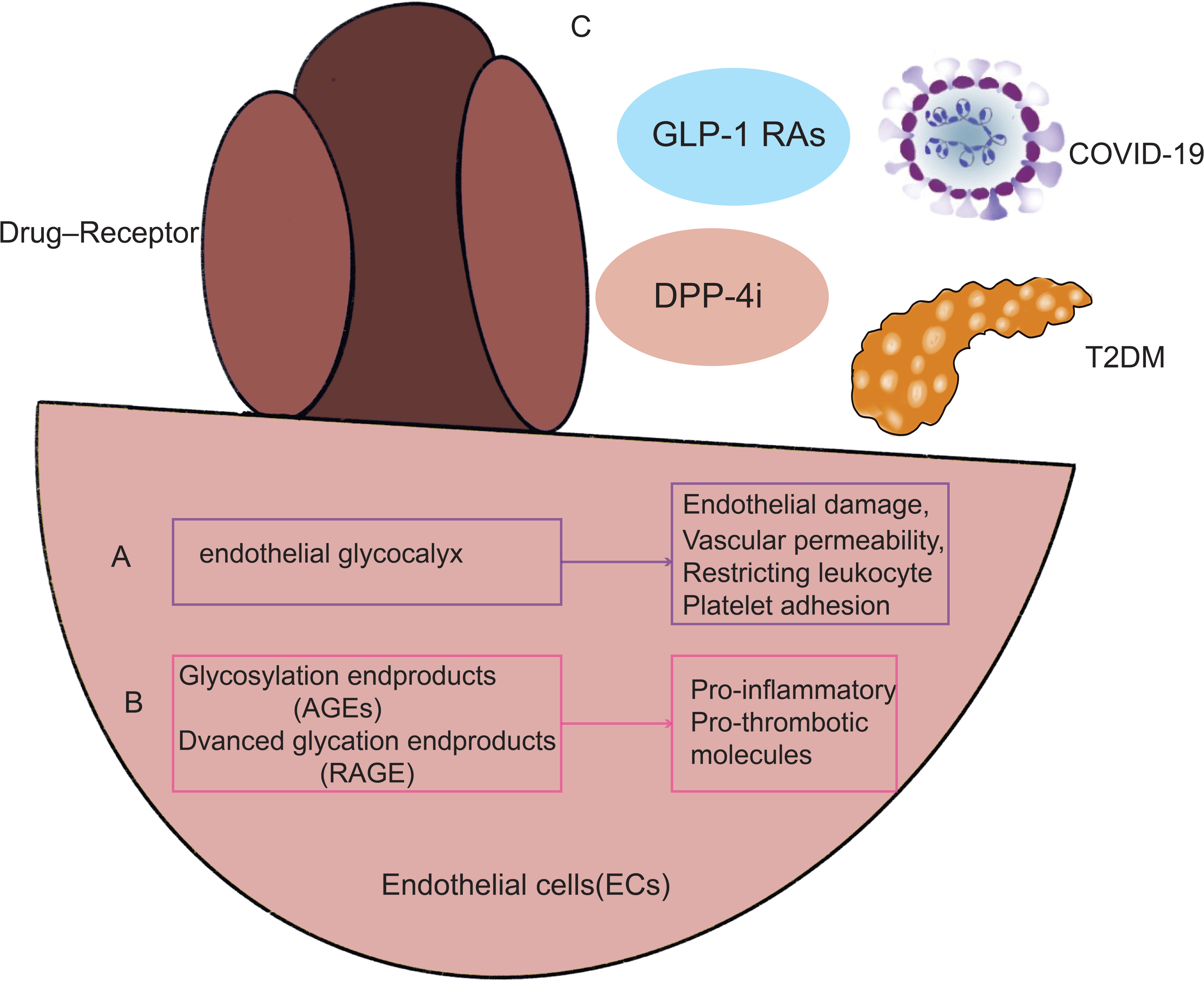
Figure 2 illustrates how endothelial cell damage from T2DM and COVID-19 infection are related. As a result, T2DM paired with COVID-19 infection may trigger the same signaling pathways and processes via endothelial cells, including endothelial glycocalyx (A), RAGE, and AGEs (B). Additionally, maintaining good management of diabetes and using glucose-lowering medicines (C) by acting on endothelial cells may lessen the likelihood that COVID-19 infection may result in decreased pulmonary function. GLP-1 RAs, GLP-1 receptor agonists; DPP-4i, Dipeptidyl peptidase 4 inhibitors.
The vascular EG may provide an explanation for the traits of COVID-19 patients who are severely unwell. The vascular EG is disturbed by inflammation brought on by SARS-CoV-2 infection, as well as in people with hypertension, obesity, diabetes, cardiovascular disease, and current smokers. The elderly may be more susceptible to SARS-CoV-2 infection than younger people, and males are more likely to get the virus than females (11). The pulmonary interstitial develops aberrant shadows (multiple speckled shadows with a ground glass interstitial appearance) as a result of microvascular leaks caused by vascular glycocalyx failure. It also triggers the coagulation cascade, which may result in multi-organ failure and thrombosis. Notably, the virus that causes COVID-19, SARS-CoV-2, interacts to angiotensin converting enzyme-2(ACE2), which is abundantly present in human lung and small intestine epithelial cells as well as vascular endothelial cells and arterial smooth muscle cells. EG abnormalities may be linked to persistent inflammation during endotoxemia in a diabetic mouse model (47). According to Lambadiari et al., persistent cardiovascular symptoms after 4 months were associated with markers of EG and vascular function (50).
Endothelial markers of COVID-19 invasive ventilation increase in correlation with the outcome of vascular damage, whereas endothelial injury indicators upregulate later (51). RAGE is abundantly expressed in the membranes and cytoplasm of pneumocytes in the lung, where it interacts with AGEs in patients with combined diabetes in COVID-19, activating downstream signaling pathways and inducing an inflammatory response (Figure 2B). In consequence of this, vascular wall cells generate more cytokines and have greater endothelial permeability (52). RAGE mediates cell migration and the activation of pro-inflammatory and pro-thrombotic molecules on cells including endothelial cells by interacting with ligand-RAGE on those cells (53). Targeting this mechanism, particularly in individuals with combined Diabetes, may help prevent cytokine storm and thrombotic symptoms related to dysregulated immunological response to SARS-CoV-2 infection (54).
The anti-inflammatory features of incretin-based treatments, diabetes treatment medications were suggested to improve the prognosis of COVID-19 (Figure 2C). Despite the older age and typically more severe disease of DPP4i users, the evidence demonstrates that pre-morbid usage of GLP1-RA is linked with decreased mortality and other deleterious outcomes compared to DPP-4i use in COVID-19 patients (55). According to study, DPP-4i users had an increased adjusted risk ratio for 30-day mortality (56). Patients with T2DM with COVID-19 have a reduced risk of death if they take metformin and sulfonylureas. However, mortality is higher in T2DM patients with COVID-19 and on insulin (57, 58). However, there are also reports in the literature that treatment with sulfonylureas is not associated with mortality (59).
In conclusion, individuals with COVID-19 infection, in addition to diabetes, have worse lung function than those with diabetes alone. One of the key mechanisms for this is the activation of endothelial cells, including endothelial glycocalyx, RAGE, and AGEs. Treatment for diabetes may also delay the decline in lung function brought on by COVID-19 infection since the medications used to manage diabetes may also have effects on the lungs, delaying the progression of pulmonary disease.
COPD and T2DM
Patients with COPD are more likely than the general population to have T2DM.Additionally, T2DM patients are more likely than non-diabetic individuals to have concomitant COPD (60, 61). The increasing incidence of diabetes in COPD has been linked to several variables, such as rising obesity, declining physical activity, rising smoking, exposure to corticosteroids, disease-related inflammation, oxidative stress, and hypoxia. Smoking, inflammation, or obesity by individually, however, do not completely explain the link between T2DM and reduced lung function. Despite controlling for other risk variables, airflow restriction also seems to be an independent predictor of death in persons with T2DM.
Among the hypothesized mechanisms to contribute for the decline in lung function associated with diabetes include microangiopathy of the alveolar capillaries and pulmonary arterioles, chronic low-grade tissue inflammation, autonomic neuropathy affecting the respiratory muscles, and loss of elastic recoil secondary to collagen glycosylation of lung parenchyma (62–65). The accumulation of glycosylated proteins, which have pro-inflammatory tendencies, in persistent hyperglycemia is the underlying cause of diabetes’ microvascular sequelae. The interstitium widened and the alveolar gap closed as a result of increased pulmonary vascular permeability, mononuclear cell infiltration, cell proliferation, hypertrophy of interstitial cells, and accelerated fibrosis, as depicted in studies on the lungs of diabetic rats (66, 67). In human autopsies (68) and trans-bronchial biopsy investigations (69), alterations in the thickness of the alveolar-capillary basement membrane and nodular fibrosis in the alveolar walls have been linked to diabetes. Given the alveolar-capillary basement membrane has undergone fibrotic modifications and an enlarged interstitium inhibits lung expansion, these changes may clinically translate to lower spirometry scores and decreased diffusion (Figure 3A).
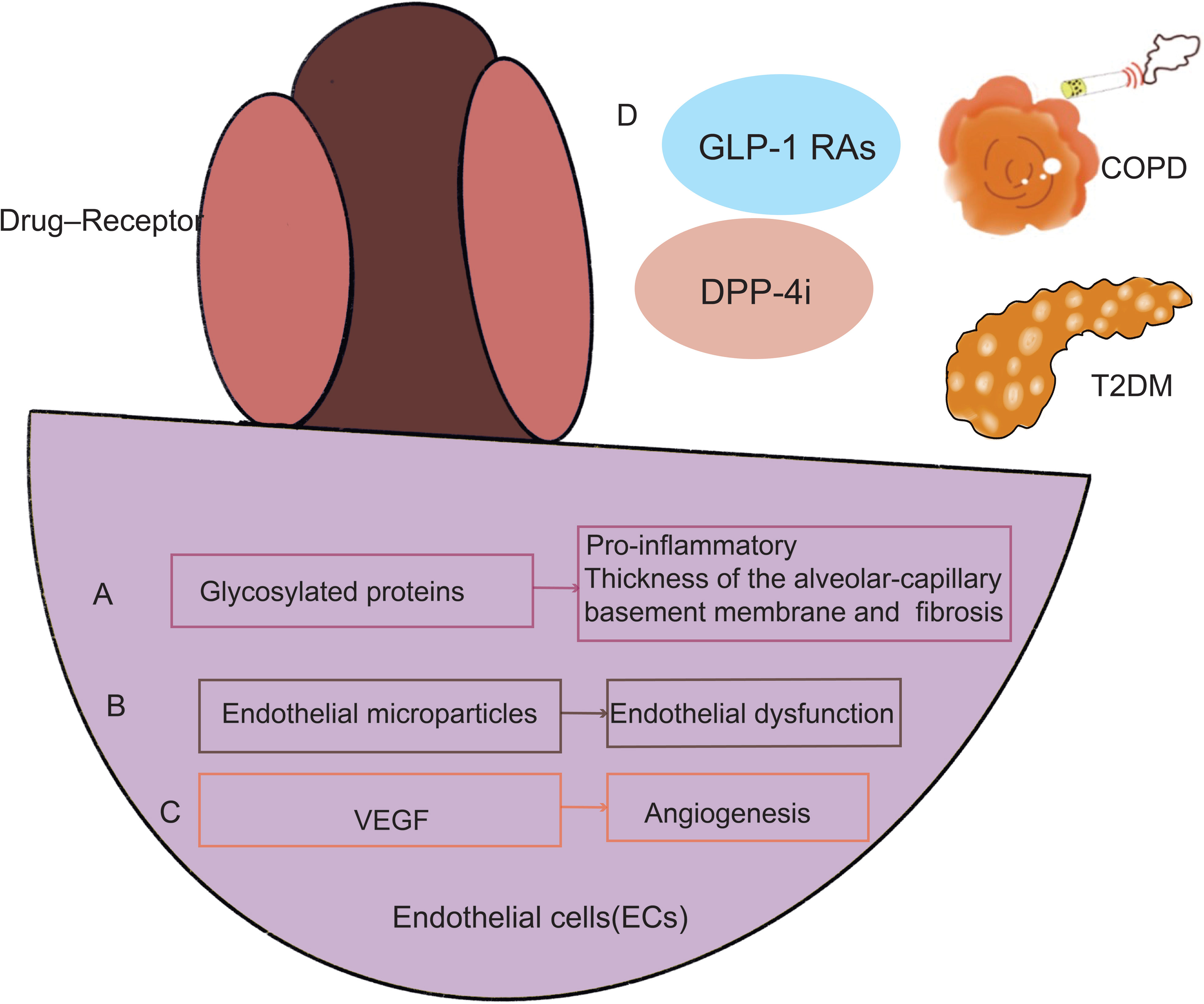
Figure 3 In endothelial cells, COPD and T2DM create a similar pathogenic pathway. Endothelial cells in T2DM may impact how a lung disease develops in the end. Endothelial cells become dysfunctional in patients who have T2DM and COPD because of the accumulation of glycosylated proteins (A), a rise in endothelial microparticles (EMPs) (B), and an increase in VEGF (C). GLP-1 RAs, GLP-1 RAs and DPP-4i can treat COPD and diabetes simultaneously (D). GLP-1 RAs, GLP-1 receptor agonists; DPP-4i, Dipeptidyl peptidase 4 inhibitors; VEGF, Vascular endothelial growth factor.
Clinical data suggest that the vascular area in the airways of COPD patients is increased and may lead to narrowing of the airways (70). Endothelial dysfunction is defined as disturbed endothelium-dependent vasodilation. Endothelial microparticles (EMPs) in blood can also be used as a measurement of endothelial dysfunction. Endothelial dysfunction correlates with the severity of COPD and is associated with forced expiratory volume in 1 second (FEV1) (71–73). EMPs levels are elevated in patients with frequently worsening COPD (74) and also predict a rapid decline in patient FEV1 (75). EMPs are positively correlated with the severity of emphysema in COPD patients, again suggesting that endothelial cells may be a potential mechanism for emphysema (75). EMPs are also associated with T2DM (76–81) (Figure 3B).
VEGF is a highly specific growth factor that targets endothelial cells and is produced in response to hypoxia (82). VEGF levels may be reduced in such patients because the major transcription factor of VEGF, hypoxia-inducible factor 1α (HIF-1α), which mediates cellular and systemic responses to hypoxia and binds to the hypoxia response element (HRE) on VEGF (83). T2DM-associated endothelial angiogenesis and rising HIF-1 levels are tightly connected. VEGF expression was reduced, and angiogenesis was prevented by HIF-1a inhibition. New treatment options for diabetic retinopathy were revealed by VEGF of endothelial cells (84–89).Further exploration of molecular mechanisms of COPD suggests that increased bronchial vascular distribution is associated with higher cellular expression of VEGF-A (90, 91). Levels of HIF-1α and VEGF may correlate with predicted FEV1 percentages in COPD patients (83, 92) (82). Excessive stimulation, including cigarette smoke (55), hypoxia (93) and cytokines) (94) increase VEGF-A production (95). Perfusion of isolated lungs under hypoxic conditions increases tissue VEGF-A and VEGFR1 (96, 97). The above studies confirm that endothelial cells initiate and participate in the pathogenesis of COPD, T2DM which induces vascular inflammation with proinflammatory, and remodeling activities (98). As research progresses, there is increasing evidence that molecular biological mechanisms such as endothelial cells play an important role in regulating endothelial cell dysregulation in COPD and T2DM (Figure 3C).
GLP-1RAs may enhance endothelial function via a variety of pathways, some of which are independent of insulin signaling or glucose homeostasis. GLP-1RAs is a potential novel therapeutic target for controlling COPD and T2DM together (69–71). GLP-1 has anti-inflammatory and surfactant-releasing effects, so studies showed that GLP-1Ras reduce the frequency of acute exacerbations by decreasing their severity and that GLP-1RAs may have therapeutic potential for the treatment of COPD (99–102). Endothelial cells express DPP-4, which has a broad variety of biological roles in glucose metabolism, cancer biology, and immunological control. DPP-4i is detrimental in respiratory diseases. Slowly but surely, its biological processes, important molecular pathways, connections, and linkages are being revealed. Respiratory diseases may be affected significantly by DPP-4i and subsequent development may be regulated (103) (Figure 3D). As diabetes may lead to an increased prevalence of COPD, and in turn COPD may lead to an increase in diabetes, future research should take advantage of the mechanisms of endothelial cell dysfunction to improve clinical outcomes in patients with diabetes combined with COPD.
Asthma and T2DM
Both early-onset diabetes and asthma are becoming more common, and adolescents who have active asthma have an increased chance of developing T2DM (104). A prospective cohort of 38,570 women, both asthma and COPD were separately and independently linked to a higher risk of developing T2DM, suggesting that chronic airway inflammation may play a role in the development of diabetes (105). In a significant nested case-control research, patients with asthma or COPD who used inhaled corticosteroids (ICS) saw a 34% higher incidence of diabetes mellitus during 5.5 years of follow-up compared to age-matched controls who did not get ICS (106). The findings are consistent with the notion that metabolic traits linked to prediabetes and diabetes, such as metabolic syndrome and insulin resistance, might affect asthma morbidity (107). Patients with concurrent DM who were hospitalized for asthma had longer hospital stays, higher costs, and a higher risk of readmission. In patients with coexisting DM and asthma, interventions are urgently required to lower the risk of hospital admission and readmission (108, 109).
The parameter of the induced sequential vascular response was used to study several metabolite (sugars and L-histidine) and pharmacological (aspirin, insulin, and glucagon) challenges. There was also a correlation with a common oral glucose tolerance test. The findings showed a range of glucose, insulin, glucagon, L-histidine, and aspirin challenges produced both comparable and different behavioral responses (Figure 4A). New therapies might improve airflow through better regulation of vessel growth, dilatation, and leakage in the airway wall (110). One mechanism that endothelial cells act in asthma is by transendothelial migration (TEM). L-selectin and Intercellular Adhesion Molecule 1(ICAM-1)are critical TEM regulating genes, and animal asthma model lacking these cell adhesion molecules have decreased lung inflammation and lower airway hyperresponsiveness (AHR) in response to ovalbumin challenge (111–113), and allergic asthma patients also have increased endothelial adhesion molecules in bronchial biopsies (114). Elevated levels of adhesion molecules have been linked to asthmatics’ inflammation and underlying endothelial dysfunction (115, 116). Increased vascular permeability brought on by alterations to the blood-retinal barrier (BRB) is one of the primary effects of early diabetes (117). VE-cadherin proteolytic degradation has been suggested by observations as a possible mechanism by which diabetes promotes BRB deterioration (Figure 4B).
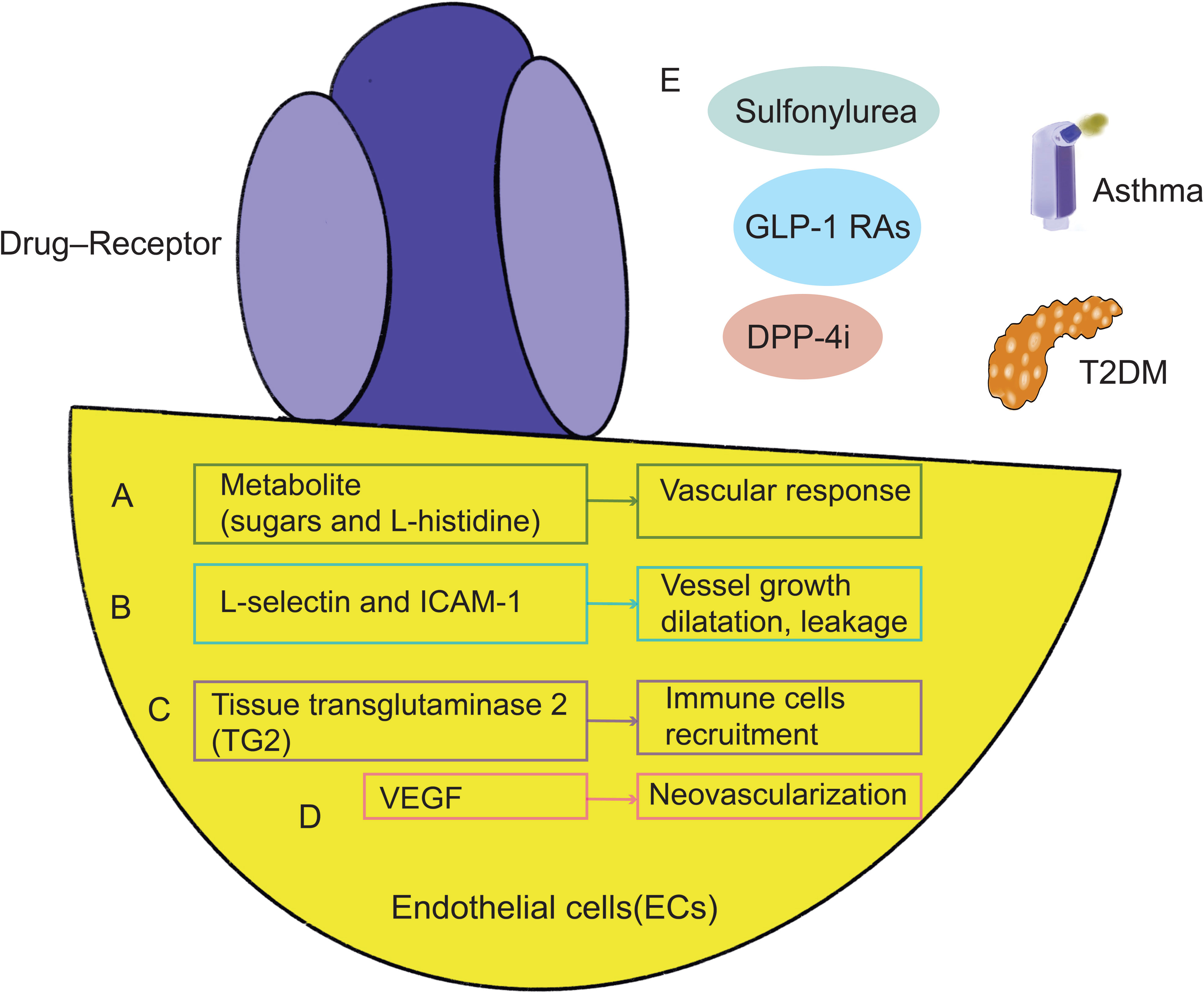
Figure 4 Mechanisms shared by asthma and T2DM in endothelial cells. Metabolite (A), L-selectin, ICAM-1 (B), tissue transglutaminase 2 (TG2) (C), and VEGF (D) are all intimately connected to and have an impact on each other in endothelial cells, which may represent a common mechanism of lung function decline. GLP-1 RAs targeting endothelial cells, DPP-4i and other drugs can treat asthma and diabetes (E). GLP-1 RAs, GLP-1 receptor agonists; DPP-4i, Dipeptidyl peptidase 4 inhibitors; VEGF, Vascular endothelial growth factor; ICAM-1, Adhesion molecule 1.
High glucose stimulated tissue transglutaminase 2 (TG2) expression and TG2 silencing prevented Aβ-induced mitochondrial calcium influx, mtROS accumulation, and cell death in neuronal cells. In clinical treatment, the prevalence of tissue transglutaminase antibodies and diabetes with their first-degree relatives (118–122). Chemokine overexpression is one more potential explanation for the rise in asthma. In order to attract and activate circulating eosinophils, endothelial cells in asthma seem to release more chemokines (123). Mice with endothelial deficit of TG2 display lower numbers of pulmonary eosinophils in response to allergen challenge, while pulmonary endothelium TG2 is increased in asthma and seems to be necessary for eosinophil recruitment to the lung (124) (Figure 4C).
The neovascularization of the tracheal and bronchial arteries that develops in asthmatics with hyperreactive airways disease. This study’s findings were corroborated by bronchoscopy samples taken from the airways of asthmatic patients and healthy control volunteers, which revealed an increase in vessel size and density (125–127). By comparing the expression of VEGF, its receptors, and angiopoietin-1 in biopsy samples and bronchoalveolar lavage fluid between asthmatic sufferers and controls, researchers were able to better understand the processes causing this neovascularization (128). Endothelial markers as CD31 or VWF used in histological analysis of parenchymal units could not distinguish between bronchial and pulmonary arteries. As a result, these models have been inaccurate in their appraisal of angiogenesis (129). Additionally elevated in asthmatics’ produced sputum samples, VEGF has a negative correlation with FEV1 (130). Additionally, several VEGF polymorphisms (such as rs4711750 and rs3025038) seem to enhance the chance of developing asthma and are linked to lung function (131).Further research is required to determine the processes behind the development of new vascular beds in adults. However, it is still challenging to distinguish the growth factors and molecular processes of lung angiogenesis from underlying diseases consequences (110, 130, 132–142) (Figure 4D).
GLP-1R is expressed in endothelial cells (143). The usage of GLP-1RAs in the management of T2DM is recommended. GLP-1RAs may also lessen airway inflammation and hyperresponsiveness at the same time. GLP-1RAs prevent adult asthmatics’ symptoms from deteriorating as much. GLP-1RAs could provide a fresh method for treating asthma brought by metabolic malfunction (144–149). As a result of increased expression of DPP-4, which is induced by the type 2 cytokines interleukin (IL)-4 and IL-13, in diseases of asthma and type 2 inflammation, such as atopic dermatitis and chronic rhinitis, DPP-4i may help with the management of asthma (103, 150–154). Glibenclamide (targeting sulfonylurea receptor 1) may lessen airway inflammation because it inhibits ATP-sensitive potassium (KATP) channels. Glibenclamide significantly decreased the AHR, airway inflammation, and T-helper type 2 (Th2) cytokines in a mouse model of asthma caused by ovalbumin (OVA). Additionally, Glibenclamide decreased the lung’s phosphorylated signal transducer and activator of transcription 6 (p-STAT6) and vascular cell adhesion molecule 1 (VCAM-1) expression that was brought on by OVA. These findings provide evidence for the safety of prescribing Glibenclamide in diabetic patients with comorbid asthma, as well as suggest a potential new therapeutic role for asthma through a pathway related to targeting the control medications for diabetes. These findings suggest that GLP-1R, DPP-4, and the sulfonylurea Glipalamides play an important role in the development of asthma (155–158) (Figure 4E). However, fewer studies have been undertaken on the combination of asthma and diabetes to yet, and endothelial cell dysfunction may become a prominent study area. Future research should focus more on this process to optimize the usage of medications and enhance patients’ lung function and prognosis to a greater extent.
IPF and T2DM
As was already indicated, the lung has a dense network of connective tissue and alveolar capillaries, indicating that it can be one of the targets of diabetic microvascular damage. Recent research has shown that having high blood sugar levels might cause interstitial fibrotic alterations and alveolar microangiopathy (159, 160). The three main stages of alveolar microangiopathy, in which hyperglycemia may cause interstitial fibrosis, are: (1) oxidative stress injury, which initiates IPF; (2) alveolar inflammation, immune cell activation, secretion of numerous pro-inflammatory factors, which activates endothelial cells; and (3) pro-fibrotic cytokines secreted by endothelial cells, which mark the end of the disease (25, 161–163).
The common pathways of endothelium damage-related lung injury in diabetes are listed below. Endothelial cells exhibit higher levels of expression of AGEs, RAGE, and sirtuin (SIRT), which are also changed in diabetes and are strongly linked to the onset of the disease (164, 165) (Figures 5A, B). Under typical circumstances, diabetes causes AGEs to accumulate in the lungs (166). Researchers discovered significant elevated AGEs in diabetic patients, which resulted in pulmonary fibroblast aggregation (167). RAGE is thought to stimulate the production of TGF-β and to inhibit the activation of Smad2, ERK, and JNK signaling (168, 169). TGF-β (170) is a key player in tissue remodeling and fibrosis and is a mediator of pro-fibrotic characteristics. SIRT has a possible involvement in the treatment of IPF by mediating the processes of insulin secretion, cell cycle, and apoptosis (171, 172). SIRT is crucial in the development of diabetic microangiopathy (173, 174). SIRT blocks oxidative stress, the IL-1, and TGF-1/Smad3 signaling pathways, which are pro-inflammatory cytokines, mitochondrial DNA damage, and fibronectin. Additionally, pro-inflammatory or pro-fibrotic substances, including fibronectin, angiotensin II (Ang II) that are heavily implicated in the development of the diabetic lung were also shown to be elevated in the lung tissue of diabetic mice (175) (Figure 5C). As a consequence of several pathways being triggered in a high-glucose environment, intracellular stress and abnormal cytokine production occur. Damage to the structural lungs and pathological pulmonary fibrosis result from the failure of re-endothelialization and consequent loss of the alveolar-capillary barrier basal layer’s integrity.
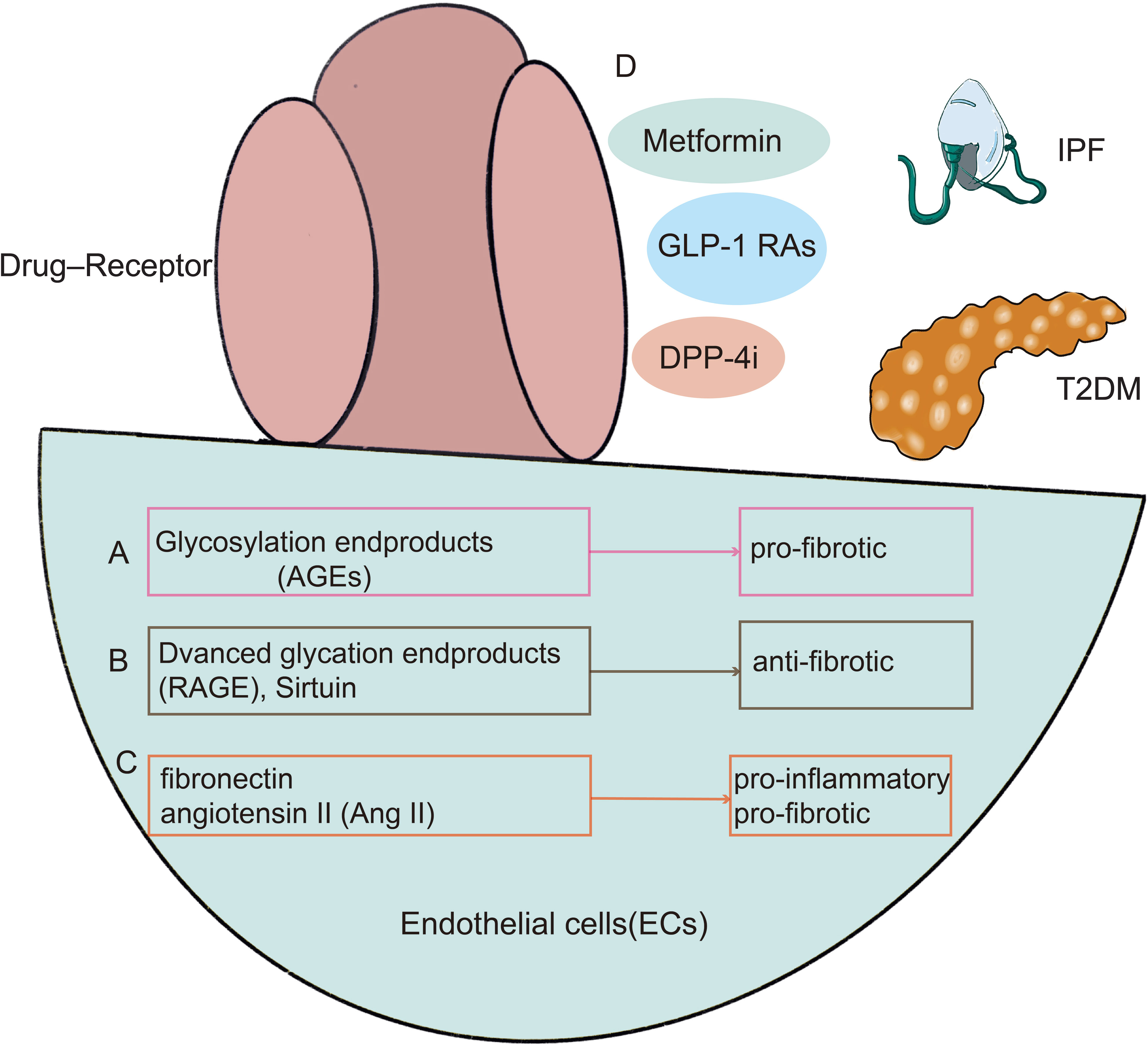
Figure 5 There are shared mechanisms in endothelial cell dysfunction in T2DM with IPF, with existing research focused on AGEs (A), RAGE (B), and Ang II (C). In the meanwhile, diabetic therapies such as metformin, GLP-1 receptor agonists, DPP-4 inhibitors, and PPAR-agonists may alleviate pulmonary function decline by enhancing endothelial function (D). GLP-1 RAs, GLP-1 receptor agonists; DPP-4i, Dipeptidyl peptidase 4 inhibitors.
In the lungs of IPF patients, metformin has potent antifibrotic effects via metabolic pathway modulation, inhibition of TGF-1 activity, inhibition of collagen synthesis, activation of PPAR signaling, and induction of lipogenic differentiation of fibroblasts (176–181). By activating AMPK and enhancing the TGF- signaling pathway, metformin induces the inactivation and apoptosis of myofibroblasts, reversing the progression of pulmonary fibrosis (180). Gu et al. studied that metformin-activated AMPK can downregulate Forkhead Box M1 (FOXM1) and alleviate BLM-induced IPF model in mice (177). GLP-1 is a crucial hormone that regulates hunger, glucose metabolism, and insulin secretion. A 39 amino acid agonist of GLP-1receptor, exendin-4. By deactivating NF-κB, GLP-1 receptor agonists greatly reduce bleomycin (BLM)-induced lung damage and fibrosis in mice (182).Exendin-4, meanwhile, reduced hyperglycemia-related lung damage by lowering oxidative stress and promoting cell growth (183).DPP-4 inhibitors and PPAR-γ agonists have proven useful in the treatment of pulmonary fibrosis. In a mouse model of pulmonary fibrosis after systemic endotoxin damage, endothelial to mesenchymal transition (EndMT) was shown in endothelial cells overexpressing DPP-4(Vildagliptin), which has been reported to alleviate pulmonary fibrosis. Vildagliptin, which inhibits EndMT, may be helpful in treating pulmonary fibrosis (184). PPAR- is a member of the nuclear hormone receptor superfamily, and its effects include modifying metabolic and inflammatory responses, among others. Recent research has also shown the effectiveness of PPAR- agonists in treating BLM-induced lung fibrosis (185, 186). In a mouse model, rosiglitazone and selegiline inhibited TGF-1-mediated fibrotic alterations in alveolar epithelial EMT, differentiation of myoblasts, and collagen synthesis, indicating a therapeutic efficacy of PPAR- ligands in fibrotic lung injury (187, 188) (Figure 5D).
In conclusion, endothelial cells are critical in the development of pulmonary fibrosis brought on by hyperglycemia. Additionally, endothelial cells must be the primary focus of therapeutic intervention; thus, determining whether endothelial cells are involved in the regulation of lung fibers might be a promising therapeutic avenue for the investigation of diabetes-related pulmonary fibrosis.
Conclusion
Future studies should focus on the mechanism and clinical management of respiratory diseases and diabetes, especially due to the COVID-19 pandemic when severe lung consequences. It is possible to improve the treatment of patients with diabetes and pulmonary diseases by understanding the interactions between the two disorders and developing an effective treatment strategy. In addition, further research is essential to pave the way for potential treatments by using endothelial cells in discussions of co-morbidities underlying mechanisms.
Author contributions
LZ, FJ, and CL, conception or design of the work and critical revision of the article. LZ, YX, and YM, drafting the article and data collection. XZ, drafting the manuscript and revision of the article. All authors contributed to the article and approved the submitted version.
Funding
Postdoctoral Funding of the Personnel Office-Special Fund for Postdoctoral New Crown Epidemic Prevention and Control (0040204153344), National Science Foundation for Young Scientists of China (81900065), Project supported by the Natural Science Foundation of Sichuan province (2022NSFSC1394), the Youth Innovation Project of Sichuan Medical Association (Q21018), and Chengdu Medical Research Projects (2022516).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kirpichnikov D, Sowers JR. Diabetes mellitus and diabetes-associated vascular disease. Trends Endocrinol Metab (2001) 12(5):225–30. doi: 10.1016/s1043-2760(01)00391-5
2. Visca D, Pignatti P, Spanevello A, Lucini E, La Rocca E. Relationship between diabetes and respiratory diseases-clinical and therapeutic aspects. Pharmacol Res (2018) 137:230–5. doi: 10.1016/j.phrs.2018.10.008
3. Khateeb J, Fuchs E, Khamaisi M. Diabetes and lung disease: A neglected relationship. Rev Diabetes Stud (2019) 15:1–15. doi: 10.1900/rds.2019.15.1
4. Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol (1993) 143(6):1699–712.
5. Stehouwer CD, Lambert J, Donker AJ, van Hinsbergh VW. Endothelial dysfunction and pathogenesis of diabetic angiopathy. Cardiovasc Res (1997) 34(1):55–68. doi: 10.1016/s0008-6363(96)00272-6
6. Nakagawa T, Tanabe K, Croker BP, Johnson RJ, Grant MB, Kosugi T, et al. Endothelial dysfunction as a potential contributor in diabetic nephropathy. Nat Rev Nephrol (2011) 7(1):36–44. doi: 10.1038/nrneph.2010.152
7. Ugusman A, Kumar J, Aminuddin A. Endothelial function and dysfunction: Impact of sodium-glucose cotransporter 2 inhibitors. Pharmacol Ther (2021) 224:107832. doi: 10.1016/j.pharmthera.2021.107832
8. Cintra RMR, Soares AAS, Breder I, Munhoz DB, Barreto J, Kimura-Medorima ST, et al. Assessment of dapagliflozin effect on diabetic endothelial dysfunction of brachial artery (Addenda-Bhs2 trial): Rationale, design, and baseline characteristics of a randomized controlled trial. Diabetol Metab Syndr (2019) 11:62. doi: 10.1186/s13098-019-0457-3
9. Pitocco D, Fuso L, Conte EG, Zaccardi F, Condoluci C, Scavone G, et al. The diabetic lung–a new target organ? Rev Diabetes Stud (2012) 9(1):23–35. doi: 10.1900/rds.2012.9.23
10. Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in covid-19. Nat Rev Immunol (2021) 21(5):319–29. doi: 10.1038/s41577-021-00536-9
11. Zhang L, Tang C, Zhang M, Tong X, Xie Y, Yan R, et al. Single cell meta-analysis of endmt and emt state in covid-19. Front Immunol (2022) 13:976512. doi: 10.3389/fimmu.2022.976512
12. Hisata S, Racanelli AC, Kermani P, Schreiner R, Houghton S, Palikuqi B, et al. Reversal of emphysema by restoration of pulmonary endothelial cells. J Exp Med (2021) 218(8):e20200938. doi: 10.1084/jem.20200938
13. Wanner A, Mendes ES. Airway endothelial dysfunction in asthma and chronic obstructive pulmonary disease: A challenge for future research. Am J Respir Crit Care Med (2010) 182(11):1344–51. doi: 10.1164/rccm.201001-0038PP
14. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res (2015) 116(7):1231–44. doi: 10.1161/circresaha.116.302855
15. Shi Y, Vanhoutte PM. Macro- and microvascular endothelial dysfunction in diabetes. J Diabetes (2017) 9(5):434–49. doi: 10.1111/1753-0407.12521
16. Irfan M, Jabbar A, Haque AS, Awan S, Hussain SF. Pulmonary functions in patients with diabetes mellitus. Lung India (2011) 28(2):89–92. doi: 10.4103/0970-2113.80314
17. Díez-Manglano J, Asìn Samper U. Pulmonary function tests in type 2 diabetes: A meta-analysis. ERJ Open Res (2021) 7(1):00371–2020. doi: 10.1183/23120541.00371-2020
18. Mauricio D, Alonso N, Gratacòs M. Chronic diabetes complications: The need to move beyond classical concepts. Trends Endocrinol Metab (2020) 31(4):287–95. doi: 10.1016/j.tem.2020.01.007
19. Zheng H, Wu J, Jin Z, Yan LJ. Potential biochemical mechanisms of lung injury in diabetes. Aging Dis (2017) 8(1):7–16. doi: 10.14336/ad.2016.0627
20. Huang C, Feng F, Shi Y, Li W, Wang Z, Zhu Y, et al. Protein Kinase C Inhibitors Reduce Sars-Cov-2 Replication in Cultured Cells. Microbiol Spectr (2022) 10(5):e0105622. doi: 10.1128/spectrum.01056-22
21. Beeson M, Sajan MP, Dizon M, Grebenev D, Gomez-Daspet J, Miura A, et al. Activation of protein kinase c-zeta by insulin and phosphatidylinositol-3,4,5-(po4)3 is defective in muscle in type 2 diabetes and impaired glucose tolerance: amelioration by rosiglitazone and exercise. Diabetes (2023) 52(8):1926–34 doi: 10.2337/diabetes.52.8.1926
22. Hsu RJ, Yu WC, Peng GR, Ye CH, Hu S, Chong PCT, et al. The role of cytokines and chemokines in severe acute respiratory syndrome coronavirus 2 infections. Front Immunol (2022) 13:832394. doi: 10.3389/fimmu.2022.832394
23. Makowski LM, Leffers M, Waltenberger J, Pardali E. Transforming growth factor-Beta1 signalling triggers vascular endothelial growth factor resistance and monocyte dysfunction in type 2 diabetes mellitus. J Cell Mol Med (2021) 25(11):5316–25. doi: 10.1111/jcmm.16543
24. Suhail S, Zajac J, Fossum C, Lowater H, McCracken C, Severson N, et al. Role of oxidative stress on sars-cov (Sars) and sars-Cov-2 (Covid-19) infection: A review. Protein J (2020) 39(6):644–56. doi: 10.1007/s10930-020-09935-8
25. Apostolova N, Iannantuoni F, Gruevska A, Muntane J, Rocha M, Victor VM. Mechanisms of action of metformin in type 2 diabetes: Effects on mitochondria and leukocyte-endothelium interactions. Redox Biol (2020) 34:101517. doi: 10.1016/j.redox.2020.101517
26. Barnes PJ. Oxidative stress-based therapeutics in copd. Redox Biol (2020) 33:101544. doi: 10.1016/j.redox.2020.101544
27. Feng Q, Yu YZ, Meng QH. Blocking tumor necrosis factor-alpha delays progression of chronic obstructive pulmonary disease in rats through inhibiting mapk signaling pathway and activating Socs3/Traf1. Exp Ther Med (2021) 22(5):1311. doi: 10.3892/etm.2021.10746
28. Lu Y, Huang Y, Li J, Huang J, Zhang L, Feng J, et al. Eosinophil extracellular traps drive asthma progression through neuro-immune signals. Nat Cell Biol (2021) 23(10):1060–72. doi: 10.1038/s41556-021-00762-2
29. Michaeloudes C, Abubakar-Waziri H, Lakhdar R, Raby K, Dixey P, Adcock IM, et al. Molecular mechanisms of oxidative stress in asthma. Mol aspects Med (2022) 85:101026. doi: 10.1016/j.mam.2021.101026
30. Stock CJ, Sato H, Fonseca C, Banya WA, Molyneaux PL, Adamali H, et al. Mucin 5b promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax (2013) 68(5):436–41. doi: 10.1136/thoraxjnl-2012-201786
31. Chen G, Zhang Z, Adebamowo SN, Liu G, Adeyemo A, Zhou Y, et al. Common and rare exonic Muc5b variants associated with type 2 diabetes in han Chinese. PloS One (2017) 12(3):e0173784. doi: 10.1371/journal.pone.0173784
32. Chanda D, Kurundkar A, Rangarajan S, Locy M, Bernard K, Sharma NS, et al. Developmental reprogramming in mesenchymal stromal cells of human subjects with idiopathic pulmonary fibrosis. Sci Rep (2016) 6:37445. doi: 10.1038/srep37445
33. Joannes A, Brayer S, Besnard V, Marchal-Sommé J, Jaillet M, Mordant P, et al. Fgf9 and Fgf18 in idiopathic pulmonary fibrosis promote survival and migration and inhibit myofibroblast differentiation of human lung fibroblasts in vitro. Am J Physiol Lung Cell Mol Physiol (2016) 310(7):L615–29. doi: 10.1152/ajplung.00185.2015
34. Königshoff M, Balsara N, Pfaff EM, Kramer M, Chrobak I, Seeger W, et al. Functional wnt signaling is increased in idiopathic pulmonary fibrosis. PloS One (2008) 3(5):e2142. doi: 10.1371/journal.pone.0002142
35. Selman M, Pardo A, Kaminski N. Idiopathic pulmonary fibrosis: Aberrant recapitulation of developmental programs? PloS Med (2008) 5(3):e62. doi: 10.1371/journal.pmed.0050062
36. Zhou T, Li HY, Zhong H, Zhong Z. Relationship between transforming growth factor-B1 and type 2 diabetic nephropathy risk in Chinese population. BMC Med Genet (2018) 19(1):201. doi: 10.1186/s12881-018-0717-3
37. Gonzalez-Gonzalez FJ, Chandel NS, Jain M, Budinger GRS. Reactive oxygen species as signaling molecules in the development of lung fibrosis. Transl Res (2017) 190:61–8. doi: 10.1016/j.trsl.2017.09.005
38. Wang J, Sun L, Nie Y, Duan S, Zhang T, Wang W, et al. Protein kinase c Δ (Pkcδ) attenuates bleomycin induced pulmonary fibrosis Via inhibiting nf-Kb signaling pathway. Front Physiol (2020) 11:367. doi: 10.3389/fphys.2020.00367
39. Meza CA, La Favor JD, Kim DH, Hickner RC. Endothelial dysfunction: Is there a hyperglycemia-induced imbalance of nox and nos? Int J Mol Sci (2019) 20(15):3775. doi: 10.3390/ijms20153775
40. Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, et al. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes (2010) 59(4):1030–7. doi: 10.2337/db09-1694
41. Oeseburg H, de Boer RA, Buikema H, van der Harst P, van Gilst WH, Silljé HHW. Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence through the activation of protein kinase a. Arterioscler Thromb Vasc Biol (2010) 30(7):1407–14. doi: 10.1161/ATVBAHA.110.206425
42. Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S-I. Glucagon-like peptide-1 (Glp-1) inhibits advanced glycation end product (Age)-induced up-regulation of vcam-1 mrna levels in endothelial cells by suppressing age receptor (Rage) expression. Biochem Biophys Res Commun (2010) 391(3):1405–8. doi: 10.1016/j.bbrc.2009.12.075
43. Shiraki A, Oyama J-i, Komoda H, Asaka M, Komatsu A, Sakuma M, et al. The glucagon-like peptide 1 analog liraglutide reduces tnf-A-Induced oxidative stress and inflammation in endothelial cells. Atherosclerosis (2012) 221(2):375–82. doi: 10.1016/j.atherosclerosis.2011.12.039
44. Krasner NM, Ido Y, Ruderman NB, Cacicedo JM. Glucagon-like peptide-1 (Glp-1) analog liraglutide inhibits endothelial cell inflammation through a calcium and ampk dependent mechanism. PloS One (2014) 9(5):e97554. doi: 10.1371/journal.pone.0097554
45. Dai Y, Mehta JL, Chen M. Glucagon-like peptide-1 receptor agonist liraglutide inhibits endothelin-1 in endothelial cell by repressing nuclear factor-kappa b activation. Cardiovasc Drugs Ther (2013) 27(5):371–80. doi: 10.1007/s10557-013-6463-z
46. Stahl K, Gronski PA, Kiyan Y, Seeliger B, Bertram A, Pape T, et al. Injury to the endothelial glycocalyx in critically ill patients with covid-19. Am J Respir Crit Care Med (2020) 202(8):1178–81. doi: 10.1164/rccm.202007-2676LE
47. Sampei S, Okada H, Tomita H, Takada C, Suzuki K, Kinoshita T, et al. Endothelial glycocalyx disorders may be associated with extended inflammation during endotoxemia in a diabetic mouse model. Front Cell Dev Biol (2021) 9:623582. doi: 10.3389/fcell.2021.623582
48. Dogné S, Rath G, Jouret F, Caron N, Dessy C, Flamion B. Hyaluronidase 1 deficiency preserves endothelial function and glycocalyx integrity in early streptozotocin-induced diabetes. Diabetes (2016) 65(9):2742–53. doi: 10.2337/db15-1662
49. Dogné S, Flamion B, Caron N. Endothelial glycocalyx as a shield against diabetic vascular complications: Involvement of hyaluronan and hyaluronidases. Arterioscler Thromb Vasc Biol (2018) 38(7):1427–39. doi: 10.1161/atvbaha.118.310839
50. Lambadiari V, Mitrakou A, Kountouri A, Thymis J, Katogiannis K, Korakas E, et al. Association of covid-19 with impaired endothelial glycocalyx, vascular function and myocardial deformation 4 months after infection. Eur J Heart Fail (2021) 23(11):1916–26. doi: 10.1002/ejhf.2326
51. Leisman DE, Mehta A, Thompson BT, Charland NC, Gonye ALK, Gushterova I, et al. Alveolar, endothelial, and organ injury marker dynamics in severe covid-19. Am J Respir Crit Care Med (2022) 205(5):507–19. doi: 10.1164/rccm.202106-1514OC
52. Dozio E, Sitzia C, Pistelli L, Cardani R, Rigolini R, Ranucci M, et al. Soluble receptor for advanced glycation end products and its forms in covid-19 patients with and without diabetes mellitus: A pilot study on their role as disease biomarkers. J Clin Med (2020) 9(11):3785. doi: 10.3390/jcm9113785
53. Ramasamy R, Yan SF, Herold K, Clynes R, Schmidt AM. Receptor for advanced glycation end products: Fundamental roles in the inflammatory response: Winding the way to the pathogenesis of endothelial dysfunction and atherosclerosis. Ann N Y Acad Sci (2008) 1126:7–13. doi: 10.1196/annals.1433.056
54. De Francesco EM, Vella V, Belfiore A. Covid-19 and diabetes: The importance of controlling rage. Front Endocrinol (Lausanne) (2020) 11:526. doi: 10.3389/fendo.2020.00526
55. Kahkoska AR, Abrahamsen TJ, Alexander GC, Bennett TD, Chute CG, Haendel MA, et al. Association between glucagon-like peptide 1 receptor agonist and sodium-glucose cotransporter 2 inhibitor use and covid-19 outcomes. Diabetes Care (2021) 44(7):1564–72. doi: 10.2337/dc21-0065
56. Israelsen SB, Pottegård A, Sandholdt H, Madsbad S, Thomsen RW, Benfield T. Comparable covid-19 outcomes with current use of glp-1 receptor agonists, dpp-4 inhibitors or sglt-2 inhibitors among patients with diabetes who tested positive for sars-Cov-2. Diabetes Obes Metab (2021) 23(6):1397–401. doi: 10.1111/dom.14329
57. Kan C, Zhang Y, Han F, Xu Q, Ye T, Hou N, et al. Mortality risk of antidiabetic agents for type 2 diabetes with covid-19: A systematic review and meta-analysis. Front Endocrinol (Lausanne) (2021) 12:708494. doi: 10.3389/fendo.2021.708494
58. Nguyen NN, Ho DS, Nguyen HS, Ho DKN, Li HY, Lin CY, et al. Preadmission use of antidiabetic medications and mortality among patients with covid-19 having type 2 diabetes: A meta-analysis. Metabolism (2022) 131:155196. doi: 10.1016/j.metabol.2022.155196
59. Chen Y, Lv X, Lin S, Arshad M, Dai M. The association between antidiabetic agents and clinical outcomes of covid-19 patients with diabetes: A Bayesian network meta-analysis. Front Endocrinol (Lausanne) (2022) 13:895458. doi: 10.3389/fendo.2022.895458
60. Gayle A, Dickinson S, Poole C, Pang M, Fauconnot O, Quint JK. Incidence of type ii diabetes in chronic obstructive pulmonary disease: A nested case-control study. NPJ Prim Care Respir Med (2019) 29(1):28. doi: 10.1038/s41533-019-0138-6
61. Mannino DM, Thorn D, Swensen A, Holguin F. Prevalence and outcomes of diabetes, hypertension and cardiovascular disease in copd. Eur Respir J (2008) 32(4):962–9. doi: 10.1183/09031936.00012408
62. Sharif S, van der Graaf Y, Cramer MJ, Kapelle LJ, de Borst GJ, Visseren FLJ, et al. Low-grade inflammation as a risk factor for cardiovascular events and all-cause mortality in patients with type 2 diabetes. Cardiovasc Diabetol (2021) 20(1):220. doi: 10.1186/s12933-021-01409-0
63. Kaminski DM, Schaan BD, da Silva AM, Soares PP, Plentz RD, Dall’Ago P. Inspiratory muscle weakness is associated with autonomic cardiovascular dysfunction in patients with type 2 diabetes mellitus. Clin Auton Res (2011) 21(1):29–35. doi: 10.1007/s10286-010-0087-1
64. Kinney GL, Black-Shinn JL, Wan ES, Make B, Regan E, Lutz S, et al. Pulmonary function reduction in diabetes with and without chronic obstructive pulmonary disease. Diabetes Care (2014) 37(2):389–95. doi: 10.2337/dc13-1435
65. Klein OL, Krishnan JA, Glick S, Smith LJ. Systematic review of the association between lung function and type 2 diabetes mellitus. Diabetes Med (2010) 27(9):977–87. doi: 10.1111/j.1464-5491.2010.03073.x
66. Ofulue AF, Thurlbeck WM. Experimental diabetes and the lung. ii. In vivo connective tissue metabolism. Am Rev Respir Dis (1988) 138(2):284–9. doi: 10.1164/ajrccm/138.2.284
67. Popov D, Hasu M, Costache G, Stern D, Simionescu M. Capillary and aortic endothelia interact in situ with nonenzymatically glycated albumin and develop specific alterations in early experimental diabetes. Acta Diabetol (1997) 34(4):285–93. doi: 10.1007/s005920050090
68. Weynand B, Jonckheere A, Frans A, Rahier J. Diabetes mellitus induces a thickening of the pulmonary basal lamina. Respiration (1999) 66(1):14–9. doi: 10.1159/000029331
69. Watanabe K, Senju S, Toyoshima H, Yoshida M. Thickness of the basement membrane of bronchial epithelial cells in lung diseases as determined by transbronchial biopsy. Respir Med (1997) 91(7):406–10. doi: 10.1016/s0954-6111(97)90254-7
70. Zanini A, Chetta A, Imperatori AS, Spanevello A, Olivieri D. The role of the bronchial microvasculature in the airway remodelling in asthma and copd. Respir Res (2010) 11(1):132. doi: 10.1186/1465-9921-11-132
71. Eickhoff P, Valipour A, Kiss D, Schreder M, Cekici L, Geyer K, et al. Determinants of systemic vascular function in patients with stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2008) 178(12):1211–8. doi: 10.1164/rccm.200709-1412OC
72. Barr RG, Mesia-Vela S, Austin JH, Basner RC, Keller BM, Reeves AP, et al. Impaired flow-mediated dilation is associated with low pulmonary function and emphysema in ex-smokers: The emphysema and cancer action project (Emcap) study. Am J Respir Crit Care Med (2007) 176(12):1200–7. doi: 10.1164/rccm.200707-980OC
73. Anderson TJ, Uehata A, Gerhard MD, Meredith IT, Knab S, Delagrange D, et al. Close relation of endothelial function in the human coronary and peripheral circulations. J Am Coll Cardiol (1995) 26(5):1235–41. doi: 10.1016/0735-1097(95)00327-4
74. Minet C, Vivodtzev I, Tamisier R, Arbib F, Wuyam B, Timsit JF, et al. Reduced six-minute walking distance, high fat-Free-Mass index and hypercapnia are associated with endothelial dysfunction in copd. Respir Physiol Neurobiol (2012) 183(2):128–34. doi: 10.1016/j.resp.2012.06.017
75. Thomashow MA, Shimbo D, Parikh MA, Hoffman EA, Vogel-Claussen J, Hueper K, et al. Endothelial microparticles in mild chronic obstructive pulmonary disease and emphysema. the multi-ethnic study of atherosclerosis chronic obstructive pulmonary disease study. Am J Respir Crit Care Med (2013) 188(1):60–8. doi: 10.1164/rccm.201209-1697OC
76. Deng F, Wang S, Zhang L. Endothelial microparticles act as novel diagnostic and therapeutic biomarkers of diabetes and its complications: A literature review. BioMed Res Int (2016) 2016:9802026. doi: 10.1155/2016/9802026
77. Pernomian L, Moreira JD, Gomes MS. In the view of endothelial microparticles: Novel perspectives for diagnostic and pharmacological management of cardiovascular risk during diabetes distress. J Diabetes Res (2018) 2018:9685205. doi: 10.1155/2018/9685205
78. Zhang Y, Ma KL, Gong YX, Wang GH, Hu ZB, Liu L, et al. Platelet microparticles mediate glomerular endothelial injury in early diabetic nephropathy. J Am Soc Nephrol (2018) 29(11):2671–95. doi: 10.1681/asn.2018040368
79. Al-Qaissi A, Papageorgiou M, Deshmukh H, Madden LA, Rigby A, Kilpatrick ES, et al. Effects of acute insulin-induced hypoglycaemia on endothelial microparticles in adults with and without type 2 diabetes. Diabetes Obes Metab (2019) 21(3):533–40. doi: 10.1111/dom.13548
80. Dec-Gilowska M, Trojnar M, Makaruk B, Czop M, Przybylska-Kuc S, Mosiewicz-Madejska B, et al. Circulating endothelial microparticles and aortic stiffness in patients with type 2 diabetes mellitus. Medicina (Kaunas) (2019) 55(9):596. doi: 10.3390/medicina55090596
81. Wang GH, Ma KL, Zhang Y, Hu ZB, Liu L, Lu J, et al. Platelet microparticles contribute to aortic vascular endothelial injury in diabetes Via the Mtorc1 pathway. Acta Pharmacol Sin (2019) 40(4):468–76. doi: 10.1038/s41401-018-0186-4
82. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (Vegf) and its receptors. FASEB J (1999) 13(1):9–22. doi: 10.1096/fasebj.13.1.9
83. Yasuo M, Mizuno S, Kraskauskas D, Bogaard HJ, Natarajan R, Cool CD, et al. Hypoxia inducible factor-1α in human emphysema lung tissue. Eur Respir J (2011) 37(4):775–83. doi: 10.1183/09031936.00022910
84. Zhang D, Lv FL, Wang GH. Effects of hif-1α on diabetic retinopathy angiogenesis and vegf expression. Eur Rev Med Pharmacol Sci (2018) 22(16):5071–6. doi: 10.26355/eurrev_201808_15699
85. Zhu Y, Wang Y, Jia Y, Xu J, Chai Y. Roxadustat promotes angiogenesis through hif-1α/Vegf/Vegfr2 signaling and accelerates cutaneous wound healing in diabetic rats. Wound Repair Regener (2019) 27(4):324–34. doi: 10.1111/wrr.12708
86. Arima M, Nakao S, Yamaguchi M, Feng H, Fujii Y, Shibata K, et al. Claudin-5 redistribution induced by inflammation leads to anti-Vegf-Resistant diabetic macular edema. Diabetes (2020) 69(5):981–99. doi: 10.2337/db19-1121
87. Huang X, Liang P, Jiang B, Zhang P, Yu W, Duan M, et al. Hyperbaric oxygen potentiates diabetic wound healing by promoting fibroblast cell proliferation and endothelial cell angiogenesis. Life Sci (2020) 259:118246. doi: 10.1016/j.lfs.2020.118246
88. Pont C, Ascaso FJ, Grzybowski A, Huerva V. Corneal endothelial cell density during diabetes mellitus and ocular diabetes complications treatment. J Fr Ophtalmol (2020) 43(8):794–8. doi: 10.1016/j.jfo.2019.12.003
89. Xiang E, Han B, Zhang Q, Rao W, Wang Z, Chang C, et al. Human umbilical cord-derived mesenchymal stem cells prevent the progression of early diabetic nephropathy through inhibiting inflammation and fibrosis. Stem Cell Res Ther (2020) 11(1):336. doi: 10.1186/s13287-020-01852-y
90. Calabrese C, Bocchino V, Vatrella A, Marzo C, Guarino C, Mascitti S, et al. Evidence of angiogenesis in bronchial biopsies of smokers with and without airway obstruction. Respir Med (2006) 100(8):1415–22. doi: 10.1016/j.rmed.2005.11.009
91. Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, et al. Inhibition of vegf receptors causes lung cell apoptosis and emphysema. J Clin Invest (2000) 106(11):1311–9. doi: 10.1172/jci10259
92. Hashimoto M, Tanaka H, Abe S. Quantitative analysis of bronchial wall vascularity in the medium and small airways of patients with asthma and copd. Chest (2005) 127(3):965–72. doi: 10.1378/chest.127.3.965
93. Green CE, Clarke J, Bicknell R, Turner AM. Pulmonary microrna changes alter angiogenesis in chronic obstructive pulmonary disease and lung cancer. Biomedicines (2021) 9(7):830. doi: 10.3390/biomedicines9070830
94. Wen F-Q, Liu X, Manda W, Terasaki Y, Kobayashi T, Abe S, et al. Th2 cytokine-enhanced and tgf-Beta-Enhanced vascular endothelial growth factor production by cultured human airway smooth muscle cells is attenuated by ifn-gamma and corticosteroids. J Allergy Clin Immunol (2003) 111(6):1307–18. doi: 10.1067/mai.2003.1455
95. Taraseviciene-Stewart L, Scerbavicius R, Choe K-H, Moore M, Sullivan A, Nicolls MR, et al. An animal model of autoimmune emphysema. Am J Respir Crit Care Med (2005) 171(7):734–42. doi: 10.1164/rccm.200409-1275OC
96. Tuder RM, Flook BE, Voelkel NF. Increased gene expression for vegf and the vegf receptors Kdr/Flk and flt in lungs exposed to acute or to chronic hypoxia. modulation of gene expression by nitric oxide. J Clin Invest (1995) 95(4):1798–807. doi: 10.1172/JCI117858
97. Tuder RM, Zhen L, Cho CY, Taraseviciene-Stewart L, Kasahara Y, Salvemini D, et al. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol (2003) 29(1):88–97. doi: 10.1165/rcmb.2002-0228OC
98. Polverino F, Celli BR, Owen CA. Copd as an endothelial disorder: Endothelial injury linking lesions in the lungs and other organs? (2017 grover conference series). Pulm Circ (2018) 8(1):2045894018758528. doi: 10.1177/2045894018758528
99. Balk-Møller E, Windeløv JA, Svendsen B, Hunt J, Ghiasi SM, Sørensen CM, et al. Glucagon-like peptide 1 and atrial natriuretic peptide in a female mouse model of obstructive pulmonary disease. J Endocr Soc (2020) 4(1):bvz034. doi: 10.1210/jendso/bvz034
100. Huang J, Yi H, Zhao C, Zhang Y, Zhu L, Liu B, et al. Glucagon-like peptide-1 receptor (Glp-1r) signaling ameliorates dysfunctional immunity in copd patients. Int J Chron Obstruct Pulmon Dis (2018) 13:3191–202. doi: 10.2147/copd.S175145
101. Sun YH, He L, Yan MY, Zhao RQ, Li B, Wang F, et al. Overexpression of glp-1 receptors suppresses proliferation and cytokine release by airway smooth muscle cells of patients with chronic obstructive pulmonary disease Via activation of Abca1. Mol Med Rep (2017) 16(1):929–36. doi: 10.3892/mmr.2017.6618
102. Viby NE, Isidor MS, Buggeskov KB, Poulsen SS, Hansen JB, Kissow H. Glucagon-like peptide-1 (Glp-1) reduces mortality and improves lung function in a model of experimental obstructive lung disease in female mice. Endocrinology (2013) 154(12):4503–11. doi: 10.1210/en.2013-1666
103. Zou H, Zhu N, Li S. The emerging role of dipeptidyl-Peptidase-4 as a therapeutic target in lung disease. Expert Opin Ther Targets (2020) 24(2):147–53. doi: 10.1080/14728222.2020.1721468
104. Shapiro M, Arbel C, Zucker I, Balmor GR, Lutski M, Derazne E, et al. Asthma in youth and early-onset type 2 diabetes: A nationwide study of 1.72 million Israeli adolescents. J Clin Endocrinol Metab (2021) 106(12):e5043–e53. doi: 10.1210/clinem/dgab542
105. Song Y, Klevak A, Manson JE, Buring JE, Liu S. Asthma, chronic obstructive pulmonary disease, and type 2 diabetes in the women’s health study. Diabetes Res Clin Pract (2010) 90(3):365–71. doi: 10.1016/j.diabres.2010.09.010
106. Suissa S, Kezouh A, Ernst P. Inhaled corticosteroids and the risks of diabetes onset and progression. Am J Med (2010) 123(11):1001–6. doi: 10.1016/j.amjmed.2010.06.019
107. Greiner B, Hartwell M. Prevalence and associations between metabolically unhealthy obesity and asthma exacerbations and emergency department usage. Ann Allergy Asthma Immunol (2022) 129(5):580–4.e2. doi: 10.1016/j.anai.2022.08.640
108. Zhang P, Lopez R, Attaway AH, Georas SN, Khatri SB, Abi-Saleh S, et al. Diabetes mellitus is associated with worse outcome in patients hospitalized for asthma. J Allergy Clin Immunol: In Pract (2021) 9(4):1562–9.e1. doi: 10.1016/j.jaip.2020.10.054
109. Waasdorp M, Duitman J, Florquin S, Spek CA, Spek AC. Protease activated receptor 2 in diabetic nephropathy: A double edged sword. Am J Transl Res (2017) 9(10):4512–20.
110. Wilson JW, Kotsimbos T. Airway vascular remodeling in asthma. Curr Allergy Asthma Rep (2003) 3(2):153–8. doi: 10.1007/s11882-003-0028-3
111. Johansson MW, Kruger SJ, Schiebler ML, Evans MD, Sorkness RL, Denlinger LC, et al. Markers of vascular perturbation correlate with airway structural change in asthma. Am J Respir Crit Care Med (2013) 188(2):167–78. doi: 10.1164/rccm.201301-0185OC
112. Tang ML, Fiscus LC. Important roles for l-selectin and icam-1 in the development of allergic airway inflammation in asthma. Pulm Pharmacol Ther (2001) 14(3):203–10. doi: 10.1006/pupt.2001.0293
113. Hirata N, Kohrogi H, Iwagoe H, Goto E, Hamamoto J, Fujii K, et al. Allergen exposure induces the expression of endothelial adhesion molecules in passively sensitized human bronchus: Time course and the role of cytokines. Am J Respir Cell Mol Biol (1998) 18(1):12–20. doi: 10.1165/ajrcmb.18.1.2704
114. Gosset P, Tillie-Leblond I, Janin A, Marquette CH, Copin MC, Wallaert B, et al. Expression of e-selectin, icam-1 and vcam-1 on bronchial biopsies from allergic and non-allergic asthmatic patients. Int Arch Allergy Immunol (1995) 106(1):69–77. doi: 10.1159/000236892
115. Liao JK. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest (2013) 123(2):540–1. doi: 10.1172/JCI66843
116. Sorkness RL, Mehta H, Kaplan MR, Miyasaka M, Hefle SL, Lemanske RF. Effect of icam-1 blockade on lung inflammation and physiology during acute viral bronchiolitis in rats. Pediatr Res (2000) 47(6):819–24. doi: 10.1203/00006450-200006000-00023
117. Navaratna D, McGuire PG, Menicucci G, Das A. Proteolytic degradation of ve-cadherin alters the blood-retinal barrier in diabetes. Diabetes (2007) 56(9):2380–7. doi: 10.2337/db06-1694
118. Parkkola A, Härkönen T, Ryhänen SJ, Uibo R, Ilonen J, Knip M. Transglutaminase antibodies and celiac disease in children with type 1 diabetes and in their family members. Pediatr Diabetes (2018) 19(2):305–13. doi: 10.1111/pedi.12563
119. Guandalini S. Type 1 diabetes and celiac disease: Can (and should) we raise the cut-off of tissue transglutaminase immunoglobulin a to decide whether to biopsy? J Pediatr (2020) 223:8–10. doi: 10.1016/j.jpeds.2020.03.063
120. Lee HJ, Jung YH, Choi GE, Kim JS, Chae CW, Lim JR, et al. Urolithin a suppresses high glucose-induced neuronal amyloidogenesis by modulating Tgm2-dependent er-mitochondria contacts and calcium homeostasis. Cell Death Differ (2021) 28(1):184–202. doi: 10.1038/s41418-020-0593-1
121. Muzslay E, Hámory E, Herczeg V, Tóth-Heyn P, Körner A, Madácsy L, et al. Transitional elevation of anti-tissue transglutaminase antibodies in children with type 1 diabetes mellitus without coeliac disease. Orv Hetil (2021) 162(48):1924–30. doi: 10.1556/650.2021.32287
122. Prat-Duran J, Pinilla E, Nørregaard R, Simonsen U, Buus NH. Transglutaminase 2 as a novel target in chronic kidney disease - methods, mechanisms and pharmacological inhibition. Pharmacol Ther (2021) 222:107787. doi: 10.1016/j.pharmthera.2020.107787
123. Ying S, Robinson DS, Meng Q, Rottman J, Kennedy R, Ringler DJ, et al. Enhanced expression of eotaxin and Ccr3 mrna and protein in atopic asthma. association with airway hyperresponsiveness and predominant Co-localization of eotaxin mrna to bronchial epithelial and endothelial cells. Eur J Immunol (1997) 27(12):3507–16. doi: 10.1002/eji.1830271252
124. Soveg F, Abdala-Valencia H, Campbell J, Morales-Nebreda L, Mutlu GM, Cook-Mills JM. Regulation of allergic lung inflammation by endothelial cell transglutaminase 2. Am J Physiol Lung Cell Mol Physiol (2015) 309(6):L573–L83. doi: 10.1152/ajplung.00199.2015
125. Li X, Wilson JW. Increased vascularity of the bronchial mucosa in mild asthma. Am J Respir Crit Care Med (1997) 156(1):229–33. doi: 10.1164/ajrccm.156.1.9607066
126. Barbato A, Turato G, Baraldo S, Bazzan E, Calabrese F, Panizzolo C, et al. Epithelial damage and angiogenesis in the airways of children with asthma. Am J Respir Crit Care Med (2006) 174(9):975–81. doi: 10.1164/rccm.200602-189OC
127. Tanaka H, Yamada G, Saikai T, Hashimoto M, Tanaka S, Suzuki K, et al. Increased airway vascularity in newly diagnosed asthma using a high-magnification bronchovideoscope. Am J Respir Crit Care Med (2003) 168(12):1495–9. doi: 10.1164/rccm.200306-727OC
128. Feltis BN, Wignarajah D, Reid DW, Ward C, Harding R, Walters EH. Effects of inhaled fluticasone on angiogenesis and vascular endothelial growth factor in asthma. Thorax (2007) 62(4):314–9. doi: 10.1136/thx.2006.069229
129. Asosingh K, Vasanji A, Tipton A, Queisser K, Wanner N, Janocha A, et al. Eotaxin-rich proangiogenic hematopoietic progenitor cells and Ccr3+ endothelium in the atopic asthmatic response. J Immunol (2016) 196(5):2377–87. doi: 10.4049/jimmunol.1500770
130. Hoshino M, Nakamura Y, Hamid QA. Gene expression of vascular endothelial growth factor and its receptors and angiogenesis in bronchial asthma. J Allergy Clin Immunol (2001) 107(6):1034–8. doi: 10.1067/mai.2001.115626
131. Simpson A, Custovic A, Tepper R, Graves P, Stern DA, Jones M, et al. Genetic variation in vascular endothelial growth factor-a and lung function. Am J Respir Crit Care Med (2012) 185(11):1197–204. doi: 10.1164/rccm.201112-2191OC
132. Lassalle P, Delneste Y, Gosset P, Tonnel A, Capron A. Potential implication of endothelial cells in bronchial asthma. Int Arch Allergy Immunol (1991) 94(1-4):233–8. doi: 10.1159/000235368
133. Ribatti D, Puxeddu I, Crivellato E, Nico B, Vacca A, Levi-Schaffer F. Angiogenesis in asthma. Clin Exp Allergy (2009) 39(12):1815–21. doi: 10.1111/j.1365-2222.2009.03385.x
134. Asosingh K, Weiss K, Queisser K, Wanner N, Yin M, Aronica M, et al. Endothelial cells in the innate response to allergens and initiation of atopic asthma. J Clin Invest (2018) 128(7):3116–28. doi: 10.1172/JCI97720
135. Keyhanmanesh R, Rahbarghazi R, Ahmadi M. Systemic transplantation of mesenchymal stem cells modulates endothelial cell adhesion molecules induced by ovalbumin in rat model of asthma. Inflammation (2018) 41(6):2236–45. doi: 10.1007/s10753-018-0866-8
136. Harkness LM, Kanabar V, Sharma HS, Westergren-Thorsson G, Larsson-Callerfelt A-K. Pulmonary vascular changes in asthma and copd. Pulmon Pharmacol Ther (2014) 29(2):144–55. doi: 10.1016/j.pupt.2014.09.003
137. Asosingh K, Cheng G, Xu W, Savasky BM, Aronica MA, Li X, et al. Nascent endothelium initiates Th2 polarization of asthma. J Immunol (2013) 190(7):3458–65. doi: 10.4049/jimmunol.1202095
138. Asosingh K, Swaidani S, Aronica M, Erzurum SC. Th1-and Th2-dependent endothelial progenitor cell recruitment and angiogenic switch in asthma. J Immunol (2007) 178(10):6482–94. doi: 10.4049/jimmunol.178.10.6482
139. Rahbarghazi R, Keyhanmanesh R, Aslani MR, Hassanpour M, Ahmadi M. Bone marrow mesenchymal stem cells and condition media diminish inflammatory adhesion molecules of pulmonary endothelial cells in an ovalbumin-induced asthmatic rat model. Microvascular Res (2019) 121:63–70. doi: 10.1016/j.mvr.2018.10.005
140. Chetta A, Zanini A, Foresi A, D’Ippolito R, Tipa A, Castagnaro A, et al. Vascular endothelial growth factor up-regulation and bronchial wall remodelling in asthma. Clin Exp Allergy (2005) 35(11):1437–42. doi: 10.1111/j.1365-2222.2005.02360.x
141. Hoshino M, Takahashi M, Aoike N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J Allergy Clin Immunol (2001) 107(2):295–301. doi: 10.1067/mai.2001.111928
142. Nadi E, Hajilooi M, Babakhani D, Rafiei A. Platelet endothelial cell adhesion molecule-1 polymorphism in patients with bronchial asthma. Iranian J Allergy Asthma Immunol (2012) 11(4):276–81.
143. McLean BA, Wong CK, Kaur KD, Seeley RJ, Drucker DJ. Differential importance of endothelial and hematopoietic cell glp-1rs for cardiometabolic versus hepatic actions of semaglutide. JCI Insight (2021) 6(22):e153732. doi: 10.1172/jci.insight.153732
144. Foer D, Beeler PE, Cui J, Karlson EW, Bates DW, Cahill KN. Asthma exacerbations in patients with type 2 diabetes and asthma on glucagon-like peptide-1 receptor agonists. Am J Respir Crit Care Med (2021) 203(7):831–40. doi: 10.1164/rccm.202004-0993OC
145. Wu AY, Cahill KN, Toki S, Peebles RS Jr. Evaluating the glucagon-like peptide-1 receptor in managing asthma. Curr Opin Allergy Clin Immunol (2022) 22(1):36–41. doi: 10.1097/aci.0000000000000797
146. Toki S, Newcomb DC, Printz RL, Cahill KN, Boyd KL, Niswender KD, et al. Glucagon-like peptide-1 receptor agonist inhibits aeroallergen-induced activation of Ilc2 and neutrophilic airway inflammation in obese mice. Allergy (2021) 76(11):3433–45. doi: 10.1111/all.14879
147. Hur J, Kang JY, Kim YK, Lee SY, Lee HY. Glucagon-like peptide 1 receptor (Glp-1r) agonist relieved asthmatic airway inflammation Via suppression of Nlrp3 inflammasome activation in obese asthma mice model. Pulm Pharmacol Ther (2021) 67:102003. doi: 10.1016/j.pupt.2021.102003
148. Toki S, Goleniewska K, Reiss S, Zhang J, Bloodworth MH, Stier MT, et al. Glucagon-like peptide 1 signaling inhibits allergen-induced lung il-33 release and reduces group 2 innate lymphoid cell cytokine production In vivo. J Allergy Clin Immunol (2018) 142(5):1515–28.e8. doi: 10.1016/j.jaci.2017.11.043
149. Mitchell PD, Salter BM, Oliveria JP, El-Gammal A, Tworek D, Smith SG, et al. Glucagon-like peptide-1 receptor expression on human eosinophils and its regulation of eosinophil activation. Clin Exp Allergy (2017) 47(3):331–8. doi: 10.1111/cea.12860
150. Zorampari C, Prakash A, Rehan HS, Gupta LK. Serum dipeptidyl peptidase-4 and eosinophil cationic protein levels in patients of bronchial asthma. Pulm Pharmacol Ther (2022) 72:102109. doi: 10.1016/j.pupt.2021.102109
151. Wang A, Tang H, Zhang N, Feng X. Association between novel glucose-lowering drugs and risk of asthma: A network meta-analysis of cardiorenal outcome trials. Diabetes Res Clin Pract (2022) 183:109080. doi: 10.1016/j.diabres.2021.109080
152. Helal MG, Megahed NA, Abd Elhameed AG. Saxagliptin mitigates airway inflammation in a mouse model of acute asthma Via modulation of nf-kb and Tlr4. Life Sci (2019) 239:117017. doi: 10.1016/j.lfs.2019.117017
153. Emson C, Pham TH, Manetz S, Newbold P. Periostin and dipeptidyl peptidase-4: Potential biomarkers of interleukin 13 pathway activation in asthma and allergy. Immunol Allergy Clin North Am (2018) 38(4):611–28. doi: 10.1016/j.iac.2018.06.004
154. Nader MA. Inhibition of airway inflammation and remodeling by sitagliptin in murine chronic asthma. Int Immunopharmacol (2015) 29(2):761–9. doi: 10.1016/j.intimp.2015.08.043
155. Cui W, Zhang S, Cai Z, Hu X, Zhang R, Wang Y, et al. The antidiabetic agent glibenclamide protects airway hyperresponsiveness and inflammation in mice. Inflammation (2015) 38(2):835–45. doi: 10.1007/s10753-014-9993-z
156. Tsukamoto K, Ohta K, Suzuki R, Hara M, Osuga J, Tobe K, et al. Glimepiride-induced bronchial asthma: Case report. Diabetes Care (2008) 31(12):e91. doi: 10.2337/dc08-1536
157. Bankers-Fulbright JL, Kephart GM, Loegering DA, Bradford AL, Okada S, Kita H, et al. Sulfonylureas inhibit cytokine-induced eosinophil survival and activation. J Immunol (1998) 160(11):5546–53. doi: 10.4049/jimmunol.160.11.5546
158. Szczeklik A, Pietoń R, Sieradzki J. Alternation in both insulin release and its hypoglycemic effects in atopic bronchial asthma. J Allergy Clin Immunol (1980) 66(5):424–7. doi: 10.1016/0091-6749(80)90123-2
159. Kopf S, Kumar V, Kender Z, Han Z, Fleming T, Herzig S, et al. Diabetic pneumopathy-a new diabetes-associated complication: Mechanisms, consequences and treatment considerations. Front Endocrinol (Lausanne) (2021) 12:765201. doi: 10.3389/fendo.2021.765201
160. Wang D, Ma Y, Tong X, Zhang Y, Fan H. Diabetes mellitus contributes to idiopathic pulmonary fibrosis: A review from clinical appearance to possible pathogenesis. Front Public Health (2020) 8:196. doi: 10.3389/fpubh.2020.00196
161. Jagadapillai R, Rane MJ, Lin X, Roberts AM, Hoyle GW, Cai L, et al. Diabetic microvascular disease and pulmonary fibrosis: The contribution of platelets and systemic inflammation. Int J Mol Sci (2016) 17(11):1853. doi: 10.3390/ijms17111853
162. Goldman MD. Lung dysfunction in diabetes. Diabetes Care (2003) 26(6):1915–8. doi: 10.2337/diacare.26.6.1915
163. Mirrakhimov AE. Chronic obstructive pulmonary disease and glucose metabolism: A bitter sweet symphony. Cardiovasc Diabetol (2012) 11:132. doi: 10.1186/1475-2840-11-132
164. Vlassara H, Uribarri J. Advanced glycation end products (Age) and diabetes: Cause, effect, or both? Curr Diabetes Rep (2014) 14(1):453. doi: 10.1007/s11892-013-0453-1
165. Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Eur Heart J (2013) 34(31):2436–43. doi: 10.1093/eurheartj/eht149
166. Rojas A, Lindner C, Gonzàlez I, Morales MA. Advanced-glycation end-products axis: A contributor to the risk of severe illness from covid-19 in diabetes patients. World J Diabetes (2021) 12(5):590–602. doi: 10.4239/wjd.v12.i5.590
167. Hunt WR, Helfman BR, McCarty NA, Hansen JM. Advanced glycation end products are elevated in cystic fibrosis-related diabetes and correlate with worse lung function. J Cyst Fibros (2016) 15(5):681–8. doi: 10.1016/j.jcf.2015.12.011
168. Machahua C, Montes-Worboys A, Llatjos R, Escobar I, Dorca J, Molina-Molina M, et al. Increased age-rage ratio in idiopathic pulmonary fibrosis. Respir Res (2016) 17(1):144. doi: 10.1186/s12931-016-0460-2
169. Oczypok EA, Perkins TN, Oury TD. All the “Rage” in lung disease: The receptor for advanced glycation endproducts (Rage) is a major mediator of pulmonary inflammatory responses. Paediatr Respir Rev (2017) 23:40–9. doi: 10.1016/j.prrv.2017.03.012
170. Talakatta G, Sarikhani M, Muhamed J, Dhanya K, Somashekar BS, Mahesh PA, et al. Diabetes induces fibrotic changes in the lung through the activation of tgf-B signaling pathways. Sci Rep (2018) 8(1):11920. doi: 10.1038/s41598-018-30449-y
171. Han X, Ding C, Sang X, Peng M, Yang Q, Ning Y, et al. Targeting Sirtuin1 to treat aging-related tissue fibrosis: From prevention to therapy. Pharmacol Ther (2022) 229:107983. doi: 10.1016/j.pharmthera.2021.107983
172. Liang J, Huang G, Liu X, Taghavifar F, Liu N, Wang Y, et al. The Zip8/Sirt1 axis regulates alveolar progenitor cell renewal in aging and idiopathic pulmonary fibrosis. J Clin Invest (2022) 132(11):e157338. doi: 10.1172/jci157338
173. Hammer SS, Vieira CP, McFarland D, Sandler M, Levitsky Y, Dorweiler TF, et al. Fasting and fasting-mimicking treatment activate Sirt1/Lxrα and alleviate diabetes-induced systemic and microvascular dysfunction. Diabetologia (2021) 64(7):1674–89. doi: 10.1007/s00125-021-05431-5
174. Wu X, Liu H, Brooks A, Xu S, Luo J, Steiner R, et al. Sirt6 mitigates heart failure with preserved ejection fraction in diabetes. Circ Res (2022) 131(11):926–43. doi: 10.1161/circresaha.121.318988
175. Yang J, Tan Y, Zhao F, Ma Z, Wang Y, Zheng S, et al. Angiotensin ii plays a critical role in diabetic pulmonary fibrosis most likely Via activation of nadph oxidase-mediated nitrosative damage. Am J Physiol Endocrinol Metab (2011) 301(1):E132–44. doi: 10.1152/ajpendo.00629.2010
176. Cheng D, Xu Q, Wang Y, Li G, Sun W, Ma D, et al. Metformin attenuates silica-induced pulmonary fibrosis Via ampk signaling. J Transl Med (2021) 19(1):349. doi: 10.1186/s12967-021-03036-5
177. Gu X, Han YY, Yang CY, Ji HM, Lan YJ, Bi YQ, et al. Activated ampk by metformin protects against fibroblast proliferation during pulmonary fibrosis by suppressing Foxm1. Pharmacol Res (2021) 173:105844. doi: 10.1016/j.phrs.2021.105844
178. Kheirollahi V, Wasnick RM, Biasin V, Vazquez-Armendariz AI, Chu X, Moiseenko A, et al. Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat Commun (2019) 10(1):2987. doi: 10.1038/s41467-019-10839-0
179. Li SX, Li C, Pang XR, Zhang J, Yu GC, Yeo AJ, et al. Metformin attenuates silica-induced pulmonary fibrosis by activating autophagy Via the ampk-mtor signaling pathway. Front Pharmacol (2021) 12:719589. doi: 10.3389/fphar.2021.719589
180. Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K, et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat Med (2018) 24(8):1121–7. doi: 10.1038/s41591-018-0087-6
181. Sato N, Takasaka N, Yoshida M, Tsubouchi K, Minagawa S, Araya J, et al. Metformin attenuates lung fibrosis development Via Nox4 suppression. Respir Res (2016) 17(1):107. doi: 10.1186/s12931-016-0420-x
182. Gou S, Zhu T, Wang W, Xiao M, Wang XC, Chen ZH. Glucagon like peptide-1 attenuates bleomycin-induced pulmonary fibrosis, involving the inactivation of nf-Kb in mice. Int Immunopharmacol (2014) 22(2):498–504. doi: 10.1016/j.intimp.2014.07.010
183. Oztay F, Sancar-Bas S, Gezginci-Oktayoglu S, Ercin M, Bolkent S. Exendin-4 partly ameliorates - hyperglycemia-mediated tissue damage in lungs of streptozotocin-induced diabetic mice. Peptides (2018) 99:99–107. doi: 10.1016/j.peptides.2017.12.007
184. Suzuki T, Tada Y, Gladson S, Nishimura R, Shimomura I, Karasawa S, et al. Vildagliptin ameliorates pulmonary fibrosis in lipopolysaccharide-induced lung injury by inhibiting endothelial-to-Mesenchymal transition. Respir Res (2017) 18(1):177. doi: 10.1186/s12931-017-0660-4
185. Jin GY, Bok SM, Han YM, Chung MJ, Yoon K-H, Kim SR, et al. Effectiveness of rosiglitazone on bleomycin-induced lung fibrosis: Assessed by micro-computed tomography and pathologic scores. Eur J Radiol (2012) 81(8):1901–6. doi: 10.1016/j.ejrad.2010.12.061
186. Yu W, Mi L, Long T. Efficacies of rosiglitazone and retinoin on bleomycin-induced pulmonary fibrosis in rats. Exp Ther Med (2017) 14(1):609–15. doi: 10.3892/etm.2017.4555
187. Milam JE, Keshamouni VG, Phan SH, Hu B, Gangireddy SR, Hogaboam CM, et al. Ppar-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol (2008) 294(5):L891–901. doi: 10.1152/ajplung.00333.2007
Keywords: endothelial cells, COVID-19, asthma, COPD, type-2 diabetes
Citation: Zhang L, Jiang F, Xie Y, Mo Y, Zhang X and Liu C (2023) Diabetic endothelial microangiopathy and pulmonary dysfunction. Front. Endocrinol. 14:1073878. doi: 10.3389/fendo.2023.1073878
Received: 19 October 2022; Accepted: 17 February 2023;
Published: 21 March 2023.
Edited by:
Liang-Jun Yan, University of North Texas Health Science Center, United StatesReviewed by:
Chuanbin Yang, First Affiliated Hospital of Southern University of Science and Technology, ChinaSaeed Kolahian, University of Marburg, Germany
Junqing Yang, Guangdong Provincial People’s Hospital, China
Copyright © 2023 Zhang, Jiang, Xie, Mo, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lanlan Zhang, bGx6aGFuZ0BzY3UuZWR1LmNu; Xin Zhang, eGluemhhbmcyMDE2MThAZ21haWwuY29t; Chuntao Liu, dGFvc2VuNjY2OTk5QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship