 Yueyue Zhu
Yueyue Zhu Xiaorui Liu
Xiaorui Liu Yichi Xu2
Yichi Xu2 Yi Lin
Yi Lin- 1Reproductive Medicine Center, Shanghai Sixth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2The International Peace Maternity and Child Health Hospital, School of Medicine, Shanghai JiaoTong University, Shanghai, China
Uterine spiral artery remodeling is necessary for fetal growth and development as well as pregnancy outcomes. During remodeling, trophoblasts invade the arteries, replace the endothelium and disrupt the vascular smooth muscle, and are strictly regulated by the local microenvironment. Elevated glucose levels at the fetal-maternal interface are associated with disorganized placental villi and poor placental blood flow. Hyperglycemia disturbs trophoblast proliferation and invasion via inhibiting the epithelial-mesenchymal transition, altering the protein expression of related proteases (MMP9, MMP2, and uPA) and angiogenic factors (VEGF, PIGF). Besides, hyperglycemia influences the cellular crosstalk between immune cells, trophoblast, and vascular cells, leading to the failure of spiral artery remodeling. This review provides insight into molecular mechanisms and signaling pathways of hyperglycemia that influence trophoblast functions and uterine spiral artery remodeling.
1 Introduction
Hyperglycemia (HG) is a common metabolic imbalance in pregnant women with Type 1 diabetes mellitus (T1DM), Type 2 diabetes mellitus (T2DM), or combined with pregnancy and gestational diabetes mellitus (GDM) (1). Abnormally high blood glucose levels in the pregnancy may lead to abnormal uterine glucose concentration (2). Unhealthy dietary habits, such as a high glucose intake, are prevalent nowadays. Both HG and HG-induced cytokines releasing affect trophoblast function and uterine spiral arteries (SAs) remodeling, which can in turn increase the incidence of pregnancy complications, such as pre-eclampsia, malformations and miscarriage, and thus endanger the health of pregnant women and fetuses (3, 4).
SAs facilitate the exchange of nutrients, gases, and waste between mother and fetus (5). During SA remodeling, the original uterine SA converts into low-resistance and highly dilated vessels to meet the pregnancy blood requirements and prevent damage to the villi (6). Uterine SA remodeling has four stages. Firstly, trophoblasts and leukocytes in the vessel wall are not yet invasion, the endothelial cells (ECs) and smooth muscle cell layers of the vessel wall are intact. Secondly, the vascular structure begins to be destroyed by decidual NK (dNK) cells and macrophages in the vessel wall before trophoblast invasion (7). Thirdly, extravillous trophoblasts (EVTs) appear in the vessel wall and lumen. Finally, vascular smooth muscle cells (VSMCs) and ECs are completely lost and replaced by intravascular EVTs, and the wall matrix is replaced by fibrin-like substances. In addition, cytokines, angiogenic factors, enzymes, and extracellular matrix (ECM) also participate in regulating SA remodeling (8, 9). Trophoblasts are exposed to the maternal circulation and are influenced by the maternal endocrine, metabolic, and inflammatory environments. Hence, the influence of various maternal microenvironmental factors, such as high fat, high sugar diets, obesity and diabetes may affect trophoblast functions and the SA remodeling (10).
There are fewer blood vessels and villi in placenta of diabetic women with unexplained stillbirths than those with live births (11). Different from being replaced by trophoblasts in normal pregnancy, VSMCs in the placenta of biobreeding diabetes-prone rat were almost complete while SA remodeling was failure (12). Taken together, HG may cause insufficient SA remodeling via impaired trophoblasts. Herein, we will summarize the current knowledge on the possible effects of HG on trophoblast function as well as their role in uterine SA remodeling.
2 Trophoblasts and uterine SA remodeling
Trophoblasts are the first cell type to differentiate during embryogenesis. In this process, trophoblast stem cells are derived from embryonic trophectoderm. They can differentiate into various trophoblast cell lines and acquire many specialized functions, including invasion potential and endocrine activity (13). Cytotrophoblasts (CTBs) are stem cells that proliferate rapidly once embedded in maternal decidua. The outer layer of CTBs fuses into primitive syncytiotrophoblasts (STBs), which erode surrounding decidua and generate lacunae filled with blood (14). Placental villi bathed in maternal blood are floating villi in charge of placenta material transport, whereas villi, anchored in the placental basal plate, differentiate into EVTs (15).
During differentiation, trophoblasts lose adherent epithelial phenotype and acquire a mesenchymal phenotype and invasion ability through epithelial-mesenchymal transition (EMT). The E-cadherin/β-catenin complex is a calcium-dependent transmembrane protein distributing in epithelial tissues which form cell tight junctions, inhibit cell movement and maintain epithelial integrity (16). Decreased E-cadherin expression results in the upregulation of integrin α1β1, α5β1 and αVβ3, VE-cadherin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1(VCAM-1) (17, 18). EVTs invade the decidual stroma to form the interstitial extravillous trophoblasts (iEVTs) to promote the muscular layer of vessel wall degradation (19). EVTs invade the decidual blood vessels to form the endovascular extravillous trophoblasts (enEVTs) to replace ECs and VSMCs (20). VSMCs undergo morphological changes while EVTs penetrate the vessel wall via intravascular or interstitial pathways. They shift to a synthetic phenotype, migrate from the vessel wall, and undergo apoptosis. Trophoblasts secrete platelet derived growth factor BB (PDGF-BB) to bind the PDGF receptor β (PDGFR-β) of VSMCs to activate the PDGF signaling pathway and induce de-differentiation of VSMCs (21). EVT secretes tumor necrosis factor α (TNF-α), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and Fas ligand to induce apoptosis of VSMCs (22) (Figure 1A).
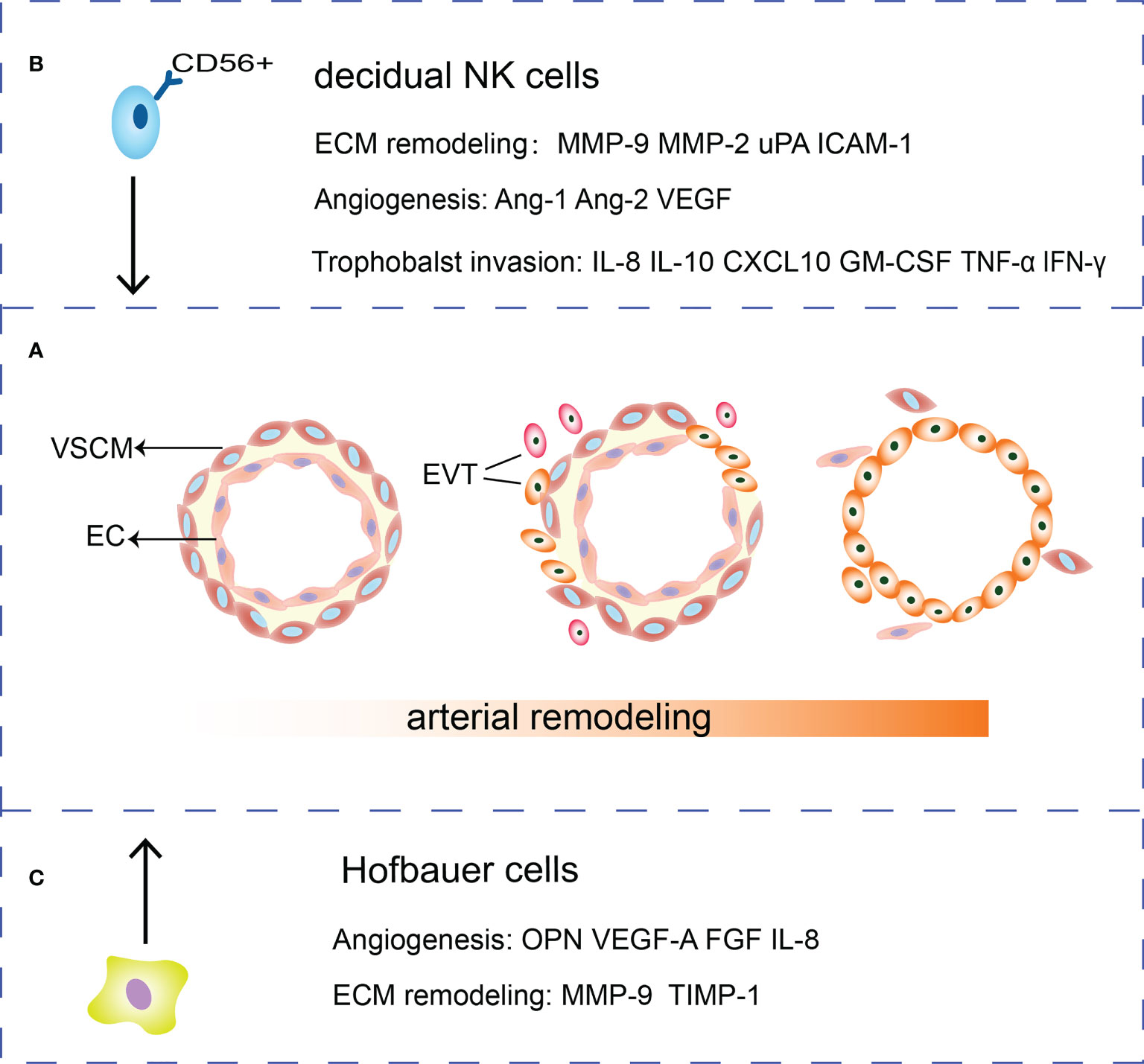
Figure 1 Roles of HBCs, EVTs and dNK cells in SA remodeling. (A) During the SA remodeling, VSMCs surrounding the arteries are removed and ECs are gradually replaced by EVTs. (B) dNK cells secrete cytokines such as IL-10, GM-CSF, TNF-α, and IFN-γ and chemokine IL-8, CXCL10 to regulate trophoblasts invasion. Meanwhile, dNK cells are potent sources of angiogenic factors such as Ang-1, Ang-2, VEGF. dNK cells secrete MMP-2, MMP-9, uPA and ICAM-1 that participate in ECM remodeling. (C) HBCs can secrete MMP and TIMP to remodel the extracellular matrix. Besides, HBCs secrete a range of factors that play a role in remodeling vessels such as OPN, FGF, VEGF-A and IL-8.
In addition, trophoblast-immune cell-vascular interactions are important determinants of adequate SA remodeling. The dNK cells, macrophages, trophoblasts, and their crosstalk are important for adequate SA remodeling, and any dysregulation may lead to remodeling obstacles (23, 24).
3 HG affects the biological functions of trophoblasts in uterine SA remodeling
Proliferation, signaling disorders, impaired placenta blood flow, and increased vascular resistance were observed in streptozotocin-induced GDM rat model (25–27). And decreased VSMC apoptosis was observed in placentas of mice with GDM (28). In addition, HG impaired the differentiation of trophoblast stem cell into an invasion phenotype and inhibited the trophoblast invasion, which further demonstrated that HG directly alters trophoblast lineage development (10). In the following paragraphs, we discuss in depth how HG influenced trophoblasts proliferation and invasion.
3.1 HG damages trophoblasts proliferation involved in uterine SA remodeling
Highly proliferating trophoblasts are necessary for placenta formation. And cell cycle control is very important in the proliferation process. In vitro studies have demonstrated that HG induced cell cycle arrest at G0/G1 in human trophoblast BeWo, JAR and HTR-8 cells, indicating that HG has the potential to inhibit trophoblasts proliferation (29, 30). Transcriptome and metabolome analysis showed that HG perturbed the phosphatidylinositol phosphate signaling pathways that involved in cell proliferation in BeWo cells (31). HG may inhibit cell proliferation by regulating the process of translation via epigenetic modifications, such as non-coding small molecule RNAs (32). HG upregulated the expression of miR-137, resulting in a negative modulatory effect on AMP-activated protein kinase, which ultimately stimulated the expression of IL-6 to inhibit cell proliferation (33). HG also promoted the expression of miR-136, inhibited the trophoblasts proliferation by suppressing E2F1 which is an important cell cycle regulator mediating the G1/S transition (34). MiR-362-5p was downregulated under HG conditions and inhibited the PI3K/AKT pathway by upregulating glutathione-disulfide reductase (GSR) directly, ultimately leading the inhibition of HTR-8 cells proliferation (35). MiR-520h was upregulated and inhibited cell proliferation by downregulating mTOR expression in HG-treated HTR-8 cells (36).
In vivo, immature villi in human diabetic placentas in term pregnancies suggested that HG provided more nutrition for continuous cell growth but delayed the cell differentiation and maturation (37). Upregulated Ki67 was observed in the term placenta of patients with GDM (38). However, Ki67 was downregulated in first-trimester human placental tissue with T1DM (39). In rat, the proliferative capacity of trophoblasts was weakened and the number of Ki67 positive cells decreased as the gestational day increases. At day 17 of pregnancy, Ki67 positive cells was higher in diabetic rat placentas than normal control (40). The different effects of HG on the proliferation of trophoblasts depend on gestational periods. HG provided excess nutrients for cell growth at the end of pregnancy, which also could explain why women with diabetes had higher placenta weight. Conversely, HG inhibited trophoblast proliferation in the first trimester because this period of placental development is particularly susceptible to environmental perturbations and any changes in the microenvironment may lead to impairment of trophoblast function (41).
3.2 HG damages trophoblasts invasion involved in uterine SA remodeling
In the progress of trophoblasts invasion into decidual tissue, EVTs produce proteases, such as fibrinogen activation system enzymes, matrix metalloproteinases (MMPs), and tissue inhibitor of metalloproteinases (TIMPs), to regulate the ECM remodeling and trophoblast invasion (42). The fibrinogen activation system comprises fibrinogen activators, such as urokinase-type plasminogen activator (uPA), and enzyme inhibitors such as fibrinogen activator inhibitor type 1 (PAI-1) (43). MMPs are a family of more than 23 zinc-binding enzymes, exhibiting proteolytic activity promoting trophoblast invasion to the uterine wall. MMP activity is mainly regulated by TIMP. MMP2 and MMP9 are the most important MMP enzymes involved in trophoblasts during the first trimester (44). Pro-uPA zymogen activates uPA after binding uPAR. Activated uPA in turn cleaves and activates MMPs as well as degrades local matrix protein (45). Exposure to excess glucose may lead to shallow trophoblast migration and invasion, leading to abnormal uterine SA remodeling.
In vitro, trophoblast cell invasion is adversely disturbed under HG condition (46–51). Belkacemi et al. showed that trophoblasts invasion was reduced by approximately 62% and the activity of uPA was lower when HTR-8 cells were treated with 10mM of glucose (52). Furthermore, uPA in human early pregnancy trophoblast cell Sw.71 also decreased with the increasing glucose concentration (53). The increased E-cadherin, decreased Twist1 and Vimentin in HTR-8 cells under HG indicated a failure of EMT. EMT not only participates in EVT invasion but also balances the CTB-EVT differentiation (54). It was reported that MiR-137 was elevated in HG-treated HTR-8 cells, and the upregulation of miR-137 decreased the expression of fibronectin type III domain-containing 5, thereby inhibited the viability and migration of HTR-8 cells (55).
Some in vitro studies has shown that HG promoted the invasion of trophoblasts. HG induced proteoglycans alterations in 3A-Sub-E cells which is isolated from human full-term placenta, followed the increased MMP-2 and MMP-9 and decreased TIMP-2 (56). However, the HG altered proteoglycans on the surface of trophoblasts can lead to ECM deposition and complications in diabetic placenta (57). Normally, physiological levels of reactive oxygen species (ROS) promote angiogenesis, and the placental antioxidant system prevents ROS overproduction (58). HG induced the expression of the Cytochrome P450 enzyme family 1, subfamily B, polypeptide 1 (CYP1B1) which promoted trophoblast migration via MMP2. Inhibition of CYP1B1 may suppress ROS production under HG condition, which may provide a new method for diabetic complications caused by ROS overload (59). In placentas of diabetic rats at mid-gestation, increased ROS triggers trophoblast spreading with the increased expression of MMP-2 and MMP-9 (60).
Collectively, it is not difficult to suppose that HG inhibits the invasion and migration of trophoblasts derived from the first trimester, but promotes the invasion and migration of trophoblasts derived from the third trimester. Primary trophoblasts isolated from human placentas culture under HG could further verify our inference.
3.3 HG alters oxygen tension in placenta during uterine SA remodeling
Before the first 10 weeks of gestation, EVT forms a trophoblast plug to prevent maternal blood from entering the intervillous space and creates a physiologically hypoxic environment (2%–3% O2) (61). The hypoxia-inducible factor 1 (HIF-1) plays a transcriptional regulatory role in hypoxic environment. There is an increased expression of TGF-β under hypoxia, thereby inhibiting trophoblasts differentiation (62). At the 12th week of gestation, the trophoblast plug dissolves and uterine SA begins to remodel, following a gradually increased oxygen concentration (8% O2) around the trophoblast (63). Both HIF-1α and TGF-β expression decreases with increasing oxygen concentration, enabling trophoblast differentiation and ensuring extensive EVTs invasion into SA with increased MMP9 (64). However, after trophoblast differentiation into mature EVT, hypoxia and elevated HIF can promote EVT invasion (65, 66).
HG increased the thickness of trophoblast membranes and the massive collagen deposition, resulting in altered oxygen gradients in placenta and local hypoxia at the maternal-fetal interface (40). Downregulation of miR-29b in placenta with GDM promoted trophoblast invasion by upregulating the expression of HIF3A (67). Hypoxia promotes the invasion of mature EVTs. It is reasonable to suppose that HG promotes the invasion of trophoblasts in the third trimester placenta. It is also suggested that mild HG increased capillaries through negative feedback regulation of ischemia and hypoxia, however, sustained severe HG triggered hypoxia/ischemia and inhibited vascular endothelial growth factor (VEGF)/VEGFR-2 binding, thereby reducing excessive capillary formation (68, 69). What is more, HG can alter trophoblasts development by blunting trophoblast stem cell responses to low oxygen levels (10).
3.4 HG disrupts trophoblasts releasing angiogenic factors
Trophoblasts secrete angiogenic factors during uterine SA remodeling. VEGF disrupts the VSMC and ECs. Placental growth factor (PlGF), prominently expressed in villous CTBs and STBs, promotes angiogenesis under hypoxic conditions (70). Angiopoietins (Ang1, Ang2) and their receptor Tie-2 play an important role in stabilization or breakdown of blood vessel (71). Fibroblast growth factor (FGF) and PDGF-BB are involved in vasculogenesis and angiogenesis (72). Anti-angiogenic factors, such as soluble fms-like tyrosine kinase-1 (sFlt-1) and soluble endoglin (sEng), are secreted. SFlt-1 is the soluble form of VEGFR-1, with a high affinity for VEGF, but no signal transduction function (73). Besides, sEng interferes with transforming growth factor β (TGF-β) and inhibits endothelial nitric oxide synthase activation, thereby disrupting angiogenesis (74).
In the first trimester trophoblasts HTR-8 and SW.71, HG decreased the secretions of VEGF, PlGF and uPA, while increased the secretions of anti-angiogenic factors sFlt-1 and sEng to inhibit artery remodeling (48, 52, 53, 75, 76). In placenta of women with GDM, increased VEGF, Ang, Eng and endothelin may lead to a collapse between angiogenic and anti-angiogenic factors (77). However, mild HG did not change the expression of VEGF (78). Persistent HG might thicken the placenta, increased the expression of HIF, thereby promoting the expression of angiogenic factors, such as VEGF and PIGF (79). FK506-binding protein like, acting as an anti-angiogenic protein and a regulator of inflammation, decreased in T1MD placenta and trophoblast cell line ACH-3P treated with HG under hypoxia condition (80). HG also promoted MT1-MMP and angiogenesis via PI3k signaling in GDM placenta (81). Despite these conflicting reports, it is certain that the balance between angiogenic and anti-angiogenic factors is disrupted under HG. HG in the first trimester inhibits uterine SA remodeling by inhibiting the proliferation, invasion, and migration of trophoblasts. HG may cause hypercapillarization of villi due to collagen deposition caused by hypoxia and abnormal trophoblasts migrations in the third trimester, but these vessels are immature (82).
4 HG affects the crosstalk between immune cells and trophoblasts
Pregnant uteri are colonized by large number of immune cells, the most abundant cells of which are dNK cells and macrophages, followed by T cells and dendritic cells. Approximately 75% of decidual leukocytes are CD56brightCD16- dNK cells and are not cytotoxic (83). Decidual macrophages, recruited from the maternal circulation, are polarized toward M1 macrophages during peri-implantation period while a profile of a mixed M1 and M2 type during EVTs invading the SA (84). Different from decidual macrophages, Hofbauer cells (HBCs) are the villous macrophages in the stroma of the first-trimester placenta arising from hematopoietic stem cells and are characterized as CD14+ CD68+ cells (85).
Chemokines and their receptors also play important roles in trophoblast migration and immune cells recruitment at the maternal-fetal interface (86). The dNK cells secrete IL8, CXCL10, TNF, interferon (INF) γ, TGF-b, and angiogenic factors such as VEGF-A, VEGF-C and PlGF (15, 87). Trophoblasts express the IL8 receptor CXCR1, the CXCL10 receptor CXCR3, TNF receptor TNFR1, as well as VEGFR-1 and VEGFR-3. Trophoblasts produce human leukocyte antigen to increase the levels of inhibitory receptors in dNK cells, maintaining their inactive phenotype (CD16−CD56+) (88). Meanwhile, macrophages secret IL-33, granulocyte colony-stimulating factor (G-CSF), CXCL1, TGF-b, TNF-α and Wnt5a to regulate trophoblasts invasion and migration (89). Immune cells, interacting with ECs, fibroblasts, and trophoblasts, promote the SA remodeling and placental growth. Any dysregulation of these factors may lead to remodeling obstacles (15).
In vitro, HG could mediate trophoblast releasing inflammatory factors IL-1β, IL-4, IL-8, and IL-6, IFN-γ, TNF-β, CXCL1 and G-CSF, indicating that HG created a pro-inflammatory environment at the maternal-fetal interface (76, 90, 91). High level of pro-inflammatory TNF-α was found both in GDM and TD2M placenta. Decreased IL-4 was found in T2DM and MGH placenta, promoting NK cell into active phenotype (92). This also reminds us that maternal HG caused by diabetes mellitus can lead to a disturbance in the balance of pro-inflammatory and anti-inflammatory factors at the maternal-fetal interface. In the following sections, we discuss in depth how HG disturbed immune cells and lead to the failure of SA remodeling.
4.1 HG affects crosstalk between dNK cells and trophoblasts
The dNK cells and EVTs interact with vascular ECs to promote SA remodeling (93). Firstly, dNK cells induce the apoptosis of VSMC and ECs, destruct blood vessel and secrete Ang-1, Ang-2, VEGF and MMPs to mediate angiogenesis (94, 95). The dNK cells secrete MMP-2, MMP-9, uPA, adhesion molecules such as ICAM-1 to regulate ECM remodeling (96–98). DNK cells express killer immunoglobulin receptor (KIR), CD94/NKG2A, and immunoglobulin like transcripts 2 (ILT2). These three receptors can interact with HLA-C, HLA-E and HLA-G on trophoblasts respectively, regulating trophoblast invasion (87). Cytokines, such as IL-10 and granulocyte-macrophage colony-stimulating factor (GM-CSF) and chemokines IL-8, CXCL10 produced by dNK cells could promote EVTs invasion while the cytokines TNFα and IFN-γ inhibited trophoblast invasion by upregulating PAI expression (87, 99). IL-8 can also increase trophoblast expressing integrins α1 and β5 to gain an invasive phenotype (100) (Figure 1B).
A test for GDM peripheral blood showed a higher percent of cytotoxic NK cells (CD16+CD56dim) in the GDM group than in controls (101). Some scholars believe that dNK cells are derived from the recruitment of peripheral CD56bright NK cells, which acquire dNK cells phenotype under the influence of a specific decidual microenvironment (102). Thus, changes in peripheral blood NK cells may lead to changes in the decidual NK cells. Fewer CD56+cells adhere to decidual endothelium, while more diabetic CD56+ cells adhere to pancreatic endothelium in pregnant women with T1DM and T2DM, indicating that HG impairs egression of CD56+ cells into the decidua (103). Cytotoxic CD16+ CD56−NK cell both increased in maternal blood and placenta extravilli of GDM and T2MD. Placental CD16-CD56+ NK cells were higher in GDM and lower in T2DM, irrespective of region (92). GDM and T2MD are characterized by excessive insulin resistance, followed by maternal HG, triggering a “glucose stress” response and concurrent systemic inflammation (104). This response involves altered infiltration, differentiation, and activation of maternal innate and adaptive immune cells, which may explain increased CD16+ NK cells. The control of blood sugar affects the expression of cytokines. Besides, cytokines differ in recent-onset DM and long-standing DM (105, 106). Thus, we don not exclude glycemic control conditions and duration of maternal HG are responsible for CD56+ NK cells percentage and cytokine levels different in GDM and T2DM. In addition to the phenotypic changes of NK cells, the cytokines secreted by NK cells also change. Simultaneously, CD56+ cells producing TGF-β and VEGF decreased significantly in patients with GDM (107). This secretory change may affect the regulation of trophoblast migration and invasion by dNK cells in high-risk pregnancy (108). Above all, HG may decrease dNK in the uterine wall, leading to a diminished interstitial trophoblast invasion and less SA remodeling.
4.2 HG affects crosstalk between macrophages and trophoblasts
Adopting an M2 polarity phenotype, HBCs express TIMP-1, MMP9, VEGF-A, osteopontin (OPN), and FGF to affect ECM and vascular remodeling (109–111). HBCs also secrete inflammatory factors such as IL-8, CCL-2, CCL-3, and CCL-4 with proangiogenic properties (109) (Figure 1C). Moreover, CD14+ macrophages in early pregnancy decidua induce the breakdown of ECM and phagocytose apoptotic VSMCs to remodel the uterine SA (112).
HBCs treated by HG switched their M2 polarity profile towards M1 phenotype, which is not conducive to angiogenesis (113). M2 macrophages involve in anti-inflammatory processes and promote angiogenesis and tumor progression, which can produce protease to degrade the ECM (114). Thus, a reduction in the number of M2 phenotype cells may lead to impaired vascular remodeling. However, Schliefsteiner et al. showed HBCs maintain their M2 polarization to maintain a successful pregnancy, even in inflammatory states such as GDM. The co-cultivation of HBCs from GDM placentas and placental arterial endothelial cells (pAECs) did not alter ECs activation (115). Zhang et al. reported that M2a macrophages, majoring in tissue repair, increased in villi and more collagen was deposited in uncontrolled T2DM group compared with the healthy group (116). All in all, HG can disturb the balance between pro-inflammatory and anti-inflammatory subtypes of HBCs, which may cause adverse pregnancy outcomes.
5 Limitation and future direction
A recent study isolated SA from 12 to 23 weeks of gestation and found that the vascular remodeling was not complete until 23 weeks of gestation (117). Previous studies have mainly focused on pregnant women with GDM with few studies focusing on pregnant women with T1DM or T2DM. GDM is mainly screened at 24-28 weeks of gestation, but hyperglycemia occurs before 24 weeks. In addition, early GDM may has worse pregnancy outcomes (118). Therefore, the molecular and signaling pathway changes in the placenta of patients with GDM are also valuable for understanding the effect of HG on the uterine SA.
However, previous in vivo studies also had some limitations. First, the number of placenta cases in these studies is relatively small and the individual differences in patients are large. Secondly, no information was discussed on medication of women in the case group. Another limitation is lack of protein involved in ECM remodeling and angiogenesis such as MMP2, MMP9, uPA, PIGF and VEGF. SA remodeling occurs mainly before 24 weeks, therefore, staining of above protein in the first and second trimester placenta villi is more indicative of the effect of HG on trophoblast function.
All previous in vitro studies differed in terms of glucose dose, treatment time and cell line. Different or opposite conclusions have been drawn regarding the effect of HG on trophoblast function and uterine SA remodeling. For example, Basak et al. reported tube formation substantially increased at 25-30mM glucose and decreased at 40mM glucose in HTR-8 cells (119). In addition, McLeese et al. pointed out that HTR-8 cells did not survive in 5 mmol/L glucose over 48h, possibly due to the rapid glucose consumption (120). Inadera et al. pointed out that when BeWo cells were cultured at physiological levels of 5 mM glucose, the cells detached from dishes (31). Thus, the above researches remind us choosing appropriate glucose concentration is necessary to study the effects of HG on trophoblasts biological behavior and SA remodeling. The in vitro experiments used in this review are summarized in Table 1.
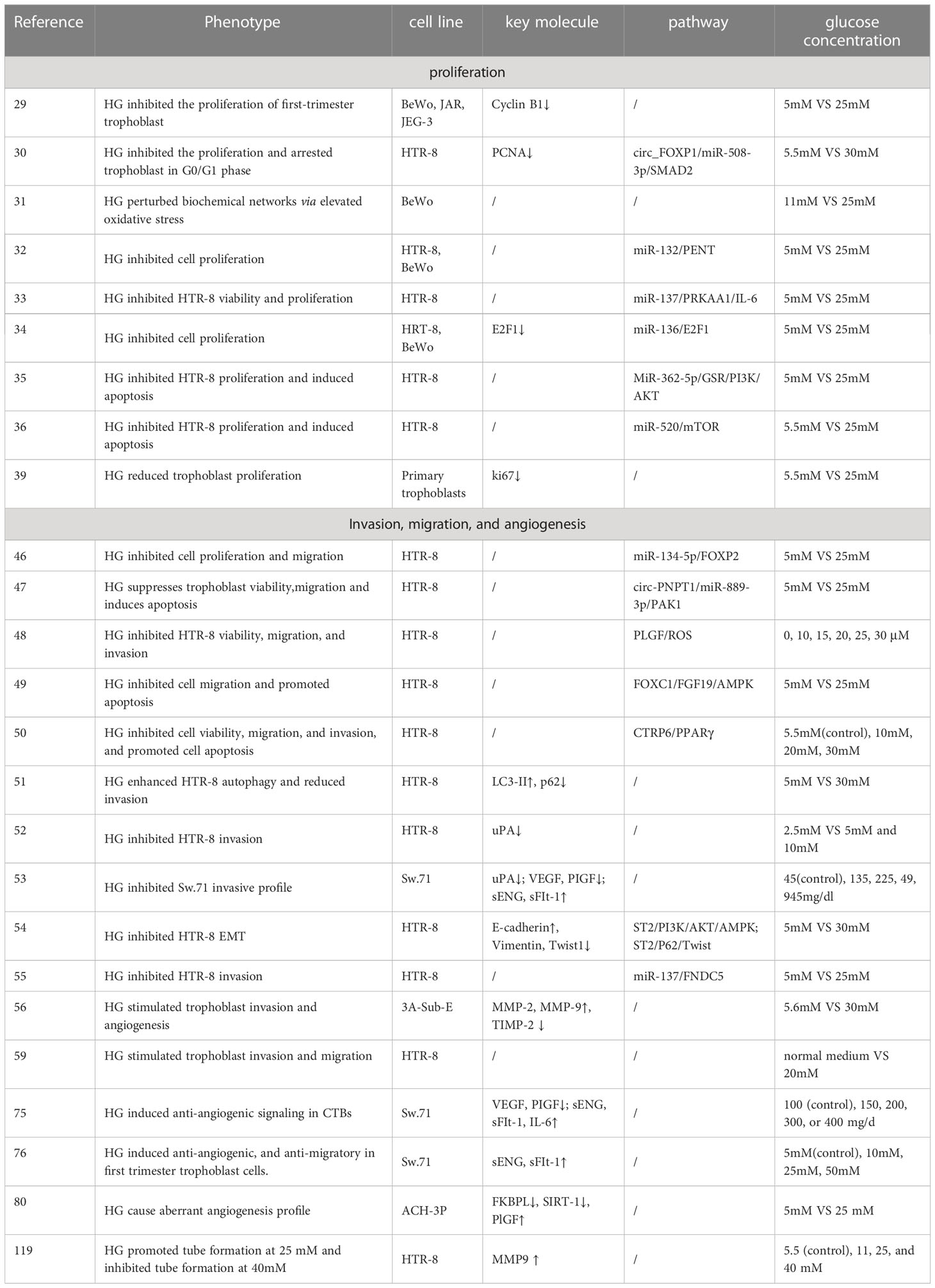
Table 1 The in vitro experiments used in this study are summarized.
Although the relevant molecular mechanisms and signaling pathways of HG influencing trophoblast functions have been reported in these studies, the studies have mostly focused on cell lines and animal models. However, cell lines do not truly reflect in vivo conditions. Information obtained from animal models is also limited because SA remodeling differs between human and rats (121, 122). Therefore, it is crucial to establish a suitable model with appropriate sugar concentration for further study. Ex vivo model, such as human placenta-decidua co-culture, can also be used to quantify the extent of SA remodeling (123). In addition, human trophoblast organoids show similar cellular composition and biological behavior to those of immature human placentas. Despite the lack of such studies, we believe that human trophoblast organoid models cultured under HG conditions will help us understand the effect of HG on the remodeling of uterine SAs (124).
6 Conclusion
Uterine SA remodeling requires appropriate trophoblast proliferation, invasion, and tissue remodeling, which involves a balanced MMP, TIMP and uPA. Meanwhile, trophoblasts, dNK cells and HBCs can secrete serious cytokines and angiogenic factors to regulate SA remodeling. Crosstalk between immunity cells and both trophoblast and vascular cells at maternal-fetal interface is also a part of the remodeling process. Inappropriate glucose concentrations may lead to abnormal trophoblast proliferation, migration, and invasion by disrupting the balance between MMP and TIMP. In addition, HG disrupts the balance between angiogenic factors Ang-1, VEGF, PlGF and anti-angiogenic factors sFlt-1 and sEng. Furthermore, an impaired immune cell profile under HG conditions influences SA remodeling (Figure 2). Understanding how HG affects SA remodeling by influencing trophoblast function is crucial for revealing the mechanisms by which diabetes leads to pregnancy complications and adverse pregnancy outcomes. This review may provide a theoretical basis for future foundation and clinical research.
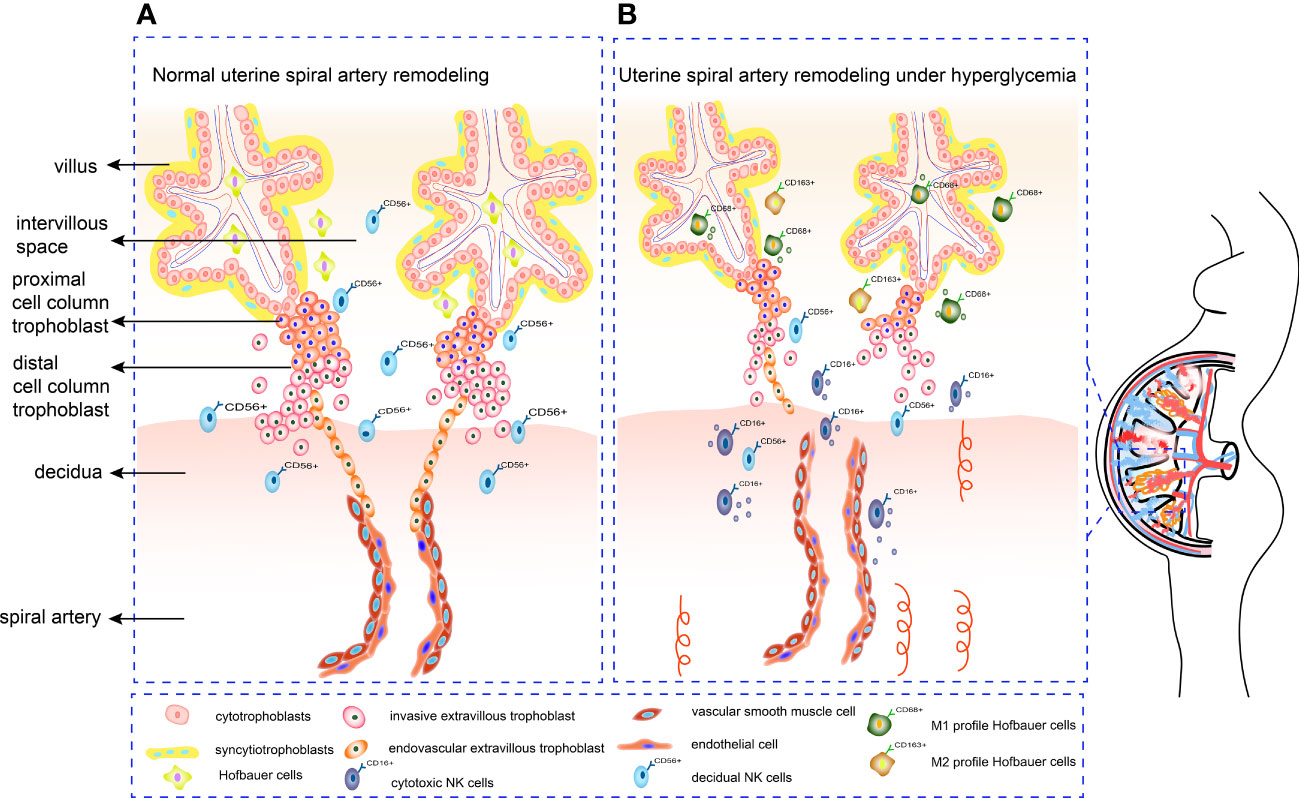
Figure 2 Mechanism of SA remodeling in normal pregnancy and hyperglycemia pregnancy. (A) CTBs proliferate rapidly once embedded in maternal decidua. The outer layer of CTBs fuses into primitive STBs, which can form proliferative proximal cell column trophoblasts. EVTs differentiate from distal cell column and break through the overlying STB layer, detaching from distal cell columns, migrating into the decidual stroma, and remodeling the SA. (B) Under hyperglycemia conditions, the proliferation and invasion ability of trophoblasts alters. Increased cytotoxic CD16+CD56dim NK cells can form an inflammatory environment. Meanwhile, HBCs switch their M2 polarity profile towards M1 phenotype, which is not conducive to angiogenesis. Deficient artery transformation and immature new blood vessels can be observed in hyperglycemic placenta.
Author contributions
YZ: Search literature and write original draft. XL: Review, editing, project administration. YZ and XL contributed equally to this work. YX: Review and editing. YL: Funding acquisition, Resources, Supervision, Writing – review and editing. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by grants from the National Key Research and Development Program of China (2018YFC1002800), the Innovative research team of high-level local universities in Shanghai (SHSMU-ZLCX20210202), the National Natural Science Foundation of China (81401274, 81971403 and 82171669), the Shanghai Jiao Tong University Trans-Med Awards Research (20210201), and Funds for Outstanding Newcomers, Shanghai Sixth People’s Hospital (X-3664), the Shanghai Science and Technology Commission (22dz1202303).
Acknowledgments
I am grateful for the contribution to language help and writing assistance from Fuju Tian, Fan Wu and Jianing Hu at The International Peace Maternity and Child Health Hospital during the research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. McIntyre HD, Fuglsang J, Kampmann U, Knorr S, Ovesen P. Hyperglycemia in Pregnancy and Women's Health in the 21st Century. Int J Environ Res Public Health (2022) 19(24):16827. doi: 10.3390/ijerph192416827
2. McCance DR, Casey C. Type 1 diabetes in pregnancy. Endocrinol Metab Clin North Am (2019) 48:495–509. doi: 10.1016/j.ecl.2019.05.008
3. Shou C, Wei YM, Wang C, Yang HX. Updates in long-term maternal and fetal adverse effects of gestational diabetes mellitus. Maternal-Fetal Med (2019) 1(2):91–4. doi: 10.1097/FM9.0000000000000019
4. Dietl J. Maternal obesity and complications during pregnancy. J Perinat Med (2005) 33:100–5. doi: 10.1515/JPM.2005.018
5. James JL, Boss AL, Sun C, Allerkamp HH, Clark AR. From stem cells to spiral arteries: A journey through early placental development. Placenta (2022) 125:68–77. doi: 10.1016/j.placenta.2021.11.004
6. Albrecht ED, Pepe GJ. Regulation of uterine spiral artery remodeling: A review. Reprod Sci (2020) 27:1932–42. doi: 10.1007/s43032-020-00212-8
7. Ander SE, Diamond MS, Coyne CB. Immune responses at the maternal-fetal interface. Sci Immunol (2019) 4(31):eaat6114. doi: 10.1126/sciimmunol.aat6114
8. Du MR, Wang SC, Li DJ. The integrative roles of chemokines at the maternal-fetal interface in early pregnancy. Cell Mol Immunol (2014) 11:438–48. doi: 10.1038/cmi.2014.68
9. Shi JW, Lai ZZ, Yang HL, Yang SL, Wang CJ, Ao D, et al. Collagen at the maternal-fetal interface in human pregnancy. Int J Biol Sci (2020) 16:2220–34. doi: 10.7150/ijbs.45586
10. Nteeba J, Varberg KM, Scott RL, Simon ME, Iqbal K, Soares MJ. Poorly controlled diabetes mellitus alters placental structure, efficiency, and plasticity. BMJ Open Diabetes Res Care (2020) 8(1):e001243. doi: 10.1136/bmjdrc-2020-001243
11. Kerby A, Shingleton D, Batra G, Sharps MC, Baker BC, Heazell A. Placental morphology and cellular characteristics in stillbirths in women with diabetes and unexplained stillbirths. Arch Pathol Lab Med (2021) 145:82–9. doi: 10.5858/arpa.2019-0524-OA
12. Groen B, Uuldriks GA, de Vos P, Visser JT, Links TP, Faas MM. Impaired trophoblast invasion and increased numbers of immune cells at day 18 of pregnancy in the mesometrial triangle of type 1 diabetic rats. Placenta (2015) 36:142–9. doi: 10.1016/j.placenta.2014.12.004
13. Hemberger M, Hanna CW, Dean W. Mechanisms of early placental development in mouse and humans. Nat Rev Genet (2020) 21:27–43. doi: 10.1038/s41576-019-0169-4
14. Red-Horse K, Zhou Y, Genbacev O, Prakobphol A, Foulk R, McMaster M, et al. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest (2004) 114:744–54. doi: 10.1172/JCI22991
15. Pollheimer J, Vondra S, Baltayeva J, Beristain AG, Knöfler M. Regulation of placental extravillous trophoblasts by the maternal uterine environment. Front Immunol (2018) 9:2597. doi: 10.3389/fimmu.2018.02597
16. Kokkinos MI, Murthi P, Wafai R, Thompson EW, Newgreen DF. Cadherins in the human placenta–epithelial-mesenchymal transition (EMT) and placental development. Placenta (2010) 31:747–55. doi: 10.1016/j.placenta.2010.06.017
17. Arimoto-Ishida E, Sakata M, Sawada K, Nakayama M, Nishimoto F, Mabuchi S, et al. Up-regulation of alpha5-integrin by e-cadherin loss in hypoxia and its key role in the migration of extravillous trophoblast cells during early implantation. Endocrinology (2009) 150:4306–15. doi: 10.1210/en.2008-1662
18. Damsky CH, Fitzgerald ML, Fisher SJ. Distribution patterns of extracellular matrix components and adhesion receptors are intricately modulated during first trimester cytotrophoblast differentiation along the invasive pathway, in vivo. J Clin Invest (1992) 89:210–22. doi: 10.1172/JCI115565
19. Pijnenborg R, Dixon G, Robertson WB, Brosens I. Trophoblastic invasion of human decidua from 8 to 18 weeks of pregnancy. Placenta (1980) 1:3–19. doi: 10.1016/s0143-4004(80)80012-9
20. Sato Y. Endovascular trophoblast and spiral artery remodeling. Mol Cell Endocrinol (2020) 503:110699. doi: 10.1016/j.mce.2019.110699
21. Nandy D, Das S, Islam S, Ain R. Molecular regulation of vascular smooth muscle cell phenotype switching by trophoblast cells at the maternal-fetal interface. Placenta (2020) 93:64–73. doi: 10.1016/j.placenta.2020.02.017
22. Whitley GS, Cartwright JE. Trophoblast-mediated spiral artery remodelling: A role for apoptosis. J Anat (2009) 215:21–6. doi: 10.1111/j.1469-7580.2008.01039.x
23. Acar N, Ustunel I, Demir R. Uterine natural killer (uNK) cells and their missions during pregnancy: A review. Acta Histochem (2011) 113:82–91. doi: 10.1016/j.acthis.2009.12.001
24. Hunt JS, Robertson SA. Uterine macrophages and environmental programming for pregnancy success. J Reprod Immunol (1996) 32:1–25. doi: 10.1016/s0165-0378(96)88352-5
25. Meng R, Gao Q, Liang R, Guan L, Liu S, Zhu Q, et al. Changes in gene expression in rat placenta at gestational day 16.5 in response to hyperglycemia. Gen Comp Endocrinol (2022) 320:113999. doi: 10.1016/j.ygcen.2022.113999
26. Phillips JK, Vance AM, Raj RS, Mandalà M, Linder EA, Gokina NI. Impact of experimental diabetes on the maternal uterine vascular remodeling during rat pregnancy. Reprod Sci (2012) 19:322–31. doi: 10.1177/1933719111424435
27. Ozmen A, Unek G, Kipmen-Korgun D, Korgun ET. The PI3K/Akt and MAPK-ERK1/2 pathways are altered in STZ induced diabetic rat placentas. Histol Histopathol (2014) 29:743–56. doi: 10.14670/HH-29.743
28. Qin Y, McCauley N, Ding Z, Lawless L, Liu Z, Zhang K, et al. Hyperglycemia results in significant pathophysiological changes of placental spiral artery remodeling and angiogenesis, further contributing to congenital defects. Front Biosci (Landmark Ed). (2021) 26:965–76. doi: 10.52586/5001
29. Weiss U, Cervar M, Puerstner P, Schmut O, Haas J, Mauschitz R, et al. Hyperglycaemia in vitro alters the proliferation and mitochondrial activity of the choriocarcinoma cell lines BeWo, JAR and JEG-3 as models for human first-trimester trophoblast. Diabetologia (2001) 44:209–19. doi: 10.1007/s001250051601
30. Li M, Huang Y, Xi H, Zhang W, Xiang Z, Wang L, et al. Circ_FOXP1 promotes the growth and survival of high glucose-treated human trophoblast cells through the regulation of miR-508-3p/SMAD family member 2 pathway. Endocr J (2022) 69(9):1067–78. doi: 10.1507/endocrj.EJ21-0528
31. Inadera H, Tachibana S, Takasaki I, Tatematsu M, Shimomura A. Hyperglycemia perturbs biochemical networks in human trophoblast BeWo cells. Endocr J (2010) 57:567–77. doi: 10.1507/endocrj.k10e-045
32. Zhou X, Xiang C, Zheng X. miR-132 serves as a diagnostic biomarker in gestational diabetes mellitus and its regulatory effect on trophoblast cell viability. Diagn Pathol (2019) 14(1):119. doi: 10.1186/s13000-019-0899-9
33. Peng HY, Li MQ, Li HP. High glucose suppresses the viability and proliferation of HTR−8/SVneo cells through regulation of the miR−137/PRKAA1/IL−6 axis. Int J Mol Med (2018) 42:799–810. doi: 10.3892/ijmm.2018.3686
34. Zhang C, Wang L, Chen J, Song F, Guo Y. Differential expression of miR-136 in gestational diabetes mellitus mediates the high-Glucose-Induced trophoblast cell injury through targeting E2F1. Int J Genomics (2020) 2020:3645371. doi: 10.1155/2020/3645371
35. Zhang C, Zhao D. MicroRNA-362-5p promotes the proliferation and inhibits apoptosis of trophoblast cells via targeting glutathione-disulfide reductase. Bioengineered (2021) 12:2410–9. doi: 10.1080/21655979.2021.1933678
36. Wen J, Bai X. miR-520h inhibits cell survival by targeting mTOR in gestational diabetes mellitus. Acta Biochim Pol (2021) 68:65–70. doi: 10.18388/abp.2020_5389
37. Daskalakis G, Marinopoulos S, Krielesi V, Papapanagiotou A, Papantoniou N, Mesogitis S, et al. Placental pathology in women with gestational diabetes. Acta Obstet Gynecol Scand (2008) 87:403–7. doi: 10.1080/00016340801908783
38. Unek G, Ozmen A, Mendilcioglu I, Simsek M, Korgun ET. Immunohistochemical distribution of cell cycle proteins p27, p57, cyclin D3, PCNA and Ki67 in normal and diabetic human placentas. J Mol Histol (2014) 45:21–34. doi: 10.1007/s10735-013-9534-3
39. Majali-Martinez A, Weiss-Fuchs U, Miedl H, Forstner D, Bandres-Meriz J, Hoch D, et al. Type 1 diabetes mellitus and the first trimester placenta: Hyperglycemia-induced effects on trophoblast proliferation, cell cycle regulators, and invasion. Int J Mol Sci (2021) 22(20):10989. doi: 10.3390/ijms222010989
40. Zorn TM, Zúñiga M, Madrid E, Tostes R, Fortes Z, Giachini F, et al. Maternal diabetes affects cell proliferation in developing rat placenta. Histol Histopathol (2011) 26:1049–56. doi: 10.14670/HH-26.1049
41. Hoch D, Gauster M, Hauguel-de Mouzon S, Desoye G. Diabesity-associated oxidative and inflammatory stress signalling in the early human placenta. Mol Aspects Med (2019) 66:21–30. doi: 10.1016/j.mam.2018.11.002
42. Chakraborty C, Gleeson LM, McKinnon T, Lala PK. Regulation of human trophoblast migration and invasiveness. Can J Physiol Pharmacol (2002) 80:116–24. doi: 10.1139/y02-016
43. Behrendt N, Rønne E, Danø K. The structure and function of the urokinase receptor, a membrane protein governing plasminogen activation on the cell surface. Biol Chem Hoppe Seyler (1995) 376:269–79.
44. Isaka K, Usuda S, Ito H, Sagawa Y, Nakamura H, Nishi H, et al. Expression and activity of matrix metalloproteinase 2 and 9 in human trophoblasts. Placenta (2003) 24:53–64. doi: 10.1053/plac.2002.0867
45. Martínez-Hernández MG, Baiza-Gutman LA, Castillo-Trápala A, Armant DR. Regulation of proteinases during mouse peri-implantation development: Urokinase-type plasminogen activator expression and cross talk with matrix metalloproteinase 9. Reproduction (2011) 141:227–39. doi: 10.1530/REP-10-0334
46. Ke W, Chen Y, Zheng L, Zhang Y, Wu Y, Li L. miR-134-5p promotes inflammation and apoptosis of trophoblast cells via regulating FOXP2 transcription in gestational diabetes mellitus. Bioengineered (2022) 13:319–30. doi: 10.1080/21655979.2021.2001219
47. Zhang L, Zeng M, Tang F, Chen J, Cao D, Tang ZN. Circ-PNPT1 contributes to gestational diabetes mellitus (GDM) by regulating the function of trophoblast cells through miR-889-3p/PAK1 axis. Diabetol Metab Syndr (2021) 13:58. doi: 10.1186/s13098-021-00678-9
48. Tao J, Xia LZ, Chen JJ, Zeng JF, Meng J, Wu S, et al. High glucose condition inhibits trophoblast proliferation, migration and invasion by downregulating placental growth factor expression. J Obstet Gynaecol Res (2020) 46:1690–701. doi: 10.1111/jog.14341
49. Cao S, Zhang S. Forkhead-box C1 attenuates high glucose-induced trophoblast cell injury during gestational diabetes mellitus via activating adenosine monophosphate-activated protein kinase through regulating fibroblast growth factor 19. Bioengineered (2022) 13:1174–84. doi: 10.1080/21655979.2021.2018094
50. Zhang J, Bai WP. C1q/tumor necrosis factor related protein 6 (CTRP6) regulates the phenotypes of high glucose-induced gestational trophoblast cells via peroxisome proliferator-activated receptor gamma (PPARγ) signaling. Bioengineered (2022) 13:206–16. doi: 10.1080/21655979.2021.2012906
51. Ji L, Chen Z, Xu Y, Xiong G, Liu R, Wu C, et al. Systematic characterization of autophagy in gestational diabetes mellitus. Endocrinology (2017) 158:2522–32. doi: 10.1210/en.2016-1922
52. Belkacemi L, Lash GE, Macdonald-Goodfellow SK, Caldwell JD, Graham CH. Inhibition of human trophoblast invasiveness by high glucose concentrations. J Clin Endocrinol Metab (2005) 90:4846–51. doi: 10.1210/jc.2004-2242
53. Cawyer CR, Horvat D, Leonard D, Allen SR, Jones RO, Zawieja DC, et al. Hyperglycemia impairs cytotrophoblast function via stress signaling. Am J Obstet Gynecol (2014) 211:541.e1–8. doi: 10.1016/j.ajog.2014.04.033
54. Lai R, Ji L, Zhang X, Xu Y, Zhong Y, Chen L, et al. Stanniocalcin2 inhibits the epithelial-mesenchymal transition and invasion of trophoblasts via activation of autophagy under high-glucose conditions. Mol Cell Endocrinol (2022) 547:111598. doi: 10.1016/j.mce.2022.111598
55. Peng HY, Li MQ, Li HP. MiR-137 restricts the viability and migration of HTR-8/SVneo cells by downregulating FNDC5 in gestational diabetes mellitus. Curr Mol Med (2019) 19(7):494–505. doi: 10.2174/1566524019666190520100422
56. Chang SC, Vivian Yang WC. Hyperglycemia induces altered expressions of angiogenesis associated molecules in the trophoblast. Evid Based Complement Alternat Med (2013) 2013:457971. doi: 10.1155/2013/457971
57. Chen CP, Chang SC, Vivian Yang WC. High glucose alters proteoglycan expression and the glycosaminoglycan composition in placentas of women with gestational diabetes mellitus and in cultured trophoblasts. Placenta (2007) 28:97–106. doi: 10.1016/j.placenta.2006.02.009
58. Wu F, Tian FJ, Lin Y, Xu WM. Oxidative stress: Placenta function and dysfunction. Am J Reprod Immunol (2016) 76:258–71. doi: 10.1111/aji.12454
59. Wu Z, Mao W, Yang Z, Lei D, Huang J, Fan C, et al. Knockdown of CYP1B1 suppresses the behavior of the extravillous trophoblast cell line HTR-8/SVneo under hyperglycemic condition. J Maternal-Fetal Neonatal Med (2021) 34(4):500–11. doi: 10.1080/14767058.2019.1610379
60. Pustovrh MC, Jawerbaum A, Capobianco E, White V, Martínez N, López-Costa JJ, et al. Oxidative stress promotes the increase of matrix metalloproteinases-2 and -9 activities in the feto-placental unit of diabetic rats. Free Radic Res (2005) 39:1285–93. doi: 10.1080/10715760500188796
61. Soares MJ, Iqbal K, Kozai K. Hypoxia and placental development. Birth Defects Res (2017) 109:1309–29. doi: 10.1002/bdr2.1135
62. Caniggia I, Winter J, Lye SJ, Post M. Oxygen and placental development during the first trimester: implications for the pathophysiology of pre-eclampsia. Placenta (2000) 21(Suppl A):S25–30. doi: 10.1053/plac.1999.0522
63. Weiss G, Sundl M, Glasner A, Huppertz B, Moser G. The trophoblast plug during early pregnancy: a deeper insight. Histochem Cell Biol (2016) 146:749–56. doi: 10.1007/s00418-016-1474-z
64. Caniggia I, Mostachfi H, Winter J, Gassmann M, Lye SJ, Kuliszewski M, et al. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J Clin Invest (2000) 105:577–87. doi: 10.1172/JCI8316
65. Zhao H, Wong RJ, Stevenson DK. The impact of hypoxia in early pregnancy on placental cells. Int J Mol Sci (2021) 22(18):9675. doi: 10.3390/ijms22189675
66. Rosario GX, Konno T, Soares MJ. Maternal hypoxia activates endovascular trophoblast cell invasion. Dev Biol (2008) 314:362–75. doi: 10.1016/j.ydbio.2007.12.007
67. Sun DG, Tian S, Zhang L, Hu Y, Guan CY, Ma X, et al. The miRNA-29b is downregulated in placenta during gestational diabetes mellitus and may alter placenta development by regulating trophoblast migration and invasion through a HIF3A-dependent mechanism. Front Endocrinol (Lausanne) (2020) 11:169. doi: 10.3389/fendo.2020.00169
68. Kumazaki K, Nakayama M, Suehara N, Wada Y. Expression of vascular endothelial growth factor, placental growth factor, and their receptors flt-1 and KDR in human placenta under pathologic conditions. Hum Pathol (2002) 33:1069–77. doi: 10.1053/hupa.2002.129420
69. Prager GW, Breuss JM, Steurer S, Mihaly J, Binder BR. Vascular endothelial growth factor (VEGF) induces rapid prourokinase (pro-uPA) activation on the surface of endothelial cells. Blood (2004) 103:955–62. doi: 10.1182/blood-2003-07-2214
70. Gobble RM, Groesch KA, Chang M, Torry RJ, Torry DS. Differential regulation of human PlGF gene expression in trophoblast and nontrophoblast cells by oxygen tension. Placenta (2009) 30:869–75. doi: 10.1016/j.placenta.2009.08.003
71. Schiessl B, Innes BA, Bulmer JN, Otun HA, Chadwick TJ, Robson SC, et al. Localization of angiogenic growth factors and their receptors in the human placental bed throughout normal human pregnancy. Placenta (2009) 30:79–87. doi: 10.1016/j.placenta.2008.10.004
72. Demir R, Seval Y, Huppertz B. Vasculogenesis and angiogenesis in the early human placenta. Acta Histochem (2007) 109:257–65. doi: 10.1016/j.acthis.2007.02.008
73. Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci USA (1993) 90:10705–9. doi: 10.1073/pnas.90.22.10705
74. Gregory AL, Xu G, Sotov V, Letarte M. Review: the enigmatic role of endoglin in the placenta. Placenta (2014) 35(Suppl):S93–9. doi: 10.1016/j.placenta.2013.10.020
75. Cawyer C, Afroze SH, Drever N, Allen S, Jones R, Zawieja DC, et al. Attenuation of hyperglycemia-induced apoptotic signaling and anti-angiogenic milieu in cultured cytotrophoblast cells. Hypertens Pregnancy (2016) 35:159–69. doi: 10.3109/10641955.2015.1122035
76. Han CS, Herrin MA, Pitruzzello MC, Mulla MJ, Werner EF, Pettker CM, et al. Glucose and metformin modulate human first trimester trophoblast function: a model and potential therapy for diabetes-associated uteroplacental insufficiency. Am J Reprod Immunol (2015) 73:362–71. doi: 10.1111/aji.12339
77. Al-Ofi E, Alrafiah A, Maidi S, Almaghrabi S, Hakami N. Altered expression of angiogenic biomarkers in pregnancy associated with gestational diabetes. Int J Gen Med (2021) 14:3367–75. doi: 10.2147/IJGM.S316670
78. Bhattacharjee D, Mondal SK, Garain P, Mandal P, Dey G. Histopathological study with immunohistochemical expression of vascular endothelial growth factor in placentas of hyperglycemic and diabetic women. J Lab Phys (2017) 9:227. doi: 10.4103/JLP.JLP_148_16
79. Tirpe AA, Gulei D, Ciortea SM, Crivii C, Berindan-Neagoe I. Hypoxia: Overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int J Mol Sci (2019) 20(24):6140. doi: 10.3390/ijms20246140
80. Alqudah A, Eastwood KA, Jerotic D, Todd N, Hoch D, McNally R, et al. FKBPL and SIRT-1 are downregulated by diabetes in pregnancy impacting on angiogenesis and endothelial function. Front Endocrinol (Lausanne) (2021) 12:650328. doi: 10.3389/fendo.2021.650328
81. Hiden U, Lassance L, Tabrizi NG, Miedl H, Tam-Amersdorfer C, Cetin I, et al. Fetal insulin and IGF-II contribute to gestational diabetes mellitus (GDM)-associated up-regulation of membrane-type matrix metalloproteinase 1 (MT1-MMP) in the human feto-placental endothelium. J Clin Endocrinol Metab (2012) 97:3613–21. doi: 10.1210/jc.2012-1212
82. Pietro L, Daher S, Rudge MV, Calderon IM, Damasceno DC, Sinzato YK, et al. Vascular endothelial growth factor (VEGF) and VEGF-receptor expression in placenta of hyperglycemic pregnant women. Placenta (2010) 31:770–80. doi: 10.1016/j.placenta.2010.07.003
83. Guo W, Fang L, Li B, Xiao X, Chen S, Wang J, et al. Decreased human leukocyte antigen-G expression by miR-133a contributes to impairment of proinvasion and proangiogenesis functions of decidual NK cells. Front Immunol (2017) 8:741. doi: 10.3389/fimmu.2017.00741
84. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci (2008) 13:453–61. doi: 10.2741/2692
85. Reyes L, Wolfe B, Golos T. Hofbauer Cells: Placental Macrophages of Fetal Origin. Results Probl Cell Differ (2017) 62:45–60. doi: 10.1007/978-3-319-54090-0_3
86. Salamonsen LA, Hannan NJ, Dimitriadis E. Cytokines and chemokines during human embryo implantation: roles in implantation and early placentation. Semin Reprod Med (2007) 25:437–44. doi: 10.1055/s-2007-991041
87. Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med (2006) 12:1065–74. doi: 10.1038/nm1452
88. Rouas-Freiss N, Gonçalves RM, Menier C, Dausset J, Carosella ED. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc Natl Acad Sci USA (1997) 94:11520–5. doi: 10.1073/pnas.94.21.11520
89. Ding J, Zhang Y, Cai X, Diao L, Yang C, Yang J. Crosstalk between trophoblast and macrophage at the maternal-fetal interface: Current status and future perspectives. Front Immunol (2021) 12:758281. doi: 10.3389/fimmu.2021.758281
90. Heim KR, Mulla MJ, Potter JA, Han CS, Guller S, Abrahams VM. Excess glucose induce trophoblast inflammation and limit cell migration through HMGB1 activation of toll-like receptor 4. Am J Reprod Immunol (2018) 80:e13044. doi: 10.1111/aji.13044
91. Rice GE, Scholz-Romero K, Sweeney E, Peiris H, Kobayashi M, Duncombe G, et al. The effect of glucose on the release and bioactivity of exosomes from first trimester trophoblast cells. J Clin Endocrinol Metab (2015) 100:E1280–8. doi: 10.1210/jc.2015-2270
92. Hara Cde C, França EL, Fagundes DL, de Queiroz AA, Rudge MV, Honorio-França AC, et al. Characterization of natural killer cells and cytokines in maternal placenta and fetus of diabetic mothers. J Immunol Res (2016) 2016:7154524. doi: 10.1155/2016/7154524
93. Hazan AD, Smith SD, Jones RL, Whittle W, Lye SJ, Dunk CE. Vascular-leukocyte interactions: mechanisms of human decidual spiral artery remodeling in vitro. Am J Pathol (2010) 177:1017–30. doi: 10.2353/ajpath.2010.091105
94. Robson A, Harris LK, Innes BA, Lash GE, Aljunaidy MM, Aplin JD, et al. Uterine natural killer cells initiate spiral artery remodeling in human pregnancy. FASEB J (2012) 26:4876–85. doi: 10.1096/fj.12-210310
95. Fraser R, Whitley GS, Johnstone AP, Host AJ, Sebire NJ, Thilaganathan B, et al. Impaired decidual natural killer cell regulation of vascular remodelling in early human pregnancies with high uterine artery resistance. J Pathol (2012) 228:322–32. doi: 10.1002/path.4057
96. Lash GE, Otun HA, Innes BA, Percival K, Searle RF, Robson SC, et al. Regulation of extravillous trophoblast invasion by uterine natural killer cells is dependent on gestational age. Hum Reprod (2010) 25:1137–45. doi: 10.1093/humrep/deq050
97. Naruse K, Lash GE, Innes BA, Otun HA, Searle RF, Robson SC, et al. Localization of matrix metalloproteinase (MMP)-2, MMP-9 and tissue inhibitors for MMPs (TIMPs) in uterine natural killer cells in early human pregnancy. Hum Reprod (2009) 24:553–61. doi: 10.1093/humrep/den408
98. Hu Y, Eastabrook G, Tan R, MacCalman CD, Dutz JP, von Dadelszen P. Decidual NK cell-derived conditioned medium enhances capillary tube and network organization in an extravillous cytotrophoblast cell line. Placenta (2010) 31:213–21. doi: 10.1016/j.placenta.2009.12.011
99. Bauer S, Pollheimer J, Hartmann J, Husslein P, Aplin JD, Knöfler M. Tumor necrosis factor-alpha inhibits trophoblast migration through elevation of plasminogen activator inhibitor-1 in first-trimester villous explant cultures. J Clin Endocrinol Metab (2004) 89:812–22. doi: 10.1210/jc.2003-031351
100. Jovanović M, Stefanoska I, Radojcić L, Vićovac L. Interleukin-8 (CXCL8) stimulates trophoblast cell migration and invasion by increasing levels of matrix metalloproteinase (MMP)2 and MMP9 and integrins alpha5 and beta1. Reproduction (2010) 139:789–98. doi: 10.1530/REP-09-0341
101. Lobo TF, Borges CM, Mattar R, Gomes CP, de Angelo A, Pendeloski K, et al. Impaired treg and NK cells profile in overweight women with gestational diabetes mellitus. Am J Reprod Immunol (2018) 79:3–7. doi: 10.1111/aji.12810
102. Tao Y, Li YH, Piao HL, Zhou WJ, Zhang D, Fu Q, et al. CD56(bright)CD25+ NK cells are preferentially recruited to the maternal/fetal interface in early human pregnancy. Cell Mol Immunol (2015) 12:77–86. doi: 10.1038/cmi.2014.26
103. Seaward AV, Burke SD, Ramshaw H, Smith GN, Croy BA. Circulating CD56+ cells of diabetic women show deviated homing potential for specific tissues during and following pregnancy. Hum Reprod (2011) 26:1675–84. doi: 10.1093/humrep/der114
104. McElwain CJ, McCarthy FP, McCarthy CM. Gestational diabetes mellitus and maternal immune dysregulation: What we know so far. Int J Mol Sci (2021) 22:4–24. doi: 10.3390/ijms22084261
105. Rodacki M, Svoren B, Butty V, Besse W, Laffel L, Benoist C, et al. Altered natural killer cells in type 1 diabetic patients. Diabetes (2007) 56:177–85. doi: 10.2337/db06-0493
106. Ruszkowska-Ciastek B, Sokup A, Wernik T, Rhone P, Góralczyk K, Bielawski K, et al. Low-grade risk of hypercoagulable state in patients suffering from diabetes mellitus type 2. J Zhejiang Univ Sci B (2015) 16:788–95. doi: 10.1631/jzus.B1500066
107. Chiba H, Fukui A, Fuchinoue K, Funamizu A, Tanaka K, Mizunuma H. Expression of natural cytotoxicity receptors on and intracellular cytokine production by NK cells in women with gestational diabetes mellitus. Am J Reprod Immunol (2016) 75:529–38. doi: 10.1111/aji.12491
108. Wallace AE, Fraser R, Cartwright JE. Extravillous trophoblast and decidual natural killer cells: a remodelling partnership. Hum Reprod Update (2012) 18:458–71. doi: 10.1093/humupd/dms015
109. Thomas JR, Appios A, Zhao X, Dutkiewicz R, Donde M, Lee C, et al. Phenotypic and functional characterization of first-trimester human placental macrophages, hofbauer cells. J Exp Med (2021) 218(1):e20200891. doi: 10.1084/jem.20200891
110. Thomas JR, Naidu P, Appios A, McGovern N. The ontogeny and function of placental macrophages. Front Immunol (2021) 12:771054. doi: 10.3389/fimmu.2021.771054
111. Pan Y, Chen M, Lash GE. Role of osteopontin (OPN) in uterine spiral artery remodeling. Placenta (2022) 126:70–5. doi: 10.1016/j.placenta.2022.06.014
112. Lash GE, Pitman H, Morgan HL, Innes BA, Agwu CN, Bulmer JN. Decidual macrophages: key regulators of vascular remodeling in human pregnancy. J Leukoc Biol (2016) 100:315–25. doi: 10.1189/jlb.1A0815-351R
113. Sisino G, Bouckenooghe T, Aurientis S, Fontaine P, Storme L, Vambergue A. Diabetes during pregnancy influences hofbauer cells, a subtype of placental macrophages, to acquire a pro-inflammatory phenotype. Biochim Biophys Acta (2013) 1832:1959–68. doi: 10.1016/j.bbadis.2013.07.009
114. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233:6425–40. doi: 10.1002/jcp.26429
115. Schliefsteiner C, Peinhaupt M, Kopp S, Lögl J, Lang-Olip I, Hiden U, et al. Human placental hofbauer cells maintain an anti-inflammatory M2 phenotype despite the presence of gestational diabetes mellitus. Front Immunol (2017) 8:888. doi: 10.3389/fimmu.2017.00888
116. Zhang M, Cui D, Yang H. The distributional characteristics of M2 macrophages in the placental chorionic villi are altered among the term pregnant women with uncontrolled type 2 diabetes mellitus. Front Immunol (2022) 13:837391. doi: 10.3389/fimmu.2022.837391
117. Olaya CM, Garrido M, Franco JA, Rodríguez JL, Vargas MJ, et al. Spiral arteries in second trimester of pregnancy: When is it possible to define expected physiological remodeling as abnormal. Reprod Sci (2021) 28:1185–93. doi: 10.1007/s43032-020-00403-3
118. Mustafa M, Bogdanet D, Khattak A, Carmody LA, Kirwan B, Gaffney G, et al. Early gestational diabetes mellitus (GDM) is associated with worse pregnancy outcomes compared with GDM diagnosed at 24-28 weeks gestation despite early treatment. QJM (2021) 114:17–24. doi: 10.1093/qjmed/hcaa167
119. Basak S, Das MK, Srinivas V, Duttaroy AK. The interplay between glucose and fatty acids on tube formation and fatty acid uptake in the first trimester trophoblast cells, HTR8/SVneo. Mol Cell Biochem (2015) 401:11–9. doi: 10.1007/s11010-014-2287-9
120. McLeese RH, Zhao J, Fu D, Yu JY, Brazil DP, Lyons TJ. Effects of modified lipoproteins on human trophoblast cells: a role in pre-eclampsia in pregnancies complicated by diabetes. BMJ Open Diabetes Res Care (2021) 9(1):e001696. doi: 10.1136/bmjdrc-2020-001696
121. Chakraborty D, Rumi MA, Konno T, Soares MJ. Natural killer cells direct hemochorial placentation by regulating hypoxia-inducible factor dependent trophoblast lineage decisions. Proc Natl Acad Sci USA (2011) 108:16295–300. doi: 10.1073/pnas.1109478108
122. Shukla V, Soares MJ. Modeling trophoblast cell-guided uterine spiral artery transformation in the rat. Int J Mol Sci (2022) 23(6):2947. doi: 10.3390/ijms23062947
123. Brkić J, Dunk C, O'Brien J, Fu G, Nadeem L, Wang YL, et al. MicroRNA-218-5p promotes endovascular trophoblast differentiation and spiral artery remodeling. Mol Ther (2018) 26:2189–205. doi: 10.1016/j.ymthe.2018.07.009
Keywords: hyperglycemia, trophoblast, decidual NK cells, Hofbauer cells, uterine spiral artery remodeling
Citation: Zhu Y, Liu X, Xu Y and Lin Y (2023) Hyperglycemia disturbs trophoblast functions and subsequently leads to failure of uterine spiral artery remodeling. Front. Endocrinol. 14:1060253. doi: 10.3389/fendo.2023.1060253
Received: 03 October 2022; Accepted: 20 March 2023;
Published: 05 April 2023.
Edited by:
Richard Ivell, University of Nottingham, United KingdomReviewed by:
Yu-Chin Lien, University of Pennsylvania, United StatesYan Li, Shandong University, China
Copyright © 2023 Zhu, Liu, Xu and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Lin, eWlsaW5vbmxpbmVAMTI2LmNvbQ==
†These authors have contributed equally to this work and share first authorship